 Dalia Ali
Dalia Ali Michaela Tencerova
Michaela Tencerova Florence Figeac
Florence Figeac Moustapha Kassem1
Moustapha Kassem1 Abbas Jafari
Abbas Jafari- 1Department of Molecular Endocrinology, KMEB, University of Southern Denmark and Odense University Hospital, Odense, Denmark
- 2Laboratory of Molecular Physiology of Bone, Institute of Physiology of the Czech Academy of Sciences, Prague, Czechia
- 3Department of Cellular and Molecular Medicine, University of Copenhagen, Copenhagen, Denmark
Osteoporosis is defined as a systemic skeletal disease characterized by decreased bone mass and micro-architectural deterioration leading to increased fracture risk. Osteoporosis incidence increases with age in both post-menopausal women and aging men. Among other important contributing factors to bone fragility observed in osteoporosis, that also affect the elderly population, are metabolic disturbances observed in obesity and Type 2 Diabetes (T2D). These metabolic complications are associated with impaired bone homeostasis and a higher fracture risk. Expansion of the Bone Marrow Adipose Tissue (BMAT), at the expense of decreased bone formation, is thought to be one of the key pathogenic mechanisms underlying osteoporosis and bone fragility in obesity and T2D. Our review provides a summary of mechanisms behind increased Bone Marrow Adiposity (BMA) during aging and highlights the pre-clinical and clinical studies connecting obesity and T2D, to BMA and bone fragility in aging osteoporotic women and men.
Osteoporosis
Osteoporosis is a chronic-skeletal disorder characterized by low bone mass, and deterioration in the microarchitecture of the bone tissue that results in bone fragility and increased fracture risk (1). It is the most common bone disease worldwide, affecting more than 200 million people and causing more than 9 million fractures in the year 2000 (2). In 2016, a European study estimated that 6.6% of men and 22.1% of women over the age of 50 years were diagnosed with osteoporosis, and around 3.5 million with fragility fractures. Osteoporosis causes vertebral and hip fractures, as well as chronic pain, that in many cases, are associated with disability and reduced life quality. Fractures, due to osteoporosis, require hospitalization and increase risk of mortality by 20%, and in 50% of the cases, result in chronic disability (3). The World Health Organization (WHO) has defined osteoporosis as a disease of low bone mass as measured by Dual-Energy X-ray Absorptiometry (DEXA) with Bone Mineral Density (BMD) equal or less than -2.5 standard deviations of the average value for young healthy persons (known as (T-score≤-2.5) (4, 5).
Pathophysiology of bone loss in age-related osteoporosis
Osteoporosis is primarily attributed to aging and sex-steroid deficiency, which at the cellular level leads to increased bone resorption by osteoclasts and decreased bone formation by osteoblasts (6, 7). A number of underlying pathogenic mechanisms of osteoporosis have been proposed, such as estrogen deficiency-associated Bone Marrow (BM) inflammation (8) or decreased levels of anabolic hormones (9, 10). However, increasing evidence has suggested a possible role of Bone Marrow Adiposity (BMA) in pathogenesis of bone loss in osteoporosis.
BMA and bone loss in age-related osteoporosis
Aging is defined as a gradual loss of normal tissue homeostasis and progressive deterioration of the organ functions, due to accumulation of cellular/DNA damage and senescence throughout aging (11). Aging is associated with significant bone loss and structural bone damage, due to accelerated trabecular thinning and disconnection, cortical thinning, and porosity (12), leading to increased fracture risk. Thus, osteoporosis is an exaggerated expression of aging process in the bone tissue (13). One of the key underlying mechanisms of bone loss in age-related osteoporosis is the altered lineage allocation of Bone Marrow Stromal Cells (BMSCs), associated with enhanced differentiation towards adipocyte lineage, leading to expansion of the Bone Marrow Adipose Tissue (BMAT), at the expense of compromised osteoblast differentiation and bone formation (13). Comprehensive characterization of the epigenomic and transcriptional changes associated with osteoblast and adipocyte differentiation of BMSCs has shown that the inverse correlation between fate specification of BMSCs into osteoblast versus adipocyte lineages is regulated by a large and diverse network of transcription factors (14).
Expansion of BMAT is caused by the increase in adipocyte size and/or number and is defined as the proportion of the BM cavity occupied by the adipocytes (15). The expansion of BMAT in aging can lead to space limitation for other cells that are required for normal skeletal homeostasis, such as BMSCs, osteoblastic, or hematopoietic cells (16). In addition, factors that are secreted by Bone Marrow Adipocytes (BMAds) can also contribute to altered skeletal homeostasis after BMAT expansion, such as adipokines, RANKL, immune-regulatory and pro-inflammatory cytokines. Examples of BMAd-secreted factors that can regulate bone cell function include RANKL, adiponectin, leptin, legumain, and chemerin (17–19).
Age-related expansion of BMAT and osteoporotic bone loss are mediated through intrinsic and extrinsic mechanisms, such as cellular senescence within the bone microenvironment or age-related endocrine dysfunction.
Intrinsic mechanisms
Accumulation of senescent cells
Aging is associated with accumulation of senescent cells, due to diverse stress stimuli (e.g. shortening of telomeres, oncogenic or metabolic insults), causing the cell to enter a state of irreversible growth arrest (20). Senescence is also associated with different cellular alterations including changes in chromatin organization, gene expression (e.g. increased expression of cell cycle regulators/tumor suppressors such as p16Ink4a and p53), mitochondrial dysfunction, and resistance to apoptosis, reviewed in (21, 22). In addition, a key feature of senescent cells is development of a distinctive secretome, known as Senescence-Associated Secretory Phenotype (SASP), characterized by secreting high levels of pro-inflammatory cytokines, immune modulators, growth factors, and proteases, that can spread throughout the tissue, and further exacerbate the senescence and tissue dysfunction (23–29).
Murine studies using the SAMP6 model had shown that accelerated senescence is associated with enhanced adipogenesis and inhibited osteoblastogenesis within the bone microenvironment, leading to compromised bone formation and decreased bone mass (30). Analysis of human bone specimens had shown presence of senescence microenvironment in the bone tissue obtained from old individuals compared to young control subjects, evidenced by increased expression of p16Ink4a and p21 (31). Furthermore genetic depletion of senescent cells, using the inducible INK-ATTAC ‘suicide’ transgene that eliminates the p16Ink4a expressing cells, in a murine pre-clinical model of age-related osteoporosis, exhibited anti-resorptive and anabolic impact on bone, leading to decreased BMAT and increased bone mass (32).
Aging is also accompanied with increased levels of Reactive Oxygen Species (ROS) and oxidative stress in the bone microenvironment, leading to impaired osteoblast differentiation and function, and enhanced adipocyte differentiation (33). It was shown that presence of the antioxidant agent Resveratrol in the cultures of human BMSCs obtained from old individuals enhances differentiation and function of osteoblasts and inhibits formation of BMAds (34). In addition, in vivo murine studies using irradiation or physiological aging models, in which increased levels of ROS within the bone microenvironment are associated with expansion of BMAT and compromised skeletal homeostasis, indicated that antioxidant agents Dasatinib and Quercetin reduced BMAT and enhanced bone formation, by decreasing the ROS levels and senescent cells (35). These studies provide strong evidence for use of antioxidants as a promising approach for preventing age-related bone loss and osteoporosis.
Few studies have reported that senescence can be regulated via transcription factors (36, 37). Forkhead box rotein P1 (FOXP1) is involved in transcriptional control of BMSC senescence, and its expression levels had been shown to decline with aging, and inversely correlate with p16Ink4a expression. Mouse genetic studies had shown that conditional depletion of Foxp1 in BMSCs leads to premature aging, bone loss, and increased BMA via Foxp1 interactions with different members of C/EBP family proteins, such as C/EBP β/δ, which are key modulators of adipogenesis (38). Another study in aged mice revealed that senescence is associated with down regulation of expression of osteoblast transcription factors such as Runx2 and Dlx5, leading to impaired osteoblastogenesis and enhanced adipogenesis of BMSCs via up-regulation of adipocyte-specific transcription factor PPAR-γ (39). Furthermore, an in vivo study using murine irradiation model indicated the increased burden of senescence in bone cells from day 1 to day 7 after irradiation (evidenced by increased expression of p21), followed by expansion of BMAT starting at day 7 and continuing until day 42 post irradiation (35). This study also indicated a direct correlation between the increased senescence burden in the bone tissue and up-regulation of mir-27a, which is known to be involved in obesity and regulation of adipose-tissue related transcription factors (40). These studies provide strong evidence for role of senescent microenvironment in expansion of BMAT.
DNA damage
DNA damage can be caused by exogenous factors such as Ionizing Radiation (IR), chemotherapeutic agents, and Ultraviolet (UV) light exposure, as well as endogenous factors such as ROS that are generated by mitochondria in the process of ATP production (41). These factors have been shown to cause impairment of osteoblastogenesis, and enhanced adipogenesis within the bone microenvironment. Pre-clinical and clinical studies have shown that cancer treatment using IR or chemotherapy leads to significant expansion of BMAT, which may contribute to the progressive bone loss in cancer survivors (42–44). In addition, it is shown that aging is associated with an intrinsic defect in osteoblasts, due to accumulation of DNA damage, leading to decreased osteoblast number, compromised osteoblast function, and induction of osteoclast formation (45). This study employed young (6 months) and old (20-24months) Osx1-Cre; TdRFP-mice and showed that the number of TdRFP-Osx1 cells are decreased by 50% in the BM of old male and female mice as compared to young mice. The TdRFP-Osx1 cells obtained from old mice also exhibited increased levels of DNA damage and senescence markers, such as formation of γ-H2AX foci, phosphorylation of p53, and G1 cell cycle arrest. BMSCs obtained from old mice also exhibited increased expression of SASP markers, leading to increased osteoclast formation, as well as increased expression of the adipogenic transcription factor PPAR-γ (45).
Murine studies using radiation-induced osteoporosis had indicated that accumulation of DNA damage leads to significant increase in the percentage of adipocytes within the BM. In addition, proteosome inhibitors or sclerostin neutralizing antibody reduced the BMA and prevented the trabecular bone structural deterioration post-radiation, via induction of DNA-repair (at least partially) (46, 47).
Although these studies provide evidence for the role of DNA damage in expansion of BMAT, the exact underlying mechanisms are not fully understood. However, induction of senescence (35) and the altered expression of genes involved in regulating the fate specification of BMSCs toward osteoblast and adipocyte lineages are among possible mechanisms. In addition, these studies suggest that mitigation of DNA damage can be employed as an approach for reducing BMA and improving the bone architecture post radiation.
Extrinsic mechanisms involved in age-related bone loss and BMAT expansion
Extrinsic factors such as disrupted hormonal status, malnutrition, and reduced physical activity, have an important role in disturbed skeletal homeostasis in aging, leading to inhibited bone formation and enhanced bone resorption (48).
Endocrine Aging
Bone growth and maintenance of the skeleton is regulated via different endocrine factors (hormones) such as estrogens and androgens (49, 50). Fuller Albright proposed 70 years ago, that osteoporosis in women was caused by estrogen deficiency (51) and that bone loss induced via ovariectomy in women can be reversed by estrogen therapy (52). Estrogen is involved in molecular signaling in bone and regulation of BMAds (53–55) as estrogen is a positive regulator of osteogenesis through activation of estrogen receptor (ER)-dependent cytoplasmic kinases, BMPs and Wnt signaling (56, 57).
Testosterone exhibits an anabolic impact on bone formation in the periosteal surface in mice (58), and that oral administration of testosterone reduces the accelerated bone loss in orchiectomized mice (59). Gradual reduction in the testosterone levels in aging men are associated with reduction in bone mass (60, 61), and osteoblast dysfunction (62). In a clinical study of 350 men between the ages of 20-90 years, the levels of bioavailable testosterone were decreased by 64%, and bioavailable estrogen by 47% in old versus young individuals. This study also indicated that age-related bone loss in men is associated with decline in testosterone as well as estrogen (63). Another human study investigated the impact of endogenous estrogen and testosterone production on bone, using 59 elderly men and demonstrated that estrogen is the dominant sex-steroid hormone protecting men from age-related bone resorption, whereas both hormones of estrogen and testosterone are critical for bone formation (64).
Several mechanisms have been proposed to be involved in mediating the direct/indirect roles of estrogen deficiency in accelerated bone loss. Estrogen deficiency is involved in increased expression of pro-inflammatory cytokines in the bone microenvironment such as TNFα, IL1, IL-6 and receptor activator of RANKL/OPG/RANK system that regulates osteoclast differentiation (65–67). Estrogen deficiency also leads to decreased production of endogenous antioxidants (68, 69), which together with the elevated ROS production and the senescence microenvironment observed during aging or in obese mice and human, can lead to significantly increased levels of oxidative stress in the bone microenvironment (70, 71).
Studies using ovariectomized rats and postmenopausal women revealed increased bone turnover in response to estrogen deficiency, i.e. increased bone resorption and bone formation, manifested by increased number of osteoblast precursors, osteoblast proliferation and increased osteoblast number (72, 73). However, the increased bone formation levels are not enough to account for the bone loss due to elevated bone resorption, thereby leading to bone loss.
An interesting question that has been raised is whether the biological sex has an impact on the expansion of BMAT during aging? Griffith et al, investigated BMD and BMAT in the lumbar spine of 259 healthy subjects (145 females, 114 males; age range: 62-90 years) using MR spectroscopy of L3 vertebral body, and revealed that in males, BMAT increases gradually throughout life, whereas in females, BMAT increased between 55 and 65 years (74). BMAT in the vertebral bones in females with the age of more than 60 years increased by nearly 10% higher compared to age-matched males, indicating the positive impact of estrogen deficiency on BMAT expansion. In addition, human studies have shown that aging and loss of steroid hormones make women more vulnerable to the negative impacts of BMAT on loss of trabecular bone at the spine and femoral neck, and greater loss of spine strength (75), while in men vertebral BMAT is significantly increased with osteoporosis (76). Table 1 provides a summary of several clinical and preclinical studies related to association of endocrine aging and BMAT.

Table 1 Summary of several clinical and preclinical studies related to investigating the association of endocrine aging and BMAT expansion .
Another question that has been raised recently is whether gender-affirming interventions have an impact on bone microstructure and BMA?
Gender-affirming interventions using hormone therapy or surgery aim to align the physical characteristics with an individual’s gender identity (85). Bretherton et al. recently employed high-resolution peripheral quantitative computed tomography of the distal radial and tibial microarchitecture and showed that trans men have normal bone microarchitecture as compared to cis female controls, whereas trans women had deteriorated bone microarchitecture compared to cis male controls (86). In addition, Nasomyont et al. showed lower bone mass acquisition and greater increases in BMAT indices in Transgender and Gender Non-Conforming (TGNC) youth after 12 months of pubertal suppression with gonadotropin-releasing hormone agonists (87). Additional investigations are required to establish the impacts of gender-affirming interventions on bone and BMA, and the related underlying mechanisms, also in the context of aging, obesity, and diabetes.
Obesity and diabetes
Bone fragility and microstructural changes in BM associated with T2D
Aging, osteoporosis and metabolic diseases such as obesity and diabetes robustly affect microstructural changes in the BM and contribute to impairment of bone homeostasis, which leads to higher risk of bone fractures (88, 89). The altered cellular landscape and molecular networks within the bone microenvironment in obesity and diabetes induce changes in trabecular and cortical bone volume or increased amount of BMAT, leading to a detrimental effect on bone quality and strength.
Osteoporosis and aging are characterized with decreased BMD, along with increased BMAT and accompanied by increased resorption activity in the BM in mice and humans (90, 91). Metabolic complications associated with impairment of glucose homeostasis have been shown to contribute to higher risk of bone fractures and accelerate the manifestation of bone fragility and osteoporosis. Previous studies in mice and humans under High Fat Diet (HFD) condition have reported increased BMAT formation evaluated by measurement of BMAT volume, BMAd size and number (70, 92–98), which was correlated with increased, unchanged, or decreased BMD in mice depending on the employed diabetic model (T1D or T2D) (70, 71, 94, 95, 99), but associated with a higher risk of fractures. Therefore, recent studies have focused on the design of experiments to include measurement of BMAT parameters in relation to bone fragility. Indeed, our recent study using T2D diabetic animal model (HFD in combination with streptozotocin) confirmed the expansion of BMAT in T2D mice along with decreased bone volume, which contributed to the delayed bone healing in monocortical fracture model (100).
Interestingly in humans, obesity is associated with increased BMD in children and adults (89, 101, 102). Along with this finding, several clinical studies including ours have noted reduced bone turnover in patients with metabolic diseases (71, 103). Paradoxically, there are also clinical studies suggesting that women and men with high BMI and T2D may be protected from osteoporosis, due to increased BMD (104–109). These clinical studies are summarized in Table 2. The mechanisms underlying the higher bone mass but reduced bone turnover in patients with obesity and T2D were partially explained by our clinical study in obese subjects that revealed hypermetabolic status of BMSCs and accelerated senescent BM microenvironment contributing to the bone fragility (71). These changes lead to impairment of the bone material properties and increased cortical porosity in T2D (126). However, further follow up studies are needed to collect more clinical data on bone microstructural parameters along with BMAT volume in relation to bone quality in specific target groups of patients, to better predict the fracture risk.

Table 2 Clinical studies investigating effect of obesity and diabetes on bone in men & women.
Human studies had shown that expansion of visceral fat is associated with enhanced adipocyte formation in the BM microenvironment in osteoporotic obese women (97). Furthermore, BMAT was shown to be expanded in the bone biopsies from overweight and obese subjects compared to healthy age-matched individuals (127). Similarly, BMSCs obtained from obese men exhibited enhanced adipocyte differentiation and accelerated senescence phenotype that would contribute to the skeletal fragility in obesity (71). T2D is another risk factor in skeletal fragility and osteoporosis in aging men and postmenopausal women (128–130), despite the increase in BMD or independent of BMD (131, 132). The combination of obesity and T2D were shown to exhibit higher serum insulin levels and BMA at the lumbar spine and femoral metaphysis compared to the subjects without T2D. In addition, it is shown that lumbar spine BMD is inversely associated with the lumbar adiposity in adults with morbid obesity, and that morbid obesity and T2D exhibits a higher BMA than non-diabetic controls of similar weight (98). In agreement with these observations, Sheu et al. reported that in 38 old diabetic men there was a higher vertebral BMA, spine and hip BMD and a higher fracture risk when compared to control men without diabetes (133).
Fazeli et al. examined how human BMAT responds to acute nutrient changes by employing 10 days of high calorie protocol followed by 10 days fasting protocol. This study indicated a significant increase in vertebral BMAT after high calorie feeding and fasting, and that high calorie feeding up-regulated the inflammatory marker TNFα in BMAds, which was decreased upon fasting (134).
Kim et al., reported that BMAT levels were reduced after bariatric surgery in obese T2D patients compared to the obese non-diabetic individuals, and that there was an inverse correlation between the changes in BMAT in lumbar spine and femoral neck and spine BMD (135). In addition, 6 weeks of low calorie diet was reported to induce weight loss in obese T2D subjects (19 women and 10 men) together with decrease in vertebral BMA (136).
Pre-clinical studies using murine models have also provided evidence for impact of obesity and diabetes on BMAT and bone mass. HFD-induced-obesity using C57Bl/6J male mice, was reported to lead to enhanced adipocyte differentiation of BMSCs, associated with increased in vivo BMAT volume and decreased trabecular and cortical bone mass (70). Another in vivo study in male C57Bl/6J mice reported that HFD-induced obesity and T2D compromised the skeletal macro- and micro-architecture of the bone (137). In addition, increased bone resorption is also shown to contribute to the reduced bone mass and strength in a pre-clinical obese-diabetic mouse model (138).
Transition of metabolic status from obesity to insulin resistance and T2D is accompanied with changes in glucose, insulin and circulating lipid levels, as well as changes in inflammatory pathways and hormonal alterations. The relative contribution of each of these factors to the expansion of BMAT, compromised bone quality, and skeletal fragility remain unclear. In addition, the length of exposure to obesogenic and diabetic conditions has an impact on the effect of these conditions on the bone microstructure in mice and humans. Therefore, it is important to include more longitudinal studies to investigate the changes of bone and BMAT parameters measured at several timepoints, in order to better understand the impact of different elements of obesity and T2D on the bone quality and strength and to determine the parameters that can be employed for predicting the risk of fracture.
Role of BMSC dysfunction and BMAds in pathogenesis of increased bone fragility in obesity and T2D
Although different mechanisms have been proposed to be involved in pathogenesis of bone fragility in obesity and T2D, the underlying cellular and molecular events are not yet fully understood. Impaired BMSC function and differentiation is one of the key mechanisms that is suggested to have role in expansion of BMAT and decreased bone mass and quality in obesity and T2D, thereby leading to increased skeletal fragility during aging.
Increased levels of oxidative stress and senescence within the bone microenvironment in obesity and T2D are among factors that can contribute to BMSC dysfunction, and a shift in BMSC differentiation phenotype, favoring adipogenesis than osteogenesis (139–142).
BMSCs from obese subjects are shown to exhibit a hypermetabolic state and a shift of molecular phenotype towards adipocyte progenitors, together with altered expression of genes involved in metabolic regulation, such as glycolysis and oxidoreductase activity. The hyper-metabolic state of BMSCs in obesity is associated with increased abundance of leptin receptor and insulin receptor positive cells and an accelerated senescence phenotype and increased ROS production (71). Leptin and leptin receptor signaling promotes adipogenesis in the BM, in response to HFD (143). In addition, insulin receptor signaling is reported to be increased under oxidative stress condition, associated with cellular senescence phenotype (144).
The above-mentioned studies provide evidence for the role of cell autonomous defects in BMSCs in pathogenesis of bone fragility in obesity and T2D. However, Devlin et al. employed the male TALLYHO/JngJ murine model of T2D and indicated that in the context of early onset T2D, impaired bone formation and the consequent skeletal deficits are due to the altered bone microenvironment, but not the cell autonomous defects in BMSCs (145).
Increasing evidence has shown that BMAds may also play an important role in altered bone microenvironment in obesity and T2D (146–148). BMAds secrete a number of adipokines and cytokines that affect bone cell functions directly or indirectly, e.g. through modulation of inflammation within the bone microenvironment (149).
IL-6 is one of the cytokines that is secreted by BMAds and increases osteoclast formation and bone resorption, thereby leading to decreased BMD in men and women (150, 151). IL-6 enhances osteoclastogenesis by stimulating the expression of RANKL on stromal/osteoblastic cells (152), and also by direct support of osteoclast formation through RANKL-independent mechanisms (153).In addition, Li et al. revealed that IL-6 KO mice are protected against HFD-induced trabecular bone loss, and the associated BMSC senescence and BMAT expansion (154). BMAd-derived factors that can affect skeletal homeostasis are reviewed by Aaron et al. (155).
Can the deleterious effects of obesity and T2D on the skeleton be accelerated by postmenopausal estrogen deficiency?
In post-menopausal women, obesity is another accompanying complication besides estrogen deficiency that affects energy homeostasis and metabolism of bone and adipose tissue (156). Menopause is associated with shift in BMSCs phenotype towards adipogenesis, reduced osteogenesis, increase in the osteoporotic bone phenotype and fracture risk (157–160). Possible mechanisms of deleterious effects of estrogen deficiency on bone metabolism is discussed above (Endocrine aging and Table 1). Combination of estrogen deficiency and obesity includes a chain of pathologic events, leadingto disturbed skeletal homeostasis that further contribute to the accelerated aging of BMSCs and compromised bone regeneration and increased fracture risk (161). Several clinical studies in postmenopausal women (presented in Table 2) have however shown that obese postmenopausal women exhibit increased BMD, that is paradoxically associated with increased bone fragility and fracture risk, whereas the status of BMA is not evaluated in these studies (116, 162).
Interventions to reduce BMA, enhance bone mass, and reduce bone fragility
To reverse the detrimental effects of metabolic complications on bone fragility, various interventions have been applied including diet, exercise, or pharmacological treatment. However, in many of these studies there is limited information on BMAT changes in relation to bone and energy metabolism, especially in human studies, as the evaluation of BMAT (fat content in BM) is not a common clinical practice.
Nutritional intervention
A “Bone Healthy Diet” should contain the nutritional components that are required for normal skeletal homeostasis, such as calcium, vitamin D, vitamin C, vitamin K, vitamin A, protein, and phytoestrogens, that help enhancing BMD and reduce bone loss (163).
1,25 dihydroxy vitamin D is the active form of vitamin D, and its main function in bone is to modulate osteoblast proliferation, differentiation, and providing suitable microenvironment for bone mineralization. Presence of calcitriol in aging osteoblast cultures revealed enhanced osteoblastic function (164). Different mechanisms of action have been proposed for the effect of vitamin D on osteoblast differentiation and bone formation (165, 166). Many studies have revealed the significance of Vitamin D deficiency in inducing bone loss, osteoporosis, and affecting the function of skeletal muscle. It is shown that vitamin D restricted diet (for 28 days) leads to decreased bone mineral content and muscle mass in ovariectomized rats challenged with HFD (167). Vitamin D deficiency in the elderly people causes secondary hyperparathyroidism and increased risk of hip fracture (168), as low circulating levels of vitamin D increases parathyroid hormone (PTH) levels, which induce bone resorption and bone loss in elderly subjects (169–171). Vitamin D combined with calcium is also used to prevent osteoporosis (172).
Is there a connection between vitamin D and BMA regulation?
Calcitriol has shown anti-adipogenic effects in cultures of 3T3-L1 cells (173, 174) as it inhibits the transactivation capacity of PPARγ, through Vitamin D Receptor (VDR) signaling. VDR could inhibit PPARγ transactivation activity, by competing for binding to their common heterodimer partner RXR (175). In VDR null mice, the BMSCs exhibited high expression of PPARγ, along with higher expression of DKK1 and SFRP2, that are inhibitors of the pro-osteogenic canonical Wnt signaling pathway, and the expression of these Wnt inhibitors was downregulated by calcitriol in wild-type BMSCs, in the absence or presence of adipogenic inducers (176). Similar results were observed in human BMSCs (177).
Anti-adipogenic effect of vitamin D is also observed in femur-derived mouse BMSCs and is mediated through inhibiting the expression of aP2 and adipsin (178). Vitamin D3 supplementation is recommended upon aging (179) and leads to increasing the circulating IGF-1 levels (180). Increasing the levels of circulating IGF-1 may help in counteracting the age associated BMAT expansion (181, 182). Thus, these in vitro and in vivo studies suggest that vitamin D signaling may contribute to the regulation of BMA. However, more clinical investigations are required to directly examine the effect of vitamin D supplementation on BMA in human, and to assess the correlation between vitamin D levels, BMAT volume, and fracture risk.
Lifestyle factors
Creating awareness and understanding of the lifestyle factors that may delay the aging bone phenotype is crucial. One of the most important lifestyle factors is the physical activity (114). Physical activity leads to strengthening of the hip and spine due to skeletal loading and inducing bone formation at the stressed skeletal sites (183).
Styner et al. employed a murine model (4 weeks old female C57BL/6 mice) to investigate the impact of running exercise on the obesity associated BMAT and whether this is associated with increased bone quantity and quality (184). The study showed that BMAT was increased by 44% in diet-induced obesity measured by osmium-µCT, whereas exercise was associated with reduced BMAT (–48%), as well as increased trabecular bone volume (+19%), and higher bone stiffness in obese mice
The anti-diabetic drug, rosiglitazone, which is a PPARγ-agonist, is known to significantly increase BMA and fracture risk. It is shown that physical exercise significantly lowers BMA in rosiglitazone-treated male C57Bl/6J mice (185).
A randomized clinical trial investigated the effect of exercise on BMA in forty patients with chronic non-specific low back pain, and revealed that lumbar vertebral fat fraction was lower post exercise in these patients compared to the baseline (186). Chronic alcohol consumption is another risk factor in osteoporotic-bone loss associated with increased BMA (187). It is shown that 3 months of chronic alcohol consumption in the diet of 4-week-old male Sprague-Dawley rats leads to skeletal abnormalities together with increased BMA.
Pharmacological treatment
Several drugs have been developed to improve bone parameters in metabolic bone diseases. Additional challenge in treatment of bone diseases is now to reduce BMAT volume, which may lead to increased bone mass and strength, together with decreased risk of bone fracture.
Several antidiabetic drugs have been tested in clinical settings for treatment of metabolic bone disease, including insulin-sensitizers such metformin, modified TZD analogs (PPARγ-independent), inhibitors of inflammatory molecules such as DPP4 (dipeptidyl peptidase-4). Ambrosi et al. has reported that DDP4 inhibition (secreted from adipocytes and increased with obesity in bones) in mouse model of obesity improved bone parameters and decreased BMAT (146). Similar results have been demonstrated in OVX female mice treated with DPP4 inhibitor (188). Clinical studies using DDP4 inhibitor, sitagliptin, showed improvement of bone turnover markers and decreased fracture risk in diabetic patients (189, 190).
Studies using metformin treatment in rodents and humans showed positive results or no changes in bone parameters suggesting that metformin might have a positive effect on bone metabolism in diabetic conditions, depending on the other medical complications associated with diabetes and length of disease manifestation. However, BMAT parameters were not evaluated in these conditions (191). Human studies of the Rochester cohort suggest that metformin decreases fracture risk in T2D patients (hazard ratio 0.7) (192). Although the ADOPT studies did not demonstrate beneficial effects of metformin on fracture risk (193), they showed decreased levels of bone resorption marker CTX and, contrary to the animal studies, decreased levels of bone formation marker P1NP (194). In a large case-control study metformin utilization was also associated with a reduction in the risk of fractures (195). In contrast, there are case control studies in which no association was observed between treatment with the insulin-sensitizing drug metformin and incidence of bone fractures in T2D patients (196).
In order to decrease the negative side effects of TZDs, novel TZD analogs with PPARγ independent effect on glucose metabolism have been developed. Stechschulte et al., reported that post-translational modifications of PPARγ at S112 and S273, which influence PPARγ pro-adipocytic and insulin sensitizing activities, improved bone parameters and decreased BMAT in lean and obese mice (197). Blocking PPARγ only at S273 by SR10171 had a beneficial effect on trabecular and cortical bone while maintaining its metabolic effect on glucose metabolism.
Another TZD analogue, with PPARγ independent affinity, MSDC-0602 has been reported to have a beneficial effect on bone parameters by decreasing osteoclast activation and BMAT formation in lean aged mice, while keeping its insulin sensitizing features (198). This drug is in clinical trial 2b (199).
Another category of pharmacological treatment used as anti-aging drugs has been tested to improve bone parameters and decreased bone fragility (e.g. resveratrol). Our recent study showed that resveratrol has anti-adipogenic effect along with decreased senescence in primary BMSCs isolated from patients with osteoporosis (34), which was also confirmed in animal studies with OVX rats (200, 201). However, the clinical relevance of these findings has not yet been investigated.
Sclerostin (SOST), osteocyte-secreted biomolecule with a negative impact on bone homeostasis (as it is an inhibitor of Wnt signaling), has become a potential drug target for anabolic treatment of diverse bone loss states (202). SOST circulating levels are increased with obesity, aging, and osteoporosis (203). Thus, neutralizing antibodies against sclerostin (SostAb) have been developed for testing its potential in clinical use to decrease bone fragility in osteoporosis and obesity. Using SostAb in animals showed improvement in fracture healing in diabetic mice (204) and OVX rats (205, 206). SostAb treatment in T2D rat models has also been shown to improve bone mass and strength (207). Clinical trials with SostAb have shown promising results with improvement of bone density and bone strength in postmenopausal women (208–210) Therefore, SostAb seems to be a promising osteoanabolic drug for treatment of skeletal fragility observed in osteoporosis, obesity and T2D (211). However, these clinical studies did not evaluate the effects of SostAb on BMAT. Recent studies in animal models suggest that SostAb may revert negative effect of rosiglitazone-induced increased BMAT in BM (212) and thus confirms its impact on adipogenesis of BMSCs (213).
Antiresorptive treatments
Several antiresorptive agents including bisphosphonates, or RANKL antagonist, denosumab have been proven to safely reduce fracture risk in various high-risk populations (214, 215). They are recommended as first-line therapy for patients with osteoporosis. However, using antiresorptive drugs in treatment of osteoporosis in T2D patients might not be the perfect choice as patients with metabolic diseases (obesity, insulin resistance or T2D) have decreased bone turnover and their use would just further diminish the bone homeostasis. In addition, a recent clinical study showed that denosumab-treated patients had improved HbA1c similar to the effect of other anti-diabetic drugs, suggesting its possible insulin sensitizing effect (216). Clinical and animal studies have indicated beneficial effect of denosumab on insulin sensitivity, bone formation and muscle strength (217). However, its impact on BMAT volume has not been evaluated yet and more clinical investigations are needed to include this parameter to measure in their outcomes.
Conclusion and future perspectives
Increased accumulation of BMAT in bones has been recognized as a feature of aging bones associated with increased fracture risk. However, endocrine, and metabolic disturbances in organism such as hormone deficiency, obesity, and T2D can accelerate the detrimental changes in bone homeostasis and contribute to the early onset of osteoporosis and microstructural changes that compromise the bone strength (Figure 1.). Several mechanisms underlie these symptoms including transcription factors, DNA damage, ROS production, accumulation of senescent cells in BM microenvironment and secretion of bioactive molecules regulating function of BMSCs directing their fate towards BMAds. Using different animal models of osteoporosis and metabolic disorders, it has been shown that targeting senescent cells and regulation of hormonal levels can modulate the negative impact of expanded BMAT on bone loss fragility. More importantly, recent clinical interventions including patients with a broad range of age and complications reported that lifestyle modifications such as a special diet, caloric restriction, and physical activity may modulate BMAand improve bone parameters. However, more follow up studies are needed to evaluate the changes on bone structure and cellular modifications in real time at different timepoints, to establish the impact of such interventions on bone remodeling. Finally, including more imaging methods to evaluate bone and BMAT parameters in future clinical studies can facilitate further examination of the BMAT and its roles in skeletal fragility in the context of obesity and T2D in aging population, thereby providing novel therapeutic possibilities and also possibly, development of better approaches for estimation of fracture risk.
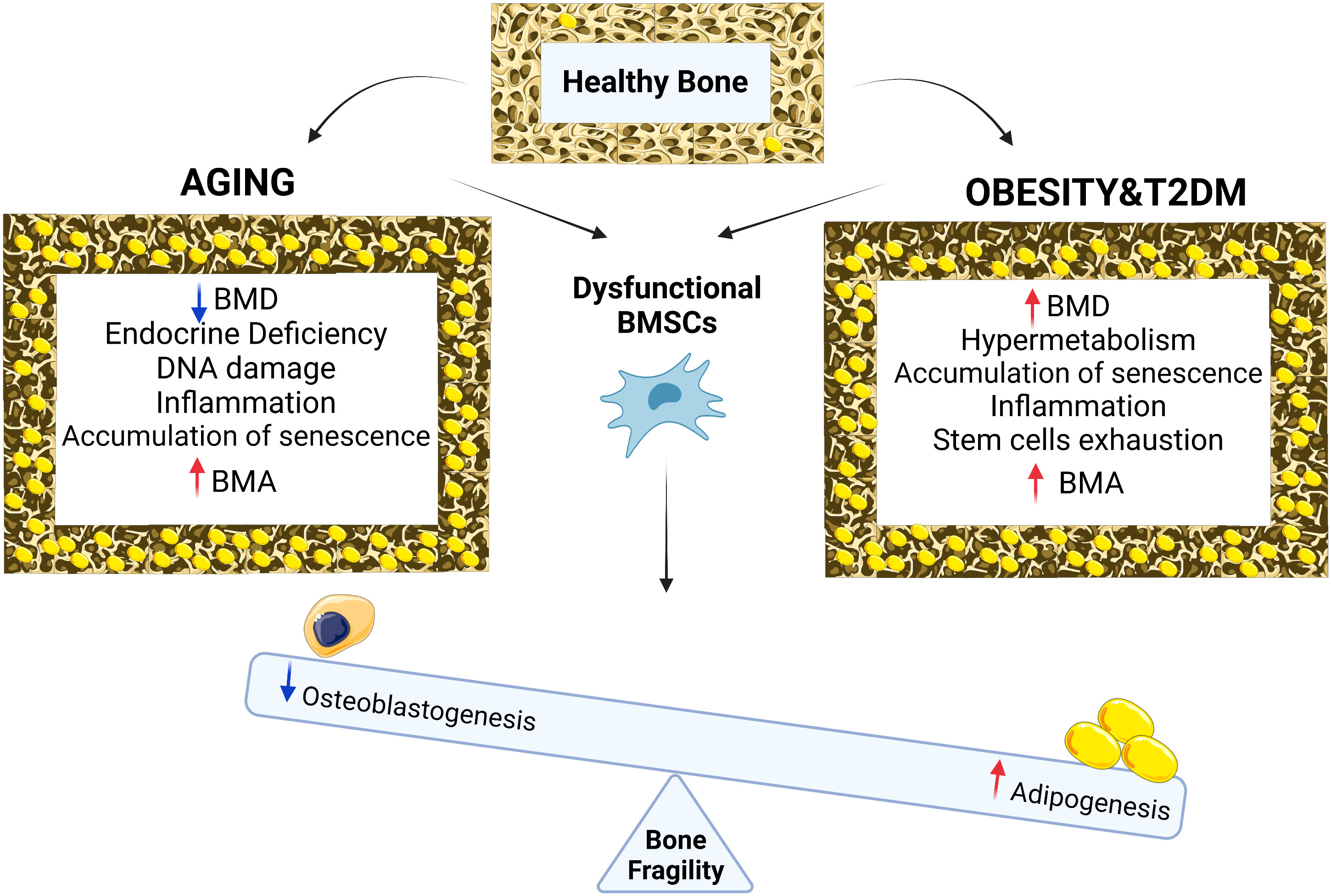
Figure 1 Chronological aging in bone is associated with reduction in BMD, endocrine deficiency, DNA damage, inflammation, accumulation of senescence, BMA and osteoporotic bone phenotype while with metabolic diseases such as obesity and type 2 diabetes, bone phenotype is associated with increased in BMD, cellular hypermetabolism, stem cell exhaustion, accumulation of senescence, inflammation, BMA and osteoporotic bone phenotype. Both conditions (aging vs metabolic diseases of obesity and T2D) result in BMSCs dysfunction leading to differentiation imbalance decreasing osteogenesis, increasing adipogenesis, BMA and bone fragility.
Authors contributions
DA, MT, FF and AJ researched data, wrote and reviewed the manuscript. MK contributed to the conceptual framing of the manuscript. DA and AJ edited the manuscript. All authors contributed to the article and approved the submitted version
Funding
(MT) START UP Research Program from IPHYS, the Czech Science Foundation GACR 20-03586S, GACR 22-12243S, EFSD/NovoNordisk foundation Future leaders award (NNF20SA0066174). (AJ, MK) Olav Thon Foundation (2019).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Abbreviations
BM, Bone Marrow; BMA, Bone marrow adiposity; BMAT, Bone marrow adipose tissue; BMD, Bone mineral density; BMSCS, Bone Marrow Stromal Stem Cells; BMPs, Bone morphogenic proteins; DEXA, Dual energy X-ray absorptiometry; HFD, High fat diet; IGF-1, Insulin like growth factor-1; IL-6, Interleukin 6; OVX, Ovariectomy; P1NP, Procollagen 1Intact N-Terminal Propeptide; PPARγ, Peroxisome proliferation-activated receptor gamma; RANKL, Receptor activator of nuclear factor kappa β ligand; RUNX2, Runt related transcription factor 2; SASP, Senescence associated secretory phenotype markers; TNFα, Tumor necrosis factor alpha.
References
1. Armas LA, Recker RR. Pathophysiology of osteoporosis: New mechanistic insights. Endocrinol Metab Clin North Am (2012) 41:475–86. doi: 10.1016/j.ecl.2012.04.006
2. Johnell O, Kanis JA. An estimate of the worldwide prevalence and disability associated with osteoporotic fractures. Osteoporos Int (2006) 17:1726–33. doi: 10.1007/s00198-006-0172-4
3. Liu J, Curtis EM, Cooper C, Harvey NC. State of the art in osteoporosis risk assessment and treatment. J Endocrinol Invest (2019) 42:1149–64. doi: 10.1007/s40618-019-01041-6
4. Kanis JA. Assessment of fracture risk and its application to screening for postmenopausal osteoporosis: synopsis of a WHO report. WHO Study Group Osteoporos Int (1994) 4:368–81. doi: 10.1007/BF01622200
5. Kanis JA, Melton LJ 3rd, Christiansen C, Johnston CC, Khaltaev N. The diagnosis of osteoporosis. J Bone Miner Res (1994) 9:1137–41. doi: 10.1002/jbmr.5650090802
6. Kassem M, Marie PJ. Senescence-associated intrinsic mechanisms of osteoblast dysfunctions. Aging Cell (2011) 10:191–7. doi: 10.1111/j.1474-9726.2011.00669.x
7. Marie PJ, Kassem M. Osteoblasts in osteoporosis: Past, emerging, and future anabolic targets. Eur J Endocrinol (2011) 165:1–10. doi: 10.1530/EJE-11-0132
8. Charatcharoenwitthaya N, Khosla S, Atkinson EJ, McCready LK, Riggs BL. Effect of blockade of TNF-alpha and interleukin-1 action on bone resorption in early postmenopausal women. J Bone Miner Res (2007) 22:724–9. doi: 10.1359/jbmr.070207
9. Boonen S, Mohan S, Dequeker J, Aerssens J, Vanderschueren D, Verbeke G, et al. Down-regulation of the serum stimulatory components of the insulin-like growth factor (IGF) system (IGF-I, IGF-II, IGF binding protein [BP]-3, and IGFBP-5) in age-related (type II) femoral neck osteoporosis. J Bone Miner Res (1999) 14:2150–8. doi: 10.1359/jbmr.1999.14.12.2150
10. Locatelli V, Bianchi VE. Effect of GH/IGF-1 on Bone Metabolism and Osteoporsosis. Int J Endocrinol (2014) 2014:235060. doi: 10.1155/2014/235060
11. Lopez-Otin C, Blasco MA, Partridge L, Serrano M, Kroemer G. The hallmarks of aging. Cell (2013) 153:1194–217. doi: 10.1016/j.cell.2013.05.039
12. Seeman E. Pathogenesis of bone fragility in women and men. Lancet (2002) 359:1841–50. doi: 10.1016/S0140-6736(02)08706-8
13. Khosla S, Riggs BL. Pathophysiology of age-related bone loss and osteoporosis. Endocrinol Metab Clin North Am (2005) 34:1015–30. doi: 10.1016/j.ecl.2005.07.009
14. Rauch A, Haakonsson AK, Madsen JGS, Larsen M, Forss I, Madsen MR, et al. Osteogenesis depends on commissioning of a network of stem cell transcription factors that act as repressors of adipogenesis. Nat Genet (2019) 51:716–27. doi: 10.1038/s41588-019-0359-1
15. Rozman C, Feliu E, Berga L, Reverter JC, Climent C, Ferran MJ. Age-related variations of fat tissue fraction in normal human bone marrow depend both on size and number of adipocytes: a stereological study. Exp Hematol (1989) 17:34–7.
16. Reinisch A, Etchart N, Thomas D, Hofmann NA, Fruehwirth M, Sinha S, et al. Epigenetic and in vivo comparison of diverse MSC sources reveals an endochondral signature for human hematopoietic niche formation. Blood (2015) 125:249–60. doi: 10.1182/blood-2014-04-572255
17. Han L, Zhang Y, Wan S, Wei Q, Shang W, Huang G, et al. Loss of chemerin triggers bone remodeling in vivo and in vitro. Mol Metab (2021) 53:101322. doi: 10.1016/j.molmet.2021.101322
18. Herrmann M. Marrow Fat-Secreted Factors as Biomarkers for Osteoporosis. Curr Osteoporos Rep (2019) 17:429–37. doi: 10.1007/s11914-019-00550-w
19. Jafari A, Qanie D, Andersen TL, Zhang Y, Chen L, Postert B, et al. Legumain Regulates Differentiation Fate of Human Bone Marrow Stromal Cells and Is Altered in Postmenopausal Osteoporosis. Stem Cell Rep (2017) 8:373–86. doi: 10.1016/j.stemcr.2017.01.003
20. de Magalhães JP, Passos JF. Stress, cell senescence and organismal ageing. Mech Ageing Dev (2018) 170:2–9. doi: 10.1016/j.mad.2017.07.001
21. Roger L, Tomas F, Gire V. Mechanisms and regulation of cellular senescence. Int J Mol Sci (2021) 22. doi: 10.3390/ijms222313173
22. Zhu X, Chen Z, Shen W, Huang G, Sedivy JM, Wang H, et al. Inflammation, epigenetics, and metabolism converge to cell senescence and ageing: the regulation and intervention. Signal Transduct Target Ther (2021) 6:245. doi: 10.1038/s41392-021-00646-9
23. Childs BG, Durik M, Baker DJ, van Deursen JM. Cellular senescence in aging and age-related disease: from mechanisms to therapy. Nat Med (2015) 21:1424–35. doi: 10.1038/nm.4000
24. Coppe JP, Desprez PY, Krtolica A, Campisi J. The senescence-associated secretory phenotype: the dark side of tumor suppression. Annu Rev Pathol (2010) 5:99–118. doi: 10.1146/annurev-pathol-121808-102144
25. Coppe JP, Patil CK, Rodier F, Sun Y, Munoz DP, Goldstein J, et al. Senescence-associated secretory phenotypes reveal cell-nonautonomous functions of oncogenic RAS and the p53 tumor suppressor. PloS Biol (2008) 6:2853–68. doi: 10.1371/journal.pbio.0060301
26. Swanson EC, Manning B, Zhang H, Lawrence JB. Higher-order unfolding of satellite heterochromatin is a consistent and early event in cell senescence. J Cell Biol (2013) 203:929–42. doi: 10.1083/jcb.201306073
27. Tchkonia T, Zhu Y, van Deursen J, Campisi J, Kirkland JL. Cellular senescence and the senescent secretory phenotype: therapeutic opportunities. J Clin Invest (2013) 123:966–72. doi: 10.1172/JCI64098
28. von Zglinicki T, Saretzki G, Ladhoff J, d’Adda di Fagagna F, Jackson SP. Human cell senescence as a DNA damage response. Mech Ageing Dev (2005) 126:111–7. doi: 10.1016/j.mad.2004.09.034
29. Ziegler DV, Wiley CD, Velarde MC. Mitochondrial effectors of cellular senescence: beyond the free radical theory of aging. Aging Cell (2015) 14:1–7. doi: 10.1111/acel.12287
30. Kajkenova O, Lecka-Czernik B, Gubrij I, Hauser SP, Takahashi K, Parfitt AM, et al. Increased adipogenesis and myelopoiesis in the bone marrow of SAMP6, a murine model of defective osteoblastogenesis and low turnover osteopenia. J Bone Miner Res (1997) 12:1772–9. doi: 10.1359/jbmr.1997.12.11.1772
31. Farr JN, Fraser DG, Wang H, Jaehn K, Ogrodnik MB, Weivoda MM, et al. Identification of Senescent Cells in the Bone Microenvironment. J Bone Miner Res (2016) 31:1920–9. doi: 10.1002/jbmr.2892
32. Farr JN, Xu M, Weivoda MM, Monroe DG, Fraser DG, Onken JL, et al. Targeting cellular senescence prevents age-related bone loss in mice. Nat Med (2017) 23:1072–9. doi: 10.1038/nm.4385
33. Atashi F, Modarressi A, Pepper MS. The role of reactive oxygen species in mesenchymal stem cell adipogenic and osteogenic differentiation: A review. Stem Cells Dev (2015) 24:1150–63. doi: 10.1089/scd.2014.0484
34. Ali D, Chen L, Kowal JM, Okla M, Manikandan M, AlShehri M, et al. Resveratrol inhibits adipocyte differentiation and cellular senescence of human bone marrow stromal stem cells. Bone (2020) 133:115252. doi: 10.1016/j.bone.2020.115252
35. Chandra A, Lagnado AB, Farr JN, Schleusner M, Monroe DG, Saul D, et al. Bone Marrow Adiposity in Models of Radiation- and Aging-Related Bone Loss Is Dependent on Cellular Senescence. J Bone Miner Res (2022) 37:997–1011. doi: 10.1002/jbmr.4537
36. Dimri GP, Itahana K, Acosta M, Campisi J. Regulation of a senescence checkpoint response by the E2F1 transcription factor and p14(ARF) tumor suppressor. Mol Cell Biol (2000) 20:273–85. doi: 10.1128/MCB.20.1.273-285.2000
37. Zhou X, Sen I, Lin XX, Riedel CG. Regulation of age-related decline by transcription factors and their crosstalk with the epigenome. Curr Genomics (2018) 19:464–82. doi: 10.2174/1389202919666180503125850
38. Li H, Liu P, Xu S, Li Y, Dekker JD, Li B, et al. FOXP1 controls mesenchymal stem cell commitment and senescence during skeletal aging. J Clin Invest (2017) 127:1241–53. doi: 10.1172/JCI89511
39. Moerman EJ, Teng K, Lipschitz DA, Lecka-Czernik B. Aging activates adipogenic and suppresses osteogenic programs in mesenchymal marrow stroma/stem cells: the role of PPAR-gamma2 transcription factor and TGF-beta/BMP signaling pathways. Aging Cell (2004) 3:379–89. doi: 10.1111/j.1474-9728.2004.00127.x
40. Lin Q, Gao Z, Alarcon RM, Ye J, Yun Z. A role of miR-27 in the regulation of adipogenesis. FEBS J (2009) 276:2348–58. doi: 10.1111/j.1742-4658.2009.06967.x
41. Peterson CL, Cote J. Cellular machineries for chromosomal DNA repair. Genes Dev (2004) 18:602–16. doi: 10.1101/gad.1182704
42. Masamoto Y, Arai S, Sato T, Kubota N, Takamoto I, Kadowaki T, et al. Adiponectin Enhances Quiescence Exit of Murine Hematopoietic Stem Cells and Hematopoietic Recovery Through mTORC1 Potentiation. Stem Cells (2017) 35:1835–48. doi: 10.1002/stem.2640
43. Zhou BO, Yu H, Yue R, Zhao Z, Rios JJ, Naveiras O, et al. Bone marrow adipocytes promote the regeneration of stem cells and haematopoiesis by secreting SCF. Nat Cell Biol (2017) 19:891–903. doi: 10.1038/ncb3570
44. Bolan PJ, Arentsen L, Sueblinvong T, Zhang Y, Moeller S, Carter JS, et al. Water-fat MRI for assessing changes in bone marrow composition due to radiation and chemotherapy in gynecologic cancer patients. J Magn Reson Imaging (2013) 38:1578–84. doi: 10.1002/jmri.24071
45. Kim HN, Chang J, Shao L, Han L, Iyer S, Manolagas SC, et al. DNA damage and senescence in osteoprogenitors expressing Osx1 may cause their decrease with age. Aging Cell (2017) 16:693–703. doi: 10.1111/acel.12597
46. Chandra A, Wang L, Young T, Zhong L, Tseng WJ, Levine MA, et al. Proteasome inhibitor bortezomib is a novel therapeutic agent for focal radiation-induced osteoporosis. FASEB J (2018) 32:52–62. doi: 10.1096/fj.201700375R
47. Chandra A, Lin T, Young T, Tong W, Ma X, Tseng WJ, et al. Suppression of Sclerostin Alleviates Radiation-Induced Bone Loss by Protecting Bone-Forming Cells and Their Progenitors Through Distinct Mechanisms. J Bone Miner Res (2017) 32:360–72. doi: 10.1002/jbmr.2996
48. Marie PJ, Kassem M. Extrinsic mechanisms involved in age-related defective bone formation. J Clin Endocrinol Metab (2011) 96:600–9. doi: 10.1210/jc.2010-2113
49. Vanderschueren D, Vandenput L, Boonen S, Lindberg MK, Bouillon R, Ohlsson C. Androgens and bone. Endocr Rev (2004) 25:389–425. doi: 10.1210/er.2003-0003
50. Khosla S, Melton LJ 3rd, Riggs BL. Clinical review 144: Estrogen and the male skeleton. J Clin Endocrinol Metab (2002) 87:1443–50. doi: 10.1210/jcem.87.4.8417
51. Forbes AP, Albright F. His concept of postmenopausal osteoporosis and what came of it. Clin Orthop Relat Res (1991), 128–41.
52. Lindsay R, Hart DM, Aitken JM, MacDonald EB, Anderson JB, Clarke AC. Long-term prevention of postmenopausal osteoporosis by oestrogen Evidence for an increased bone mass after delayed onset of oestrogen treatment. Lancet (1976) 1:1038–41. doi: 10.1016/s0140-6736(76)92217-0
53. Abdallah BM, Bay-Jensen AC, Srinivasan B, Tabassi NC, Garnero P, Delaisse JM, et al. Estrogen inhibits Dlk1/FA1 production: A potential mechanism for estrogen effects on bone turnover. J Bone Miner Res (2011) 26:2548–51. doi: 10.1002/jbmr.444
54. Abdallah BM, Ditzel N, Mahmood A, Isa A, Traustadottir GA, Schilling AF, et al. DLK1 is a novel regulator of bone mass that mediates estrogen deficiency-induced bone loss in mice. J Bone Miner Res (2011) 26:1457–71. doi: 10.1002/jbmr.346
55. Jacobsen BK, Bonaa KH. The reproducibility of dietary data from a self-administered questionnaire. The Tromso Study. Int J Epidemiol (1990) 19:349–53. doi: 10.1093/ije/19.2.349
56. Almeida M, Martin-Millan M, Ambrogini E, Bradsher R 3rd, Han L, Chen XD, et al. Estrogens attenuate oxidative stress and the differentiation and apoptosis of osteoblasts by DNA-binding-independent actions of the ERalpha. J Bone Miner Res (2010) 25:769–81. doi: 10.1359/jbmr.091017
57. Kousteni S, Almeida M, Han L, Bellido T, Jilka RL, Manolagas SC. Induction of osteoblast differentiation by selective activation of kinase-mediated actions of the estrogen receptor. Mol Cell Biol (2007) 27:1516–30. doi: 10.1128/MCB.01550-06
58. Wiren KM, Semirale AA, Zhang XW, Woo A, Tommasini SM, Price C, et al. Targeting of androgen receptor in bone reveals a lack of androgen anabolic action and inhibition of osteogenesis: a model for compartment-specific androgen action in the skeleton. Bone (2008) 43:440–51. doi: 10.1016/j.bone.2008.04.026
59. Prakasam G, Yeh JK, Chen MM, Castro-Magana M, Liang CT, Aloia JF. Effects of growth hormone and testosterone on cortical bone formation and bone density in aged orchiectomized rats. Bone (1999) 24:491–7. doi: 10.1016/s8756-3282(99)00018-6
60. Orwoll ES. Androgens as anabolic agents for bone. Trends Endocrinol Metab (1996) 7:77–84. doi: 10.1016/1043-2760(96)00024-0
61. Ebeling PR. Clinical practice. Osteoporosis in men. N Engl J Med (2008) 358:1474–82. doi: 10.1056/NEJMcp0707217
62. Marie PJ, de Vernejoul MC, Connes D, Hott M. Decreased DNA synthesis by cultured osteoblastic cells in eugonadal osteoporotic men with defective bone formation. J Clin Invest (1991) 88:1167–72. doi: 10.1172/JCI115418
63. Khosla S, Melton LJ 3rd, Atkinson EJ, O’Fallon WM, Klee GG, Riggs BL. Relationship of serum sex steroid levels and bone turnover markers with bone mineral density in men and women: a key role for bioavailable estrogen. J Clin Endocrinol Metab (1998) 83:2266–74. doi: 10.1210/jcem.83.7.4924
64. Falahati-Nini A, Riggs BL, Atkinson EJ, O’Fallon WM, Eastell R, Khosla S. Relative contributions of testosterone and estrogen in regulating bone resorption and formation in normal elderly men. J Clin Invest (2000) 106:1553–60. doi: 10.1172/JCI10942
65. Kearns AE, Khosla S, Kostenuik PJ. Receptor activator of nuclear factor kappaB ligand and osteoprotegerin regulation of bone remodeling in health and disease. Endocr Rev (2008) 29:155–92. doi: 10.1210/er.2007-0014
66. Pacifici R. Cytokines, estrogen, and postmenopausal osteoporosis–the second decade. Endocrinology (1998) 139:2659–61. doi: 10.1210/endo.139.6.6087
67. Zallone A. Direct and indirect estrogen actions on osteoblasts and osteoclasts. Ann N Y Acad Sci (2006) 1068:173–9. doi: 10.1196/annals.1346.019
68. Ha H, Kwak HB, Lee SW, Jin HM, Kim HM, Kim HH, et al. Reactive oxygen species mediate RANK signaling in osteoclasts. Exp Cell Res (2004) 301:119–27. doi: 10.1016/j.yexcr.2004.07.035
69. Lean JM, Davies JT, Fuller K, Jagger CJ, Kirstein B, Partington GA, et al. A crucial role for thiol antioxidants in estrogen-deficiency bone loss. J Clin Invest (2003) 112:915–23. doi: 10.1172/JCI18859
70. Tencerova M, Figeac F, Ditzel N, Taipaleenmaki H, Nielsen TK, Kassem M. High-fat diet-induced obesity promotes expansion of bone marrow adipose tissue and impairs skeletal stem cell functions in mice. J Bone Miner Res (2018) 33:1154–65. doi: 10.1002/jbmr.3408
71. Tencerova M, Frost M, Figeac F, Nielsen TK, Ali D, Lauterlein JL, et al. Obesity-associated hypermetabolism and accelerated senescence of bone marrow stromal stem cells suggest a potential mechanism for bone fragility. Cell Rep (2019) 27:2050–2062.e6. doi: 10.1016/j.celrep.2019.04.066
72. Marie PJ, Sabbagh A, de Vernejoul MC, Lomri A. Osteocalcin and deoxyribonucleic acid synthesis in vitro and histomorphometric indices of bone formation in postmenopausal osteoporosis. J Clin Endocrinol Metab (1989) 69:272–9. doi: 10.1210/jcem-69-2-272
73. Modrowski D, Miravet L, Feuga M, Marie PJ. Increased proliferation of osteoblast precursor cells in estrogen-deficient rats. Am J Physiol (1993) 264:E190–6. doi: 10.1152/ajpendo.1993.264.2.E190
74. Griffith JF, Yeung DK, Ma HT, Leung JC, Kwok TC, Leung PC. Bone marrow fat content in the elderly: A reversal of sex difference seen in younger subjects. J Magn Reson Imaging (2012) 36:225–30. doi: 10.1002/jmri.23619
75. Woods GN, Ewing SK, Sigurdsson S, Kado DM, Eiriksdottir G, Gudnason V, et al. Greater bone marrow adiposity predicts bone loss in older women. J Bone Miner Res (2020) 35:326–32. doi: 10.1002/jbmr.3895
76. Griffith JF, Yeung DK, Antonio GE, Lee FK, Hong AW, Wong SY, et al. Vertebral bone mineral density, marrow perfusion, and fat content in healthy men and men with osteoporosis: Ddynamic contrast-enhanced MR imaging and MR spectroscopy. Radiology (2005) 236:945–51. doi: 10.1148/radiol.2363041425
77. Rothman MS, Iwamoto SJ. Bone Health in the Transgender Population. Clin Rev Bone Mineral Metab (2019) 17:77–85. doi: 10.1007/s12018-019-09261-3
78. Bretherton I, Ghasem-Zadeh A, Leemaqz SY, Seeman E, Wang X, McFarlane T, et al. Bone microarchitecture in transgender adults: A cross-sectional study. J Bone Miner Res (2022) 37:643–8. doi: 10.1002/jbmr.4497
79. Nasomyont N, Meisman AR, Ecklund K, Vajapeyam S, Cecil KM, Tkach JA, et al. Changes in bone marrow adipose tissue in transgender and gender non-conforming youth undergoing pubertal suppression: A pilot study. J Clin Densitometry (2022). doi: 10.1016/j.jocd.2022.06.006
80. Khosla S, Samakkarnthai P, Monroe DG, Farr JN. Update on the pathogenesis and treatment of skeletal fragility in type 2 diabetes mellitus. Nat Rev Endocrinol (2021) 17:685–97. doi: 10.1038/s41574-021-00555-5
81. Napoli N, Chandran M, Pierroz DD, Abrahamsen B, Schwartz AV, Ferrari SL, et al. Mechanisms of diabetes mellitus-induced bone fragility. Nat Rev Endocrinol (2017) 13:208–19. doi: 10.1038/nrendo.2016.153
82. Morris JA, Kemp JP, Youlten SE, Laurent L, Logan JG, Chai RC, et al. An atlas of genetic influences on osteoporosis in humans and mice. Nat Genet (2019) 51:258–66. doi: 10.1038/s41588-018-0302-x
83. Riggs BL, Wahner HW, Seeman E, Offord KP, Dunn WL, Mazess RB, et al. Changes in bone mineral density of the proximal femur and spine with aging. Differences between the postmenopausal and senile osteoporosis syndromes. J Clin Invest (1982) 70:716–23. doi: 10.1172/jci110667
84. Lecka-Czernik B, Stechschulte LA, Czernik PJ, Dowling AR. High bone mass in adult mice with diet-induced obesity results from a combination of initial increase in bone mass followed by attenuation in bone formation; implications for high bone mass and decreased bone quality in obesity. Mol Cell Endocrinol (2015) 410:35–41. doi: 10.1016/j.mce.2015.01.001
85. Halade GV, Rahman MM, Williams PJ, Fernandes G. High fat diet-induced animal model of age-associated obesity and osteoporosis. J Nutr Biochem (2010) 21:1162–9. doi: 10.1016/j.jnutbio.2009.10.002
86. Doucette CR, Horowitz MC, Berry R, MacDougald OA, Anunciado-Koza R, Koza RA, et al. A high fat diet increases bone marrow adipose tissue (mat) but does not alter trabecular or cortical bone mass in c57bl/6j mice. J Cell Physiol (2015) 230:2032–7. doi: 10.1002/jcp.24954
87. Scheller EL, Khoury B, Moller KL, Wee NK, Khandaker S, Kozloff KM, et al. Changes in skeletal integrity and marrow adiposity during high-fat diet and after weight loss. Front Endocrinol (Lausanne) (2016) 7:102. doi: 10.3389/fendo.2016.00102
88. Bredella MA, Gill CM, Keating LK, Torriani M, Anderson EJ, Punyanitya M, et al. Assessment of abdominal fat compartments using DXA in premenopausal women from anorexia nervosa to morbid obesity. Obes (Silver Spring) (2013) 21:2458–64. doi: 10.1002/oby.20424
89. Bredella MA, Torriani M, Ghomi RH, Thomas BJ, Brick DJ, Gerweck AV, et al. Vertebral bone marrow fat is positively associated with visceral fat and inversely associated with IGF-1 in obese women. Obes (Silver Spring) (2011) 19:49–53. doi: 10.1038/oby.2010.106
90. Yu EW, Greenblatt L, Eajazi A, Torriani M, Bredella MA. Marrow adipose tissue composition in adults with morbid obesity. Bone (2017) 97:38–42. doi: 10.1016/j.bone.2016.12.018
91. Bilha SC, Leustean L, Preda C, Branisteanu DD, Mihalache L, Ungureanu MC. Bone mineral density predictors in long-standing type 1 and type 2 diabetes mellitus. BMC Endocr Disord (2021) 21:156. doi: 10.1186/s12902-021-00815-5
92. Figeac F, Tencerova M, Ali D, Andersen TL, Appadoo DRC, Kerckhofs G, et al. Impaired Bone Fracture Healing in Type 2 Diabetes Is Caused by Defective Functions of Skeletal Progenitor Cells. Stem Cells (2022) 40:149–64. doi: 10.1093/stmcls/sxab011
93. Fintini D, Cianfarani S, Cofini M, Andreoletti A, Ubertini GM, Cappa M, et al. The Bones of Children With Obesity. Front Endocrinol (Lausanne) (2020) 11:200. doi: 10.3389/fendo.2020.00200
94. Walsh JS, Vilaca T. Obesity, Type 2 Diabetes and Bone in Adults. Calcif Tissue Int (2017) 100:528–35. doi: 10.1007/s00223-016-0229-0
95. Tonks KT, White CP, Center JR, Samocha-Bonet D, Greenfield JR. Bone Turnover Is Suppressed in Insulin Resistance, Independent of Adiposity. J Clin Endocrinol Metab (2017) 102:1112–21. doi: 10.1210/jc.2016-3282
96. Migliaccio S, Greco EA, Fornari R, Donini LM, Lenzi A. Is obesity in women protective against osteoporosis? Diabetes Metab Syndr Obes (2011) 4:273–82. doi: 10.2147/DMSO.S11920
97. Kin K, Kushida K, Yamazaki K, Okamoto S, Inoue T. Bone mineral density of the spine in normal Japanese subjects using dual-energy X-ray absorptiometry: effect of obesity and menopausal status. Calcif Tissue Int (1991) 49:101–6. doi: 10.1007/BF02565129
98. Ribot C, Tremollieres F, Pouilles JM, Bonneu M, Germain F, Louvet JP. Obesity and postmenopausal bone loss: The influence of obesity on vertebral density and bone turnover in postmenopausal women. Bone (1987) 8:327–31. doi: 10.1016/8756-3282(87)90062-7
99. Jang M, Kim H, Lea S, Oh S, Kim JS, Oh B. Effect of duration of diabetes on bone mineral density: A population study on East Asian males. BMC Endocr Disord (2018) 18:61. doi: 10.1186/s12902-018-0290-y
100. Bonds DE, Larson JC, Schwartz AV, Strotmeyer ES, Robbins J, Rodriguez BL, et al. Risk of fracture in women with type 2 diabetes: The Women’s Health Initiative Observational Study. J Clin Endocrinol Metab (2006) 91:3404–10. doi: 10.1210/jc.2006-0614
101. Schwartz AV, Vittinghoff E, Bauer DC, Hillier TA, Strotmeyer ES, Ensrud KE, et al. Association of BMD and FRAX score with risk of fracture in older adults with type 2 diabetes. JAMA (2011) 305:2184–92. doi: 10.1001/jama.2011.715
102. Farr JN, Drake MT, Amin S, Melton LJ 3rd, McCready LK, Khosla S. In vivo assessment of bone quality in postmenopausal women with type 2 diabetes. J Bone Miner Res (2014) 29:787–95. doi: 10.1002/jbmr.2106
103. Cohen A, Dempster DW, Recker RR, Lappe JM, Zhou H, Zwahlen A, et al. Abdominal fat is associated with lower bone formation and inferior bone quality in healthy premenopausal women: A transiliac bone biopsy study. J Clin Endocrinol Metab (2013) 98:2562–72. doi: 10.1210/jc.2013-1047
104. Carnevale V, Romagnoli E, D’Erasmo L, D’Erasmo E. Bone damage in type 2 diabetes mellitus. Nutr Metab Cardiovasc Dis (2014) 24:1151–7. doi: 10.1016/j.numecd.2014.06.013
105. Lee RH, Sloane R, Pieper C, Lyles KW, Adler RA, Van Houtven C, et al. Glycemic control and insulin treatment alter fracture risk in older men with type 2 diabetes mellitus. J Bone Miner Res (2019) 34:2045–51. doi: 10.1002/jbmr.3826
106. Vestergaard P. Diabetes and bone fracture: risk factors for old and young. Diabetologia (2014) 57:2007–8. doi: 10.1007/s00125-014-3338-1
107. Janghorbani M, Van Dam RM, Willett WC, Hu FB. Systematic review of type 1 and type 2 diabetes mellitus and risk of fracture. Am J Epidemiol (2007) 166:495–505. doi: 10.1093/aje/kwm106
108. Schwartz AV, Sellmeyer DE, Ensrud KE, Cauley JA, Tabor HK, Schreiner PJ, et al. Older women with diabetes have an increased risk of fracture: a prospective study. J Clin Endocrinol Metab (2001) 86:32–8. doi: 10.1210/jcem.86.1.7139
109. Sheu Y, Amati F, Schwartz AV, Danielson ME, Li X, Boudreau R, et al. Vertebral bone marrow fat, bone mineral density and diabetes: The Osteoporotic Fractures in Men (MrOS) study. Bone (2017) 97:299–305. doi: 10.1016/j.bone.2017.02.001
110. Fazeli PK, Bredella MA, Pachon-Peña G, Zhao W, Zhang X, Faje AT, et al. The dynamics of human bone marrow adipose tissue in response to feeding and fasting. JCI Insight (2021) 6. doi: 10.1172/jci.insight.138636
111. Kim TY, Schwartz AV, Li X, Xu K, Black DM, Petrenko DM, et al. Bone marrow fat changes after gastric bypass surgery are associated with loss of bone mass. J Bone Miner Res (2017) 32:2239–47. doi: 10.1002/jbmr.3212
112. Vogt LJ, Steveling A, Meffert PJ, Kromrey ML, Kessler R, Hosten N, et al. Magnetic resonance imaging of changes in abdominal compartments in obese diabetics during a low-calorie weight-loss program. PloS One (2016) 11:e0153595. doi: 10.1371/journal.pone.0153595
113. Kerckhofs G, Durand M, Vangoitsenhoven R, Marin C, van der Schueren B, Carmeliet G, et al. Changes in bone macro- and microstructure in diabetic obese mice revealed by high resolution microfocus X-ray computed tomography. Sci Rep (2016) 6:35517. doi: 10.1038/srep35517
114. Tanaka H, Yamashita T, Yoneda M, Takagi S, Miura T. Characteristics of bone strength and metabolism in type 2 diabetic model Tsumura, Suzuki, Obese Diabetes mice. Bone Rep (2018) 9:74–83. doi: 10.1016/j.bonr.2018.07.004
115. Wellen KE, Hotamisligil GS. Obesity-induced inflammatory changes in adipose tissue. J Clin Invest (2003) 112:1785–8. doi: 10.1172/JCI20514
116. Mundy GR. Osteoporosis and inflammation. Nutr Rev (2007) 65:S147–51. doi: 10.1111/j.1753-4887.2007.tb00353.x
117. Wright E Jr., Scism-Bacon JL, Glass LC. Oxidative stress in type 2 diabetes: The role of fasting and postprandial glycaemia. Int J Clin Pract (2006) 60:308–14. doi: 10.1111/j.1368-5031.2006.00825.x
118. Dos Santos JM, Tewari S, Mendes RH. The role of oxidative stress in the development of diabetes mellitus and its complications. J Diabetes Res (2019) 2019:4189813. doi: 10.1155/2019/4189813
119. Yue R, Zhou BO, Shimada IS, Zhao Z, Morrison SJ. Leptin receptor promotes adipogenesis and reduces osteogenesis by regulating mesenchymal stromal cells in adult bone marrow. Cell Stem Cell (2016) 18:782–96. doi: 10.1016/j.stem.2016.02.015
120. Droge W. Oxidative aging and insulin receptor signaling. J Gerontol A Biol Sci Med Sci (2005) 60:1378–85. doi: 10.1093/gerona/60.11.1378
121. Devlin MJ, Van Vliet M, Motyl K, Karim L, Brooks DJ, Louis L, et al. Early-onset type 2 diabetes impairs skeletal acquisition in the male TALLYHO/JngJ mouse. Endocrinology (2014) 155:3806–16. doi: 10.1210/en.2014-1041
122. Ambrosi TH, Scialdone A, Graja A, Gohlke S, Jank AM, Bocian C, et al. Adipocyte accumulation in the bone marrow during obesity and aging impairs stem cell-based hematopoietic and bone regeneration. Cell Stem Cell (2017) 20:771–784.e6. doi: 10.1016/j.stem.2017.02.009
123. Boskey AL, Imbert L. Bone quality changes associated with aging and disease: a review. Ann N Y Acad Sci (2017) 1410:93–106. doi: 10.1111/nyas.13572
124. Naveiras O, Nardi V, Wenzel PL, Hauschka PV, Fahey F, Daley GQ. Bone-marrow adipocytes as negative regulators of the haematopoietic microenvironment. Nature (2009) 460:259–63.
125. Fantuzzi G, tissue A. adipokines, and inflammation. J Allergy Clin Immunol (2005) 115:911–9. doi: 10.1016/j.jaci.2005.02.023
126. Barbour KE, Zmuda JM, Boudreau R, Strotmeyer ES, Horwitz MJ, Evans RW, et al. The effects of adiponectin and leptin on changes in bone mineral density. Osteoporos Int (2012) 23:1699–710. doi: 10.1007/s00198-011-1768-x
127. Basurto L, Galvan R, Cordova N, Saucedo R, Vargas C, Campos S, et al. Adiponectin is associated with low bone mineral density in elderly men. Eur J Endocrinol (2009) 160:289–93. doi: 10.1530/EJE-08-0569
128. O’Brien CA, Gubrij I, Lin SC, Saylors RL, Manolagas SC. STAT3 activation in stromal/osteoblastic cells is required for induction of the receptor activator of NF-kappaB ligand and stimulation of osteoclastogenesis by gp130-utilizing cytokines or interleukin-1 but not 1,25-dihydroxyvitamin D3 or parathyroid hormone. J Biol Chem (1999) 274:19301–8. doi: 10.1074/jbc.274.27.19301
129. Kudo O, Sabokbar A, Pocock A, Itonaga I, Fujikawa Y, Athanasou NA. Interleukin-6 and interleukin-11 support human osteoclast formation by a RANKL-independent mechanism. Bone (2003) 32:1–7. doi: 10.1016/s8756-3282(02)00915-8
130. Li Y, Lu L, Xie Y, Chen X, Tian L, Liang Y, et al. Interleukin-6 Knockout Inhibits Senescence of Bone Mesenchymal Stem Cells in High-Fat Diet-Induced Bone Loss. Front Endocrinol (Lausanne) (2020) 11:622950. doi: 10.3389/fendo.2020.622950
131. Aaron N, Costa S, Rosen CJ, Qiang L. The Implications of Bone Marrow Adipose Tissue on Inflammaging. Front Endocrinol (Lausanne) (2022) 13:853765. doi: 10.3389/fendo.2022.853765
132. Kozakowski J, Gietka-Czernel M, Leszczynska D, Majos A. Obesity in menopause - our negligence or an unfortunate inevitability? Prz Menopauzalny (2017) 16:61–5. doi: 10.5114/pm.2017.68594
133. Justesen J, Stenderup K, Ebbesen EN, Mosekilde L, Steiniche T, Kassem M. Adipocyte tissue volume in bone marrow is increased with aging and in patients with osteoporosis. Biogerontology (2001) 2:165–71. doi: 10.1023/a:1011513223894
134. Li G, Xu Z, Fan J, Yuan W, Zhang L, Hou L, et al. To assess differential features of marrow adiposity between postmenopausal women with osteoarthritis and osteoporosis using water/fat MRI. Menopause (2017) 24:105–11. doi: 10.1097/GME.0000000000000732
135. Milisic L, Vegar-Zubovic S, Valjevac A. Bone marrow adiposity is inversely associated with bone mineral density in postmenopausal females. Med Glas (Zenica) (2020) 17:15–21. doi: 10.17392/1053-20
136. Sekiya I, Larson BL, Vuoristo JT, Cui JG, Prockop DJ. Adipogenic differentiation of human adult stem cells from bone marrow stroma (MSCs). J Bone Miner Res (2004) 19:256–64. doi: 10.1359/JBMR.0301220
137. Nehlin JO, Jafari A, Tencerova M, Kassem M. Aging and lineage allocation changes of bone marrow skeletal (stromal) stem cells. Bone (2019) 123:265–73. doi: 10.1016/j.bone.2019.03.041
138. Salamat MR, Salamat AH, Janghorbani M. Association between obesity and bone mineral density by gender and menopausal status. Endocrinol Metab (Seoul) (2016) 31:547–58. doi: 10.3803/EnM.2016.31.4.547
139. Silva HG, Mendonca LM, Conceicao FL, Zahar SE, Farias ML. Influence of obesity on bone density in postmenopausal women. Arq Bras Endocrinol Metabol (2007) 51:943–9. doi: 10.1590/s0004-27302007000600008
140. Bonjour JP. Dietary protein: An essential nutrient for bone health. J Am Coll Nutr (2005) 24:526S–36S. doi: 10.1080/07315724.2005.10719501
141. Kveiborg M, Rattan SI, Clark BF, Eriksen EF, Kassem M. Treatment with 1,25-dihydroxyvitamin D3 reduces impairment of human osteoblast functions during cellular aging in culture. J Cell Physiol (2001) 186:298–306. doi: 10.1002/1097-4652(200002)186:2<298::AID-JCP1030>3.0.CO;2-H
142. Fretz JA, Zella LA, Kim S, Shevde NK, Pike JW. 1,25-Dihydroxyvitamin D3 regulates the expression of low-density lipoprotein receptor-related protein 5 via deoxyribonucleic acid sequence elements located downstream of the start site of transcription. Mol Endocrinol (2006) 20:2215–30. doi: 10.1210/me.2006-0102
143. van Driel M, van Leeuwen JP. Vitamin D endocrine system and osteoblasts. Bonekey Rep (2014) 3:493. doi: 10.1038/bonekey.2013.227
144. Nakaoka K, Yamada A, Noda S, Goseki-Sone M. Influence of dietary vitamin D deficiency on bone strength, body composition, and muscle in ovariectomized rats fed a high-fat diet. Nutrition (2019) 60:87–93. doi: 10.1016/j.nut.2018.09.001
145. Lips P. Vitamin D deficiency and secondary hyperparathyroidism in the elderly: consequences for bone loss and fractures and therapeutic implications. Endocr Rev (2001) 22:477–501. doi: 10.1210/edrv.22.4.0437
146. Boonen S, Lips P, Bouillon R, Bischoff-Ferrari HA, Vanderschueren D, Haentjens P. Need for additional calcium to reduce the risk of hip fracture with vitamin d supplementation: Evidence from a comparative metaanalysis of randomized controlled trials. J Clin Endocrinol Metab (2007) 92:1415–23. doi: 10.1210/jc.2006-1404
147. Chapuy MC, Pamphile R, Paris E, Kempf C, Schlichting M, Arnaud S, et al. Combined calcium and vitamin D3 supplementation in elderly women: confirmation of reversal of secondary hyperparathyroidism and hip fracture risk: the Decalyos II study. Osteoporos Int (2002) 13:257–64. doi: 10.1007/s001980200023
148. Sahota O, Mundey MK, San P, Godber IM, Lawson N, Hosking DJ. The relationship between vitamin D and parathyroid hormone: Calcium homeostasis, bone turnover, and bone mineral density in postmenopausal women with established osteoporosis. Bone (2004) 35:312–9. doi: 10.1016/j.bone.2004.02.003
149. Tang BM, Eslick GD, Nowson C, Smith C, Bensoussan A. Use of calcium or calcium in combination with vitamin D supplementation to prevent fractures and bone loss in people aged 50 years and older: A meta-analysis. Lancet (2007) 370:657–66. doi: 10.1016/S0140-6736(07)61342-7
150. Ding J, Nagai K, Woo JT. Insulin-dependent adipogenesis in stromal ST2 cells derived from murine bone marrow. Biosci Biotechnol Biochem (2003) 67:314–21. doi: 10.1271/bbb.67.314
151. Kong J, Li YC. Molecular mechanism of 1,25-dihydroxyvitamin D3 inhibition of adipogenesis in 3T3-L1 cells. Am J Physiol Endocrinol Metab (2006) 290:E916–24. doi: 10.1152/ajpendo.00410.2005
152. Whitfield GK, Hsieh JC, Nakajima S, MacDonald PN, Thompson PD, Jurutka PW, et al. A highly conserved region in the hormone-binding domain of the human vitamin D receptor contains residues vital for heterodimerization with retinoid X receptor and for transcriptional activation. Mol Endocrinol (1995) 9:1166–79. doi: 10.1210/mend.9.9.7491109
153. Cianferotti L, Demay MB. VDR-mediated inhibition of DKK1 and SFRP2 suppresses adipogenic differentiation of murine bone marrow stromal cells. J Cell Biochem (2007) 101:80–8. doi: 10.1002/jcb.21151
154. Duque G, Rivas D. Alendronate has an anabolic effect on bone through the differentiation of mesenchymal stem cells. J Bone Miner Res (2007) 22:1603–11. doi: 10.1359/jbmr.070701
155. Kelly KA, Gimble JM. 1,25-Dihydroxy vitamin D3 inhibits adipocyte differentiation and gene expression in murine bone marrow stromal cell clones and primary cultures. Endocrinology (1998) 139:2622–8. doi: 10.1210/endo.139.5.5970
156. Meehan M, Penckofer S. The role of vitamin d in the aging adult. J Aging Gerontol (2014) 2:60–71. doi: 10.12974/2309-6128.2014.02.02.1
157. Ameri P, Giusti A, Boschetti M, Bovio M, Teti C, Leoncini G, et al. Vitamin D increases circulating IGF1 in adults: Potential implication for the treatment of GH deficiency. Eur J Endocrinol (2013) 169:767–72. doi: 10.1530/EJE-13-0510
158. Gomez JM. The role of insulin-like growth factor I components in the regulation of vitamin D. Curr Pharm Biotechnol (2006) 7:125–32. doi: 10.2174/138920106776597621
159. Sadie-Van Gijsen H, Crowther NJ, Hough FS, Ferris WF. The interrelationship between bone and fat: From cellular see-saw to endocrine reciprocity. Cell Mol Life Sci (2013) 70:2331–49. doi: 10.1007/s00018-012-1211-2
160. Baxter-Jones AD, Kontulainen SA, Faulkner RA, Bailey DA. A longitudinal study of the relationship of physical activity to bone mineral accrual from adolescence to young adulthood. Bone (2008) 43:1101–7. doi: 10.1016/j.bone.2008.07.245
162. Styner M, Pagnotti GM, McGrath C, Wu X, Sen B, Uzer G, et al. Exercise decreases marrow adipose tissue through ss-oxidation in obese running mice. J Bone Miner Res (2017) 32:1692–702. doi: 10.1002/jbmr.3159
163. Pagnotti GM, Styner M. Exercise Regulation of Marrow Adipose Tissue. Front Endocrinol (Lausanne) (2016) 7:94. doi: 10.3389/fendo.2016.00094
164. Belavy DL, Miller CT, Owen PJ, Rantalainen T, Connell D, Hahne AJ, et al. Exercise may impact on lumbar vertebrae marrow adipose tissue: Randomised controlled trial. Bone (2022) 157:116338. doi: 10.1016/j.bone.2022.116338
165. Maddalozzo GF, Turner RT, Edwards CH, Howe KS, Widrick JJ, Rosen CJ, et al. Alcohol alters whole body composition, inhibits bone formation, and increases bone marrow adiposity in rats. Osteoporos Int (2009) 20:1529–38. doi: 10.1007/s00198-009-0836-y
166. Kyle KA, Willett TL, Baggio LL, Drucker DJ, Grynpas MD. Differential effects of PPAR-{gamma} activation versus chemical or genetic reduction of DPP-4 activity on bone quality in mice. Endocrinology (2011) 152:457–67. doi: 10.1210/en.2010-1098
167. Dombrowski S, Kostev K, Jacob L. Use of dipeptidyl peptidase-4 inhibitors and risk of bone fracture in patients with type 2 diabetes in Germany-A retrospective analysis of real-world data. Osteoporos Int (2017) 28:2421–8. doi: 10.1007/s00198-017-4051-y
168. Monami M, Dicembrini I, Antenore A, Mannucci E. Dipeptidyl peptidase-4 inhibitors and bone fractures: A meta-analysis of randomized clinical trials. Diabetes Care (2011) 34:2474–6. doi: 10.2337/dc11-1099
169. Sedlinsky C, Molinuevo MS, Cortizo AM, Tolosa MJ, Felice JI, Sbaraglini ML, et al. Metformin prevents anti-osteogenic in vivo and ex vivo effects of rosiglitazone in rats. Eur J Pharmacol (2011) 668:477–85. doi: 10.1016/j.ejphar.2011.07.033
170. Melton LJ 3rd, Leibson CL, Achenbach SJ, Therneau TM, Khosla S. Fracture risk in type 2 diabetes: Update of a population-based study. J Bone Miner Res (2008) 23:1334–42. doi: 10.1359/jbmr.080323
171. Kahn SE, Zinman B, Lachin JM, Haffner SM, Herman WH, Holman RR, et al. Rosiglitazone-associated fractures in type 2 diabetes: An Analysis from A Diabetes Outcome Progression Trial (ADOPT). Diabetes Care (2008) 31:845–51. doi: 10.2337/dc07-2270
172. Zinman B, Haffner SM, Herman WH, Holman RR, Lachin JM, Kravitz BG, et al. Effect of rosiglitazone, metformin, and glyburide on bone biomarkers in patients with type 2 diabetes. J Clin Endocrinol Metab (2010) 95:134–42. doi: 10.1210/jc.2009-0572
173. Vestergaard P, Rejnmark L, Mosekilde L. Relative fracture risk in patients with diabetes mellitus, and the impact of insulin and oral antidiabetic medication on relative fracture risk. Diabetologia (2005) 48:1292–9. doi: 10.1007/s00125-005-1786-3
174. Monami M, Cresci B, Colombini A, Pala L, Balzi D, Gori F, et al. Bone fractures and hypoglycemic treatment in type 2 diabetic patients: A case-control study. Diabetes Care (2008) 31:199–203. doi: 10.2337/dc07-1736
175. Stechschulte LA, Czernik PJ, Rotter ZC, Tausif FN, Corzo CA, Marciano DP, et al. PPARG post-translational modifications regulate bone formation and bone resorption. EBioMedicine (2016) 10:174–84. doi: 10.1016/j.ebiom.2016.06.040
176. Fukunaga T, Zou W, Rohatgi N, Colca JR, Teitelbaum SL, thiazolidinedione Ani-s. which minimally activates PPARgamma, does not cause bone loss. J Bone Miner Res (2015) 30:481–8. doi: 10.1002/jbmr.2364
177. Harrison SA, Alkhouri N, Davison BA, Sanyal A, Edwards C, Colca JR, et al. Insulin sensitizer MSDC-0602K in non-alcoholic steatohepatitis: A randomized, double-blind, placebo-controlled phase IIb study. J Hepatol (2020) 72:613–26. doi: 10.1016/j.jhep.2019.10.023
178. Lin Q, Huang YM, Xiao BX, Ren GF. Effects of resveratrol on bone mineral density in ovarectomized rats. Int J BioMed Sci (2005) 1:76–81.
179. Yang X, Jiang T, Wang Y, Guo L. The role and mechanism of sirt1 in resveratrol-regulated osteoblast autophagy in osteoporosis rats. Sci Rep (2019) 9:18424. doi: 10.1038/s41598-019-44766-3
180. Rios H, Koushik SV, Wang H, Wang J, Zhou HM, Lindsley A, et al. periostin null mice exhibit dwarfism, incisor enamel defects, and an early-onset periodontal disease-like phenotype. Mol.Cell Biol (2005) 25:11131–44.
181. Suen PK, Qin L. Sclerostin, an emerging therapeutic target for treating osteoporosis and osteoporotic fracture: A general review. J Orthop Translat (2016) 4:1–13. doi: 10.1016/j.jot.2015.08.004
182. Yee CS, Xie L, Hatsell S, Hum N, Murugesh D, Economides AN, et al. Sclerostin antibody treatment improves fracture outcomes in a Type I diabetic mouse model. Bone (2016) 82:122–34. doi: 10.1016/j.bone.2015.04.048
183. McDonald MM, Morse A, Mikulec K, Peacock L, Yu N, Baldock PA, et al. Inhibition of sclerostin by systemic treatment with sclerostin antibody enhances healing of proximal tibial defects in ovariectomized rats. J Orthop Res (2012) 30:1541–8. doi: 10.1002/jor.22109
184. Suen PK, He YX, Chow DH, Huang L, Li C, Ke HZ, et al. Sclerostin monoclonal antibody enhanced bone fracture healing in an open osteotomy model in rats. J Orthop Res (2014) 32:997–1005. doi: 10.1002/jor.22636
185. Hamann C, Rauner M, Hohna Y, Bernhardt R, Mettelsiefen J, Goettsch C, et al. Sclerostin antibody treatment improves bone mass, bone strength, and bone defect regeneration in rats with type 2 diabetes mellitus. J Bone Miner Res (2013) 28:627–38. doi: 10.1002/jbmr.1803
186. McColm J, Hu L, Womack T, Tang CC, Chiang AY. Single- and multiple-dose randomized studies of blosozumab, a monoclonal antibody against sclerostin, in healthy postmenopausal women. J Bone Miner Res (2014) 29:935–43. doi: 10.1002/jbmr.2092
187. Recker RR, Benson CT, Matsumoto T, Bolognese MA, Robins DA, Alam J, et al. A randomized, double-blind phase 2 clinical trial of blosozumab, a sclerostin antibody, in postmenopausal women with low bone mineral density. J Bone Miner Res (2015) 30:216–24. doi: 10.1002/jbmr.2351
188. Recknor CP, Recker RR, Benson CT, Robins DA, Chiang AY, Alam J, et al. The effect of discontinuing treatment with blosozumab: follow-up results of a phase 2 randomized clinical trial in postmenopausal women with low bone mineral density. J Bone Miner Res (2015) 30:1717–25. doi: 10.1002/jbmr.2489
189. Rauner M, Taipaleenmaki H, Tsourdi E, Winter EM. Osteoporosis treatment with anti-sclerostin antibodies-mechanisms of action and clinical application. J Clin Med (2021) 10. doi: 10.3390/jcm10040787
190. Farrell M, Fairfield H, Costa S, D’Amico A, Falank C, Brooks DJ, et al. Sclerostin-neutralizing antibody treatment rescues negative effects of rosiglitazone on mouse bone parameters. J Bone Miner Res (2021) 36:158–69. doi: 10.1002/jbmr.4170
191. Fairfield H, Falank C, Harris E, Demambro V, McDonald M, Pettitt JA, et al. The skeletal cell-derived molecule sclerostin drives bone marrow adipogenesis. J Cell Physiol (2018) 233:1156–67. doi: 10.1002/jcp.25976
192. Jackuliak P, Kuzma M, Payer J. Effect of antidiabetic treatment on bone. Physiol Res (2019) 68:S107–20. doi: 10.33549/physiolres.934297
193. Chen JS, Sambrook PN. Antiresorptive therapies for osteoporosis: a clinical overview. Nat Rev Endocrinol (2011) 8:81–91. doi: 10.1038/nrendo.2011.146
194. Weivoda MM, Chew CK, Monroe DG, Farr JN, Atkinson EJ, Geske JR, et al. Identification of osteoclast-osteoblast coupling factors in humans reveals links between bone and energy metabolism. Nat Commun (2020) 11:87. doi: 10.1038/s41467-019-14003-6
195. Bonnet N, Bourgoin L, Biver E, Douni E, Ferrari S. RANKL inhibition improves muscle strength and insulin sensitivity and restores bone mass. J Clin Invest (2019) 129:3214–23. doi: 10.1172/JCI125915
196. Beekman KM, Akkerman EM, Streekstra GJ, Veldhuis-Vlug AG, Acherman Y, Gerdes VE, et al. The effect of roux-en-y gastric bypass on bone marrow adipose tissue and bone mineral density in postmenopausal, nondiabetic women. Obes (Silver Spring) (2021) 29:1120–7. doi: 10.1002/oby.23171
197. Limonard EJ, Veldhuis-Vlug AG, van Dussen L, Runge JH, Tanck MW, Endert E, et al. Short-term effect of estrogen on human bone marrow fat. J Bone Miner Res (2015) 30:2058–66. doi: 10.1002/jbmr.2557
198. Syed FA, Oursler MJ, Hefferanm TE, Peterson JM, Riggs BL, Khosla S. Effects of estrogen therapy on bone marrow adipocytes in postmenopausal osteoporotic women. Osteoporos Int (2008) 19:1323–30. doi: 10.1007/s00198-008-0574-6
199. Schwartz AV, Sigurdsson S, Hue TF, Lang TF, Harris TB, Rosen CJ, et al. Vertebral bone marrow fat associated with lower trabecular BMD and prevalent vertebral fracture in older adults. J Clin Endocrinol Metab (2013) 98:2294–300. doi: 10.1210/jc.2012-3949
200. Baum T, Yap SP, Karampinos DC, Nardo L, Kuo D, Burghardt AJ, et al. Does vertebral bone marrow fat content correlate with abdominal adipose tissue, lumbar spine bone mineral density, and blood biomarkers in women with type 2 diabetes mellitus? J Magn Reson Imaging (2012) 35:117–24. doi: 10.1002/jmri.22757
201. Duque G, Li W, Adams M, Xu S, Phipps R. Effects of risedronate on bone marrow adipocytes in postmenopausal women. Osteoporos Int (2011) 22:1547–53. doi: 10.1007/s00198-010-1353-8
202. Elbaz A, Rivas D, Duque G. Effect of estrogens on bone marrow adipogenesis and Sirt1 in aging C57BL/6J mice. Biogerontology (2009) 10:747–55. doi: 10.1007/s10522-009-9221-7
203. Martin RB, Chow BD, Lucas PA. Bone marrow fat content in relation to bone remodeling and serum chemistry in intact and ovariectomized dogs. Calcif Tissue Int (1990) 46:189–94. doi: 10.1007/BF02555043
204. Edelstein SL, Barrett-Connor E. Relation between body size and bone mineral density in elderly men and women. Am J Epidemiol (1993) 138:160–9. doi: 10.1093/oxfordjournals.aje.a116842
205. Jankowska EA, Rogucka E, Medras M. Are general obesity and visceral adiposity in men linked to reduced bone mineral content resulting from normal ageing? A population-based study. Andrologia (2001) 33:384–9. doi: 10.1046/j.1439-0272.2001.00469.x
206. Yamaguchi T, Kanazawa I, Yamamoto M, Kurioka S, Yamauchi M, Yano S, et al. Associations between components of the metabolic syndrome versus bone mineral density and vertebral fractures in patients with type 2 diabetes. Bone (2009) 45:174–9. doi: 10.1016/j.bone.2009.05.003
207. Greco EA, Fornari R, Rossi F, Santiemma V, Prossomariti G, Annoscia C, et al. Is obesity protective for osteoporosis? Evaluation of bone mineral density in individuals with high body mass index. Int J Clin Pract (2010) 64:817–20. doi: 10.1111/j.1742-1241.2009.02301.x
208. Reid IR, Ames R, Evans MC, Sharpe S, Gamble G, France JT, et al. Determinants of total body and regional bone mineral density in normal postmenopausal women–a key role for fat mass. J Clin Endocrinol Metab (1992) 75:45–51. doi: 10.1210/jcem.75.1.1619030
209. Slemenda CW, Hui SL, Longcope C, Johnston CC Jr. Cigarette smoking, obesity, and bone mass. J Bone Miner Res (1989) 4:737–41. doi: 10.1002/jbmr.5650040513
210. Goulding A, Taylor RW. Plasma leptin values in relation to bone mass and density and to dynamic biochemical markers of bone resorption and formation in postmenopausal women. Calcif Tissue Int (1998) 63:456–8. doi: 10.1007/s002239900557
211. Ricci TA, Chowdhury HA, Heymsfield SB, Stahl T, Pierson RN Jr., Shapses SA. Calcium supplementation suppresses bone turnover during weight reduction in postmenopausal women. J Bone Miner Res (1998) 13:1045–50. doi: 10.1359/jbmr.1998.13.6.1045
212. Heidari B, Hosseini R, Javadian Y, Bijani A, Sateri MH, Nouroddini HG. Factors affecting bone mineral density in postmenopausal women. Arch Osteoporos (2015) 10:15. doi: 10.1007/s11657-015-0217-4
213. El Maghraoui A, Sadni S, El Maataoui A, Majjad A, Rezqi A, Ouzzif Z, et al. Influence of obesity on vertebral fracture prevalence and vitamin D status in postmenopausal women. Nutr Metab (Lond) (2015) 12:44. doi: 10.1186/s12986-015-0041-2
214. Andreoli A, Bazzocchi A, Celi M, Lauro D, Sorge R, Tarantino U, et al. Relationship between body composition, body mass index and bone mineral density in a large population of normal, osteopenic and osteoporotic women. Radiol Med (2011) 116:1115–23. doi: 10.1007/s11547-011-0689-2
215. Karimifar M, Pasha MA, Salari A, Zamani A, Salesi M, Motaghi P. Evaluation of bone loss in diabetic postmenopausal women. J Res Med Sci (2012) 17:1033–8.
216. Yanik B, Ayrim A, Ozol D, Koktener A, Gokmen D. Influence of obesity on bone mineral density in postmenopausal asthma patients undergoing treatment with inhaled corticosteroids. Clinics (Sao Paulo) (2009) 64:313–8. doi: 10.1590/s1807-59322009000400008
Keywords: aging, osteoporosis, bone marrow adiposity, bone fragility, type 2 diabetes (T2D), obesity
Citation: Ali D, Tencerova M, Figeac F, Kassem M and Jafari A (2022) The pathophysiology of osteoporosis in obesity and type 2 diabetes in aging women and men: The mechanisms and roles of increased bone marrow adiposity. Front. Endocrinol. 13:981487. doi: 10.3389/fendo.2022.981487
Received: 29 June 2022; Accepted: 29 August 2022;
Published: 15 September 2022.
Edited by:
Jason Horton, Upstate Medical University, United StatesReviewed by:
Elizabeth Rendina-Ruedy, Vanderbilt University Medical Center, United StatesDrenka Trivanović, University Hospital Würzburg, Germany
Copyright © 2022 Ali, Tencerova, Figeac, Kassem and Jafari. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Dalia Ali, ZGF5ZXNoQGhlYWx0aC5zZHUuZGs=; Abbas Jafari, YWphZmFyaUBzdW5kLmt1LmRr