
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Endocrinol. , 30 September 2022
Sec. Cellular Endocrinology
Volume 13 - 2022 | https://doi.org/10.3389/fendo.2022.997304
This article is part of the Research Topic Sexual Dimorphisms on Central and Peripheral Control of Energy Balance View all 5 articles
 Elena V. Kozlova1,2†
Elena V. Kozlova1,2† Maximillian E. Denys1†Jonathan Benedum1
Maximillian E. Denys1†Jonathan Benedum1 Matthew C. Valdez1Dave Enriquez1
Matthew C. Valdez1Dave Enriquez1 Anthony E. Bishay1†Bhuvaneswari D. Chinthirla1
Anthony E. Bishay1†Bhuvaneswari D. Chinthirla1 Edward Truong1Julia M. Krum1Nicholas V. DiPatrizio3
Edward Truong1Julia M. Krum1Nicholas V. DiPatrizio3 Poonamjot Deol1
Poonamjot Deol1 Manuela Martins-Green1Margarita C. Curras-Collazo1*†
Manuela Martins-Green1Margarita C. Curras-Collazo1*†Polybrominated diphenyl ethers (PBDEs) are a class of flame-retardant organohalogen pollutants that act as endocrine/neuroendocrine disrupting chemicals (EDCs). In humans, exposure to brominated flame retardants (BFR) or other environmentally persistent organic pollutants (POPs) such as polychlorinated biphenyls (PCBs) and novel organophosphate flame retardants has been associated with increasing trends of diabetes and metabolic disease. However, the effects of PBDEs on metabolic processes and their associated sex-dependent features are poorly understood. The metabolic-disrupting effects of perinatal exposure to industrial penta-PBDE mixture, DE-71, on male and female progeny of C57BL/6N mouse dams were examined in adulthood. Dams were exposed to environmentally relevant doses of PBDEs daily for 10 weeks (p.o.): 0.1 (L-DE-71) and 0.4 mg/kg/d (H-DE-71) and offspring parameters were compared to corn oil vehicle controls (VEH/CON). The following lipid metabolism indices were measured: plasma cholesterol, triglycerides, adiponectin, leptin, and liver lipids. L-DE-71 female offspring were particularly affected, showing hypercholesterolemia, elevated liver lipids and fasting plasma leptin as compared to same-sex VEH/CON, while L- and H-DE-71 male F1 only showed reduced plasma adiponectin. Using the quantitative Folch method, we found that mean liver lipid content was significantly elevated in L-DE-71 female offspring compared to controls. Oil Red O staining revealed fatty liver in female offspring and dams. General measures of adiposity, body weight, white and brown adipose tissue (BAT), and lean and fat mass were weighed or measured using EchoMRI. DE-71 did not produce abnormal adiposity, but decreased BAT depots in L-DE-71 females and males relative to same-sex VEH/CON. To begin to address potential central mechanisms of deregulated lipid metabolism, we used RT-qPCR to quantitate expression of hypothalamic genes in energy-regulating circuits that control lipid homeostasis. Both doses of DE-71 sex-dependently downregulated hypothalamic expression of Lepr, Stat3, Mc4r, Agrp, Gshr in female offspring while H-DE-71 downregulated Npy in exposed females relative to VEH/CON. In contrast, exposed male offspring displayed upregulated Stat3 and Mc4r. Intestinal barrier integrity was measured using FITC-dextran since it can lead to systemic inflammation that leads to liver damage and metabolic disease, but was not affected by DE-71 exposure. These findings indicate that maternal transfer of PBDEs disproportionately endangers female offspring to lipid metabolic reprogramming that may exaggerate risk for adult metabolic disease.
Metabolic syndrome (MetS) is a cluster of several conditions defined by the International Diabetes Federation to include: obesity, hypertension, dyslipidemia and hyperglycemia (1). MetS is increasing in an epidemic-like fashion, affecting 20-25% of the adult population worldwide, placing a significant economic burden on individuals and society (2, 3). While the prevalence of MetS and type II diabetes (T2D) has been attributed to the sedentary lifestyles and unhealthy diets of industrialized societies in the 20th century, other factors may be contributing to the etiology and pathophysiology of this epidemic, including exposure to endocrine disrupting chemicals (EDCs). EDC exposure during highly vulnerable life stages such as early development may increase the risk of disease later in life and in subsequent generations (4).
One class of compounds with endocrine disrupting properties are polybrominated diphenyl ethers (PBDEs), which have been increasingly used as additives in household products, furniture, clothing, toys and electronics since the 1970’s. After their adverse effects became known, three commercial mixtures of PBDEs, the penta-, octa-, and deca-BDEs were banned by the European Union and all PBDEs were voluntarily withdrawn by manufacturers in the US by 2013. In 2021, an unprecedented action by the US Environmental Protection Agency (US-EPA), formally banned deca-BDEs, Despite these bans, PBDEs continue to be released from existing products and environmental PBDE levels may be increasing due to their persistence, inadvertent recycling and e-waste (5, 6), PBDEs are ubiquitous and still being detected in various tissue samples (7–9). Therefore, PBDEs remain a public health concern and the long-term health consequences of exposure warrant further study.
Of the 75+ brominated flame retardants (BFRs) in circulation, PBDEs are of particular concern for diabetes and MetS due to their disproportionate developmental efficacy and lipid accumulating properties (10–15). Developmental PBDE exposure has been shown to produce marked and long-lasting effects on diabetic markers but these studies were mostly performed in male rodents. Recently, we reported that DE-71, a penta mixture of PBDEs, maternally transferred to offspring at 0.1 mg/kg/d, produces a diabetogenic phenotype characterized by several clinically used parameters, in females (16).
The potential effects of BFRs on adiposity and weight gain in developmentally exposed children are just becoming known. A screen of POPs and BFRs in human serum samples showed a significant association with T2D and MetS with two (PBB-153 and PBDE-153) out of six BFRs tested (17), suggesting that brominated POPs, stored in adipose tissue, may be involved in the pathogenesis of diabetes and MetS. Sexually dimorphic findings related to maternal serum BDE-153 levels during pregnancy were associated with high body mass index (BMI) in 7-year-old boys, but lower scores in 7-year-old girls (18). An inverse association was also found between the child’s own PBDE-153 concentration and BMI at 7 years of age, with no difference by sex.
The discovery that PBDEs activate and/or interact with signaling components involved in obesogenic pathways has spurred numerous studies on the effects of PBDEs on adipogenesis or lipogenesis outcomes in rodent and in vitro models (19–23). Evidence linking early-life exposure to PBDEs (BDE-47), with other MetS parameters is supported by a limited number of studies reporting exaggerated liver triglycerides and lipids and altered transcriptome related to lipid and glucose metabolism in male rats (10). Using a double hit HFD and BDE-47 model Wang (2018) found impaired lipid metabolism in male mice. However, there is limited information available about the effects of developmental PBDE exposure on lipid balance of females (24). Therefore, the purpose of this study was to compare the susceptibility of male and female offspring to the effect of developmental PBDE exposure. We measured peripheral parameters controlling lipid metabolism such as adipokines and plasma and liver markers of lipid metabolism.
Homeostatic regulation of feeding behaviors and energy balance is predominantly controlled by nutrient-sensing and endocrine-responsive signaling pathways in the hypothalamus (25, 26). The hypothalamus consists of a group of nuclei that communicate with each other to coordinate the organism’s energy state via metabolic hormones originating in white adipose tissue (WAT), stomach and pancreas (leptin, insulin, ghrelin) and nutrients (glucose, fatty acids) (27). For example, the arcuate nucleus, located near the blood brain barrier, detects internal and peripheral energy signals such as glucose, insulin, leptin and ghrelin via specific receptors for these molecules (28). Circuits in the arcuate work with those in the lateral hypothalamus and hypothalamic ventromedial nucleus and autonomic brainstem to control feeding, autonomic regulation of metabolism and energy expenditure (28, 29). Two hypothalamic regulatory pathways perform opposing functions: one consisting of neurons co-expressing Agouti-related protein (AgRP) and Neuropeptide Y (NPY), which stimulate food intake, and the other set of neurons co-expressing proopiomelanocortin (POMC) and cocaine- and amphetamine-regulated transcript (CART), which suppress food intake with presumed additional effects on energy expenditure and adipocyte lipolysis (30). Reduced feeding and increased energy expenditure is stimulated by peripheral leptin, a satiety hormone, secreted by WAT via activation of POMC/CART and inhibition of NPY/AgRP neurons. Together with the lipogenic hormone ghrelin, leptin targets the central melanocortin system, including its receptor, Mc4r, to modulate adipocyte and liver metabolism (31). The hypothalamus interacts with the autonomic nervous system which helps maintain energy balance in response to such signals, by regulating lipid metabolism in WAT, thermogenesis in brown adipose tissue (BAT), liver glucose production, pancreatic insulin secretion, and glucose uptake in skeletal muscle. Because hypothalamic control of energy homeostasis is regulated by these hormones signaling through receptors found here, these may be vulnerable to disruption by EDCs. PBDEs may interact with hypothalamic energy-sensing indicators/receptors and/or peripheral metabolic tissues and disrupt their complex balance leading to pathological states such as T2D and MetS. In the current study, we investigated the potential effect of perinatal exposure to DE-71 on plasma lipids and adipokines, hepatic lipid storage, and adiposity. We also studied putative central mechanisms involving hypothalamic markers of energy homeostasis as well as potential changes in gut barrier integrity. Our experimental design included males and female offspring as well as their DE-71 exposed mothers. We have attempted to integrate our findings in to an adverse outcomes pathway.
C57Bl/6 N mice were generated using breeders obtained from Charles River Labs (West Sacramento, CA, USA) and were maintained in accordance with the guidelines in the National Institutes of Health Guide for the Care and Use of Laboratory Animals (32). Mice were group-housed 3-4 per cage in standard polycarbonate plastic cages with corn cob bedding unless otherwise noted. Food pellets (Laboratory Rodent Diet 5001; LabDiet, Quakertown, PA, USA) and municipal tap water were provided ad libitum except during the experimental period. Temperature was maintained at 21.1–22.8°C and relative humidity fluctuated between 20–70% under a 12/12 h photoperiod (lights on from 07:00–19:00 h). All experiments were approved by the IACUC on animal care and use at the University of California, Riverside.
Offspring were exposed to the penta-brominated PBDE commercial mixture, DE-71, daily via maternal transfer using a 10-week dosing regimen as described previously (16). In brief, mice were randomly assigned to one of the three exposure groups: corn oil vehicle control (VEH/CON), L-DE-71 or H-DE-71. DE-71 dosing solutions were prepared in corn oil vehicle to yield two doses: 0.1 mg/kg/d (L-DE-71) and 0.4 mg/kg/d (H-DE-71) using 2 mL of stock solution/kg body weight. Dams were fed oral treats, (Kellogg’s Corn Flakes) infused with dosing solution daily, except on PND 0 and 1. Consumption was visually confirmed and offspring co-housed with dams were never observed to ingest cornflakes. The DE-71 doses were selected to contain the same molar concentrations of BDE-47 used in other mouse studies (24, 33). This exposure regimen involving maternal transfer to offspring yields accumulation of BDE congeners in offspring at ppm concentrations in liver (.2-1.0) and brain (.07-.3) (16, 34). In toddlers, ∑PBDE values in plasma have been reported to range from .1-.5 ppm. (35, 36). The published EPA reference dose for penta mixture of PBDEs (DE-71), is 2x10-3 mg/kg/day or 2 ppb (37).
Dams were dosed daily for a duration of 12 (70-80 d) weeks, which encompassed 3 weeks of gestation and 3 weeks of lactation until pup weaning at postnatal day (PND) 21. Female and male offspring were exposed during the in utero (GD 0-18) and lactational periods (PND 0-21) for approximately 39 d. Body composition measurements were performed at PND 54-80 in offspring and PND 160-180 in dams. Intestinal permeability assay was performed at PND 140-190 in offspring and PND 110-300 in dams. At necropsy, fat depots were excised and weights recorded and blood and organ tissue were collected for further analysis: liver lipids and histology, blood plasma hormones via ELISAs, liver enzymatic activity and hypothalamic gene markers via RT-qPCR.
Whole body composition was determined in live, unanesthetized mice by use of quantitative magnetic resonance (QMR) system, which relies on nuclear magnetic resonance (NMR) technology (EchoMRI; Echo Medical Systems, Houston, TX, USA) (38). Using various pulse sequences, the QMR system provides estimates of fat and lean tissue mass, which were expressed at % body weight. Duplicate QMR scans with accumulation times of 2 min were performed by placing previously-weighed mice into a well-ventilated plastic cylinder (1.5 mm thick, 4.7 cm inner diameter), with a cylindrical plastic insert added to limit movement. While in the tube, animals were briefly subjected to a low-intensity (0.05 Tesla) electromagnetic field.
On the day of sacrifice, mice were anesthetized using brief CO2 inhalation followed by isoflurane inhalation, and blood was collected via cardiac puncture within 2-3 min. Plasma was isolated from blood, stored at -80°C and later used for immunoassays. At necropsy the following fat tissue depots were excised and weighed: interscapular BAT, mesenteric and inguinal WAT. Sample weights were normalized to body weight.
Animals were subjected to a 12 h ON fast on wood chip bedding (SaniChip, P.J. Murphy Forest Products Corp., Montville, NJ, USA). Plasma collected from tail blood was analyzed for leptin and adiponectin using commercially available kits according to manufacturer’s instructions. Plasma leptin was measured using a commercial ELISA kit (ALPCO, Cat. #22-LEPMS-E01, Salem, NH, USA) having a standard range of 0.025-1.6 ng/mL. Therefore, we considered the accurate sensitivity to be the lowest standard, 0.025 ng/mL. Plasma Adiponectin was measured using a kit from Raybiotech (Cat. #ELM-Adiponectin, Norcross, GA, USA). The kit had an analytical sensitivity of 49 pg/mL in a standard range of 49-12,000 pg/mL. The colorimetric reaction products were read as optical density at 450 nm on a microplate reader (Molecular Devices, San Jose, CA, USA). Plasma leptin and adiponectin concentrations were determined by interpolating absorbance values using a linear standard curve.
Blood collected via cardiac puncture at sacrifice (ad libitum fed state) was assayed for plasma total cholesterol (high- and low-density lipoprotein) and triglyceride levels using commercially available fluorometric (total cholesterol) and colorimetric (triglyceride) kits (Cayman Cat. #10007640 & 10010303, Ann Arbor, MI, USA) according to the manufacturer’s instructions. The cholesterol assay had a sensitivity of 1.1 uM in a standard range of 2-20 uM. The triglyceride kit had a sensitivity of 0.5 mg/dL in a standard range of 3.125-200 mg/dL. Plasma cholesterol and triglyceride concentrations were determined by interpolating absorbance values using a linear standard curve.
A modified Folch method was used to quantify total hepatic lipid content. Flash-frozen tissue samples (75-100 mg) were weighed, homogenized in 1 mL of methanol and vortexed followed by the addition of 2 mL of chloroform, 1 ml of water, and centrifugation at 3000 rpm for 15 min at 4°C in pre-weighed glass vials. The lower organic phase was transferred to a clean vial and the upper phase subjected to another chloroform extraction. The pooled lower phases were dried under N2 gas for 30 min at 37°C and the final lyophilized mass weighed and normalized to starting wet weight.
At harvest, tissues were rinsed in phosphate-buffered saline (PBS) and immersion-fixed in 4% paraformaldehyde for 24 h at 4°C. After fixation, tissues were cryoprotected by two successive incubations in 20 and 30% sucrose (in 1X phosphate buffer) and flash-frozen in Tissue-Tek embedding medium over dry ice. Frozen liver tissue was cryosectioned at 7-12 μm. Liver sections were rehydrated with 60% isopropanol, stained for 15 min in freshly made 0.5% Oil-Red-O (ORO) stain (Sigma, St. Louis, MO, USA; Cat# 09755) in 3:2 solution of isopropanol:ddH2O). The slides were rinsed with 60% isopropanol, counter-stained to identify hepatocyte nuclei with modified Harris hematoxylin (Ricca Chemical, Cat# 3530-16, Arlington, TX, USA) for 90 sec, submerged in tap water for 3 min, followed by rinsing for 30 sec in distilled water and mounted with glycerin jelly consisting of gelatin, glycerol and phenol (IHC world). ORO lysochrome stains neutral lipids- triglycerides and cholesterol ethers (but not biological membranes) represented as red droplets (objects) in spaces between hepatocytes (39, 40). Images were captured at 40X using bright field optics (Nikon TMS, Tokyo, Japan) and digital camera (Spot Insight U3.2).
For each animal, images were acquired of 3-5 microscope fields from each liver with a 40X objective using brightfield optics. All images to be compared were acquired using identical microscope settings and were used to quantify the ORO positive signal using open source software [QuPath v0.2.3 (41)]. For each sample, 3-5 subfields were selected, each designated as a region of interest (ROI). The total ORO positive area was summed for each subfield and normalized to ROI area. The normalized ORO positive area values were averaged and reported as mean ± sem.
Mice were fasted overnight (14-16 h) on wood chip bedding. Mice were gavaged with 60 mg/100 g b.w. of a 100 mg/mL of dextran conjugated to fluorescein isothiocyanate (FITC; 60 mg/100 g b.w.; 3-5 kDa; Sigma-Aldrich, Cat#: FD4) solution prepared in sterile 1X PBS. Twenty percent more of the calculated individual dose was given to each mouse to compensate for the volume the gavage needle retains. After 4 h, mice were euthanized under isoflurane anesthesia and blood (300–400 μL) collected via cardiac puncture into sterile tubes on ice. Serum was collected after cold-centrifugation at 2000 g for 15 min and diluted 1:5 in double-distilled water. Plasma FITC was measured in duplicate or triplicate spectrophotometrically (GloMax Promega, USA) in 96-well plates (excitation: 490/510–570 nm, 525/580–640 nm). FITC-dextran concentration of samples was determined from a standard curve generated using a diluted fluorophore stock gavage solution in the plasma matrix from fasted untreated mice. Three technical replicates of each sample were run.
Dextran sodium sulfate (DSS), a water-soluble, negatively charged sulfated polysaccharide containing ~19% sulfur with a highly variable molecular weight ranging from (MW = 36,000–50,000 Da) is often used to induce breakdown of gut integrity leading a form of mouse colitis that mimics the clinical and histological features of irritable bowel disease (IBD) including ulcerative colitis (42). Colitis was established by the daily oral administration of 2-3% DSS (colitis grade, MP Biosciences) via drinking water, leading to inflammation and leaky gut in the mid-distal colon within 5-8 days of treatment. The solution was prepared fresh every 4 d. DSS mice were used as positive controls for gut permeability measurements in FITC-dextran experiments described above after 6-7 d post-treatment.
At sacrifice, using isoflurane anesthesia and cervical dislocation, whole hypothalami were rapidly dissected and snap-frozen in 2-methylbutane over dry ice. The hypothalamus was hemissected and one-half portions immediately homogenized in TRIzol Reagent (Ambion by Life Technologies, Carlsbad, CA, USA) using a hand-held homogenizer. Total RNA was prepared via a guanidinium thiocyanate–phenol–chloroform extraction followed by manufacturer’s instructions, including DNAase1 treatment (Monarch Total RNA Miniprep Kit, #T2010, New England Biolabs, Ipswich, MA, USA and QIAGEN RNeasyMini, #74104, Dusseldorf, Germany). Purity and quantity of RNA were assessed by determining the optical density (OD) photometrically using 260/280 and 260/230 nm ratios (NanoDrop ND-2000, Thermo-Fisher Scientific Inc., Waltham, MA, USA).
RT-qPCR was used to quantitate mRNA transcripts for genes: Npy, Lepr, Gshr, Stat3, Mc4r, Agrp. Custom- or pre-designed DNA oligonucleotide PCR primers were obtained from Integrated DNA Technologies (Coralville, IA, USA). Primers were designed to meet several criteria using NCBI Primer Blast and then optimized by testing against complementary DNA generated using RT-PCR and gel electrophoresis. Only primers that gave single-band amplicons in the presence of reverse transcriptase (RT) and that matched the base length of the predicted target were selected. In addition, primers selected yielded 91.5 to 107.8% efficiency by RT-qPCR validation (Table 1). A temperature gradient was used to capture ideal annealing temperatures for custom designed primer pairs. For all primers, intercalating dye (SYBR green) chemistry was used. The primer concentration ranged from 200-500 nM.
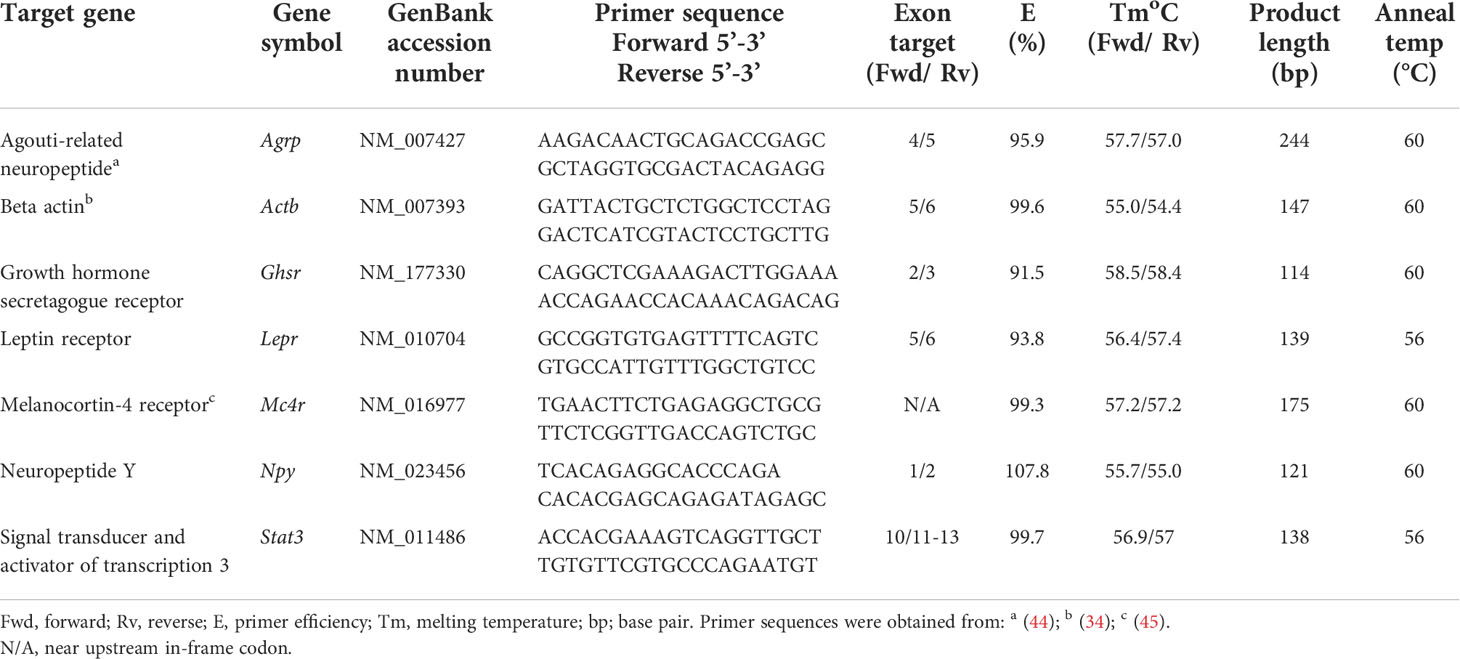
Table 1 RT-qPCR primer sequences and efficiencies and PCR products.
RT-qPCR was performed on RNA samples (40 ng), run in triplicate, on a CFX Connect or CFX96 thermocycler (Bio-Rad, Irvine, CA, USA) with the Luna Universal one-step qPCR Master Mix (E3005; New England Biolabs, Ispwich, MA, USA). Amplification reactions for genes of interest were performed in 50 cycles of the following cycling protocol: reverse transcription 55°C/10 min; initial denaturation 95°C/1 min; per cycle: 95°C/10 s denaturation, 60°C or 55°C/30 s extension; 65-95°C in 0.5°C, 5s increments melt curve analysis. In each experiment, no-template controls (NTCs) were run without mRNA to rule out extraneous nucleic acid contamination and primer dimer formation. Negative RT controls, which contained the complete set of RNA synthesis reaction components without the addition of the enzyme reverse transcriptase were used to rule out presence of genomic DNA (gDNA). Relative gene expression was measured relative to the reference gene, ActB, and differential gene expression was determined compared to the null group (VEH/CON) using the Pfaffl method (43). We have previously determined that DE-71 does not interfere with the expression of ActB.
Normal distribution and homogeneity of variance of the data were tested. All statistical analyses were conducted using GraphPad Prism (GraphPad Software version 9.4.1, San Diego, CA, USA). All values are expressed as mean ± s.e.m. One-Way ANOVA was used to compare the effects of exposure. Brown-Forsythe ANOVA was used when variances were significantly different. Where the F ratio was significant, post-hoc comparisons were completed using Tukey’s, Dunnet’s or Sidak’s post-hoc test. Differences were considered statistically significant at p<0.05.
In order to ascertain the effects of DE-71 on body composition, Echo MRI was used to measure total fat and lean mass and body weight in exposed and control mice. The results are summarized in Table 2. There were no differences in adult male or female offspring nor dams. However, there was an apparent decrease in relative lean mass in H-DE-71 female offspring relative to VEH/CON (One-way ANOVA F(2,41)=2.72, p=0.08, Dunnett’s post-hoc VEH/CON vs H-DE-71, p=0.06). No changes were observed in this cohort, although we found a decrease in body weight in L-DE-71 female offspring previously (16).
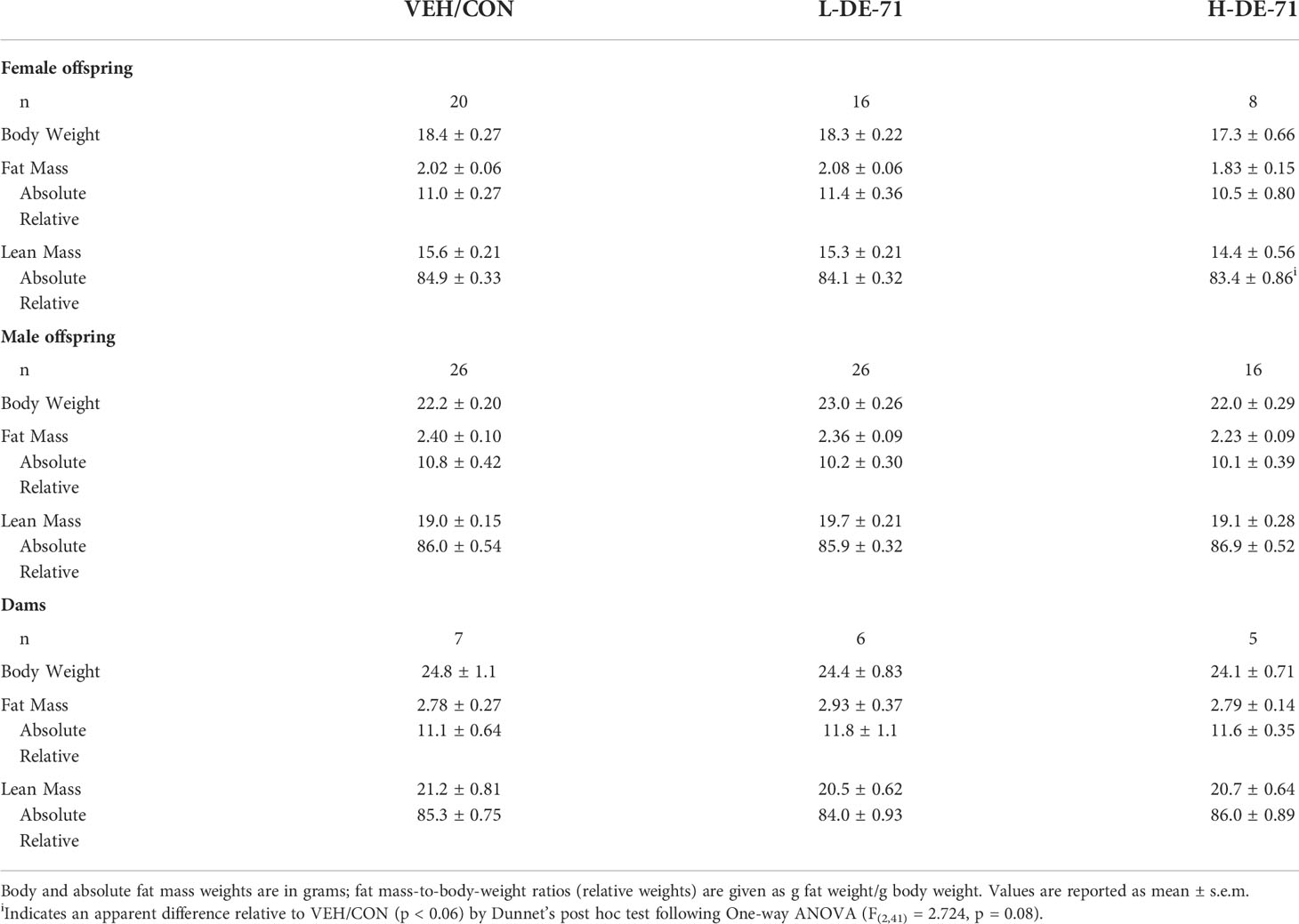
Table 2 Chronic DE-71 exposure has minimal effects on body composition.
Obesity is defined largely by body mass index (BMI); however, physiological evidence indicates that body fat distribution, irrespective of BMI, most strongly predicts the risk of obesity-associated disease (46). Therefore, we examined the fat stores in interscapular BAT, inguinal WAT and mesenteric WAT. Figure 2 shows decreased BAT in L-DE-71 female and L-DE-71 male offspring (Figures 1A, B) (Females: Brown-Forsythe ANOVA F(2,21.2)=7.448, p=0.0035, Dunnett’s post-hoc VEH/CON vs L-DE-71, p=0.02; Males: One-Way ANOVA F(2,41)=6.82, p=0.0028, Tukey’s post-hoc VEH/CON vs L-DE-71, p=0.01, L-DE-71 vs H-DE-71, p=0.01. No group differences were observed in dams (Figure 1C).
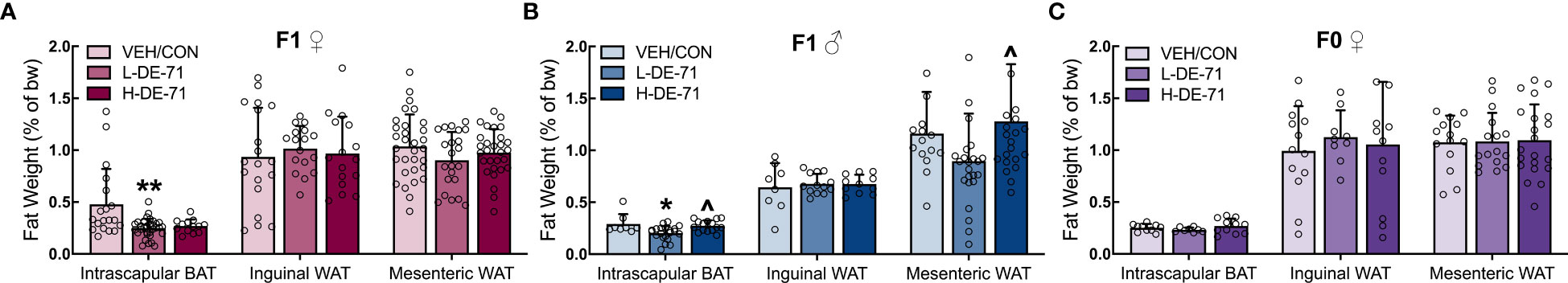
Figure 1 Brown adipose tissue (BAT) in DE-71-exposed male and female offspring. (A–C) Normalized weight of fat depots (WAT and BAT) were compared across treatment groups: 0.1 mg/kg/d (L-DE-71), 0.4 mg/kg/d (H-DE-71) and oil vehicle controls (VEH/CON) of female (A), male (B), and dams (C). *p<0.05, **p<0.01 compared to VEH/CON; ^p<0.05 compared to L-DE-71. Sample size (n/group): (A) female offspring 16-31/group; (B) male offspring 8-24/group; (C) dams 8-21/group. Bars and error bars represent mean ± s.e.m. BAT, brown adipose tissue. WAT, white adipose tissue [Permission obtained for reuse of left panel in Figure 2A and left panel in 2C, Kozlova et al, 2020, Sci. Rep. 10:18102].
Leptin, as a single factor, is associated with increased cardiometabolic risk and predicts MetS (47). Adiponectin, secreted from WAT, decreases liver lipids and deficiency of adiponectin is associated with insulin resistance, T2D and cardiac mortality (48). Therefore, we measured plasma levels of adiponectin and leptin in a fasted state using commercial immunoassay kits. Our results show that, relative to VEH/CON, L-DE-71 female offspring display elevated plasma leptin: Brown-Forsythe ANOVA: F(2,18.8)=5.8, p<0.05; Dunnet’s post-hoc VEH/CON vs L-DE-71 p=0.04 (Figure 2A). No group differences were observed in male offspring or dams (Figures 2B, C). For adiponectin L- and H-DE-71 male offspring displayed significantly lower levels as compared to VEH/CON: One way ANOVA: F(2,19)=8.224, p<.0.01; Tukey’s post-hoc VEH/CON vs L-DE-71 p=0.05, vs H-DE-71 p<.01 (Figure 2E). No group differences were observed in female offspring or dams (Figures 2D, F).
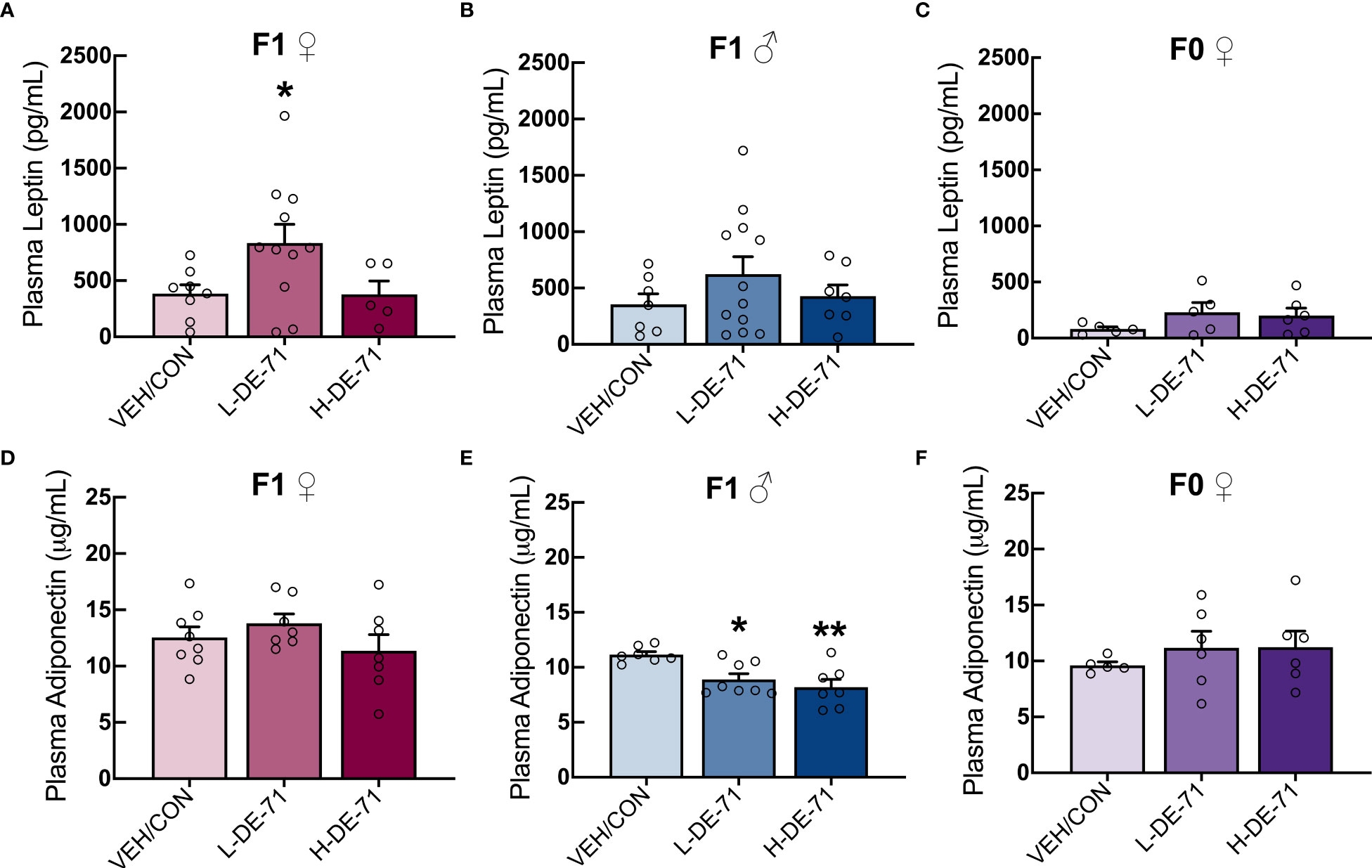
Figure 2 Plasma levels of leptin and adiponectin in DE-71-exposed male and female offspring. Female and male offspring were fasted for 12 h and plasma collected and analyzed using commercial ELISA kits. Samples were also processed for dams in ad libitum fed state. (A–C) Mean plasma leptin levels in DE-71-exposed and VEH/CON groups. (D–F) Mean plasma adiponectin levels DE-71-exposed and VEH/CON groups. Asterisk(s) indicate compared to VEH/CON, *p<0.05, **p<0.01. Sample size (n/group): (A) female offspring, 6-11, (B) male offspring, 7-12, (C) dams, 5-6, (D) female offspring, 7-8, (E) male offspring, 7-8, (F) dams, 5-6.
In order to evaluate lipid metabolism, we examined plasma levels of total cholesterol and triglyceride since high total of LDL cholesterol, low HDL cholesterol and high triglycerides are indicators of dyslipidemia and risk factors for cardiovascular disease and other comorbidities (49). A significant elevation in plasma total cholesterol was observed in female L-DE-71 offspring compared to VEH/CON (One-Way ANOVA F(2,41)=6.818, p=0.0028, Tukey’s post-hoc VEH/CON vs L-DE-71, p=0.0112) (Figure 3A). No group differences were observed in male offspring nor dams (Figures 3B, C). Triglyceride levels were similar across DE-71 exposure groups in male and female offspring and dams (Figures 3D–F) as previously reported for BDE-47 exposed male offspring (10). These results reveal a sexually dimorphic effect of PBDEs on cholesterol levels, with exposed female offspring showing a unique susceptibility to L-DE-71.
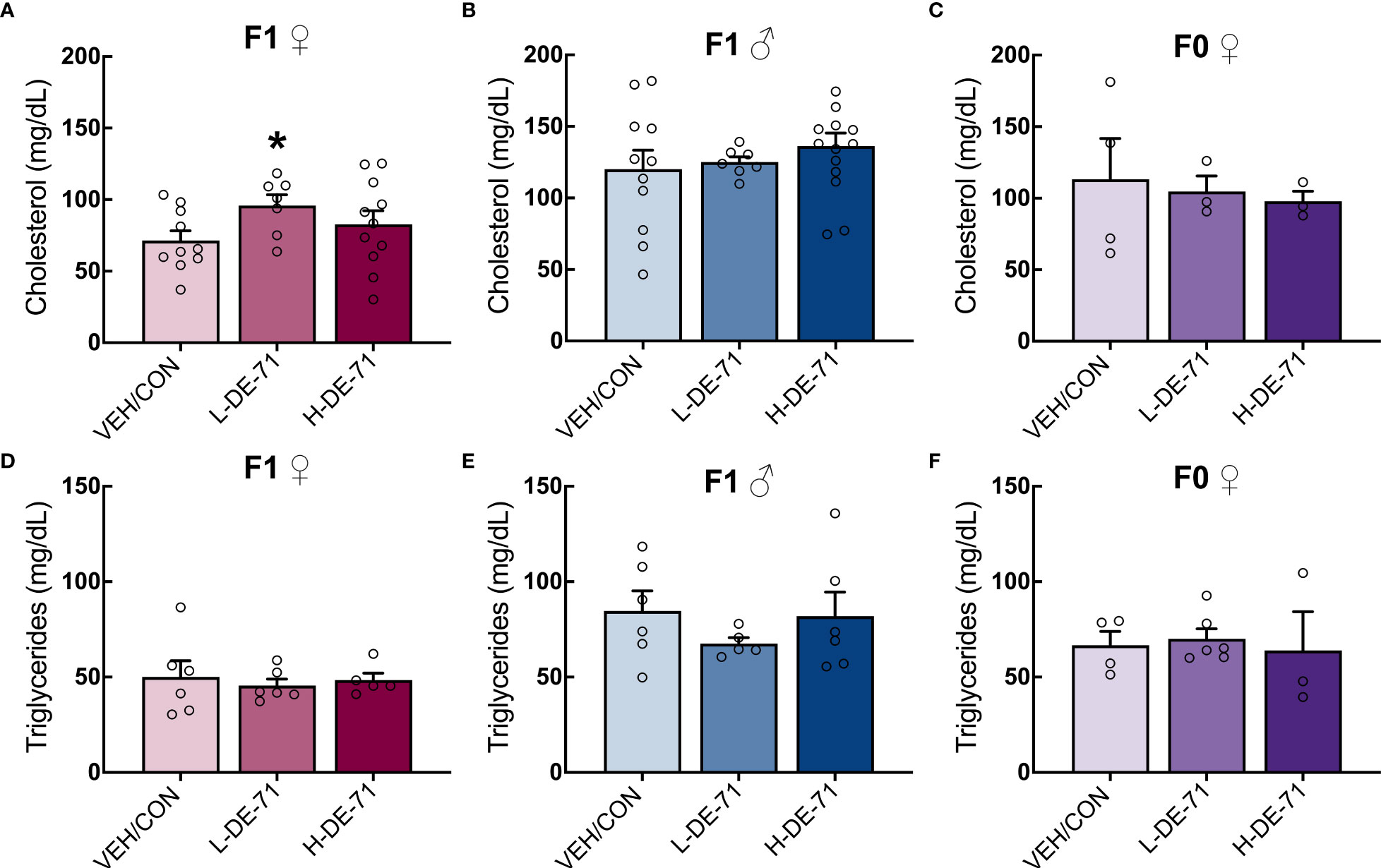
Figure 3 Effect of perinatal L-DE-71 exposure on plasma total cholesterol and triglycerides in DE-71-exposed male and female offspring. (A–C) Plasma total cholesterol in female (A) and male (B) offspring and dams (C). (D–F) Plasma triglyceride levels in female (A) and male (B) offspring and dams (C). *p<0.05 compared to VEH/CON; Bars and error bars represent mean ± s.e.m. Sample size (n/group): (A–C) females, 7-11; males, 7-14; dams, 3-4. (D–F) females, 5-6; males, 5-6; dams, 3-6.
The excessive accumulation of lipids in the liver may be a risk factor for the development of liver steatohepatitis, the most prevalent chronic liver disease with a widespread prevalence of 25% in the adult population (50). Liver steatohepatitis is also a risk factor for markedly greater mortality and morbidities like MetS (51), T2D, obesity, dyslipidemia, hypertension and cardiovascular disease (51). We, therefore, examined the effects of DE-71 on liver lipid content by processing snap frozen liver tissue using the Folch method. Results in Figure 4 show mean normalized lipid levels are significantly elevated in L-DE-71 exposed female offspring as compared to VEH/CON (Figure 4A). Mean raw lipid concentrations were 204, 249, 231 mg lipid/g tissue for VEH/CON, L-DE-71 and H-DE-71, respectively. A Brown-Forsythe ANOVA revealed a statistically significant effect of exposure (F(2,14.7) =11.08), p=0.001. Dunnett’s T3 post-hoc showed differences between L-DE-71 and VEH/CON females (p=0.01) as well as between L-DE71 and H-DE-71 females (p=0.02). No differences were observed in males or dams (Figure 4A).
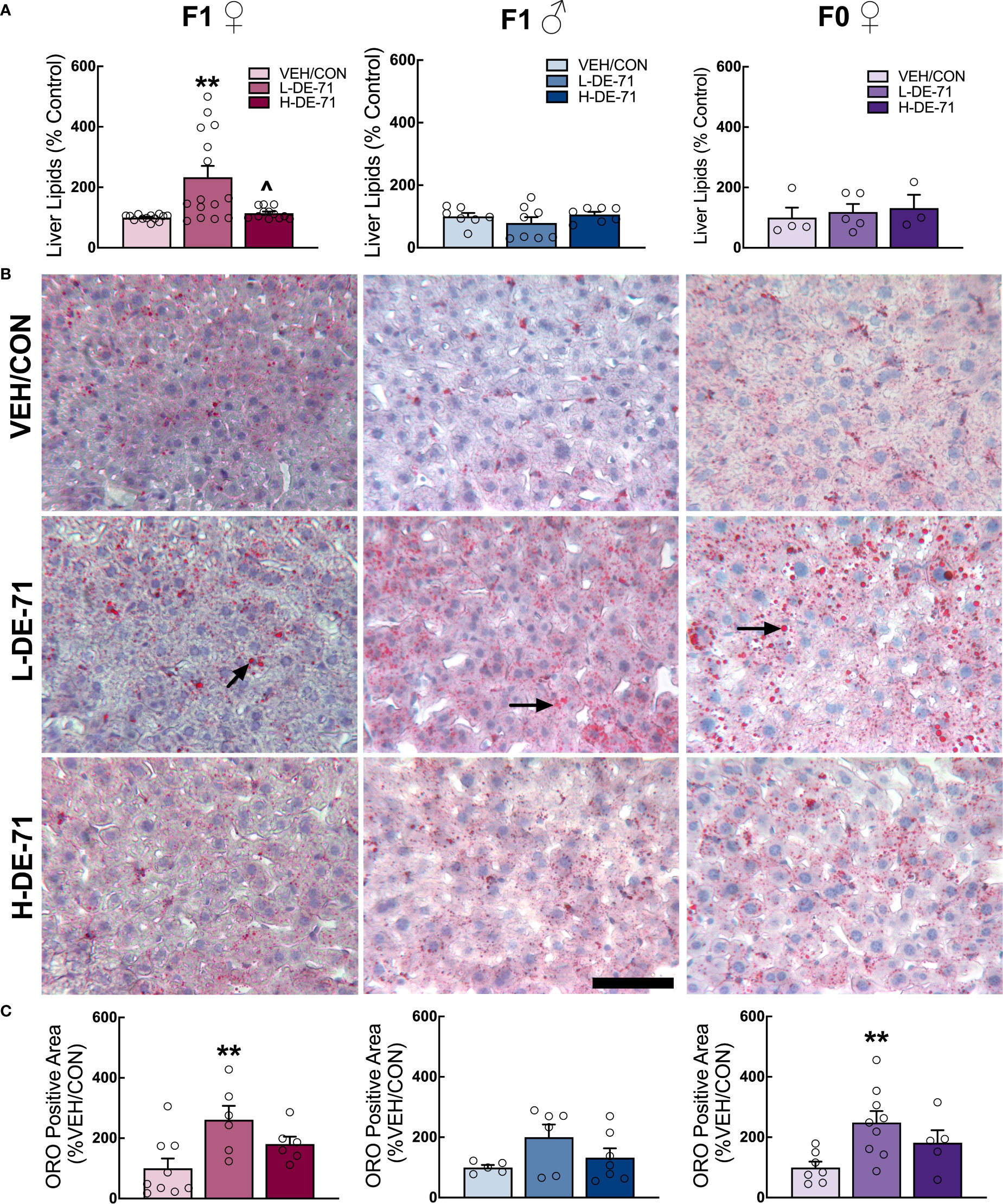
Figure 4 Sex-dependent effects of perinatal exposure to L-DE-71 on liver lipids in male and female offspring. (A) Total hepatic liver lipids expressed as percent VEH/CON. (B) Representative micrographs of liver samples processed for Oil Red O staining show lipid globules (black arrows) on hematoxylin stained hepatocytes. (C) Densitometric quantification in ORO-stained livers. **compared to VEH/CON, p<0.01. Bars and error bars represent mean ± s.e.m. Sample size (n/group): (A) female offspring, 12-15; male offspring, 5-8; dams, 5-9. (C) female offspring, 6-9; male offspring, 5-6; dams, 4-8. Scale bar, 100 microns.
In another subset of mice, we performed ORO histochemistry on cryosectioned liver tissue sections. Microscope analysis revealed bright red circular-stained structures, indicating fat deposits (Figure 4B). Densitometric quantification indicated that L-DE-71 produced additional fat deposits in exposed female but not male offspring as compared to their respective VEH/CON group (Figure 4C). Although studied at different exposure conditions, previous reports indicate increased ORO staining in developmentally exposed male rodent offspring when exposed to higher doses of DE-71 (50 mg/kg, Wistar Han rats) (52); or to a specific BDE congener, BDE-47 (1 mg/kg, CD-1 mice) (10). L-DE-71 dams also showed more numerous lipid globules as compared to VEH/CON (Figure 4C). In agreement with adult susceptibility to altered hepatic fat storage after direct PBDE exposure (BDE-47, 1 mg/kg), greater ORO staining was reported for adult male rats (11). With regard to Folch measured lipids and ORO staining, previous reports on females and quantitative analysis of PBDE-induced liver lipids are lacking.
Intestinal permeability was also examined, since intestinal-derived bacterial products entering the bloodstream may cause inflammation and promote liver disease that may contribute to obesity and diabetes (53). To explore whether BDEs in DE-71 produce metabolic effects in parallel with gut inflammation, we evaluated gut barrier integrity by subjecting mice to an oral dextran-FITC permeability test. Plasma FITC levels were analyzed 4 h later using fluorimetry. Our results, shown in Figures 5A–C, indicate normal gut permeability across all experimental groups (Male: Brown-Forsythe ANOVA, F(2,23.2)=2.97, p=0.07; Female offspring: F(2,29.5)=0.48, p=0.62; Dam: One-way ANOVA, F(2,38)=2.5, p=0.09). H-DE-71 male offspring showed an apparent increase in intestinal leakage (p=0.07). We included an experimental group exposed to dextran sodium sulfate (DSS), a chemical colitogen with anticoagulant properties, to induce disease, that was used as a positive control and expectedly found a 154 and 356% greater FITC leakage into plasma than VEH/CON in male and female offspring, respectively (unpaired t-test, p<0.01).
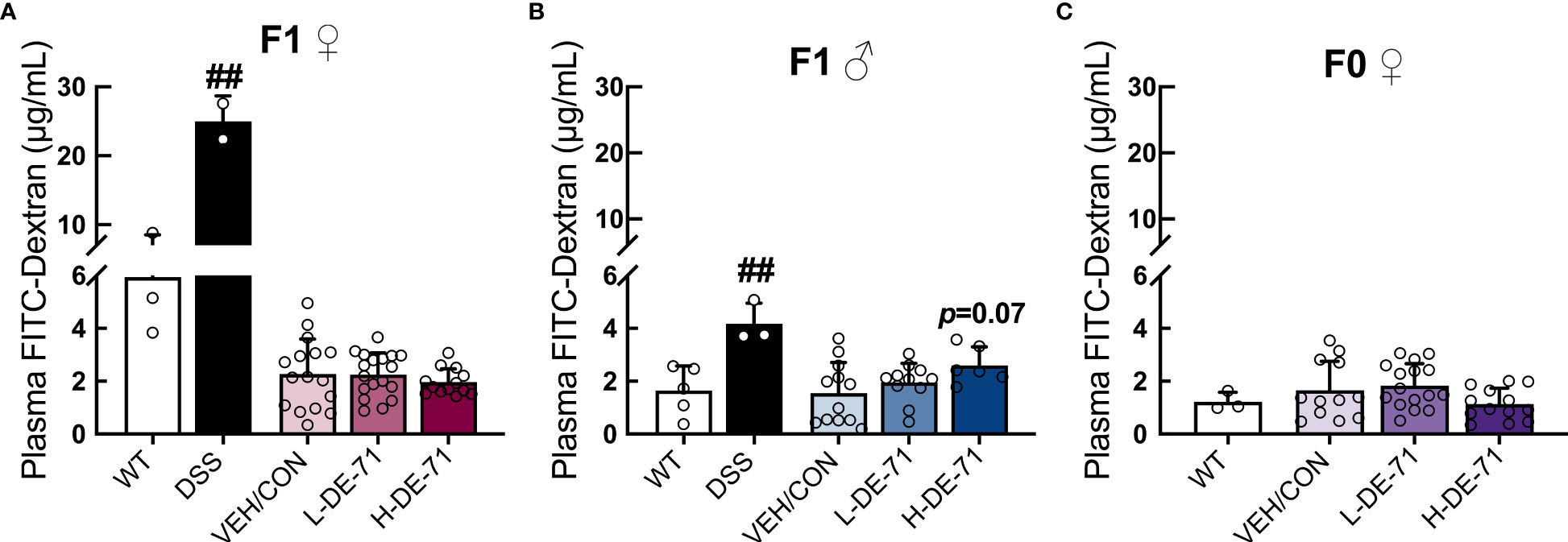
Figure 5 Gut barrier permeability in DE-71 exposed dams and offspring. Mice were fasted 14 h before gavage with 60mg/kg of 4 kDa FITC-dextran and serum samples collected 4 h later were read for fluorescence intensity at 490 nm using a Promega GloMax spectrometer. (A–C) Plasma FITC levels in exposed and VEH/CON groups. Bars and error bars represent mean ± s.e.m. Exposure had no main effect as indicated by One-way ANOVA. DSS treatment was used as a positive control; ##significantly different compared to VEH/CON, p<0.01, Student’s t-test. Sample size (n/group): (A) female offspring 2-16. (B) male offspring: 3-12. (C) dams 3-16.
The regulation of energy balance and body composition is a complex process that involves interaction between the central and peripheral systems. We explored the effects of developmental exposure on hypothalamic gene markers for energy-sensing and signaling important for regulation of energy balance. Figures 6A–F shows the mean fold change in hypothalamic RNA samples of exposed offspring relative to VEH/CON. Overall, DE-71-exposed female offspring showed abnormal downregulation of all genes tested. Specifically, exposed females showed reduced Lepr, Stat3, Agrp, Npy, Mc4r and Gshr. One-way ANOVA values ranged between p<0.05 to p<0.001. The greatest reduction occurred in Agrp in L-DE-71 (49.8 ± 4.3%) and H-DE-71 (66.9 ± 10.0%) in female offspring (p<0.001, n=8-10/group). In contrast, males showed changes in only two genes (Stat3 and Mc4r) and these were both upregulated (p<0.05 in H-DE-71 and p<0.01 in L-DE-71, respectively, n=9-10/group). This is consistent with the more pronounced lipid metabolic reprogramming characterized in L-DE-71 exposed female offspring. However, abnormal gene expression occurred in both L-DE-71 and H-DE-71 mice, indicating a lack of dependency on DE-71 dose.
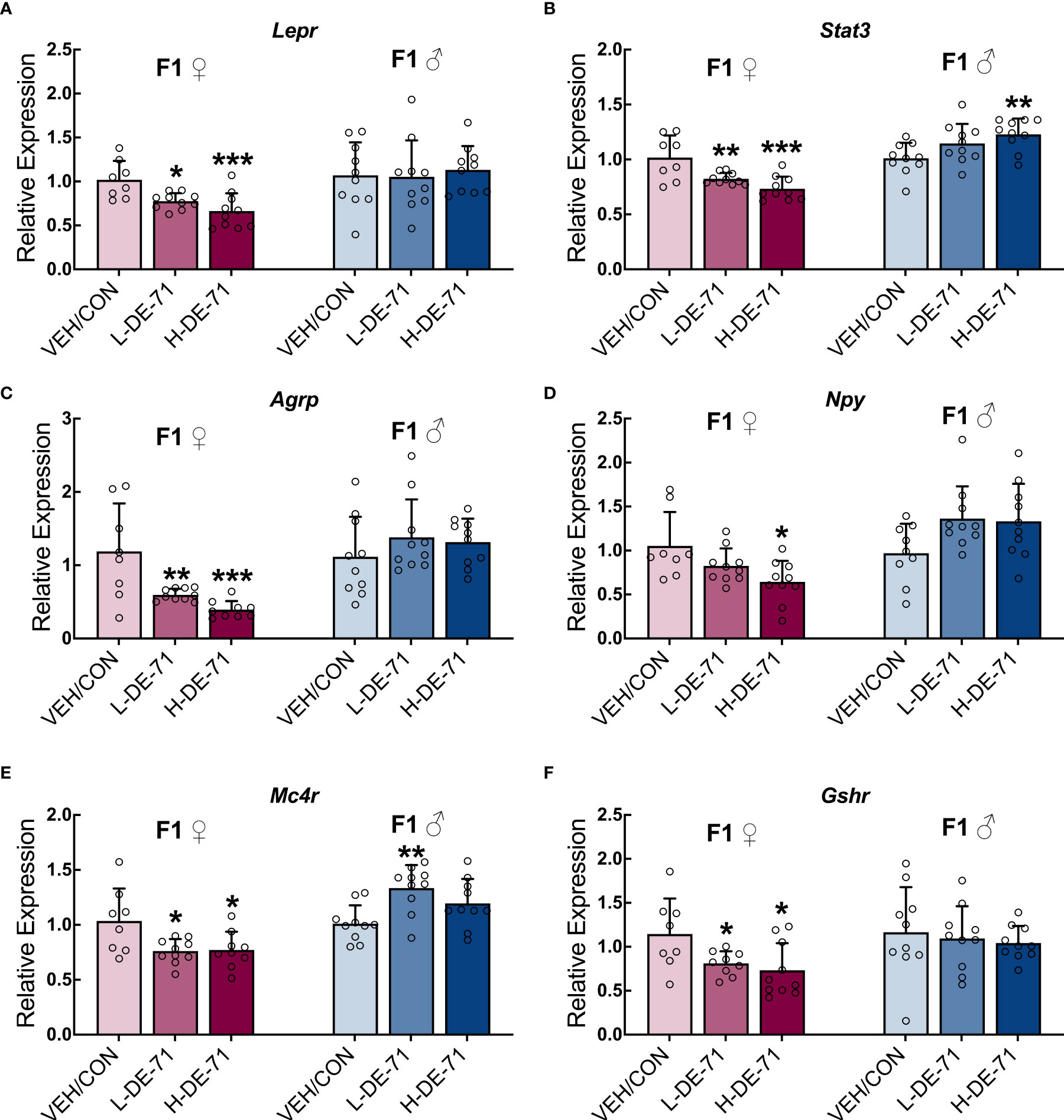
Figure 6 Sex-dependent alterations of hypothalamic energy balance genes in DE-71-exposed F1 offspring. Relative gene expression in hypothalamic samples of adult male and female offspring from dams orally dosed with vehicle or DE-71 was measured using RT-qPCR and mean values normalized to Actb and expressed as fold-change relative to the mean of respective VEH/CON samples. (A) Leptin receptor, Lepr (B) Signal transducer and activator of transcription 3, Stat3 (C) Agouti-related peptide, Agrp (D) Neuropeptide Y, Npy (E) Melanocortin-4 receptor, Mc4r (F) Growth hormone secretagogue receptor, Gshr. Data are represented mean ± s.e.m. Sample size (n/group): Females (8-10) and males (9-10). Asterisks indicates significant difference compared to VEH/CON, *p<0.05, **p<0.01, ***p<0.001.
The present study provides the first evidence of an environmentally relevant PBDE mixture on lipid metabolism and potential associated central mechanisms (34). Specifically, lipid parameters in exposed female offspring were compared to those obtained from their male counterparts and their mothers. Female offspring of dams fed L-DE-71 (0.1 mg/kg/d) are susceptible to develop hypercholesterolemia, fatty liver, elevated plasma leptin and altered central markers of energy homeostatis relative to same-sex vehicle controls. Notably, female mice exposed to the greater dose of DE-71 (0.4 mg/kg/day) were not symptomatic. This is in agreement with the non-monotonic dose response reported for other EDCs (54). In contrast, female adult dams lacked this phenotype, suggesting that developing females are more vulnerable to the endocrine- and metabolic-disrupting effects of PBDEs. Taken together, our findings indicate that central mechanisms involving hypothalamic markers of energy homeostasis as well as hepatic lipid storage, may, in combination, pose a unique risk to EDC-exposed female offspring for metabolic disorders.
Perinatal exposure to L-DE-71 produced elevated plasma total cholesterol in female but not male offspring, relative to their respective vehicle controls (Figure 3), in spite of the fact that males display greater basal levels of cholesterol than female mice (55). Abnormal plasma cholesterol or triglycerides are one criteria of MetS, along with abdominal a diposity (56), elevated glycemia/diabetes and high blood pressure (47). While we did not detect changes in mesenteric fat (Figure 1), we have previously reported diabetogenic (increased fasting glycemia and glucose intolerance, insulin insensitivity) and exaggerated pressor responses in adult offspring of DE-71 exposed dams (16, 57). Hypercholesterolemia may follow from fatty liver phenotypic reprogramming (58) produced by PBDEs in DE-71 as speculated in the AOP. Recently, Tartu (2017), used metabolome and lipidome approaches to establish that PBDEs (along with PCBs, chlordanes and perfluoroalkyl substances) were significantly related to cholesterol homeostasis and other biomarkers involved in lipid accumulation, FA metabolism, and insulin utilization in Norwegian polar bears (59). In addition, altered adipocyte function may also contribute (23). For example, penta-BDE mixture promotes stimulated lipolysis and reduced insulin-stimulated glucose oxidation in adipocytes in vitro (60). Further study is needed to determine the source of elevated cholesterol and the initiating events triggered by PBDEs.
Our previous mass spectroscopy results indicated persistent accumulation of BDE-28/33 and -153 in adult livers of DE-71 exposed female offspring, suggesting that livers are a target of BFRs such as PBDEs (16). The hypercholesterolemia observed in L-DE-71 females led us to quantify lipid content in the livers of all mice using the Folch method. Elevated hepatic lipid content was only displayed by L-DE-71 female offspring (Figure 4). This group also showed elevated ORO staining, which represents a partial set of lipids. Other studies in male mice have shown that perinatal/postnatal exposure to BDE-47 produces excess ORO staining and altered transcriptomics in liver pathways related to lipid and carbohydrate metabolism, insulin signaling and T2D (10, 61). Notably, upregulated cluster of differentiation (Cd36) membrane protein may indicate that PBDEs alter fatty acid uptake since Cd36 has been associated with non-alcoholic fatty liver disease (NAFLD) in humans. In combination, our findings emphasize the risk for fatty liver in humans with high body burdens of PBDEs. NAFLD is the most common liver disease in world, affecting adults at epidemic proportions (25%), with risk factors ranging from age and sex to MetS and insulin resistance (62). Genetics alone cannot explain the large increase in the prevalence of NAFLD during the past 2 decades and the increase that is projected for the next decades. Developmental exposure to EDCs such as PBDEs may contribute added risk; a recently identified form of NAFLD is toxicant-associated fatty liver disease (TAFLD) in occupationally exposed workers (63). Unlike BDE-47, our findings show that DE-71 produces minor effects on liver lipids of developmentally exposed males compared to females. In our hands, exposed females but not males also show hallmarks of T2D such as elevated fasting glycemia, glucose intolerance, insulin resistance and reduced plasma insulin (16). Indeed, fatty liver and T2D share insulin resistance as their chief pathogenic determinant (64).
The abnormal hepatic lipid accumulation in L-DE-71 female phenotype occurred in the absence of excess fat depots, although lean mass showed an apparent decrease (Table 2). At first, this may appear inconsistent with fatty liver disease despite being the most common risk factor for NAFLD. In fact, up to 20% of Americans with normal BMI have NAFLD. Similarly, null associations were noted between concurrent PBDEs and BMI among adolescents from a cohort in the Netherlands (65) and between prenatal PBDEs and BMI in children of the Center for the Health Assessment of Mothers and Children of Salinas (CHAMACOS) study (15). Other epidemiological studies have reported inverse relationships with postnatal PBDE congeners −47, −100, −153, and −154 and adiposity in girls aged 6–8 years (66).
Our immunoassay results demonstrate elevated fasting plasma leptin in L-DE-71 female offspring (Figure 2). Our findings of augmented circulating leptin, and fatty liver without increased adiposity are in agreement with previous studies. Leptin levels are positively associated with severity of NAFLD (47, 67), that cannot be ascribed to a corresponding elevation in WAT (Figure 1). The importance of our finding is further emphasized since elevated fasting leptin, as a single factor, is associated with increased cardiometabolic risk and predicts MetS. Leptin is also an important regulator of blood glucose levels (68). In contrast, a longitudinal study on a small sample of children in the Netherlands failed to show any correlation between leptin and toxicants (PBDEs, PCBs or dioxins) (65). Adiponectin was also studied because it is inversely linked with insulin resistance, lipid accumulation, inflammation and NAFLD and increasing plasma adiponectin is being tested as a new therapy for NAFLD (69). PBDE-exposed male offspring showed reduced plasma adiponectin but their liver lipids and plasma cholesterol were normal (Figure 2). In humans, a positive association has been found between ΣPBDE and fasting levels of adiponectin in older persons (70). More studies are needed to determine the endocrine disrupting effects of PBDEs and plasma adipokines.
The role of the hypothalamus is also physiologically relevant to the abnormal lipid metabolic phenotype (excess hepatic lipid storage and hypercholesterolemia). We found a widespread effect of early-life exposure to PBDEs on energy-regulating gene markers in the hypothalamus (Figure 6). Notably, female offspring exposed to L-DE-71 display reduced Lepr, and its signaling partners Stat3 and Mc4r. Together with chronically elevated fasting levels of leptin (Figure 2), the central changes may represent a form of leptin resistance that has not been previously reported EDC action of PBDEs. Downregulated Lepr, Stat3 and Mc4r (see below) may alter effectiveness of peripheral leptin’s anorexigenic and lipid balancing homeostatic actions (lipolysis, decrease WAT mass and lipid accumulation). PBDE-induced elevation in circulating leptin levels may impair cholesterol removal in the diabetogenic phenotype of L-DE-71 females (16) as can happen to non-exposed animals under conditions of high blood glucose (71). Therefore, the aberrant lipid metabolic profile of L-DE-71 in female offspring (Figure 4) may be caused, in part, by abnormal leptin actions.
Leptin actions are, in part, mediated via inhibition of NPY/AgRP neurons (30, 72). Notably, neuropeptide gene expression of Agrp was also downregulated in L-DE-71 (and Npy in H-DE-71) female offspring. Female Agrp expression is also susceptible (opposite change) to perinatal organophosphate flame retardants (OPFR) exposure when combined with a high fat diet (73). In contrast to the results obtained on females, DE-71 exposed males showed upregulated Stat3 and Mc4r without altered Npy, Agrp, Ghsr, or Lepr. For reference, others have reported downregulated hypothalamic NPY and POMC systems in male C57BJ/6 mice by adult exposure to other EDCs such as bisphenol A, diethylstilbestrol and tributyltin (74).
L-DE-71 female offspring also displayed downregulated expression of growth hormone secretagogue receptor (GHSR) (Figure 6) to which peripheral ghrelin binds and produces orexigenic and lipogenic actions (25, 75, 76). Both ghrelin and leptin target the central melanocortin system, including its receptor, MC4R, to modulate adipocyte and liver metabolism (77). Therefore, downregulated central levels of Mc4r, seen in L-DE-71 female offspring, may play a role in deregulating hypothalamic-mediated metabolic homeostasis (Figure 6). The central melanocortin system also modulates hepatic lipid metabolism; animals with reduced MC4R expression or function show increased lipogenesis rate and triglyceride content in the liver (78, 79). Therefore, changes seen in Mc4r may participate in the exaggerated liver lipid content, and hypercholesterolemia produced in females by developmental exposure to L-DE-71 (Figure 3). Other EDCs such as OPFRs that have replaced PBDEs in home environment, disrupt energy homeostasis through toxic actions on the hypothalamic melanocortin circuitry in a sex-dependent manner (80).
Developmental abnormalities of hypothalamic neural circuits can lead to body weight and metabolic imbalance (81) from neuroinflammation, as reported after air pollutant exposure (82). Whether organohalogens that have access to hypothalamus (83) produce their actions via similar neuroinflammation is still unresolved. One study has shown that perinatal polychlorinated biphenyls (0.5 mg/kg) can produce persistent changes in glial fibrillary acidic protein, a measure of neuroinflammatory response in brainstem and cerebellum, but hypothalamus was not tested (84). It should be noted that PBDEs have prominent disruptive effects on the thyroid hormone system, which controls lipid metabolism and inflammatory processes, and responds to leptin receptor activation (85).
The lipid metabolic profile detected in female offspring perinatally exposed to L-DE-71 may indicate a propensity for MetS. In agreement with this broader assignment, we have previously shown that L-DE-71- exposed female offspring show a combination of diabetogenic features including fasting hyperglycemia, insulin insensitivity, glucose intolerance and altered glucoregulatory hormones. Female dams do not display altered hypercholesterolemia nor diabetogenic phenotype thus, adults are less susceptible to DE-71 exposure relative to offspring. Our previous findings also indicate elevated plasma vasopressin in L-DE-71 female offspring (34); elevated vasopressin (as measured by its stable surrogate marker copeptin) is positively associated with hepatic steatosis and MetS (86). The effects produced by developmental exposure to DE-71 have been incorporated into a possible adverse outcome pathway that describes key events leading to physiological and organismal outcomes such as MetS, T2D, fatty liver disease and cardiovascular disease (Figure 7). Exposure to DE-71, with primary constituents BDE-47 and BDE-99, triggers changes in liver gene expression with patterns matching that of metabolic syndrome (52, 90). BDE-47 concentrations have been associated with liver enzymes indicating hepatobiliary dysfunction (11, 14). While we did not investigate the possible participatory MIE(s) in Figure 7, a focus on nuclear receptors and aryl hydrocarbon signaling has been recommended on the basis of involvement in endocrine disruption by BFRs (91). There is evidence that BFRs like PBDEs activate the nuclear hormone receptors, Pregnane X receptor (PXR) and peroxisome proliferator-activated receptors (PPARγ), both transcriptional regulators of hepatic uptake/deposit of lipids (92–94). Indeed, PBDE-99 stimulates adipogenesis and increases PPARγ expression in other metabolic organs such as murine and human preadipocytes (20).
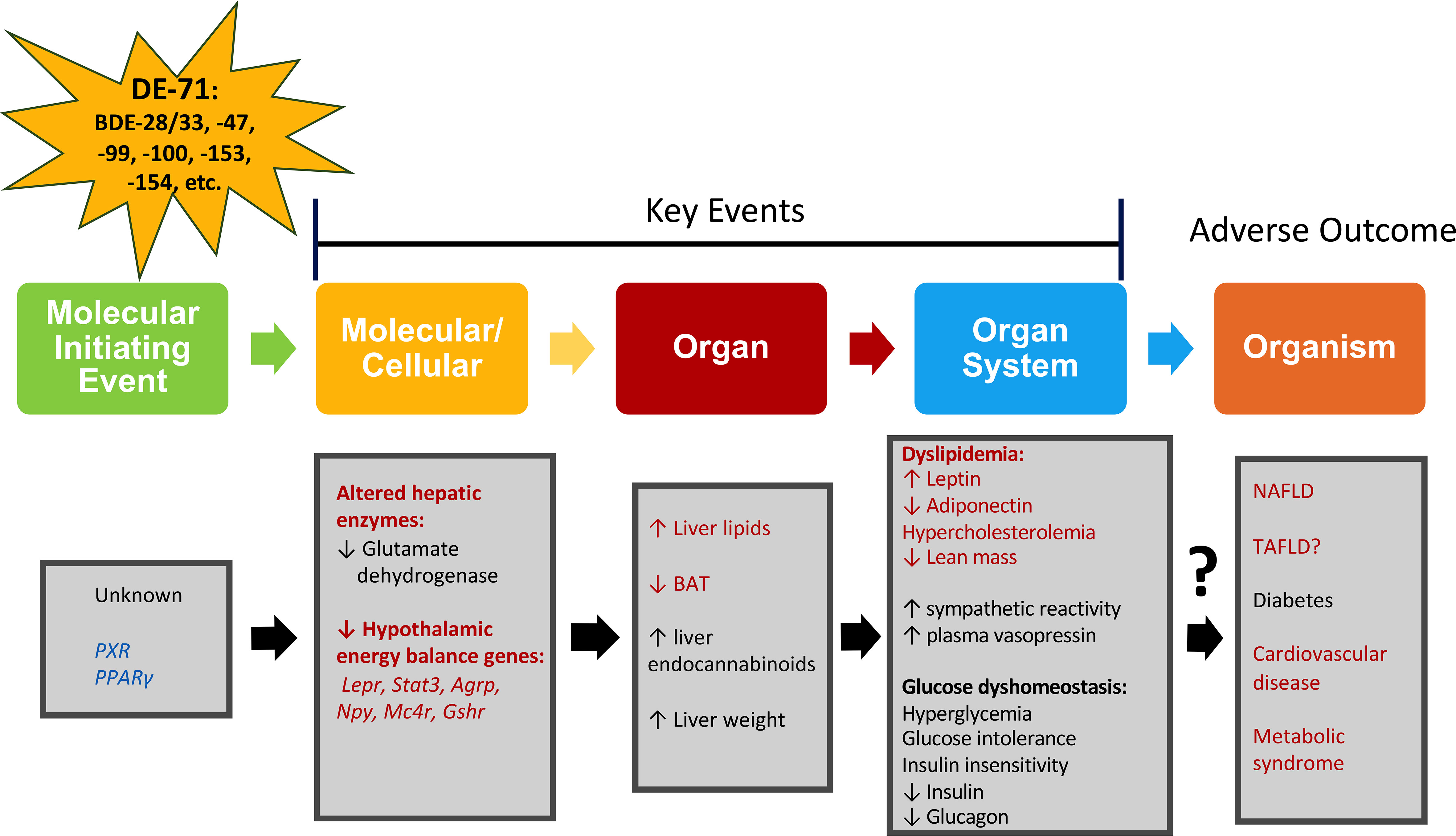
Figure 7 Adverse outcomes pathway (AOP) for metabolic alterations produced by DE-71. An AOP network portrays the plausible pathway from an initial perturbation by a penetrating toxicant called a molecular initiating event (MIE) to an adverse outcome, through different intermediate events called key events (KE) taking place at diverse levels of biological organization such as molecular, cellular, organ and organ system (93). Metrics evaluated are indicated in red (current study) and black (previously reported in (16, 57, 87). Four KEs leading to liver steatosis, diabetes, and metabolic syndrome are proposed: elevated hepatic lipid content, reduced expression of energy balance genes, increased plasma leptin, decreased plasma adiponectin. Blue text: MIE has not been determined but activation of pregnane X receptor (PXR) and peroxisome proliferator-activated receptors (PPARγ) has been suggested as part of drug-induced liver injury and steatosis (88, 89, 93). Hypothalamic energy balance genes: Leptin receptor, Lepr; Signal transducer and activator of transcription 3, Stat3; Agouti-related peptide, Agrp; Neuropeptide Y, Npy; Melanocortin-4 receptor, Mc4r; Growth hormone secretagogue receptor, Gshr; toxicant-associated fatty liver disease, TAFLD.
An aggravated inflammatory response in the liver may contribute to the elevated lipid content and dyslipidemic phenotype displayed by L-DE-71 exposed female offspring. In NAFLD livers, lipid elevation is associated with inflammation; however, the precise mechanism by which hepatic lipid accumulation and inflammation contribute to NAFLD remains poorly understood (95). We have previously shown elevated hepatic endocannabinoids in response to perinatal exposure to DE-71 in female offspring that may participate in lipid dyshomeostasis and inflammation (16). Notably, elevated N-arachidonoylethanolamine (AEA) can serve as a source of arachidonic acid but can also be metabolized by most eicosanoid biosynthetic enzymes, yielding bioactive lipids with pro-inflammatory actions (96). Although the release of proinflammatory arachidonic acid in response to PBDE exposure has not been studied in metabolic organs like liver or adipose tissue, DE-71, a mostly pentabromodiphenyl ether mixture, but not DE-79, a mostly octabromodiphenyl ether mixture, causes a dose-dependent increase in arachidonic acid release from rat neuronal cultures (97). Moreover, livers of DE-71 exposed females and dams show reduced glutamate dehydrogenase, which normally offsets fatty liver characteristics such as enhanced lipogenesis (and gluconeogenesis) (98). Mechanistic studies will be necessary to determine the cellular and biochemical targets of PBDEs that may predispose mammals to abnormal metabolic phenotypes (99).
Our findings suggest that, in contrast to male offspring, the normal lipid balance of female offspring is uniquely susceptible to environmentally relevant doses and composition of PBDEs congeners. Given that the sex steroid environment and receptor distribution in metabolic tissue are distinct between males and females, it is conceivable that PBDEs’ mechanistic actions vary by sex (100). It is plausible that PBDEs have sexually dimorphic actions on biological processes since they are agonists of estrogen receptors and antagonists of androgen receptors (101, 102). Alternatively, it has been suggested that the findings of metabolic effect modification by sex of PBDEs may be partially explained by body fat and lean mass percentage rather than sexual dimorphism. For example, a case has been made that higher gains in fat mass (in females) relative to greater lean mass (in males) may yield disproportionate sex-dependent consequences of PBDEs (103). In our hands, DE-71 did not produce increased adiposity (fat mass, increased WAT or body weight) in any of the experimental groups studied (Figure 1; Table 2).
In this study, we did not detect elevated adiposity such as fat mass, increased WAT or body weight. In contrast, the specific effect of L-DE-71 on BAT (Figure 1) in female offspring is especially significant since the latter is now recognized as playing a significant role in controlling obesity and body glucose homeostasis (104). The significantly lower BAT measured in L-DE-71 female offspring relative to vehicle controls may secondarily result from significantly elevated catecholamine levels reported by our group (16). Indeed, increased catecholamine levels and catecholamine resistance resulted in reduced BAT (105), a state that clinical trials have been designed to oppose in order to alleviate metabolic diseases. The effects of PBDEs on BAT may be secondary to PBDE-triggered upregulation of sympathetic activity; the latter is supported by previous reports from our lab and others (57, 106). Elevated sympathetic activity could also contribute to changes in glycolipid metabolism measured here and in previous works (16). The adverse lipid metabolic implications of low developmental PBDE exposure seen in adulthood warrant further study into the underlying mechanisms that could be protected or suppressed by novel approaches. Since PBDEs impact metabolomic profiles related to lipid, carbohydrate and energy metabolism in association with gut dysbiosis (107), one such treatment could involve an anti-obesogenic probiotic therapy with lipolytic consequences (108).
In conclusion, perinatal exposure to an environmentally relevant mixture/concentration of PBDEs during early life produces elevated fasting leptin, hypercholesterolemia and fatty liver in adult female offspring. In combination with the PBDE-induced diabetogenic phenotype characterized in our previous report (16), current findings suggest increased risk of MetS posed by BFRs like PBDEs. More research is critically needed to understand the molecular mechanisms and long-term adverse outcomes of early-life exposure to BFRs. Further concern is warranted by their sex-dependent effects, with developing females having greater susceptibility for metabolic disturbances, and the widespread environmental occurrence of legacy and emerging environmental flame retardants. The unique abnormal glucose and lipid metabolic phenotypes of exposed female offspring are not likely caused by gut inflammation as suggested by gut integrity experiments (Figure 5), but may result, in part, from abnormal expression of central gene markers in hypothalamic energy-regulating circuits. In particular, reduced hypothalamic Lepr and downstream STAT3 signaling with elevated fasting plasma levels of leptin may represent deregulation of the central control of lipid (and glucose) metabolism. Interestingly, L-DE-71-exposed females also show autism-relevant deficient social recognition and deregulated neuromolecular markers (34) indicating potential interactions between metabolic and behavioral disorders as speculated previously (109).
The original contributions presented in the study are included in the article/supplementary materials. Further inquiries can be directed to the corresponding author.
The animal study was reviewed and approved by The University of California, Riverside IACUC.
Conceptualization, EK and MC-C; Methodology, EK, MD, MC-C, MV, ND, and PD; Validation, EK, MV, MD, and MC-C; Formal Analysis, AB, EK, MV, MD, and MC-C; Investigation, AB, BC, DE, EK, ET, JB, JK, MV, MD, MM-G, MC-C, and PD; Writing – Original Draft, EK, AB, MD, and MC-C; Writing – Reviewing and Editing, EK and MC-C; Visualization, EK and MC-C; Resources, MM-G, MC-C, and ND; Data Curation, AB, BC, EK, MD, and MC-C; Supervision, EK, MV, and MC-C; Project Administration, EK, MD, AB, and MC-C; Funding Acquisition, EK, MM-G, MC-C, and ND. All authors reviewed and approved the final manuscript.
This work was supported by a University of California President’s Pre-Professoriate Fellowship and UCR CNAS HSI Undergraduate Research Program Fellowship to EK and UC Riverside Omnibus and Committee on Research (CoR) Grants, UCMEXUS and Academic Senate Committee on Research grants to MC-C.
We thank Drs. Prue Talbot and David Carter for their help with microscopy. We also thank Karthik Basappa, Richard Martirosian, Dr. Clara Berdasco, Crystal Luna (Curras-Collazo Lab), Drs. Donovan Argueta and Pedro Perez (DiPatrizio lab), Anthony Castro, Yagnika Patel and Dr. Cristina Flores (Martins-Green lab), Dr. Anica Sayok, Rocio Alvarez and Dr. McCole (McCole lab), Dr. Hashini Batugedara and Dr. Meera Nair (Nair Lab) for their technical assistance. We are grateful to Great Lakes Corporation for the gift of DE-71 and to Drs. F. Sladek and I. Ethell for the gift of C57BL6 mice. We thank Dr. Djurdjica Coss for the use of plate reader (Promega Glo-Max Pro, Madison, WI, USA) and Dr. Sachiko Haga-Yamanaka for use of plate shaker. We thank D. Marvin (ALPCO) for adiponectin EIA kit.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
JK is now a 2nd Lieutenant at the Uniformed Services University, Department of Defense. Her work was performed at the University of California, Riverside before becoming a military officer. However, we want to emphasize that the opinions and assertions expressed herein are those of the authors and do not necessarily reflect the official policy or position of the Uniformed Services University or the Department of Defense.
1. Alberti KG, Eckel RH, Grundy SM, Zimmet PZ, Cleeman JI, Donato KA, et al. Harmonizing the metabolic syndrome: a joint interim statement of the International Diabetes Federation Task Force on Epidemiology and Prevention; National Heart, Lung, and Blood Institute; American Heart Association; World HeartFederation; International Atherosclerosis Society; and International Association for the Study of Obesity. Obes Metab (2010) 7:63–5. doi: 10.14341/2071-8713-5281
2. Boudreau DM, Malone DC, Raebel MA, Fishman PA, Nichols GA, Feldstein AC, et al. Health care utilization and costs by metabolic syndrome risk factors. Metab Syndr Relat Disord (2009) 7:305–14. doi: 10.1089/met.2008.0070
3. Moler N. Abstract 18103: Medical Costs Associated with All Combinations of Metabolic Syndrome Components. Circulation. doi: 10.1161/circ.122.suppl_21.A18103
4. Tudurí E, Marroqui L, Dos Santos RS, Quesada I, Fuentes E, Alonso-Magdalena P. Timing of Exposure and Bisphenol-A: Implications for Diabetes Development. Front Endocrinol (2018) 9:648. doi: 10.3389/fendo.2018.00648
5. Abbasi G, Li L, Breivik K. Global Historical Stocks and Emissions of PBDEs. Environ Sci Technol (2019) 53:6330–40. doi: 10.1021/acs.est.8b07032
6. Cai K, Song Q, Yuan W, Ruan J, Duan H, Li Y, et al. Human exposure to PBDEs in e-waste areas: A review. Environ pollut (2020) 267:115634. doi: 10.1016/j.envpol.2020.115634
7. Hurley S, Goldberg D, Nelson DO, Guo W, Wang Y, Baek H-G, et al. Temporal Evaluation of Polybrominated Diphenyl Ether (PBDE) Serum Levels in Middle-Aged and Older California Women, 2011-2015. Environ Sci Technol (2017) 51:4697–704. doi: 10.1021/acs.est.7b00565
8. Darrow LA, Jacobson MH, Preston EV, Lee GE, Panuwet P, Hunter RE, et al. Predictors of Serum Polybrominated Diphenyl Ether (PBDE) Concentrations among Children Aged 1–5 Years. Environ Sci Technol (2017) 51:645–54. doi: 10.1021/acs.est.6b04696
9. Lyche JL, Rosseland C, Berge G, Polder A. Human health risk associated with brominated flame-retardants (BFRs). Environ Int (2015) 74:170–80. doi: 10.1016/j.envint.2014.09.006
10. Khalil A, Cevik SE, Hung S, Kolla S, Roy MA, Suvorov A. Developmental Exposure to 2,2′,4,4′-Tetrabromodiphenyl Ether Permanently Alters Blood-Liver Balance of Lipids in Male Mice. Front Endocrinol (2018) 9:548. doi: 10.3389/fendo.2018.00548
11. Zhang Z, Li S, Liu L, Wang L, Xiao X, Sun Z, et al. Environmental exposure to BDE47 is associated with increased diabetes prevalence: Evidence from community-based case-control studies and an animal experiment. Sci Rep (2016) 6:27854. doi: 10.1038/srep27854
12. Scoville DK, Li CY, Wang D, Dempsey JL, Raftery D, Mani S, et al. Polybrominated Diphenyl Ethers and Gut Microbiome Modulate Metabolic Syndrome–Related Aqueous Metabolites in Mice. Drug Metab Dispos (2019) 47:928–40. doi: 10.1124/dmd.119.086538
13. Helaleh M, Diboun I, Al-Tamimi N, Al-Sulaiti H, Al-Emadi M, Madani A, et al. Association of polybrominated diphenyl ethers in two fat compartments with increased risk of insulin resistance in obese individuals. Chemosphere (2018) 209:268–76. doi: 10.1016/j.chemosphere.2018.06.108
14. McIntyre RL, Kenerson HL, Subramanian S, Wang SA, Kazami M, Stapleton HM, et al. Polybrominated diphenyl ether congener, BDE-47, impairs insulin sensitivity in mice with liver-specific Pten deficiency. BMC Obes (2015) 2:3. doi: 10.1186/s40608-014-0031-3
15. Dunnick JK, Brix A, Cunny H, Vallant M, Shockley KR. Characterization of polybrominated diphenyl ether toxicity in Wistar Han rats and use of liver microarray data for predicting disease susceptibilities. Toxicol Pathol (2012) 40:93–106. doi: 10.1177/0192623311429973
16. Kozlova EV, Chinthirla BD, Pérez PA, DiPatrizio NV, Argueta DA, Phillips AL, et al. Maternal transfer of environmentally relevant polybrominated diphenyl ethers (PBDEs) produces a diabetic phenotype and disrupts glucoregulatory hormones and hepatic endocannabinoids in adult mouse female offspring. Sci Rep (2020) 10:1–17. doi: 10.1038/s41598-020-74853-9
17. Lim J-S, Lee D-H, Jacobs DR. Association of Brominated Flame Retardants With Diabetes and Metabolic Syndrome in the U.S. Population, 2003–2004. Diabetes Care (2008) 31:1802–7. doi: 10.2337/dc08-0850
18. Erkin-Cakmak A, Harley KG, Chevrier J, Bradman A, Kogut K, Huen K, et al. In utero and childhood polybrominated diphenyl ether exposures and body mass at age 7 years: the CHAMACOS study. Environ Health Perspect (2015) 123:636–42. doi: 10.1289/ehp.1408417
19. Kamstra JH, Hruba E, Blumberg B, Janesick A, Mandrup S, Hamers T, et al. Transcriptional and epigenetic mechanisms underlying enhanced in vitro adipocyte differentiation by the brominated flame retardant BDE-47. Environ Sci Technol (2014) 48:4110–9. doi: 10.1021/es405524b
20. Wen Q, Xie X, Zhao C, Ren Q, Zhang X, Wei D, et al. The brominated flame retardant PBDE 99 promotes adipogenesis via regulating mitotic clonal expansion and PPARγ expression. Sci Total Environ (2019) 670:67–77. doi: 10.1016/j.scitotenv.2019.03.201
21. Yang C, Wong C-M, Wei J, Chung ACK, Cai Z. The brominated flame retardant BDE 47 upregulates purine metabolism and mitochondrial respiration to promote adipocyte differentiation. Sci Total Environ (2018) 644:1312–22. doi: 10.1016/j.scitotenv.2018.07.087
22. Tung EWY, Boudreau A, Wade MG, Atlas E. Induction of adipocyte differentiation by polybrominated diphenyl ethers (PBDEs) in 3T3-L1 cells. PloS One (2014) 9:e94583. doi: 10.1371/journal.pone.0094583
23. Armstrong LE, Akinbo S, Slitt AL. 2,2’,4,4’,5-Pentabromodiphenyl ether induces lipid accumulation throughout differentiation in 3T3-L1 and human preadipocytes. vitro. J Biochem Mol Toxicol (2020) 34:e22485. doi: 10.1002/jbt.22485
24. Wang D, Yan J, Teng M, Yan S, Zhou Z, Zhu W. In utero and lactational exposure to BDE-47 promotes obesity development in mouse offspring fed a high-fat diet: impaired lipid metabolism and intestinal dysbiosis. Arch Toxicol (2018) 92:1847–60. doi: 10.1007/s00204-018-2177-0
25. Timper K, Brüning JC. Hypothalamic circuits regulating appetite and energy homeostasis: pathways to obesity. Dis Model Mech (2017) 10:679–89. doi: 10.1242/dmm.026609
26. Caron A, Richard D. Neuronal systems and circuits involved in the control of food intake and adaptive thermogenesis. Ann N Y Acad Sci (2017) 1391:35–53. doi: 10.1111/nyas.13263
27. Ruigrok SR, Stöberl N, Yam K-Y, de Lucia C, Lucassen PJ, Thuret S, et al. Modulation of the Hypothalamic Nutrient Sensing Pathways by Sex and Early-Life Stress. Front Neurosci (2021) 15:695367. doi: 10.3389/fnins.2021.695367
28. Burdakov D, Gerasimenko O, Verkhratsky A. Physiological changes in glucose differentially modulate the excitability of hypothalamic melanin-concentrating hormone and orexin neurons in situ. J Neurosci (2005) 25:2429–33. doi: 10.1523/JNEUROSCI.4925-04.2005
29. Contreras C, González-García I, Martínez-Sánchez N, Seoane-Collazo P, Jacas J, Morgan DA, et al. Central ceramide-induced hypothalamic lipotoxicity and ER stress regulate energy balance. Cell Rep (2014) 9:366–77. doi: 10.1016/j.celrep.2014.08.057
30. Diéguez C, Vazquez MJ, Romero A, López M, Nogueiras R. Hypothalamic control of lipid metabolism: focus on leptin, ghrelin and melanocortins. Neuroendocrinology (2011) 94:1–11. doi: 10.1159/000328122
31. Fan W, Morrison SF, Cao W-H, Yu P. Thermogenesis activated by central melanocortin signaling is dependent on neurons in the rostral raphe pallidus (rRPa) area. Brain Res (2007) 1179:61–9. doi: 10.1016/j.brainres.2007.04.006
32. National Research Council, Division on Earth and Life Studies, Institute for Laboratory Animal Research, Committee for the Update of the Guide for the Care and Use of Laboratory Animals. Guide for the Care and Use of Laboratory Animals. Eighth Edition. National Academies Press (2011). doi: 10.17226/12910
33. Woods R, Vallero RO, Golub MS, Suarez JK, Ta TA, Yasui DH, et al. Long-lived epigenetic interactions between perinatal PBDE exposure and Mecp2308 mutation. Hum Mol Genet (2012) 21:2399–411. doi: 10.1093/hmg/dds046
34. Kozlova EV, Valdez MC, Denys ME, Bishay AE, Krum JM, Rabbani KM, et al. Persistent autism-relevant behavioral phenotype and social neuropeptide alterations in female mice offspring induced by maternal transfer of PBDE congeners in the commercial mixture DE-71. Arch Toxicol (2022) 96, 335–365. doi: 10.1007/s00204-021-03163-4
35. Stapleton HM, Eagle S, Sjödin A, Webster TF. Serum PBDEs in a North Carolina toddler cohort: associations with hand wipes, house dust, and socioeconomic variables. Environ Health Perspect (2012) 120:1049–54. doi: 10.1289/ehp.1104802
36. Fischer D, Hooper K, Athanasiadou M, Athanassiadis I, Bergman A. Children show highest levels of polybrominated diphenyl ethers in a California family of four: a case study. Environ Health Perspect (2006) 114:1581–4. doi: 10.1289/ehp.8554
37. Iris E. Pentabromodiphenyl ether (CASRN 32534-81-9) (2014). Available at: https://iris.epa.gov/ChemicalLanding/&substance_nmbr=184.
38. Tinsley FC, Taicher GZ, Heiman ML. Evaluation of a quantitative magnetic resonance method for mouse whole body composition analysis. Obes Res (2004) 12:150–60. doi: 10.1038/oby.2004.20
39. Mehlem A, Hagberg CE, Muhl L, Eriksson U, Falkevall A. Imaging of neutral lipids by oil red O for analyzing the metabolic status in health and disease. Nat Protoc (2013) 8:1149–54. doi: 10.1038/nprot.2013.055
40. Ramírez-Zacarías JL, Castro-Muñozledo F, Kuri-Harcuch W. Quantitation of adipose conversion and triglycerides by staining intracytoplasmic lipids with Oil red O. Histochemistry (1992) 97:493–7. doi: 10.1007/BF00316069
41. Bankhead P, Loughrey MB, Fernández JA, Dombrowski Y, McArt DG, Dunne PD, et al. QuPath: Open source software for digital pathology image analysis. Sci Rep (2017) 7:16878. doi: 10.1038/s41598-017-17204-5
42. Zhao L, Xiao H-T, Mu H-X, Huang T, Lin Z-S, Zhong L, et al. Magnolol, a Natural Polyphenol, Attenuates Dextran Sulfate Sodium-Induced Colitis in Mice. Molecules (2017) 22:1218. doi: 10.3390/molecules22071218
43. Pfaffl MW. A new mathematical model for relative quantification in real-time RT–PCR. Nucleic Acids Res (2001) 29:e45–5. doi: 10.1093/nar/29.9.e45
44. Nguyen TT, Kambe Y, Kurihara T, Nakamachi T, Shintani N, Hashimoto H, et al. Pituitary Adenylate Cyclase-Activating Polypeptide in the Ventromedial Hypothalamus Is Responsible for Food Intake Behavior by Modulating the Expression of Agouti-Related Peptide in Mice. Mol Neurobiol (2020) 57:2101–14. doi: 10.1007/s12035-019-01864-7
45. Zheng J, Xiao X, Zhang Q, Yu M, Xu J, Wang Z, et al. Maternal and post-weaning high-fat, high-sucrose diet modulates glucose homeostasis and hypothalamic POMC promoter methylation in mouse offspring. Metab Brain Dis (2015) 30:1129–37. doi: 10.1007/s11011-015-9678-9
46. Grundy SM, Adams-Huet B, Vega GL. Variable contributions of fat content and distribution to metabolic syndrome risk factors. Metab Syndr Relat Disord (2008) 6:281–8. doi: 10.1089/met.2008.0026
47. Li W-C, Hsiao K-Y, Chen I-C, Chang Y-C, Wang S-H, Wu K-H. Serum leptin is associated with cardiometabolic risk and predicts metabolic syndrome in Taiwanese adults. Cardiovasc Diabetol (2011) 10:36. doi: 10.1186/1475-2840-10-36
48. Lee S, Kwak H-B. Role of adiponectin in metabolic and cardiovascular disease. J Exerc Rehabil (2014) 10:54–9. doi: 10.12965/jer.140100
49. Jeppesen J, Hein HO, Suadicani P, Gyntelberg F. High triglycerides and low HDL cholesterol and blood pressure and risk of ischemic heart disease. Hypertension (2000) 36:226–32. doi: 10.1161/01.hyp.36.2.226
50. Cotter TG, Rinella M. Nonalcoholic Fatty Liver Disease 2020: The State of the Disease. Gastroenterology (2020) 158:1851–64. doi: 10.1053/j.gastro.2020.01.052
51. Marchesini G, Bugianesi E, Forlani G, Cerrelli F, Lenzi M, Manini R, et al. Nonalcoholic fatty liver, steatohepatitis, and the metabolic syndrome. Hepatology (2003) 37:917–23. doi: 10.1053/jhep.2003.50161
52. Dunnick JK, Shockley KR, Pandiri AR, Kissling GE, Gerrish KE, Ton TV, et al. PBDE-47 and PBDE mixture (DE-71) toxicities and liver transcriptomic changes at PND 22 after in utero/postnatal exposure in the rat. Arch Toxicol (2018) 92:3415–33. doi: 10.1007/s00204-018-2292-y
53. Turner JR. Intestinal mucosal barrier function in health and disease. Nat Rev Immunol (2009) 9:799–809. doi: 10.1038/nri2653
54. Vandenberg LN, Colborn T, Hayes TB, Heindel JJ, Jacobs DR Jr, Lee D-H, et al. Hormones and endocrine-disrupting chemicals: low-dose effects and nonmonotonic dose responses. Endocr Rev (2012) 33:378–455. doi: 10.1210/er.2011-1050
55. Bruell JH, Daroczy AF, Hellerstein HK. Strain and sex differences in serum cholesterol levels of mice. Science (1962) 135:1071–2. doi: 10.1126/science.135.3508.1071
56. Metabolic syndrome - what is metabolic syndrome? . Available at: https://www.nhlbi.nih.gov/health/metabolic-syndrome (Accessed July 15, 2022).
57. Shah A, Coburn CG, Watson-Siriboe A, Whitley R, Shahidzadeh A, Gillard ER, et al. Altered cardiovascular reactivity and osmoregulation during hyperosmotic stress in adult rats developmentally exposed to polybrominated diphenyl ethers (PBDEs). Toxicol Appl Pharmacol (2011) 256:103–13. doi: 10.1016/j.taap.2011.07.014
58. Sanyal D, Mukherjee P, Raychaudhuri M, Ghosh S, Mukherjee S, Chowdhury S. Profile of liver enzymes in non-alcoholic fatty liver disease in patients with impaired glucose tolerance and newly detected untreated type 2 diabetes. Indian J Endocrinol Metab (2015) 19:597–601. doi: 10.4103/2230-8210.163172
59. Tartu S, Lille-Langøy R, Størseth TR, Bourgeon S, Brunsvik A, Aars J, et al. Multiple-stressor effects in an apex predator: combined influence of pollutants and sea ice decline on lipid metabolism in polar bears. Sci Rep (2017) 7:16487. doi: 10.1038/s41598-017-16820-5
60. Hoppe AA, Carey GB. Polybrominated diphenyl ethers as endocrine disruptors of adipocyte metabolism. Obesity (2007) 15:2942–50. doi: 10.1038/oby.2007.351
61. Suvorov A, Naumov V, Shtratnikova V, Logacheva M, Shershebnev A, Wu H, et al. Rat liver epigenome programing by perinatal exposure to 2,2’,4’4’-tetrabromodiphenyl ether. Epigenomics (2020) 12:235–49. doi: 10.2217/epi-2019-0315
62. Lonardo A, Nascimbeni F, Ballestri S, Fairweather D, Win S, Than TA, et al. Sex Differences in Nonalcoholic Fatty Liver Disease: State of the Art and Identification of Research Gaps. Hepatology (2019) 70:1457–69. doi: 10.1002/hep.30626
63. Wahlang B, Beier JI, Clair HB, Bellis-Jones HJ, Falkner KC, McClain CJ, et al. Toxicant-associated steatohepatitis. Toxicol Pathol (2013) 41:343–60. doi: 10.1177/0192623312468517
64. Loria P, Lonardo A, Anania F. Liver and diabetes. A vicious circle. Hepatol Res (2013) 43:51–64. doi: 10.1111/j.1872-034X.2012.01031.x
65. Leijs MM, Koppe JG, Vulsma T, Olie K, van Aalderen WMC, de Voogt P, et al. Alterations in the programming of energy metabolism in adolescents with background exposure to dioxins, dl-PCBs and PBDEs. PloS One (2017) 12:e0184006. doi: 10.1371/journal.pone.0184006
66. Windham GC, Pinney SM, Sjodin A, Lum R, Jones RS, Needham LL, et al. Body burdens of brominated flame retardants and other persistent organo-halogenated compounds and their descriptors in US girls. Environ Res (2010) 110:251–7. doi: 10.1016/j.envres.2010.01.004
67. Rotundo L, Persaud A, Feurdean M, Ahlawat S, Kim H-S. The Association of leptin with severity of non-alcoholic fatty liver disease: A population-based study. Clin Mol Hepatol (2018) 24:392–401. doi: 10.3350/cmh.2018.0011
68. D’souza AM, Neumann UH, Glavas MM, Kieffer TJ. The glucoregulatory actions of leptin. Mol Metab (2017) 6:1052–65. doi: 10.1016/j.molmet.2017.04.011
69. Shabalala SC, Dludla PV, Mabasa L, Kappo AP, Basson AK, Pheiffer C, et al. The effect of adiponectin in the pathogenesis of non-alcoholic fatty liver disease (NAFLD) and the potential role of polyphenols in the modulation of adiponectin signaling. BioMed Pharmacother (2020) 131:110785. doi: 10.1016/j.biopha.2020.110785
70. Turyk M, Fantuzzi G, Persky V, Freels S, Lambertino A, Pini M, et al. Persistent organic pollutants and biomarkers of diabetes risk in a cohort of Great Lakes sport caught fish consumers. Environ Res (2015) 140:335–44. doi: 10.1016/j.envres.2015.03.037
71. Koh KK, Park SM, Quon MJ. Leptin and cardiovascular disease: response to therapeutic interventions. Circulation (2008) 117:3238–49. doi: 10.1161/CIRCULATIONAHA.107.741645
72. Buettner C, Muse ED, Cheng A, Chen L, Scherer T, Pocai A, et al. Leptin controls adipose tissue lipogenesis via central, STAT3-independent mechanisms. Nat Med (2008) 14:667–75. doi: 10.1038/nm1775
73. Walley SN, Krumm EA, Yasrebi A, Wiersielis KR, O’Leary S, Tillery T, et al. Maternal organophosphate flame-retardant exposure alters offspring feeding, locomotor and exploratory behaviors in a sexually-dimorphic manner in mice. J Appl Toxicol (2021) 41:442–57. doi: 10.1002/jat.4056
74. Marraudino M, Bo E, Carlini E, Farinetti A, Ponti G, Zanella I, et al. Hypothalamic Expression of Neuropeptide Y (NPY) and Pro-OpioMelanoCortin (POMC) in Adult Male Mice Is Affected by Chronic Exposure to Endocrine Disruptors. Metabolites (2021) 11. doi: 10.3390/metabo11060368
75. Gil-Campos M, Aguilera CM, Cañete R, Gil A. Ghrelin: a hormone regulating food intake and energy homeostasis. Br J Nutr (2006) 96:201–26. doi: 10.1079/bjn20061787
76. Wren AM, Small CJ, Abbott CR, Dhillo WS, Seal LJ, Cohen MA, et al. Ghrelin causes hyperphagia and obesity in rats. Diabetes (2001) 50:2540–7. doi: 10.2337/diabetes.50.11.2540
77. Fan W, Boston BA, Kesterson RA, Hruby VJ, Cone RD. Role of melanocortinergic neurons in feeding and the agouti obesity syndrome. Nature (1997) 385:165–8. doi: 10.1038/385165a0
78. Albarado DC, McClaine J, Stephens JM, Mynatt RL, Ye J, Bannon AW, et al. Impaired coordination of nutrient intake and substrate oxidation in melanocortin-4 receptor knockout mice. Endocrinology (2004) 145:243–52. doi: 10.1210/en.2003-0452
79. Li G, Zhang Y, Rodrigues E, Zheng D, Matheny M, Cheng K-Y, et al. Melanocortin activation of nucleus of the solitary tract avoids anorectic tachyphylaxis and induces prolonged weight loss. Am J Physiol Endocrinol Metab (2007) 293:E252–8. doi: 10.1152/ajpendo.00451.2006
80. Vail GM, Roepke TA. Organophosphate Flame Retardants Excite Arcuate Melanocortin Circuitry and Increase Neuronal Sensitivity to Ghrelin in Adult Mice. Endocrinology (2020) 161. doi: 10.1210/endocr/bqaa168
81. Michaud JL. The developmental program of the hypothalamus and its disorders. Clin Genet (2001) 60:255–63. doi: 10.1034/j.1399-0004.2001.600402.x
82. Snow SJ, Henriquez AR, Costa DL, Kodavanti UP. Neuroendocrine Regulation of Air Pollution Health Effects: Emerging Insights. Toxicol Sci (2018) 164:9–20. doi: 10.1093/toxsci/kfy129
83. Sharma R, Kodavanti PRS. In vitro effects of polychlorinated biphenyls and hydroxy metabolites on nitric oxide synthases in rat brain. Toxicol Appl Pharmacol (2002) 178:127–36. doi: 10.1006/taap.2001.9328
84. Morse DC, Plug A, Wesseling W, van den Berg KJ, Brouwer A. Persistent alterations in regional brain glial fibrillary acidic protein and synaptophysin levels following pre- and postnatal polychlorinated biphenyl exposure. Toxicology and applied pharmacology (1996) 139(2):252–61. doi: 10.1006/taap.1996.0164
85. Toniazzo AP, Arcego D M, Lazzaretti C, Lampert C, N Weis S, Proto-Siqueira R, et al. Sex-specific effects of prepubertal stress and high-fat diet on leptin signaling in rats. Nutrition (2018) 50:18–25. doi: 10.1016/j.nut.2017.10.018
86. Taveau C, Chollet C, Waeckel L, Desposito D, Bichet DG, Arthus M-F, et al. Vasopressin and hydration play a major role in the development of glucose intolerance and hepatic steatosis in obese rats. Diabetologia (2015) 58:1081–1090. doi: 10.1007/s00125-015-3496-9
87. Gutierrez R, Spurgin K, Prien A, Curras-Collazo M. Developmental Exposure to Indoor Flame Retardants Disrupts Sympathetic and Hypothalamic-Pituitary-Adrenal (HPA) Axis Activity in Osmotic Challenged Rats. FASEB J (2015) 29. doi: 10.1096/fasebj.29.1_supplement.650.1
88. Mellor CL, Steinmetz FP, Cronin MTD. The identification of nuclear receptors associated with hepatic steatosis to develop and extend adverse outcome pathways. Crit Rev Toxicol (2016) 46:138–52. doi: 10.3109/10408444.2015.1089471
89. Vinken M. Adverse Outcome Pathways and Drug-Induced Liver Injury Testing. Chem Res Toxicol (2015) 28:1391–7. doi: 10.1021/acs.chemrestox.5b00208
90. Sanders JM, Burka LT, Smith CS, Black W, James R, Cunningham ML. Differential expression of CYP1A, 2B, and 3A genes in the F344 rat following exposure to a polybrominated diphenyl ether mixture or individual components. Toxicol Sci (2005) 88:127–33. doi: 10.1093/toxsci/kfi288
91. Swedenborg E, Rüegg J, Mäkelä S, Pongratz I. Endocrine disruptive chemicals: mechanisms of action and involvement in metabolic disorders. J Mol Endocrinol (2009) 43:1–10. doi: 10.1677/JME-08-0132
92. Nguyen P, Leray V, Diez M, Serisier S, Le Bloc’h J, Siliart B, et al. Liver lipid metabolism. J Anim Physiol Anim Nutr (2008) 92:272–83. doi: 10.1111/j.1439-0396.2007.00752.x
93. Negi CK, Bajard L, Kohoutek J, Blaha L. An adverse outcome pathway based in vitro characterization of novel flame retardants-induced hepatic steatosis. Environ pollut (2021) 289:117855. doi: 10.1016/j.envpol.2021.117855
94. Luthe G, Jacobus JA, Robertson LW. Receptor interactions by polybrominated diphenyl ethers versus polychlorinated biphenyls: A theoretical structure–activity assessment. Environ Toxicol Pharmacol (2008) 25:202–10. doi: 10.1016/j.etap.2007.10.017
95. Tsuru H, Osaka M, Hiraoka Y, Yoshida M. HFD-induced hepatic lipid accumulation and inflammation are decreased in Factor D deficient mouse. Sci Rep (2020) 10:17593. doi: 10.1038/s41598-020-74617-5
96. Turcotte C, Chouinard F, Lefebvre JS, Flamand N. Regulation of inflammation by cannabinoids, the endocannabinoids 2-arachidonoyl-glycerol and arachidonoyl-ethanolamide, and their metabolites. J Leukoc Biol (2015) 97:1049–70. doi: 10.1189/jlb.3RU0115-021R
97. Kodavanti PRS, Derr-Yellin EC. Differential effects of polybrominated diphenyl ethers and polychlorinated biphenyls on [3H]arachidonic acid release in rat cerebellar granule neurons. Toxicol Sci (2002) 68:451–7. doi: 10.1093/toxsci/68.2.451
98. Han H-S, Kang G, Kim JS, Choi BH, Koo S-H. Regulation of glucose metabolism from a liver-centric perspective. Exp Mol Med (2016) 48:e218. doi: 10.1038/emm.2015.122
99. Casals-Casas C, Desvergne B. Endocrine Disruptors: From Endocrine to Metabolic Disruption. Annu Rev Physiol (2011) 73:135–62. doi: 10.1146/annurev-physiol-012110-142200
100. Grün F, Blumberg B. Perturbed nuclear receptor signaling by environmental obesogens as emerging factors in the obesity crisis. Rev Endocr Metab Disord (2007) 8:161–71. doi: 10.1007/s11154-007-9049-x
101. Meerts IA, Letcher RJ, Hoving S, Marsh G, Bergman A, Lemmen JG, et al. In vitro estrogenicity of polybrominated diphenyl ethers, hydroxylated PDBEs, and polybrominated bisphenol A compounds. Environ Health Perspect (2001) 109:399–407. doi: 10.1289/ehp.01109399
102. Stoker TE, Cooper RL, Lambright CS, Wilson VS, Furr J, Gray LE. In vivo and in vitro anti-androgenic effects of DE-71, a commercial polybrominated diphenyl ether (PBDE) mixture. Toxicol Appl Pharmacol (2005) 207:78–88. doi: 10.1016/j.taap.2005.05.010
103. Vuong AM, Braun JM, Wang Z, Yolton K, Xie C, Sjodin A, et al. Exposure to polybrominated diphenyl ethers (PBDEs) during childhood and adiposity measures at age 8 years. Environ Int (2019) 123:148–55. doi: 10.1016/j.envint.2018.11.050
104. Sponton CH, Hosono T, Taura J, Jedrychowski MP, Yoneshiro T, Wang Q, et al. The regulation of glucose and lipid homeostasis via PLTP as a mediator of BAT-liver communication. EMBO Rep (2020) 21:e49828. doi: 10.15252/embr.201949828
105. Alcalá M, Calderon-Dominguez M, Serra D, Herrero L, Viana M. Mechanisms of Impaired Brown Adipose Tissue Recruitment in Obesity. Front Physiol (2019) 10:94. doi: 10.3389/fphys.2019.00094
106. Gump BB, Yun S, Kannan K. Polybrominated diphenyl ether (PBDE) exposure in children: possible associations with cardiovascular and psychological functions. Environ Res (2014) 132:244–50. doi: 10.1016/j.envres.2014.04.009
107. Gao H, Wan X, Xiao B, Yang K, Wang Y, Zhang C, et al. Impacts of PBDE-47 exposure before, during and after pregnancy on the maternal gut microbiome and its association with host metabolism. Ecotoxicol Environ Saf (2021) 222:112530. doi: 10.1016/j.ecoenv.2021.112530
108. Guha D, Mukherjee R, Aich P. Effects of two potential probiotic Lactobacillus bacteria on adipogenesis. vitro. Life Sci (2021) 278:119538. doi: 10.1016/j.lfs.2021.119538
Keywords: metabolic syndrome & type II diabetes, polybrominated diphenyl ethers (PBDEs), maternal, leptin - adiponectin, fatty liver, endocrine-disrupting chemical (EDC), hypothalamus, dyslipidemia (DLP)
Citation: Kozlova EV, Denys ME, Benedum J, Valdez MC, Enriquez D, Bishay AE, Chinthirla BD, Truong E, Krum JM, DiPatrizio NV, Deol P, Martins-Green M and Curras-Collazo MC (2022) Developmental exposure to indoor flame retardants and hypothalamic molecular signatures: Sex-dependent reprogramming of lipid homeostasis. Front. Endocrinol. 13:997304. doi: 10.3389/fendo.2022.997304
Received: 18 July 2022; Accepted: 25 August 2022;
Published: 30 September 2022.
Edited by:
Edith Sánchez-Jaramillo, National Institute of Psychiatry Ramon de la Fuente Muñiz (INPRFM), MexicoReviewed by:
Tomohiko Isobe, National Institute for Environmental Studies (NIES), JapanCopyright © 2022 Kozlova, Denys, Benedum, Valdez, Enriquez, Bishay, Chinthirla, Truong, Krum, DiPatrizio, Deol, Martins-Green and Curras-Collazo. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Margarita C. Curras-Collazo, bWN1ckB1Y3IuZWR1
†ORCID ID: Elena V. Kozlova, orcid.org/0000-0002-4691-6618
Maximilian E. Denys, orcid.org/0000-0002-7423-6265
Matthew C. Valdez, orcid.org/0000-0002-8227-7921
Anthony E. Bishay, orcid.org/0000-0002-6057-3770
Poonamjot Deol, orcid.org/0000-0002-7109-0619
Margarita C. Curras-Collazo, orcid.org/0000-0002-0189-4179
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.