 Yingyu Chen
Yingyu Chen An Song
An Song Min Nie
Min Nie Yan Jiang
Yan Jiang Mei Li
Mei Li Weibo Xia
Weibo Xia- Department of Endocrinology, Key Laboratory of Endocrinology, National Health Commission, Peking Union Medical College Hospital, Chinese Academy of Medical Science & Peking Union Medical College, Beijing, China
Context: The malignant potential and molecular signature of atypical parathyroid adenoma (APA) remain elusive. Data from Asia are still lacking.
Design and setting: This was a retrospective study on a large APA cohort in a single center from mainland China.
Methods: A total of 320 patients with primary hyperparathyroidism (PHPT), containing 79 APA, 79 Parathyroid cancer (PC) and 162 benign lesions cases, were enrolled after surgery for collection of clinical data and genetic analysis.
Results: APA patients showed earlier mean onset age than benign group (46.9 ± 17.1 vs. 52.0 ± 14.3 yrs). Less bone involvement and gastrointestinal symptoms were presented in APA compared to PC (35.4% vs. 62.0%, and 17.7% vs. 41.8%), while more urolithiasis was seen in APA than in benign lesions (57.0% vs. 29.6%). The APA group had moderate hypercalcemia (mean 3.02 ± 0.44mmol/L) with elevated serum PTH (median 593.0pg/ml) and proportion of hypercalcemic crisis as 22.8%, all higher than those of benign lesions but lower than those of PC group. The recurrence/no remission rate of the APA group was significantly lower than that of the PC and similar to the benign group (5.1% vs. 31.6% vs. 3.1%). Germline CDC73 mutation was the most common molecular abnormality in both PC and APA subjects. APA patients with nonsynonymous germline variants showed earlier onset age (28.5 ± 16.9 vs. 48.1 ± 17.7 yrs) and more cases developing no remission/recurrence (25.0% vs. 0.0%).
Conclusions: Patients with APA presented clinical and biochemical characteristics much less severe than PC and resembling the benign neoplasms, with a relatively good prognosis. Germline gene variations were associated with earlier onset and probably more recurrence of PHPT in APA.
1 Introduction
Primary hyperparathyroidism (PHPT) is the third most common endocrine disorder in western countries. In PHPT, primary abnormal parathyroid tumors inappropriately secret excessive parathyroid hormone (PTH) and thus lead to hypercalcaemia and a series of symptoms.
According to the existing data, almost all PHPT cases are caused by benign parathyroid lesions. Parathyroid adenoma (PA) is the most common type (~85%), with mostly single parathyroid gland being involved. Parathyroid hyperplasia (PH) arises in 10-15% PHPT patients (1, 2). Atypical parathyroid adenoma (APA) and parathyroid carcinoma (PC) occur rarer with PC being the rarest type (~1% in the western world and up to 8% in Asia) (3–5). Up to now, a total of less than 1, 000 cases of APA have been reported in different cohorts with only several studies including over 50 patients (6–11). Due to its rarity, there is still no general consensus on treatment and follow-up.
The term ‘APA’ or ‘atypical parathyroid neoplasm (APN)’ has been used to describe a subgroup of parathyroid tumors with uncertain malignant potential, having histological features suspicious for PC (i.e., solid growth pattern, prominent fibrous bands, questionable capsular invasion, adherence to surrounding tissues, marked cellular/nuclear atypia and increased mitotic figures) but lacking evidence of unequivocal invasion and/or metastasis which are the key morphological features of PC (12). It is sometimes difficult to make the differential diagnosis between APA and PC, which may be a big challenge even for an experienced pathologist. In some cases, the initial diagnosis of APA may be revised to carcinoma, because of the local reoccurrence or metastasis (13).
PHPT can be sporadic or familial/syndrome-related type, accounting for 90% and 10% of the disease, respectively. Familial PHPT consists of multiple endocrine neoplasia (MEN) type 1 (MEN1), type 2A (MEN2A), type 4 (MEN4) and hyperparathyroidism-jaw tumor syndrome (HPT-JT), caused by known germline genetic mutations (14). PC is commonly sporadic, but it may occur in familial/syndrome-related PHPT, including HPT-JT and, very rarely, MEN1 and MEN2A (15, 16). Up to 70% of sporadic PC carry a somatic mutation of the CDC73 gene, with one-third of apparently sporadic PC having germline CDC73 mutations. Our knowledge of the molecular pathogenesis of parathyroid carcinoma and benign adenoma has largely increased over the last decades. On the contrary, understanding of the molecular mechanisms underlying atypical parathyroid adenoma still lacks progress.
This study aimed to investigate the differences of clinical characteristics, biochemistry, outcomes and genetic features between APA and PC, in order to help construct treatment and follow-up strategies.
2 Subjects and methods
2.1 Subjects
From November 1992 to November 2019, 79 APA and 79 PC patients with relative complete medical records diagnosed and treated at Peking Union Medical College Hospital (PUMCH) were consecutively included in this study. A total of 162 patients with benign parathyroid lesions hospitalized in PUMCH during the same period were randomly selected according to the ratio of 2:1 to APA cases, including 135 parathyroid adenoma (PA) and 27 parathyroid hyperplasia (PH) patients. All cases included in this study met the criteria for PHPT and were confirmed by histopathology after surgery.
Diagnosis of PHPT was defined biochemically as increased serum calcium (SCa) or albumin-corrected calcium (CCa, CCa = SCa + [0.02 × (40 − albumin)]) level (>2.70 mmol/L) and/or serum ionized calcium (iCa) level (>1.28 mmol/L) combined with unsuppressed PTH level without other causes of hypercalcemia (1).
Histopathology diagnoses were confirmed by the pathologists in our institution. PC was diagnosed on the basis of finding lesions with vascular or perineural invasion, capsular penetration, and/or documented metastases (12). The diagnostic criterion for APA was finding parathyroid tumors partially sharing some of the atypical features in PC, but not enough histopathological criteria for diagnosing PC (6, 12). For patients with multi-glandular involvement, if the pathological findings were different for different lesions, the more malignant areas were chosen for diagnosis.
This study was approved by the Ethics Committee of PUMCH and conducted according to the principles in the Declaration of Helsinki. Written informed consent was obtained from the subjects or from the parents for subjects ≤18 years for genetic analysis.
2.2 Clinical and laboratory investigation
The clinical data were retrospectively obtained from medical records reserved at PUMCH, including demographics, clinical manifestations (e.g., bone involvement, urinary system damage, gastrointestinal symptoms, pancreatitis and hypercalcemia crisis), pre-operative biochemical indices, radiographic findings, pathology, follow-up and outcomes (postoperative recurrence and metastasis). Bone involvement included pathological fracture, osteomalacia, and the typical X-ray features of bone resorption (such as subperiosteal absorption and osteitis fibrosa cystica). The definition of asymptomatic PHPT was hyperparathyroidism lacking traditionally specific symptoms or signs associated with hypercalcemia or PTH excess (17). Hypercalcemia crisis was defined as a SCa level of ≥3.5 mmol/L, and usually associated with acute signs and symptoms of hypercalcemia (18). Recurrence was defined as hypercalcemia with high or inappropriately normal serum parathyroid hormone (PTH) level recurring after a disease-free period of at least 6 months after parathyroid operation (19).
Bone mineral density (BMD) was measured by dual-energy X-ray absorptiometry (DXA; GE-Lunar, USA) at the femoral neck (FN, CV: 1.73%) and lumbar spine (L2-L4, CV: 1.70%). Z-scores and T-scores of BMD were calculated using database of normal subjects in our institution (20). Urolithiasis or renal calcification was assessed using ultrasound application. Ultrasonography and 99m-sestamibi-scintigraphy (MIBI) were conducted in all PHPT patients for pre-operative localization.
The serum and urinary biochemical indices were measured in the Department of the Clinical Laboratory of PUMCH. iCa level was measured with a blood-gas analyzer radiometer (ABL800 FLEX; Denmark). SCa, phosphorus (P), alkaline phosphatase (ALP), and 24h urinary calcium (24hUCa) were measured with the Beckman Automatic Biochemical Analyzer (AU5800; Beckman Coulter). The serum PTH level was measured by chemiluminescence (ADVIA Centaur; Siemens, Germany). Serum 25-dihydroxyvitamin D (25OHD) value was measured using an electrochemiluminescence immunoassay (e601; Roche Cobas, Germany).
2.3 DNA isolation and gene-mutation analysis
Genomic DNA was extracted from the peripheral blood lymphocytes of 69 patients from PC and APA groups, using the QIAamp Blood DNA Kit (Qiagen; Hilden, Germany) according to the manufacturer’s protocol. A custom-designed gene panel was conducted by Nimblegen SeqCap EZ system (Novogene, Beijing, China) to capture all exons and 10-basepairs (bps) flanking intron sequences of the eight candidate genes for PHPT, i.e., GCM2, MEN1, RET, CDKN1B, CASR, HRPT2/CDC73, GNA11, and AP2S1. The Targeted Next-Generation Sequencing (NGS) was performed at Novogene Corporation (Beijing, China) and sequenced on the HiSeq 2500 Sequencing System (Illumina, San Diego, CA, USA) as previously described (21). The coverage of target regions of all samples was > 99.9%. The average sequencing depth on target of all samples was > 700×.
All coding exons and exon-intron boundaries of the CDC73/HRPT2, GCM2, MEN1, CDKN1B and CASR genes and exons 8, 10-11 and 13-16 of the RET gene were amplified via polymerase chain reaction (PCR), followed by Sanger sequencing which was performed as previously prescribed (22–26). For those patients with no CDC73 gene mutation as detected by routine PCR, multiplex ligation-dependent probe amplification (MLPA) was further performed to screen for large deletions in the CDC73 gene.
In order to uncover genotype-phenotype correlation, the PC and APA patients who had accepted genetic analysis were each divided into two subgroups according to whether they carried nonsynonymous genetic variations or not. Subjects in subgroup 1 and subgroup A carried missense, nonsense, frameshift, gross deletion, and splice site variations. Subjects in subgroup 2 and subgroup B carried synonymous or no variations. Subgroup 1 (A) was compared with subgroup 2 (B) in aspect of clinical characteristics and follow-up outcomes.
2.4 Statistical analysis
Statistical analysis of original data was conducted using SPSS Statistics 26.0 (Chicago, Illinois, USA). Normality of distributions was checked. Results were described as percentages for categorical variables, mean ± standard deviation (SD) for normally distributed variables, and median and 25th and 75th interquartile ranges (Q25, Q75) for non-normally distributed variables.
Between-group differences were analyzed using One-Way ANOVA for normally distributed variables and Kruskal-Wallis H Test for non-normally distributed variables. Categorical variables were compared by Pearson chi-square test or Fisher’s exact test, as appropriate. For all analyses, P-value <.05 was considered to be statistically significant.
3 Results
3.1 Demographics and clinical characteristics
A total of 320 subjects were included in this study, containing 79 cases pathologically diagnosed with APA, 79 cases with PC, 162 cases with benign neoplasms consisting of 135 PA and 27 PH. Comparisons of the demographics, clinical manifestations, biochemical indices, radiographic and densitometric data of the APA, PC and benign groups are shown in Table 1.
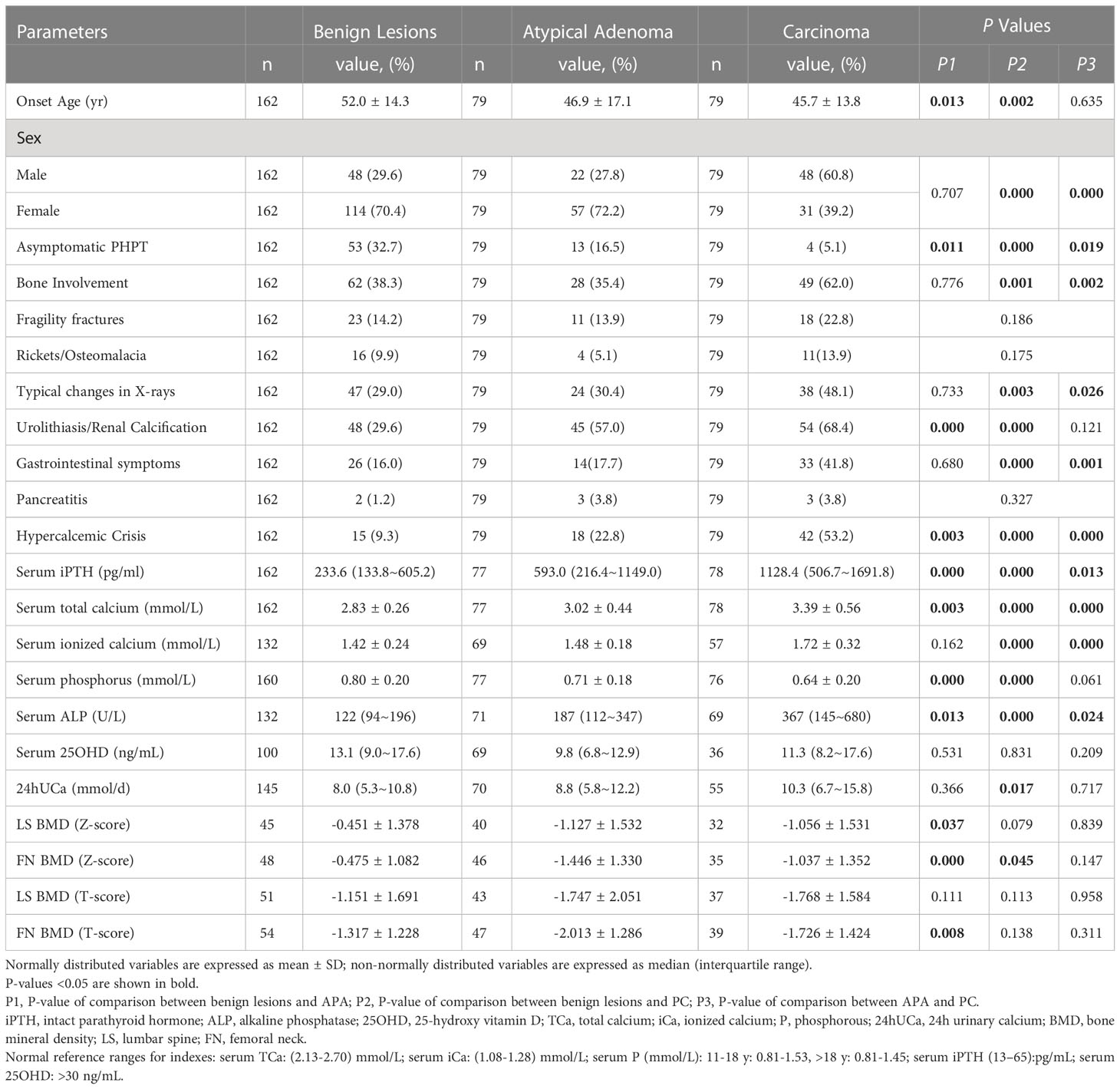
Table 1 Demographics, clinical presentation, biochemical and densitomatric parameters of patients with pathologically confirmed primary hyperparathyroidism.
The onset ages of PHPT in the APA and PC groups were not different, while both of them were younger than that of the benign group (46.9 ± 17.1 and 45.7 ± 13.8 vs. 52.0 ± 14.3 yrs). There were 48 males (60.8%) and 31 females (39.2%) in PC patients, with a male to female ratio of 1.55:1. In contrast, females accounted for the majority of both APA (72.2%) and benign lesions group (70.4%), with the female to male ratio being 2.59:1 and 2.38:1, respectively.
Bone involvement, typical X-ray features of PHPT and gastrointestinal symptoms occurred in 28 (35.4%), 24 (30.4%) and 14 (17.7%) patients in the APA group, resembling those of the benign group and significantly less than the PC group (Table 1). Urolithiasis/renal calcification was less in patients with benign lesions compared with APA and PC patients (29.6% vs. 57.0% and 68.4%, P<0.001), while no significant difference of that was found in the last two groups. In contrast with the benign lesions group, the proportion of hypercalcemic crisis was significantly higher and the percentage of asymptomatic PHPT was much lower in the PC group [53.2% vs. 9.3% (P=0.000) and 5.1% vs. 32.7% (P=0.000)], while these two proportions of the APA group (22.8% and 16.5%, respectively) lay exactly right between those of the benign lesions and the PC group (P<0.05). The mean serum total calcium and median PTH levels were 3.02 ± 0.44mmol/L and 593.0pg/ml in the APA group. Significant between-group differences were found in serum PTH, serum calcium and ALP levels, with the highest levels being in the PC group and the second being in the APA group (p<0.05). Patients of PC had the lowest level of serum phosphorus and the highest level of 24-hours urinary calcium among the three groups. Z scores at both femoral neck and lumbar spine (L2-L4) of APA and PC patients were lower in contrast to those of patients with benign neoplasms. No remarkable distinctions were found in other clinical, biochemical and densitometric parameters of the three groups (Table 1).
3.2 Surgery outcomes and follow-up
Details regarding the operations and follow-ups are shown in Table 2. The PC group had a significantly higher proportion of parathyroidectomies (PTXs) combined with thyroid lobectomy and central neck dissect (CND) when compared with the benign group, and the APA group was in between. According to the preoperative localization and intra-operative exploration, the majority of the patients in all three groups had a single lesion. The patients with PC had the largest size of parathyroid tumor, with that of APA being the second largest (3.18 ± 1.06 and 2.77 ± 1.03 cm, respectively). Remission was defined as postoperative normalization of serum calcium and PTH levels during the whole follow-up (at least 6 months). No remission (i.e., persistence) was defined as PHPT persistent within 6 months after surgery. Recurrence meant that PHPT occurred again after surgery for at least 6 months.
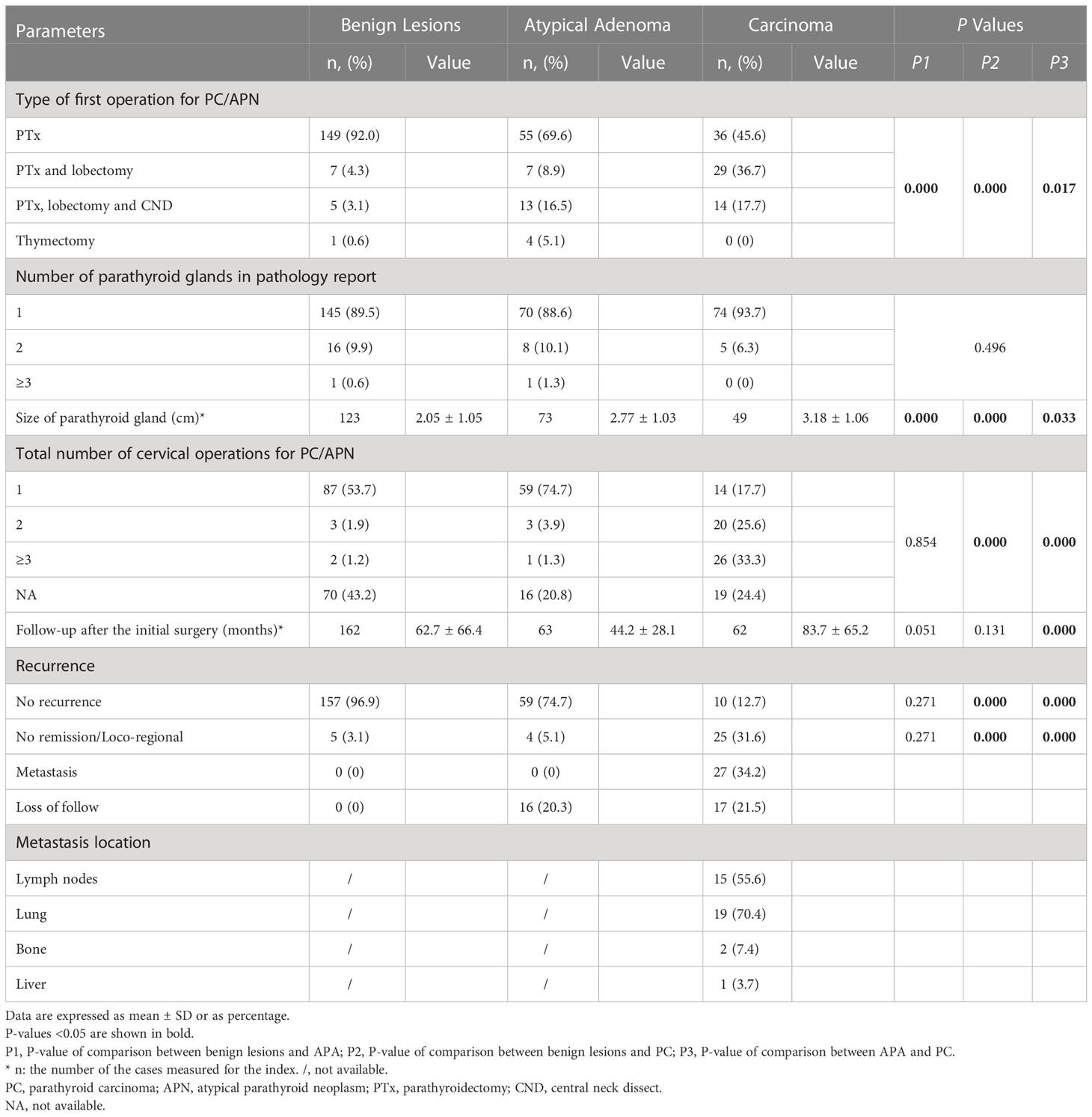
Table 2 Surgery-related variables and outcomes of patients with pathologically confirmed primary hyperparathyroidism.
The mean periods of follow-up after the initial parathyroid surgery of the PC, APA and benign lesions groups were 83.7 ± 65.2, 44.2 ± 28.1 and 62.7 ± 66.4 months, respectively. Meanwhile, the number of patients loss to follow-up in the APA and PC were 16 (20.3%) and 17 (21.5%), respectively. Patients with PC had a significantly higher rate of cervical reoperations than the APA and benign groups (p = 0.000). Four patients in the PC group and one in the APA did not obtain remission after their first operations. The regional recurrence occurred in 5 (3.1%), 3 (3.8%) and 21 (26.6%) cases of the benign lesions, APA and PC groups, respectively (P=0.000). A total of 27 PC patients developed distant metastasis. The metastasis locations contained lymph nodes, lung, bone and liver, with the numbers of cases being 15 (55.6%), 19 (70.4%), 2 (7.4%) and 1 (3.7%), respectively. Furthermore, 5 and 10 cases from the PC group had been diagnosed as atypical adenoma and benign adenoma, respectively, according to the histopathology at their first operations. And two APA patients had been diagnosed with benign adenoma by first surgery.
3.3 Genetic analysis
Among the 38 PC patients and 31 APA patients who agreed to genetic analysis, except for one PC case carrying single nucleotide synonymous variant of AP2S1 gene, 8 APA patients (25.8%, 8/31) and 17 PC patients (44.7%, 17/38) were found to carry germline nonsynonymous rare variants of the candidate genes, including 14 CDC73, 4 GCM2, 3 MEN1, 2 RET, 1 GNA11 and 1 CDKN1B gene variants (Table 3). No alterations in CASR gene were found. The CDC73 gene accounted for the majority of all altered genes in both PC (64.7%, 11/17) and APA groups (37.5%, 3/8). Among all the 14 CDC73 gene mutations, there were seven frameshift mutations (50.0%), five nonsense mutations (35.7%), one splice site mutations (7.1%), and one gross deletion mutation (7.1%). Mutations in exon 7 were observed more frequently (57.1%, 8/14) than those in other exons, but hot spot mutation region was not found. One PC patient and one APA patient were confirmed to have family history of PHPT. The overall recurrence/no remission/metastasis rate was as high as 72.0% (18/25) among the nonsynonymous variants carriers regardless of whether or not they had family history of PHPT, while there were only two APA patients (25.0%, 2/8) among them and all the rest were PC patients (43.2%, 16/37). Only two of the four APA patients presenting recurrence or no remission accepted genetic analysis and carried a nonsense mutation in exon 7 of the CDC73 gene and a missense MEN1 variants.
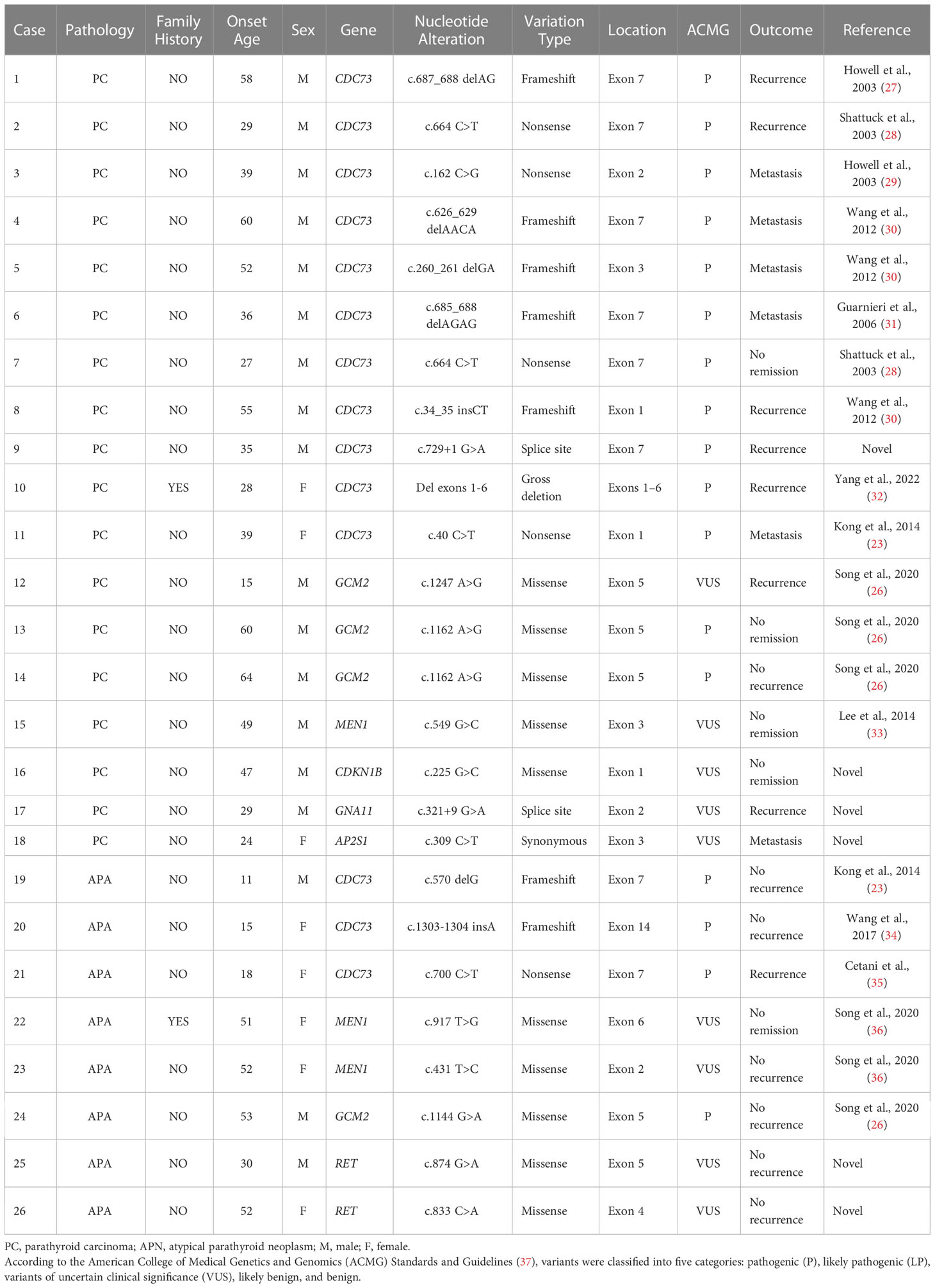
Table 3 Germline variants of candidate genes for primary hyperparathyroidism in studied APA and PC patients.
3.4 Genotype-phenotype correlations
The 38 PC patients and 31 APA patients were each divided into two subgroups, to assess the influence of nonsynonymous variants of those PHPT-related genes on the patients’ clinical characteristics and prognosis (Table 4). The subgroup 1 of the APA subjects, consisting of 8 patients having nonsynonymous variants, showed an earlier onset age for PHPT in comparison with subgroup 2, that contained 23 patients with no variations or synonymous variants (28.5 ± 16.9 vs. 48.1 ± 17.7 yrs, P<0.01). As for the clinical, biochemical and radiological characteristics, neither the APA group nor the PC group presented statistically significant distinctions between their two subgroups, except for a higher serum ALP concentration of the APA subgroup 1 and more elevated serum iPTH level of the PC subgroup b. The recurrence rate of the APA subgroup 1 had reached 25% (2/8) but it turned out to have no statistical significance compared to that of the subgroup 2. Those PC patients with nonsynonymous variations (i.e., the PC subgroup A) showed a higher proportion of no remission and regional reoccurrence.
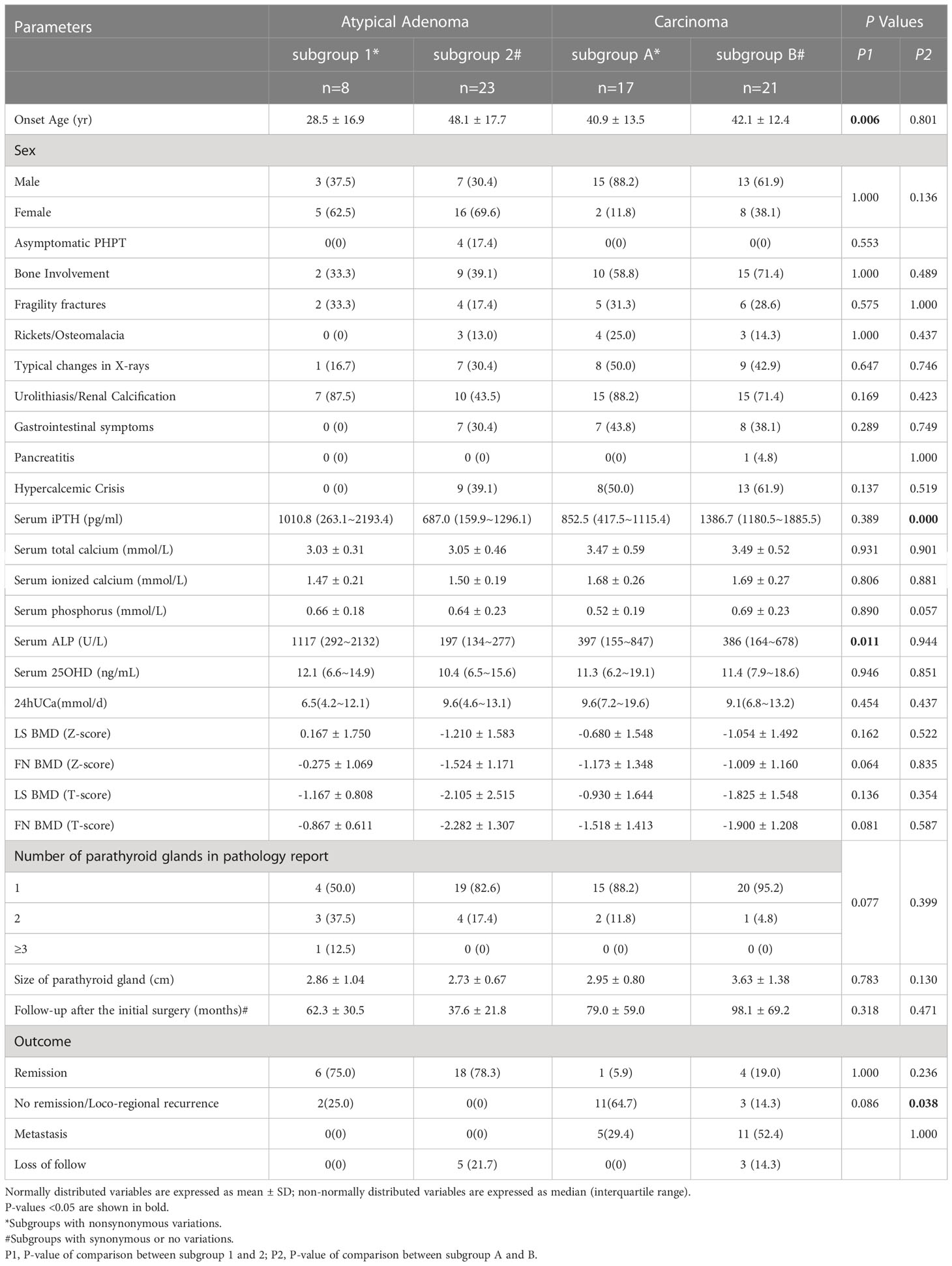
Table 4 Comparison of clinical characteristics and outcomes between patients with nonsynonymous variations and without such variations.
4 Discussion
Atypical adenoma and carcinoma only account for a very small proportion of primary hyperparathyroidism. The clinical and genetic data are still limited. Here we report the largest single-center cohort of patients with APA (n=79) followed by a mean time of approximately 3.7 years, comparing the clinical characteristics as well as outcomes after surgeries with patients with PC (n=79) and benign parathyroid tumors (n=162). Furthermore, genetic screening for candidate genes of PHPT were performed in about half of APA and PC patients to provide a genotype-phenotype analysis.
The mean onset age of APA was comparable to that of PC in our study, while both of them were 5-6 years younger than that of the benign controls. A review lately summed up 672 APA patients from available research and found the mean age at diagnosis of APA being 43.3 ± 18.9 years, which was, as in PC, a decade earlier than the typical age of benign disease (6). Besides, this review also provided a female to male ratio in APA patients as about 1.5: 1 (6), relatively lower than that in our cohort (2.6: 1) and both lower than that reported in the benign counterpart, who showed a female predominance with a ratio of 3–4: 1 (6, 15, 38, 39). As for PC, there was no gender preference (15, 40).
Our study demonstrated that APA had a relatively benign clinical behavior. Similar to the benign group, the APA patients showed less bone involvement and typical changes in X-rays (bone resorption) than the PC group. But the results of BMD suggested that APA might be more similar to PC as for osteoporosis. For the incidence of urolithiasis, the APA patients were alike the PC patients. But for the gastrointestinal symptoms and asymptomatic PHPT, the APA patients behaved closer to the benign controls. In the present study, the APA group had a ratio of bone involvement within the range reported by the previous literatures from western countries as 35.4% vs. 20.0~67.9%, and a relatively higher ratio of kidney manifestations as 57.0% vs. 32.1~50.0% (6, 7, 41–43). The two largest cohorts in existing studies, reported by Saponaro et al. (7) and Schneider et al. (10), contained 58 APA patients with 57 PA controls and 68 APA with 15 PC controls, respectively. Saponaro et al. (7) reported that the ratio of nephrolithiasis of the whole APA group was 34.5% (20/58), being higher than the PA controls (28.1%, 16/57). Schneider et al. (10) found that bone alterations of APA was significantly less than that of PC (34% vs. 60%, P=0.01), but their ratios of renal stones were equivalent (34% vs. 20%, P=0.37). Although our results suggested that the APA patients had more benign clinical presentations than PC, none of these symptoms or signs were specific for them.
The levels of serum calcium and PTH of our benign lesions and PC groups were comparable to those reported in the largest cohort of PHPT in Chinese people (39). Moreover, our study suggested that the biochemical profile of APA was less severe than PC’s though not as mild as that of benign neoplasms. The APA group had serum calcium level, serum PTH level and proportion of hypercalcemic crisis all higher than those of benign lesions but lower than those of PC group. According to available data, the ranges of median serum PTH and mean SCa levels were 133~1833ng/L and 11.7~15.8mg/dL in APA patients (6, 7, 41, 43, 44). A systematic review concluded that patients with APA were characterized by moderate hypercalcemia (mean 13.4 ± 2.4mg/dL) associated with elevated PTH level [median 430 (73–3242) ng/L], 6.6 times above the upper value of normal range (6). Our biochemical results, as well as current data from the literature, had shown an overlap among series of parathyroid typical and atypical adenoma and carcinoma (6). Thus, no reliable biochemical marker could help identifying APA patients carrying the risks of malignant behaviors.
In our study, the recurrence/no remission rate of the APA group, being 3.9%, was significantly lower than that of the PC group and almost the same as the benign group. Four patients in the PC group and one in the APA group did not obtain remission after their first operation, with the former all carrying nonsynonymous variations of candidate genes for PHPT and the latter finally being diagnosed with MEN1. The rate of recurrence/persistence of atypical parathyroid adenomas ranged from 0% to 3.7% in most literatures (6), however, much higher rates were also reported by three studies as 10.3% (6/58) (7), 24% (4/17) (42) and 28.6% (2/7) (45). Our study emphasized that the prognosis of APA was rather better than PC. But it should be taken into account that the mean follow-up of APA patients was 1.3 years shorter than that of PC patients. A relatively high percentage of pathologic diagnosis as adenoma at the first operation in PC (12.7%) and APA (2.5%) patients further confirmed the dilemma of the differentiation between benign and malignant lesions. Besides, a total of 5 PC cases (6.3%) in this study had been misdiagnosed as APA for the pathohistology of their first surgery, suggesting the necessity for APA patients to be continuously monitored. McCoy et al. (46) observed that none of the 51 APA patients recurred over a mean follow-up of 5 years (range, 0.5-18). Nevertheless, Schulte et al. (9) reported that 23.5% (4/17) APA patients developed recurrence over an average follow-up of 107.2 ± 82.7 months, with two of them diagnosed as PC and one suspicious for PC. Christakis et al. (43) provided a 5-year recurrence-free survival rate as 90.91% (95% CI 50.81–98.67) of the APA patients, whose time from initial surgery to first recurrence was longer than that of the PC group, although it did not achieve statistical significance. While it’s known that PC patients should be followed-up lifelong, the duration and frequency of follow-up for APA patients remain to be decided depending on more long-term data.
Germline variations of the candidate genes for PHPT were screened in our study, and the CDC73 gene was the most frequent mutated gene among all the candidate genes detected not only in PC as expected, but also in APA subjects. The molecular pathogenic mechanisms underlying the origin of atypical adenomas had been investigated only in a few studies until now. Loss-of-function mutations of the CDC73 gene were the most frequent genetic anomaly for PC, with germline mutations of the CDC73 gene being presented in one-third of apparently sporadic PC and 90% of HPT-JT patients (28). Oppositely, CDC73 mutations were very rare in benign neoplasms (27, 47). A latest review of APA summarized the inactivating CDC73 mutations in various studies, finding 36.5% (23/63) cases carrying germline mutations, including two large deletions spanning exons 1–10 of the gene, most of them being found in familial cases (11 patients with FIHP and 4 with HPT-JT) and 7 in apparently sporadic PHPT (6). Saponaro et al. (7) had screened for the germline CDC73 mutations in APA patients and found the mutation rate to be four out of 56 (~7%) with two FIHP and two sporadic cases, similar to that displayed in our study (3/31, ~9.7%) and lower than the number concluded in that review. The reason might be the distinction of the proportion of family-related cases. Apparently, the probability of carrying CDC73 mutations in APA patients was lower than that in PC patients. As mentioned before, two patients with APA and another with PC in our cohort were found out all carrying a missense variation of MEN1 gene (36). PC occurred very rarely in MEN1-HPT, whose parathyroid lesions were almost exclusively benign (48). Ten APA patients from several studies had been searched for MEN1 loss-of-function mutations and only one germline mutation (c.253A>T, p.127S) in exon 2 was detected (29, 49–51). Two GCM2 variants (c.1162A>G and c.1247A>G) detected in our PC patients and one (c.1144 G>A) in this cohort of APA patients were reported in our earlier study (26). The frequency of germline GCM2 variants in clinically sporadic PHPT cases was less than 5% in almost all populations (26). Existing research suggested that mutations in GCM2 could increase the risk of familial or sporadic PHPT (26, 52, 53) and had a tendency for malignancy (26, 53–55), indicating that it might be beneficial for PC and APA patients without CDC73 mutations to screen for GCM2 mutations.
The APA subjects, carrying nonsynonymous variants, showed earlier onset age but no other distinctions for clinical characteristics and outcomes, in comparison with the other subgroup without gene alterations. Although with no statistical significance (P=0.086), two out of 8 (25%) APA cases with nonsynonymous variants developed recurrence or no remission, whereas all cases of no-variation subgroup showed no relapse. As we expected, gene variations were associated with more recurrence/no remission of PHPT in parathyroid carcinoma, although they seemed to have not so much influence on the pre-surgical clinical characteristics of PC. These results required further demonstration due to the limited sample size. Until now, only a few studies have investigated the effects of molecular alterations on APA patients’ clinical outcome and concentrated on CDC73 gene. Cetani et al. (6) concluded that after a time of follow-up ranged from 23 to 252 months (mean 110 ± 71 months), 42% of APA patients carrying germline CDC73 mutations had recurrence/persistence of disease, while 95% CDC73 mutation-negative patients were cured by operation. The study of Saponaro et al. (7)showed that 50% (2/4) APA patients harboring CDC73 germline mutations and 7.7% (4/52) of those without CDC73 mutation had persistent/recurrent disease. From these facts we could draw a conclusion that closer and prolonged surveillance should be beneficial to APA patients carrying germline variations, especially of CDC73 gene. Hence, genetic analysis is probably useful and recommended for APA patients.
The current study has several limitations. First, it is a retrospective study and data missing is unavoidable. About 20% subjects were lost to follow-up. Second, the sample size for genetic screening is restricted. However, considering the rarity of APA and PC, these results are still valuable for clinicians and investigators. Third, somatic mutation detection of parathyroid lesions was not conducted, due to lacking of the surgical specimens.
In summary, our study shows that patients with APA present clinical and biochemical characteristics much less severe than PC and resembling the benign neoplasms, as well as having a relatively good prognosis. The molecular signature of APA remains unclear and the germline CDC73 mutations appears to be the most common abnormality in this series. Patients with APA carrying germline gene variations may have an earlier onset and greater possibility of recurrence for PHPT, probably needing more positive follow-up strategies. More evidence and longer follow-up studies are necessary.
Data availability statement
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found in the article/Supplementary Material.
Ethics statement
The studies involving human participants were reviewed and approved by The Institutional Review Board of Peking Union Medical College Hospital (PUMCH). Written informed consent to participate in this study was provided by the participants’ legal guardian/next of kin.
Author contributions
OW, XX, MN, YJ, ML, and WX contributed to conception and design of the study. YC and AS performed research, analyzed data, and contributed to the discussion and writing of the manuscript. OW and XX contributed to the discussion and edition of the manuscript. All authors contributed to the article and approved the submitted version.
Funding
This work was supported by the Chinese Academy of Medical Sciences (CAMS) Innovation Fund for Medical Sciences (CIFMS) (2017-I2M-1-001) and the National Natural Science Foundation of China (No.81873641).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fendo.2023.1027598/full#supplementary-material
References
1. Fraser WD. Hyperparathyroidism. Lancet. (2009) 374(9684):145–58. doi: 10.1016/S0140-6736(09)60507-9
2. Clark OH. How should patients with primary hyperparathyroidism be treated? J Clin Endocrinol Metab (2003) 88(7):3011–4. doi: 10.1210/jc.2003-030588
3. Xue S, Chen H, Lv C, Shen X, Ding J, Liu J, et al. Preoperative diagnosis and prognosis in 40 parathyroid carcinoma patients. Clin Endocrinol (Oxf). (2016) 85(1):29–36. doi: 10.1111/cen.13055
4. Lee PK, Jarosek SL, Virnig BA, Evasovich M, Tuttle TM. Trends in the incidence and treatment of parathyroid cancer in the united states. Cancer (2007) 109(9):1736–41. doi: 10.1002/cncr.22599
5. Harari A, Waring A, Fernandez-Ranvier G, Hwang J, Suh I, Mitmaker E, et al. Parathyroid carcinoma: a 43-year outcome and survival analysis. J Clin Endocrinol Metab (2011) 96(12):3679–86. doi: 10.1210/jc.2011-1571
6. Cetani F, Marcocci C, Torregrossa L, Pardi E. Atypical parathyroid adenomas: challenging lesions in the differential diagnosis of endocrine tumors. Endocr Relat Cancer (2019) 26(7):R441–r464. doi: 10.1530/ERC-19-0135
7. Saponaro F, Pardi E, Mazoni L, Borsari S, Torregrossa L, Apicella M, et al. Do patients with atypical parathyroid adenoma need a close follow-up? J Clin Endocrinol Metab (2021) 106(11):e4565–79. doi: 10.1210/clinem/dgab452
8. Galani A, Morandi R, Dimko M, Molfino S, Baronchelli C, Lai S, et al. Atypical parathyroid adenoma: clinical and anatomical pathologic features. World J Surg Oncol (2021) 19(1):19. doi: 10.1186/s12957-021-02123-7
9. Schulte JJ, Pease G, Taxy JB, Hall C, Cipriani NA. Distinguishing parathyromatosis, atypical parathyroid adenomas, and parathyroid carcinomas utilizing histologic and clinical features. Head Neck Pathol (2021) 15(3):727–36. doi: 10.1007/s12105-020-01281-6
10. Schneider R, Bartsch-Herzog S, Ramaswamy A, Bartsch DK, Karakas E. Immunohistochemical expression of e-cadherin in atypical parathyroid adenoma. World J Surg (2015) 39(10):2477–83. doi: 10.1007/s00268-015-3149-7
11. Saraydaroglu O, Ozsen M, Narter S, Kirdak T, Erturk E. A close look at our cases with parathyroidectomy: 11 years of experience. Minerva Endocrinol (2022) 47(2):160–6. doi: 10.23736/S0391-1977.20.03171-5
12. DeLellis R LC, Arnold A, Lloy R, Bilezikian J, Mete O, Eng C. Tumors of the parathyroid glands. In: Lloyd RV, Osamura RY, Klöppel G, Rosai J, editors. WHO classification of tumours of endocrine organs, 4th ed, vol. 2017. Lyon: IARC (2017). p. 145–59.
13. Sandelin K, Tullgren O, Farnebo LO. Clinical course of metastatic parathyroid cancer. World J Surg (1994) 18(4):594–8. discussion 599. doi: 10.1007/BF00353773
14. Cardoso L, Stevenson M, Thakker RV. Molecular genetics of syndromic and non-syndromic forms of parathyroid carcinoma. Hum Mutat (2017) 38(12):1621–48. doi: 10.1002/humu.23337
15. Cetani F, Pardi E, Marcocci C. Update on parathyroid carcinoma. J endocrinological Invest (2016) 39(6):595–606. doi: 10.1007/s40618-016-0447-3
16. Cinque L, Pugliese F, Salcuni AS, Scillitani A, Guarnieri V. Molecular pathogenesis of parathyroid tumours. Best Pract Res Clin Endocrinol Metab (2018) 32(6):891–908. doi: 10.1016/j.beem.2018.11.001
17. Silverberg SJ, Walker MD, Bilezikian JP. Asymptomatic primary hyperparathyroidism. J Clin Densitom. (2013) 16(1):14–21. doi: 10.1016/j.jocd.2012.11.005
18. Ahmad S, Kuraganti G, Steenkamp D. Hypercalcemic crisis: a clinical review. Am J Med (2015) 128(3):239–45. doi: 10.1016/j.amjmed.2014.09.030
19. Lou I, Schneider DF, Sippel RS, Chen H, Elfenbein DM. The changing pattern of diagnosing primary hyperparathyroidism in young patients. Am J Surg (2017) 213(1):146–50. doi: 10.1016/j.amjsurg.2016.03.019
20. Qin MW, Yu W, Xu L, Tian JP, Xing XP, Meng XW, et al. [Bone mineral and body composition analysis of whole body in 292 normal subjects assessed by dual X-ray absorptiometry]. Zhongguo Yi Xue Ke Xue Yuan Xue Bao. (2003) 25(1):66–9.
21. Wang Y, Nie M, Wang O, Li Y, Jiang Y, Li M, et al. Genetic screening in a Large Chinese cohort of childhood onset hypoparathyroidism by next-generation sequencing combined with TBX1-MLPA. J Bone Miner Res (2019) 34(12):2254–63. doi: 10.1002/jbmr.3854
22. Kong J, Wang O, Nie M, Shi J, Hu Y, Jiang Y, et al. Clinical and genetic analysis of multiple endocrine neoplasia type 1-related primary hyperparathyroidism in Chinese. PLoS One (2016) 11(11):e0166634. doi: 10.1371/journal.pone.0166634
23. Kong J, Wang O, Nie M, Shi J, Hu Y, Jiang Y, et al. Familial isolated primary hyperparathyroidism/hyperparathyroidism-jaw tumour syndrome caused by germline gross deletion or point mutations of CDC73 gene in Chinese. Clin Endocrinol (Oxf). (2014) 81(2):222–30. doi: 10.1111/cen.12461
24. Da Silva AM, Maciel RM, Da Silva MR, Toledo SR, De Carvalho MB, Cerutti JM. A novel germ-line point mutation in RET exon 8 (Gly(533)Cys) in a large kindred with familial medullary thyroid carcinoma. J Clin Endocrinol Metab (2003) 88(11):5438–43. doi: 10.1210/jc.2003-030997
25. Song A, Wang W, Chen S, Wang Y, Liu S, Nie M, et al. Primary hyperparathyroidism during pregnancy: A case series of 8 patients. Endocr Pract (2019) 25(11):1127–36. doi: 10.4158/EP-2019-0035
26. Song A, Yang Y, Wang Y, Liu S, Nie M, Jiang Y, et al. Germline GCM2 mutation screening in Chinese primary hyperparathyroidism patients. Endocr Pract (2020) 26(10):1093–104. doi: 10.4158/EP-2020-0132
27. Howell VM, Haven CJ, Kahnoski K, Khoo SK, Petillo D, Chen J, et al. HRPT2 mutations are associated with malignancy in sporadic parathyroid tumours. J Med Genet (2003) 40(9):657–63. doi: 10.1136/jmg.40.9.657
28. Shattuck TM, Välimäki S, Obara T, Gaz RD, Clark OH, Shoback D, et al. Somatic and germ-line mutations of the HRPT2 gene in sporadic parathyroid carcinoma. N Engl J Med (2003) 349(18):1722–9. doi: 10.1056/NEJMoa031237
29. Mizusawa N, Uchino S, Iwata T, Tsuyuguchi T, Suzuki Y, Mizukoshi M, et al. Genetic analyses in patients with familial isolated hyperparathyroidism and hyperparathyroidism-jaw tumour syndrome. Clin Endocrinol (Oxf). (2006) 65(1):9–16. doi: 10.1111/j.1365-2265.2006.02534.x
30. Wang O, Wang C, Nie M, Cui Q, Guan H, Jiang Y, et al. Novel HRPT2/CDC73 gene mutations and loss of expression of parafibromin in Chinese patients with clinically sporadic parathyroid carcinomas. PLoS One (2012) 7(9):e45567. doi: 10.1371/journal.pone.0045567
31. Guarnieri V, Scillitani A, Muscarella LA, Battista C, Bonfitto N, Bisceglia M, et al. Diagnosis of parathyroid tumors in familial isolated hyperparathyroidism with HRPT2 mutation: implications for cancer surveillance. J Clin Endocrinol Metab (2006) 91(8):2827–32. doi: 10.1210/jc.2005-1239
32. Yang Y, Song A, Nie M, Jiang Y, Li M, Xia W, et al. A novel long-range deletion spanning CDC73 and upper-stream genes discovered in a kindred of familial primary hyperparathyroidism. Endocrine (2022) 75(3):907–15. doi: 10.1007/s12020-021-02917-5
33. Lee KM, Kim EJ, Choi WS, Park WS, Kim SW. Intrathyroidal parathyroid carcinoma mimicking a thyroid nodule in a MEN type 1 patient. J Clin Ultrasound (2014) 42(4):212–4. doi: 10.1002/jcu.22090
34. Wang W, Kong J, Nie M, Jiang Y, Li M, Xia W, et al. Primary hyperparathyroidism in Chinese children and adolescents: A single-centre experience at Peking union medical college hospital. Clin Endocrinol (Oxf) (2017) 87(6):865–73. doi: 10.1111/cen.13453
35. Cetani F, Pardi E, Borsari S, Agarwal S, Viacava P, Dipollina G, Cianferotti L, et al. Genetic analyses of the HRPT2 gene in primary hyperparathyroidism: germline and somatic mutations in familial and sporadic parathyroid tumors. J Clin Endocrinol Metab. (2004) 89(11):5583–91. doi: 10.1210/jc.2004-0294
36. Song A, Yang Y, Liu S, Nie M, Jiang Y, Li M, et al. Prevalence of parathyroid carcinoma and atypical parathyroid neoplasms in 153 patients with multiple endocrine neoplasia type 1: Case series and literature review. Front Endocrinol (2020) 11:557050–0. doi: 10.3389/fendo.2020.557050
37. Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American college of medical genetics and genomics and the association for molecular pathology. Genet Med (2015) 17(5):405–24. doi: 10.1038/gim.2015.30
38. Marcocci C, Cetani F, Rubin MR, Silverberg SJ, Pinchera A, Bilezikian JP. Parathyroid carcinoma. J Bone Miner Res (2008) 23(12):1869–80. doi: 10.1359/jbmr.081018
39. Lin X, Fan Y, Zhang Z, Yue H. Clinical characteristics of primary hyperparathyroidism: 15-year experience of 457 patients in a single center in China. Front Endocrinol (Lausanne) (2021) 12:602221. doi: 10.3389/fendo.2021.602221
40. Rodrigo JP, Hernandez-Prera JC, Randolph GW, Zafereo ME, Hartl DM, Silver CE, et al. Parathyroid cancer: An update. Cancer Treat Rev (2020) 86:102012. doi: 10.1016/j.ctrv.2020.102012
41. Quinn CE, Healy J, Lebastchi AH, Brown TC, Stein JE, Prasad ML, et al. Modern experience with aggressive parathyroid tumors in a high-volume new England referral center. J Am Coll Surg (2015) 220(6):1054–62. doi: 10.1016/j.jamcollsurg.2014.10.007
42. Christakis I, Busaidy NL, Cote GJ, Williams MD, Hyde SM, Silva Figueroa AM, et al. Parathyroid carcinoma and atypical parathyroid neoplasms in MEN1 patients; a clinico-pathologic challenge. the MD Anderson case series and review of the literature. Int J Surg (2016) 31:10–6. doi: 10.1016/j.ijsu.2016.05.035
43. Christakis I, Bussaidy N, Clarke C, Kwatampora LJ, Warneke CL, Silva AM, et al. Differentiating atypical parathyroid neoplasm from parathyroid cancer. Ann Surg Oncol (2016) 23(9):2889–97. doi: 10.1245/s10434-016-5248-6
44. Cakir B, Polat SB, Kilic M, Ozdemir D, Aydin C, Süngü N, et al. Evaluation of preoperative ultrasonographic and biochemical features of patients with aggressive parathyroid disease: Is there a reliable predictive marker? Arch Endocrinol Metab (2016) 60(6):537–44. doi: 10.1590/2359-3997000000224
45. Fernandez-Ranvier GG, Khanafshar E, Jensen K, Zarnegar R, Lee J, Kebebew E, et al. Parathyroid carcinoma, atypical parathyroid adenoma, or parathyromatosis? Cancer. (2007) 110(2):255–64. doi: 10.1002/cncr.22790
46. McCoy KL, Seethala RR, Armstrong MJ, Nikiforova MN, Stang MT, Carty SE, et al. The clinical importance of parathyroid atypia: Is long-term surveillance necessary? Surgery. (2015) 158(4):929–35. discussion 935-6. doi: 10.1016/j.surg.2015.06.022
47. Haven C, Howell V, Eilers P, Dunne R, Takahashi M, van Puijenbroek M, et al. Gene expression of parathyroid tumors: Molecular subclassification and identification of the potential malignant phenotype. Cancer Res (2004) 64(20):7405–11. doi: 10.1158/0008-5472.CAN-04-2063
48. Brewer K, Costa-Guda J, Arnold A. Molecular genetic insights into sporadic primary hyperparathyroidism. Endocr Relat Cancer (2019) 26(2):R53–r72. doi: 10.1530/ERC-18-0304
49. Cetani F, Pardi E, Giovannetti A, Vignali E, Borsari S, Golia F, et al. Genetic analysis of the MEN1 gene and HPRT2 locus in two Italian kindreds with familial isolated hyperparathyroidism. Clin Endocrinol (Oxf). (2002) 56(4):457–64. doi: 10.1046/j.1365-2265.2002.01502.x
50. Juhlin C, Larsson C, Yakoleva T, Leibiger I, Leibiger B, Alimov A, et al. Loss of parafibromin expression in a subset of parathyroid adenomas. Endocr Relat Cancer. (2006) 13(2):509–23. doi: 10.1677/erc.1.01058
51. Sulaiman L, Nilsson IL, Juhlin CC, Haglund F, Höög A, Larsson C, et al. Genetic characterization of large parathyroid adenomas. Endocr Relat Cancer. (2012) 19(3):389–407. doi: 10.1530/ERC-11-0140
52. Maret A, Ding C, Kornfield SL, Levine MA. Analysis of the GCM2 gene in isolated hypoparathyroidism: a molecular and biochemical study. J Clin Endocrinol Metab (2008) 93(4):1426–32. doi: 10.1210/jc.2007-1783
53. Canaff L, Guarnieri V, Kim Y, Wong BYL, Nolin-Lapalme A, Cole DEC, et al. Novel glial cells missing-2 (GCM2) variants in parathyroid disorders. Eur J Endocrinol (2022) 186(3):351–66. doi: 10.1530/EJE-21-0433
54. Marchiori E, Pelizzo MR, Herten M, Townsend DM, Rubello D, Boschin IM. Specifying the molecular pattern of sporadic parathyroid tumorigenesis-the Y282D variant of the GCM2 gene. Biomedicine pharmacotherapy = Biomedecine pharmacotherapie. (2017) 92:843–8. doi: 10.1016/j.biopha.2017.05.028
Keywords: primary hyperparathyroidism, atypical parathyroid adenoma, parathyroid carcinoma, parathyroid adenoma, parathyroid hyperplasia, genetic analysis
Citation: Chen Y, Song A, Nie M, Jiang Y, Li M, Xia W, Wang O and Xing X (2023) Clinical and genetic analysis of atypical parathyroid adenoma compared with parathyroid carcinoma and benign lesions in a Chinese cohort. Front. Endocrinol. 14:1027598. doi: 10.3389/fendo.2023.1027598
Received: 25 August 2022; Accepted: 06 January 2023;
Published: 26 January 2023.
Edited by:
Laura Gianotti, Azienda Sanitaria Ospedaliera S.Croce e Carle Cuneo, ItalyReviewed by:
Alfredo Scillitani, Home for Relief of Suffering (IRCCS), ItalyHao Zhang, Shanghai Jiao Tong University, China
Ling-Qing Yuan, Second Xiangya Hospital, Central South University, China
Copyright © 2023 Chen, Song, Nie, Jiang, Li, Xia, Wang and Xing. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Ou Wang, d2FuZ19vdTIwMTBAMTI2LmNvbQ==; Xiaoping Xing, eGlhb3Bpbmd4aW5nQDEyNi5jb20=
†These authors have contributed equally to this work and share first authorship
‡ORCID: Yingyu Chen, orcid.org.0000-0001-6687-1464
An Song, orcid.org.0000-0001-6572-1827
Ou Wang, orcid.org.0000-0002-0395-8789
Xiaoping Xing, orcid.org.0000-0002-2759-5177