 Adriana C. H. Neven
Adriana C. H. Neven Aya Mousa
Aya Mousa Jacqueline A. Boyle
Jacqueline A. Boyle Helena J. Teede
Helena J. Teede- 1Monash Centre for Health Research and Implementation (MCHRI), School of Public Health and Preventive Medicine, Monash University, Clayton, VIC, Australia
- 2Monash Department of Obstetrics and Gynaecology, Monash Health, Clayton, VIC, Australia
- 3Eastern Health Clinical School, Monash University, Box Hill, VIC, Australia
During pregnancy, the fetoplacental unit is key in the pronounced physiological endocrine changes which support pregnancy, fetal development and survival, birth and lactation. In healthy women, pregnancy is characterized by changes in insulin sensitivity and increased maternal androgen levels. These are accompanied by a suite of mechanisms that support fetal growth, maintain glucose homeostasis and protect both mother and fetus from adverse effects of pregnancy induced insulin and androgen excess. In pregnancies affected by endocrine, metabolic disorders such as polycystic ovary syndrome (PCOS), diabetes and obesity, there is an imbalance of beneficial and adverse impacts of pregnancy induced endocrine changes. These inter-related conditions are characterized by an interplay of hyperinsulinemia and hyperandrogenism which influence fetoplacental function and are associated with adverse pregnancy outcomes including hypertensive disorders of pregnancy, macrosomia, preterm delivery and caesarean section. However, the exact underlying mechanisms and relationships of the endocrine and metabolic milieu in these disorders and the impact they have on the prenatal endocrine environment and developing fetus remain poorly understood. Here we aim to review the complex endocrine and metabolic interactions in healthy women during normal pregnancies and those in pregnancies complicated by hyperinsulinemic disorders (PCOS, diabetes and obesity). We also explore the relationships between these endocrine and metabolic differences and the fetoplacental unit, pregnancy outcomes and the developing fetus.
1. Introduction
During pregnancy, pronounced physiological endocrine changes support maintenance of pregnancy, fetal development and survival, birth and lactation. Hormonal factors including progesterone, estrogens, androgens, and glucocorticoids are important during critical developmental windows of pregnancy (1). Increasingly, links between maternal endocrine metabolic conditions and pregnancy outcomes are recognized, with insulin resistance and hyperandrogenism central in these interactions. During normal progression of pregnancy, insulin resistance (IR) increases in the mother to provide energy for the growing fetus and (2) androgens increase, regulating key processes during pregnancy and parturition (3). These changes are balanced by pregnancy-specific mechanisms that are activated to maintain glucose homeostasis and to protect both the mother and fetus from pregnancy-induced insulin and androgen excess (4, 5).
Endocrine metabolic disorders such as polycystic ovary syndrome (PCOS), type 1 and type 2 diabetes (T1D, T2D), gestational diabetes (GDM) and maternal obesity are common and the prevalence of these conditions is rising globally, in line with increasing obesity. These conditions influence the endocrine environment and impact of the balance of beneficial and protective mechanisms in pregnancy. PCOS, the most common endocrinopathy in reproductive age women, affects 8 to 21% of this population. (6) PCOS is diagnosed (Rotterdam criteria) based on the presence of two of three features: oligo- or anovulation, hyperandrogenism (clinical or biochemical), and polycystic ovary morphology on ultrasound (7). Pre-existing diabetes is estimated to affect around 2% of pregnancies, doubling from 1990 to 2020 (8), while diabetes in pregnancy (DIP), either from pre-existing diabetes or GDM, affects approximately 16% of pregnancies globally (9). Overweight and obesity, characterized by an elevated body mass index (BMI) of 25 kg/m (2) and 30 kg/m (2) respectively, currently affects 26% and 25% of US women of reproductive age (20-39 years) (10). Globally, 18% and 20% of pregnant women start pregnancy overweight or obese and 47% gain gestational weight above that recommended by guidelines (11). These conditions are interrelated, with GDM prevalence closely associated with maternal obesity (12) and with PCOS (13).
According to the Barker hypothesis, the health and development of children is directly affected by fetal programming in utero during critical stages of development in early embryonic and fetal life (14). Critically, the presence of PCOS, diabetes or obesity is associated with higher rates of hypertensive disorders of pregnancy, macrosomia, pre-term delivery and caesarean sections, among other adverse impacts (15–19). These conditions may also lead to unfavorable long-term developmental programming in offspring and manifest as higher rates of metabolic disorders in adulthood, adverse cardiometabolic profiles and even mortality (16, 20). However, the exact underlying mechanisms and relationships between the endocrine and metabolic milieu in these disorders and the mechanisms and impacts they may have on the developing fetus are not fully understood.
A complex interplay of hyperinsulinemia and hyperandrogenism alters the endocrine milieu in pregnant women with PCOS, diabetes and obesity. In non-pregnant women with PCOS, insulin resistance (IR) and compensatory hyperinsulinemia contribute to hyperandrogenic traits as insulin facilitates androgen secretion (21). In turn, androgen excess itself facilitates metabolic dysfunction (21). This persists in pregnancy when women with PCOS are reported to have higher IR, insulin and androgens compared to women without PCOS (22–26). Systemic hyperinsulinemia in pregnancy also occurs with endogenous factors such as obesity (27), GDM (28) and T2D or exogenous factors (such as insulin use in type 1 diabetes or GDM) (29). Associations between maternal hyperinsulinemia and adverse neonatal outcomes are poorly understood, but some studies point to a role of hyperinsulinemia in macrosomia, neurological disorders and endothelial dysfunction in the neonate and impaired glucose tolerance in childhood (30, 31). These endocrine metabolic conditions are also characterized by hyperandrogenism. Fetal exposure to high androgen concentrations is associated with virilization (32–34), intra-uterine growth restriction (IUGR) (34, 35), placental differentiation (36), reproductive and metabolic dysfunction (37–42), adverse cardiac programming (43–45), and behavioral outcomes later in life (46–48). Hyperandrogenism is also associated with high levels of Anti-Müllerian hormone (AMH) in women with PCOS (49). AMH is a member of the transforming growth factor beta (TGFβ) family, produced by granulosa cells of the ovarian (pre-) antral follicles. Most non-pregnant women with PCOS exhibit higher levels of AMH than women without PCOS, with a positive correlation with androstenedione (A4) and testosterone (T) (49). Further, a positive correlation between AMH levels and HOMA-IR has been reported in non-obese women with PCOS (50). Elevated AMH concentrations have also been seen in prepubescent girls with T1D, suggesting a stimulatory effect of insulin therapy on granulosa cells (51). Recently, a number of cohort studies have reported elevated levels of AMH in pregnant women with PCOS versus controls (22, 23, 25, 52, 53), and positive correlations between AMH and maternal total T levels (53). Altered levels of AMH are associated with increased risk of miscarriage (53), lower live birth rates in women with PCOS undergoing assisted reproductive technology (54), milder forms of Mullerian anomalies (22), preterm delivery (55–57) and PCOS features in adulthood (53).
With the rising incidence of these endocrine metabolic disorders in pregnancy, an improved understanding of their impact on pregnancy and on the developing fetus and child is pertinent. Whilst this is a broad topic with other factors involved such as lipid metabolism, growth factors and cytokines, here we will focus on core hormonal factors linking endocrine and metabolic dysfunction - hyperinsulinemia, hyperandrogenism and AMH. This review aims to explore the complex endocrine and metabolic interactions in healthy women during normal pregnancies and those in pregnancies complicated by hyperinsulinemic disorders (PCOS, diabetes and obesity). We also explore the relationships between these endocrine and metabolic differences and the fetoplacental unit, the developing fetus and child health.
2. Endocrine and metabolic interactions in healthy pregnant women
During pregnancy, pronounced physiological endocrine changes occur to support fetal development and survival, birth and lactation. Effective exchange of nutritive and metabolic products is essential for intrauterine life. Although the endocrine systems are compartmentalized, processes within the fetal, placental and maternal compartments, complement each other, functioning as a unit that utilizes building materials from the maternal compartment for steroidogenic activities (58, 59). The fetus influences maternal adaptations, and thereby its own growth and development. Moreover, mechanisms involved in the regulation and metabolism of sex steroids during pregnancy could be dependent on adaptations in the maternal/placental compartment, determined by the fetal gender (60). Relative contributions of sex steroids shift from maternal ovaries and adrenals to the fetoplacental unit. The fetoplacental unit also acts as a barrier to some substances crossing to the fetus, with maternal hormones larger than 0.7 kDA barely passing the placenta. This ensures that the fetal endocrine environment is largely independent of maternal hormones. Whilst steroids are highly lipophilic and cross the placenta in both directions, most of them are metabolized en route (59). Although the placenta functions as a hypothalamic-pituitary-end organ-like entity, placental function is more complex.
In the sections below, we will describe the endocrine actions of the fetoplacental unit in relation to reproductive and steroid hormones and the impact of insulin, androgens and AMH on these hormones in healthy pregnant women.
2.1. Actions of the fetoplacental unit
Taking over from LH eight days after ovulation, hCG supports survival of the corpus luteum. A maximum level is reached at 8-10 weeks of gestation, then decreases to 10,000-20,000 IU/L by 18-20 weeks and remains at that level to term. Similar to the pituitary secretion of gonadotropins, inhibin and progesterone are inhibiting factors and estrogen and activin are enhancing factors of GnRH-hCG regulation. Maternal serum FSH concentrations are almost undetectable, and LH concentrations slowly decline until birth (3). Elevated levels of hCG in the second trimester are associated with miscarriage, small-for-gestational age infants, pre-eclampsia and preterm delivery (58).
2.1.1. Progesterone
Progesterone prepares and maintains the endometrium to allow implantation, is important in maternal immunologic responses to fetal antigens and has a role in parturition. Initially, the corpus luteum is responsible for its production until about 10 weeks of gestation. After a transition period, the placental unit functions as the major source of progesterone synthesis and maternal circulating levels increase across pregnancy to reach a peak concentration (in order of 130 ng/ml) in the third trimester (59) whilst the fetal contribution is negligible (58). The precursor for progesterone derives from maternal cholesterol and production is therefore independent of uteroplacental perfusion or fetal wellbeing. Firstly, cholesterol is converted to pregnenolone by cytochrome P450scc (CYP11A1). Secondly, Pregnenolone is converted to progesterone by type 1 3β-hydroxysteroid dehydrogenase (HSD3B1) (59). The fetus uses progesterone to synthesize biologically important corticosteroids such as cortisol and aldosterone (58).
2.1.2. Estrogens
Three forms of estrogen are produced in women: estrone (E1), estradiol (E2) and estriol (E3) (61). During pregnancy, E1 and E2 production is increased about a 100-fold, and E3 secretion by about a 1000-fold. This increase is due mainly to estrogen production in the placenta (59). Estrogens contribute to progesterone production, maternal cardiovascular adaptations, blood volume and uteroplacental blood flow. They also regulate genes involved in cholesterol supply to the placenta which is important for fetal and placental steroid hormone production (62). Additionally, estrogens exert effects on the developing fetus by maintaining intrauterine homeostasis, promoting maturation of fetal organs, regulating the fetal neuroendocrine system and regulating timing of parturition (63).
Human placental estrogen synthesis depends on DHEA and its sulfated form (DHEAS), produced from maternal androgens in the early months of gestation and derived from fetal androgens by the 20th week of pregnancy (64). Further conversion of DHEAS to E1 and E2 requires four key enzymes. Placental sulfatases convert DHEAS into DHEA. Placental type I 3β-hydroxysteroid dehydrogenase (HSD)/Δ5Δ4isomerase (HSD3B1), converts DHEA into Δ4-androstenedione. Aromatase irreversibly converts Δ4-androstenedione into estrone (E1). Finally, E1 is converted into E2 by 17-β-HSD type 1 (HSD17B1) and then delivered to the maternal circulation (64). Placental E3 formation is carried out by 16a hydroxylation in the fetal liver (58), and then metabolized by placental sulfate, 3β-hydroxysteroid dehydrogenase, 17-hydroxysteroid oxidoreductase and maternal P450 aromatase to yield estriol (59). Moreover, the human fetal liver responds by increasing production of SHBG in order to bind estrogen in the circulation and limit levels of bioactive estrogens (65). These processes all ensure that biological effects of potent steroids are blocked. After birthing this ability disappears and estrogen concentrations decline rapidly in the postpartum period (3). Because estrogen production is dependent on fetal and placental steroidogenic cooperation, the amount of estrogen (especially estriol) present in the maternal blood or urine reflects fetal and placental enzymatic capability and, hence, well-being (58). In fact, Wan et al. reported lower levels of estrogen in preeclampsia and speculate that this reduction may be due to the impairment of placental function in preeclampsia (66).
2.1.3. Activin and inhibin
Inhibin, produced by the placenta, shows increased levels during pregnancy (3). It peaks at 8 weeks gestation, then decreases (67), and increases again in the third trimester to 100-fold more than that during the normal menstrual cycle (3). Inhibin and estrogen account for the suppression of maternal gonadotropins during pregnancy (58). Activin A is also increased during pregnancy, with stable levels from 8 to 24 weeks and then increasing to a 100-fold of that during the normal menstrual cycle (58). Inhibin and Activin act as regulators within the placenta for the production of GnRH, hCG and steroids; inhibin is inhibitory and activin is stimulatory. Abnormal concentrations of Inhibin A have been associated with miscarriage, fetal growth restriction, gestational diabetes, and pre-eclampsia (68, 69). Elevated levels of Activin-A are associated with preeclampsia (70–72).
A summary of steroid synthesis during pregnancy is provided in Figure 1 with details in Table 1.
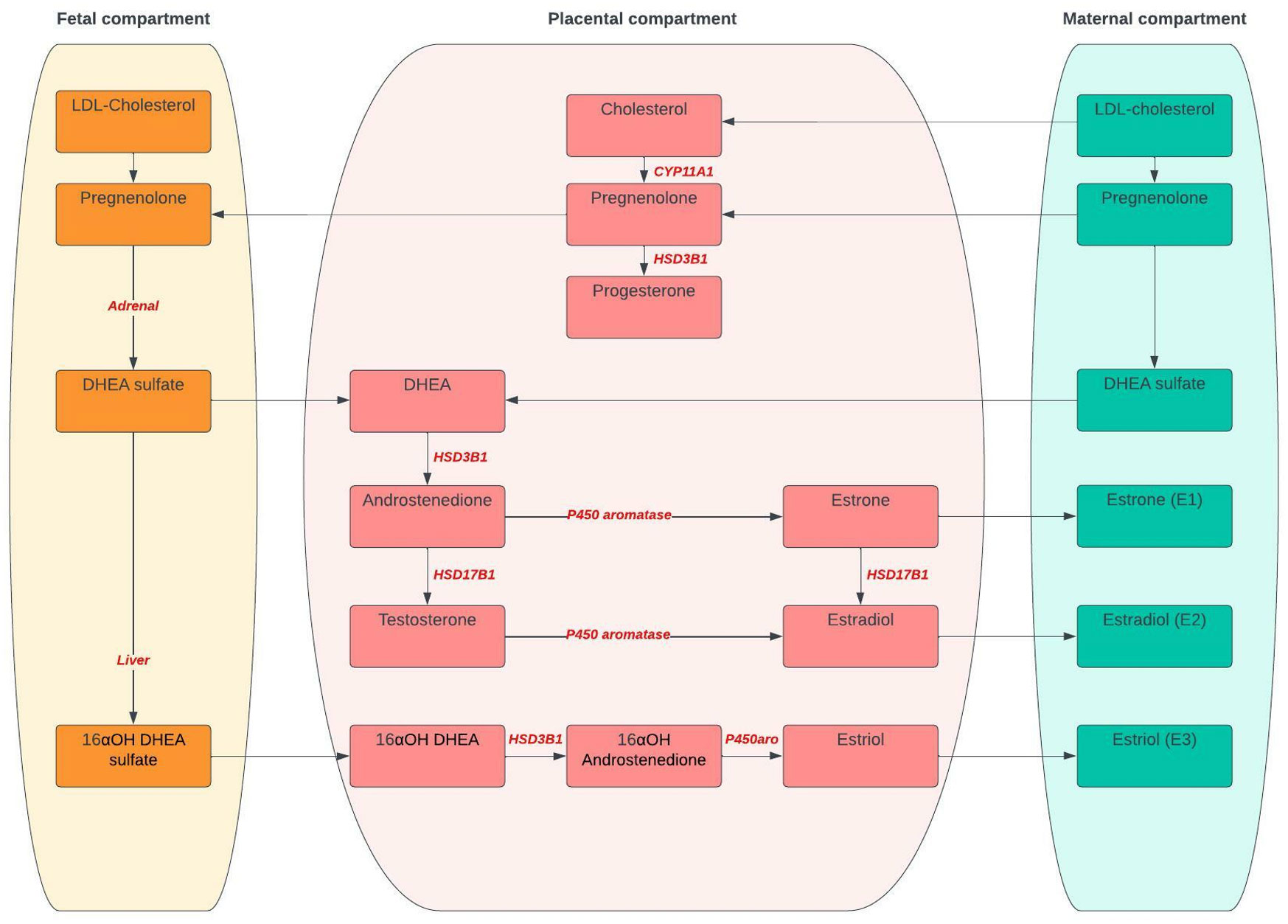
Figure 1 Steroid synthesis during pregnancy. CYP11A1, Cytochrome P450 side-change cleavage enzyme; HSD3B1, 3β-hydroxysteroid dehydrogenase 1; HSD17B1, 17-β-HSD type 1; DHEA, Dehydroepiandrosterone; 16αOH; 16α-hydroxydehydro.
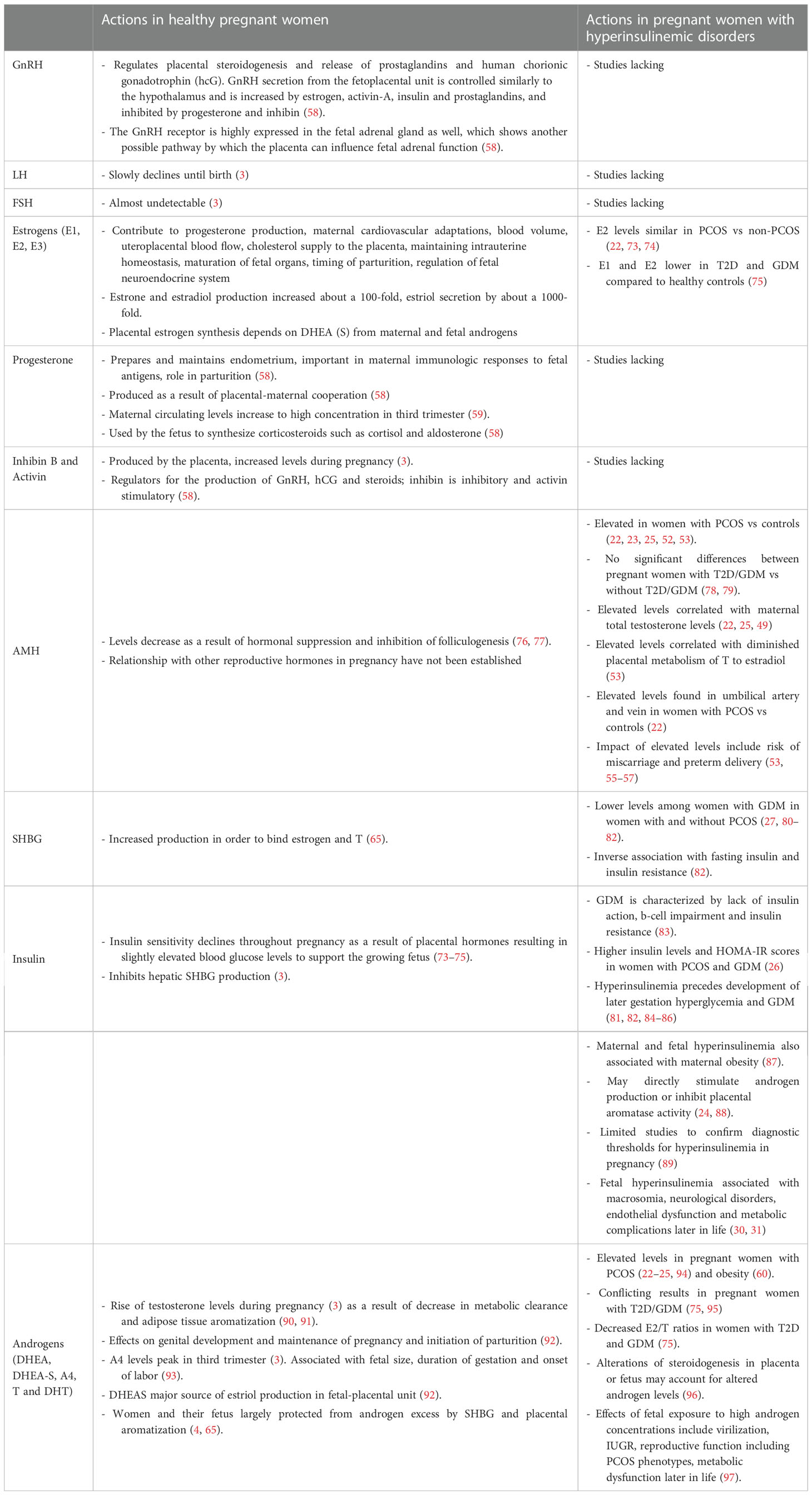
Table 1 Physiology of the feto-placental unit and the influence of metabolic factors in healthy pregnant women vs pregnant women with hyperinsulinemic disorders.
2.2. Metabolic factors influencing actions of the feto-placental unit
To understand the potential impacts of high levels of insulin, androgens and AMH, an understanding of their actions in normal pregnancies is critical. The sections below describe the normal actions of insulin, androgens and AMH and their relation to the hormones described in the previous sections.
2.2.1. Insulin
Depending on the requirements of pregnancy, insulin sensitivity shifts. At 12–14 weeks’ gestation, insulin sensitivity is somewhat increased to promote the uptake of glucose into adipose stores in preparation for the energy demands later in pregnancy. However, insulin sensitivity then declines throughout pregnancy as a result of local and placental hormones, including estrogen, progesterone, leptin, cortisol, placental cytokines (especially TNF-α), placental lactogen (hPL) and placental growth hormone that promote IR (98, 99). This results in slightly elevated blood glucose levels as a means to provide energy to support the growing fetus. The state of IR also promotes endogenous glucose production and breakdown of fat stores with further increase in blood glucose concentrations (100). Hypertrophy and hyperplasia of pancreatic β-cells and increased glucose-stimulated insulin secretion compensate for these changes to maintain glucose homeostasis, as shown in animal studies (101). Insulin has been found to inhibit hepatic SHBG production (3) and, low SHBG concentrations might even reflect IR better than fasting glucose or insulin levels (102, 103).
2.2.2. Androgens
The fetoplacental unit, the adrenals and potentially adipose tissue have been assumed to be responsible for maternal androgen levels in pregnancy (90, 97). During normal progression of pregnancy, levels of total testosterone (T) increase by 70%, with peak levels in the third trimester of pregnancy (3). Among factors contributing to the rise of T are the decrease in metabolic clearance of T (91) and adipose tissue aromatization (90). Among others, T exerts effects on genital development during early gestation (3) and on the maintenance of pregnancy and initiation of parturition (92). Maternal serum A4 concentrations start to increase in the later part of pregnancy, with peak levels in the third trimester (3). A4 levels have been associated with nausea, fetal size, duration of gestation and onset of labour (93). DHEAS plays a role in cervical ripening through activation of collagenase activity induced by the enhanced conversion to E2 (104). It is also a major source of E3 production in the fetal-placental unit (92). It has been shown to decline during pregnancy (105). However, recent studies suggest a more fluctuating pattern of DHEAS throughout gestation (3).
Pregnant women and their infants remain largely unaffected by the changes in androgen concentrations due to pregnancy-specific mechanisms that are activated to protect both the mother and fetus from this androgen excess. The increased levels of SHBG bind T, while the cytochrome P450 aromatase enzyme converts androgens into E2. The fetal liver subsequently converts E2 to E3, which is then excreted in maternal urine. Placental aromatization is so efficient, that little androgen presented to the placenta escapes (4). It has been proposed that the increase in SHBG during pregnancy protects the fetus from exposure to maternal androgens, however, reciprocally, maternal SHBG may protect mothers from androgens originating from the fetus (65). This has been demonstrated in a clinical case report that showed profound and transient maternal virilization resulting from very low plasma SHBG, whereas the twin daughters did not show any evidence of excessive androgen exposure (106). Something similar has been demonstrated in patients with a fetus with a defective P450 aromatase gene, which resulted in a spillover of fetal androgens into the maternal circulation (107). This highlights the importance of SHBG and placental aromatase in protecting the mother from fetal androgens.
2.2.3. Anti-Müllerian hormone
During pregnancies of women without hyperinsulinemic disorders, AMH levels significantly drop during the course of pregnancy and immediately after birth, (first trimester: 1.69 ng/ml (IQR 0.71-3.10), second trimester: 0.8 ng/ml (IQR 0.48-1.41), third trimester: 0.5 ng/ml (IQR 0.18-1.00)) but subsequently increases over the first four days postpartum (76). This decrease throughout the three trimesters of pregnancy most likely results in the inhibition of folliculogenesis in pregnancy (78). Hormones such as estrogen may regulate AMH production (77), however some studies have not found associations between AMH levels and E1 in pregnancies in women with or without T2D and GDM (78, 108). The relationship between other reproductive hormones (e.g. LH, activin, inhibin) and AMH production has not been established. Although possible involvement of these factors in regulating gravid AMH levels cannot be excluded, other factors, such as Follistatin (109) may also regulate AMH levels during pregnancy.
In summary, in normal pregnancies, insulin sensitivity declines as a result of local and placental hormones resulting in slightly elevated blood glucose levels to support the growing fetus, with a concomitant inhibition of hepatic SHBG production. Androgen levels increase due to decreased metabolic clearance and adipose tissue aromatization to regulate key processes during pregnancy and parturition. Mechanisms such as altered SHBG that binds T and conversion of androgens into E2 by cytochrome P450 are in place to protect both the mother and fetus from androgen excess. Levels of AMH decrease as a result of hormonal suppression and inhibition of folliculogenesis. Relationships between AMH and other reproductive hormones in pregnancy are not extensively studied.
3. Hormonal and metabolic interactions in pregnant women with hyperinsulinemic disorders
The interrelationships between hyperinsulinemia, androgen excess, and AMH are potentially reflected in pregnancies complicated by hyperinsulinemic disorders. The preconception state of hyperinsulinemia and/or androgen and AMH excess might drive the mechanisms underlying fetoplacental endocrine dysfunction. However, because hyperinsulinemic conditions including PCOS, diabetes (GDM, T1D and T2D), and obesity often appear concurrently, no one condition explains the altered endocrine milieu and eventual adverse maternal and neonatal outcomes in these women. The following section aims to assess the endocrine and metabolic factors, focusing on insulin, androgens and AMH (and their interactions) in women with PCOS, diabetes and obesity and how they impact the fetoplacental unit.
3.1. Actions of the fetoplacental unit and the influence of metabolic factors in women with hyperinsulinemic pregnancies
Clinical studies on placental GnRH, hCG, estrogens, progesterone, activin and inhibin in women with hyperinsulinemic conditions in pregnancy are scarce. Studies have reported differences in some hormones in pregnant women with and without PCOS, but do not specifically compare the fetoplacental unit between these groups or address interactions between androgens, estrogens, AMH and pituitary hormones in the mothers and the fetus (22). Maternal E2 levels appear similar in women with and without PCOS (22, 73, 74). A recent study found that E1 and E2 levels were lower in women with T2D and GDM than in healthy controls during the second half of pregnancy (75). Data on cord blood estrogens show conflicting outcomes. Lower E1 concentrations were found in cord blood from girls and women with PCOS compared with controls, but there were no differences in E2 and E3 in these studies (74, 88). Anderson et al. (73) reported decreased E2 levels in female PCOS cord blood samples compared with controls, and lower E2 levels have been noted in cord blood compared to maternal levels (22, 110). Few studies report on estrogens in relation with other reproductive and metabolic hormones.
3.1.1. Hyperinsulinemia
Whereas increased IR during pregnancy is an expected physiological state, complications arise when these changes are augmented by pre-existing hyperinsulinemia and related metabolic dysfunction. When there is a relative lack of insulin action in body tissues to overcome IR, GDM ensues (83). Critical to the pathophysiology of GDM is β-cell impairment aggravated by tissue IR during pregnancy. These impairments usually exist prior to pregnancy and can be progressive. Women with a history of GDM were almost 10 times more likely to develop T2DM than those with a normoglycemic pregnancy with pooled cumulative incidence estimated to be around 16% for studies with more than 10 years of follow up (111). Reduced insulin-stimulated glucose uptake further contributes to hyperglycemia which overburdens the β-cell, producing an additional insulin response and worsening β-cell dysfunction (5). IR, which includes a failure of insulin signaling, further exacerbates β-cell dysfunction (5).
The mechanisms underlying GDM are thought to be similar to other disorders of insulin sensitivity such as T2DM, prediabetes, obesity and PCOS. De Wilde et al. (26) demonstrated that in 22 women with PCOS and GDM, insulin levels and homeostasis model assessment of insulin resistance (HOMA-IR) scores were higher before conception and at each sampling point in pregnancy compared to women with PCOS who did not develop GDM, independently of BMI. Insulin sensitivity in women with PCOS who develop GDM is potentially not as high as in normal glucose-tolerant women in the first trimester resulting in hyperinsulinemia to keep glucose concentrations within normal ranges. Additionally, insulin levels and HOMA-IR levels were already significantly increased prior to conception in the GDM group compared with the non-GDM group. These findings support the hypothesis that the risk of developing GDM is already present early in pregnancy. A growing body of evidence suggests that hyperinsulinemia in early gestation precedes later gestation hyperglycemia and GDM development (89, 112–114). Associations have also been found between maternal obesity and maternal and fetal hyperinsulinemia (87).
Insulin interacts with other important metabolic markers. High levels of insulin may contribute to fetal hyperandrogenism exposure by directly stimulating androgen production (24). Furthermore, insulin, Insulin-like growth factor 1 (IGF-I) and IGF-II have been shown to inhibit placental aromatase activity (88). This is important, given the exacerbated IR and increased levels of insulin in pregnant women with PCOS. In turn, a decrease in P450 aromatase activity has been described in GDM and pre-eclampsia (115, 116). Importantly, a recent meta-analysis of prospective studies found an inverse association of SHBG with fasting insulin and IR (80). SHBG levels were lower among women who subsequently developed GDM compared to those who did not, independent of adiposity (80). Similarly, lower preconception SHBG concentrations are also associated with GDM in women with PCOS (81, 82). The study by de Wilde et al. (26) also demonstrated that in 22 women with PCOS and GDM, SHBG levels were significantly lower before conception and in the second trimester. Low plasma SHBG in pregnancy was also associated with hyperandrogenism (106). We therefore hypothesize that like outside pregnancy, hyperinsulinemia in these disorders and subsequent direct stimulation of androgen production, and inhibition of SHBG (that binds androgens), contributes to hyperandrogenism. In pregnancy, inhibition of placental aromatase activity is likely to exacerbate this phenomenon.
Hyperinsulinemia in pregnancy may impact maternal and fetal health. Routine assessments and standardized diagnostics of hyperinsulinemia in pregnancy are lacking. Therefore, associations between pathological hyperinsulinemia and maternal and neonatal outcomes are not well understood. However, it is proposed that hyperinsulinemia has effects on maternal hemodynamic adaptations, potentially increasing the risk of pre-eclampsia, stillbirth and intrauterine growth restriction (84). Studies have reported increased insulin in cord blood of babies born to mothers with T2D and GDM compared to controls (85, 86). Fetal hyperinsulinism is associated with increased risk of later metabolic complications (30). Further, some studies point to a role of hyperinsulinemia in fetal development such as macrosomia, neurological disorders, endothelial dysfunction in the neonate (31), offspring hyperinsulinemia and cardiac hypertrophy (84). North et al. (89) hypothesize that hyperinsulinemia in women with “borderline” glucose intolerance may explain pivotal observations from The Hyperglycemia and Adverse Pregnancy Outcome (HAPO) cohort that showed an increased risk of fetal overgrowth, primary caesarean delivery, elevated cord-blood Connecting peptide (C-peptide) and neonatal hypoglycemia in mothers with mild-to-moderately elevated glucose levels (19). Importantly, metformin use in pregnancy might enhance insulin sensitivity, reduce insulin resistance and fetal hyperinsulinemia and in turn, reduce neonatal adiposity (117). However, it’s use in pregnancy continues to be investigated to determine efficacy and safety for preventing or treating GDM (118).
Human data on the effects of exogenous insulin use in pregnancy in relation to other reproductive factors or pregnancy outcomes is scarce. Insulin does not cross the placenta and in animal studies, exogenous insulin has no effects on embryo or fetal development (119). However, insulin receptors are expressed on the placenta and may be a means through which insulin mediates effects on the fetus through action on the placenta (84). A recent meta-analysis compared outcomes between women with GDM with insulin use, GDM without insulin use and no GDM (120). In studies with no insulin use, when adjusted for confounders such as BMI, women with GDM had increased odds of caesarean section, preterm delivery, low one-minute Apgar scores, macrosomia and infant born large for gestational age compared with women without GDM. In studies with insulin use, when adjusted for confounders, women with GDM had increased odds of having an infant born large for gestational age, with respiratory distress syndrome, neonatal jaundice, and/or requiring admission to the neonatal intensive care unit compared to women without GDM (120). Furthermore, an increase in placental weight in women with GDM that used insulin versus women with GDM that were controlled with diet and exercise only was observed (121). In pregnant women with T1D, the use of insulin lispro was associated with an increased risk of macrosomia (122). Importantly, a high incidence of macrosomia was found despite overall good glycemic control, as assessed by HbA1c. It was proposed that the acute pulsatile rise and fall of maternal blood glucose levels which occur after food intake (hyperglycemia) and insulin treatment (hypoglycemia), respectively, might result in increased fetal release of endogenous insulin leading to increased placental weight and macrosomia (121, 122). However, clinical implications of hyperinsulinemia and links to perinatal and neonatal outcomes remain poorly understood. There is limited research to confirm diagnostic thresholds for hyperinsulinemia in to insulin metabolism become potentially harmful are needed (89).
Overall, studies show relative hyperinsulinemia during pregnancy in women with PCOS GDM, and maternal obesity, compared to healthy pregnancies without these conditions. Hyperinsulinemia contributes to fetal hyperandrogenism exposure by stimulating androgen production, inhibiting placental aromatase activity and inhibiting SHBG. Hyperinsulinemia may also increase risks of PE, stillbirth, IUGR, macrosomia, and neurological disorders, endothelial dysfunction and later metabolic complications in the offspring. Finally, similar to the exogenous action of insulin on androgen excess in women with T1D, we hypothesize that the use of insulin in women with GDM (with or without PCOS) might also aggravate hyperinsulinemia in pregnancy; however, this phenomenon and its potential complications await further study.
3.1.2. Hyperandrogenism
The adaptations which occur in pregnancy to protect both the mother and fetus from pregnancy-induced androgen excess might not be sufficient in pregnant women with underlying hyperinsulinemic pregnancies, as evidenced by elevated levels of androgens in pregnant women with PCOS. Several studies found elevated serum androgen levels, including T (22–25, 94), A4 (23, 24, 94), DHEAS and free androgen index (FAI) (24, 25, 94) in pregnant women with PCOS. This may be a potential source of fetal androgen excess and induce effects on the development of the fetus, even if virilization of a female fetus is not observed (24). Results regarding T and GDM are conflicting. Some report that women who develop GDM have significantly higher T concentrations compared with controls (95), while others demonstrated hyperandrogenemia in pregnant women with T2D, but not in GDM (75). Additionally, decreased estrogen and E/T ratios were found in women with T2D and women with GDM (75). In the latter study, BMI and T levels were positively associated in the T2D and GDM groups (75). Maternal obesity has also been found to be associated with elevated maternal serum T concentrations (60).
In relation to the impact of androgen excess on fetal health, some studies have used cord blood to investigate the intrauterine fetal environment in women with PCOS (22, 73, 74, 88, 123, 124). Levels of DHEA and DHEAS in fetal cord blood were generally not associated with PCOS (73, 74, 88, 95), but studies regarding T and A4 have produced conflicting results. A recent meta-analysis of seven studies (n= 570) found no significant differences in cord blood T levels between women with and without PCOS, irrespective of neonatal sex (125). The authors suggest that T may be quickly degraded and converted by placental aromatase into E2 when passing through the placenta. However, cord blood A4 levels in female newborns were significantly lower in PCOS than in the control group (125). As explained in section 2, cord blood androgens are derived from both fetal adrenal as well as placental steroidogenesis. The fetal adrenals produce DHEAS in utero, which is transformed into A4, T, and E2 by the placenta (126). Alterations of steroidogenesis in the fetus or an abnormality in placental steroidogenesis could potentially account for the decreased A4 levels (125). Similarly, Kelly et al. (96) proposed that maternal androgens may exert a programming effect on placental and/or fetal steroidogenesis to alter androgen levels within the fetal-placental unit. This concept is supported by Maliqueo et al. (88), wherein placental tissue from women with PCOS had increased 3β-hydroxysteroid dehydrogenase 1 enzymatic activity and decreased aromatase activity compared with non-PCOS controls inducing accumulation of androgenic substrate. This is in line with findings of lower E1 and E2 serum levels in women with GDM and lower E2/T ratios suggesting a lower conversion of T to estrogens (75).
Despite the fact that maternal androgen excess was not associated with elevated T concentrations in cord blood, hyperandrogenism in pregnancy is clinically relevant given the numerous reports, both from animal models and clinical studies, on the adverse effects of fetal exposure to high androgen concentrations. Importantly, umbilical cord blood is only a representation of the end of pregnancy rather than the entire gestation period. The effects of fetal exposure to high androgen concentrations have been reviewed in detail elsewhere and include virilization (32–34), IUGR (34, 35), placental differentiation culminating in low birth weight (36), reproductive function including PCOS like phenotypes (37–39), metabolic dysfunction (37, 39–42), adverse cardiac programming (43–45), and behavioral outcomes later in life (46–48).
To what extent high levels of androgens directly impact maternal and fetal health is not clear, given that androgens are converted to estrogens. In animal studies, examining direct effects of DHT, a nonaromatizable androgen, was associated with decreased placental weight and cotreatment of T with an androgen antagonist prevented placental changes, suggesting placental changes were mediated at least in part by androgenic action (96). Additionally, evidence from studies in rhesus monkeys indicates that gestational T and DHT treatment induces maternal hyperinsulinemia and insulin resistance in addition to elevating circulating androgen levels in both maternal and fetal compartments (42, 127). This may result in reprogramming of insulin target tissues in offspring such as liver and adipose tissue, leading to hyperglycemia in adulthood (128). As such, effects of hyperandrogenism on fetal development and pregnancy outcomes could be mediated via insulin and further altered by the impact of hyperinsulinemic states. Importantly, the inhibitory action of insulin and AMH on placental aromatase action might contribute to changes in sexual steroids and insulin levels during pregnancy. Placental steroidogenesis possibly follows different pathways in hyperinsulinemic pregnancies, which results in a different ratio of E/T fractions during gestation and in offspring.
In summary, studies have found relatively higher levels of androgens in pregnant women with PCOS and obesity, whereas this is less clear in T2D and GDM. Reports of cord blood levels of androgens are scarce and conflicting. Different androgen profiles within the fetal-placental unit may be reflective of abnormalities of steroidogenesis in the fetus or placenta. Hyperandrogenism in pregnancy is associated with adverse short- and long-term neonatal outcomes. Rather than a function of direct androgenic action only, effects from hyperandrogenism on fetal development and pregnancy outcomes are potentially mediated by insulin and further altered by hyperinsulinemic states.
3.1.3. Anti-Müllerian hormone
As described in section 2, AMH levels decrease successively during pregnancy, mostly between the first and second trimester. Several studies show elevated levels of AMH during pregnancy in women with PCOS versus controls (22, 23, 25, 52, 53). In fact, high AMH levels in women with PCOS are maintained throughout the entire pregnancy (23). However, studies have not found significantly different AMH concentrations between women with diabetes (T2D; GDM) and healthy pregnant women (78, 79). In relation to obesity, studies found that AMH was negatively associated with maternal BMI (22, 108, 129).
Interactions between AMH and other hormones in pregnancy have recently been investigated by Tata et al. (53) who measured AMH in a cohort of 66 pregnant women with PCOS and 63 control women, at gestational week 16-19. AMH was significantly higher in pregnant lean women with PCOS compared with the control group but not in obese women with PCOS versus obese control women. Furthermore, AMH was significantly higher in lean women with PCOS who had hyperandrogenism than in those without. No differences between obese women with PCOS with and without hyperandrogenism were detected, potentially as the impact of obesity may negate PCOS effects on AMH. An animal model that treated pregnant mice with AMH resulted in maternal neuroendocrine-driven T excess and diminished placental metabolism of T to E2. Authors suggest that this could be a viable route by which maternal and placental T can be transferred to the human fetus (53). Although not extensively studied, lower levels of E1 and E2 in pregnant women with T2D and GDM and in female cord blood of women with PCOS support this suggestion, and could be reflective of increased AMH levels leading to decreased placental metabolism of T, however a direct relationship has not been established (73, 74, 78, 88).
Several other studies report a positive correlation between elevated AMH levels and maternal total T levels (both in women with PCOS as well as controls) (22, 23, 25). It is thought that the ovaries may contribute to maternal serum concentrations of T. In addition, high AMH may influence placental T production. This is in line with findings in prenatal AMH-exposed mice, in which AMH exposure was associated with elevated T levels in the dams and offspring. Treatment of pregnant mice with AMH resulted in a masculinization of the exposed female fetus and PCOS-like phenotypic traits in adulthood (reproductive and neuroendocrine) (53). Lastly, although studies in non-pregnant adolescents with PCOS show a positive association between serum AMH and HOMA-IR, no significant associations of AMH with insulin, HOMA-IR or IGF-1 have been observed in pregnant women (78, 108).
Elevated levels of AMH potentially impact the developing fetus. Studies have found elevated serum AMH levels in the umbilical vein at time of delivery in newborns of women with PCOS compared with healthy controls (110). Importantly, higher levels of AMH were observed in neonates born from women with PCOS and maternal hyperandrogenism or maternal BMI higher than 30 kg/m2 (110). On the other hand, no significant associations were observed between maternal AMH concentrations and AMH in female offspring (110), whereas AMH was higher in male fetuses, than in the mother (22). This suggests that AMH concentrations in umbilical cord blood might differ from that in maternal serum because there is no passage of AMH from fetus to mother (22, 110). Instead, elevated AMH concentrations could be a result of placental passage from the mother and increased AMH production by the fetal compartment. Detti et al. (22) hypothesize that genetic inheritance of PCOS by the fetus causes increased AMH production by the embryonal/fetal granulosa and Sertoli cells.
Studies investigating associations between levels of AMH and pregnancy outcomes are scarce. One study showed increased risk of miscarriage in association with prenatal exposure to AMH (53). Further, high serum AMH levels were associated with lower live birth rates in women with PCOS undergoing assisted reproductive technology (54). Studies have reported no association between maternal AMH and infant birthweight (25, 79, 129, 130). Increased incidence of PCOS is observed in women with sub-separate uteri. Detti et al. (22) hypothesize that higher AMH concentrations could contribute to development of milder forms of Mullerian anomalies by delay/arrest in Mullerian duct fusion and inner wall reabsorption (22, 110). AMH may also be associated with preterm delivery (55–57). Kaing et al. (57) examined women with PCOS and high AMH who conceived after ovulation induction, and found that 62% of women who delivered preterm had AMH levels above the 75th percentile. Women with PCOS who delivered preterm had notably higher AMH than their term counterparts (11.1 vs 5.4 ng/mL). Similarly, a recent retrospective cohort study with patients with PCOS who had undergone IVF/ICSI showed that for patients with a BMI ≥ 24 kg/m (2) plus serum AMH > 6.45 ng/ml (75th percentile), the risk of preterm birth was 2.1 times that in the AMH <2.71 ng/ml group (55).Associations with other maternal and perinatal outcomes remain uncertain. Most existing studies do not show statistically increased risk of other adverse maternal and perinatal outcomes in women with elevated AMH levels (25, 55, 130). Prenatal AMH treatment in mice resulted in PCOS features in adulthood: hyperandrogenism, LH elevation, sporadic ovulation, and fertility defects. However, these mice did not show weight alterations, leading to the assumption that prenatal AMH could predispose to the lean PCOS phenotype in adulthood (53).
In summary, elevated levels of AMH during pregnancy have been associated with T excess, and diminished metabolism of T to E2. However, associations between AMH and insulin or HOMA-IR in pregnancy remains to be determined. Elevated AMH may be associated with preterm birth in women with PCOS, however associations with other maternal and neonatal outcomes remain uncertain.
4. Limitations and future directions
While an abnormal prenatal endocrine milieu could possibly reflect a deranged feto-placental unit in women with hyperinsulinemic disorders, studies specifically addressing relationships and associations between insulin, androgens, pituitary hormones, estrogens, AMH and placental steroidogenesis are scarce. Studies are often cross-sectional in design, do not adjust for confounding factors such as nutrition and physical activity and longitudinal studies on maternal hormones do not always include early pregnancy samples. Furthermore, they are often limited by small sample sizes and to increase the power, the conclusions drawn from these studies need to be further studied with larger sample sizes. Data regarding subsequent alterations in the intrauterine environment, often studied in cord blood have produced conflicting results and overall evidence is scarce. Hormonal dysregulation in women with PCOS, diabetes or obesity may impact placental development and function. However, placental studies (looking at macroscopic and microscopic changes) in pregnancies impacted by these conditions are limited.
In order to study the interrelationships between insulin, androgens and AMH, investigating placental tissue could be an important additional mode of research.
Hyperinsulinemia in pregnancies affected by PCOS, obesity or diabetes seems largely a result of pre-existing hyperinsulinemia and related metabolic dysfunction paired with IR in pregnancy exacerbating B-cell dysfunction. However, further research is warranted on the factors that promote IR more than the physiological state of IR hPL for example, contributes to IR and secretion of insulin. Pregnancies affected by obesity and diabetes show altered hPL levels: lower concentrations have been shown in obesity and increased levels in GDM. Disruptions in hPL are also thought to be associated with an increased prevalence of placental dysfunction and increased fetal growth (131). Likewise, TNF-α, a proinflammatory cytokine produced by the placenta and other placental adipokines (e.g. Chemerin, Apelin, Omentin) also likely play a role in development of IR in pregnancy (131). In order to understand the pathways by which IR occurs, factors associated with IR in women with hyperinsulinemic pregnancies should be further explored.
Studies of diagnostic thresholds of insulin levels in pregnancy are lacking. Therefore, in order to study associations between pathological hyperinsulinemia and maternal and neonatal outcomes, criteria need to be established to determine when normal hyperinsulinemia in pregnancy results in levels that result in clinically relevant adverse outcomes. In addition, data on insulin levels and IR in pregnancies affected by metabolic and endocrine disorders is notably lacking. Longitudinal measurements of insulin and IR in pregnancy would be beneficial. The possible associations between maternal hyperinsulinemia and offspring health highlight the need for further study into the interactions and mechanisms between maternal insulin and fetal development and health through pregnancy, post birth and into childhood.
Whilst studies show that hyperinsulinemia is likely to result in decreased levels of SHBG (leading to increased free T), direct stimulation of androgens and inhibition of placental aromatase activity, future research is needed to substantiate associations between insulin levels in pregnancy and the maternal and prenatal endocrine milieu. Human association studies of the various hyperinsulinemic states, coupled with animal models that ablate variables of interest could help clarify these links. Furthermore, the use of insulin in women across a range of hyperinsulinemic disorders in pregnancy, the potential concurrent worsening of hyperinsulinemia in pregnancy, related hyperandrogenism and its potential complications have not been studied previously.
The effects of hyperandrogenism in pregnancy on developmental programming have been widely studied. However, questions regarding the pathways by which androgens impact maternal and fetal health remain unanswered. Whilst evidence is clear regarding the contribution of hyperinsulinemia on hyperandrogenism, few studies examine the effects of androgens on insulin and insulin sensitivity. Observational studies of various hyperandrogenic states coupled with further explorations using animal models will enhance understanding of how hyperandrogenism interacts with other variables such as estrogens, AMH, insulin and potential other markers in pregnancy.
The hyperandrogenic phenotype is likely to be underreported in maternal samples as observational studies do not always include a full assessment of the different androgens. This will influence interpretation of gestation AMH levels across PCOS phenotypes. Future research, with bigger sample sizes and full assessment of hyperandrogenism is warranted to confirm elevated levels of AMH during pregnancy between groups, to support the hypothesis that high AMH levels lead to decreased placental metabolism of T and to assess associations between AMH and insulin and HOMA-IR. Animal studies could explore what contributes to higher levels of AMH in pregnancy. In addition, the physiologically low AMH levels observed during the second half of pregnancy may require more sensitive tests to find a significant difference between groups under study. Previous studies assayed serum AMH using the widely clinically used assay AMH/MIS ELISA kit (Immunotech-Beckman, Marseilles, France) (75, 79). Due to a lower threshold of detection, a newer commercially available AMH enzyme-linked assay (pico AMH ELISA, AnshLabs) might be more appropriate for the measurement of low AMH concentrations (132). Further, studies are needed to investigate associations between high AMH levels and pregnancy and fetal and offspring health and to elucidate the potential actions of high AMH on the maternal and placental adaptations in pregnancy. Reports so far show no conclusive results and the polycystic ovarian morphology, reflected by high AMH levels, might not drive mechanisms behind adverse outcomes in pregnant women with PCOS.
Other factors important for pregnancy, birth, lactation, child development and survival, such as hCG, activin, inhibin, hPL and placental cytokines have not been widely studied in relation to hyperinsulinemic pregnancies but might be potential markers relevant to explain altered hormonal states in pregnancies affected by hyperinsulinemic disorders. For example, while Inhibin and Activin act as regulators within the placenta for GnRH, hCG and steroids, to our knowledge no reports are currently present on their actions in hyperinsulinemic pregnancies. Additionally, hyperinsulinemia could stimulate production of leptin which could amplify inflammation (133).
Interestingly, upon analyzing T and P450 aromatase in normal weight and obese women, Maliqueo et al. (2017) only found an increment of T and lower P450 aromatase in obese women with male fetuses. The authors therefore suggest that regulation and metabolism of sex steroids in pregnancy could be dependent on maternal/placental adaptations determined by fetal gender (60). In relation to pregnancies affected by hyperinsulinemic disorders, future research is warranted to understand the mechanisms by which fetal gender may impact placental function.
5. Conclusions
This comprehensive review compares physiological changes in normal pregnancies to those in women with pre-existing hyperinsulinemic conditions including PCOS, diabetes and obesity. We highlight how the maternal, placental and fetal compartments are separate, but functionally complement each other in healthy pregnancies. Yet in pregnant women with pre-existing hyperinsulinemic disorders, it appears that abnormal levels of insulin, androgens, estrogens and AMH are reflective of a deranged fetoplacental unit, ultimately leading to adverse pregnancy and neonatal outcomes. Hyperinsulinemia in these disorders and subsequent direct stimulation of androgen production, inhibition of SHBG and inhibition of placental aromatase activity, contributes to hyperandrogenism. Effects of hyperandrogenism could be mediated via insulin and altered placental steroidogenesis. Elevated levels of AMH contribute to T excess, and diminished metabolism of T to E2. With the high prevalence of hyperinsulinemic endocrine metabolic conditions including PCOS, diabetes and obesity in pregnancy, more research, including longitudinal sampling, and consideration of severity of hyperinsulinemia and interaction across these linked conditions is needed. A focus on placental and fetal steroidogenesis and hormonal interactions in these conditions is important. Finally, further study on the impacts of exogenous insulin administered in women with T1D, T2D and GDM, is needed, in relation to important endocrine and metabolic markers, and impact on fetal health.
Author contributions
AN collated and reviewed the literature and wrote the first draft of the manuscript. AM, JB, and HT reviewed and edited the manuscript. All authors provided intellectual input in line with the ICMJE criteria for authorship and have approved the final version for publication.
Funding
AN is a PhD student supported by the Monash International Tuition Scholarship (MITS) and Research Training Program Stipend (RTP stipend). AM is supported by a Peter Doherty Biomedical Research Fellowship provided by the National Health and Medical Research Council (NHMRC) of Australia. HT is supported by an NHMRC Investigator Fellowship. This body of work is funded by an NHMRC Centre for Research Excellence on Women’s Health in Reproductive Life.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Banker M, Puttabyatappa M, O'Day P, et al. Association of maternal-neonatal steroids with early pregnancy endocrine disrupting chemicals and pregnancy outcomes. J Clin Endocrinol Metab (2021) 106(3):665–87. doi: 10.1210/clinem/dgaa909
2. Ryan EA. Hormones and insulin resistance during pregnancy. Lancet (2003) 362(9398):1777–8. doi: 10.1016/S0140-6736(03)14942-2
3. Kuijper EA, Ket JC, Caanen MR, Lambalk CB. Reproductive hormone concentrations in pregnancy and neonates: a systematic review. Reprod BioMed Online (2013) 27(1):33–63. doi: 10.1016/j.rbmo.2013.03.009
4. Thompson EA Jr., Siiteri PK. The involvement of human placental microsomal cytochrome p-450 in aromatization. J Biol Chem (1974) 249(17):5373–8. doi: 10.1016/S0021-9258(20)79736-X
5. Plows JF, Stanley JL, Baker PN, Reynolds CM, Vickers MH. The pathophysiology of gestational diabetes mellitus. Int J Mol Sci (2018) 19(11). doi: 10.3390/ijms19113342
6. Bozdag G, Mumusoglu S, Zengin D, Karabulut E, Yildiz BO. The prevalence and phenotypic features of polycystic ovary syndrome: a systematic review and meta-analysis. Hum Reprod (2016) 31(12):2841–55. doi: 10.1093/humrep/dew218
7. Revised 2003 consensus on diagnostic criteria and long-term health risks related to polycystic ovary syndrome. Fertil Steril (2004) 81(1):19–25.
8. Chivese T, Hoegfeldt CA, Werfalli M, et al. IDF diabetes atlas: The prevalence of pre-existing diabetes in pregnancy - a systematic reviewand meta-analysis of studies published during 2010-2020. Diabetes Res Clin Pract (2022) 183:109049. doi: 10.1016/j.diabres.2021.109049
9. Wang H, Li N, Chivese T, et al. IDF diabetes atlas: Estimation of global and regional gestational diabetes mellitus prevalence for 2021 by international association of diabetes in pregnancy study group's criteria. Diabetes Res Clin Pract (2022) 183:109050. doi: 10.1016/j.diabres.2021.109050
10. Shrestha A, Prowak M, Berlandi-Short V-M, Garay J, Ramalingam L. Maternal obesity: A focus on maternal interventions to improve health of offspring. Front Cardiovasc Med (2021) 8. doi: 10.3389/fcvm.2021.696812
11. Goldstein RF, Abell SK, Ranasinha S, et al. Association of gestational weight gain with maternal and infant outcomes: A systematic review and meta-analysis. JAMA (2017) 317(21):2207–25. doi: 10.1001/jama.2017.3635
12. Deputy NP, Kim SY, Conrey EJ, Bullard KM. Prevalence and changes in preexisting diabetes and gestational diabetes among women who had a live birth - united states, 2012-2016. MMWR Morb Mortal Wkly Rep (2018) 67(43):1201–7. doi: 10.15585/mmwr.mm6743a2
13. Joham AE, Ranasinha S, Zoungas S, Moran L, Teede HJ. Gestational diabetes and type 2 diabetes in reproductive-aged women with polycystic ovary syndrome. J Clin Endocrinol Metab (2014) 99(3):E447–452. doi: 10.1210/jc.2013-2007
14. Barker DJ, Osmond C, Golding J, Kuh D, Wadsworth ME. Growth in utero, blood pressure in childhood and adult life, and mortality from cardiovascular disease. BMJ (Clinical Res ed) (1989) 298(6673):564–7. doi: 10.1136/bmj.298.6673.564
15. Bahri Khomami M, Joham AE, Boyle JA, et al. Increased maternal pregnancy complications in polycystic ovary syndrome appear to be independent of obesity-a systematic review, meta-analysis, and meta-regression. Obes Rev (2019) 20(5):659–74. doi: 10.1111/obr.12829
16. Alexopoulos AS, Blair R, Peters AL. Management of preexisting diabetes in pregnancy: A review. Jama (2019) 321(18):1811–9. doi: 10.1001/jama.2019.4981
17. Kjerulff LE, Sanchez-Ramos L, Duffy D. Pregnancy outcomes in women with polycystic ovary syndrome: a metaanalysis. Am J Obstet Gynecol (2011) 204(6):558.e551–556. doi: 10.1016/j.ajog.2011.03.021
18. Marchi J, Berg M, Dencker A, Olander EK, Begley C. Risks associated with obesity in pregnancy, for the mother and baby: a systematic review of reviews. Obes Rev (2015) 16(8):621–38. doi: 10.1111/obr.12288
19. Metzger BE, Lowe LP, Dyer AR, et al. Hyperglycemia and adverse pregnancy outcomes. N Engl J Med (2008) 358(19):1991–2002.
20. Gunning MN, Sir Petermann T, Crisosto N, et al. Cardiometabolic health in offspring of women with PCOS compared to healthy controls: a systematic review and individual participant data meta-analysis. Hum Reprod update (2020) 26(1):103–17. doi: 10.1093/humupd/dmz036
21. Escobar-Morreale HF. Polycystic ovary syndrome: definition, aetiology, diagnosis and treatment. Nat Rev Endocrinol (2018) 14(5):270–84. doi: 10.1038/nrendo.2018.24
22. Detti L, Christiansen ME, Francillon L, et al. Serum anti-müllerian hormone (AMH) in mothers with polycystic ovary syndrome (PCOS) and their term fetuses. Syst Biol Reprod Med (2019) 65(2):147–54. doi: 10.1080/19396368.2018.1537385
23. Piltonen TT, Giacobini P, Edvinsson Å, et al. Circulating antimüllerian hormone and steroid hormone levels remain high in pregnant women with polycystic ovary syndrome at term. Fertil Steril (2019) 111(3):588–596.e581.
24. Sir-Petermann T, Maliqueo M, Angel B, Lara HE, Pérez-Bravo F, Recabarren SE. Maternal serum androgens in pregnant women with polycystic ovarian syndrome: possible implications in prenatal androgenization. Hum Reprod (2002) 17(10):2573–9. doi: 10.1093/humrep/17.10.2573
25. Valdimarsdottir R, Valgeirsdottir H, Wikström AK, et al. Pregnancy and neonatal complications in women with polycystic ovary syndrome in relation to second-trimester anti-müllerian hormone levels. Reprod BioMed Online (2019) 39(1):141–8. doi: 10.1016/j.rbmo.2019.02.004
26. de Wilde MA, Goverde AJ, Veltman-Verhulst SM, et al. Insulin action in women with polycystic ovary syndrome and its relation to gestational diabetes. Hum Reproduction (2015) 30(6):1447–53. doi: 10.1093/humrep/dev072
27. Alvarez-Blasco F, Botella-Carretero JI, San Millán JL, Escobar-Morreale HF. Prevalence and characteristics of the polycystic ovary syndrome in overweight and obese women. Arch Intern Med (2006) 166(19):2081–6. doi: 10.1001/archinte.166.19.2081
28. Lo JC, Feigenbaum SL, Escobar GJ, Yang J, Crites YM, Ferrara A. Increased prevalence of gestational diabetes mellitus among women with diagnosed polycystic ovary syndrome: a population-based study. Diabetes Care (2006) 29(8):1915–7. doi: 10.2337/dc06-0877
29. Escobar-Morreale HF, Roldán-Martín MB. Type 1 diabetes and polycystic ovary syndrome: Systematic review and meta-analysis. Diabetes Care (2016) 39(4):639–48. doi: 10.2337/dc15-2577
30. Silverman BL, Metzger BE, Cho NH, Loeb CA. Impaired glucose tolerance in adolescent offspring of diabetic mothers. Relationship to fetal hyperinsulinism. Diabetes Care (1995) 18(5):611–7.
31. Sobrevia L, Salsoso R, Fuenzalida B, et al. Insulin is a key modulator of fetoplacental endothelium metabolic disturbances in gestational diabetes mellitus. Front Physiol (2016) 7. doi: 10.3389/fphys.2016.00119
32. Merke DP, Bornstein SR. Congenital adrenal hyperplasia. Lancet (2005) 365(9477):2125–36. doi: 10.1016/S0140-6736(05)66736-0
33. Spitzer RF, Wherrett D, Chitayat D, et al. Maternal luteoma of pregnancy presenting with virilization of the female infant. J Obstet Gynaecol Can (2007) 29(10):835–40. doi: 10.1016/S1701-2163(16)32642-1
34. Manikkam M, Crespi EJ, Doop DD, et al. Fetal programming: prenatal testosterone excess leads to fetal growth retardation and postnatal catch-up growth in sheep. Endocrinology (2004) 145(2):790–8. doi: 10.1210/en.2003-0478
35. Wolf CJ, Hotchkiss A, Ostby JS, LeBlanc GA, Gray LE Jr.. Effects of prenatal testosterone propionate on the sexual development of male and female rats: a dose-response study. Toxicol Sci (2002) 65(1):71–86. doi: 10.1093/toxsci/65.1.71
36. Beckett EM, Astapova O, Steckler TL, Veiga-Lopez A, Padmanabhan V. Developmental programing: impact of testosterone on placental differentiation. Reproduction (2014) 148(2):199–209. doi: 10.1530/REP-14-0055
37. Padmanabhan V, Veiga-Lopez A. Animal models of the polycystic ovary syndrome phenotype. Steroids (2013) 78(8):734–40. doi: 10.1016/j.steroids.2013.05.004
38. Abbott DH, Barnett DK, Bruns CM, Dumesic DA. Androgen excess fetal programming of female reproduction: a developmental aetiology for polycystic ovary syndrome? Hum Reprod Update (2005) 11(4):357–74.
39. Padmanabhan V, Veiga-Lopez A. Sheep models of polycystic ovary syndrome phenotype. Mol Cell Endocrinol (2013) 373(1-2):8–20. doi: 10.1016/j.mce.2012.10.005
40. Speiser PW, Serrat J, New MI, Gertner JM. Insulin insensitivity in adrenal hyperplasia due to nonclassical steroid 21-hydroxylase deficiency. J Clin Endocrinol Metab (1992) 75(6):1421–4.
41. Yan X, Dai X, Wang J, Zhao N, Cui Y, Liu J. Prenatal androgen excess programs metabolic derangements in pubertal female rats. J Endocrinol (2013) 217(1):119–29. doi: 10.1530/JOE-12-0577
42. Abbott DH, Bruns CR, Barnett DK, et al. Experimentally induced gestational androgen excess disrupts glucoregulation in rhesus monkey dams and their female offspring. Am J Physiol Endocrinol Metab (2010) 299(5):E741–751. doi: 10.1152/ajpendo.00058.2010
43. King AJ, Olivier NB, Mohankumar PS, Lee JS, Padmanabhan V, Fink GD. Hypertension caused by prenatal testosterone excess in female sheep. Am J Physiol Endocrinol Metab (2007) 292(6):E1837–1841. doi: 10.1152/ajpendo.00668.2006
44. Blesson CS, Chinnathambi V, Hankins GD, Yallampalli C, Sathishkumar K. Prenatal testosterone exposure induces hypertension in adult females via androgen receptor-dependent protein kinase cδ-mediated mechanism. Hypertension (2015) 65(3):683–90. doi: 10.1161/HYPERTENSIONAHA.114.04521
45. Vyas AK, Hoang V, Padmanabhan V, Gilbreath E, Mietelka KA. Prenatal programming: adverse cardiac programming by gestational testosterone excess. Sci Rep (2016) 6:28335. doi: 10.1038/srep28335
46. Auyeung B, Ahluwalia J, Thomson L, et al. Prenatal versus postnatal sex steroid hormone effects on autistic traits in children at 18 to 24 months of age. Mol Autism (2012) 3(1):17. doi: 10.1186/2040-2392-3-17
47. Roberts EK, Flak JN, Ye W, Padmanabhan V, Lee TM. Juvenile rank can predict male-typical adult mating behavior in female sheep treated prenatally with testosterone. Biol Reprod (2009) 80(4):737–42. doi: 10.1095/biolreprod.108.073429
48. Roberts EK, Padmanabhan V, Lee TM. Differential effects of prenatal testosterone timing and duration on phenotypic and behavioral masculinization and defeminization of female sheep. Biol Reprod (2008) 79(1):43–50. doi: 10.1095/biolreprod.107.067074
49. Piltonen T, Morin-Papunen L, Koivunen R, Perheentupa A, Ruokonen A, Tapanainen JS. Serum anti-müllerian hormone levels remain high until late reproductive age and decrease during metformin therapy in women with polycystic ovary syndrome. Hum Reprod (2005) 20(7):1820–6. doi: 10.1093/humrep/deh850
50. Tokmak A, Kokanali D, Timur H, Kuntay Kokanali M, Yilmaz N. Association between anti-mullerian hormone and insulin resistance in non-obese adolescent females with polycystic ovary syndrome. Gynecol Endocrinol (2016) 32(11):926–30. doi: 10.1080/09513590.2016.1193140
51. Codner E, Iñiguez G, Hernández IM, et al. Elevated anti-müllerian hormone (AMH) and inhibin b levels in prepubertal girls with type 1 diabetes mellitus. Clin Endocrinol (Oxf) (2011) 74(1):73–8. doi: 10.1111/j.1365-2265.2010.03887.x
52. Köninger A, Kampmeier A, Schmidt B, et al. Trends in anti-müllerian hormone concentrations across different stages of pregnancy in women with polycystic ovary syndrome. Reprod BioMed Online (2018) 37(3):367–74. doi: 10.1016/j.rbmo.2018.05.011
53. Tata B, Mimouni NEH, Barbotin AL, et al. Elevated prenatal anti-müllerian hormone reprograms the fetus and induces polycystic ovary syndrome in adulthood. Nat Med (2018) 24(6):834–46. doi: 10.1038/s41591-018-0035-5
54. Tal R, Seifer CM, Khanimov M, Seifer DB, Tal O. High serum antimullerian hormone levels are associated with lower live birth rates in women with polycystic ovarian syndrome undergoing assisted reproductive technology. Reprod Biol Endocrinol (2020) 18(1):20. doi: 10.1186/s12958-020-00581-4
55. Du M, Zhang J, Yu X, Guan Y. Elevated anti-müllerian hormone is an independent risk factor for preterm birth among patients with overweight polycystic ovary syndrome. Front Endocrinology (2021) 12.
56. Hu KL, Liu FT, Xu H, Li R, Qiao J. High antimüllerian hormone levels are associated with preterm delivery in patients with polycystic ovary syndrome. Fertil Steril (2020) 113(2):444–452.e441.
57. Kaing A, Jaswa EA, Diamond MP, Legro RS, Cedars MI, Huddleston HG. Highly elevated level of antimüllerian hormone associated with preterm delivery in polycystic ovary syndrome patients who underwent ovulation induction. Fertil Steril (2020). doi: 10.1016/j.fertnstert.2020.06.015
58. Fritz MA, Speroff L. Clinical gynecologic endocrinology and infertility (8th ed.). Philadelphia: Wolters Kluwer Health/Lippincott Williams & Wilkins (2011).
59. Morel Y, Roucher F, Plotton I, Goursaud C, Tardy V, Mallet D. Evolution of steroids during pregnancy: Maternal, placental and fetal synthesis. Ann Endocrinol (Paris) (2016) 77(2):82–9. doi: 10.1016/j.ando.2016.04.023
60. Maliqueo M, Cruz G, Espina C, et al. Obesity during pregnancy affects sex steroid concentrations depending on fetal gender. Int J Obes (Lond) (2017) 41(11):1636–45. doi: 10.1038/ijo.2017.159
61. Simoni RD, Hill RL, Vaughan M. The discovery of estrone, estriol, and estradiol and the biochemical study of reproduction. work Edward Adelbert Doisy. J Biol Chem (2002) 277(28):e1–2.
62. Chatuphonprasert W, Jarukamjorn K, Ellinger I. Physiology and pathophysiology of steroid biosynthesis, transport and metabolism in the human placenta. Front Pharmacol (2018) 9(1027). doi: 10.3389/fphar.2018.01027
63. Kaludjerovic J, Ward WE. The interplay between estrogen and fetal adrenal cortex. J Nutr Metab (2012) 2012:837901. doi: 10.1155/2012/837901
64. Berkane N, Liere P, Oudinet J-P, et al. From pregnancy to preeclampsia: A key role for estrogens. Endocrine Rev (2017) 38(2):123–44. doi: 10.1210/er.2016-1065
65. Hammond GL. Diverse roles for sex hormone-binding globulin in reproduction. Biol Reprod (2011) 85(3):431–41. doi: 10.1095/biolreprod.111.092593
66. Wan J, Hu Z, Zeng K, et al. The reduction in circulating levels of estrogen and progesterone in women with preeclampsia. Pregnancy Hypertens (2018) 11:18–25. doi: 10.1016/j.preghy.2017.12.003
67. Phupong V, Hanprasertpong T, Honsawek S. First trimester serum inhibin a in normal pregnant women. Arch Gynecology Obstetrics (2008) 277(4):307–10. doi: 10.1007/s00404-007-0491-5
68. Singnoi W, Wanapirak C, Sekararithi R, Tongsong T. A cohort study of the association between maternal serum inhibin-a and adverse pregnancy outcomes: a population-based study. BMC Pregnancy Childbirth (2019) 19(1):124. doi: 10.1186/s12884-019-2266-y
69. Yue CY, Zhang CY, Ni YH, Ying CM. Are serum levels of inhibin a in second trimester predictors of adverse pregnancy outcome? PloS One (2020) 15(5):e0232634. doi: 10.1371/journal.pone.0232634
70. Tarca AL, Romero R, Benshalom-Tirosh N, et al. The prediction of early preeclampsia: Results from a longitudinal proteomics study. PloS One (2019) 14(6):e0217273. doi: 10.1371/journal.pone.0217273
71. Lai J, Pinas A, Syngelaki A, Poon LC, Nicolaides KH. Maternal serum activin-a at 30-33 weeks in the prediction of preeclampsia. J Matern Fetal Neonatal Med (2013) 26(8):733–7. doi: 10.3109/14767058.2012.755167
72. Muttukrishna S, North RA, Morris J, et al. Serum inhibin a and activin a are elevated prior to the onset of pre-eclampsia. Hum Reprod (2000) 15(7):1640–5. doi: 10.1093/humrep/15.7.1640
73. Anderson H, Fogel N, Grebe SK, Singh RJ, Taylor RL, Dunaif A. Infants of women with polycystic ovary syndrome have lower cord blood androstenedione and estradiol levels. J Clin Endocrinol Metab (2010) 95(5):2180–6. doi: 10.1210/jc.2009-2651
74. Caanen MR, Kuijper EA, Hompes PG, et al. Mass spectrometry methods measured androgen and estrogen concentrations during pregnancy and in newborns of mothers with polycystic ovary syndrome. Eur J Endocrinol (2016) 174(1):25–32. doi: 10.1530/EJE-15-0699
75. Villarroel C, Salinas A, López P, et al. Pregestational type 2 diabetes and gestational diabetes exhibit different sexual steroid profiles during pregnancy. Gynecol Endocrinol (2017) 33(3):212–7. doi: 10.1080/09513590.2016.1248933
76. Köninger A, Kauth A, Schmidt B, et al. Anti-mullerian-hormone levels during pregnancy and postpartum. Reprod Biol Endocrinol (2013) 11:60. doi: 10.1186/1477-7827-11-60
77. Pankhurst MW, Clark CA, Zarek J, Laskin CA, McLennan IS. Changes in circulating ProAMH and total AMH during healthy pregnancy and post-partum: A longitudinal study. PloS One (2016) 11(9):e0162509–e0162509. doi: 10.1371/journal.pone.0162509
78. Villarroel C, Salinas A, López P, et al. Anti-müllerian hormone in type 2 and gestational diabetes during the second half of pregnancy: relationship with sexual steroid levels and metabolic parameters. Gynecol Endocrinol (2018) 34(2):120–4. doi: 10.1080/09513590.2017.1359824
79. Gerli S, Favilli A, Brozzetti A, et al. Anti-mullerian hormone concentration during the third trimester of pregnancy and puerperium: a longitudinal case-control study in normal and diabetic pregnancy. Endocrine (2015) 50(1):250–5. doi: 10.1007/s12020-014-0515-4
80. Li M-Y, Rawal S, Hinkle SN, et al. Sex hormone-binding globulin, cardiometabolic biomarkers, and gestational diabetes: A longitudinal study and meta-analysis. Matern Fetal Med (2020) 2(1):2–9. doi: 10.1097/FM9.0000000000000037
81. Veltman-Verhulst SM, van Haeften TW, Eijkemans MJ, de Valk HW, Fauser BC, Goverde AJ. Sex hormone-binding globulin concentrations before conception as a predictor for gestational diabetes in women with polycystic ovary syndrome. Hum Reprod (2010) 25(12):3123–8. doi: 10.1093/humrep/deq272
82. Zhang YJ, Jin H, Qin ZL, et al. Predictors of gestational diabetes mellitus in chinese women with polycystic ovary syndrome: A cross-sectional study. Gynecol Obstet Invest (2016) 81(3):220–4. doi: 10.1159/000440618
83. Yamashita H, Shao J, Friedman JE. Physiologic and molecular alterations in carbohydrate metabolism during pregnancy and gestational diabetes mellitus. Clin Obstet Gynecol (2000) 43(1):87–98. doi: 10.1097/00003081-200003000-00009
84. Hufnagel A, Dearden L, Fernandez-Twinn DS, Ozanne SE. Programming of cardiometabolic health: the role of maternal and fetal hyperinsulinaemia. J Endocrinol (2022) 253(2):R47–r63. doi: 10.1530/JOE-21-0332
85. Westgate JA, Lindsay RS, Beattie J, et al. Hyperinsulinemia in cord blood in mothers with type 2 diabetes and gestational diabetes mellitus in new zealand. Diabetes Care (2006) 29(6):1345–50. doi: 10.2337/dc05-1677
86. Uebel K, Pusch K, Gedrich K, Schneider KT, Hauner H, Bader BL. Effect of maternal obesity with and without gestational diabetes on offspring subcutaneous and preperitoneal adipose tissue development from birth up to year-1. BMC Pregnancy Childbirth (2014) 14:138. doi: 10.1186/1471-2393-14-138
87. Catalano PM, Presley L, Minium J, Hauguel-de Mouzon S. Fetuses of obese mothers develop insulin resistance. utero. Diabetes Care (2009) 32(6):1076–80. doi: 10.2337/dc08-2077
88. Maliqueo M, Lara HE, Sánchez F, Echiburú B, Crisosto N, Sir-Petermann T. Placental steroidogenesis in pregnant women with polycystic ovary syndrome. Eur J Obstet Gynecol Reprod Biol (2013) 166(2):151–5. doi: 10.1016/j.ejogrb.2012.10.015
89. North S, Zinn C, Crofts C. Hyperinsulinemia during pregnancy across varying degrees of glucose tolerance: An examination of the kraft database. J Obstetrics Gynaecology Res (2021) 47(5):1719–26. doi: 10.1111/jog.14731
90. Kallak TK, Hellgren C, Skalkidou A, et al. Maternal and female fetal testosterone levels are associated with maternal age and gestational weight gain. Eur J Endocrinol (2017) 177(4):379–88. doi: 10.1530/EJE-17-0207
91. Saez JM, Forest MG, Morera AM, Bertrand J. Metabolic clearance rate and blood production rate of testosterone and dihydrotestosterone in normal subjects, during pregnancy, and in hyperthyroidism. J Clin Invest (1972) 51(5):1226–34. doi: 10.1172/JCI106917
92. Makieva S, Saunders PT, Norman JE. Androgens in pregnancy: roles in parturition. Hum Reprod Update (2014) 20(4):542–59. doi: 10.1093/humupd/dmu008
93. Jaffe RB. Fetoplacental endocrine and metabolic physiology. Clin Perinatol (1983) 10(3):669–93. doi: 10.1016/S0095-5108(18)30958-8
94. Andræ F, Abbott D, Stridsklev S, et al. Sustained maternal hyperandrogenism during PCOS pregnancy reduced by metformin in non-obese women carrying a male fetus. J Clin Endocrinol Metab (2020) 105(12):3762–70. doi: 10.1210/clinem/dgaa605
95. Thadhani R, Wolf M, Hsu-Blatman K, Sandler L, Nathan D, Ecker JL. First-trimester sex hormone binding globulin and subsequent gestational diabetes mellitus. Am J Obstet Gynecol (2003) 189(1):171–6. doi: 10.1067/mob.2003.343
96. Kelley AS, Smith YR, Padmanabhan V. A narrative review of placental contribution to adverse pregnancy outcomes in women with polycystic ovary syndrome. J Clin Endocrinol Metab (2019) 104(11):5299–315. doi: 10.1210/jc.2019-00383
97. Hakim C, Padmanabhan V, Vyas AK. Gestational hyperandrogenism in developmental programming. Endocrinology (2016) 158(2):199–212.
98. Barbour LA, McCurdy CE, Hernandez TL, Kirwan JP, Catalano PM, Friedman JE. Cellular mechanisms for insulin resistance in normal pregnancy and gestational diabetes. Diabetes Care (2007) 30(Supplement 2):S112. doi: 10.2337/dc07-s202
99. Di Cianni G, Miccoli R, Volpe L, Lencioni C, Del Prato S. Intermediate metabolism in normal pregnancy and in gestational diabetes. Diabetes Metab Res Rev (2003) 19(4):259–70. doi: 10.1002/dmrr.390
100. Phelps RL, Metzger BE, Freinkel N. Carbohydrate metabolism in pregnancy. XVII. diurnal profiles of plasma glucose, insulin, free fatty acids, triglycerides, cholesterol, and individual amino acids in late normal pregnancy. Am J Obstet Gynecol (1981) 140(7):730–6. doi: 10.1016/0002-9378(81)90731-6
101. Parsons JA, Brelje TC, Sorenson RL. Adaptation of islets of langerhans to pregnancy: increased islet cell proliferation and insulin secretion correlates with the onset of placental lactogen secretion. Endocrinology (1992) 130(3):1459–66.
102. Smirnakis KV, Plati A, Wolf M, Thadhani R, Ecker JL. Predicting gestational diabetes: choosing the optimal early serum marker. Am J Obstet Gynecol (2007) 196(4):410.e411–416. doi: 10.1016/j.ajog.2006.12.011
103. Spencer K, Yu CK, Rembouskos G, Bindra R, Nicolaides KH. First trimester sex hormone-binding globulin and subsequent development of preeclampsia or other adverse pregnancy outcomes. Hypertens Pregnancy (2005) 24(3):303–11. doi: 10.1080/10641950500281068
104. Mochizuki M, Maruo T. Effect of dehydroepiandrosterone sulfate on uterine cervical ripening in late pregnancy. Acta Physiol Hung (1985) 65(3):267–74.
105. Tagawa N, Hidaka Y, Takano T, Shimaoka Y, Kobayashi Y, Amino N. Serum concentrations of dehydroepiandrosterone and dehydroepiandrosterone sulfate and their relation to cytokine production during and after normal pregnancy. Clin Chim Acta (2004) 340(1-2):187–93. doi: 10.1016/j.cccn.2003.10.018
106. Hogeveen KN, Cousin P, Pugeat M, Dewailly D, Soudan B, Hammond GL. Human sex hormone-binding globulin variants associated with hyperandrogenism and ovarian dysfunction. J Clin Invest (2002) 109(7):973–81. doi: 10.1172/JCI0214060
107. Jones ME, Boon WC, McInnes K, Maffei L, Carani C, Simpson ER. Recognizing rare disorders: aromatase deficiency. Nat Clin Pract Endocrinol Metab (2007) 3(5):414–21. doi: 10.1038/ncpendmet0477
108. Nelson SM, Stewart F, Fleming R, Freeman DJ. Longitudinal assessment of antimüllerian hormone during pregnancy–relationship with maternal adiposity, insulin, and adiponectin. Fertility Sterility (2010) 93(4):1356–8. doi: 10.1016/j.fertnstert.2009.07.1676
109. Köninger A, Kampmeier A, Mach P, et al. Tight interplay in early pregnancy between follistatin and anti-mullerian hormone in women with polycystic ovarian syndrome (PCOS). Arch Gynecology Obstetrics (2018) 297(5):1307–16. doi: 10.1007/s00404-018-4718-4
110. Zhou S, Lu D, Wen S, Sheng Y, Kang D, Xu L. Elevated anti-müllerian hormone levels in newborns of women with polycystic ovary syndrome: a systematic review and meta-analysis based on observational studies. Reprod Sci (2022) 29(1):301–11. doi: 10.1007/s43032-021-00652-w
111. Vounzoulaki E, Khunti K, Abner SC, Tan BK, Davies MJ, Gillies CL. Progression to type 2 diabetes in women with a known history of gestational diabetes: systematic review and meta-analysis. Bmj (2020) 369:m1361.
112. Bitó T, Földesi I, Nyári T, Pál A. Prediction of gestational diabetes mellitus in a high-risk group by insulin measurement in early pregnancy. Diabetes Med (2005) 22(10):1434–9. doi: 10.1111/j.1464-5491.2005.01634.x
113. Lapolla A, Dalfrà MG, Mello G, et al. Early detection of insulin sensitivity and beta-cell function with simple tests indicates future derangements in late pregnancy. J Clin Endocrinol Metab (2008) 93(3):876–80. doi: 10.1210/jc.2007-1363
114. Zhang N-J, M-f T, Li H-P, Zhao F, Wang F-H. The relationship between patterns of insulin secretion and risks of gestational diabetes mellitus. Int J Gynecology Obstetrics (2020) 150(3):318–23. doi: 10.1002/ijgo.13200
115. Uzelac PS, Li X, Lin J, et al. Dysregulation of leptin and testosterone production and their receptor expression in the human placenta with gestational diabetes mellitus. Placenta (2010) 31(7):581–8. doi: 10.1016/j.placenta.2010.04.002
116. Hähnel ME, Martin JD, Michael CA, Hähnel R. Metabolism of androstenedione by placental microsomes in pregnancy hypertension. Clin Chim Acta (1989) 181(1):103–8. doi: 10.1016/0009-8981(89)90322-7
117. Brown FM, Wyckoff J, Rowan JA, Jovanovic L, Sacks DA, Briggs GG. Metformin in pregnancy: its time has not yet come. Diabetes Care (2006) 29(2):485–6. doi: 10.2337/diacare.29.02.06.dc05-2098
118. Mousa A, Løvvik T, Hilkka I, et al. Metformin in pregnancy study (MiPS): protocol for a systematic review with individual patient data meta-analysis. BMJ Open (2020) 10(5):e036981. doi: 10.1136/bmjopen-2020-036981
119. Blum AK. Insulin use in pregnancy: An update. Diabetes Spectr (2016) 29(2):92–7. doi: 10.2337/diaspect.29.2.92
120. Ye W, Luo C, Huang J, Li C, Liu Z, Liu F. Gestational diabetes mellitus and adverse pregnancy outcomes: systematic review and meta-analysis. BMJ (2022) 377:e067946. doi: 10.1136/bmj-2021-067946
121. Arshad R, Karim N, Ara Hasan J. Effects of insulin on placental, fetal and maternal outcomes in gestational diabetes mellitus. Pak J Med Sci (2014) 30(2):240–4.
122. Evers IM, de Valk HW, Mol BW, ter Braak EW, Visser GH. Macrosomia despite good glycaemic control in type i diabetic pregnancy; results of a nationwide study in the netherlands. Diabetologia (2002) 45(11):1484–9.
123. Daan NM, Koster MP, Steegers-Theunissen RP, Eijkemans MJ, Fauser BC. Endocrine and cardiometabolic cord blood characteristics of offspring born to mothers with and without polycystic ovary syndrome. Fertil Steril (2017) 107(1):261–268.e263. doi: 10.1016/j.fertnstert.2016.09.042
124. Mehrabian F, Kelishadi R. Comparison of the metabolic parameters and androgen level of umbilical cord blood in newborns of mothers with polycystic ovary syndrome and controls. J Res Med Sci (2012) 17(3):207–11.
125. Duan C, Pei T, Li Y, Cao Q, Chen H, Fu J. Androgen levels in the fetal cord blood of children born to women with polycystic ovary syndrome: a meta-analysis. Reprod Biol Endocrinol (2020) 18(1):81. doi: 10.1186/s12958-020-00634-8
126. Kallen CB. Steroid hormone synthesis in pregnancy. Obstetrics Gynecology Clinics North America (2004) 31(4):795–816. doi: 10.1016/j.ogc.2004.08.009
127. Padmanabhan V, Veiga-Lopez A. Developmental origin of reproductive and metabolic dysfunctions: androgenic versus estrogenic reprogramming. Semin Reprod Med (2011) 29(3):173–86. doi: 10.1055/s-0031-1275519
128. Padmanabhan V, Veiga-Lopez A, Abbott DH, Recabarren SE, Herkimer C. Developmental programming: impact of prenatal testosterone excess and postnatal weight gain on insulin sensitivity index and transfer of traits to offspring of overweight females. Endocrinology (2010) 151(2):595–605. doi: 10.1210/en.2009-1015
129. Vanky E, Carlsen SM. Androgens and antimüllerian hormone in mothers with polycystic ovary syndrome and their newborns. Fertil Steril (2012) 97(2):509–15. doi: 10.1016/j.fertnstert.2011.11.021
130. Tokmak A, Güney G, Aksoy RT, et al. May maternal anti-mullerian hormone levels predict adverse maternal and perinatal outcomes in preeclampsia? J Matern Fetal Neonatal Med (2015) 28(12):1451–6. doi: 10.3109/14767058.2014.955007
131. Parrettini S, Caroli A, Torlone E. Nutrition and metabolic adaptations in physiological and complicated pregnancy: Focus on obesity and gestational diabetes. Front Endocrinol (Lausanne) (2020) 11:611929. doi: 10.3389/fendo.2020.611929
132. Welsh P, Smith K, Nelson SM. A single-centre evaluation of two new anti-mullerian hormone assays and comparison with the current clinical standard assay. Hum Reprod (2014) 29(5):1035–41. doi: 10.1093/humrep/deu036
Keywords: hyperandrogenism, hyperinsulinemia, polycystic ovary syndrome, obesity, gestational diabetes (GDM), type 1 diabetes (T1D), type 2 diabetes, pregnancy
Citation: Neven ACH, Mousa A, Boyle JA and Teede HJ (2023) Endocrine and metabolic interactions in healthy pregnancies and hyperinsulinemic pregnancies affected by polycystic ovary syndrome, diabetes and obesity. Front. Endocrinol. 13:993619. doi: 10.3389/fendo.2022.993619
Received: 13 July 2022; Accepted: 06 December 2022;
Published: 17 January 2023.
Edited by:
Lisa Owens, St. James’s Hospital, IrelandReviewed by:
Vasantha Padmanabhan, University of Michigan, United StatesFatme Al Anouti, Zayed University, United Arab Emirates
Copyright © 2023 Neven, Mousa, Boyle and Teede. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jacqueline A. Boyle, amFjcXVlbGluZS5ib3lsZUBtb25hc2guZWR1
†These authors share senior authorship