 Zsombor Koszegi
Zsombor Koszegi Rachel Y. Cheong
Rachel Y. Cheong- 1Institute of Metabolism and Systems Research, University of Birmingham, Birmingham, United Kingdom
- 2Timeline Bioresearch AB, Medicon Village, Lund, Sweden
Estrogens can alter the biology of various tissues and organs, including the brain, and thus play an essential role in modulating homeostasis. Despite its traditional role in reproduction, it is now accepted that estrogen and its analogues can exert neuroprotective effects. Several studies have shown the beneficial effects of estrogen in ameliorating and delaying the progression of neurodegenerative diseases, including Alzheimer’s and Parkinson’s disease and various forms of brain injury disorders. While the classical effects of estrogen through intracellular receptors are more established, the impact of the non-classical pathway through receptors located at the plasma membrane as well as the rapid stimulation of intracellular signaling cascades are still under active research. Moreover, it has been suggested that the non-classical estrogen pathway plays a crucial role in neuroprotection in various brain areas. In this mini-review, we will discuss the use of compounds targeting the non-classical estrogen pathway in their potential use as treatment in neurodegenerative diseases and brain injury disorders.
Introduction
Estrogens are a group of gonadal sex hormones that exist naturally in three different forms in humans. 17β-estradiol (E2) is the most dominant biological form, followed by estrone (E1) the intermediate form, and estriol (E3), which has very low levels in the body that are only increased during pregnancy. In this mini-review, we will use the abbreviation E2 to refer to 17β-estradiol and will focus predominantly on this form as this is the most abundant and most of the research has been largely focused on studying this molecule. In addition to its role in reproductive functions, E2 has a profound influence on the central nervous system (1, 2). This has contributed to the interest generated around the impact of E2 on neuronal function in health and disease. Investigations over the past few decades have shown that E2 has the potential to prevent or counterbalance the symptoms of neurodegenerative diseases. The gender differences observed in two of the most common neurodegenerative diseases, Alzheimer’s disease (AD) and Parkinson’s disease (PD), clearly suggest this role (3–5). Although there is no conclusive evidence for E2 treatment in neurodegenerative diseases in human clinical trials, there have been several in vivo rodent and in vitro cell line models that indicate the therapeutic effects of E2. This mini-review will discuss the neuroprotective, non-classical effects of E2 in the context of some of the most typical neurodegenerative cases (that is AD and PD) as well as brain injuries that possibly lead to neurodegeneration (traumatic brain injury and stroke) and highlight the use of some of the non-classical E2 analogues to potentially prevent or treat these disorders.
Classical versus non-classical estrogen pathways
E2 regulates cellular processes by binding to specific estrogen receptors (ERs) with two distinct modes of action, broadly classified as the classical and non-classical estrogen pathway. Stimulation of the classical pathway results in direct transcriptional effects through the binding of E2 to its intracellular receptors (ERα and ERβ) and activation of the estrogen response element (ERE) (6). In contrast, the non-classical pathway involves the rapid activation of ion channels and intracellular second messenger signaling pathways. The latter is followed by the stimulation of an array of gene transcription factors, but activation via the non-classical pathway is ERE-independent. The non-classical pathway is often described as rapid, as the activation of intracellular signaling pathways can be detected in a matter of seconds, as first demonstrated by Szego and Davis, whereby E2 induced an increase in cyclic adenosine monophosphate (cAMP) levels in the uterus few seconds following administration (7). However, this rapid signaling pathway activation will also often lead to gene transcription, which can be detected at a slower rate. One of the most important transcription factors of the non-classical pathway is the cAMP response element-binding protein (CREB), which has been implicated in multiple studies (8–10).
Apart from the classical ERα and ERβ, experiments looking at the rapid signaling pathway activation by E2 highlighted that these classical intracellular receptors – mediating ERE-dependent gene transcription – might not be sufficient to account for the variety of responses observed. This led to the discovery of membrane linked receptors, which can be membrane-localized classical ERα and ERβ or other types, for example, the ER-X and the G protein coupled GPR30 (GPER1) (11–13), which are all different from the classical receptors in their structure, localization, as well as modes of action. A schematic illustration of the classical and non-classical modes of E2 action is depicted in Figure 1.
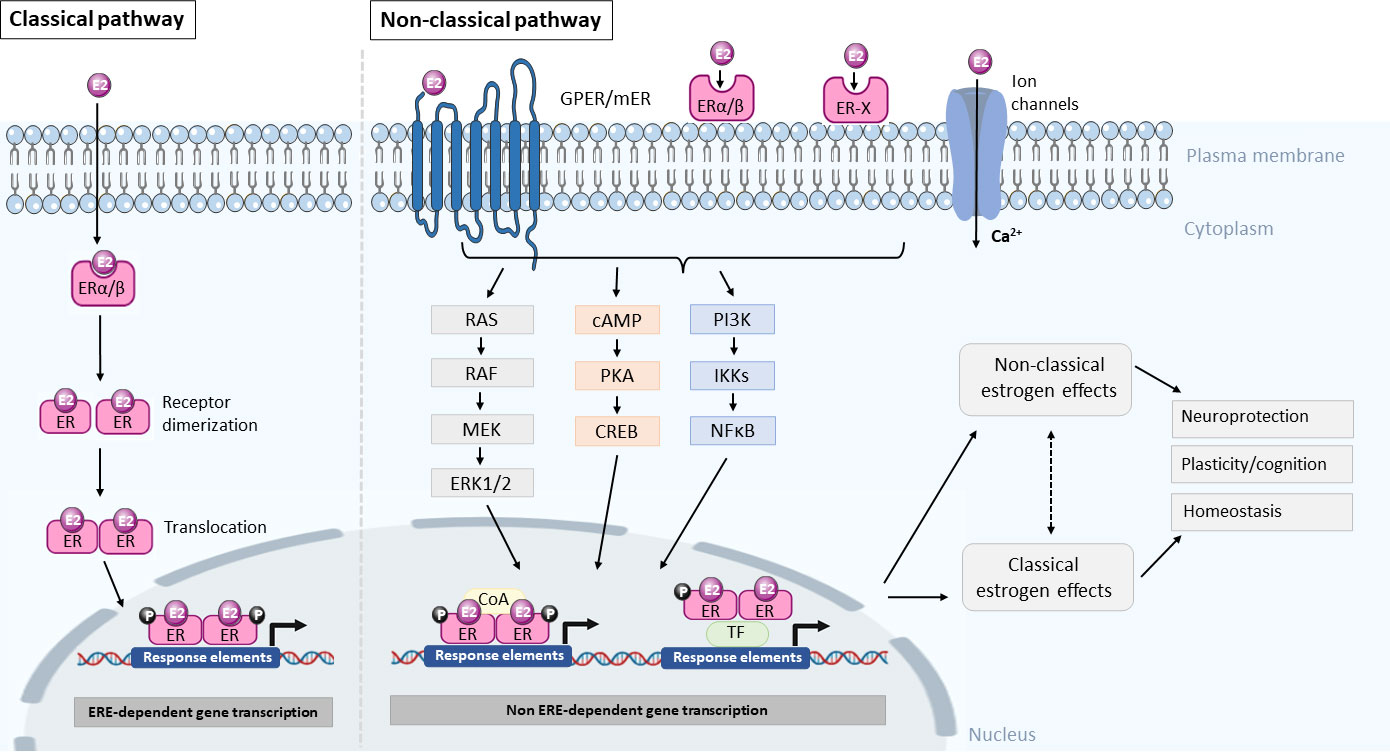
Figure 1 Summary diagram of the classical and non-classical modes of estrogen action. In the classical pathway, E2 crosses the plasma membrane by diffusion and binds to the estrogen receptor (ER) and forms an E2-receptor complex, which dimerizes and translocates to the nucleus to regulate gene transcription through an estrogen response element (ERE) dependent manner. In the non-classical pathway, E2 interacts with membrane bound estrogen receptors (mER), G-protein coupled estrogen receptors (GPER), ER-X, or classic ER (ERα/β) and activates kinases and second messenger signaling pathways to phosphorylate transcription factors (TF) or coactivators to influence gene transcription in the nucleus via a non-ERE-dependent manner. The resultant effect of activating these pathways is neuroprotection, modulating plasticity and cognition as well as maintenance of homeostasis. However, the extent to which the non-classical and classical pathways crosstalk or interact with each other is not known. It is likely that both pathways contribute to neuroprotection and homeostasis. RAS, Ras small GTPase, RAF, Raf kinase, MEK, mitogen-activated protein kinase, ERK1/2, extracellular signal-regulated kinase 1/2, cAMP, cyclic adenosine monophosphate, PKA, protein kinase A, CREB, cAMP-responsive element-binding protein, PI3K, phosphatidylinositol-3 kinase, IKKs, IκB kinases, NFκB, nuclear factor kappa-light-chain-enhancer of activated B cells, coA, coactivator.
Mechanism for non-classical E2 neuroprotection
There are several possible molecular mechanisms contributing to non-classical E2 neuroprotection, such as control of neuroinflammation, myelin protection, mitochondrial protection and control of oxidative stress, regulating autophagy as well as maintenance of synaptic transmission and plasticity. One of the important protective actions of E2 is in the control of neuroinflammation whereby E2 reduces the secretion of proinflammatory cytokines and interleukins and thereby reducing microglia activation via the inhibition of the nuclear factor kappa-light-chain-enhancer of activated B cells (NFκB) signaling pathway (14, 15). In addition, the neuroprotective effects of E2 are in part due to its protective actions on myelin and remyelination, which can be mediated by activation of the phosphoinositide 3-kinases (PI3K)/protein kinase B (Akt)/mammalian target of rapamycin (mTOR) signaling pathway (16–18). Dysfunction in the myelin sheaths is often a common feature in neurodegenerative diseases such as AD and PD as well as in other central nervous system pathologies, such as traumatic brain injury (TBI), stroke and multiple sclerosis. In these neuropathological conditions, E2 has been shown to upregulate genes involved in synaptogenesis, axonal repair and synaptic plasticity, such as Bcl2, TrkB and cadherin-2 (19–21). Another way in which E2 exerts its neuroprotective effects is against oxidative stress through the protection of mitochondrial function and by reducing the production of reactive oxygen species (22, 23). Under pathological conditions, E2 may also elicit various of the above-mentioned responses, but may also promote the release of different neurotrophic factors such as the glial cell line-derived neurotrophic factor (GDNF), insulin-like growth factor 1 (IGF-1) and brain-derived neurotrophic factor (BDNF) to protect neurons and promote reparation of injured neuronal circuits (24, 25).
Compounds targeting the non-classical estrogen pathway
Importantly, previous findings indicate that apart from the classical estrogen pathway, the non-classical pathway also plays a role in ameliorating neurodegeneration in disease models. The latter is of particular interest as E2 replacement therapy, which affects both the classical and non-classical pathways, has been shown to not only increase the risk of myocardial infarction or coronary heart disease but could potentially lead to an array of side effects, including increased risk of breast cancer and stroke (26–28). Therefore, there has been a renewed interest in developing new compounds that are able to trigger protective or restorative effects without the risk of unwanted side effects. One of these groups of such compounds is the ‘selective estrogen-receptor modulators’ (SERMs), which are non-steroidal molecules with specific mechanism of action in target tissues. They primarily act as partial ER agonists in the target tissue while being antagonists in non-target tissues. Some SERMs, for example, tamoxifen and raloxifene are already in clinical use for pre- and post-menopausal women (29), while others, such as the GPER1 agonist G-1 or the STX (a Gq-coupled membrane ER agonist) are used in preclinical animal studies (30, 31). The challenge with SERMs lies in the balance between the efficacy of the agonistic profile and, at the same time, the reduction of unwanted side effects on non-target tissues. While newer third generation SERMs, such as bazedoxifene, ospemifene and lasofoxifene, have improved efficacy, their use as SERMs in the brain is not known (32). Other important compounds are the ‘activators of non-genomic estrogen-like signaling’ (ANGELS), which is a novel group in E2 therapy that is aimed at targeting the non-classical E2 pathway. Three of these molecules are known, estren (4-estren-3alpha, 17beta-diol), compound A, and compound B, which are all capable of triggering the non-classical E2 pathway (33, 34). However, these compounds are yet to be used in clinical practice, although estren has been found to have protective effects on basal forebrain cholinergic neurons (35, 36), indicating that there is prospect for the use of these non-classical activators as treatment for neurodegenerative diseases.
Alzheimer’s disease
Pathophysiology
Alzheimer’s disease (AD) is a chronic progressive neurodegenerative disorder, characterized by distinct hallmark pathologies, such as the presence of amyloid plaques, which comprises primarily of aggregated amyloid β (Aβ) peptide, and formation of neurofibrillary tangles with hyperphosphorylated tau protein. These pathologies lead to progressive and selective neuronal loss in the hippocampus and temporal cortex, cognitive decline and eventual death. There is no curative treatment available for AD at present and current treatments only target the management of symptoms with no influence on disease progression. The pathogenesis of AD has been postulated to be due to the accumulation of Aβ as a result of altered amyloid precursor protein (APP), accumulation of tau, oxidative stress caused by mitochondrial dysfunction and persistent neuroinflammation.
Neuroprotective effects of E2 in AD
Neuroprotective effects of E2 have been proposed in experimental models of AD. Estrogen deficiency in the brain accelerates Aβ plaque formation (37–39), while E2 treatment has been shown to reduce the expression of Aβ peptide and abnormal accumulation of amyloid proteins (40–42). The reduction of Aβ following E2 administration might be linked to the alteration of the APP gene, as APP protein levels are reduced following E2 treatment (43) as well as the cleavage of APP into toxic Aβ. E2 stimulation increases the secreted APPα, which can lead to a decrease in toxic Aβ species (44, 45). This neuroprotection against β-amyloid toxicity have been shown to occur via ERα and ERβ (46). In addition, to protection against Aβ accumulation, E2 is known to also decrease tau hyperphosphorylation in experimental models of AD (47, 48).
A loss of cholinergic neurons is recognized as one of the hallmarks of AD. There is considerable evidence showing the effects of E2 on plasticity and protection of cholinergic neurons through an ERα dependent pathway (49, 50). Accordingly, E2 has been reported to upregulate fiber density of the remaining cholinergic neurons after an excitotoxic insult via the mitogen-activated protein kinase (MAPK) signaling pathway, leading to the stimulation of CREB phosphorylation (8, 35, 51). E2 has also been known to alter the dynamics of neural circuits, such as modulating the plasticity of dendritic spines and stimulating neurogenesis and synaptic contacts in numerous brain regions like the hippocampus, hypothalamus and amygdala (52–54). In experimental models of AD, such as the transgenic APP/PS1 and 3xTg AD mice, ovariectomy increased the accumulation of the Aβ peptide and decreased hippocampal-dependent behavioral performance. Treatment with E2 not only prevented the worsening of pathologies, but also reduced the accumulation of Aβ in the hippocampus, subiculum and amygdala (55, 56), suggesting a protective role of E2 in AD progression. With the potential impact of E2 on systemic tissues, there is a need to develop brain-specific therapies. Treatment with a brain-selective prodrug, DHED (10β,17β-dihydroxyestra-1,4-dien-3-one), in APP/PS1 double transgenic mice showed no systemic off-target effects in the uterine tissue, but similar improvements in APP levels, suggesting that the brain-selective treatment with DHED can be used as an early-stage intervention for AD (57).
Taken together, E2 has the potential to regenerate, restore and strengthen the formation of new synaptic networks from the remaining neurons and/or rewire neural circuits under pathological conditions.
Targeting non-classical E2 pathway as potential treatment in AD
Given the neuroprotective potential of E2 in AD, targeting the non-classical E2 pathway selectively may provide an alternative treatment strategy. Studies have shown that ANGELS compounds, such as estren, can activate the non-classical E2 pathway and rescue the survival of basal forebrain cholinergic neurons after injection of Aβ (1–42) in mice (36) and is neuroprotective against Aβ-induced injury in vitro (58). A key important feature of estren treatment is that, unlike E2, it does not increase the size of the uterus, indicating that it might not have unwanted, genomic side effects (59). Regarding cognition, E2 has consistently been reported to have the ability to enhance cognitive function via the non-classical E2 pathway involving the ERK1/2 and Akt signaling pathways (60–64). A number of clinical trials in AD have been conducted with the second generation SERM, raloxifene, with varying results, in hope of alleviating cognitive deficits. While some showed that raloxifene improved verbal memory and reduced the risk of AD and mild cognitive impairment, others showed no significant changes in cognition (65–67).
More recent studies show that targeting non-nuclear ERs, such as GPER1, or using non-classical ligands, such as STX, could ameliorate memory impairments or protect against Aβ-toxicity in experimental models of AD via activation of the ERK and PI3K/Akt signaling pathways (68–70). These studies provide evidence that activation of the membrane-bound, non-nuclear ERs can provide an alternative therapeutic target in AD. Another novel compound that is of emerging interest is the Pathway Preferential Estrogen-1 (PaPE-1), which is a selective non-nuclear ER pathway activator, which can protect neurons against Aβ-induced toxicity through a mechanism that involves inhibition of oxidative stress and apoptosis (71). This compound strongly activates the MAPK and mTOR pathways without interaction with the nuclear receptors and has a broad spectrum of utility in other neurological disorders, where it also decreases the severity of stroke (72). However, there is a clear lack of clinical trials for these newly developed compounds and more studies are warranted to determine the viability of using non-classical E2 activators as a preventive treatment alternative for AD.
Parkinson’s disease
Pathophysiology
Parkinson’s disease (PD) is one of the most common age-related neurodegenerative movement disorders. The main pathological hallmark of PD is motor symptoms consisting of resting tremor, rigidity, bradykinesia and postural imbalance, attributed primarily to the substantial loss of midbrain dopamine (DA) neurons in the substantia nigra pars compacta and the accumulation of α-synuclein cytoplasmic protein deposits, termed Lewy Bodies, in the surviving neurons. The dopaminergic system is not the only affected network in PD. Degeneration of serotonergic neurons in the raphe nucleus, noradrenergic neurons of the locus coeruleus and cholinergic neurons of the nucleus basalis of Meynert have also been reported in PD. Numerous different treatment methods have been investigated to alleviate motor deficits, but no effective clinical therapy has been found to be able to prevent or reverse the degeneration of DA neurons (73). There is currently no cure for PD and available treatments are only symptomatic. DA itself is not a suitable drug as it does not cross the blood-brain-barrier, has a short half-life and has peripheral hemodynamic side effects. Oral administration of L-DOPA remains the gold standard treatment today (74, 75). However, the challenge with L-DOPA is that it cannot be utilized as a long-term treatment for PD. As such, the development of new therapeutics and strategies with several mechanisms of action, such as neurosteroids, could provide an alternative treatment for PD.
Neuroprotective effects of E2 in PD
While E2 effects on the dopaminergic system have not been well characterized, there is some evidence of a modulatory effect of E2 in PD patients. Postmenopausal women who received hormone replacement therapy have a reduced risk of developing PD and lower disease severity in early stages of the disease (76, 77). E2 has been reported to be protective against 6-OHDA (6-hydroxy dopamine) toxicity in DA neurons (78). Similarly, in the neurotoxin MPTP (1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine) model of PD, E2 treatment improved DA release in the striatum and nucleus accumbens and could protect DA neurons (79–82). In fact, E2 treatment has been shown to increase fiber density of tyrosine hydroxylase-positive DA neurons in both 6-OHDA and MPTP-induced models (83–85). In order to determine the ER subtype regulating neuroprotection in PD, studies have used selective ER agonists and found that the activation of ERα but not ERβ rescued the depletion of DA and prevented the loss of DA transporter in the striatum and cell death in the substantia nigra in MPTP-treated mice (86–88). These studies suggest that neuroprotection of DA neurons occurs through an ERα-specific manner in experimental models of PD.
Targeting non-classical E2 pathway as potential treatment in PD
There is a lack of research on SERMs in human studies of PD. The majority of the studies have been performed in rodent models with contradictory results. In the MPTP model, raloxifene treatment prevented the MPTP-induced DA depletion, restored DA levels and prevented DA cell death (89, 90) while in other studies was proven ineffective (91). The varying results could be due to differences in the models used, dosing paradigm or pharmacological properties of the different compounds. The other new estrogen analogue, the brain-selective estrogen prodrug, DHED, was found to protect DA neurons in the MPTP-toxicity model and in 3K α-synuclein transgenic mice (mouse model that exhibits many features of PD neuropathology) (92, 93). DHED was also found to selectively increase E2 in the brain while the periphery was spared, which in turn, reduced the secondary effects of E2 on the body (94). In addition, DHED treatment significantly alleviated the neuronal pathology of PD via decreasing α-synuclein monomer accumulation and aggregation, restoring vesicle and dopaminergic fiber densities as well as improving PD-associated motor deficits (92–94). Taken together, this evidence highlights the potential for modulating E2 signaling with pharmaceutical analogues for neuroprotection in PD. More investigations into the use of these non-classical activator compounds in PD models are warranted to determine their therapeutic potential.
Brain injury disorders
Pathophysiology
Brain injuries can be classified into two main categories, traumatic and non-traumatic. Traumatic brain injury (TBI) occurs when the original function of the brain or the underlying anatomy changes due to an external force (e.g., injury). Non-traumatic brain injury, also referred to as acquired brain injury, is caused by internal factors such as lack of oxygen, exposure to toxins or infection. Examples of non-traumatic brain injury include stroke and cerebral ischemia. Although brain injury is not a neurodegenerative disease per se, it is now clear that brain injuries can trigger progressive neurodegeneration and dementia (e.g., AD) (95). As TBI and stroke are recognized as one of the leading causes of disability and death in most societies (96, 97), it is important to discuss the potential of using alternative non-surgical therapies.
Neuroprotective effects of E2 in brain injury disorders
The evidence is not clear, especially when it comes to human studies, but there is a strong indication that there is a trend for sex differences, potentially due to differing circulating E2 levels, in the incidence and mortality rate of TBI (98–100). Another indication that E2 might play a role in ameliorating neuronal damage following injury is that the activity of aromatase (a key enzyme in E2 synthesis) increases, particularly in brain astroglia cells (101). This increased aromatase activity has been reported to be neuroprotective in various animal models (102). Besides locally produced E2 in the brain, exogenous E2 application before or immediately after injury has also been shown to rescue damage following a controlled impact in ovariectomized mice (103, 104), indicating that E2 does have treatment potential following trauma in both the TBI and stroke experimental models.
Targeting non-classical E2 pathway as potential treatment in brain injury disorders
As in the case of other forms of neuronal brain damage, the non-classical estrogen pathway has been reported to have treatment potential in TBI and also in stroke. A known characteristic of TBI is that the primary injury due to the external force is often followed by a slower secondary injury. One of the most common secondary injuries is excessive glutamate release, which is followed by overactivation of NMDA (N-methyl-D-aspartate) and AMPA (α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid) receptors and consequentially intracellular ion imbalance, leading to excitatory cell death (105). In an experimental model of NMDA-induced toxicity, E2 treatment following injury ameliorated the damage in basal forebrain cholinergic fibers in mice (35). Importantly, this study highlighted the involvement of the non-classical E2 pathway via the MAPK/PKA signaling system. The non-classical pathway activator, estren (a member of the ANGELS compounds), has also been able to trigger E2-like restorative actions. And, as for the receptor dependence of the protective actions of E2 in TBI, the above-mentioned study highlighted that ERα is required for the ameliorative effects after damage (35). However, another study has shown that both ERα and ERβ helped to reduce brain edema following TBI in rats (106). It has also been shown that E2 treatment following TBI can increase ERα and restore ERβ expression in the brain (107). In addition to these classical E2 receptors, it appears that GPER1 is also involved in neuroprotection following TBI. Both E2 and treatment with the GPER1 agonist, G-1, increased neuronal survival as well as decreased neuronal degeneration and apoptotic cell death in a rodent model of TBI (108). These results were corroborated in other rat TBI studies, where G-1 was found to promote neuronal survival and improve cognitive impairment (109) as well as reduced neuronal apoptosis and increased microglia polarization (110), through the PI3K/Akt signaling pathway. Likewise, the non-classical pathway has also been implicated as an alternative treatment in other brain injury disorders. Treatment with G-1 improved neuronal survival after brain ischemia, reduced infarct size, neuronal injury and improved neuroinflammation and immunosuppression after experimentally induced stroke and cerebral ischemia (104, 111, 112). Furthermore, treatment with other non-classical pathway activators, such as PaPE-1 and the SERM bazedoxifene, protected neurons against ischemic brain damage in rodents and in neuronal culture, potentially through the MAPK/ERK1/2 signaling pathway (113, 114).
Neuroinflammation can play a key role in the secondary injury observed in TBI as well as after stroke with the activation of microglia cells, among others, and the release of inflammatory factors (115–117). Following TBI, G-1 exerts anti-inflammatory effects, but it appears that there are sex specific differences as these results were observed in males and ovariectomized females, but not in intact females. Therefore, the circulating levels of E2 in patients will likely influence any potential medical treatment following brain injury. In addition to G-1, STX has also been found to be capable of attenuating ischemia-induced neuronal loss in middle-aged rats (30). Importantly, this study showed that animals which have not been exposed to E2 for some time still maintained their responsiveness to E2 and E2-like compounds as treatment, highlighting the use of non-feminizing estrogens, that can be candidates in both males and females and at different age groups. Taken together, these results strongly suggest that the non-classical pathway can be targeted as potential treatment in traumatic and non-traumatic brain injury disorders.
Conclusions
In this mini-review, we discussed the neuroprotective role of E2 and the potential involvement of the non-classical estrogen pathway in ameliorating or alleviating disease phenotype in experimental models of AD, PD and brain injury disorders. The results from in vivo and in vitro studies with selective non-classical pathway activators, such as raloxifene, estren, STX, G-1, PaPE-1 and DHED, are very promising targets and present hopeful beneficial effects on their potential use as treatment in neurodegenerative diseases. However, as both the classical and non-classical pathways are intact in most, if not all, of these studies, it is difficult to ascertain whether the observed neuroprotective effects of E2 are solely attributed to the non-classical pathway. Some of the ongoing challenges with these selective non-classical pathway activators include how to modulate selectivity and sensitivity to ensure that the non-classical pathway is stimulated without triggering the classical pathway. Extra caution also needs to be taken in their interpretation as, at present, there is a lack of conclusive evidence for their use in the human brain. More studies are warranted to translate these neuroprotective effects in human clinical trials before they can be utilized as a novel therapeutic strategy to ameliorate, prevent the onset and/or slow down disease progression in neurodegenerative diseases.
Author contributions
Both ZK and RC developed the concept and wrote the manuscript. Both authors have made a substantial, direct and intellectual contribution to the work and approved the manuscript prior to its submission.
Conflict of interest
RC was employed by Timeline Bioresearch AB.
The remaining author declares that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. McEwen B. Estrogen actions throughout the brain. Recent Prog Horm Res (2002) 57:357–84. doi: 10.1210/rp.57.1.357
2. McEwen BS, Alves SE. Estrogen actions in the central nervous system. Endocr Rev (1999) 20(3):279–307. doi: 10.1210/edrv.20.3.0365
3. Dluzen DE, McDermott JL. Gender differences in neurotoxicity of the nigrostriatal dopaminergic system: Implications for Parkinson's disease. J Gend Specif Med (2000) 3(6):36–42.
4. Baum LW. Sex, hormones, and Alzheimer's disease. J Gerontol A Biol Sci Med Sci (2005) 60(6):736–43. doi: 10.1093/gerona/60.6.736
5. Zhu D, Montagne A, Zhao Z. Alzheimer’s pathogenic mechanisms and underlying sex difference. Cell Mol Life Sci (2021) 78(11):4907–20. doi: 10.1007/s00018-021-03830-w
6. Yaşar P, Ayaz G, User SD, Güpür G, Muyan M. Molecular mechanism of estrogen-estrogen receptor signaling. Reprod Med Biol (2017) 16(1):4–20. doi: 10.1002/rmb2.12006
7. Szego CM, Davis JS. Adenosine 3',5'-monophosphate in rat uterus: Acute elevation by estrogen. Proc Natl Acad Sci U S A (1967) 58(4):1711–8. doi: 10.1073/pnas.58.4.1711
8. Szego EM, Barabás K, Balog J, Szilágyi N, Korach KS, Juhász G, et al. Estrogen induces estrogen receptor α-dependent camp response element-binding protein phosphorylation via mitogen activated protein kinase pathway in basal forebrain cholinergic neurons in vivo. J Neurosci (2006) 26(15):4104–10. doi: 10.1523/jneurosci.0222-06.2006
9. Wade CB, Dorsa DM. Estrogen activation of cyclic adenosine 5'-monophosphate response element-mediated transcription requires the extracellularly regulated kinase/mitogen-activated protein kinase pathway. Endocrinology (2003) 144(3):832–8. doi: 10.1210/en.2002-220899
10. Cheong RY, Kwakowsky A, Barad Z, Porteous R, Herbison AE, Ábrahám IM. Estradiol acts directly and indirectly on multiple signaling pathways to phosphorylate cAMP-response element binding protein in GnRH neurons. Endocrinology (2012) 153(8):3792–803. doi: 10.1210/en.2012-1232
11. Prossnitz ER, Barton M. The G-protein-coupled estrogen receptor GPER in health and disease. Nat Rev Endocrinol (2011) 7(12):715–26. doi: 10.1038/nrendo.2011.122
12. Toran-Allerand CD, Guan X, MacLusky NJ, Horvath TL, Diano S, Singh M, et al. ER-X: A novel, plasma membrane-associated, putative estrogen receptor that is regulated during development and after ischemic brain injury. J Neurosci (2002) 22(19):8391–401. doi: 10.1523/jneurosci.22-19-08391.2002
13. Qiu J, Bosch MA, Tobias SC, Grandy DK, Scanlan TS, Ronnekleiv OK, et al. Rapid signaling of estrogen in hypothalamic neurons involves a novel G-protein-coupled estrogen receptor that activates protein kinase c. J Neurosci (2003) 23(29):9529–40. doi: 10.1523/jneurosci.23-29-09529.2003
14. Khaksari M, Abbasloo E, Dehghan F, Soltani Z, Asadikaram G. The brain cytokine levels are modulated by estrogen following traumatic brain injury: Which estrogen receptor serves as modulator? Int Immunopharmacol (2015) 28(1):279–87. doi: 10.1016/j.intimp.2015.05.046
15. Cerciat M, Unkila M, Garcia-Segura LM, Arevalo MA. Selective estrogen receptor modulators decrease the production of interleukin-6 and interferon-gamma-inducible protein-10 by astrocytes exposed to inflammatory challenge in vitro. Glia (2010) 58(1):93–102. doi: 10.1002/glia.20904
16. Xiao Q, Luo Y, Lv F, He Q, Wu H, Chao F, et al. Protective effects of 17β-estradiol on hippocampal myelinated fibers in ovariectomized middle-aged rats. Neuroscience (2018) 385:143–53. doi: 10.1016/j.neuroscience.2018.06.006
17. He Q, Luo Y, Lv F, Xiao Q, Chao F, Qiu X, et al. Effects of estrogen replacement therapy on the myelin sheath ultrastructure of myelinated fibers in the white matter of middle-aged ovariectomized rats. J Comp Neurol (2018) 526(5):790–802. doi: 10.1002/cne.24366
18. Kumar S, Patel R, Moore S, Crawford DK, Suwanna N, Mangiardi M, et al. Estrogen receptor β ligand therapy activates PI3k/Akt/mTOR signaling in oligodendrocytes and promotes remyelination in a mouse model of multiple sclerosis. Neurobiol Dis (2013) 56:131–44. doi: 10.1016/j.nbd.2013.04.005
19. Feng J, Zhang G, Hu X, Si Chen C, Qin X. Estrogen inhibits estrogen receptor α-mediated Rho-kinase expression in experimental autoimmune encephalomyelitis rats. Synapse (2013) 67(7):399–406. doi: 10.1002/syn.21650
20. Khan MM, Wakade C, de Sevilla L, Brann DW. Selective estrogen receptor modulators (SERMS) enhance neurogenesis and spine density following focal cerebral ischemia. J Steroid Biochem Mol Biol (2015) 146:38–47. doi: 10.1016/j.jsbmb.2014.05.001
21. Saraceno GE, Bellini MJ, Garcia-Segura LM, Capani F. Estradiol activates PI3k/Akt/GSK3 pathway under chronic neurodegenerative conditions triggered by perinatal asphyxia. Front Pharmacol (2018) 9:335. doi: 10.3389/fphar.2018.00335
22. Rettberg JR, Yao J, Brinton RD. Estrogen: A master regulator of bioenergetic systems in the brain and body. Front Neuroendocrinol (2014) 35(1):8–30. doi: 10.1016/j.yfrne.2013.08.001
23. Simpkins JW, Yi KD, Yang S-H, Dykens JA. Mitochondrial mechanisms of estrogen neuroprotection. BBA-GEN Subj (2010) 1800(10):1113–20. doi: 10.1016/j.bbagen.2009.11.013
24. Yuan LJ, Wang XW, Wang HT, Zhang M, Sun JW, Chen WF. G protein-coupled estrogen receptor is involved in the neuroprotective effect of IGF-1 against MPTP/MPP(+)-induced dopaminergic neuronal injury. J Steroid Biochem Mol Biol (2019) 192:105384. doi: 10.1016/j.jsbmb.2019.105384
25. Arevalo MA, Azcoitia I, Garcia-Segura LM. The neuroprotective actions of oestradiol and oestrogen receptors. Nat Rev Neurosci (2015) 16(1):17–29. doi: 10.1038/nrn3856
26. Anderson GL, Limacher M, Assaf AR, Bassford T, Beresford SA, Black H, et al. Effects of conjugated equine estrogen in postmenopausal women with hysterectomy: The women's health initiative randomized controlled trial. Jama (2004) 291(14):1701–12. doi: 10.1001/jama.291.14.1701
27. Chlebowski RT, Anderson GL, Aragaki AK, Manson JE, Stefanick ML, Pan K, et al. Association of menopausal hormone therapy with breast cancer incidence and mortality during long-term follow-up of the women's health initiative randomized clinical trials. Jama (2020) 324(4):369–80. doi: 10.1001/jama.2020.9482
28. Rossouw JE. Prescribing postmenopausal hormone therapy to women in their 50s in the post-women's health initiative era. Maturitas (2010) 65(3):179–80. doi: 10.1016/j.maturitas.2009.11.012
29. Maximov PY, Lee TM, Jordan VC. The discovery and development of selective estrogen receptor modulators (SERMS) for clinical practice. Curr Clin Pharmacol (2013) 8(2):135–55. doi: 10.2174/1574884711308020006
30. Lebesgue D, Traub M, De Butte-Smith M, Chen C, Zukin RS, Kelly MJ, et al. Acute administration of non-classical estrogen receptor agonists attenuates ischemia-induced hippocampal neuron loss in middle-aged female rats. PloS One (2010) 5(1):e8642. doi: 10.1371/journal.pone.0008642
31. Amirkhosravi L, Khaksari M, Soltani Z, Esmaeili-Mahani S, Asadi Karam G, Hoseini M. E2-BSA and G1 exert neuroprotective effects and improve behavioral abnormalities following traumatic brain injury: The role of classic and non-classic estrogen receptors. Brain Res (2021) 1750:147168. doi: 10.1016/j.brainres.2020.147168
32. Liu JH. Selective estrogen receptor modulators (SERMS): Keys to understanding their function. Menopause (2020) 27(10):1171–6. doi: 10.1097/gme.0000000000001585
33. Wessler S, Otto C, Wilck N, Stangl V, Fritzemeier KH. Identification of estrogen receptor ligands leading to activation of non-genomic signaling pathways while exhibiting only weak transcriptional activity. J Steroid Biochem Mol Biol (2006) 98(1):25–35. doi: 10.1016/j.jsbmb.2005.08.003
34. Kousteni S, Bellido T, Plotkin LI, O'Brien CA, Bodenner DL, Han L, et al. Nongenotropic, sex-nonspecific signaling through the estrogen or androgen receptors: Dissociation from transcriptional activity. Cell (2001) 104(5):719–30. doi: 10.1016/S0092-8674(01)00268-9
35. Koszegi Z, Szego EM, Cheong RY, Tolod-Kemp E, Abraham IM. Postlesion estradiol treatment increases cortical cholinergic innervations via estrogen receptor-α dependent nonclassical estrogen signaling in vivo. Endocrinology (2011) 152(9):3471–82. doi: 10.1210/en.2011-1017
36. Kwakowsky A, Potapov K, Kim S, Peppercorn K, Tate WP, Ábrahám IM. Treatment of β amyloid 1-42 (Aβ(1-42))-induced basal forebrain cholinergic damage by a non-classical estrogen signaling activator in vivo. Sci Rep (2016) 6:21101. doi: 10.1038/srep21101
37. Li R, He P, Cui J, Staufenbiel M, Harada N, Shen Y. Brain endogenous estrogen levels determine responses to estrogen replacement therapy via regulation of BACE1 and NEP in female Alzheimer's transgenic mice. Mol Neurobiol (2013) 47(3):857–67. doi: 10.1007/s12035-012-8377-3
38. Xu H, Wang R, Zhang YW, Zhang X. Estrogen, β-amyloid Metabolism/Trafficking, and Alzheimer's disease. Ann N Y Acad Sci (2006) 1089:324–42. doi: 10.1196/annals.1386.036
39. Yue X, Lu M, Lancaster T, Cao P, Honda S, Staufenbiel M, et al. Brain estrogen deficiency accelerates Aβ plaque formation in an Alzheimer's disease animal model. Proc Natl Acad Sci U S A (2005) 102(52):19198–203. doi: 10.1073/pnas.0505203102
40. Jayaraman A, Carroll JC, Morgan TE, Lin S, Zhao L, Arimoto JM, et al. 17β-estradiol and progesterone regulate expression of β-amyloid clearance factors in primary neuron cultures and female rat brain. Endocrinology (2012) 153(11):5467–79. doi: 10.1210/en.2012-1464
41. Xu H, Gouras GK, Greenfield JP, Vincent B, Naslund J, Mazzarelli L, et al. Estrogen reduces neuronal generation of Alzheimer β-amyloid peptides. Nat Med (1998) 4(4):447–51. doi: 10.1038/nm0498-447
42. Petanceska SS, Nagy V, Frail D, Gandy S. Ovariectomy and 17β-estradiol modulate the levels of Alzheimer's amyloid β peptides in brain. Exp Gerontol (2000) 35(9-10):1317–25. doi: 10.1016/s0531-5565(00)00157-1
43. Kim JY, Mo H, Kim J, Kim JW, Nam Y, Rim YA, et al. Mitigating effect of estrogen in Alzheimer's disease-mimicking cerebral organoid. Front Neurosci (2022) 16:816174. doi: 10.3389/fnins.2022.816174
44. Zhang S, Huang Y, Zhu YC, Yao T. Estrogen stimulates release of secreted amyloid precursor protein from primary rat cortical neurons via protein kinase C pathway. Acta Pharmacol Sin (2005) 26(2):171–6. doi: 10.1111/j.1745-7254.2005.00538.x
45. Manthey D, Heck S, Engert S, Behl C. Estrogen induces a rapid secretion of amyloid β precursor protein via the mitogen-activated protein kinase pathway. Eur J Biochem (2001) 268(15):4285–91. doi: 10.1046/j.1432-1327.2001.02346.x
46. Fitzpatrick JL, Mize AL, Wade CB, Harris JA, Shapiro RA, Dorsa DM. Estrogen-mediated neuroprotection against β-amyloid toxicity requires expression of estrogen receptor α or β and activation of the MAPK pathway. J Neurochem (2002) 82(3):674–82. doi: 10.1046/j.1471-4159.2002.01000.x
47. Liu XA, Zhu LQ, Zhang Q, Shi HR, Wang SH, Wang Q, et al. Estradiol attenuates tau hyperphosphorylation induced by upregulation of protein kinase-A. Neurochem Res (2008) 33(9):1811–20. doi: 10.1007/s11064-008-9638-4
48. Zhang QG, Wang R, Khan M, Mahesh V, Brann DW. Role of dickkopf-1, an antagonist of the WNT/β-catenin signaling pathway, in estrogen-induced neuroprotection and attenuation of tau phosphorylation. J Neurosci (2008) 28(34):8430–41. doi: 10.1523/JNEUROSCI.2752-08.2008
49. Shughrue PJ, Scrimo PJ, Merchenthaler I. Estrogen binding and estrogen receptor characterization (ER α and ER β) in the cholinergic neurons of the rat basal forebrain. Neuroscience (2000) 96:41–9. doi: 10.1016/S0306-4522(99)00520-5
50. Miettinen RA, Kalesnykas G, Koivisto EH. Estimation of the total number of cholinergic neurons containing estrogen receptor-α in the rat basal forebrain. J Histochem Cytochem (2002) 50(7):891–902. doi: 10.1177/002215540205000703
51. Pongrac JL, Gibbs RB, Defranco DB. Estrogen-mediated regulation of cholinergic expression in basal forebrain neurons requires extracellular-signal-regulated kinase activity. Neuroscience (2004) 124(4):809–16. doi: 10.1016/j.neuroscience.2004.01.013
52. Brinton RD. Estrogen-induced plasticity from cells to circuits: Predictions for cognitive function. Trends Pharmacol Sci (2009) 30(4):212–22. doi: 10.1016/j.tips.2008.12.006
53. Hojo Y, Munetomo A, Mukai H, Ikeda M, Sato R, Hatanaka Y, et al. Estradiol rapidly modulates spinogenesis in hippocampal dentate gyrus: Involvement of kinase networks. Hormones Behav (2015) 74:149–56. doi: 10.1016/j.yhbeh.2015.06.008
54. Sheppard PAS, Choleris E, Galea LAM. Structural plasticity of the hippocampus in response to estrogens in female rodents. Mol Brain (2019) 12(1):22. doi: 10.1186/s13041-019-0442-7
55. Carroll JC, Pike CJ. Selective estrogen receptor modulators differentially regulate Alzheimer-like changes in female 3xTg-AD mice. Endocrinology (2008) 149(5):2607–11. doi: 10.1210/en.2007-1346
56. Lai YJ, Zhu BL, Sun F, Luo D, Ma YL, Luo B, et al. Estrogen receptor α promotes Cav1.2 ubiquitination and degradation in neuronal cells and in APP/PS1 mice. Aging Cell (2019) 18(4):e12961. doi: 10.1111/acel.12961
57. Tschiffely AE, Schuh RA, Prokai-Tatrai K, Prokai L, Ottinger MA. A comparative evaluation of treatments with 17β-estradiol and its brain-selective prodrug in a double-transgenic mouse model of Alzheimer's disease. Hormones Behav (2016) 83:39–44. doi: 10.1016/j.yhbeh.2016.05.009
58. Cordey M, Gundimeda U, Gopalakrishna R, Pike CJ. The synthetic estrogen 4-Estren-3 α,17 β-diol (Estren) induces estrogen-like neuroprotection. Neurobiol Dis (2005) 19(1-2):331–9. doi: 10.1016/j.nbd.2005.01.011
59. Kwakowsky A, Koszegi Z, Cheong RY, Abraham IM. Neuroprotective effects of non-classical estrogen-like signaling activators: From mechanism to potential implications. CNS Neurol Disord Drug Targets (2013) 12(8):1219–25. doi: 10.2174/187152731131200123
60. Fernandez SM, Lewis MC, Pechenino AS, Harburger LL, Orr PT, Gresack JE, et al. Estradiol-induced enhancement of object memory consolidation involves hippocampal extracellular signal-regulated kinase activation and membrane-bound estrogen receptors. J Neurosci (2008) 28(35):8660–7. doi: 10.1523/jneurosci.1968-08.2008
61. Zhao Z, Fan L, Frick KM. Epigenetic alterations regulate estradiol-induced enhancement of memory consolidation. Proc Natl Acad Sci U.S.A. (2010) 107(12):5605–10. doi: 10.1073/pnas.0910578107
62. Fortress AM, Fan L, Orr PT, Zhao Z, Frick KM. Estradiol-induced object recognition memory consolidation is dependent on activation of mTOR signaling in the dorsal hippocampus. Learn Mem (2013) 20(3):147–55. doi: 10.1101/lm.026732.112
63. Srivastava DP, Woolfrey KM, Penzes P. Insights into rapid modulation of neuroplasticity by brain estrogens. Pharmacolo Rev (2013) 65(4):1318–50. doi: 10.1124/pr.111.005272
64. Fan L, Zhao Z, Orr PT, Chambers CH, Lewis MC, Frick KM. Estradiol-induced object memory consolidation in middle-aged female mice requires dorsal hippocampal extracellular signal-regulated kinase and phosphatidylinositol 3-kinase activation. J Neurosci (2010) 30(12):4390–400. doi: 10.1523/jneurosci.4333-09.2010
65. Jacobsen DE, Samson MM, Emmelot-Vonk MH, Verhaar HJ. Raloxifene improves verbal memory in late postmenopausal women: A randomized, double-blind, placebo-controlled trial. Menopause (2010) 17(2):309–14. doi: 10.1097/gme.0b013e3181bd54df
66. Yaffe K, Krueger K, Cummings SR, Blackwell T, Henderson VW, Sarkar S, et al. Effect of raloxifene on prevention of dementia and cognitive impairment in older women: The multiple outcomes of raloxifene evaluation (MORE) randomized trial. Am J Psychiatry (2005) 162(4):683–90. doi: 10.1176/appi.ajp.162.4.683
67. Nickelsen T, Lufkin EG, Riggs BL, Cox DA, Crook TH. Raloxifene hydrochloride, a selective estrogen receptor modulator: Safety assessment of effects on cognitive function and mood in postmenopausal women. Psychoneuroendocrinology (1999) 24(1):115–28. doi: 10.1016/s0306-4530(98)00041-9
68. Kubota T, Matsumoto H, Kirino Y. Ameliorative effect of membrane-associated estrogen receptor G protein coupled receptor 30 activation on object recognition memory in mouse models of Alzheimer's disease. J Pharmacol Sci (2016) 131(3):219–22. doi: 10.1016/j.jphs.2016.06.005
69. Gray NE, Zweig JA, Kawamoto C, Quinn JF, Copenhaver PF. STX, a novel membrane estrogen receptor ligand, protects against amyloid-β toxicity. J Alzheimers Dis (2016) 51(2):391–403. doi: 10.3233/JAD-150756
70. Deng LJ, Cheng C, Wu J, Wang CH, Zhou HB, Huang J. Oxabicycloheptene sulfonate protects against β-Amyloid-Induced toxicity by activation of PI3k/Akt and ERK signaling pathways via GPER1 in C6 cells. Neurochem Res (2017) 42(8):2246–56. doi: 10.1007/s11064-017-2237-5
71. Wnuk A, Przepiorska K, Rzemieniec J, Pietrzak B, Kajta M. Selective targeting of non-nuclear estrogen receptors with PaPE-1 as a new treatment strategy for Alzheimer's disease. Neurotox Res (2020) 38(4):957–66. doi: 10.1007/s12640-020-00289-8
72. Selvaraj UM, Zuurbier KR, Whoolery CW, Plautz EJ, Chambliss KL, Kong X, et al. Selective nonnuclear estrogen receptor activation decreases stroke severity and promotes functional recovery in female mice. Endocrinology (2018) 159(11):3848–59. doi: 10.1210/en.2018-00600
73. Stocchi F, Olanow CW. Obstacles to the development of a neuroprotective therapy for Parkinson's disease. Mov Disord (2013) 28(1):3–7. doi: 10.1002/mds.25337
74. Cotzias GC, Papavasiliou PS, Gellene R. L-DOPA in Parkinson's syndrome. N Engl J Med (1969) 281(5):272. doi: 10.1056/NEJM196907312810518
75. Olanow CW, Stocchi F. Levodopa: A new look at an old friend. Mov Disord (2018) 33(6):859–66. doi: 10.1002/mds.27216
76. Saunders-Pullman R, Gordon-Elliott J, Parides M, Fahn S, Saunders HR, Bressman S. The effect of estrogen replacement on early Parkinson's disease. Neurology (1999) 52(7):1417–21. doi: 10.1212/wnl.52.7.1417
77. Song YJ, Li SR, Li XW, Chen X, Wei ZX, Liu QS, et al. The effect of estrogen replacement therapy on Alzheimer's disease and Parkinson's disease in postmenopausal women: A meta-analysis. Front Neurosci (2020) 14:157. doi: 10.3389/fnins.2020.00157
78. Callier S, Le Saux M, Lhiaubet AM, Di Paolo T, Rostene W, Pelaprat D. Evaluation of the protective effect of oestradiol against toxicity induced by 6-hydroxydopamine and 1-Methyl-4-Phenylpyridinium ion (MPP+) towards dopaminergic mesencephalic neurones in primary culture. J Neurochem (2002) 80(2):307–16. doi: 10.1046/j.0022-3042.2001.00693.x
79. Becker JB, Rudick CN. Rapid effects of estrogen or progesterone on the amphetamine-induced increase in striatal dopamine are enhanced by estrogen priming: A microdialysis study. Pharmacol Biochem Behav (1999) 64(1):53–7. doi: 10.1016/s0091-3057(99)00091-x
80. Sawada H, Ibi M, Kihara T, Honda K, Nakamizo T, Kanki R, et al. Estradiol protects dopaminergic neurons in a MPP+Parkinson's disease model. Neuropharmacology (2002) 42(8):1056–64. doi: 10.1016/s0028-3908(02)00049-7
81. Hwang CJ, Choi DY, Jung YY, Lee YJ, Yun JS, Oh KW, et al. Inhibition of P38 pathway-dependent MPTP-induced dopaminergic neurodegeneration in estrogen receptor α knockout mice. Horm Behav (2016) 80:19–29. doi: 10.1016/j.yhbeh.2016.01.011
82. Yoest KE, Cummings JA, Becker JB. Oestradiol influences on dopamine release from the nucleus accumbens shell: Sex differences and the role of selective oestradiol receptor subtypes. Br J Pharmacol (2019) 176(21):4136–48. doi: 10.1111/bph.14531
83. Liu B, Dluzen DE. Oestrogen and nigrostriatal dopaminergic neurodegeneration: Animal models and clinical reports of Parkinson's disease. Clin Exp Pharmacol Physiol (2007) 34(7):555–65. doi: 10.1111/j.1440-1681.2007.04616.x
84. Gillies GE, McArthur S. Estrogen actions in the brain and the basis for differential action in men and women: A case for sex-specific medicines. Pharmacol Rev (2010) 62(2):155–98. doi: 10.1124/pr.109.002071
85. Gillies GE, McArthur S. Independent influences of sex steroids of systemic and central origin in a rat model of Parkinson's disease: A contribution to sex-specific neuroprotection by estrogens. Horm Behav (2010) 57(1):23–34. doi: 10.1016/j.yhbeh.2009.06.002
86. D'Astous M, Morissette M, Di Paolo T. Effect of estrogen receptor agonists treatment in MPTP mice: Evidence of neuroprotection by an ER α agonist. Neuropharmacology (2004) 47(8):1180–8. doi: 10.1016/j.neuropharm.2004.08.020
87. Baraka AM, Korish AA, Soliman GA, Kamal H. The possible role of estrogen and selective estrogen receptor modulators in a rat model of Parkinson's disease. Life Sci (2011) 88(19-20):879–85. doi: 10.1016/j.lfs.2011.03.010
88. Yadav SK, Pandey S, Singh B. Role of estrogen and levodopa in 1-Methyl-4-Pheny-L-1, 2, 3, 6-tetrahydropyridine (MPTP)-induced cognitive deficit in parkinsonian ovariectomized mice model: A comparative study. J Chem Neuroanat (2017) 85:50–9. doi: 10.1016/j.jchemneu.2017.07.002
89. Callier S, Morissette M, Grandbois M, Pélaprat D, Di Paolo T. Neuroprotective properties of 17β-estradiol, progesterone, and raloxifene in MPTP C57BL/6 mice. Synapse (2001) 41(2):131–8. doi: 10.1002/syn.1067
90. Bourque M, Morissette M, Di Paolo T. Raloxifene activates G protein-coupled estrogen receptor 1/Akt signaling to protect dopamine neurons in 1-Methyl-4-Phenyl-1,2,3,6-Tetrahydropyridine mice. Neurobiol Aging (2014) 35(10):2347–56. doi: 10.1016/j.neurobiolaging.2014.03.017
91. Ramirez AD, Liu X, Menniti FS. Repeated estradiol treatment prevents MPTP-induced dopamine depletion in male mice. Neuroendocrinology (2003) 77(4):223–31. doi: 10.1159/000070277
92. Rajsombath MM, Nam AY, Ericsson M, Nuber S. Female sex and brain-selective estrogen benefit α-synuclein tetramerization and the PD-like motor syndrome in 3K transgenic mice. J Neurosci (2019) 39(38):7628–40. doi: 10.1523/JNEUROSCI.0313-19.2019
93. Thadathil N, Xiao J, Hori R, Alway SE, Khan MM. Brain selective estrogen treatment protects dopaminergic neurons and preserves behavioral function in MPTP-induced mouse model of Parkinson's disease. J Neuroimmune Pharmacol (2021) 16(3):667–78. doi: 10.1007/s11481-020-09972-1
94. Prokai L, Nguyen V, Szarka S, Garg P, Sabnis G, Bimonte-Nelson HA, et al. The prodrug DHED selectively delivers 17beta-estradiol to the brain for treating estrogen-responsive disorders. Sci Transl Med (2015) 7(297):297ra113. doi: 10.1126/scitranslmed.aab1290
95. Graham NS, Sharp DJ. Understanding neurodegeneration after traumatic brain injury: From mechanisms to clinical trials in dementia. J Neurol Neurosurg Psychiatry (2019) 90(11):1221–33. doi: 10.1136/jnnp-2017-317557
96. Maas AIR, Stocchetti N, Bullock R. Moderate and severe traumatic brain injury in adults. Lancet Neurol (2008) 7(8):728–41. doi: 10.1016/S1474-4422(08)70164-9
97. Feigin VL, Stark BA, Johnson CO, Roth GA, Bisignano C, Abady GG, et al. Global, regional, and national burden of stroke and its risk factors, 1990–2019: A systematic analysis for the global burden of disease study 2019. Lancet Neurol (2021) 20(10):795–820. doi: 10.1016/S1474-4422(21)00252-0
98. Armstead WM, Kiessling JW, Kofke WA, Vavilala MS. Impaired cerebral blood flow autoregulation during posttraumatic arterial hypotension after fluid percussion brain injury is prevented by phenylephrine in female but exacerbated in male piglets by extracellular signal-related kinase mitogen-activated protein kinase upregulation. Crit Care Med (2010) 38(9):1868–74. doi: 10.1097/CCM.0b013e3181e8ac1a
99. Groswasser Z, Cohen M, Keren O. Female TBI patients recover better than males. Brain Inj (1998) 12(9):805–8. doi: 10.1080/026990598122197
100. Khaksari M, Soltani Z, Shahrokhi N. Effects of female sex steroids administration on pathophysiologic mechanisms in traumatic brain injury. Transl Stroke Res (2018) 9(4):393–416. doi: 10.1007/s12975-017-0588-5
101. Saldanha CJ, Duncan KA, Walters BJ. Neuroprotective actions of brain aromatase. Front Neuroendocrinol (2009) 30(2):106–18. doi: 10.1016/j.yfrne.2009.04.016
102. Garcia-Segura LM, Veiga S, Sierra A, Melcangi RC, Azcoitia I. Aromatase: A neuroprotective enzyme. Prog Neurobiol (2003) 71(1):31–41. doi: 10.1016/j.pneurobio.2003.09.005
103. Lu H, Ma K, Jin L, Zhu H, Cao R. 17β-estradiol rescues damages following traumatic brain injury from molecule to behavior in mice. J Cell Physiol (2018) 233(2):1712–22. doi: 10.1002/jcp.26083
104. Zhang B, Subramanian S, Dziennis S, Jia J, Uchida M, Akiyoshi K, et al. Estradiol and G1 reduce infarct size and improve immunosuppression after experimental stroke. J Immunol (2010) 184(8):4087–94. doi: 10.4049/jimmunol.0902339
105. Khatri N, Thakur M, Pareek V, Kumar S, Sharma S, Datusalia AK. Oxidative stress: Major threat in traumatic brain injury. CNS Neurol Disord Drug Targets (2018) 17(9):689–95. doi: 10.2174/1871527317666180627120501
106. Naderi V, Khaksari M, Abbasi R, Maghool F. Estrogen provides neuroprotection against brain edema and blood brain barrier disruption through both estrogen receptors α and β following traumatic brain injury. Iran J Basic Med Sci (2015) 18(2):138–44. doi: 10.22038/IJBMS.2015.4015
107. Khaksari M, Hajializadeh Z, Shahrokhi N, Esmaeili-Mahani S. Changes in the gene expression of estrogen receptors involved in the protective effect of estrogen in rat׳S trumatic brain injury. Brain Res (2015) 1618:1–8. doi: 10.1016/j.brainres.2015.05.017
108. Day NL, Floyd CL, D'Alessandro TL, Hubbard WJ, Chaudry IH. 17β-estradiol confers protection after traumatic brain injury in the rat and involves activation of G protein-coupled estrogen receptor 1. J Neurotrauma (2013) 30(17):1531–41. doi: 10.1089/neu.2013.2854
109. Wang ZF, Pan ZY, Xu CS, Li ZQ. Activation of G-protein coupled estrogen receptor 1 improves early-onset cognitive impairment via PI3k/Akt pathway in rats with traumatic brain injury. Biochem Biophys Res Commun (2017) 482(4):948–53. doi: 10.1016/j.bbrc.2016.11.138
110. Pan MX, Tang JC, Liu R, Feng YG, Wan Q. Effects of estrogen receptor GPR30 agonist G1 on neuronal apoptosis and microglia polarization in traumatic brain injury rats. Chin J Traumatol (2018) 21(4):224–8. doi: 10.1016/j.cjtee.2018.04.003
111. Kosaka Y, Quillinan N, Bond C, Traystman R, Hurn P, Herson P. GPER1/GPR30 activation improves neuronal survival following global cerebral ischemia induced by cardiac arrest in mice. Transl Stroke Res (2012) 3(4):500–7. doi: 10.1007/s12975-012-0211-8
112. Wang XS, Yue J, Hu LN, Tian Z, Zhang K, Yang L, et al. Activation of G protein-coupled receptor 30 protects neurons by regulating autophagy in astrocytes. Glia (2020) 68(1):27–43. doi: 10.1002/glia.23697
113. Wnuk A, Przepiórska K, Pietrzak BA, Kajta M. Posttreatment strategy against hypoxia and ischemia based on selective targeting of nonnuclear estrogen receptors with PaPE-1. Neurotox Res (2021) 39(6):2029–41. doi: 10.1007/s12640-021-00441-y
114. Jover-Mengual T, Castelló-Ruiz M, Burguete MC, Jorques M, López-Morales MA, Aliena-Valero A, et al. Molecular mechanisms mediating the neuroprotective role of the selective estrogen receptor modulator, bazedoxifene, in acute ischemic stroke: A comparative study with 17β-estradiol. J Steroid Biochem Mol Biol (2017) 171:296–304. doi: 10.1016/j.jsbmb.2017.05.001
115. Lozano D, Gonzales-Portillo GS, Acosta S, de la Pena I, Tajiri N, Kaneko Y, et al. Neuroinflammatory responses to traumatic brain injury: Etiology, clinical consequences, and therapeutic opportunities. Neuropsychiatr Dis Treat (2015) 11:97–106. doi: 10.2147/ndt.S65815
116. Witcher KG, Bray CE, Chunchai T, Zhao F, O'Neil SM, Gordillo AJ, et al. Traumatic brain injury causes chronic cortical inflammation and neuronal dysfunction mediated by microglia. J Neurosci (2021) 41(7):1597–616. doi: 10.1523/jneurosci.2469-20.2020
Keywords: estrogen, non-classical, non-genomic, neurodegeneration, neuroprotection
Citation: Koszegi Z and Cheong RY (2022) Targeting the non-classical estrogen pathway in neurodegenerative diseases and brain injury disorders. Front. Endocrinol. 13:999236. doi: 10.3389/fendo.2022.999236
Received: 20 July 2022; Accepted: 15 August 2022;
Published: 15 September 2022.
Edited by:
Andrea Kwakowsky, National University of Ireland Galway, IrelandReviewed by:
Kelli A. Duncan, Vassar College, United StatesÉva M. Szegő, Technical University of Dresden, Germany
Copyright © 2022 Koszegi and Cheong. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Rachel Y. Cheong, cnkuY2hlb25nQGdtYWlsLmNvbQ==