 Yifei Zhu
Yifei Zhu Xinyan Li1
Xinyan Li1 Lei Wang
Lei Wang Jie Yang
Jie Yang
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Endocrinol., 15 August 2022
Sec. Cancer Endocrinology
Volume 13 - 2022 | https://doi.org/10.3389/fendo.2022.988295
It is notorious that cancer cells alter their metabolism to adjust to harsh environments of hypoxia and nutritional starvation. Metabolic reprogramming most often occurs in the tumor microenvironment (TME). TME is defined as the cellular environment in which the tumor resides. This includes surrounding blood vessels, fibroblasts, immune cells, signaling molecules and the extracellular matrix (ECM). It is increasingly recognized that cancer cells, fibroblasts and immune cells within TME can regulate tumor progression through metabolic reprogramming. As the most significant proportion of cells among all the stromal cells that constitute TME, cancer-associated fibroblasts (CAFs) are closely associated with tumorigenesis and progression. Multitudinous studies have shown that CAFs participate in and promote tumor metabolic reprogramming and exert regulatory effects via the dysregulation of metabolic pathways. Previous studies have demonstrated that curbing the substance exchange between CAFs and tumor cells can dramatically restrain tumor growth. Emerging studies suggest that CAFs within the TME have emerged as important determinants of metabolic reprogramming. Metabolic reprogramming also occurs in the metabolic pattern of immune cells. In the meanwhile, immune cell phenotype and functions are metabolically regulated. Notably, immune cell functions influenced by metabolic programs may ultimately lead to alterations in tumor immunity. Despite the fact that multiple previous researches have been devoted to studying the interplays between different cells in the tumor microenvironment, the complicated relationship between CAFs and immune cells and implications of metabolic reprogramming remains unknown and requires further investigation. In this review, we discuss our current comprehension of metabolic reprogramming of CAFs and immune cells (mainly glucose, amino acid, and lipid metabolism) and crosstalk between them that induces immune responses, and we also highlight their contributions to tumorigenesis and progression. Furthermore, we underscore potential therapeutic opportunities arising from metabolism dysregulation and metabolic crosstalk, focusing on strategies targeting CAFs and immune cell metabolic crosstalk in cancer immunotherapy.
The term “metabolic reprogramming” is often used to denote a set of abnormal metabolic pathways observed in highly proliferative tumor or cancer cells (1). In cancer, malignant cells acquire metabolic adaptations through various extracellular and endocytic pathways to meet the necessary nutrients required for tumor growth and to use these nutrients to maintain survival and produce new biomass (2, 3). Some of these adaptations initiate the transformation process, and some promote the growth of malignant cells, making them vulnerable to inhibitors of key pathways (4). Cancer cells depend on a variety of different metabolic pathways and the specific nutrients used are influenced by both cancer cell genes and environmental conditions (5). The vast majority of mammalian cells use glucose as a source of energy. Glucose is metabolized by glycolysis, which undergoes multiple reaction steps to yield pyruvate. In typical cells with normal blood oxygen levels, most of the pyruvate gets access in the mitochondria. It is oxidized by the tricarboxylic acid cycle (TCA) to generate ATP to satisfy the cell’s energy needs. However, in highly proliferative cell types such as cancer cells, the vast majority of the pyruvate produced by glycolysis leaves the mitochondria. It produces lactate by lactate dehydrogenase (LDH/LDHA), a procedure that usually occurs in the hypoxic state. The product of lactic acid in the existence of oxygen is called “aerobic glycolysis” or “the Warburg effect” (2, 3, 6–11).
Tumor microenvironment (TME) is identified as the surrounding microenvironment where tumor cells reside, including surrounding blood vessels, fibroblasts, immune cells, bone marrow-derived inflammatory cells, various signaling molecules and ECM. TME is a complicated integrated system made up of the metabolic reprogramming of tumor cells with surrounding tissues and immune cells, as well as the crosstalk between them. It has long been identified that TME acts as a pivotal position in the development and progression of tumors. Although other reviews have provided a detailed examination of the role of the TME in tumorigenesis (12–15), here, we concentrate on the metabolic reprogramming in cancer-associated fibroblasts (CAFs) and immune cells along with the crosstalk between them that induces immune responses, and we also highlight the therapeutic opportunities presented by metabolic dysregulation and metabolic crosstalk, focusing on strategies that may help to precisely target alterations in tumor metabolism.
Fibroblasts play a prominent role in tissue homeostasis, tumorigenesis, and inflammatory and fibrosis progression (16). CAFs are one of the most plentiful matrix components in TME and become specific targets in the vast majority of solid tumors (17). A large number of researches have illustrated that CAFs play protruding roles in tumor pathogenesis and have important clinical significance (16, 18–21). Mechanistically and functionally, CAFs secrete various cytokines or metabolites through metabolic reprogramming to inhibit the function of immune cells and promote tumor development, invasion, and metastasis; CAFs also have the ability to shape the EMC, form a barrier for drug or therapeutic immune cell penetration and prevent the deep penetration of drugs and immune cells into tumor tissues, thus reducing the effectiveness of tumor treatment (22–25). In cancer, a greater understanding of the complexity of CAFs may have therapeutic and prognostic value. Various types of adaptive and innate immune cells exist or infiltrate into TME. The metabolic reprogramming between these immune cells and tumor cells determines the immune status of the tumor and can promote or suppress the immune response of the tumor (26). To better understand the role of this metabolic crosstalk in TME, this review discusses the metabolic reprogramming of CAFs and immune cells, as well as their interactions and further highlights the meaning of developing novel therapeutic strategies on the basis of metabolism.
Previous studies have demonstrated that CAFs activation and reprogramming can occur in TME (17, 27–29). Therefore, identifying the cellular origin of CAF subtypes is a critical issue, as it may partly confirm the functions among different CAF populations and may enlighten new therapeutic strategies.
There is growing evidence that CAFs is a complex heterogeneous cell population, which may be attributed to the diverse potential cellular origins of CAFs (30, 31) (Figure 1). Resting tissue fibroblasts can form CAFs upon activation by neighboring tumor cells. For instance, resting hepatic stellate cells (HSCs) and pancreatic stellate cells (PSCs) are separately considered as CAFs in pancreatic and hepatocellular carcinomas (32–35). Some other major cell sources of CAFs identified in different studies include mesenchymal stem cells, endothelial cells and adipocytes (36–44). Notably, although CAFs and tumor-associated mesenchymal stem cells (TA-MSCs) are derived from the same cells (primitive mesenchymal stem cells), TA- MSCs have greater self-renewal capability and low expression of markers associated with CAFs such as wave proteins, fibroblast activation protein (FAP) and fibroblast-specific proteins 1 (FSP1, or called S100A4), which warrants further mechanistic studies (45–47). Overall, the exact origin of CAFs and their subpopulations have not been fully elucidated. Probably because of these cells’ phenotypic and functional plasticity and the lack of specific genealogical biomarkers. The development of genealogical tracing techniques may help better to trace the origin of CAFs in the coming years.
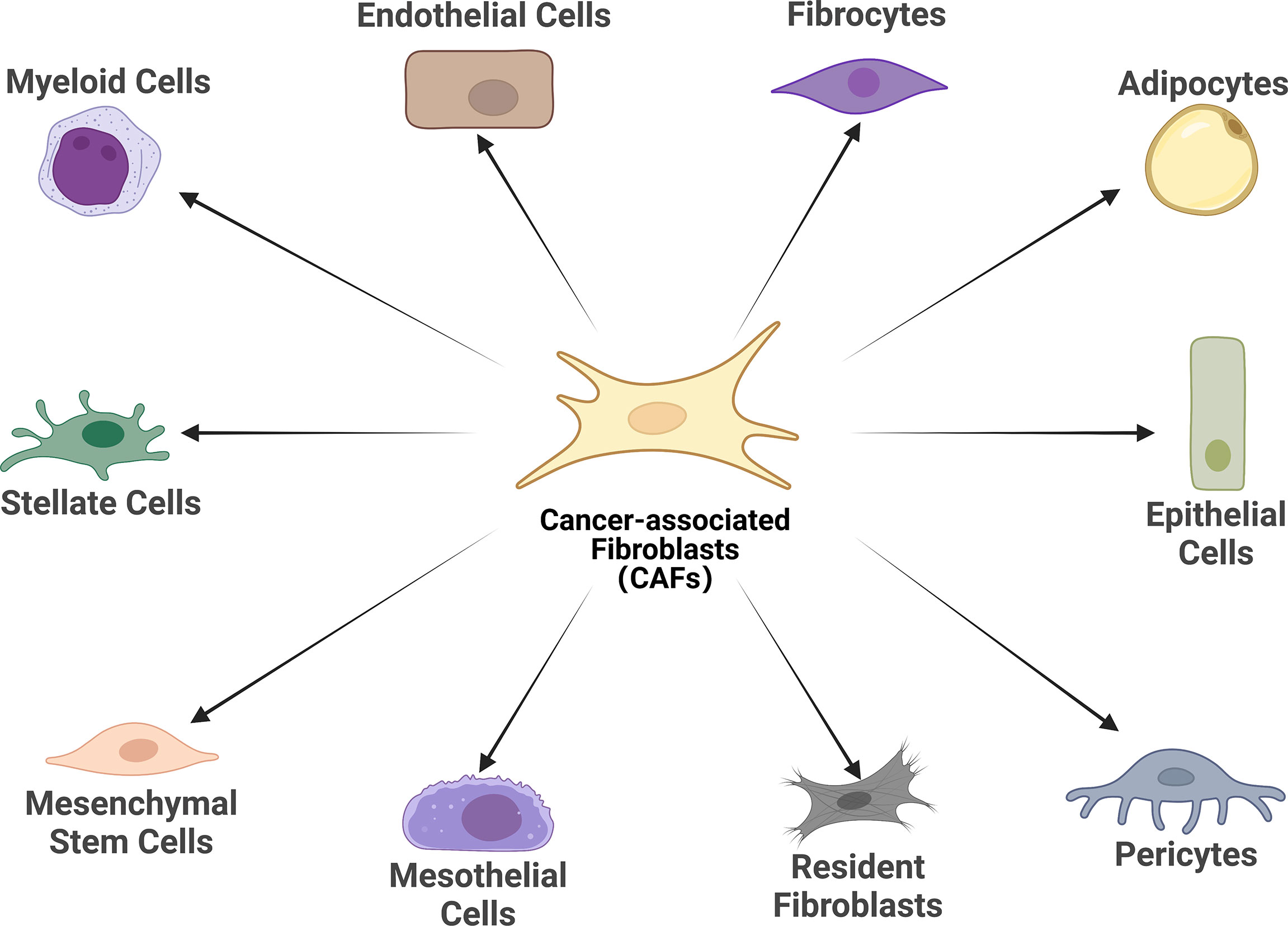
Figure 1 Cell of origin of cancer-associated fibroblasts (CAFs). Schematic representation of the cells of origin of CAFs that have been reported or in potential, including epithelial cells, mesothelial cells, resident fibroblasts, stellate cells, pericytes, adipocytes, mesenchymal stem cells, myeloid cells, fibrocytes, and endothelial cells.
Generally speaking, CAFs are considered to be genetically stable, especially when it comes to tumor cells (48, 49). Still, certain DNA damage, such as radiation, can lead to the transformation of normal fibroblasts to CAFs, which is reminiscent of tumorigenic development (50). Future studies on the role of genetic mutations in CAFs need to be more in-depth. However, to date, the origin of some CAFs, the cancer-restraining subgroups (rCAFs) in particular, remains unclear, and further exploration of rCAFs may turn into a prospective research direction.
The heterogeneity of CAFs, especially those activated from the resting state, may depend on their origin. Because of their different origins, the functions of these activated fibroblasts may be diverse (19, 30). CAFs are highly plastic and pluripotent. Activated CAFs can adapt to perivascular and vascular functions. This plasticity that leads to the heterogeneity of CAFs (19). The observed heterogeneity and plasticity of CAFs may have the following possible explanations (1): dynamic and interchangeable changes in CAFs between tumor-promoting or tumor-suppressing phenotypes, counting on the intricate background of the surrounding TME (2); the existence of a wide range of CAF subpopulations with different functions, and the diversity of CAF subpopulations may exceed the few major subpopulations defined in previous studies (51). Many MSC biomarkers can selectively identify activated CAFs in specific tumor microenvironments (28).
Over the past few years, it has been possible to identify a variety of biomarker genes that define different potential subgroups of CAFs with the advent of single-cell RNA sequencing (scRNA-seq) (52), leading to a deeper knowledge of the plasticity and heterogeneity of CAFs exhibited in various tumor types. Different biomarkers and classification approaches are often used to classify tumor types and various CAF subgroups. Here, we summarize common tumors and the corresponding CAF subgroups according to the latest studies (Table 1) (21, 53–74). The expression of surface markers varies among different subtypes of CAFs. For example, FAP is considered to be a CAFs-specific expressed protein, and therefore FAP is often used as a target of CAFs for tumor diagnosis and treatment (53, 75, 76). α-SMA has serine protease activity and can be involved in fibrillogenesis and ECM remodelling, thus enhancing the malignant behavior of activated tumor cells. α- can be found in most cancer types. α-SMA not only assists in recognizing activated CAFs, but also assumes the role of a universal marker for some mesenchymal cells. Therefore, α-SMA is usually used as a major evaluation criterion for the prognosis of targeted CAFs treatment (30, 77, 78). Another renowned marker is fibroblast-specific protein, which, as the name suggests, is expressed comparatively specifically on fibroblasts. CAFs with the expression of FSP1 have a unique tumor-protective role in immunosurveillance because such CAFs can produce collagen (79–81).
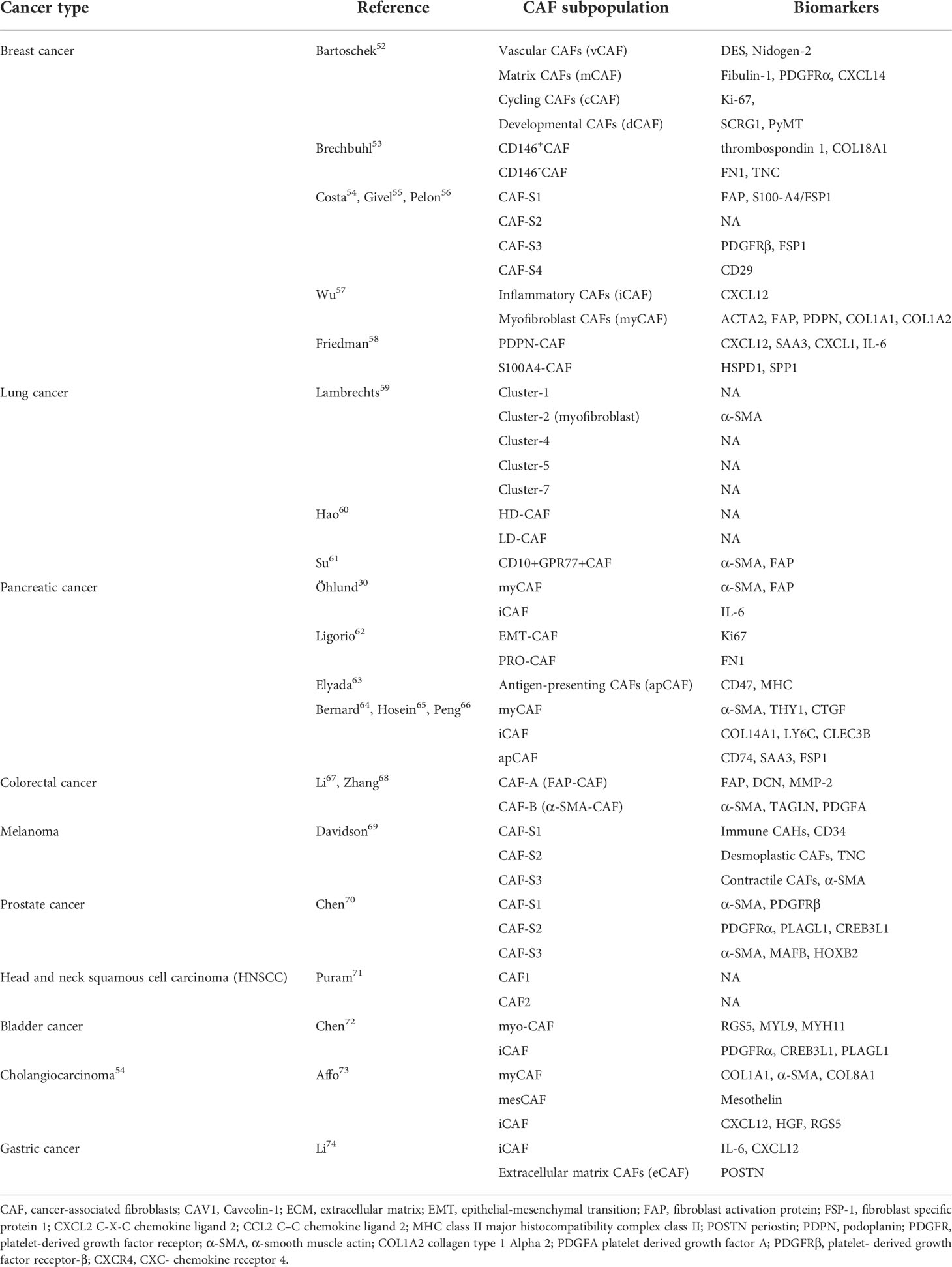
Table 1 The proposed classification of CAFs in different cancer types.
Similar to the mode of operation of T lymphocytes, CAFs consist of many cell subsets that respond to different metabolic and immune responses, form specific secretory manifestations, and perform irreplaceable biological functions in TME. Although CAF markers are diverse, identifying the functional subset of CAFs with the application of cell surface markers remains arduous. The development of in vitro CAF functional and mechanistic studies of live cell sorting together with in vivo CAF-targeted therapies have been significantly hampered by the absence of well-defined cell surface markers. It is undeniable that single-cell detection technology has made significant advancements in the past years, the variety of markers for CAFs is abundant, and the genetic signature of specific CAF subtypes may vary between cancer types, and different stages of certain types of cancer, or between patients. These pose a great challenge to differentiate CAF subtypes and identify specific and accurate cell surface markers.
In recent years, it has been increasingly recognized that malignancies can be considered not only genetic diseases but also metabolic diseases (82, 83). Metabolic reprogramming meets the high demand of tumor cells for rapid proliferation and growth and assists tumor cells in surviving in the relatively low oxygen and hostile environment (83). Malignant cells require sufficient Adenosine Triphosphate (ATP), as well as several other nutrients, including nucleic acids, lipids and proteins to sustain their growth. Previous studies have found that cancer cells tend to meet the need for additional energy through metabolic reprogramming as a way to maintain a sustained state of cell proliferation and hypo differentiation (84). As the most profoundly understood cancer metabolic modality or pathway to date, the Warburg effect, meticulously depicts the metabolic reprogramming of cancer cells. In a specific TME setting, CAFs may be involved in the metabolic balance of synthesis and catabolism in conditional cancer cells (84–87). The cellular metabolism of CAFs is very similar to that of continuously proliferating cells and both are dependent on aerobic glycolysis (7). The Warburg effect is more pronounced in CAFs and seems to be associated with increased cellular catabolic activity and cellular autophagy. The interactions between cancer cells and CAFs are inextricably linked to metabolic reprogramming, which promotes the growth of cancer cells, metastasis and absconds of immune surveillance (88). However, this interaction’s detailed mechanisms and specific processes of this interaction are still unclear. Typically, tumor cells increase the uptake and the metabolic rate of various nutrients, of which glucose and glutamine are the most prominent components. CAFs have been shown to be involved in the elaborate metabolism of tumors, mainly containing glucose, amino acid and lipid metabolism, prompting tumor cells to counteract energy depletion due to the Warburg effect. The regulation of CAFs through these metabolic switches shapes the unique code of CAFs through these metabolic switches shapes the unusual CAF behavior and leads to altered tumor cell behavior (18, 31, 89–91). Therefore, an in-depth study of the metabolic reprogramming of CAFs can help to understand tumor cell growth and metastasis better. The metabolic reprogramming of CAFs will be discussed in the following aspects (Figure 2).
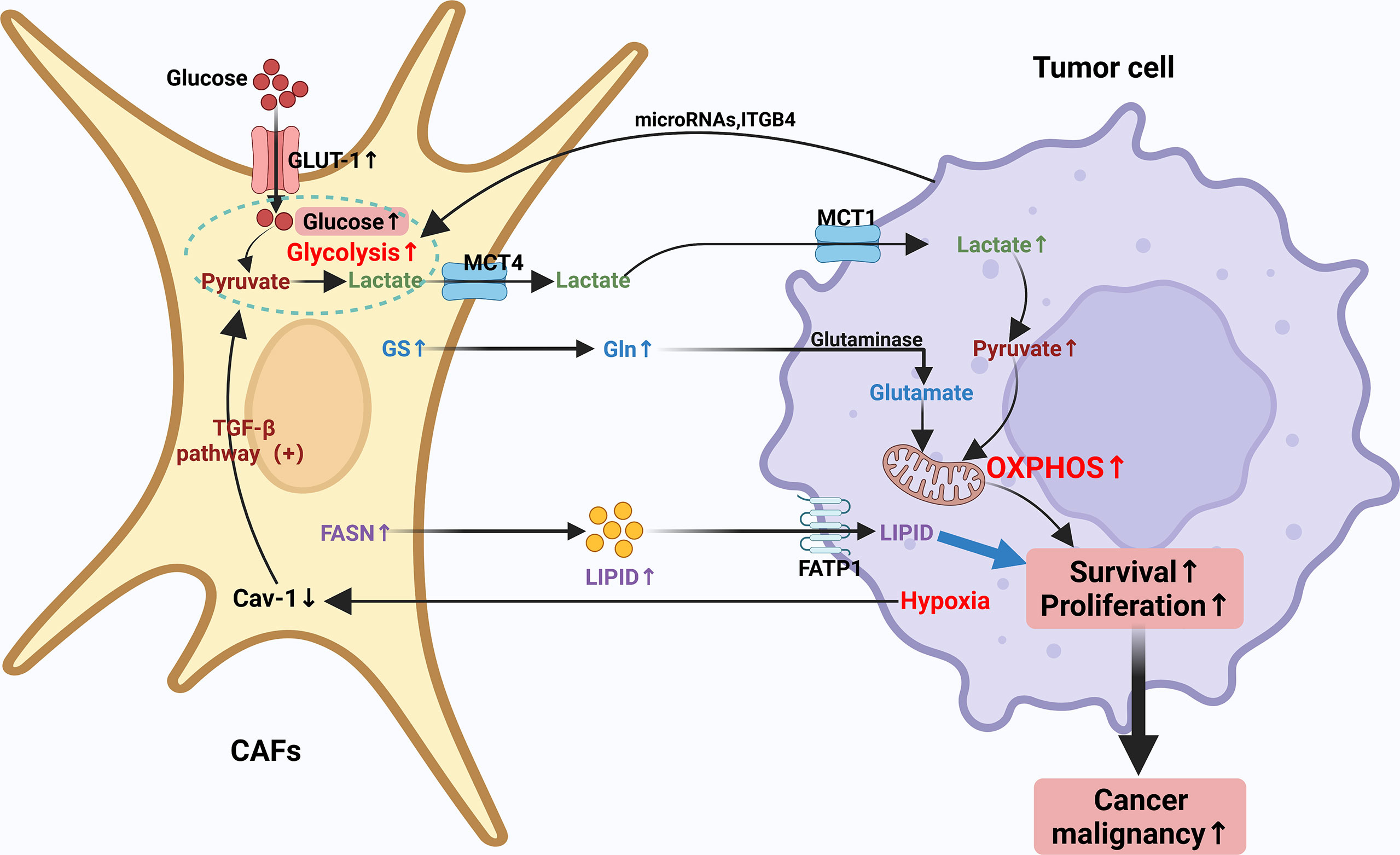
Figure 2 Crosstalk of metabolic reprogramming between CAFs and tumor cell. Both cancer cells and CAFs undergo a regulated metabolic reprogramming in the tumor microenvironment, in particular, CAFs undergo a clear Warburg effect. Typically, tumor cells increase their uptake of various nutrients and enhance their metabolic rate, with glucose and glutamine being the most prominent components. CAFs have been shown to be involved in the complex metabolism of tumors, mainly including glucose, amino acid and lipid metabolism, prompting tumor cells to counteract energy depletion due to the Warburg effect. The regulation of CAFs through these metabolic switches forms a unique CAF behavior and leads to altered tumor cell behavior through the unique regulation of CAFs through these metabolic switches.
CAFs have shifted to a more glycolysis-dependent phenotype through metabolic reprogramming, whereas normal cells count more on the mitochondrial pathway to generate energy via mitochondrial oxidative phosphorylation (OXPHOS). Moreover, it has been demonstrated that repression of glycolysis in tumor cells can recover OXPHOS (92–95). Under certain circumstances, energy supply through mitochondrial respiration is responsible for 80% of the ATP required by certain types of cancer cells such as MCF7 breast cancer (2, 96–99). However, the specific mechanism remains unclear currently, one possible explanation is that tumor cells that rely primarily on OXPHOS metabolism for energy supply, achieve resistance to oxidative stress by enhancing antioxidant responses and increasing detoxification capacity (100–102). Some of the enzymes involved in the glycolytic process, for example, HK2 and 6-phosphofructokinase liver type (PFKL) are considerably up-regulated in CAFs, which corroborates their glycolytic properties. HK2, as a fundamental glycolytic enzyme, is overexpressed in tumors, resulting in the “Warburg effect”. In the CAF model, the level of HK2 protein is apparently up-regulated during the differentiation of CAFs (103–106).
In the past, the high glycolytic rate of CAFs was often considered to be one of the main drivers for the maintenance of uninterrupted growth and proliferation of tumor cells. However, the specific biomolecular mechanisms underlying the occurrence of such high glycolytic rates in CAFs have not been fully understood to date. Encouragingly, lots of potential mechanisms have been put forward and further investigated to illustrate the metabolic reprogramming correlated with the upregulation of glycolysis in CAFs (107). One of the more accepted theories is that during contact with cancer cells, CAFs are reprogrammed to develop a glycolysis-dependent phenotype that increases glucose uptake and utilization and facilitates the transport of pyruvate and lactate (end products of glycolysis) (106). Lactate is expeditiously and effectively utilized by the cancer cells to gain energy and molecules through strengthened anabolism and to fuel OXPHOS (108). The above evidence mentioned offers further perception into the conception of metabolic reprogramming in TME.
Glucose enters cells through upregulation of glucose transporter 1 (GLUT-1) expression. Past studies have found that oncogenes like cMyc enhance glucose uptake by increasing lactate dehydrogenase a (LDHA) and GLUT-1 expression as a way to increase metabolic flux (109). It has also been documented that the promoters of pyruvate kinase (PK) and lactate dehydrogenase (LDHA) genes, key enzymes in the glycolytic process, are highly methylated in breast cancer-associated CAFs, which increases pyruvate kinase M2 (PKM2) and LDHA expression (105). Furthermore, changes in miR-186 expression during the formation of CAFs result in altered protein levels of GLUT-1. Thus, miR-186 can regulate glycolysis through changes in GLUT-1 expression (110). Decreased expression of isocitrate dehydrogenase 3 (IDH3α) α subunit has been reported to be connected with the metabolic transition from OXPHOS to glycolysis, and IDH3α overexpression prevents fibroblasts from converting to CAFs (105). Downregulation of Caveolin-1 (Cav-1) may give rise to the activation of the TGF-β pathway in CAFs, and the activated pathway allows for enhanced oxidative stress and aerobic glycolytic responses. Cav-1-deficient CAFs facilitate tumor cell development and aberrant vascular branching. Proteomic analysis of Cav-1-deficient CAFs demonstrates upregulation of glycolytic enzymes (e.g. PKM2 and LDH-B) (111–113). The investigators found that Cav-1 dysregulation brings about aberrant mitochondrial transcription factor A (TFAM) expression in fibroblasts, which in turn induces oxidative stress, mitochondrial dysfunction and aerobic glycolysis in TME (103). CAFs lacking TFAM may yield more hydrogen peroxide and L-lactic acid by paracrine means providing energy-rich metabolites for the purpose of promoting tumor growth and angiogenesis (114). Finally, it has been found that fibroblasts knocking down Cav-1 may exert an influence on cancer cell metabolism by promoting the production of lactate, a mitochondrial respiration product of synthetic cancer cells (103, 115). Overall, Cav-1 deficiency is critical in the metabolic reprogramming of CAFs. Some other markers such as integrin- 4 (ITGB4) in triple-negative breast cancer (TNBC) (113), MCT4 in nasopharyngeal carcinoma (116), MCT1 and MCT4 in breast and bladder cancer (117, 118), ITGB2 in oral squamous carcinoma (OSCC) (119), MCT1, succinate dehydrogenase (SDH) and fumarate hydratase (FH) expression levels are significantly elevated in pancreatic cancer cells (120). The overexpression of these markers further suggests metabolic crosstalk between CAFs and cancer cells.
Besides glucose, previous studies have proven that CAFs can raise the output of other nutrients required by cancer cells, such as amino acids that may function as a source of anabolic or fuel for OXPHOS, as cancer cells need more amino acids to meet their demand for rapid proliferation (121–124). Multiple studies have demonstrated that CAFs compound certain amino acids through the TCA cycle to maintain the continuous proliferation of tumor cells (125, 126). Amino acids can be separated into two classifications: essential amino acids and non-essential amino acids. Amino Acids have been widely studied for their use in the synthesis of proteins, peptides and other nitrogenous substances that are unique to the body, as well as for their role as metabolites that regulate the rapid and uninterrupted proliferation of cancer cells, with glutamine being the most studied (127, 128). Glutamine (Gln), an amide of glutamate, is one of the non-essential amino acids (which can be produced by the body without relying on diet), a major source of carbon and nitrogen, and has a role in promoting almost every biosynthetic pathway in cancer cells (122).
Previous studies have shown that exosomes derived from patients with prostate and pancreatic cancer can inhibit mitochondrial OXPHOS and compensate for increased glycolysis (129). Furthermore, it was demonstrated that exosomes are a source of metabolites carrying lactate, amino acids, TCA cycle intermediates and lipids that cancer cells utilize to proliferate and replenish levels of TCA cycle metabolites (130). Researchers found that these exosome-fed prostate cancer cells also presented a greater reliance on glutamine, with significantly raised levels of 13C-labeled m + 5 glutamate and m + 5 α-KG through 13C5-glutamine labeling experiments. This outcome demonstrates that CAFs have the ability to shift the metabolic pattern of cancer cells from mitochondria-reliable to glycolysis-reliable and up-regulate glutamine metabolism as a means of feeding the proliferating cancer cells with nutrients (129, 131, 132). In other cancers, such as ovarian cancer, CAFs produce Gln in large amounts via glutamine synthetase (GS). the Gln produced through the metabolism of CAFs is delivered to ovarian cancer cells, where Gln provides a source of nutrients for cancer cells on the one hand, and is converted to glutamate via glutaminase on the other hand, further supporting tumor cell growth by supplementing TCA metabolic pathway intermediates (133). Glutamine addiction is the process by which glutamine enters the TCA cycle to provide energy and growth for proliferating cancer cells (124). In recent studies, glutamine dependence was found to drive CAFs to migrate from nutrient-depleted regions where glutamine is about to be depleted to more glutamine-rich regions (134, 135). In a way, glutamine dependence accelerates the migration and aggression of CAFs, which boosts the migration of cancer cells to the eutrophic area in turn. The transfer of CAFs to Gln-rich areas is mediated by a polarized protein kinase B (AKT2), and the presence of polarized AKT2 recruits the aggression of CAFs and the absconds of cancer cells from original tumor sites (134). However, more studies are needed to demonstrate whether AKT2 can be a target for metabolic reprogramming therapy targeting CAFs. The specific roles of numerous amino acids in CAF metabolic reprogramming have yet to be determined. Further research is needed to paraphrase the metabolic roles of other amino acids in cancer cell growth and proliferation. These findings open up opportunities for metabolic reprogramming therapies targeting CAFs.
Lipids are defined as natural compounds that are soluble in non-polar solvents but insoluble in water (136). The most important components of lipids are fatty acids (FAs), which are not merely essential composition of cell membranes but also serve as important precursors of second messengers involved in transducing intracellular signals and as an important source of energy when the main energy donor is restricted (137–140). Few previous studies have investigated the effects of abnormal lipid metabolism of CAFs on tumor growth and metastasis. Recent studies indicate that CAFs may play a fundamental role in lipid translocation and uptake. It has been documented that CAFs induce FATP1 upregulation in human MDA-MB-231 TNBC cells, leading to increased absorption of exogenous fatty acids by TME (141). CAFs can also transport lipids into cancer cells via exosomes, which have been proven to boost cancer cell growth and proliferation (142). Previous studies have shown that CAFs can undergo reprogramming of lipidome and cumulate more fatty acids and phospholipids, thereby promoting colorectal cancer (CRC) cell migration. CAFs increase CRC cell migration even after protein deprivation, suggesting that non-protein molecular metabolites in CAFs are accountable for CRC cell migration, and these non-protein molecular metabolites may be CAFs metabolic reprogramming. These non-protein metabolites may be lipid metabolites secreted by CAFs after metabolic reprogramming (142–144). Fatty acid synthase (FASN), one of the essential enzymes in fatty acid composite, is dramatically increased in CAFs, thus increasing the production of FAs (82). Recent studies have confirmed that the CRC cell-derived exosome HSPC111 promotes CRC cell migration by reprogramming lipidomic metabolism in CAFs, suggesting that HSPC111 has the tendency to be a potential therapeutic target for the prevention of CRC cell metastasis (144, 145).
In recent years, numerous studies have demonstrated that TME plays an essential role in tumor immunosuppression, targeted therapy and other responses, and has gained widespread attention (146–148). The presence of different immune cell populations in TME, including various innate immune cells (macrophages, dendritic cells, innate lymphocytes and NK cells, etc.) and adaptive immune cells (T cells and B cells, etc.), is highly relevant to the anti-tumor immune status is highly correlated (76, 149, 150). When TME is affiliated with the function and signaling of these immune cells, it is also referred to as the tumor immune microenvironment (TIME) (151), which controls the development, evolution and metastasis of the tumor (149, 152, 153). Therefore, the use of good immune cells is of great importance in the treatment of tumors. Here, we list the functions and metabolic phenotypes of several common immune cells (Table 2).
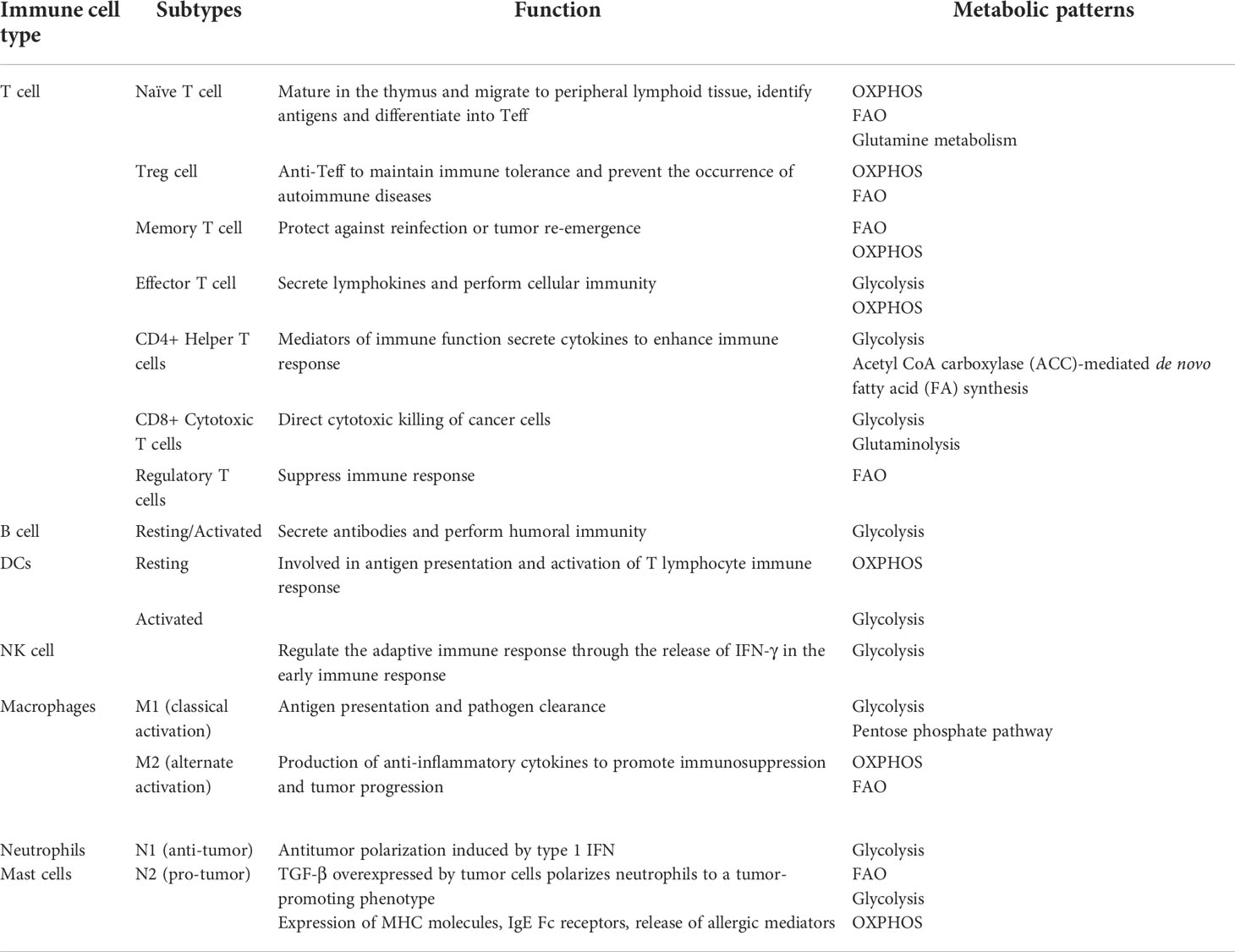
Table 2 Functions and metabolic phenotypes of immune cells.
DCs have important functions that can bridge the innate and adaptive immune responses (154). DCs acquire full antigen-presenting cell (APC) function upon maturation, phagocytose and process non-self-antigens, and take a leading antitumor role in tumor immunity (155). Once mature, DCs up-regulate their antigen-presenting and co-stimulatory molecules, and when they receive distress or activation signals, they are activated and move to secondary lymphoid organs to start activating adaptive responses, such as transforming themselves into potent T-cell activators (156). Glycolysis serves as an essential role in promoting the activation of DCs, while activation of DCs also alters lipid metabolism and affects their function.
In the early stage of tumorigenesis, macrophages from healthy tissues can inhibit the growth of tumor cells after activation. However, at the stage of tumor progression, macrophages recruited from the tumor microenvironment to the tumor area by chemokines and growth factors (157, 158), i.e., tumor-associated macrophages (TAMs), promote the growth, aggression and metastasis of tumor cells (159–161). Therefore, TAMs often represent markers of poor clinical prognosis and immunosuppression of tumors in the body (162, 163).TAMs have both classical (M1) and alternative (M2) subtypes, with M1 cells having immune-enhancing and cancer-suppressive effects and M2 the opposite (164). The above evidence implies that balancing M1 and M2 in TAMs is not negligible for immunotherapy.
NK cells, as spectral killer cells, are the first line of defense of the organism. It does not require antigen pre-sensitization or antibody involvement to kill target cells in a straightforward, sensitive and fast response (165, 166). They have an important role in antitumor because of their ability to kill almost all common cancer cell types and multi-drug resistant tumor cells (167). They also control adaptive immunity by secreting cytokines, such as IFN-γ (168), affecting dendritic cells, macrophages and neutrophils (169). Interacting with a variety of other immune cells in the body, they regulate the body’s immune status and immune function.
Neutrophils regulate the inflammatory response by secreting cytokines and chemokines and are considered to be the first line of defense against different types of microorganisms. However, neutrophils can also cause harm to the body. Neutrophils can be recruited from the circulation into tumor tissue along chemokines generated by tumor cells and immune cells, among others, and are converted into TANs in TME (170). Similar to TAMs, TANs can be classified into N1 (antitumor) or N2 (tumor-promoting) phenotype (N2) and will shift between the two depending on TME characteristics (171, 172).TANs have been reported to have important effects on promoting tumor proliferation, invasion, angiogenesis, and metastasis (173–175), and therefore tan levels can be used as an indicator of poor patient prognosis (176). Neutrophils derive most of their energy from glycolysis, and enhanced glucose consumption has been shown to be critical for enhancing neutrophil survival and function (176, 177).
CD4+ T cells and CD8+ T cells have a significant ability to suppress antitumor immune responses, recognizing and eliminating tumor cells by releasing suppressive cytokines, cytotoxic particles, and other mechanisms. Treg cells are a class of cell populations that control the autoimmune response and can form an immunosuppressive environment by secreting IL-10, TGF-β, and IL-35. These immunosuppressive properties coincide with the promotion of immune escape of tumor cells (178). Several experiments have verified that tumor patients usually have dysfunctional antitumor immune cells with a sharp rise in the amount of Treg cells (179–181). Interestingly, many tumor cells can exploit this suppressive mechanism and express multiple ligands (e.g. PD-L1, PD-L2) that help to absconds from T cells. Some articles indicate that Tregs are also heterogeneous, mainly: naturally occurring thymus-derived CD4+CD25+FOXP3+ Tregs (nTregs), and induced Treg cells (iTregs). There are three further subpopulations of iTregs: iTregs expressing FOXP3, Tr1 secreting CD4+FOXP3-IL-10, and Th3 expressing TGF-β (182, 183). iTregs exhibit completely different metabolic patterns depending on their activation status (184). iTregs, like M2 macrophages, depend mainly on fatty acid oxidation (FAO) of OXPHOS for energy (185).
Immunotherapy has brought a dramatic change in the treatment paradigm of tumors in the last decade, ensuring the reactivation of host defenses and playing an important strategic role in the fight against cancer. However, there are still problems of low efficiency and high adverse effects, the reason for which is incredibly significant is the metabolic reprogramming-mediated immunosuppression of the microenvironment (186, 187). It is well known that alterations in metabolism occur within cancer cells and that this metabolic disturbance not only affects their survival and proliferation signals, but also leads to a hyper acidic, nutrient-deficient and hypoxic TME (188, 189), which further exacerbates the metabolic reprogramming process of tumor cells and tumor ecotone immune cells. Recent studies have shown that immune cells also exhibit different metabolic patterns in various states of activation or stages of differentiation, which can regulate the phenotype, function and survival of immune cells (190–192). In addition, the environmental and metabolic state of the organism can also influence the phenotype and function of immune cells. Metabolic reprogramming mainly includes abnormalities in glucose metabolism, amino acid metabolism and lipid metabolism (193). Therefore, we will explore the impact of metabolic reprogramming of immune cells on carcinoma immunity in the carcinoma below (Figure 3).
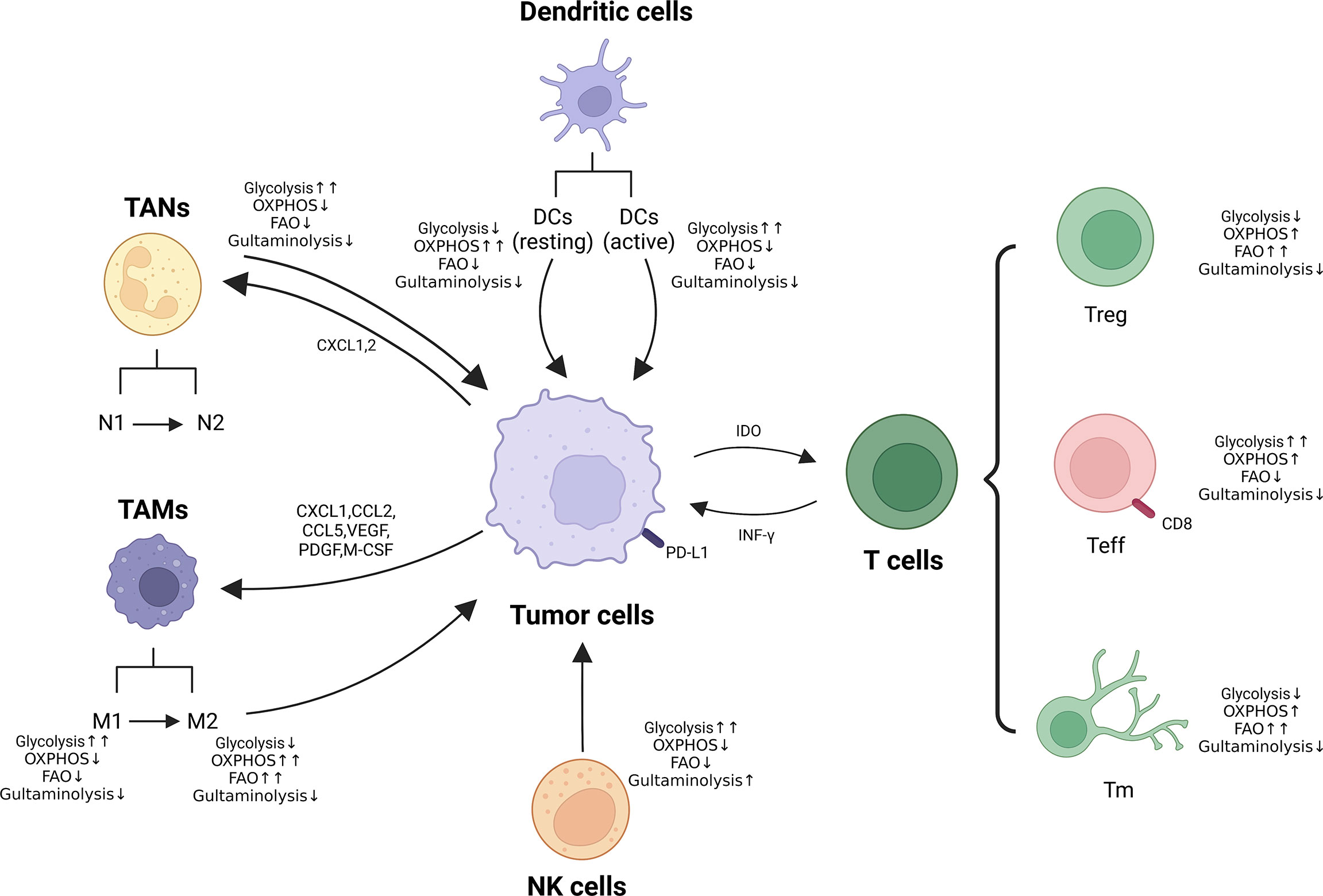
Figure 3 Metabolic reprogramming of immune cells in TME. Metabolic alterations of macrophages, neutrophils, NK cells and T cells (Treg, Teff and Tm) are presented. ↑↑: Significantly up-regulated; ↑: Up-regulated; ↓: Down-regulated.
Glucose metabolism is the principle productive mode of immune cells. Under aerobic conditions, glucose is transformed to pyruvate. It penetrates the tricarboxylic acid cycle (TCA) in the mitochondria, which then couples OXPHOS to produce large amounts of ATP, thereby providing the energy required for body metabolism (194). Under anaerobic or hypoxic conditions, glucose is transformed to pyruvate, which no longer penetrates the TCA, but synthesizes lactate in the cytoplasmic matrix to produce a small amount of ATP (195). The activation of immune cells is analogous to the Warburg effect in tumor cells, which also undergoes a metabolic reprogramming from a mitochondria-dominated oxidative phosphorylation process to aerobic glycolysis, with neutrophils, M1 macrophages, dendritic cells and other activated immune cells rapidly providing ATP and metabolic intermediates (196). The glycolytic intermediate glucose-6-phosphate also ameliorates the attunement of tumor cells to the stern microenvironment through the pentose phosphate pathway (PPP) and significantly affects the antitumor immune response of immune cells (197). Accurate information from the number of groups have shown that interference with OXPHOS, PPP of immune cells can suppress their immune function (198, 199).
It has been found that excessive glucose intake activates some helper T-cell Th17-related cytokines: TGF-β and RORγt, promoting exorbitant differentiation and stimulation of Th17 cells and triggering inflammatory responses in vivo (200). When lacking of glucose in the TME, the function of most immune cells becomes faulty. Inhibition of glycolytic capacity at depressed glucose levels activates the intracellular “energy receptor” AMPK kinase, which inhibits mTORC1 and HIF-1α, thus down-regulating the differentiation and function of DC, Teff and NK cells and promoting Treg differentiation and anti-inflammatory M2 macrophage formation (201–203). These analysis consequences intimate that glycolytic upregulation not only provides an intrinsic growth convenience for tumor cells, but also has an outward role in suppressing tumor immune surveillance.
From another perspective, the enhanced glycolysis of tumor cells is accompanied by the production of large amounts of metabolites such as lactic acid and CO2, which accumulate in the TME to form an acidic environment and further exert metabolic pressure on the infiltrating immune cells. The acidic environment: 1) impedes the extracellular transport of lactic acid in cytotoxic T lymphocytes (CTL) and NK cells, leading to intracellular acidification, which directly affects the proliferation and cytokine secretion of CTL and NK cells, leading to their lethality impaired (204, 205); 2) promotes the differentiation of initial T cells, MDSCs and TAMs toward the pro-tumor phenotype for differentiation and proliferation (205), which ultimately results in immune escape of tumors through multiple mechanisms. Recent clinical findings have shown that glycolytic activity in tumor cells is inversely correlated with host antitumor immune response and treatment outcome of immunotherapy (193). Patients with tumors that are difficult to control with peripatetic T-cell therapy have higher levels of aerobic glycolytic activity and have lower tissue TIL numbers and cytotoxic functions (206). Higher levels of lactate and concomitant acidified TME in tumors suppress immune cell function, abolish immune surveillance of the tumor, and ultimately result in immune escape.
Glutamine (Gln) is the second most important intracellular nutrient after glucose, producing glutamate and ammonia under the action of glutaminase (GLS), which is subsequently converted to α-ketoglutarate catalyzed by GLS or transaminase and enters the mitochondria, where it undergoes OXPHOS through the TCA cycle and electron transport chain, producing Most of the ATP, and participate in nucleotide, amino acid and fatty acid synthesis (124). At the same time, Gln can be converted to glutathione, which is applied to maintain intracellular reactive oxygen species homeostasis and prevent its damage to biomolecules. Therefore, amino acid metabolism acts as an essential role in maintaining the growth and proliferation of tumors. Many tumor cells undergo reprogramming of amino acid metabolism, resulting in a deficiency of the corresponding amino acids in TME, resulting in impaired immune effector cell function. Targeting at tumor cell amino acid metabolism is one of the effective strategies to restore the immune response.
Activated T cells and macrophages also present enhanced Gln metabolism to maintain cell proliferation and immune response (207). Tumor cells have the capacity to prevent T cells from proliferation, activation and secretion of related cytokines through competitive consumption of glutamate, resulting in the formation of an immunosuppressive microenvironment. For example, in Gln-deficient microenvironment, renal cancer cells induce programmed death ligand-1, PD-L1 expression through activation of EGFR/ERK/c-Jun pathway, which inhibits IFN-γsecretion by T cells and allows tumor cells to evade immune killing (208). However, restriction of Gln during T cell activation leads to differentiation to CD8+ memory T cells (209), moreover, inhibition of GLS facilitated the differentiation and effector functions of Th1 and CTL but undermined the differentiation of Th17 cells (210). The mechanisms indicate the regulation of Gln metabolism in various cells in TME and how Gln affects T cell responses need to be further elucidated.
Lipid metabolism is a fundamental mode of energy supply in addition to glycolysis. It was found that increased fatty acid content in TME facilitates Treg production and that Treg relies on exogenous fatty acid uptake for immunosuppressive functions (211). Similarly, lipid accumulation in myeloid cells infiltrating TME induces a conversion to an immunosuppressive and anti-inflammatory phenotype through metabolic reprogramming, which may be partly derived from neighboring cancer cells with enhanced fatty acid synthesis. From another perspective, CD8+ T cells with higher levels of lipids in tumor patients up-regulated the expression of programmed death-1 (PD-1) receptor, which should generally show inhibitory effects; however, the combination of PD-1 inhibitors showed efficient antigen recognition and better antitumor results.
Fatty acid oxidation (FAO), is not the primary metabolic pathway (212). Nevertheless, the role of FAO in adjusting immune cell behavior cannot be overlooked; regulatory T cells (Treg cells), M2 macrophages, and memory T cells depend mainly on FAO-derived OXPHOS to produce energy. M2-like macrophages depend on FAO to meet the bioenergetic requirements to preserve their antitumor effects; inhibition of FAO promotes M1 polarization (213, 214). In addition, FAO serves as a significant section in the origination and preservation of memory T cells (Tm, memory T cells) (215). Investigators have verified that FAO is the metabolic energy foundation for the timely response of Tm to antigenic stimuli and facilitates the maintenance of normal Tm mitochondrial function and long-term cell survival. Importantly, FAO is also essential in upholding the equilibrium between Teff and Treg (216). FAO suppresses Teff cell activation, and up-regulates the expression of repressive PD1 receptor and CPT1A, which in turn attenuates IFN-γ secretion (217). Conversely, FAO genes such as CPT1A expression are up-regulated in Treg cells and FAO levels are elevated, providing energy to promote Treg cell production (216). FAO acts as a fundamental role in the regulation of innate and adaptive immune responses, which are largely determined by the diverse principal requirements of various immune cells. Consequently, figuring out the metabolism of distinct immune cells is crucial for a broad understanding of the mechanism of immune regulation of FAO.
It has also been indicated that inhibition of the cholesterol synthesis pathway in macrophages provokes the production of type I interferon responses, which initiate antiviral immune responses (218). ACAT1 is the major enzyme that catalyzes the synthesis of cholesteryl esters in CD8+ T cells, and pharmacological or genetic inhibition of ACAT1 increases intracellular cholesterol levels in melanoma tumor-infiltrating T lymphocytes (TIL), thereby inducing a superior immune response (219). The application of ACAT1, avasimibe (a small molecule inhibitor), in combination with a PD-1 inhibitor showed synergistic effects, significantly inhibiting the growth of tumor-bearing mice (219). However, recent studies have shown that high cholesterol levels in tumors can lead to T-cell dysfunction by activating endoplasmic reticulum stress (220). Therefore, despite the vital role of cholesterol for effector T-cell proliferation and metabolism, and although cholesterol acts as a crucial part in the proliferation and metabolism of effector T cells, targeting specific cholesterol metabolism requires further research.
To date, accumulating evidence suggests that CAFs are critical for regulating the antitumor activity of immune cells in the TME, including innate and adaptive immune cells (221, 222). By secreting cytokines, chemokines, and other effector molecules, including TGF-β, CCL2, CXCL2, laminin, and MMP, CAFs can enhance the involvement of immune cells in tumorigenesis and progression (20, 223). In turn, some influences of several immune cells on CAF also attracted our attention (224, 225). In conclusion, just as abounding research has revealed that the interaction between CAFs and immune cells can modulate TIME, thereby suppressing antitumor immune responses (226, 227) (Figure 4).
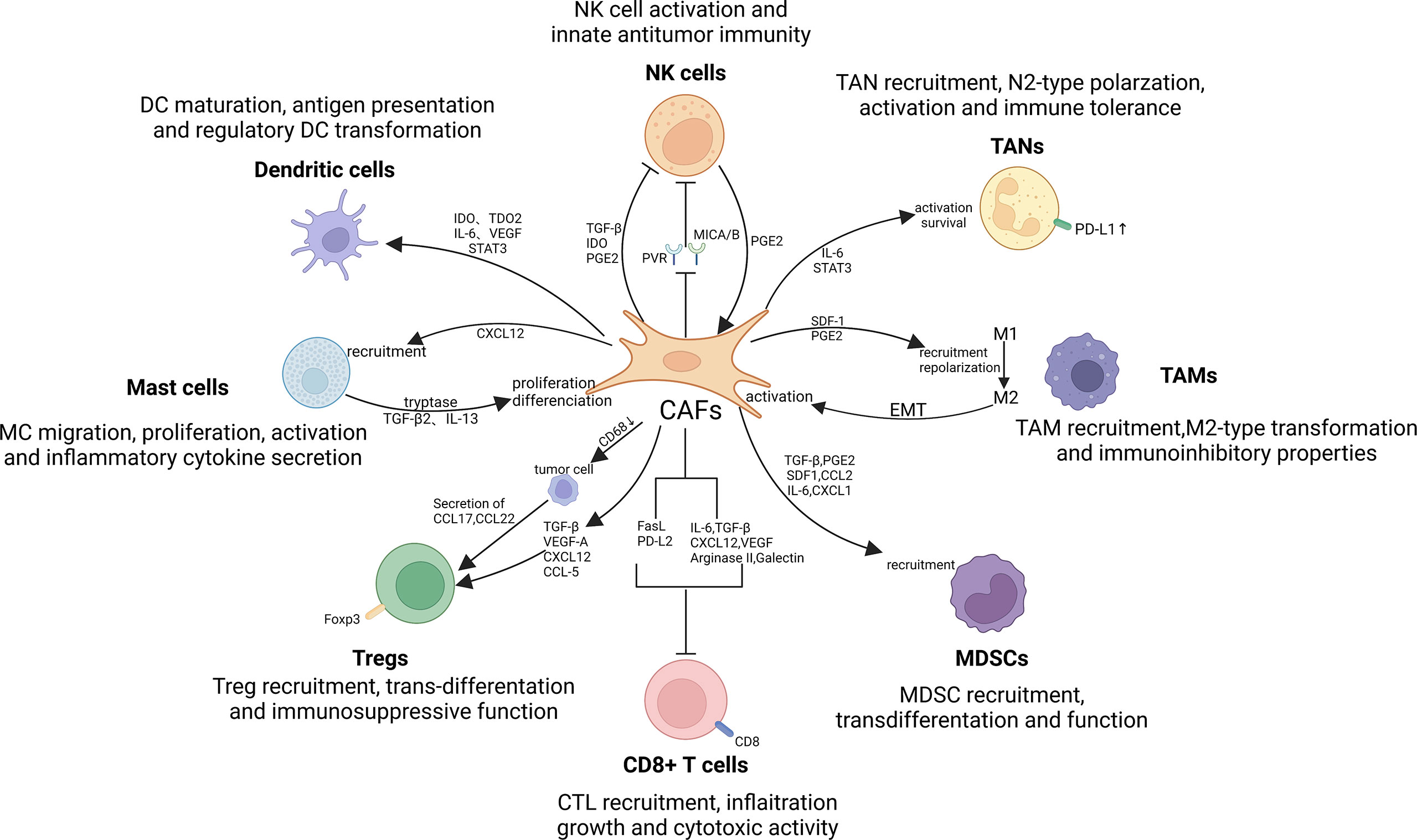
Figure 4 Crosstalk between CAFs and immune cells in the TIME. There exist significant interactions between CAFs and immune cells, such as tumor-associated macrophages (TAMs), tumor-associated neutrophils (TANs), mast cells (MCs), dendritic cells (DCs), myeloid-derived suppressor cells (MDSCs), natural killer (NK) cells and T lymphocytes. By secreting cytokines, chemokines, and other effector molecules, including TGF-β, CCL2, CXCL2, laminin, and MMP, CAFs can promote the involvement of immune cells in tumorigenesis and progression. Notably, TAMs, NK cells and MCs can in turn exert promoting effect on CAFs activation and function, thereby contributing to the formation of immune suppressive loops.
The so-called TAMs have been divided into two manifest subsets called M1 and M2 (164). Type-1 TAM exert antitumor effects principally through the mediation of antibody-dependent cytotoxicity and secretion of ROS and TNF (228). However, in sharp contrast to the former, M2 macrophages mainly exhibit tumor-promoting activity. Studies have shown that this diametrically opposite effect is closely related to the promotion of tumor angiogenesis, immunosuppression, cancer cell aggression and spreading, and ECM reconstruction (164).
CAFs are mediated by a variety of regulatory molecules that encourage the TAM precursors and monocytes to recruit and simultaneously differentiation into type-M2 TAMs. Thus, CAFs can attenuate effector T cell responses and induce immunosuppression in the TME (229). Both Ksiazkiewicz M and Cohen N found that, in breast cancer, CAF is capable of promoting monocyte chemoattractant protein-1(MCP-1), SDF-1 and chitinase 3-like 1(Chi3L1) secretion. As a result, polarization of monocyte can be migrated and enhanced into M2 phenotype (230, 231). Several cytokines secreted by CAFs, including IL-8, IL-10, TGF-β, and CCL2, just as what have been revealed that, could enhance the monocyte recruitment and type-M2 TAM transformation (232–234).
As for the fact that CAFs can stimulate the immunosuppressive properties of TAMs, many studies have found that CAF-induced up-regulate the expression of PD-1 of type-M2 TAMs. Evidence suggests that high level expression of PD-1 in TAMs can reduce the phagocytosis to cancer cells and inhibit the infiltration and proliferation of T lymphocytes, implying that CAFs can induce restraint of not only innate but also adaptive antitumor immune responses to TAMs.
Likewise, TAMs with the M2 phenotype also have a counteracting effect on CAF, which can modulate CAF activation and progression (225, 235). Comito et al. (236) found that M2 macrophages can secrete IL-6 and SDF-1, and these factors enhance the advance of epithelial-mesenchymal transition and induce CAF activation. Activated CAFs can also further enhance TAM activity. Thus, a closed-loop pathway with both cancer-promoting and immunosuppressive effects quietly play its indispensable role in the TME. In addition, there is relevant evidence that TAM affects the differentiation and activation of MSCs, known as the precursors of CAF (237). TAMs can promote the transdifferentiation of MSCs into CAFs in terms of properties and functions and at the same time acquire a pro-inflammatory phenotype to reshape the inflammatory microenvironment (238).
Similar to the classification of TAM, TAN can also be divided into antitumor phenotypes (N1) and tumor-promoting phenotypes (N2) (239–241). The main difference between N1 and N2 or the basis of classification lies in whether they can be activated by TGF-β and the different activation degrees (242). There is evidence that CXC chemokine receptor 2 (CXCR2) expressed by CAFs is a major force in mediating neutrophil recruitment to tumors. In short, this means that CAFs may promote the migration of TANs by relying on CXCR2 as a medium (243, 244). Cheng Y et al. found that the link between CAF and TAN also lies in the fact that the STAT3 signaling pathway in TAN can be activated by CAF-derived IL-6, which directly leads to PD-1/PD-L1 activation. When the expression is up-regulated, the activation of T lymphocytes can be inhibited and immune tolerance will soon be established (245). Unexpectedly, peripheral neutrophils are also regulated by CAF-secreted SDF-1α to migrate into tumor cells (245).
It should also be noticed that CAF-secreted cardiomyokine-like cytokine 1 (CLCF1) is crucial to promote the development of tumors. In advances in hepatocellular carcinoma research, polarization of TAN might be modulated by CAFs. The expression of CXCL-6 and TGF-β can be up-regulated by CLCF1, thereby inducing polarization of N2-type TAN, which contributes to tumor progression (246).
Taking prostate cancer as an example, CAFs can enhance MC proliferation and migration after estrogen overexpression and at the same time promote the secretion of inflammatory cytokines, which is a manifestation of tumor-promoting effect (247). Under the induction and catalysis of estrogen, the binding of CXCL12 to CXCR4 promotes the recruitment of MCs (247). In addition, Ma et al. (248) found their study stimulatory effects of MCs on CAFs. MC-secreted IL-13 and tryptase induce CAF proliferation in a manner distinct from TGF-β2-STAT6. Increased CAFs lead to the formation of fibrotic TMEs, with the direct consequence of suppressing antitumor immune and therapeutic responses (248). In addition, it has been reported that MCs in neurofibromas can enhance the proliferation and secretion of CAFs and promote CAF activity through the TGF-β pathway (249).
Emerging studies have indicated that CAFs induce direct or indirect restriction to NK cells via a variety of procedures, consisting of activating NK receptor, cytotoxic activity and cytokine secretion (31, 250).
Via PGE-2 and indoleamine 2,3-dioxygenase (IDO), NK cells are induced by CAF to switch to a quiescent phenotype and display a status characterized by unresponsiveness or even paralysis during antitumor immunity in hepatocellular carcinoma (251). NK cells are able to encourage the establishment of the immunosuppressive loop prompted by CAF through inducing PGE-2 discharge (252). By regulating the expression of NK cells’ energizing receptor-associated ligands, CAF could even restrain the activity and function of them. Whether the down-regulation of the MICA/B expression caused by CAF in melanoma on tumor cells (253), or CAF-mediated reduction of cell surface poliovirus receptor PVR expression, the most direct consequence is the inhibition of NK cell killing activity (254).
In the current discussion of the mechanism, TGF-β is widely recognized as the key linking CAFs to NK cells in TIME (255). Numerous investigations have demonstrated that TGF-β substantially restricts NK cell activation and cytotoxicity (256). Whether there are other related factors influence needs further research to explore and demonstrate.
Several studies have shown that CAFs of hepatocellular carcinoma are able to recruit natural DCs and stimulate their transdifferentiation into regulatory DCs (rDCs) through the activation of IL-6-STAT3 pathway. IDOs, for example, are incapacitated DCs that express low levels and present minoro antigen, but have the ability to secrete immunosuppressive effector cytokines (257). IDO is also critical to the proliferation of Treg cells, thereby suppressing T cell-mediated immune effects (258). In lung cancer, CAF releases IDO1 and tryptophan 2,3-dioxygenase (TDO2) under the induction of galectin-1. IDO1 and TDO2 are then degraded by tryptophan, which impairs the differentiation and normal immune function of DCs (259, 260). In addition, VEGF emerged by CAF participates in the anomalous differentiation of DCs and leads to damaged antigen presentation (261, 262).
Studies have shown that the level of Foxp3 expression on Treg cells is essential to restrain antitumor immune function (263). Several studies have found that CAF prompts Treg cells migration and significantly enhances the accumulation in the tumor cells (264). In regard to breast cancer, both the Karnoub AE team and the Tan W team found that the chemokine CCL5 determines the process of recruiting CD4+CD25+ Treg cells to CAFs (43, 265). Furthermore, down-regulation of CD68 expression in CAFs promoted tumor cell secretion of CCL17 and CCL22, two chemokines that directly increased Treg cell infiltration (266). It needs to be added that the growth factor released by CAFs, VEGF-A, is likely to implicate in the activation and preservation of Treg cells mediately or immediately (267, 268). Besides encouraging the migration and infiltration of Treg cells, the effects of CAFs on Treg cells also lie in inducing convention and exerting immunosuppressive effects as well. For example, CAF promotes the differentiation of naive T cells to relatively mature CD4 + CD25 + Treg cells through the induction of Foxp3 gene expression in T lymphocytes by secreting TGF-β (269).
Cytotoxic T lymphocytes (CTL) are considered to be the most critical components of antitumor immunity, also known as CD8 + T, and their well-known cytotoxic activity is mainly achieved by inducing tumor cell apoptosis (270, 271). Numerous studies have reported the crosstalk between CAFs and CTLs, especially the inhibitory effects of CAFs on CD8+ T cell infiltration, growth, and antitumor immunity (272). CAF activates pancreatic stellate cells (PSCs) through the secretion of cytokines such as CXCL12, which drives CD8+ T cells away from tumors, thereby reducing their infiltrating numbers in pancreatic tumors (273). When there is hypoxia in the microenvironment, CAFs can be stimulated to release angiogenic factors such as VEGF, resulting in a decrease in the expression of cell adhesion molecules on endothelial cells, making it difficult for circumferential CD8 + T cells to reach the tumor through the vasculature. Site exerts effects (274, 275). Hypoxic state or other physical barriers in the TME itself have also been shown to be closely related to the mediation of CAF (276). IL-6 and TGF-β secreted by CAFs can inhibit the recruitment of CD8+ T cells and counteract the cytotoxic activity of CTLs on tumor cells (277, 278). Arginase as well as galectin is important forces for CAF to restrict CD8 + T cells proliferation and destroy their immune function (279–281). Recent studies have pointed out that CAF promotes the reduction of the amount of CD8 + T cells and the improvement of tumor cell survival capability by inducing the expression of immune checkpoints in an indirect manner, thereby achieving the effect of weakening the antitumor response of effector T cells (229, 245, 282). Possible immune checkpoint molecules include factor associated suicide (FAS)/factor associated suicide ligand (FASL) and PD-1/programmed death-ligand 2 (PD-L2).
In conclusion, the effect of CAFs on T lymphocytes is relatively straightforward. It can promote the transition of naive T cells to a cancer-promoting phenotype, strengthen the function of immunosuppressive T lymphocytes, and inhibit the activity of effector T lymphocytes, thereby achieving immunosuppression. However, research on the effect of T lymphocytes on CAF is still uncharted territory, which may be a new direction for future research.
MDSCs, originating from the bone marrow, have been well known for their powerful immunosuppression of the TIME (283–285).
The infiltration and manufacturing of MDSC can be promoted by CAF through the discharge of diverse cytokines and chemokines. Thereby, antitumor activity of effector T lymphocytes is inhibited. Among them, CCL2 is a typical example, which can recruit MDSCs to migrate to tumor sites (282, 286, 287). The net result of increased M-MDSC aggregation is that the maturation of CD8+ T cell and the secretion of IFN-γ are restricted (288). IL-6 secreted by CAF instigates the differentiation of recruited monocytes into M-MDSC through STAT 3-dependent pathway, thereby inhibiting T cell proliferation and immune function (289). After interfering CAF with tranilast, Ohshio Y’s team found that the expressions of SDF-1, PGE2 and TGF-β1 secreted from CAF were significantly down-regulated, and the differentiation of primitive MDSCs showed a low-level state (290). In this way, SDF-1, PGE2 and TGF-β1 are highly likely to be involved in the differentiation of MDSC (290). Furthermore, CXCL1, secreted by CAFs, is also associated with the recruitment of PMN-MDSCs (291).
CAFs play a significant role in the profound changes in ECM structure in tumors, and changes in ECM rigidity (or “compliance”) also have extraordinary impacts on tumor development and may regulate tumor infiltration and metastasis (119, 292–296). CAFs lead to ECM remodeling probably due to the progression of fibrosis, characterized by the degradation of type IV collagen accompanied by the deposition of type I and type III collagen (116, 118). One possible mechanism by which increased ECM rigidity promotes tumor infiltration and metastasis is the enhancement of growth factor-mediated cell migration (292, 293). This CAFs-modified ECM remodeling acts as a barrier, preventing immune cells from contacting tumor cells, thereby suppressing immune cell-mediated immune responses (297, 298). Therefore, CAFs-mediated ECM remodeling has been used as a predictor of clinical outcomes in patients in several clinical models. The progressive isolation of T cells from tumor cells during ECM remodeling has been demonstrated to be the primary immunosuppressive mechanism in various types of cancer (297, 299). In some types of tumors, higher T cell motility was observed in tumor areas where fibrosis had not progressed or was not fully fibrotic, whereas T-cell migration was poorer in areas of more intact fibrosis (300, 301). In addition, the order of the fibrous tissue surrounding the tumor epithelium determines the migration trajectory and migration speed of T cells to some extent, thus limiting their contact with tumor cells to exert immune effects (302). Therefore, we can infer that CAFs can restrict the movement of CD4+ and CD8+ T cells. In addition to T cells, CAFs-mediated ECM remodeling also affects other immune cell populations, such as regulation of macrophage polarization, and effects on MDSCs and DCs, but the specific mechanisms remain elusive (20). Follow-up studies should focus on the specific mechanisms of CAFs-mediated ECM remodeling in various immune cells to investigate the therapeutic targets for enhancing immune response in TME.
Apart from its barrier function that prevents immune cells from contacting with tumor cells, the dense ECM structure shaped by CAFs can also lead to changes in the efficient utilization of oxygen, thereby inducing a hypoxic state that affects nutrient uptake and induces changes in cellular metabolism, thereby influencing the TIME (303). Hypoxia is a recognized tumor immunomodulator. There is also evidence that hypoxia induces ECM deposition in hypoxic tumor regions, suggesting a positive feedback loop between hypoxia and ECM deposition (304, 305). CAFs may be involved in aberrant angiogenesis leading to a limited number of functional vessels, thus creating hypoxic zones and promoting immunosuppressive networks within the TME (303). Possible mechanisms by which CAFs regulate aberrant angiogenesis contain recruitment of tumor endothelial progenitor cells by secreting pro-angiogenic factors and by releasing SDF-1 in TME (306). Hypoxia can inhibit the infiltration of T cells in TIME, preventing them from making contact with tumor cells to mount an immune response, while also impairing the function of T cells (307). One mechanism of hypoxia-mediated T-cell suppression is the sustained activation of HIF-1α that negatively regulates T-cell receptor signaling, in part due to increased NF-κB activation (308). Multiple studies have demonstrated that hypoxia induced by CAFs interferes with T cell effector functions and allows tumor cells to escape from immune surveillance (309–312). Hypoxia also causes immunosuppression and contributes to immune tolerance via other immune cell populations such as TAMs and MDSCs (307). Possible mechanisms involved include selective upregulation of PD-L1 on MDSCs by hypoxia through HIF-1α binding to the HRE in the proximal promoter of PD-L1 (312).
In conclusion, CAFs play multiple roles in immune cell-mediated immune responses, but the specific mechanisms remain to be further investigated, which may provide new ideas for future drug research targeting CAFs.
Considering the fact that CAFs exert their suppressive influences on tumor immunity utilizing multiple mechanisms and immune cell crosstalk, targeted therapies that target these cells are very promising. The quantity of preclinical trials to enhance or restore anti-cancer immune responses by targeting CAF therapy has augmented dramatically during recent decades. At present, CAFs-based immunotherapy mainly contains the following strategies: direct action on CAF targets to deplete CAFs, inhibition of the activation of CAFs and their functions. A brief summary of immunotherapy strategies for CAF in clinical and preclinical studies is given in the Table 3, and we will discuss part of them in the following section.
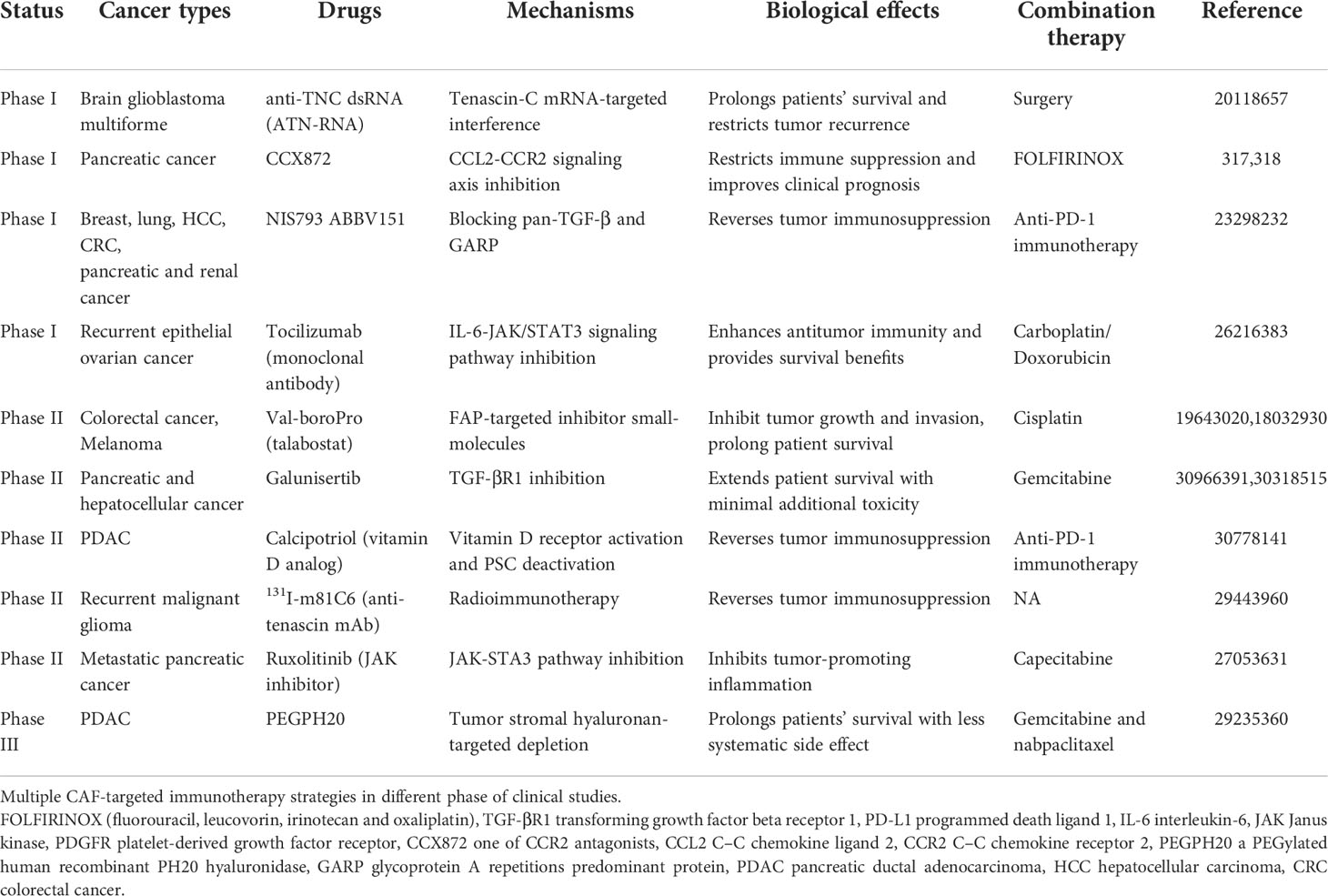
Table 3 Multiple clinical studies of CAF-targeted immunotherapy and related drugs.
Current targeted CAF therapy focuses on the CAF-specific protein FAP (21, 29, 313). Recent studies have shown that ablation by FAP-expressing cells in a transgenic mouse model can lead to quick hypoxic necrosis and immunogenic tumor stromal cells of Lewis lung cancer, a process associated with the involvement of TNF-α and IFN-γ (314, 315). Previous studies have demonstrated that reducing or inhibiting FAP expression can reduce TME mesenchymal pro-connective tissue proliferation while enhancing the metabolic effects and cytotoxicity of CD8+ T cell-reliable killing of cancer cells. Several pioneer studies found that co-targeting CAF and cancer cells to increase T cell immunotherapy in a mouse-based model of lung cancer and malignant pleural mesothelioma patients with metastatic models showed unimpressive effect (316–319). Emerging research suggests that FAP is a crucial target for chimeric antigen receptor (CAR) therapy as well. CAR-T-cell therapy harnesses the host immune system to struggle against cancer. CAR-T-cell therapy counts on artificial receptors where immune cells are modified to express cancer-specific markers and injected into patients, in which they will specifically recognize and wipe out tumor cells (320, 321). Notably, however, FAP is expressed to varying degrees in cells in other tissues, such as pluripotent bone marrow stem cells, which can lead to serious side effects of CAR-T cell therapies. One study showed that relay transport of FAP-reactive T cells into mice suffering from various subcutaneous tumors exerted only finite antitumor consequences, but induced severe lethal osteotoxicity and cachexia in different strains of the mice (322, 323). Therefore, these fatal osteotoxicity and cachexia observed after immunotherapy with CAR-T cells targeting FAP emphasize its severe side effects as a universal target and should be used with caution in specific situations. However, as mentioned above (324), CAFs lack specific markers and therefore only a small number of treatments targeting CAFs have been applied in the clinic. In order to find out more concrete molecular targets for CAFs, we call for the need for more in-depth studies on the specific typing and heterogeneity of CAFs.
Given the difficulty of removing CAFs directly, converting CAFs in the activated state to the resting state is also a promising approach. In patients with pancreatic ductal adenocarcinoma (PDAC) and colon cancer, vitamin A deficiency led to PSC activation in PDAC patients (325–327), while a previous study showed that the vitamin D receptor (VDR) was a suppressor of PSC activation (325). And the activation of PSC led to the activation of CAFs. It has been demonstrated that supplementation with vitamin A or stimulation of VDR by drugs can inactivate PSCs (328). In PDAC patients, administration of the pleiotropic agent all-trans retinoic acid (ATRA) proved to inhibit the activation of PSCs, with the possible mechanism being the restoration of retinol levels in PSCs (327). In animal experiments, reversal of the phenotype of PSCs increased the infiltration of CD8+ T cells, thus enhancing the immune response (273). Thus, metabolic reprogramming of CAFs by vitamin A and VDR could effectively inhibit the activation of CAFs from exerting their corresponding effects in tumorigenesis and progression. Therefore, in certain types of tumor treatment, inactivation of PSC rather than direct elimination of CAFs may be a better therapeutic modality.
Given the fact that crosstalk between CAFs and other immune cells serves as a crucial role in the induction of immunosuppression in TME, it seems more feasible to inhibit the activation of CAFs and limit the crosstalk between CAFs and immune cells by targeting key effector molecules associated with CAFs, for instance, growth factors, signaling pathways and cytokines (324). Immunotherapy is sometimes ineffective in some specific cancers, such as pancreatic ductal carcinoma, so researchers have tried combining CAF-derived cytokines or chemokines with immunotherapy in an attempt to enhance the efficacy of immunotherapy (20). The researchers found that after administration of AMD3100 for inhibition of chemokine (C-X-C motif) ligand 12 (CXCL12) receptor 4, T cells were rapidly recruited in cancer cells and acted synergistically with α-PD-L1 to dramatically reduce cancer cell proliferation and metastasis. This combination of immunotherapy with CAF-derived cytokines or chemokines somewhat avoids the immune evasion caused by the crosstalk between CAFs and immune cells (329, 330). Previous studies have demonstrated that TGF-β acts as a critical role in the activation of CAFs and the crosstalk between CAFs and immune cells, suggesting that inhibition of TGF-β may be able to restore the impaired immune response to TME (331, 332). The clinical importance of immunotherapy is underscored by the fact that clinical and preclinical studies of multiple TGF-β-associated immunotherapies are currently underway. Previous studies have found that in mice using specific models, combined treatment with blockade of TGF-β and anti-PD-L1 antibodies restrained signaling of TGF-β in stromal cells, promoting T cell recruitment to tumor centers and subsequently provoking a robust antitumor immune response (224, 255). Similarly, in several phase I clinical trials, it was noticed that high-dose tocilizumab assists in stimulating the activation of CD8+ T cell and raising the expression of antitumor-connected effectors (e.g. IFN-γ and TNF-α), thus strengthening antitumor immunity (333). Different patients require specific treatment strategies, so gene identification is critical. For patients with lung squamous cell carcinoma (LUSC), E2 factor-related gene signatures can help screen out high-risk patients so that they can be assigned personalized treatment strategies (334).
Some other possible therapeutic strategies include limiting CAF- incurred ECM reshaping in TME. A typical example is retinoic acid (RA), a small molecule derivative of vitamin A, which affects the immunosuppressive properties of CAFs by inhibiting their IL-6 and ECM production (335). Despite the remarkable progress made so far, more research is required to discover other possible therapeutics targeting CAFs and their crosstalk network with immune cells, which may lead to new ideas for future antitumor immunotherapy and be a boon for cancer patients.
Tumorigenesis and progression require metabolic reprogramming of TME. Furthermore, solid tumors are usually considered as metabolically heterogeneous diseases, in which metabolic reprogramming occurs in CAFs and immune cells, together with crosstalk between them ensure continuous cancer cell growth and proliferation. In this biological context, CAFs and immune cells may stand for the major cell types regulating endosmosis and interactions in cancer tissues. Currently, the prevalent metabolic circuit plasticity is considered to be the most vital limiting factor for successful metabolic inhibition, rendering single-targeted metabolic pathways ineffective. Multiple metabolic inhibition is a promising strategy to overcome this problem, and although reliable preclinical data are supporting (336), clinical efficacy assessment of multiple metabolic inhibitor strategies is still in its infancy. CAFs affect immune cell activity in numerous ways, and metabolic reprogramming in TME leads to immune effector cell dysfunction. These interactions further reinforce the immunosuppressive effects in TME, leading to rampant proliferation and metastasis of tumor cells. Future research directions may need to explore multiple metabolic inhibition in CAFs and immune cells as well as strategies related to immunotherapy. In particular, the maturing of strategies dedicated to inhibiting metabolic reprogramming of CAFs and immune cells and reducing metabolic crosstalk between them could help to eliminate proliferation in the cancer network and enhance immune cell-mediated immune responses.
YZ: Literature retrieval, writing the draft manuscript, and literature statistics and tabulation. LW: Writing, revising the manuscript, and designing the figures. XL: Writing, revising the manuscript, and designing the figures. XH: Designing the figures. JY: Concept of the study. All authors contributed to the article and approved the submitted version.
This work was supported by Social Development Science and Technology Special Project of Kunshan (KS1935).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.

1. Faubert B, Solmonson A, DeBerardinis RJ. Metabolic reprogramming and cancer progression. Sci (New York NY) (2020) 368(6487):eaaw5473. doi: 10.1126/science.aaw5473
2. Vander Heiden MG, Cantley LC, Thompson CB. Understanding the warburg effect: the metabolic requirements of cell proliferation. Sci (New York NY) (2009) 324:1029–33. doi: 10.1126/science.1160809
3. Samudio I, Fiegl M, Andreeff M. Mitochondrial uncoupling and the warburg effect: molecular basis for the reprogramming of cancer cell metabolism. Cancer Res (2009) 69:2163–66. doi: 10.1158/0008-5472.CAN-08-3722
4. Dang CV, Le A, Gao P. MYC-induced cancer cell energy metabolism and therapeutic opportunities. Clin Cancer Res (2009) 15:6479–83. doi: 10.1158/1078-0432.CCR-09-0889
5. Gogvadze V, Zhivotovsky B, Orrenius S. The warburg effect and mitochondrial stability in cancer cells. Mol Aspects Med (2010) 31:60–74. doi: 10.1016/j.mam.2009.12.004
6. Hensley CT, Wasti AT, DeBerardinis RJ. Glutamine and cancer: cell biology, physiology, and clinical opportunities. J Clin Invest (2013) 123:3678–84. doi: 10.1172/JCI69600
7. Lunt SY, Vander Heiden MG. Aerobic glycolysis: meeting the metabolic requirements of cell proliferation. Annu Rev Cell Dev Bi (2011) 27:441–64. doi: 10.1146/annurev-cellbio-092910-154237
8. Semenza GL. HIF-1 mediates metabolic responses to intratumoral hypoxia and oncogenic mutations. J Clin Invest (2013) 123:3664–71. doi: 10.1172/JCI67230
9. Tennant DA, Durán RV, Gottlieb E. Targeting metabolic transformation for cancer therapy. Nat Rev Cancer (2010) 10:267–77. doi: 10.1038/nrc2817
10. White E. Exploiting the bad eating habits of ras-driven cancers. Gene Dev (2013) 27:2065–71. doi: 10.1101/gad.228122.113
11. Kimmelman AC. Metabolic dependencies in RAS-driven cancers. Clin Cancer Res (2015) 21(8):1828–34. doi: 10.1158/1078-0432.CCR-14-2425
12. Paget S. Data from: The distribution of secondary growths in cancer of the breast. 1889. Cancer Metastasis Rev (1989) 8(2):98–101. doi: 10.1016/S0140-6736(00)49915-0
13. Ribeiro Franco PI, Rodrigues AP, de Menezes LB, Pacheco Miguel M. Tumor microenvironment components: Allies of cancer progression. Pathol Res Pract (2020) 216:152729. doi: 10.1016/j.prp.2019.152729
14. Hanahan D, Coussens LM. Accessories to the crime: functions of cells recruited to the tumor microenvironment. Cancer Cell (2012) 21:309–22. doi: 10.1016/j.ccr.2012.02.022
15. Yuan Y, Jiang Y, Sun C, Chen Q. Role of the tumor microenvironment in tumor progression and the clinical applications (Review). Oncol Rep (2016) 35:2499–515. doi: 10.3892/or.2016.4660
16. Mezawa Y, Orimo A. The roles of tumor- and metastasis-promoting carcinoma-associated fibroblasts in human carcinomas. Cell Tissue Res (2016) 365:675–89. doi: 10.1007/s00441-016-2471-1
17. Biffi G, Tuveson DA. Diversity and biology of cancer-associated fibroblasts. Physiol Rev (2021) 101:147–76. doi: 10.1152/physrev.00048.2019
18. Sahai E, Astsaturov I, Cukierman E, DeNardo DG, Egeblad M, Evans RM, et al. A framework for advancing our understanding of cancer-associated fibroblasts. Nat Rev Cancer (2020) 20:174–86. doi: 10.1038/s41568-019-0238-1
19. Kalluri R. The biology and function of fibroblasts in cancer. Nat Rev Cancer (2016) 16:582–98. doi: 10.1038/nrc.2016.73
20. Ziani L, Chouaib S, Thiery J. Alteration of the antitumor immune response by cancer-associated fibroblasts. Front Immunol (2018) 9:414. doi: 10.3389/fimmu.2018.00414
21. Lambrechts D, Wauters E, Boeckx B, Aibar S, Nittner D, Burton O, et al. Phenotype molding of stromal cells in the lung tumor microenvironment. Nat Med (2018) 24:1277–89. doi: 10.1038/s41591-018-0096-5
22. Gascard P, Tlsty TD. Carcinoma-associated fibroblasts: orchestrating the composition of malignancy. Gene Dev (2016) 30:1002–19. doi: 10.1101/gad.279737.116
23. Yoshida GJ, Azuma A, Miura Y, Orimo A. Activated fibroblast program orchestrates tumor initiation and progression; molecular mechanisms and the associated therapeutic strategies. Int J Mol Sci (2019) 20(9):2256. doi: 10.3390/ijms20092256
24. Polanska UM, Orimo A. Carcinoma-associated fibroblasts: non-neoplastic tumour-promoting mesenchymal cells. J Cell Physiol (2013) 228:1651–57. doi: 10.1002/jcp.24347
25. Kalluri R, Zeisberg M. Fibroblasts in cancer. Nat Rev Cancer (2006) 6:392–401. doi: 10.1038/nrc1877
26. Morad G, Helmink BA, Sharma P, Wargo JA. Hallmarks of response, resistance, and toxicity to immune checkpoint blockade. Cell (2022) 185(3):576. doi: 10.1016/j.cell.2022.01.008
27. Dzobo K, Dandara C. Architecture of cancer-associated fibroblasts in tumor microenvironment: Mapping their origins, heterogeneity, and role in cancer therapy resistance. Omics J Integr Biol (2020) 24:314–39. doi: 10.1089/omi.2020.0023
28. Kanzaki R, Pietras K. Heterogeneity of cancer-associated fibroblasts: Opportunities for precision medicine. Cancer Sci (2020) 111:2708–17. doi: 10.1111/cas.14537
29. Nurmik M, Ullmann P, Rodriguez F, Haan S, Letellier E. In search of definitions: Cancer-associated fibroblasts and their markers. Int J Cancer (2020) 146:895–905. doi: 10.1002/ijc.32193
30. Öhlund D, Handly-Santana A, Biffi G, Elyada E, Almeida AS, Ponz-Sarvise M, et al. Distinct populations of inflammatory fibroblasts and myofibroblasts in pancreatic cancer. J Exp Med (2017) 214:579–96. doi: 10.1084/jem.20162024
31. Chen X, Song E. Turning foes to friends: targeting cancer-associated fibroblasts. Nat Rev Drug Discov (2019) 18:99–115. doi: 10.1038/s41573-018-0004-1
32. Bachem MG, Schneider E, Gross H, Weidenbach H, Schmid RM, Menke A, et al. Identification, culture, and characterization of pancreatic stellate cells in rats and humans. Gastroenterology (1998) 115:421–32. doi: 10.1016/S0016-5085(98)70209-4
33. Apte MV, Haber PS, Darby SJ, Rodgers SC, McCaughan GW, Korsten MA, et al. Pancreatic stellate cells are activated by proinflammatory cytokines: implications for pancreatic fibrogenesis. Gut (1999) 44:534–41. doi: 10.1136/gut.44.4.534
34. Yin C, Evason KJ, Asahina K, Stainier DYR. Hepatic stellate cells in liver development, regeneration, and cancer. J Clin Invest (2013) 123:1902–10. doi: 10.1172/JCI66369
35. Omary MB, Lugea A, Lowe AW, Pandol SJ. The pancreatic stellate cell: a star on the rise in pancreatic diseases. J Clin Invest (2007) 117:50–9. doi: 10.1172/JCI30082
36. Jung Y, Kim JK, Shiozawa Y, Wang J, Mishra A, Joseph J, et al. Recruitment of mesenchymal stem cells into prostate tumours promotes metastasis. Nat Commun (2013) 4:1795. doi: 10.1038/ncomms2766
37. Mishra PJ, Mishra PJ, Humeniuk R, Medina DJ, Alexe G, Mesirov JP, et al. Carcinoma-associated fibroblast-like differentiation of human mesenchymal stem cells. Cancer Res (2008) 68:4331–39. doi: 10.1158/0008-5472.CAN-08-0943
38. Zhu Q, Zhang X, Zhang L, Li W, Wu H, Yuan X, et al. The IL-6-STAT3 axis mediates a reciprocal crosstalk between cancer-derived mesenchymal stem cells and neutrophils to synergistically prompt gastric cancer progression. Cell Death Dis (2014) 5(6):e1295. doi: 10.1038/cddis.2014.263
39. Weber CE, Kothari AN, Wai PY, Li NY, Driver J, Zapf MAC, et al. Osteopontin mediates an MZF1-TGF-β1-dependent transformation of mesenchymal stem cells into cancer-associated fibroblasts in breast cancer. Oncogene (2015) 34:4821–33. doi: 10.1038/onc.2014.410
40. Zeisberg EM, Potenta S, Xie L, Zeisberg M, Kalluri R. Discovery of endothelial to mesenchymal transition as a source for carcinoma-associated fibroblasts. Cancer Res (2007) 67:10123–28. doi: 10.1158/0008-5472.CAN-07-3127
41. Zeisberg EM, Tarnavski O, Zeisberg M, Dorfman AL, McMullen JR, Gustafsson E, et al. Endothelial-to-mesenchymal transition contributes to cardiac fibrosis. Nat Med (2007) 13:952–61. doi: 10.1038/nm1613
42. Bochet L, Lehuédé C, Dauvillier S, Wang YY, Dirat B, Laurent V, et al. Adipocyte-derived fibroblasts promote tumor progression and contribute to the desmoplastic reaction in breast cancer. Cancer Res (2013) 73:5657–68. doi: 10.1158/0008-5472.CAN-13-0530
43. Karnoub AE, Dash AB, Vo AP, Sullivan A, Brooks MW, Bell GW, et al. Mesenchymal stem cells within tumour stroma promote breast cancer metastasis. Nature (2007) 449:557–63. doi: 10.1038/nature06188
44. Raz Y, Cohen N, Shani O, Bell RE, Novitskiy SV, Abramovitz L, et al. Bone marrow-derived fibroblasts are a functionally distinct stromal cell population in breast cancer. J Exp Med (2018) 215:3075–93. doi: 10.1084/jem.20180818
45. Shi Y, Du L, Lin L, Wang Y. Tumour-associated mesenchymal stem/stromal cells: emerging therapeutic targets. Nat Rev Drug Discovery (2017) 16:35–52. doi: 10.1038/nrd.2016.193
46. Ridge SM, Sullivan FJ, Glynn SA. Mesenchymal stem cells: key players in cancer progression. Mol Cancer (2017) 16:31. doi: 10.1186/s12943-017-0597-8
47. Bianchi G, Borgonovo G, Pistoia V, Raffaghello L. Immunosuppressive cells and tumour microenvironment: focus on mesenchymal stem cells and myeloid derived suppressor cells. Histol Histopathol (2011) 26:941–51. doi: 10.14670/HH-26.941
48. Hosein AN, Wu M, Arcand SL, Lavallée S, Hébert J, Tonin PN, et al. Breast carcinoma-associated fibroblasts rarely contain p53 mutations or chromosomal aberrations. Cancer Res (2010) 70:5770–77. doi: 10.1158/0008-5472.CAN-10-0673
49. Allinen M, Beroukhim R, Cai L, Brennan C, Lahti-Domenici J, Huang H, et al. Molecular characterization of the tumor microenvironment in breast cancer. Cancer Cell (2004) 6:17–32. doi: 10.1016/j.ccr.2004.06.010
50. Haviv I, Polyak K, Qiu W, Hu M, Campbell I. Origin of carcinoma associated fibroblasts. Cell Cycle (Georgetown Tex.) (2009) 8:589–95. doi: 10.4161/cc.8.4.7669
51. Cirri P, Chiarugi P. Cancer-associated-fibroblasts and tumour cells: a diabolic liaison driving cancer progression. Cancer metastasis Rev (2012) 31:195–208. doi: 10.1007/s10555-011-9340-x
52. Dai Z, Gu X, Xiang S, Gong D, Man C, Fan Y. Research and application of single-cell sequencing in tumor heterogeneity and drug resistance of circulating tumor cells. biomark Res (2020) 8:60. doi: 10.1186/s40364-020-00240-1
53. Bartoschek M, Oskolkov N, Bocci M, Lövrot J, Larsson C, Sommarin M, et al. Spatially and functionally distinct subclasses of breast cancer-associated fibroblasts revealed by single cell RNA sequencing. Nat Commun (2018) 9:5150. doi: 10.1038/s41467-018-07582-3
54. Brechbuhl HM, Barrett AS, Kopin E, Hagen JC, Han AL, Gillen AE, et al. Fibroblast subtypes define a metastatic matrisome in breast cancer. JCI Insight (2020) 5(4)e130751. doi: 10.1172/jci.insight.130751
55. Costa A, Kieffer Y, Scholer-Dahirel A, Pelon F, Bourachot B, Cardon M, et al. Fibroblast heterogeneity and immunosuppressive environment in human breast cancer. Cancer Cell (2018) 33:463–79. doi: 10.1016/j.ccell.2018.01.011
56. Givel A, Kieffer Y, Scholer-Dahirel A, Sirven P, Cardon M, Pelon F, et al. miR200-regulated CXCL12β promotes fibroblast heterogeneity and immunosuppression in ovarian cancers. Nat Commun (2018) 9:1056. doi: 10.1038/s41467-018-03348-z
57. Pelon F, Bourachot B, Kieffer Y, Magagna I, Mermet-Meillon F, Bonnet I, et al. Cancer-associated fibroblast heterogeneity in axillary lymph nodes drives metastases in breast cancer through complementary mechanisms. Nat Commun (2020) 11:404. doi: 10.1038/s41467-019-14134-w
58. Wu SZ, Roden DL, Wang C, Holliday H, Harvey K, Cazet AS, et al. Stromal cell diversity associated with immune evasion in human triple-negative breast cancer. EMBO J (2020) 39:e104063. doi: 10.15252/embj.2019104063
59. Friedman G, Levi-Galibov O, David E, Bornstein C, Giladi A, Dadiani M, et al. Cancer-associated fibroblast compositions change with breast cancer progression linking the ratio of S100A4(+) and PDPN(+) CAFs to clinical outcome. Nat Cancer (2020) 1:692–708. doi: 10.1038/s43018-020-0082-y
60. Hao J, Zeltz C, Pintilie M, Li Q, Sakashita S, Wang T, et al. Characterization of distinct populations of carcinoma-associated fibroblasts from non-small cell lung carcinoma reveals a role for ST8SIA2 in cancer cell invasion. Neoplasia (2019) 21:482–93. doi: 10.1016/j.neo.2019.03.009
61. Su S, Chen J, Yao H, Liu J, Yu S, Lao L, et al. CD10(+)GPR77(+) cancer-associated fibroblasts promote cancer formation and chemoresistance by sustaining cancer stemness. Cell (2018) 172:841–56. doi: 10.1016/j.cell.2018.01.009
62. Ligorio M, Sil S, Malagon-Lopez J, Nieman LT, Misale S, Di Pilato M, et al. Stromal microenvironment shapes the intratumoral architecture of pancreatic cancer. Cell (2019) 178:160–75. doi: 10.1016/j.cell.2019.05.012
63. Elyada E, Bolisetty M, Laise P, Flynn WF, Courtois ET, Burkhart RA, et al. Cross-species single-cell analysis of pancreatic ductal adenocarcinoma reveals antigen-presenting cancer-associated fibroblasts. Cancer Discov (2019) 9:1102–23. doi: 10.1158/2159-8290.CD-19-0094
64. Bernard V, Semaan A, Huang J, San Lucas FA, Mulu FC, Stephens BM, et al. Single-cell transcriptomics of pancreatic cancer precursors demonstrates epithelial and microenvironmental heterogeneity as an early event in neoplastic progression. Clin Cancer Res (2019) 25:2194–205. doi: 10.1158/1078-0432.CCR-18-1955
65. Hosein AN, Huang H, Wang Z, Parmar K, Du W, Huang J, et al. Cellular heterogeneity during mouse pancreatic ductal adenocarcinoma progression at single-cell resolution. JCI Insight (2019) 5(16):e129212. doi: 10.1172/jci.insight.129212
66. Peng J, Sun B, Chen C, Zhou J, Chen Y, Chen H, et al. Single-cell RNA-seq highlights intra-tumoral heterogeneity and malignant progression in pancreatic ductal adenocarcinoma. Cell Res (2019) 29:725–38. doi: 10.1038/s41422-019-0195-y
67. Li H, Courtois ET, Sengupta D, Tan Y, Chen KH, Goh JJL, et al. Reference component analysis of single-cell transcriptomes elucidates cellular heterogeneity in human colorectal tumors. Nat Genet (2017) 49:708–18. doi: 10.1038/ng.3818
68. Zhang L, Li Z, Skrzypczynska KM, Fang Q, Zhang W, O'Brien SA, et al. Single-cell analyses inform mechanisms of myeloid-targeted therapies in colon cancer. Cell (2020) 181:442–59. doi: 10.1016/j.cell.2020.03.048
69. Davidson S, Efremova M, Riedel A, Mahata B, Pramanik J, Huuhtanen J, et al. Single-cell RNA sequencing reveals a dynamic stromal niche that supports tumor growth. Cell Rep (2020) 31:107628. doi: 10.1016/j.celrep.2020.107628
70. Chen S, Zhu G, Yang Y, Wang F, Xiao Y, Zhang N, et al. Single-cell analysis reveals transcriptomic remodellings in distinct cell types that contribute to human prostate cancer progression. Nat Cell Biol (2021) 23:87–98. doi: 10.1038/s41556-020-00613-6
71. Kourtidou S, Slee AE, Bruce ME, Wren H, Mangione-Smith RM, Portman MA. Kawasaki Disease substantially impacts health-related quality of life. J Pediatr (2018) 193:155–63. doi: 10.1016/j.jpeds.2017.09.070
72. Chen Z, Zhou L, Liu L, Hou Y, Xiong M, Yang Y, et al. Single-cell RNA sequencing highlights the role of inflammatory cancer-associated fibroblasts in bladder urothelial carcinoma. Nat Commun (2020) 11:5077. doi: 10.1038/s41467-020-18916-5
73. Affo S, Nair A, Brundu F, Ravichandra A, Bhattacharjee S, Matsuda M, et al. Promotion of cholangiocarcinoma growth by diverse cancer-associated fibroblast subpopulations. Cancer Cell (2021) 39:866–82. doi: 10.1016/j.ccell.2021.03.012
74. Li X, Sun Z, Peng G, Xiao Y, Guo J, Wu B, et al. Single-cell RNA sequencing reveals a pro-invasive cancer-associated fibroblast subgroup associated with poor clinical outcomes in patients with gastric cancer. Theranostics (2022) 12:620–38. doi: 10.7150/thno.60540
75. Zhou Y, Bian S, Zhou X, Cui Y, Wang W, Wen L, et al. Single-cell multiomics sequencing reveals prevalent genomic alterations in tumor stromal cells of human colorectal cancer. Cancer Cell (2020) 38:818–28. doi: 10.1016/j.ccell.2020.09.015
76. Quail DF, Joyce JA. Microenvironmental regulation of tumor progression and metastasis. Nat Med (2013) 19:1423–37. doi: 10.1038/nm.3394
77. Ayala G, Tuxhorn JA, Wheeler TM, Frolov A, Scardino PT, Ohori M, et al. Reactive stroma as a predictor of biochemical-free recurrence in prostate cancer. Clin Cancer Res (2003) 9:4792–801.
78. Pietras K, Ostman A. Hallmarks of cancer: interactions with the tumor stroma. Exp Cell Res (2010) 316:1324–31. doi: 10.1016/j.yexcr.2010.02.045
79. Zhang J, Chen L, Liu X, Kammertoens T, Blankenstein T, Qin Z. Fibroblast-specific protein 1/S100A4-positive cells prevent carcinoma through collagen production and encapsulation of carcinogens. Cancer Res (2013) 73:2770–81. doi: 10.1158/0008-5472.CAN-12-3022
80. Le Hir M, Hegyi I, Cueni-Loffing D, Loffing J, Kaissling B. Characterization of renal interstitial fibroblast-specific protein 1/S100A4-positive cells in healthy and inflamed rodent kidneys. Histochem Cell Biol (2005) 123(4–5):335–46. doi: 10.1007/s00418-005-0788-z
81. Qian L, Gong J, Ma W, Sun Y, Hong J, Xu D, et al. Circulating S100A4 as a prognostic biomarker for patients with nonparoxysmal atrial fibrillation after catheter ablation. Ann Trans Med (2021) 9:1400. doi: 10.21037/atm-21-1101
82. Li Z, Sun C, Qin Z. Metabolic reprogramming of cancer-associated fibroblasts and its effect on cancer cell reprogramming. Theranostics (2021) 11:8322–36. doi: 10.7150/thno.62378
83. Zhang W, Bouchard G, Yu A, Shafiq M, Jamali M, Shrager JB, et al. GFPT2-expressing cancer-associated fibroblasts mediate metabolic reprogramming in human lung adenocarcinoma. Cancer Res (2018) 78:3445–57. doi: 10.1158/0008-5472.CAN-17-2928
84. Vander Heiden MG, DeBerardinis RJ. Understanding the intersections between metabolism and cancer biology. Cell (2017) 168:657–69. doi: 10.1016/j.cell.2016.12.039
85. Potter M, Newport E, Morten KJ. The warburg effect: 80 years on. Biochem Soc T (2016) 44:1499–505. doi: 10.1042/BST20160094
86. Vaupel P, Schmidberger H, Mayer A. The warburg effect: essential part of metabolic reprogramming and central contributor to cancer progression. Int J Radiat Biol (2019) 95:912–19. doi: 10.1080/09553002.2019.1589653
87. Lu J, Tan M, Cai Q. The warburg effect in tumor progression: mitochondrial oxidative metabolism as an anti-metastasis mechanism. Cancer Lett (2015) 356:156–64. doi: 10.1016/j.canlet.2014.04.001
88. Arcucci A, Ruocco MR, Granato G, Sacco AM, Montagnani S. Cancer: An oxidative crosstalk between solid tumor cells and cancer associated fibroblasts. BioMed Res Int (2016) 2016:4502846. doi: 10.1155/2016/4502846
89. Ishii G, Ochiai A, Neri S. Phenotypic and functional heterogeneity of cancer-associated fibroblast within the tumor microenvironment. Adv Drug Deliver Rev (2016) 99:186–96. doi: 10.1016/j.addr.2015.07.007
90. Comito G, Ippolito L, Chiarugi P, Cirri P. Nutritional exchanges within tumor microenvironment: Impact for cancer aggressiveness. Front Oncol (2020) 10:396. doi: 10.3389/fonc.2020.00396
91. Sanford-Crane H, Abrego J, Sherman MH. Fibroblasts as modulators of local and systemic cancer metabolism. Cancers (2019) 11(5):619. doi: 10.3390/cancers11050619
92. Bonnet S, Archer SL, Allalunis-Turner J, Haromy A, Beaulieu C, Thompson R, et al. A mitochondria-k+ channel axis is suppressed in cancer and its normalization promotes apoptosis and inhibits cancer growth. Cancer Cell (2007) 11:37–51. doi: 10.1016/j.ccr.2006.10.020
93. Fantin VR, St-Pierre J, Leder P. Attenuation of LDH-a expression uncovers a link between glycolysis, mitochondrial physiology, and tumor maintenance. Cancer Cell (2006) 9:425–34. doi: 10.1016/j.ccr.2006.04.023
94. Michelakis ED, Sutendra G, Dromparis P, Webster L, Haromy A, Niven E, et al. Metabolic modulation of glioblastoma with dichloroacetate. Sci Transl Med (2010) 2:31r–4r. doi: 10.1126/scitranslmed.3000677
95. Moreno-Sánchez R, Rodríguez-Enríquez S, Marín-Hernández A, Saavedra E. Energy metabolism in tumor cells. FEBS J (2007) 274:1393–418. doi: 10.1111/j.1742-4658.2007.05686.x
96. Martinez-Outschoorn UE, Lin Z, Trimmer C, Flomenberg N, Wang C, Pavlides S, et al. Cancer cells metabolically "fertilize" the tumor microenvironment with hydrogen peroxide, driving the warburg effect: implications for PET imaging of human tumors. Cell Cycle (2011) 10:2504–20. doi: 10.4161/cc.10.15.16585
97. Gentric G, Mieulet V, Mechta-Grigoriou F. Heterogeneity in cancer metabolism: New concepts in an old field. Antioxid Redox Sign (2017) 26:462–85. doi: 10.1089/ars.2016.6750
98. Xing Y, Zhao S, Zhou BP, Mi J. Metabolic reprogramming of the tumour microenvironment. FEBS J (2015) 282:3892–98. doi: 10.1111/febs.13402
99. Guppy M, Leedman P, Zu X, Russell V. Contribution by different fuels and metabolic pathways to the total ATP turnover of proliferating MCF-7 breast cancer cells. Biochem J (2002) 364:309–15. doi: 10.1042/bj3640309
100. Caro P, Kishan AU, Norberg E, Stanley IA, Chapuy B, Ficarro SB, et al. Metabolic signatures uncover distinct targets in molecular subsets of diffuse large b cell lymphoma. Cancer Cell (2012) 22:547–60. doi: 10.1016/j.ccr.2012.08.014
101. Lim J, Luo C, Vazquez F, Puigserver P. Targeting mitochondrial oxidative metabolism in melanoma causes metabolic compensation through glucose and glutamine utilization. Cancer Res (2014) 74:3535–45. doi: 10.1158/0008-5472.CAN-13-2893-T
102. Vazquez F, Lim J, Chim H, Bhalla K, Girnun G, Pierce K, et al. PGC1α expression defines a subset of human melanoma tumors with increased mitochondrial capacity and resistance to oxidative stress. Cancer Cell (2013) 23:287–301. doi: 10.1016/j.ccr.2012.11.020
103. Pavlides S, Whitaker-Menezes D, Castello-Cros R, Flomenberg N, Witkiewicz AK, Frank PG, et al. The reverse warburg effect: aerobic glycolysis in cancer associated fibroblasts and the tumor stroma. Cell Cycle (2009) 8:3984–4001. doi: 10.4161/cc.8.23.10238
104. Roy A, Bera S. CAF cellular glycolysis: linking cancer cells with the microenvironment. Tumour Biol (2016) 37:8503–14. doi: 10.1007/s13277-016-5049-3
105. Zhang D, Wang Y, Shi Z, Liu J, Sun P, Hou X, et al. Metabolic reprogramming of cancer-associated fibroblasts by IDH3α downregulation. Cell Rep (2015) 10:1335–48. doi: 10.1016/j.celrep.2015.02.006
106. Hu J, Sun P, Zhang D, Xiong W, Mi J. Hexokinase 2 regulates G1/S checkpoint through CDK2 in cancer-associated fibroblasts. Cell Signal (2014) 26:2210–16. doi: 10.1016/j.cellsig.2014.04.015
107. Chiarugi P, Cirri P. Metabolic exchanges within tumor microenvironment. Cancer Lett (2016) 380:272–80. doi: 10.1016/j.canlet.2015.10.027
108. Baltazar F, Pinheiro C, Morais-Santos F, Azevedo-Silva J, Queirós O, Preto A, et al. Monocarboxylate transporters as targets and mediators in cancer therapy response. Histol Histopathol (2014) 29:1511–24. doi: 10.14670/HH-29.1511
109. Shim H, Dolde C, Lewis BC, Wu CS, Dang G, Jungmann RA, et al. C-myc transactivation of LDH-a: implications for tumor metabolism and growth. P Natl Acad Sci USA (1997) 94:6658–63. doi: 10.1073/pnas.94.13.6658
110. Sun P, Hu J, Xiong W, Mi J. miR-186 regulates glycolysis through Glut1 during the formation of cancer-associated fibroblasts. Asian Pacific J Cancer Prev (2014) 15:4245–50. doi: 10.7314/APJCP.2014.15.10.4245
111. Guido C, Whitaker-Menezes D, Capparelli C, Balliet R, Lin Z, Pestell RG, et al. Metabolic reprogramming of cancer-associated fibroblasts by TGF-β drives tumor growth: connecting TGF-β signaling with "Warburg-like" cancer metabolism and l-lactate production. Cell Cycle (2012) 11:3019–35. doi: 10.4161/cc.21384
112. Sotgia F, Martinez-Outschoorn UE, Howell A, Pestell RG, Pavlides S, Lisanti MP. Caveolin-1 and cancer metabolism in the tumor microenvironment: markers, models, and mechanisms. Annu Rev Pathol (2012) 7:423–67. doi: 10.1146/annurev-pathol-011811-120856
113. Sung JS, Kang CW, Kang S, Jang Y, Chae YC, Kim BG, et al. ITGB4-mediated metabolic reprogramming of cancer-associated fibroblasts. Oncogene (2020) 39:664–76. doi: 10.1038/s41388-019-1014-0
114. Capparelli C, Whitaker-Menezes D, Guido C, Balliet R, Pestell TG, Howell A, et al. CTGF drives autophagy, glycolysis and senescence in cancer-associated fibroblasts via HIF1 activation, metabolically promoting tumor growth. Cell Cycle (2012) 11:2272–84. doi: 10.4161/cc.20717
115. Wu D, Zhuo L, Wang X. Metabolic reprogramming of carcinoma-associated fibroblasts and its impact on metabolic heterogeneity of tumors. Semin Cell Dev Biol (2017) 64:125–31. doi: 10.1016/j.semcdb.2016.11.003
116. Kauppila S, Stenbäck F, Risteli J, Jukkola A, Risteli L. Aberrant type I and type III collagen gene expression in human breast cancer in vivo. J Pathol (1998) 186:262–68. doi: 10.1002/(SICI)1096-9896(1998110)186:3<262::AID-PATH191>3.0.CO;2-3
117. Chun T, Hotary KB, Sabeh F, Saltiel AR, Allen ED, Weiss SJ. A pericellular collagenase directs the 3-dimensional development of white adipose tissue. Cell (2006) 125:577–91. doi: 10.1016/j.cell.2006.02.050
118. Huijbers IJ, Iravani M, Popov S, Robertson D, Al-Sarraj S, Jones C, et al. A role for fibrillar collagen deposition and the collagen internalization receptor endo180 in glioma invasion. PloS One (2010) 5:e9808. doi: 10.1371/journal.pone.0009808
119. Assoian RK, Klein EA. Growth control by intracellular tension and extracellular stiffness. Trends Cell Biol (2008) 18:347–52. doi: 10.1016/j.tcb.2008.05.002
120. Shan T, Chen S, Chen X, Lin WR, Li W, Ma J, et al. Cancer-associated fibroblasts enhance pancreatic cancer cell invasion by remodeling the metabolic conversion mechanism. Oncol Rep (2017) 37:1971–79. doi: 10.3892/or.2017.5479
121. Bertero T, Oldham WM, Grasset EM, Bourget I, Boulter E, Pisano S, et al. Tumor-stroma mechanics coordinate amino acid availability to sustain tumor growth and malignancy. Cell Metab (2019) 29:124–40. doi: 10.1016/j.cmet.2018.09.012
122. Leone RD, Zhao L, Englert JM, Sun I, Oh M, Sun I, et al. Glutamine blockade induces divergent metabolic programs to overcome tumor immune evasion. Science (2019) 366:1013–21. doi: 10.1126/science.aav2588
123. Wang Y, Bai C, Ruan Y, Liu M, Chu Q, Qiu L, et al. Coordinative metabolism of glutamine carbon and nitrogen in proliferating cancer cells under hypoxia. Nat Commun (2019) 10:201. doi: 10.1038/s41467-018-08033-9
124. Bose S, Le A. Glucose metabolism in cancer. Adv Exp Med Biol (2018) 1063:3–12. doi: 10.1007/978-3-319-77736-8_1
125. DeBerardinis RJ, Cheng T. Q's next: the diverse functions of glutamine in metabolism, cell biology and cancer. Oncogene (2010) 29:313–24. doi: 10.1038/onc.2009.358
126. Still ER, Yuneva MO. Hopefully devoted to q: targeting glutamine addiction in cancer. Brit J Cancer (2017) 116:1375–81. doi: 10.1038/bjc.2017.113
127. Wu G. Amino acids: metabolism, functions, and nutrition. Amino Acids (2009) 37:1–17. doi: 10.1007/s00726-009-0269-0
128. Wu G. Functional amino acids in nutrition and health. Amino Acids (2013) 45(3):407–11. doi: 10.1007/s00726-013-1500-6
129. Zhao H, Yang L, Baddour J, Achreja A, Bernard V, Moss T, et al. Tumor microenvironment derived exosomes pleiotropically modulate cancer cell metabolism. Elife (2016) 5:e10250. doi: 10.7554/eLife.10250
130. Commisso C, Davidson SM, Soydaner-Azeloglu RG, Parker SJ, Kamphorst JJ, Hackett S, et al. Macropinocytosis of protein is an amino acid supply route in ras-transformed cells. Nature (2013) 497:633–37. doi: 10.1038/nature12138
131. Sazeides C, Le A. Metabolic relationship between cancer-associated fibroblasts and cancer cells. Adv Exp Med Biol (2021) 1311:189–204. doi: 10.1007/978-3-030-65768-0_14
132. Jung JG, Le A. Targeting metabolic cross talk between cancer cells and cancer-associated fibroblasts. Adv Exp Med Biol (2018) 1063:167–78. doi: 10.1007/978-3-319-77736-8_12
133. Yang L, Achreja A, Yeung T, Mangala LS, Jiang D, Han C, et al. Targeting stromal glutamine synthetase in tumors disrupts tumor microenvironment-regulated cancer cell growth. Cell Metab (2016) 24:685–700. doi: 10.1016/j.cmet.2016.10.011
134. Mestre-Farrera A, Bruch-Oms M, Peña R, Rodríguez-Morató J, Alba-Castellón L, Comerma L, et al. Glutamine-directed migration of cancer-activated fibroblasts facilitates epithelial tumor invasion. Cancer Res (2021) 81:438–51. doi: 10.1158/0008-5472.CAN-20-0622
135. Li X, Zhu H, Sun W, Yang X, Nie Q, Fang X. Role of glutamine and its metabolite ammonia in crosstalk of cancer-associated fibroblasts and cancer cells. Cancer Cell Int (2021) 21:479. doi: 10.1186/s12935-021-02121-5
136. Munir R, Lisec J, Swinnen JV, Zaidi N. Lipid metabolism in cancer cells under metabolic stress. Brit J Cancer (2019) 120:1090–98. doi: 10.1038/s41416-019-0451-4
137. Koundouros N, Poulogiannis G. Reprogramming of fatty acid metabolism in cancer. Brit J Cancer (2020) 122:4–22. doi: 10.1038/s41416-019-0650-z
138. Huang C, Freter C. Lipid metabolism, apoptosis and cancer therapy. Int J Mol Sci (2015) 16:924–49. doi: 10.3390/ijms16010924
139. Cullis PR, de Kruijff B. Lipid polymorphism and the functional roles of lipids in biological membranes. Biochim Biophys Acta (1979) 559:399–420. doi: 10.1016/0304-4157(79)90012-1
140. van Meer G, Voelker DR, Feigenson GW. Membrane lipids: where they are and how they behave. Nat Rev Mol Cell Biol (2008) 9:112–24. doi: 10.1038/nrm2330
141. Lopes-Coelho F, André S, Félix A, Serpa J. Breast cancer metabolic cross-talk: Fibroblasts are hubs and breast cancer cells are gatherers of lipids. Mol Cell Endocrinol (2018) 462:93–106. doi: 10.1016/j.mce.2017.01.031
142. Santi A, Caselli A, Ranaldi F, Paoli P, Mugnaioni C, Michelucci E, et al. Cancer associated fibroblasts transfer lipids and proteins to cancer cells through cargo vesicles supporting tumor growth. Biochim Biophys Acta (2015) 1853:3211–23. doi: 10.1016/j.bbamcr.2015.09.013
143. Gong J, Lin Y, Zhang H, Liu C, Cheng Z, Yang X, et al. Reprogramming of lipid metabolism in cancer-associated fibroblasts potentiates migration of colorectal cancer cells. Cell Death Dis (2020) 11:267. doi: 10.1038/s41419-020-2434-z
144. Hu JL, Wang W, Lan XL, Zeng ZC, Liang YS, Yan YR, et al. CAFs secreted exosomes promote metastasis and chemotherapy resistance by enhancing cell stemness and epithelial-mesenchymal transition in colorectal cancer. Mol Cancer (2019) 18:91. doi: 10.1186/s12943-019-1019-x
145. Ilkhani K, Bastami M, Delgir S, Safi A, Talebian S, Alivand M. The engaged role of tumor microenvironment in cancer metabolism: Focusing on cancer-associated fibroblast and exosome mediators. Anti-Cancer Agent Me (2021) 21:254–66. doi: 10.2174/1871520620666200910123428
146. Chen F, Zhuang X, Lin L, Yu P, Wang Y, Shi Y, et al. New horizons in tumor microenvironment biology: challenges and opportunities. BMC Med (2015) 13:45. doi: 10.1186/s12916-015-0278-7
147. Paluskievicz CM, Cao X, Abdi R, Zheng P, Liu Y, Bromberg JS. T Regulatory cells and priming the suppressive tumor microenvironment. Front Immunol (2019) 10:2453. doi: 10.3389/fimmu.2019.02453
148. Schulz M, Salamero-Boix A, Niesel K, Alekseeva T, Sevenich L. Microenvironmental regulation of tumor progression and therapeutic response in brain metastasis. Front Immunol (2019) 10:1713. doi: 10.3389/fimmu.2019.01713
149. Baghban R, Roshangar L, Jahanban-Esfahlan R, Seidi K, Ebrahimi-Kalan A, Jaymand M, et al. Tumor microenvironment complexity and therapeutic implications at a glance. Cell Commun Signal (2020) 18:59. doi: 10.1186/s12964-020-0530-4
150. Gajewski TF, Schreiber H, Fu Y. Innate and adaptive immune cells in the tumor microenvironment. Nat Immunol (2013) 14:1014–22. doi: 10.1038/ni.2703
151. Binnewies M, Roberts EW, Kersten K, Chan V, Fearon DF, Merad M, et al. Understanding the tumor immune microenvironment (TIME) for effective therapy. Nat Med (2018) 24:541–50. doi: 10.1038/s41591-018-0014-x
152. Worzfeld T, Pogge von Strandmann E, Huber M, Adhikary T, Wagner U, Reinartz S, et al. The unique molecular and cellular microenvironment of ovarian cancer. Front Oncol (2017) 7:24. doi: 10.3389/fonc.2017.00024
153. Cicco S, Cicco G, Racanelli V, Vacca A. Neutrophil extracellular traps (NETs) and damage-associated molecular patterns (DAMPs): Two potential targets for COVID-19 treatment. Mediat Inflamm (2020) 2020:7527953. doi: 10.1155/2020/7527953
154. Haas L, Obenauf AC. Allies or enemies-the multifaceted role of myeloid cells in the tumor microenvironment. Front Immunol (2019) 10:2746. doi: 10.3389/fimmu.2019.02746
155. Gardner A, Ruffell B. Dendritic cells and cancer immunity. Trends Immunol (2016) 37:855–65. doi: 10.1016/j.it.2016.09.006
156. Bol KF, Schreibelt G, Gerritsen WR, de Vries IJM, Figdor CG. Dendritic cell-based immunotherapy: State of the art and beyond. Clin Cancer Res (2016) 22:1897–906. doi: 10.1158/1078-0432.CCR-15-1399
157. Larionova I, Cherdyntseva N, Liu T, Patysheva M, Rakina M, Kzhyshkowska J. Interaction of tumor-associated macrophages and cancer chemotherapy. Oncoimmunology (2019) 8:1596004. doi: 10.1080/2162402X.2019.1596004
158. Laviron M, Boissonnas A. Ontogeny of tumor-associated macrophages. Front Immunol (2019) 10:1799. doi: 10.3389/fimmu.2019.01799
159. Komohara Y, Fujiwara Y, Ohnishi K, Takeya M. Tumor-associated macrophages: Potential therapeutic targets for anti-cancer therapy. Adv Drug Deliv Rev (2016) 99:180–85. doi: 10.1016/j.addr.2015.11.009
160. Van Overmeire E, Stijlemans B, Heymann F, Keirsse J, Morias Y, Elkrim Y, et al. M-CSF and GM-CSF receptor signaling differentially regulate monocyte maturation and macrophage polarization in the tumor microenvironment. Cancer Res (2016) 76:35–42. doi: 10.1158/0008-5472.CAN-15-0869
161. Chanmee T, Ontong P, Konno K, Itano N. Tumor-associated macrophages as major players in the tumor microenvironment. Cancers (2014) 6:1670–90. doi: 10.3390/cancers6031670
162. Noy R, Pollard JW. Tumor-associated macrophages: from mechanisms to therapy. Immunity (2014) 41:49–61. doi: 10.1016/j.immuni.2014.06.010
163. Kuang D, Zhao Q, Peng C, Xu J, Zhang J, Wu C, et al. Activated monocytes in peritumoral stroma of hepatocellular carcinoma foster immune privilege and disease progression through PD-L1. J Exp Med (2009) 206:1327–37. doi: 10.1084/jem.20082173
164. Mantovani A, Sica A, Sozzani S, Allavena P, Vecchi A, Locati M. The chemokine system in diverse forms of macrophage activation and polarization. Trends Immunol (2004) 25:677–86. doi: 10.1016/j.it.2004.09.015
165. Kiessling R, Petranyi G, Klein G, Wigzel H. Genetic variation of in vitro cytolytic activity and in vivo rejection potential of non-immunized semi-syngeneic mice against a mouse lymphoma line. Int J Cancer (1975) 15:933–40. doi: 10.1002/ijc.2910150608
166. Kiessling R, Klein E, Wigzell H. "Natural" killer cells in the mouse. i. cytotoxic cells with specificity for mouse moloney leukemia cells. specificity and distribution according to genotype. Eur J Immunol (1975) 5:112–17. doi: 10.1002/eji.1830050208
167. Gazit R, Gruda R, Elboim M, Arnon TI, Katz G, Achdout H, et al. Lethal influenza infection in the absence of the natural killer cell receptor gene Ncr1. Nat Immunol (2006) 7:517–23. doi: 10.1038/ni1322
168. Keppel MP, Saucier N, Mah AY, Vogel TP, Cooper MA. Activation-specific metabolic requirements for NK cell IFN-γ production. J Immunol (2015) 194:1954–62. doi: 10.4049/jimmunol.1402099
169. Orr MT, Lanier LL. Natural killer cell education and tolerance. Cell (2010) 142:847–56. doi: 10.1016/j.cell.2010.08.031
170. Coffelt SB, Wellenstein MD, de Visser KE. Neutrophils in cancer: neutral no more. Nat Rev Cancer (2016) 16:431–46. doi: 10.1038/nrc.2016.52
171. Andzinski L, Kasnitz N, Stahnke S, Wu C, Gereke M, von Köckritz-Blickwede M, et al. Type I IFNs induce anti-tumor polarization of tumor associated neutrophils in mice and human. Int J Cancer (2016) 138:1982–93. doi: 10.1002/ijc.29945
172. Hajizadeh F, Aghebati Maleki L, Alexander M, Mikhailova MV, Masjedi A, Ahmadpour M, et al. Tumor-associated neutrophils as new players in immunosuppressive process of the tumor microenvironment in breast cancer. Life Sci (2021) 264:118699. doi: 10.1016/j.lfs.2020.118699
173. Ustyanovska Avtenyuk N, Visser N, Bremer E, Wiersma VR. The neutrophil: The underdog that packs a punch in the fight against cancer. Int J Mol Sci (2020) 21(21):7820. doi: 10.3390/ijms21217820
174. Granot Z, Jablonska J. Distinct functions of neutrophil in cancer and its regulation. Mediat Inflamm (2015) 2015:701067. doi: 10.1155/2015/701067
175. Zhang B, Jiang T, Ling L, Cao Z, Zhao J, Tuo Y, et al. Enhanced antitumor activity of EGFP-EGF1-Conjugated nanoparticles by a multitargeting strategy. ACS Appl Mater Inter (2016) 8:8918–27. doi: 10.1021/acsami.6b00036
176. Kuang D, Zhao Q, Wu Y, Peng C, Wang J, Xu Z, et al. Peritumoral neutrophils link inflammatory response to disease progression by fostering angiogenesis in hepatocellular carcinoma. J Hepatol (2011) 54:948–55. doi: 10.1016/j.jhep.2010.08.041
177. Fossati G, Moulding DA, Spiller DG, Moots RJ, White MRH, Edwards SW. The mitochondrial network of human neutrophils: role in chemotaxis, phagocytosis, respiratory burst activation, and commitment to apoptosis. J Immunol (2003) 170:1964–72. doi: 10.4049/jimmunol.170.4.1964
178. Frydrychowicz M, Boruczkowski M, Kolecka-Bednarczyk A, Dworacki G. The dual role of treg in cancer. Scand J Immunol (2017) 86:436–43. doi: 10.1111/sji.12615
179. van den Hout MFCM, Sluijter BJR, Santegoets SJAM, van Leeuwen PAM, van den Tol MP, van den Eertwegh AJM, et al. Local delivery of CpG-b and GM-CSF induces concerted activation of effector and regulatory T cells in the human melanoma sentinel lymph node. Cancer Immunol Immunother (2016) 65(4):405–15. doi: 10.1007/s00262-016-1811-z
180. Engels CC, Charehbili A, van de Velde CJH, Bastiaannet E, Sajet A, Putter H, et al. The prognostic and predictive value of tregs and tumor immune subtypes in postmenopausal, hormone receptor-positive breast cancer patients treated with adjuvant endocrine therapy: a Dutch TEAM study analysis. Breast Cancer Res Tr (2015) 149:587–96. doi: 10.1007/s10549-015-3269-7
181. Chen C, Chen D, Zhang Y, Chen Z, Zhu W, Zhang B, et al. Changes of CD4+CD25+FOXP3+ and CD8+CD28- regulatory T cells in non-small cell lung cancer patients undergoing surgery. Int Immunopharmacol (2014) 18:255–61. doi: 10.1016/j.intimp.2013.12.004
182. Saito T, Nishikawa H, Wada H, Nagano Y, Sugiyama D, Atarashi K, et al. Two FOXP3(+)CD4(+) T cell subpopulations distinctly control the prognosis of colorectal cancers. Nat Med (2016) 22:679–84. doi: 10.1038/nm.4086
183. Boonpiyathad T, Satitsuksanoa P, Akdis M, Akdis CA. Il-10 producing T and b cells in allergy. Semin Immunol (2019) 44:101326. doi: 10.1016/j.smim.2019.101326
184. Ricciardi S, Manfrini N, Alfieri R, Calamita P, Crosti MC, Gallo S, et al. The translational machinery of human CD4(+) T cells is poised for activation and controls the switch from quiescence to metabolic remodeling. Cell Metab (2018) 28:895–906. doi: 10.1016/j.cmet.2018.08.009
185. Pearce EL, Pearce EJ. Metabolic pathways in immune cell activation and quiescence. Immunity (2013) 38:633–43. doi: 10.1016/j.immuni.2013.04.005
186. Li X, Wenes M, Romero P, Huang SC, Fendt S, Ho P. Navigating metabolic pathways to enhance antitumour immunity and immunotherapy. Nat Rev Clin Oncol (2019) 16:425–41. doi: 10.1038/s41571-019-0203-7
187. Vanpouille-Box C, Lhuillier C, Bezu L, Aranda F, Yamazaki T, Kepp O, et al. Trial watch: Immune checkpoint blockers for cancer therapy. Oncoimmunology (2017) 6:e1373237. doi: 10.1080/2162402X.2017.1373237
188. Brown ZJ, Fu Q, Ma C, Kruhlak M, Zhang H, Luo J, et al. Carnitine palmitoyltransferase gene upregulation by linoleic acid induces CD4(+) T cell apoptosis promoting HCC development. Cell Death Dis (2018) 9:620. doi: 10.1038/s41419-018-0687-6
189. Biswas SK. Metabolic reprogramming of immune cells in cancer progression. Immunity (2015) 43:435–49. doi: 10.1016/j.immuni.2015.09.001
190. Brown ZJ, Yu SJ, Heinrich B, Ma C, Fu Q, Sandhu M, et al. Indoleamine 2,3-dioxygenase provides adaptive resistance to immune checkpoint inhibitors in hepatocellular carcinoma. Cancer Immunol Immunother (2018) 67:1305–15. doi: 10.1007/s00262-018-2190-4
191. Biswas SK, Mantovani A. Orchestration of metabolism by macrophages. Cell Metab (2012) 15:432–37. doi: 10.1016/j.cmet.2011.11.013
192. Ghesquière B, Wong BW, Kuchnio A, Carmeliet P. Metabolism of stromal and immune cells in health and disease. Nature (2014) 511:167–76. doi: 10.1038/nature13312
193. Brand A, Singer K, Koehl GE, Kolitzus M, Schoenhammer G, Thiel A, et al. LDHA-associated lactic acid production blunts tumor immunosurveillance by T and NK cells. Cell Metab (2016) 24:657–71. doi: 10.1016/j.cmet.2016.08.011
194. Kedia-Mehta N, Finlay DK. Competition for nutrients and its role in controlling immune responses. Nat Commun (2019) 10:2123. doi: 10.1038/s41467-019-10015-4
195. Cheng S, Quintin J, Cramer RA, Shepardson KM, Saeed S, Kumar V, et al. mTOR- and HIF-1α-mediated aerobic glycolysis as metabolic basis for trained immunity. Science (2014) 345:1250684. doi: 10.1126/science.1250684
196. Pearce EL, Poffenberger MC, Chang C, Jones RG. Fueling immunity: insights into metabolism and lymphocyte function. Science (2013) 342:1242454. doi: 10.1126/science.1242454
197. Elf SE, Chen J. Targeting glucose metabolism in patients with cancer. Cancer-Am Cancer Soc (2014) 120:774–80. doi: 10.1002/cncr.28501
198. Terrén I, Orrantia A, Vitallé J, Zenarruzabeitia O, Borrego F. NK cell metabolism and tumor microenvironment. Front Immunol (2019) 10:2278. doi: 10.3389/fimmu.2019.02278
199. Takeshima Y, Iwasaki Y, Fujio K, Yamamoto K. Metabolism as a key regulator in the pathogenesis of systemic lupus erythematosus. Semin Arthritis Rheu (2019) 48:1142–45. doi: 10.1016/j.semarthrit.2019.04.006
200. Zhang D, Jin W, Wu R, Li J, Park S, Tu E, et al. High glucose intake exacerbates autoimmunity through reactive-Oxygen-Species-Mediated TGF-β cytokine activation. Immunity (2019) 51:671–81. doi: 10.1016/j.immuni.2019.08.001
201. Lin S, Hardie DG. AMPK: Sensing glucose as well as cellular energy status. Cell Metab (2018) 27:299–313. doi: 10.1016/j.cmet.2017.10.009
202. Blagih J, Coulombe F, Vincent EE, Dupuy F, Galicia-Vázquez G, Yurchenko E, et al. The energy sensor AMPK regulates T cell metabolic adaptation and effector responses in vivo. Immunity (2015) 42:41–54. doi: 10.1016/j.immuni.2014.12.030
203. Chang C, Qiu J, O'Sullivan D, Buck MD, Noguchi T, Curtis JD, et al. Metabolic competition in the tumor microenvironment is a driver of cancer progression. Cell (2015) 162:1229–41. doi: 10.1016/j.cell.2015.08.016
204. Dodard G, Tata A, Erick TK, Jaime D, Miah SMS, Quatrini L, et al. Inflammation-induced lactate leads to rapid loss of hepatic tissue-resident NK cells. Cell Rep (2020) 32:107855. doi: 10.1016/j.celrep.2020.107855
205. Xia C, Liu C, He Z, Cai Y, Chen J. Metformin inhibits cervical cancer cell proliferation by modulating PI3K/Akt-induced major histocompatibility complex class I-related chain a gene expression. J Exp Clin Cancer Res CR (2020) 39:127. doi: 10.1186/s13046-020-01627-6
206. Cascone T, McKenzie JA, Mbofung RM, Punt S, Wang Z, Xu C, et al. Increased tumor glycolysis characterizes immune resistance to adoptive T cell therapy. Cell Metab (2018) 27:977–87. doi: 10.1016/j.cmet.2018.02.024
207. Liu P, Wang H, Li X, Chao T, Teav T, Christen S, et al. α-ketoglutarate orchestrates macrophage activation through metabolic and epigenetic reprogramming. Nat Immunol (2017) 18:985–94. doi: 10.1038/ni.3796
208. Ma G, Liang Y, Chen Y, Wang L, Li D, Liang Z, et al. Glutamine deprivation induces PD-L1 expression via activation of EGFR/ERK/c-jun signaling in renal cancer. Mol Cancer Res MCR (2020) 18:324–39. doi: 10.1158/1541-7786.MCR-19-0517
209. Nabe S, Yamada T, Suzuki J, Toriyama K, Yasuoka T, Kuwahara M, et al. Reinforce the antitumor activity of CD8(+) T cells via glutamine restriction. Cancer Sci (2018) 109:3737–50. doi: 10.1111/cas.13827
210. Johnson MO, Wolf MM, Madden MZ, Andrejeva G, Sugiura A, Contreras DC, et al. Distinct regulation of Th17 and Th1 cell differentiation by glutaminase-dependent metabolism. Cell (2018) 175:1780–95. doi: 10.1016/j.cell.2018.10.001
211. Pacella I, Procaccini C, Focaccetti C, Miacci S, Timperi E, Faicchia D, et al. Fatty acid metabolism complements glycolysis in the selective regulatory T cell expansion during tumor growth. P Natl Acad Sci USA (2018) 115:E6546–55. doi: 10.1073/pnas.1720113115
212. Lu H, Liu F, Li Y, Wang J, Ma M, Gao J, et al. Chromatin accessibility of CD8 T cell differentiation and metabolic regulation. Cell Biol Toxicol (2021) 37:367–78. doi: 10.1007/s10565-020-09546-0
213. Shang C, Huang J, Guo H. Identification of an metabolic related risk signature predicts prognosis in cervical cancer and correlates with immune infiltration. Front Cell Dev Biol (2021) 9:677831. doi: 10.3389/fcell.2021.677831
214. Van den Bossche J, van der Windt GJW. Fatty acid oxidation in macrophages and T cells: Time for reassessment? Cell Metab (2018) 28(4):538–40. doi: 10.1016/j.cmet.2018.09.018
215. Saibil SD, St Paul M, Laister RC, Garcia-Batres CR, Israni-Winger K, Elford AR, et al. Activation of peroxisome proliferator-activated receptors α and δ synergizes with inflammatory signals to enhance adoptive cell therapy. Cancer Res (2019) 79:445–51. doi: 10.1158/0008-5472.CAN-17-3053
216. Zhang C, Yue C, Herrmann A, Song J, Egelston C, Wang T, et al. STAT3 activation-induced fatty acid oxidation in CD8(+) T effector cells is critical for obesity-promoted breast tumor growth. Cell Metab (2020) 31:148–61. doi: 10.1016/j.cmet.2019.10.013
217. Amersfoort J, Schaftenaar FH, Douna H, van Santbrink PJ, van Puijvelde GHM, Slütter B, et al. Diet-induced dyslipidemia induces metabolic and migratory adaptations in regulatory T cells. Cardiovasc Res (2021) 117:1309–24. doi: 10.1093/cvr/cvaa208
218. York AG, Williams KJ, Argus JP, Zhou QD, Brar G, Vergnes L, et al. Limiting cholesterol biosynthetic flux spontaneously engages type I IFN signaling. Cell (2015) 163:1716–29. doi: 10.1016/j.cell.2015.11.045
219. Yang W, Bai Y, Xiong Y, Zhang J, Chen S, Zheng X, et al. Potentiating the antitumour response of CD8(+) T cells by modulating cholesterol metabolism. Nature (2016) 531:651–55. doi: 10.1038/nature17412
220. Ma X, Bi E, Lu Y, Su P, Huang C, Liu L, et al. Cholesterol induces CD8(+) T cell exhaustion in the tumor microenvironment. Cell Metab (2019) 30:143–56. doi: 10.1016/j.cmet.2019.04.002
221. Liu T, Han C, Wang S, Fang P, Ma Z, Xu L, et al. Cancer-associated fibroblasts: an emerging target of anti-cancer immunotherapy. J Hematol Oncol (2019) 12:86. doi: 10.1186/s13045-019-0770-1
222. Harper J, Sainson RCA. Regulation of the anti-tumour immune response by cancer-associated fibroblasts. Semin Cancer Biol (2014) 25:69–77. doi: 10.1016/j.semcancer.2013.12.005
223. Kim R, Emi M, Tanabe K. Cancer immunosuppression and autoimmune disease: beyond immunosuppressive networks for tumour immunity. Immunology (2006) 119:254–64. doi: 10.1111/j.1365-2567.2006.02430.x
224. Erez N, Truitt M, Olson P, Arron ST, Hanahan D. Cancer-associated fibroblasts are activated in incipient neoplasia to orchestrate tumor-promoting inflammation in an NF-kappaB-Dependent manner. Cancer Cell (2010) 17:135–47. doi: 10.1016/j.ccr.2009.12.041
225. Ueshima E, Fujimori M, Kodama H, Felsen D, Chen J, Durack JC, et al. Macrophage-secreted TGF-β(1) contributes to fibroblast activation and ureteral stricture after ablation injury. Am J Physiol Renal Physiol (2019) 317:F52–64. doi: 10.1152/ajprenal.00260.2018
226. Sun Q, Zhang B, Hu Q, Qin Y, Xu W, Liu W, et al. The impact of cancer-associated fibroblasts on major hallmarks of pancreatic cancer. Theranostics (2018) 8:5072–87. doi: 10.7150/thno.26546
227. Shiga K, Hara M, Nagasaki T, Sato T, Takahashi H, Takeyama H. Cancer-associated fibroblasts: Their characteristics and their roles in tumor growth. Cancers (2015) 7:2443–58. doi: 10.3390/cancers7040902
228. Shapouri-Moghaddam A, Mohammadian S, Vazini H, Taghadosi M, Esmaeili S, Mardani F, et al. Macrophage plasticity, polarization, and function in health and disease. J Cell Physiol (2018) 233:6425–40. doi: 10.1002/jcp.26429
229. Tan B, Shi X, Zhang J, Qin J, Zhang N, Ren H, et al. Inhibition of rspo-Lgr4 facilitates checkpoint blockade therapy by switching macrophage polarization. Cancer Res (2018) 78:4929–42. doi: 10.1158/0008-5472.CAN-18-0152
230. Ksiazkiewicz M, Gottfried E, Kreutz M, Mack M, Hofstaedter F, Kunz-Schughart LA. Importance of CCL2-CCR2A/2B signaling for monocyte migration into spheroids of breast cancer-derived fibroblasts. Immunobiology (2010) 215:737–47. doi: 10.1016/j.imbio.2010.05.019
231. Cohen N, Shani O, Raz Y, Sharon Y, Hoffman D, Abramovitz L, et al. Fibroblasts drive an immunosuppressive and growth-promoting microenvironment in breast cancer via secretion of chitinase 3-like 1. Oncogene (2017) 36:4457–68. doi: 10.1038/onc.2017.65
232. Nagarsheth N, Wicha MS, Zou W. Chemokines in the cancer microenvironment and their relevance in cancer immunotherapy. Nat Rev Immunol (2017) 17:559–72. doi: 10.1038/nri.2017.49
233. Zhang R, Qi F, Zhao F, Li G, Shao S, Zhang X, et al. Cancer-associated fibroblasts enhance tumor-associated macrophages enrichment and suppress NK cells function in colorectal cancer. Cell Death Dis (2019) 10:273. doi: 10.1038/s41419-019-1435-2
234. Zhang J, Chen L, Xiao M, Wang C, Qin Z. FSP1+ fibroblasts promote skin carcinogenesis by maintaining MCP-1-mediated macrophage infiltration and chronic inflammation. Am J Pathol (2011) 178:382–90. doi: 10.1016/j.ajpath.2010.11.017
235. Takahashi H, Sakakura K, Kudo T, Toyoda M, Kaira K, Oyama T, et al. Cancer-associated fibroblasts promote an immunosuppressive microenvironment through the induction and accumulation of protumoral macrophages. Oncotarget (2017) 8:8633–47. doi: 10.18632/oncotarget.14374
236. Comito G, Giannoni E, Segura CP, Barcellos-de-Souza P, Raspollini MR, Baroni G, et al. Cancer-associated fibroblasts and M2-polarized macrophages synergize during prostate carcinoma progression. Oncogene (2014) 33:2423–31. doi: 10.1038/onc.2013.191
237. Hashimoto O, Yoshida M, Koma Y, Yanai T, Hasegawa D, Kosaka Y, et al. Collaboration of cancer-associated fibroblasts and tumour-associated macrophages for neuroblastoma development. J Pathol (2016) 240:211–23. doi: 10.1002/path.4769
238. Zhang Q, Chai S, Wang W, Wan C, Zhang F, Li Y, et al. Macrophages activate mesenchymal stem cells to acquire cancer-associated fibroblast-like features resulting in gastric epithelial cell lesions and malignant transformation in vitro. Oncol Lett (2019) 17:747–56. doi: 10.3892/ol.2018.9703
239. Fridlender ZG, Sun J, Kim S, Kapoor V, Cheng G, Ling L, et al. Polarization of tumor-associated neutrophil phenotype by TGF-beta: "N1" versus "N2" TAN. Cancer Cell (2009) 16:183–94. doi: 10.1016/j.ccr.2009.06.017
240. Fridlender ZG, Sun J, Mishalian I, Singhal S, Cheng G, Kapoor V, et al. Transcriptomic analysis comparing tumor-associated neutrophils with granulocytic myeloid-derived suppressor cells and normal neutrophils. PLoS One (2012) 7(2):e31524. doi: 10.1371/journal.pone.0031524
241. Jablonska J, Leschner S, Westphal K, Lienenklaus S, Weiss S. Neutrophils responsive to endogenous IFN-beta regulate tumor angiogenesis and growth in a mouse tumor model. J Clin Invest (2010) 120:1151–64. doi: 10.1172/JCI37223
242. Piccard H, Muschel RJ, Opdenakker G. On the dual roles and polarized phenotypes of neutrophils in tumor development and progression. Crit Rev Oncol Hematol (2012) 82:296–309. doi: 10.1016/j.critrevonc.2011.06.004
243. Fridlender ZG, Albelda SM. Tumor-associated neutrophils: friend or foe? Carcinogenesis (2012) 33:949–55. doi: 10.1093/carcin/bgs123
244. Raman D, Baugher PJ, Thu YM, Richmond A. Role of chemokines in tumor growth. Cancer Lett (2007) 256:137–65. doi: 10.1016/j.canlet.2007.05.013
245. Cheng Y, Li H, Deng Y, Tai Y, Zeng K, Zhang Y, et al. Cancer-associated fibroblasts induce PDL1+ neutrophils through the IL6-STAT3 pathway that foster immune suppression in hepatocellular carcinoma. Cell Death Dis (2018) 9:422. doi: 10.1038/s41419-018-0458-4
246. Song M, He J, Pan Q, Yang J, Zhao J, Zhang Y, et al. Cancer-associated fibroblast-mediated cellular crosstalk supports hepatocellular carcinoma progression. Hepatol (Baltimore Md.) (2021) 73:1717–35. doi: 10.1002/hep.31792
247. Ellem SJ, Taylor RA, Furic L, Larsson O, Frydenberg M, Pook D, et al. A pro-tumourigenic loop at the human prostate tumour interface orchestrated by oestrogen, CXCL12 and mast cell recruitment. J Pathol (2014) 234:86–98. doi: 10.1002/path.4386
248. Ma Y, Hwang RF, Logsdon CD, Ullrich SE. Dynamic mast cell-stromal cell interactions promote growth of pancreatic cancer. Cancer Res (2013) 73:3927–37. doi: 10.1158/0008-5472.CAN-12-4479
249. Yang F, Chen S, Clegg T, Li X, Morgan T, Estwick SA, et al. Nf1+/- mast cells induce neurofibroma like phenotypes through secreted TGF-beta signaling. Hum Mol Genet (2006) 15:2421–37. doi: 10.1093/hmg/ddl165
250. Turley SJ, Cremasco V, Astarita JL. Immunological hallmarks of stromal cells in the tumour microenvironment. Nat Rev Immunol (2015) 15:669–82. doi: 10.1038/nri3902
251. Li T, Yang Y, Hua X, Wang G, Liu W, Jia C, et al. Hepatocellular carcinoma-associated fibroblasts trigger NK cell dysfunction via PGE2 and IDO. Cancer Lett (2012) 318:154–61. doi: 10.1016/j.canlet.2011.12.020
252. Li T, Yi S, Liu W, Jia C, Wang G, Hua X, et al. Colorectal carcinoma-derived fibroblasts modulate natural killer cell phenotype and antitumor cytotoxicity. Med Oncol (2013) 30:663. doi: 10.1007/s12032-013-0663-z
253. Balsamo M, Scordamaglia F, Pietra G, Manzini C, Cantoni C, Boitano M, et al. Melanoma-associated fibroblasts modulate NK cell phenotype and antitumor cytotoxicity. P Natl Acad Sci USA (2009) 106:20847–52. doi: 10.1073/pnas.0906481106
254. Inoue T, Adachi K, Kawana K, Taguchi A, Nagamatsu T, Fujimoto A, et al. Cancer-associated fibroblast suppresses killing activity of natural killer cells through downregulation of poliovirus receptor (PVR/CD155), a ligand of activating NK receptor. Int J Oncol (2016) 49:1297–304. doi: 10.3892/ijo.2016.3631
255. Flavell RA, Sanjabi S, Wrzesinski SH, Licona-Limón P. The polarization of immune cells in the tumour environment by TGFbeta. Nat Rev Immunol (2010) 10:554–67. doi: 10.1038/nri2808
256. Batlle E, Massagué J. Transforming growth factor-β signaling in immunity and cancer. Immunity (2019) 50:924–40. doi: 10.1016/j.immuni.2019.03.024
257. Cheng J, Deng Y, Yi H, Wang G, Fu B, Chen W, et al. Hepatic carcinoma-associated fibroblasts induce IDO-producing regulatory dendritic cells through IL-6-mediated STAT3 activation. Oncogenesis (2016) 5:e198. doi: 10.1038/oncsis.2016.7
258. Suciu-Foca N, Berloco P, Cortesini R. Tolerogenic dendritic cells in cancer, transplantation, and autoimmune diseases. Hum Immunol (2009) 70:277–80. doi: 10.1016/j.humimm.2009.03.003
259. Kuo P, Hung J, Huang S, Chou S, Cheng D, Jong Y, et al. Lung cancer-derived galectin-1 mediates dendritic cell anergy through inhibitor of DNA binding 3/IL-10 signaling pathway. J Immunol (2011) 186:1521–30. doi: 10.4049/jimmunol.1002940
260. Hsu Y, Hung J, Chiang S, Jian S, Wu C, Lin Y, et al. Lung cancer-derived galectin-1 contributes to cancer associated fibroblast-mediated cancer progression and immune suppression through TDO2/kynurenine axis. Oncotarget (2016) 7:27584–98. doi: 10.18632/oncotarget.8488
261. Oyama T, Ran S, Ishida T, Nadaf S, Kerr L, Carbone DP, et al. Vascular endothelial growth factor affects dendritic cell maturation through the inhibition of nuclear factor-kappa b activation in hemopoietic progenitor cells. J Immunol (1998) 160:1224–32.
262. Rahma OE, Hodi FS. The intersection between tumor angiogenesis and immune suppression. Clin Cancer Res (2019) 25:5449–57. doi: 10.1158/1078-0432.CCR-18-1543
263. Tanaka A, Sakaguchi S. Regulatory T cells in cancer immunotherapy. Cell Res (2017) 27:109–18. doi: 10.1038/cr.2016.151
264. Jacobs J, Deschoolmeester V, Zwaenepoel K, Flieswasser T, Deben C, Van den Bossche J, et al. Unveiling a CD70-positive subset of cancer-associated fibroblasts marked by pro-migratory activity and thriving regulatory T cell accumulation. Oncoimmunology (2018) 7:e1440167. doi: 10.1080/2162402X.2018.1440167
265. Tan W, Zhang W, Strasner A, Grivennikov S, Cheng JQ, Hoffman RM, et al. Tumour-infiltrating regulatory T cells stimulate mammary cancer metastasis through RANKL-RANK signalling. Nature (2011) 470:548–53. doi: 10.1038/nature09707
266. Zhao X, Ding L, Lu Z, Huang X, Jing Y, Yang Y, et al. Diminished CD68(+) cancer-associated fibroblast subset induces regulatory T-cell (Treg) infiltration and predicts poor prognosis of oral squamous cell carcinoma patients. Am J Pathol (2020) 190:886–99. doi: 10.1016/j.ajpath.2019.12.007
267. Bourhis M, Palle J, Galy-Fauroux I, Terme M. Direct and indirect modulation of T cells by VEGF-a counteracted by anti-angiogenic treatment. Front Immunol (2021) 12:616837. doi: 10.3389/fimmu.2021.616837
268. Wada J, Suzuki H, Fuchino R, Yamasaki A, Nagai S, Yanai K, et al. The contribution of vascular endothelial growth factor to the induction of regulatory T-cells in malignant effusions. Anticancer Res (2009) 29(3):881–8.
269. Chen W, Jin W, Hardegen N, Lei K, Li L, Marinos N, et al. Conversion of peripheral CD4+CD25- naive T cells to CD4+CD25+ regulatory T cells by TGF-beta induction of transcription factor Foxp3. J Exp Med (2003) 198:1875–86. doi: 10.1084/jem.20030152
270. Farhood B, Najafi M, Mortezaee K. CD8(+) cytotoxic T lymphocytes in cancer immunotherapy: A review. J Cell Physiol (2019) 234:8509–21. doi: 10.1002/jcp.27782
271. Uzhachenko RV, Shanker A. CD8(+) T lymphocyte and NK cell network: Circuitry in the cytotoxic domain of immunity. Front Immunol (2019) 10:1906. doi: 10.3389/fimmu.2019.01906
272. Freeman P, Mielgo A. Cancer-associated fibroblast mediated inhibition of CD8+ cytotoxic T cell accumulation in tumours: Mechanisms and therapeutic opportunities. Cancers (2020) 12(9):2687. doi: 10.3390/cancers12092687
273. Ene-Obong A, Clear AJ, Watt J, Wang J, Fatah R, Riches JC, et al. Activated pancreatic stellate cells sequester CD8+ T cells to reduce their infiltration of the juxtatumoral compartment of pancreatic ductal adenocarcinoma. Gastroenterology (2013) 145:1121–32. doi: 10.1053/j.gastro.2013.07.025
274. De Francesco EM, Lappano R, Santolla MF, Marsico S, Caruso A, Maggiolini M. HIF-1α/GPER signaling mediates the expression of VEGF induced by hypoxia in breast cancer associated fibroblasts (CAFs). Breast Cancer Res (2013) 15:R64. doi: 10.1186/bcr3458
275. Bellone M, Calcinotto A. Ways to enhance lymphocyte trafficking into tumors and fitness of tumor infiltrating lymphocytes. Front Oncol (2013) 3:231. doi: 10.3389/fonc.2013.00231
276. Henke E, Nandigama R, Ergün S. Extracellular matrix in the tumor microenvironment and its impact on cancer therapy. Front Mol Biosci (2019) 6:160. doi: 10.3389/fmolb.2019.00160
277. Thomas DA, Massagué J. TGF-beta directly targets cytotoxic T cell functions during tumor evasion of immune surveillance. Cancer Cell (2005) 8:369–80. doi: 10.1016/j.ccr.2005.10.012
278. Kato T, Noma K, Ohara T, Kashima H, Katsura Y, Sato H, et al. Cancer-associated fibroblasts affect intratumoral CD8(+) and FoxP3(+) T cells Via IL6 in the tumor microenvironment. Clin Cancer Res (2018) 24:4820–33. doi: 10.1158/1078-0432.CCR-18-0205
279. Ino Y, Yamazaki-Itoh R, Oguro S, Shimada K, Kosuge T, Zavada J, et al. Arginase II expressed in cancer-associated fibroblasts indicates tissue hypoxia and predicts poor outcome in patients with pancreatic cancer. PloS One (2013) 8:e55146. doi: 10.1371/journal.pone.0055146
280. Rabinovich GA, Toscano MA. Turning 'sweet' on immunity: galectin-glycan interactions in immune tolerance and inflammation. Nat Rev Immunol (2009) 9:338–52. doi: 10.1038/nri2536
281. Valach J, Fík Z, Strnad H, Chovanec M, Plzák J, Cada Z, et al. Smooth muscle actin-expressing stromal fibroblasts in head and neck squamous cell carcinoma: increased expression of galectin-1 and induction of poor prognosis factors. Int J Cancer (2012) 131:2499–508. doi: 10.1002/ijc.27550
282. Lakins MA, Ghorani E, Munir H, Martins CP, Shields JD. Cancer-associated fibroblasts induce antigen-specific deletion of CD8 (+) T cells to protect tumour cells. Nat Commun (2018) 9:948. doi: 10.1038/s41467-018-03347-0
283. Gabrilovich DI. Myeloid-derived suppressor cells. Cancer Immunol Res (2017) 5:3–08. doi: 10.1158/2326-6066.CIR-16-0297
284. Talmadge JE, Gabrilovich DI. History of myeloid-derived suppressor cells. Nat Rev Cancer (2013) 13(10):739–52. doi: 10.1038/nrc3581
285. Ugel S, De Sanctis F, Mandruzzato S, Bronte V. Tumor-induced myeloid deviation: when myeloid-derived suppressor cells meet tumor-associated macrophages. J Clin Invest (2015) 125:3365–76. doi: 10.1172/JCI80006
286. Qian B, Li J, Zhang H, Kitamura T, Zhang J, Campion LR, et al. CCL2 recruits inflammatory monocytes to facilitate breast-tumour metastasis. Nature (2011) 475:222–25. doi: 10.1038/nature10138
287. Yang X, Lin Y, Shi Y, Li B, Liu W, Yin W, et al. FAP promotes immunosuppression by cancer-associated fibroblasts in the tumor microenvironment via STAT3-CCL2 signaling. Cancer Res (2016) 76:4124–35. doi: 10.1158/0008-5472.CAN-15-2973
288. Xiang H, Ramil CP, Hai J, Zhang C, Wang H, Watkins AA, et al. Cancer-associated fibroblasts promote immunosuppression by inducing ROS-generating monocytic MDSCs in lung squamous cell carcinoma. Cancer Immunol Res (2020) 8:436–50. doi: 10.1158/2326-6066.CIR-19-0507
289. Deng Y, Cheng J, Fu B, Liu W, Chen G, Zhang Q, et al. Hepatic carcinoma-associated fibroblasts enhance immune suppression by facilitating the generation of myeloid-derived suppressor cells. Oncogene (2017) 36:1090–101. doi: 10.1038/onc.2016.273
290. Ohshio Y, Hanaoka J, Kontani K, Teramoto K. Tranilast inhibits the function of cancer-associated fibroblasts responsible for the induction of immune suppressor cell types. Scand J Immunol (2014) 80:408–16. doi: 10.1111/sji.12242
291. Kumar V, Donthireddy L, Marvel D, Condamine T, Wang F, Lavilla-Alonso S, et al. Cancer-associated fibroblasts neutralize the anti-tumor effect of CSF1 receptor blockade by inducing PMN-MDSC infiltration of tumors. Cancer Cell (2017) 32:654–68. doi: 10.1016/j.ccell.2017.10.005
292. Levental KR, Yu H, Kass L, Lakins JN, Egeblad M, Erler JT, et al. Matrix crosslinking forces tumor progression by enhancing integrin signaling. Cell (2009) 139:891–906. doi: 10.1016/j.cell.2009.10.027
293. Zaman MH, Trapani LM, Sieminski AL, Mackellar D, Gong H, Kamm RD, et al. Migration of tumor cells in 3D matrices is governed by matrix stiffness along with cell-matrix adhesion and proteolysis. Proc Natl Acad Sci USA (2006) 103:10889–94. doi: 10.1073/pnas.0604460103
294. Dumont N, Liu B, Defilippis RA, Chang H, Rabban JT, Karnezis AN, et al. Breast fibroblasts modulate early dissemination, tumorigenesis, and metastasis through alteration of extracellular matrix characteristics. Neoplasia (2013) 15:249–62. doi: 10.1593/neo.121950
295. Lu P, Weaver VM, Werb Z. The extracellular matrix: a dynamic niche in cancer progression. J Cell Biol (2012) 196:395–406. doi: 10.1083/jcb.201102147
296. Sangaletti S, Chiodoni C, Tripodo C, Colombo MP. The good and bad of targeting cancer-associated extracellular matrix. Curr Opin Pharmacol (2017) 35:75–82. doi: 10.1016/j.coph.2017.06.003
297. Joyce JA, Fearon DT. T Cell exclusion, immune privilege, and the tumor microenvironment. Science (2015) 348:74–80. doi: 10.1126/science.aaa6204
298. Sorokin L. The impact of the extracellular matrix on inflammation. Nat Rev Immunol (2010) 10:712–23. doi: 10.1038/nri2852
299. Hartmann N, Giese NA, Giese T, Poschke I, Offringa R, Werner J, et al. Prevailing role of contact guidance in intrastromal T-cell trapping in human pancreatic cancer. Clin Cancer Res (2014) 20:3422–33. doi: 10.1158/1078-0432.CCR-13-2972
300. Salmon H, Donnadieu E. Within tumors, interactions between T cells and tumor cells are impeded by the extracellular matrix. Oncoimmunology (2012) 1:992–94. doi: 10.4161/onci.20239
301. Salmon H, Franciszkiewicz K, Damotte D, Dieu-Nosjean M, Validire P, Trautmann A, et al. Matrix architecture defines the preferential localization and migration of T cells into the stroma of human lung tumors. J Clin Invest (2012) 122:899–910. doi: 10.1172/JCI45817
302. Jiang H, Hegde S, Knolhoff BL, Zhu Y, Herndon JM, Meyer MA, et al. Targeting focal adhesion kinase renders pancreatic cancers responsive to checkpoint immunotherapy. Nat Med (2016) 22:851–60. doi: 10.1038/nm.4123
303. Jiang H, Hegde S, DeNardo DG. Tumor-associated fibrosis as a regulator of tumor immunity and response to immunotherapy. Cancer Immunol Immunother CII (2017) 66:1037–48. doi: 10.1007/s00262-017-2003-1
304. Moon J, Welch TP, Gonzalez FJ, Copple BL. Reduced liver fibrosis in hypoxia-inducible factor-1alpha-deficient mice. Am J Physiol Gastrointest Liver Physiol (2009) 296:G582–92. doi: 10.1152/ajpgi.90368.2008
305. Higgins DF, Kimura K, Bernhardt WM, Shrimanker N, Akai Y, Hohenstein B, et al. Hypoxia promotes fibrogenesis in vivo via HIF-1 stimulation of epithelial-to-mesenchymal transition. J Clin Invest (2007) 117:3810–20. doi: 10.1172/JCI30487
306. Orimo A, Gupta PB, Sgroi DC, Arenzana-Seisdedos F, Delaunay T, Naeem R, et al. Stromal fibroblasts present in invasive human breast carcinomas promote tumor growth and angiogenesis through elevated SDF-1/CXCL12 secretion. Cell (2005) 121:335–48. doi: 10.1016/j.cell.2005.02.034
307. Chouaib S, Noman MZ, Kosmatopoulos K, Curran MA. Hypoxic stress: obstacles and opportunities for innovative immunotherapy of cancer. Oncogene (2017) 36:439–45. doi: 10.1038/onc.2016.225
308. Jiang C, Kim J, Li F, Qu A, Gavrilova O, Shah YM, et al. Hypoxia-inducible factor 1α regulates a SOCS3-STAT3-adiponectin signal transduction pathway in adipocytes. J Biol Chem (2013) 288:3844–57. doi: 10.1074/jbc.M112.426338
309. Sitkovsky MV, Kjaergaard J, Lukashev D, Ohta A. Hypoxia-adenosinergic immunosuppression: tumor protection by T regulatory cells and cancerous tissue hypoxia. Clin Cancer Res (2008) 14:5947–52. doi: 10.1158/1078-0432.CCR-08-0229
310. Hasmim M, Messai Y, Ziani L, Thiery J, Bouhris J, Noman MZ, et al. Critical role of tumor microenvironment in shaping NK cell functions: Implication of hypoxic stress. Front Immunol (2015) 6:482. doi: 10.3389/fimmu.2015.00482
311. Kouidhi S, Elgaaied AB, Chouaib S. Impact of metabolism on T-cell differentiation and function and cross talk with tumor microenvironment. Front Immunol (2017) 8:270. doi: 10.3389/fimmu.2017.00270
312. Noman MZ, Hasmim M, Messai Y, Terry S, Kieda C, Janji B, et al. Hypoxia: a key player in antitumor immune response. a review in the theme: Cellular responses to hypoxia. Am J Physiol Cell Physiol (2015) 309:C569–79. doi: 10.1152/ajpcell.00207.2015
313. Jiang G, Xu W, Du J, Zhang K, Zhang Q, Wang X, et al. The application of the fibroblast activation protein α-targeted immunotherapy strategy. Oncotarget (2016) 7:33472–82. doi: 10.18632/oncotarget.8098
314. Kraman M, Bambrough PJ, Arnold JN, Roberts EW, Magiera L, Jones JO, et al. Suppression of antitumor immunity by stromal cells expressing fibroblast activation protein-alpha. Science (2010) 330:827–30. doi: 10.1126/science.1195300
315. Gunderson AJ, Yamazaki T, McCarty K, Phillips M, Alice A, Bambina S, et al. Blockade of fibroblast activation protein in combination with radiation treatment in murine models of pancreatic adenocarcinoma. PloS One (2019) 14:e211117. doi: 10.1371/journal.pone.0211117
316. Loeffler M, Krüger JA, Niethammer AG, Reisfeld RA. Targeting tumor-associated fibroblasts improves cancer chemotherapy by increasing intratumoral drug uptake. J Clin Invest (2006) 116:1955–62. doi: 10.1172/JCI26532
317. Wang LS, Lo A, Scholler J, Sun J, Majumdar RS, Kapoor V, et al. Targeting fibroblast activation protein in tumor stroma with chimeric antigen receptor T cells can inhibit tumor growth and augment host immunity without severe toxicity. Cancer Immunol Res (2014) 2(2):154–66. doi: 10.1158/2326-6066.CIR-13-0027
318. Zhang Y, Ertl HCJ. Depletion of FAP+ cells reduces immunosuppressive cells and improves metabolism and functions CD8+T cells within tumors. Oncotarget (2016) 7:23282–99. doi: 10.18632/oncotarget.7818
319. Lo A, Wang LS, Scholler J, Monslow J, Avery D, Newick K, et al. Tumor-promoting desmoplasia is disrupted by depleting FAP-expressing stromal cells. Cancer Res (2015) 75:2800–10. doi: 10.1158/0008-5472.CAN-14-3041
320. Schuberth PC, Hagedorn C, Jensen SM, Gulati P, van den Broek M, Mischo A, et al. Treatment of malignant pleural mesothelioma by fibroblast activation protein-specific re-directed T cells. J Transl Med (2013) 11:187. doi: 10.1186/1479-5876-11-187
321. Kakarla S, Chow KKH, Mata M, Shaffer DR, Song X, Wu M, et al. Antitumor effects of chimeric receptor engineered human T cells directed to tumor stroma. Mol Ther (2013) 21:1611–20. doi: 10.1038/mt.2013.110
322. Tran E, Chinnasamy D, Yu Z, Morgan RA, Lee CR, Restifo NP, et al. Immune targeting of fibroblast activation protein triggers recognition of multipotent bone marrow stromal cells and cachexia. J Exp Med (2013) 210:1125–35. doi: 10.1084/jem.20130110
323. Roberts EW, Deonarine A, Jones JO, Denton AE, Feig C, Lyons SK, et al. Depletion of stromal cells expressing fibroblast activation protein-α from skeletal muscle and bone marrow results in cachexia and anemia. J Exp Med (2013) 210:1137–51. doi: 10.1084/jem.20122344
324. Mao X, Xu J, Wang W, Liang C, Hua J, Liu J, et al. Crosstalk between cancer-associated fibroblasts and immune cells in the tumor microenvironment: new findings and future perspectives. Mol Cancer (2021) 20:131. doi: 10.1186/s12943-021-01428-1
325. Sherman MH, Yu RT, Engle DD, Ding N, Atkins AR, Tiriac H, et al. Vitamin d receptor-mediated stromal reprogramming suppresses pancreatitis and enhances pancreatic cancer therapy. Cell (2014) 159:80–93. doi: 10.1016/j.cell.2014.08.007
326. Ferrer-Mayorga G, Gómez-López G, Barbáchano A, Fernández-Barral A, Peña C, Pisano DG, et al. Vitamin d receptor expression and associated gene signature in tumour stromal fibroblasts predict clinical outcome in colorectal cancer. Gut (2017) 66:1449–62. doi: 10.1136/gutjnl-2015-310977
327. Froeling FEM, Feig C, Chelala C, Dobson R, Mein CE, Tuveson DA, et al. Retinoic acid-induced pancreatic stellate cell quiescence reduces paracrine wnt-β-catenin signaling to slow tumor progression. Gastroenterology (2011) 141:1486–97. doi: 10.1053/j.gastro.2011.06.047
328. Ishihara S, Inman DR, Li W, Ponik SM, Keely PJ. Mechano-signal transduction in mesenchymal stem cells induces prosaposin secretion to drive the proliferation of breast cancer cells. Cancer Res (2017) 77:6179–89. doi: 10.1158/0008-5472.CAN-17-0569
329. Kobayashi H, Enomoto A, Woods SL, Burt AD, Takahashi M, Worthley DL. Cancer-associated fibroblasts in gastrointestinal cancer. Nat Rev Gastroenterol Hepatol (2019) 16:282–95. doi: 10.1038/s41575-019-0115-0
330. Feig C, Jones JO, Kraman M, Wells RJB, Deonarine A, Chan DS, et al. Targeting CXCL12 from FAP-expressing carcinoma-associated fibroblasts synergizes with anti-PD-L1 immunotherapy in pancreatic cancer. Proc Natl Acad Sci USA (2013) 110:20212–17. doi: 10.1073/pnas.1320318110
331. Mariathasan S, Turley SJ, Nickles D, Castiglioni A, Yuen K, Wang Y, et al. TGFβ attenuates tumour response to PD-L1 blockade by contributing to exclusion of T cells. Nature (2018) 554:544–8. doi: 10.1038/nature25501
332. Wei Y, Kim TJ, Peng DH, Duan D, Gibbons DL, Yamauchi M, et al. Fibroblast-specific inhibition of TGF-β1 signaling attenuates lung and tumor fibrosis. J Clin Invest (2017) 127:3675–88. doi: 10.1172/JCI94624
333. Dijkgraaf EM, Santegoets SJAM, Reyners AKL, Goedemans R, Wouters MCA, Kenter GG, et al. A phase I trial combining carboplatin/doxorubicin with tocilizumab, an anti-IL-6R monoclonal antibody, and interferon-α2b in patients with recurrent epithelial ovarian cancer. Ann Oncol (2015) 26(10):2141–9. doi: 10.1093/annonc/mdv309
334. Wang C, Gu X, Zhang X, Zhou M, Chen Y. Development and validation of an E2F-related gene signature to predict prognosis of patients with lung squamous cell carcinoma. Front Oncol (2021) 11:756096. doi: 10.3389/fonc.2021.756096
335. Guan J, Zhang H, Wen Z, Gu Y, Cheng Y, Sun Y, et al. Retinoic acid inhibits pancreatic cancer cell migration and EMT through the downregulation of IL-6 in cancer associated fibroblast cells. Cancer Lett (2014) 345:132–9. doi: 10.1016/j.canlet.2013.12.006
Keywords: Tumor microenvironment, cancer-associated fibroblasts, immune cells, metabolic reprogramming, immunotherapy
Citation: Zhu Y, Li X, Wang L, Hong X and Yang J (2022) Metabolic reprogramming and crosstalk of cancer-related fibroblasts and immune cells in the tumor microenvironment. Front. Endocrinol. 13:988295. doi: 10.3389/fendo.2022.988295
Received: 07 July 2022; Accepted: 25 July 2022;
Published: 15 August 2022.
Edited by:
Huashan Shi, Sichuan University, ChinaReviewed by:
Yaying Sun, Fudan University, ChinaCopyright © 2022 Zhu, Li, Wang, Hong and Yang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jie Yang, eWFuZ2ppZV9ob3NAMTYzLmNvbQ==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.