 Zemin Lv
Zemin Lv Yunyun Yu1
Yunyun Yu1 Xiaowen Mao
Xiaowen Mao Shigang Cheng
Shigang Cheng
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Endocrinol., 12 September 2022
Sec. Pediatric Endocrinology
Volume 13 - 2022 | https://doi.org/10.3389/fendo.2022.977105
This article is part of the Research TopicPediatric Adrenal NeoplasmsView all 12 articles
Objective: To investigate the clinicopathological characteristics and long-term survival outcomes of pediatric adrenal malignancies.
Method: This study retrospectively analyzed children with pathologically confirmed pediatric adrenal malignancies from Surveillance, Epidemiology, and End Results Database from 2000 to 2019. Kaplan-Meier curve was used to assess the overall survival (OS) and cancer-special survival (CSS), and the Log-Rank method was used to calculate statistical differences. Cox proportional hazards model and Fine-and-Grey model were used to calculate the hazard ratio (HR) of all-cause mortality risk and the sub-distribution HR (sHR) of disease-specific mortality risk, respectively, and their corresponding 95% confidence intervals (CI).
Results: 1601 children were included in the study in which 1335 (83.4%) neuroblastoma, 151 (9.4%) ganglioneuroblastoma, 89 (5.6%) adrenocortical carcinoma, and 26 (1.6%) were diagnosed with other types malignancies. Metastatic disease accounted for the largest proportion (69.3%), and the proportion of metastases diagnosed by neuroblastoma was higher than that of adrenocortical carcinoma and ganglioneuroblastoma (73.9% vs. 45.7% vs. 47.2%). The 5-year OS and CSS of all cohort were 69.5% and 70.5%, respectively. Adrenal cortical carcinoma had the worst prognosis, with 5-year OS and CSS of 52.5% and 53.1%, respectively. Patients in recent years had no better OS and CSS than in previous years at diagnosis. The tumor stage remained the main prognostic predictor. Compared to metastatic adrenal tumors, the risk of all-cause mortality (adjusted HR: 0.12, 95% CI: 0.06-0.25, P < 0.001) and the risk of disease-specific mortality (adjusted sHR: 0.11, 95% CI: 0.05-0.25, P<0.001) was significantly lower for patients with localized diseases. Additionally, higher age, adrenal cortical carcinoma, and lack of complete tumor resection are independent risk factors for poor prognosis. Furthermore, it was found that the prognosis of patients who received chemotherapy was worse than those who did not, mainly because the former mostly had metastasis at the presentation and complete resection of the tumor cannot be achieved.
Conclusion: The clinicopathological characteristics of pediatric adrenal malignancies have not changed significantly in the past two decades, while the prognosis of patients has improved. Early diagnosis of disease and complete resection of local tumors are the keys to improving prognosis.
Pediatric malignancies of adrenal glands are rare but have become one of the leading causes of death in pediatrics (1, 2). Their onset is usually stealthy, discovered incidentally, or identified by recognizing symptoms associated with excess hormone secretion (3, 4). Neuroblastoma, ganglioneuroblastoma, and adrenal cortical cancer account for most pediatric adrenal malignancies. Adrenal cortical cancer rarely occurs and has a poor prognosis (5–8). Pediatric malignant patients have benefited from the rapid progress in tumor treatment as the optimization of multimodality therapy and early tumor screening has dramatically improved the survival rate of pediatric malignant tumors (9–13). Given the low incidence of pediatric adrenal tumors, the small sample size, and the short follow-up time of prior studies on pediatric adrenal malignancies, the clinicopathological characteristics, treatment status, and prognosis are not well understood (2, 4, 11–15). Here, we used the Surveillance, Epidemiology, and End Results (SEER) database to study pediatric malignancies of adrenal glands to explore the long-term follow-up survival outcomes and prognostic risk analysis of pediatric adrenal malignancies.
All data were obtained from the SEER database of 18 registries (https://seer.cancer.gov/). Since SEER databases were anonymized and were not associated with human research, therefore, the need for ethics approval was waived by the Ethics Review Board of our institute. As a retrospective study, the patients diagnosed with primary adrenal malignancy from 2000 to 2019 were identified. Patients ≤19 years and having adrenal cortical carcinoma, adrenal ganglioneuroblastoma, adrenal neuroblastoma, and other non-common tumors were identified. All diagnoses were confirmed by histology and not by autopsy or death certification, and all patients had a detailed cause of death and follow-up data. Cases with missing values were excluded.
The study mainly included the following variables for data analysis: year of diagnosis (2000-2004, 2005-2009, 2010-2014, and 2015-2019), age at diagnosis (0-4 years, 5-9 years, 10-14 year and 15+ year), gender (male and female), race (white, black and other), median household income ($0-$59999, $60000-$69999 and $70000+), residence locality (metropolitan and non-metropolitan), tumor size, tumor stage (distant, localized and regional), metastatic site (bone, brain, liver, lung), surgery and approaches (none, local tumor destruction/excision, radical surgery with or without other organs and simple/partial surgical removal), metastatic surgery Treatment (yes or no), chemoradiation (yes or no).
The continuous variables were described as mean ± standard deviation (SD) for data with a normal distribution and compared with the student t-test. For non-normal distribution, the Wilcoxon rank-sum test was used to compare the data, and the data were described as median and interquartile range (IQR). Classification variables were represented by frequency (%) and compared by the chi-square test. OS and CSS were the primary endpoints of interest for this study. Kaplan-Meier was used to calculate OS and CSS. The hazard ratio (HR), the Sub-distribution hazard ratio (sHR), and the corresponding 95% confidence Interval (95% CI) of all-cause death and adrenal tumor-specific death were calculated by Cox proportional hazards model and Fine and Grey model, respectively. Logistic regression analysis was done to calculate odds ratios (OR) and their corresponding 95% CI for factors associated with treatment choice. All analyses were conducted using R (Version 42.0). P < 0.05 (two-sided) was considered statistically significant.
Our study included 1601 patients with adrenal-derived malignant tumors. The primary histological type was neuroblastoma (1335 cases, 83.4%). Ganglioneuroblastoma and adrenal cortical carcinoma were the other two main pathological types, accounting for 9.4% and 5.6%, respectively, whereas other rare cancers accounted for only 1.6% (Table 1 and Figure 1A). The incidence of neuroblastoma did not change much from 2000 to 2019 (Figure 1B). Most cases had an onset in the age group of 0-4 years, accounting for 79.9% of the cases. Neuroblastoma accounted for the largest proportion (86.4%) in 0-4 years old. However, ganglioneuroblastoma and adrenal cortical carcinoma accounted for 55.6% and 41.5%, respectively; furthermore, the proportion of other rare cases was 19.2% in the older age groups (Table 1).
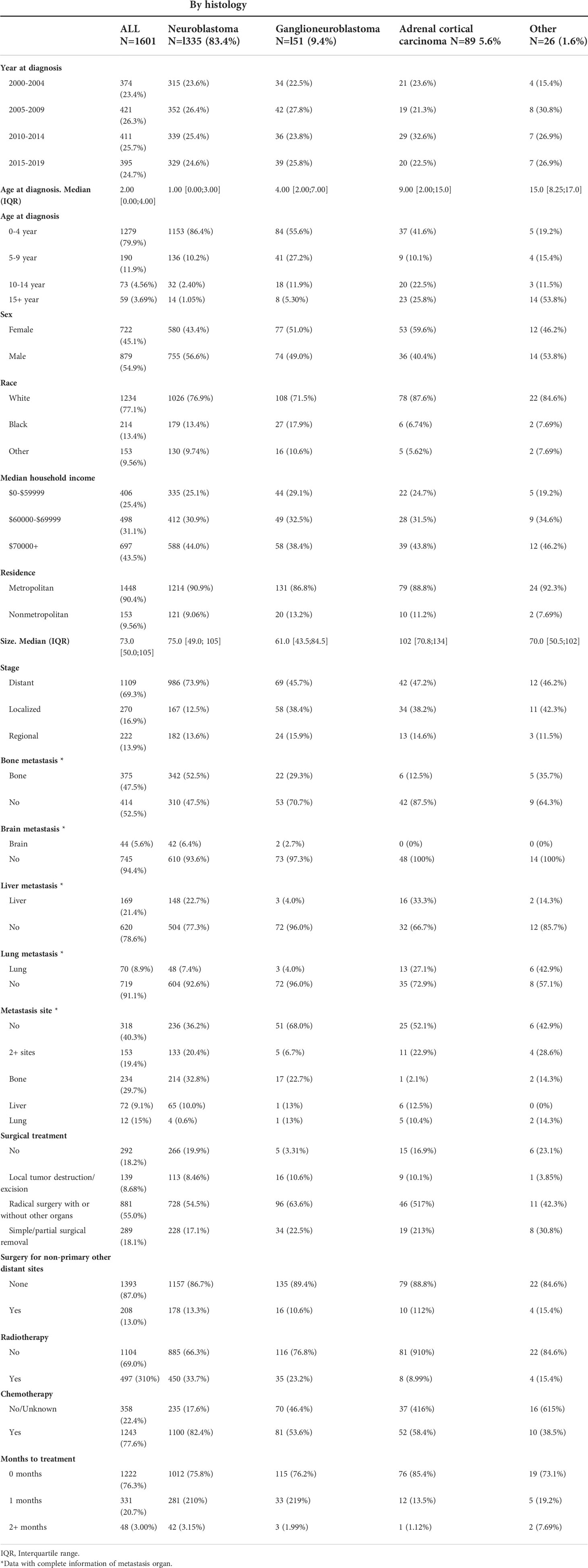
Table 1 Baseline characteristics of the study cohort.
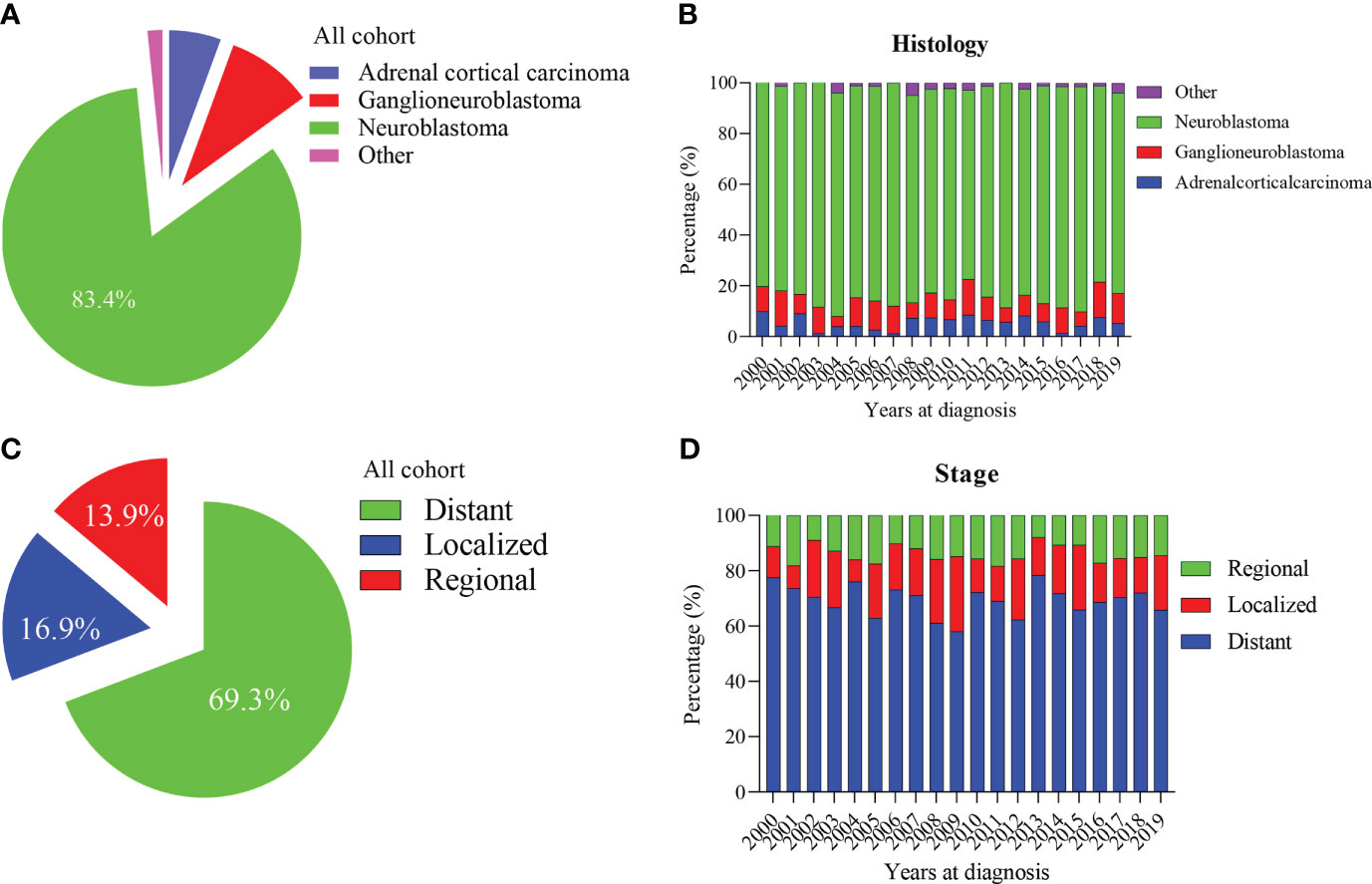
Figure 1 Percentage of pediatric adrenal malignancies by histology and tumor stage. (A, B) Percentage of histology in all cohorts and stratified by year at diagnosis. (C, D) Percentage of tumor stage in all cohorts and stratified by year at diagnosis.
The median tumor size (102mm) of adrenal cortical carcinoma was larger than other tumor types (neuroblastoma- 75mm, ganglioneuroblastoma- 61mm and others- 70mm). Most of the children had metastatic diseases at the time of diagnosis (Figure 1C). This condition has not changed much in these two decades (Figure 1D). Patients with neuroblastoma present diststant metastasis accounted for 73.9%, while Ganglioneuroblastoma and adrenal cortical carcinoma accounted for 45.7% and 47.2%, respectively. Bone was the most common metastasis site (about 80.0% of all metastasis cases, data did not present here). Figure 2A shows that about 70% of children under 9 years old had disease metastasis at the time of treatment (0 – 4 years old: 70.1%, 5 – 9 years old: 71.1%); although the proportion of children over 10 years old with metastasis decreased, it still more than the general children with disease metastasis (10-14 years old: 60.3%; 15-19 years old: 55.9%). However, we found that with the increase in age, the proportion of adrenal cortical carcinoma patients with metastasis increased (Figure 2B). The proportion of metastasis from adrenal neuroblastoma is higher in all age stages. In contrast, the proportion of metastatic lesions in Ganglioneuroblastoma is lower (Figures 2C, D).
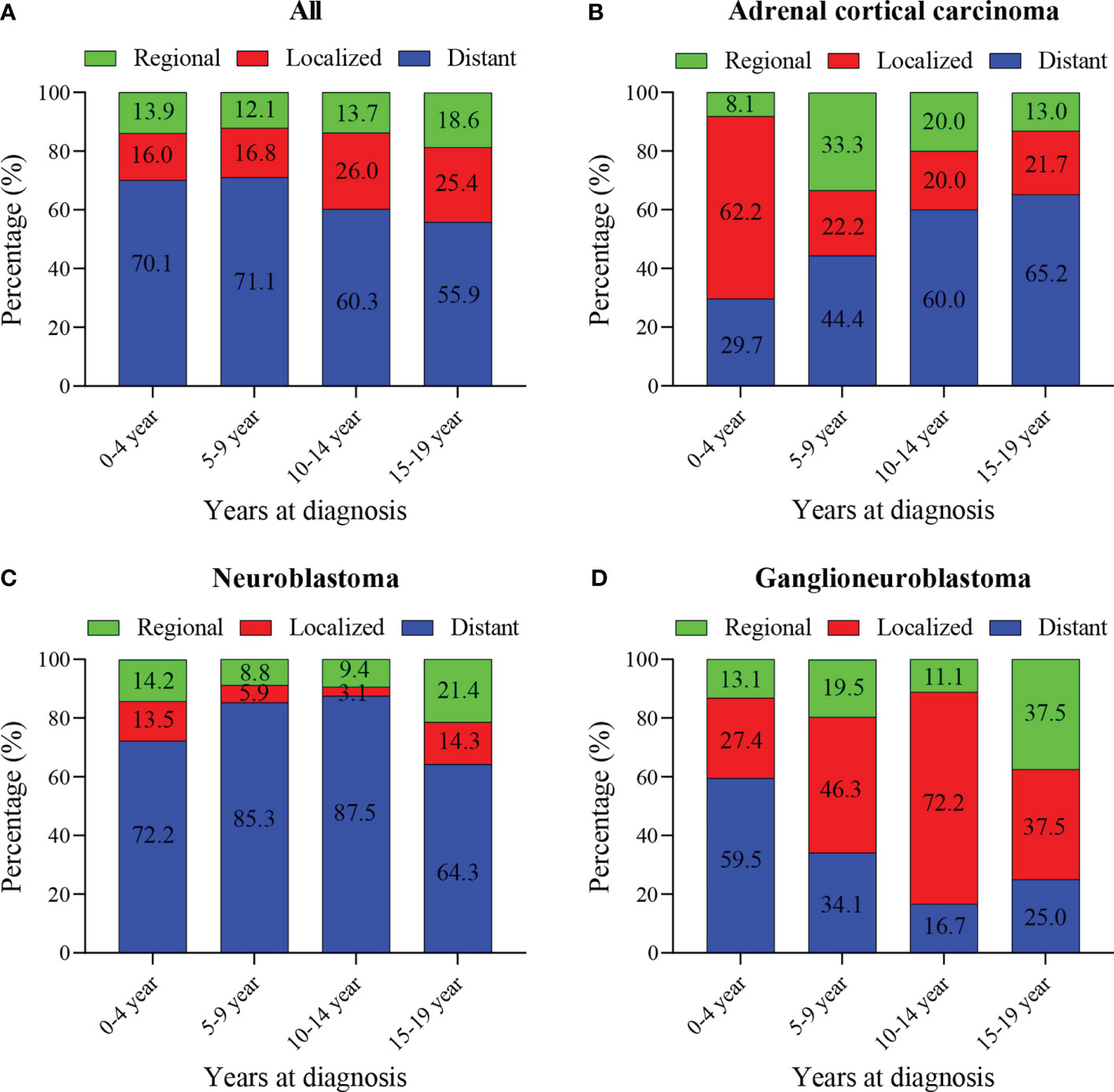
Figure 2 Percentage of pediatric adrenal malignancies by age at diagnosis, tumor stage and histology. (A) Percentage of stage in all cohorts and stratified by age at diagnosis. (B) Percentage of stage in Adrenal cortical carcinoma cohorts and stratified by age at diagnosis. (C) Percentage of stage in neuroblastoma cohorts and stratified by age at diagnosis. (D) Percentage of stage in ganglioneuroblastoma cohorts and stratified by age at diagnosis.
Most children received surgical treatment (8.68% local tumor destruction/excision, 55.0% radical surgery with or without other organs, and 18.1% simple/partial surgical removal), and a few did not (18.2%). At the same time, 77.6% and 31.0% of patients received chemotherapy and radiotherapy, respectively. Additionally, 13.0% of patients also received surgical treatment for distant lymph node or organ metastasis, and most (76.3%) patients underwent either surgery or radiotherapy and chemotherapy within 1 month after diagnosis (Table 1).
Our results showed that OS and CSS didn’t change over the years (200-2019) (Figures 3A, B). The OS and CSS of neuroblastoma and ganglioneuroblastoma was better than adrenal cortical carcinoma (Figures 3C, D). The 5-year OS of the total population was 69.5% (95% CI: 67.1% – 72.0%, Table 2). The 5-year OS of metastatic patients was 59.3%, while that of local tumor patients was 89.3%. (Table 2). Table 3 summarizes the risk factors of disease-specific death in 5-year CSS of different subgroups. Our results show that the overall 5-year CSS was 70.5% (95% CI: 68.1% -73.0%). The multivariate COX model showed that the age, histological type, tumor size, tumor stage, and treatment were independent risk factors for both, all-cause mortality and disease-specific death. Although in the recent year of the diagnosis had a lower risk of mortality compared to the prior years, the year of diagnosis was neither a significant risk predictor of all-cause mortality (Figure 4A) nor disease-cause mortality (Figure 4B).
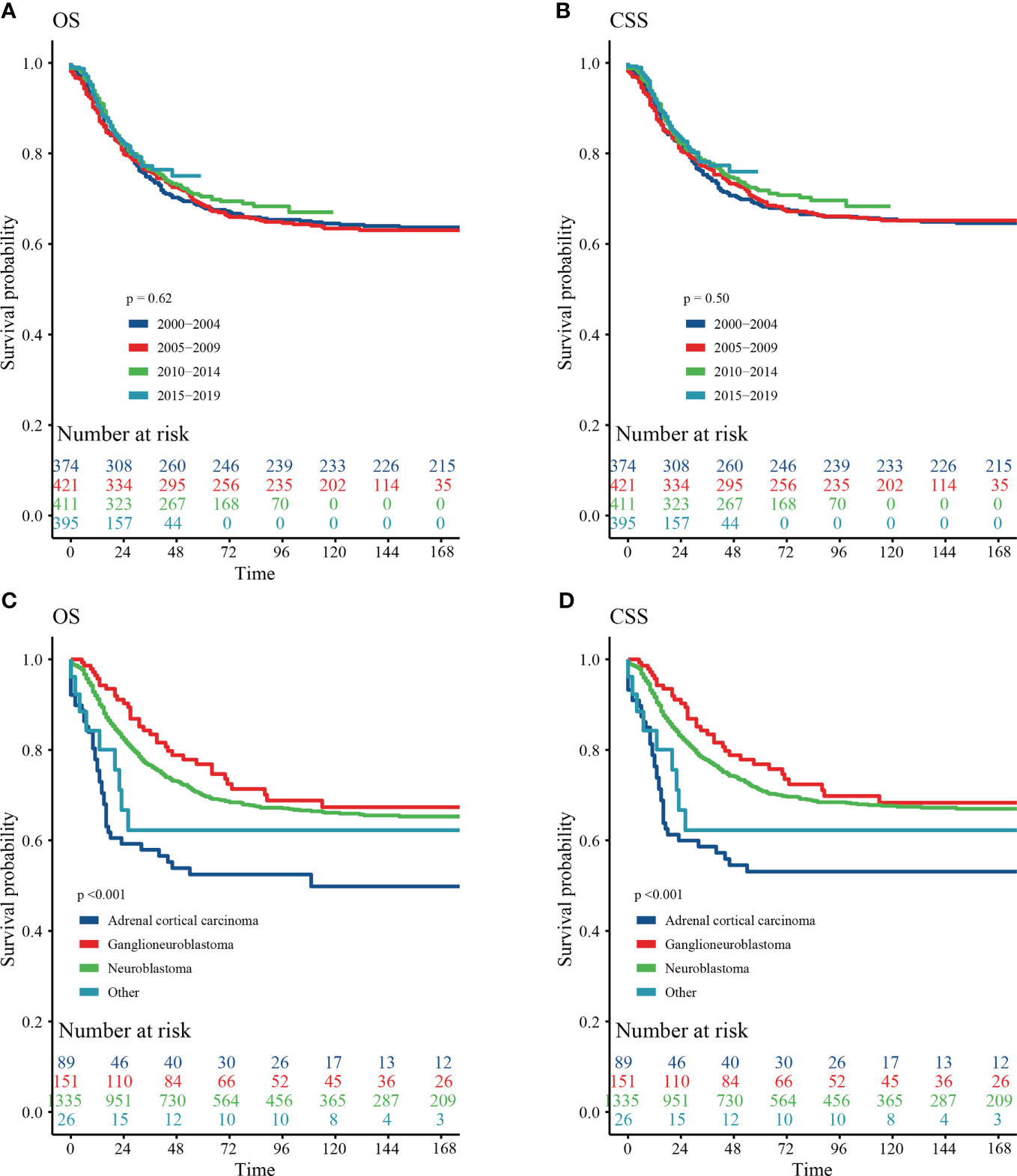
Figure 3 Survival curve of analyzed by Kaplan–Meier method. (A) Overall survival (OS) stratified by year at diagnosis. (B) Cancer-special survival (CSS) stratified by year at diagnosis. (C) OS stratified by histology. (D) CSS stratified by histology.
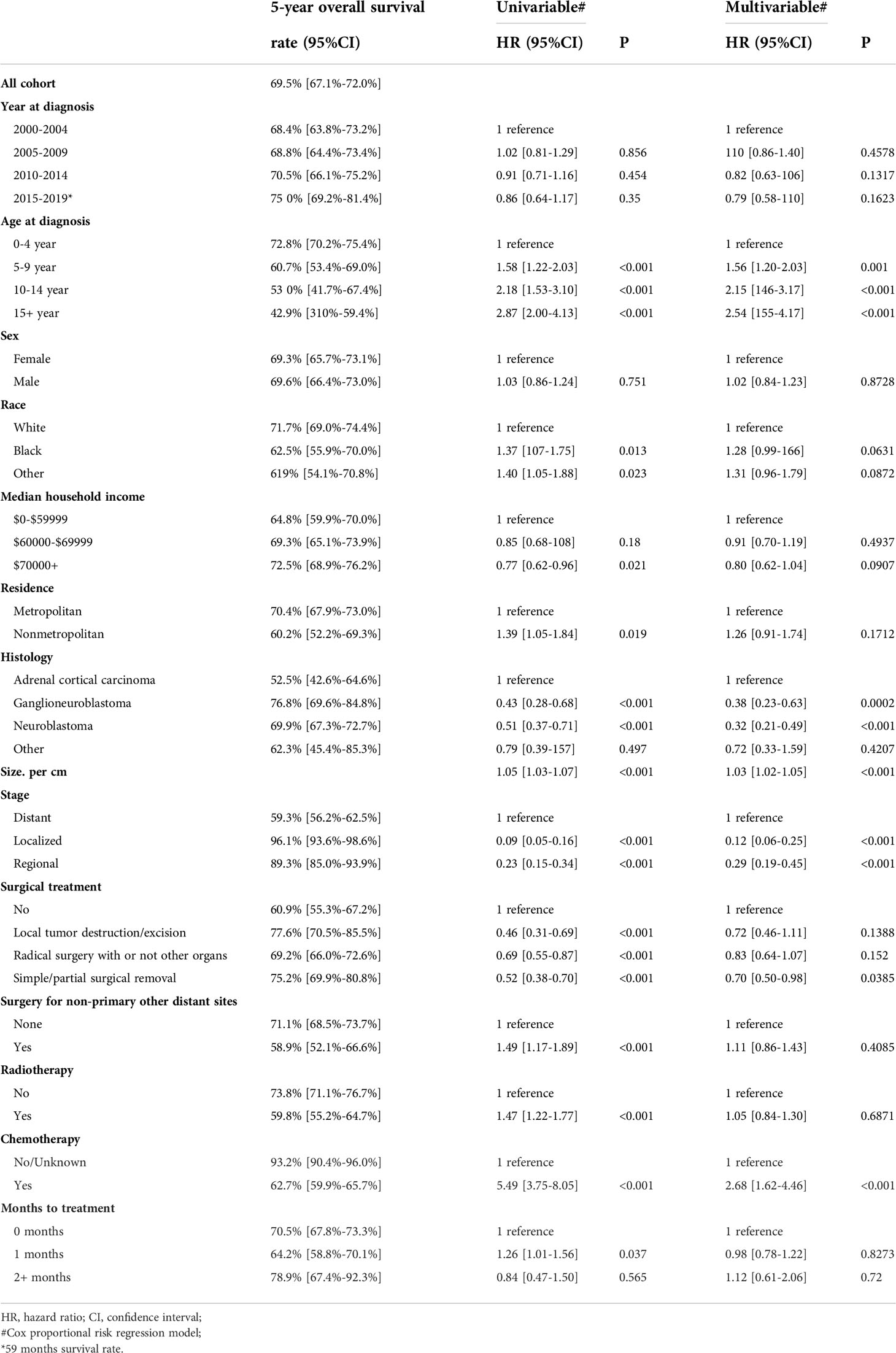
Table 2 Five-year overall survival and predictors of all-cause mortality.
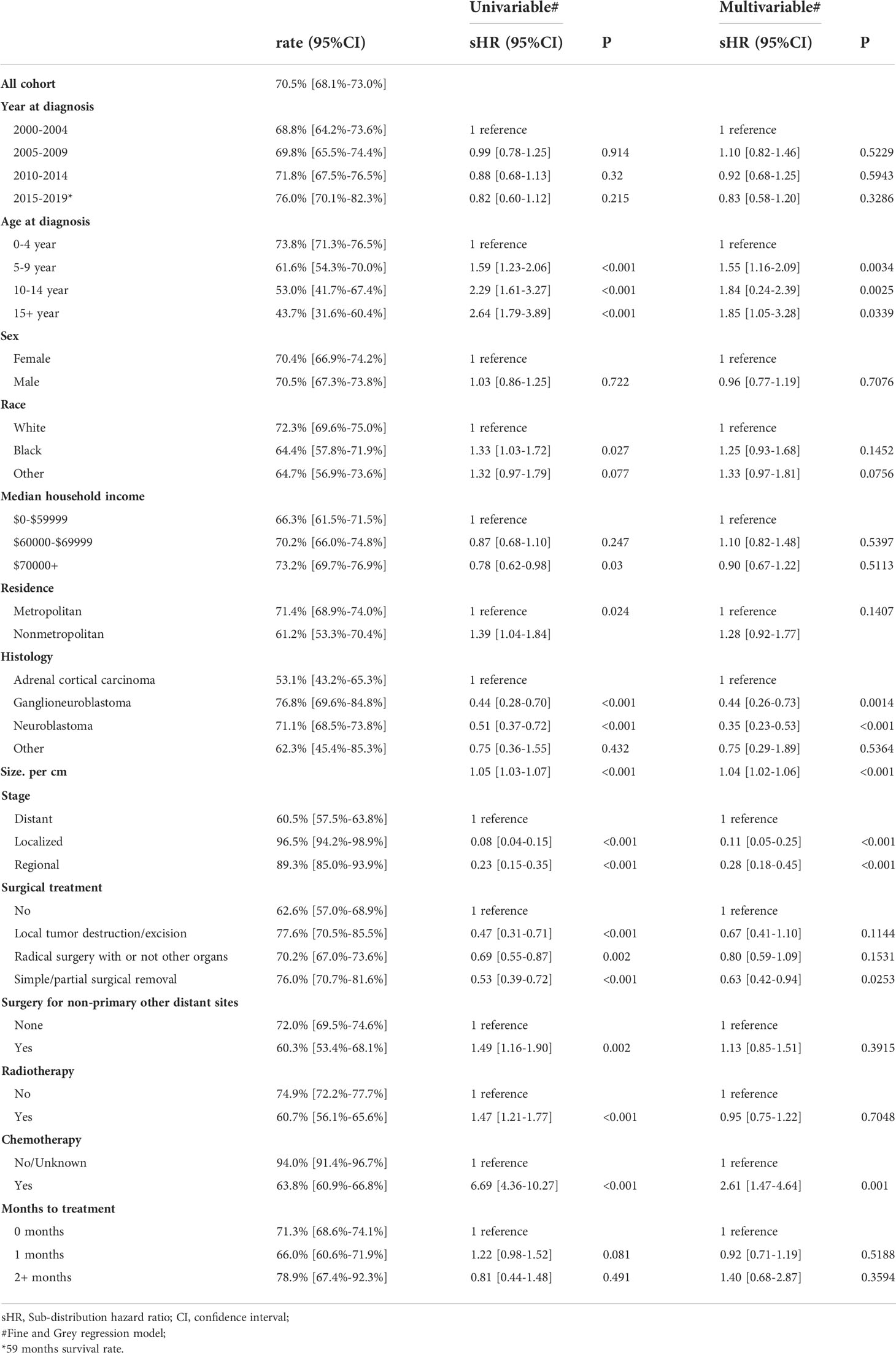
Table 3 Five-year cancer-sQecial survival and Qredictors of mortality by adrenal cancer.
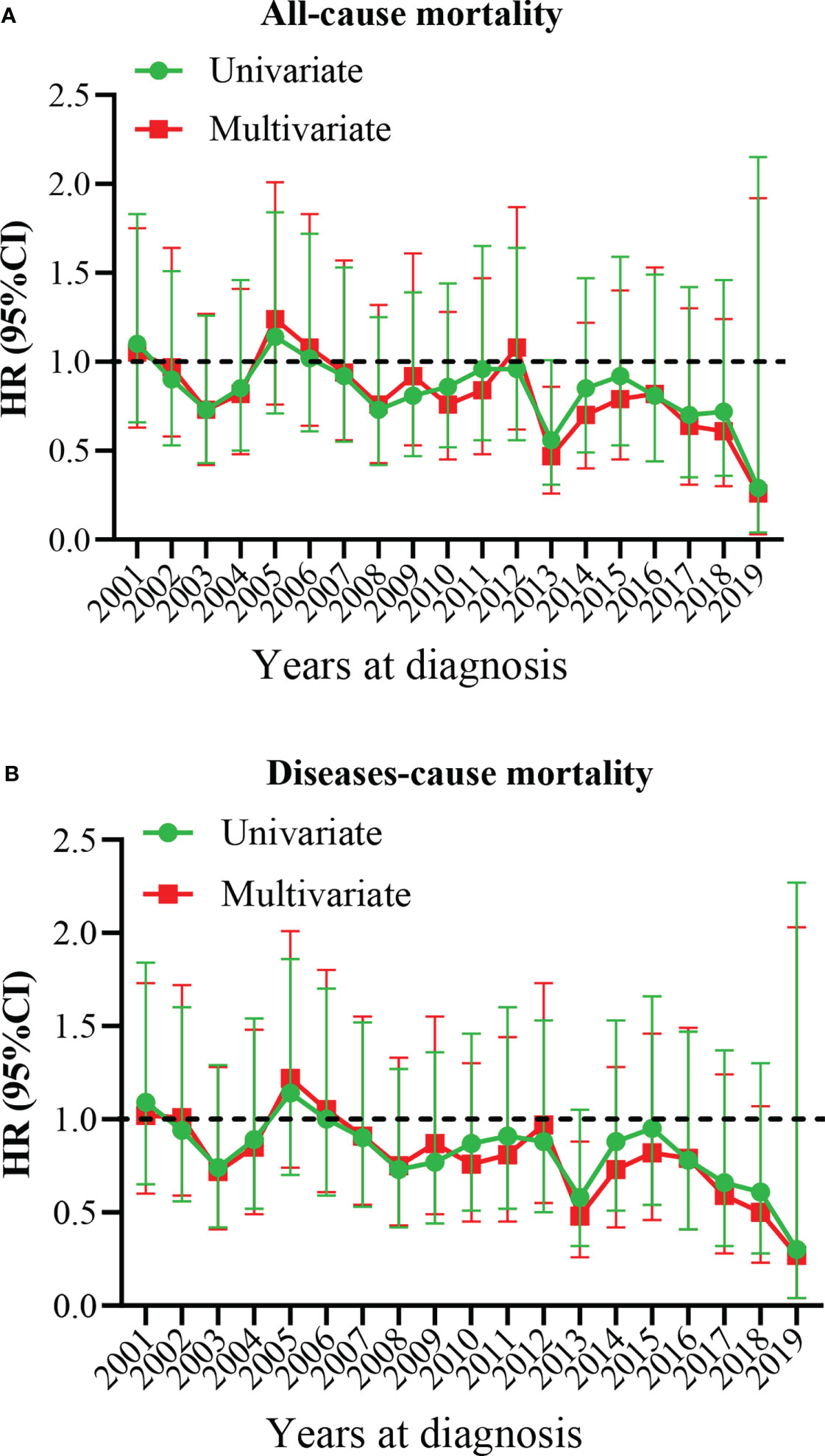
Figure 4 Multivariate hazard ratio (HR) analysis for all-cause mortality and diseases-cause mortality. (A) All-cause mortality. (B) Diseases-cause mortality. The year 2000 at diagnosis was considered as a reference.
It’s puzzling that the survival analysis showed that chemotherapy did not benefit patients but increased the risk of death. However, it should be noted that chemotherapy was significantly correlated with the recent year of diagnosis, younger age of patients at diagnosis, metropolitan patient’s, larger tumor size, tumor with distant disease, nonsurgical treatment performed (Supplementary Table 1).
Considering the differences in treatment and prognosis between adrenocortical carcinoma and adrenal neuroblastoma/ganglioneuroblastoma, we will perform survival analysis for the two subgroups separately. We found that OS in the adrenal neuroblastoma/ganglioneuroblastoma subgroups correlated with patient age (the older the age, the worse the OS), race (worse in blacks than whites), and tumor stage (worse in metastatic cases than in patients without metastases) (Supplementary Table 2). However, only tumor stage and complete tumor resection were independent prognostic factors for adrenocortical carcinoma (Supplementary Table 3).
Neuroblastoma is the most common pathological type of adrenal malignancy in children (7, 16), and approximately 46% of neuroblastomas arise from the adrenal gland (17). The primary site of the adrenal neuroblastoma is often concealed and extensive, but its biological behavior shows high malignancy and rapid growth and is prone to multiple early metases (18). Our study found that ganglioneuroblastoma and neuroblastoma accounted for more than 80% of metastatic lesions. Since the clinical symptoms and signs of pediatric neuroblastoma are not specific, they are hard to diagnose. Hence, diagnosis is often missed, delaying treatment and losing the best time for the treatment. Therefore, delayed diagnosis is a major factor for the poor prognosis of neuroblastoma. With the improvement of multidisciplinary comprehensive treatment, the survival rate of patients has improved to a certain extent. However, our findings found that there was no statistically significant reduction in the risk of death in patients diagnosed recently compared with patients with pediatric adrenal malignancies diagnosed in the past.
Neuroblastoma is an embryonal tumor originating from primitive neural crest cells of the sympathetic nervous system. It affects the normal development of the adrenal medulla and paravertebral sympathetic ganglia, although its exact pathogenesis is still unclear (19). MYCN amplification is associated with the development of neuroblastoma. Numerous studies have shown that MYCN amplification correlates with neuroblastoma progressing rapidly, leading to a worse prognosis (20, 21). Therefore, drugs inhibiting MYCN amplification, such as retinoic acid, are used for maintenance therapy in high-risk children (22, 23).
Furthermore, the 1p, 11q loss, and 17q gain are also common chromosomal abnormalities in neuroblastoma (24–26). 11q aberration is reported in 20 to 45% of neuroblastoma, and 1p and 11q loss can render neuroblastomas in an undifferentiated or poorly differentiated state, increasing the risk of recurrence and leading to a poor prognosis (21, 25, 27, 28). Most children with neuroblastoma develop a 17q gain (80.4%), which, together with other unfavorable prognostic factors, contributes to the poor prognosis of neuroblastoma (21, 24).
Neuroblastoma may show biochemical abnormalities in blood and urine. Neuron-specific enolase (NSE) is an acid protease unique to neurons and neuroendocrine cells and a specific marker of neuroendocrine tumors (29, 30). NSE is expressed in neurons and is highly sensitive and specific to neuroblastoma and ganglioneuroblastoma. Its elevation often indicates advanced disease progression and poor prognosis. Studies have shown that NSE has a high positive expression rate in undifferentiated neuroblastoma and has high sensitivity and accuracy in monitoring tumor recurrence, especially in metastatic tumors. Urinary vanillylmandelic acid (VMA) is also an important indicator of early diagnosis of neuroblastoma (30, 31). A vast majority of neuroblastomas can also be accompanied by abnormal metabolism of catecholamines in the body. It can directly secrete the precursor substances of VMA, increasing its concentration in the blood and the urine. Lactate dehydrogenase (LDH) is an essential enzyme in the glycolytic pathway. The tumor tissue has the characteristics of high metabolism, and the serum LDH level can be used as an important indicator representing the tumor cell burden in the whole body, which is of great significance for the prognosis of neuroblastoma (29, 32, 33).
Surgically resecting the whole tumor tissue is the key to treating pediatric adrenal tumors (17). Our study showed that achieving complete tumor resection improves patient outcomes. However, when complete tumor resection cannot be performed, local surgery did not lead to better outcomes than patients without surgery. Without adjuvant radiotherapy and chemotherapy, children with localized diseases can achieve long-term survival after complete tumor resection and no surgical complications alone. However, in the diagnosis of pediatric adrenal malignancies, the tumor is often large, adheres tightly to the surrounding tissues, and even surrounds important blood vessels such as the inferior vena cava, making it difficult to operate on the tumor and remove it altogether. Blind pursuit of complete resection may also lead to more severe complications and affect the patient’s survival.
Patients with intermediate-risk tumors may receive chemotherapy to shrink the tumor before surgical removal (17, 34). Preoperative chemotherapy can significantly shrink the tumor, thicken the capsule, reduce the clinical stage of the tumor, and inhibit distant metastasis. The treatment model of neoadjuvant chemotherapy, surgical resection, adjuvant high-dose chemotherapy with hematopoietic stem cell rescue, and radiation therapy has become a classic mode of treatment for high-risk neuroblastoma (17, 35, 36). Chemotherapy can easily aggravate the degree of adhesion between the tumor and surrounding tissues and increase the difficulty of surgical resection. Moreover, chemotherapy can confuse or change the pathological staging, and special attention should be paid to the timing of surgery and chemotherapy. Prior studies have also found that surgical resection is vital in predicting outcomes for high-risk neuroblastomas that do not show a clinical response to induction treatment. However, gross total resection versus subtotal resection did not affect these outcomes (37).
Neuroblastoma is more sensitive to chemotherapy, but at the same time, it is necessary to pay attention to the toxic side effects of chemotherapy on children. Although high-intensity chemotherapy is used only for high-risk groups of neuroblastomata, the toxic side effects on children are significant and need vigilant analysis (38). An international, randomized, multi-arm, open-label, phase 3 trial found that Busulfan and melphalan improved event-free survival in children with high-risk neuroblastoma with an adequate response to induction treatment and caused fewer severe adverse events than carboplatin, etoposide, and melphalan treatment (38). A reasonable grasp of the intensity of chemotherapy is critical to avoid the effects of insufficient intensity in high-risk children and excessive intensity in low-risk children on prognosis.
In addition to surgical resection, radiotherapy can be a crucial treatment for neuroblastoma tumors (34). Local radiotherapy has an inhibitory effect on the recurrence of neuroblastoma. For high-risk patients, radiotherapy for primary and metastatic lesions can control the patient’s condition. Also, Iodine-131 metaiodobenzylguanidine (131I-MIBG) and radiolabeled DOTA-conjugated peptides have been reported to be useful for neuroblastoma treatment (39, 40). Because neuroblastoma cells highly express ganglioglyceride GD2 antibodies, they can be used as a better target for immunotherapy. Immunotherapy with an anti-GD2 antibody with GM-CSF, interleukin-2, and isotretinoin was associated with a significantly improved outcome compared with standard therapy in patients with high-risk neuroblastoma (41).
Adrenal cortical carcinoma in children is a rare adrenal malignancy, and its incidence is lower than that of neuroblastoma and ganglioneuroblastoma (10). In our study, adrenal cortical carcinoma patients only accounted for 5.6% of total patients. However, the degree of malignancy was very high in adrenal cortical carcinoma; the disease developed rapidly and had an infiltrative growth. The diagnosis is that the tumor volume is large and more likely to invade surrounding organs and tissues, making them hard to operate on (42). However, our study found that metastases in adrenocortical carcinoma are less common at the time of diagnosis than in neuroblastoma. The pathogenesis is unclear and can be associated with epigenetic alterations, manifesting as germline TP53 mutations or chromosome 11p abnormalities (8, 43). Compared to adult adrenal cortical carcinoma, pediatric adrenal cortical carcinoma has distinct features. It is closely associated with germline TP53 mutations, which are present in 53% of cases but only in < 10% in adults’ adrenal cortical carcinoma (44). At the time of onset, there may be indicators of excessive secretion of adrenal corticosteroids, such as hypertension, precocious puberty in childhood (manifested as male feminization, female virilization, penis enlargement, pubic hair development, deepening of the voice, breast development, etc.) and Cushing syndrome (manifested as a full moon face, bloody appearance, central obesity, acne, purple striae, hypertension, secondary diabetes, and osteoporosis, etc.). For pediatric adrenocortical carcinoma, 90% of patients presented with evidence of hormonal hypersecretion, and there was an association between the endocrine phenotype and stage (13). The above symptoms may be considered as a warning sign of adrenocortical carcinomas and can be further studied as prognosis indicators.
Currently, the preferred criteria for the diagnosis of pediatric adrenocortical carcinoma are those described by Wieneke et al (45). It mainly includes the following 9 criteria: tumor weight > 400 g; tumor diameter > 10.5 cm; Extension into the periadrenal soft tissue or adjacent organs; invasion into vena cava; vascular invasion; capsular invasion; the presence of tumor necrosis; mitoses > 15 per 20 high-power fields (4 mm2); and presence of atypical mitosis. Adrenocortical carcinoma can be diagnosed if 4 or more of the 9 items are satisfied. If less than or equal to 2 items, it is diagnosed as benign, and if any 3 items are satisfied, it is diagnosed as uncertain malignant potential. In addition, Ki67 has been proposed as an auxiliary biomarker to differentiate childhood adrenal adenoma from adrenal carcinoma and to predict tumor behavior. A Ki67 index of less than 10% is associated with benign disease, and a marker index of greater than 15% is associated with a higher risk of malignancy or adverse outcomes. The p53 gene is an important tumor suppressor gene in the human body. Its wild type makes cancer cells apoptotic, thereby preventing canceration. It has the function of helping cells to repair defects in genes (5, 46). The mutant type of p53 will increase canceration. Multiple studies have found that p53 is overexpressed in adrenocortical carcinomas, whereas it is normal in adrenal adenomas. Adrenocortical carcinomas are present in excess among carriers of germline p53 mutations, and, p53-associated Adrenocortical carcinomas occur predominantly in the pediatric age group (47). Unfortunately, the specific diagnostic criteria, Ki67 index, and p53 mutation of adrenocortical carcinoma could not be provided in our study. This is a limitation of this study that needs to be pointed out. However, the pediatric adrenocortical carcinoma patients included in our study were all pathologically diagnosed under rigorous scrutiny.
Like neuroblastoma, the treatment of adrenocortical carcinoma is still a surgery-based comprehensive treatment. Complete tumor resection to achieve negative margins is an important prognostic factor (8). Stage I patients are curable with surgery alone (48). Retroperitoneal lymph node dissection has failed to improve outcomes in patients with larger tumors (stage II), and its role as an independent treatment strategy is uncertain (13). Combination of surgery and chemotherapy shows good outcomes in patients with stage III adrenocortical carcinoma, but mitotane- and cisplatin-based regimens lead to higher toxicity in patients with metastatic disease, and efficacy in patients with metastatic disease was still poor and should be revised to maximize risk-benefit (13).
Drug therapy is often required to control tumor growth and excessive hormone secretion for patients who cannot achieve complete tumor resection and for patients with recurrence. Mitotane is currently the most effective drug to control adrenal hormone secretion, and it is used alternately with steroid hormones during use. It can be used as a single drug for adjuvant therapy after complete resection of an early tumor, or it can be used in combination with chemotherapy drugs for advanced childhood adrenal cortical carcinoma (13). Mitotane combined with chemotherapy improves the prognosis of patients with advanced adrenocortical carcinoma. However, not all patients can bear the drug’s side effects and can complete all the courses of the treatment as prescribed due to its severe toxicity and side effects (8, 13, 49). The common toxicities of mitotane are nausea, vomiting, diarrhea, and abdominal pain. Drowsiness, lethargy, ataxic gait, depression, and vertigo have also been reported in a few cases. Common chemotherapeutic agents used alone or in combination to treat adrenocortical carcinoma include 5-FU, etoposide, cisplatin, carboplatin, cyclophosphamide, doxorubicin, and streptozotocin. Chemotherapy regimens used to treat childhood adrenocortical carcinoma consist primarily of etoposide and cisplatin, with or without doxorubicin and mitotane. Despite the diverse treatment modalities of chemotherapy, the prognosis for advanced cases remains poor (8). Unfortunately, new therapeutic strategies targeting tumor-specific aberrant pathways have not been studied in pediatric adrenocortical carcinoma. And Radiation therapy in pediatric adrenocortical carcinoma needs further exploration (50).
According to The Children’s Oncology Group ARAR0332 Protocol (13), which is a prospective single-arm risk-stratified interventional study to describe the outcome of stage III or IV pediatric adrenocortical carcinoma patients treated with mitotane and chemotherapy. In the study, eight cycles of chemotherapy, and mitotane for 8 months were used for stages III and IV treatment; at the same time, according to clinical practice, surgical treatment of primary tumors and metastases may be considered as appropriate. The chemotherapy regimen was cisplatin 50 mg/m2/dose for 1-2 days, etoposide 100 mg/m2/dose for 1-3 days, and doxorubicin 25 mg/m2/dose for 4-5 days per cycle. Filgrastim was administered daily at 5 mcg/kg/dose starting on day 6 until neutrophil recovery. Treatment with mitotane is administered daily to adjust the plasma concentration of mitotane to 14-20 μg/mL. During the treatment, nearly one-third of patients were unable to complete their scheduled treatment. Finally, 38 patients have evaluated for toxicity or feasibility analysis and found that 4 patients had mitotane feasibility event (10.5%), and 12 patients had chemotherapy feasibility event (31.6%). It can be seen that it is necessary to modify the treatment plan to improve the tolerability of the treatment. What’s more, combining surgery and chemotherapy has a good prognosis in stage III patients and a poor prognosis in stage IV patients; with a median follow-up for OS of 60 months, the 5-year OS estimates for stages III, and IV were 94.7%, and 15.6%, respectively.
Note that mitotane produces multiple severe side effects on the metabolic and endocrine systems in the treatment of adrenocortical carcinoma, although this effect appears to be treatable and partially reversible. One study analyzed lipid profiles, thyroid hormones, sex hormones, and adrenal function in 50 patients from the first year of mitotane treatment and after discontinuation (51). In their study, they found levels of total cholesterol, LDL, HDL, and triglycerides increased after 6 months of mitotane treatment, and total cholesterol and LDL levels were reduced when statins were given concomitantly, and mitotane was discontinued Lipid can be further reduced; at the same time, it was also found that plasma free thyroxine decreased in the mitotane treatment group, but thyroid-stimulating hormone remained unchanged. The total amount of T4 increased when mitotane was discontinued. Mitotane increases plasma sex hormone-binding globulin and luteinizing hormone and increases the level of testosterone in male patients; in addition, the adrenal function can be recovered after six months of discontinuation of mitotane. Therefore, during the treatment of adrenocortical carcinoma with mitotane, in addition to causing adrenal insufficiency, special attention should be paid to the interference of mitotane on lipid metabolism and endocrine, and it should be corrected to normal levels in time. Patients taking mitotane may require high-dose hydrocortisone replacement therapy, and reducing mitotane can interfere with steroid metabolism (52).
Because of the heterogeneity and rarity of pediatric adrenocortical carcinoma, the short follow-up time, and the fact that most patients are diagnosed at an advanced stage of the disease, it is often challenging to identify prognostic factors. Multiple studies have found that age and tumor stage are two independent predictors (4, 13, 53, 54). Other risk factors include tumor size/tumor volume/weight, surgical treatment, presence of virilization, Cushing syndrome, and hypertension. Germline TP53 status and the presence of a somatic ATRX mutation were also associated with the outcome (8, 13).
This study was retrospective and had a few limitations. Firstly, the research data comes from the SEER database, which has not recorded specific radiotherapy, chemotherapy, or targeted treatment plans and lacks laboratory test results (such as NSE, VMA, LDH, cortisol, etc.) and medical data (mitotane dosage, side effects, need for substitutive treatment with cortisone, fludrocortisone and/or levothyroxine). Additionally, due to missing values, there may be some selection bias in the process of screening cases in the study.
This study retrospectively performed a prognostic analysis of pediatric adrenal malignancies using a large sample size. Neuroblastoma, ganglioneuroblastoma, and adrenocortical carcinoma were the three most common pediatric adrenal malignancies. Patients with neuroblastoma and ganglioneuroblastoma often accompany metastatic lesions at presentation, making treatment challenging. Complete surgical resection of the tumor is the key to ensuring a good prognosis. Radiation and chemotherapy should be given to patients whose tumors cannot be entirely removed by surgery. The prognosis of pediatric adrenal malignancies is related to the age of the patient (older children have a poorer prognosis), type of the pathology (cortical cancer has a poor prognosis), tumor size (the larger the tumor, the worse the prognosis), and the tumor stage (the higher the stage of the patient). Notably, our study demonstrated that chemotherapy patients had a worse prognosis than those who did not. This was mainly related to the fact that patients receiving chemotherapy tend to reside in non-metropolitan regions, the pathological type was neuroblastoma, the tumor diameter was larger, the stage was higher, and the patients had not received surgery. Although the field of oncology have progressed with respect to treatment of pediatric adrenal malignancies, there still no statistically significant reduction in the risk of all-cause mortality and tumor-specific mortality in patients with recently diagnosed pediatric adrenal malignancies compared with patients in the past diagnosis period. Therefore, more experimental studies are needed in this regard urgently and efficiently to save lives
The original contributions presented in the study are included in the article/Supplementary Material. Further inquiries can be directed to the corresponding authors.
ZL, XM, and SC conceived the research. ZL wrote the manuscript. ZL, YY, and YL analyzed the data and prepared the figures and tables. All authors contributed to the article and approved the submitted version.
We thank the SEER database supported by the Surveillance Research Program in the National Cancer Institute’s Division of Cancer Control and Population Sciences. And we thank Bullet Edits Limited for the linguistic editing and proofreading of the manuscript.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fendo.2022.977105/full#supplementary-material
1. Xu X, Sergi C. Pediatric adrenal cortical carcinomas: Histopathological criteria and clinical trials. A systematic review Contemp Clin Trials (2016) 50:37–44. doi: 10.1016/j.cct.2016.07.011
2. Lin X, Wu D, Chen C, Zheng N. Clinical characteristics of adrenal tumors in children: a retrospective review of a 15-year single-center experience. Int Urol Nephrol (2017) 49(3):381–5. doi: 10.1007/s11255-016-1480-z
3. Abib SCV, Weldon CB. Management of adrenal tumors in pediatric patients. Surg Oncol Clin N Am (2021) 30(2):275–90. doi: 10.1016/j.soc.2020.11.012
4. Miele E, Di Giannatale A, Crocoli A, Cozza R, Serra A, Castellano A, et al. Clinical, genetic, and prognostic features of adrenocortical tumors in children: A 10-year single-center experience. Front Oncol (2020) 10:554388. doi: 10.3389/fonc.2020.554388
5. Mete O, Erickson LA, Juhlin CC, de Krijger RR, Sasano H, Volante M, et al. Overview of the 2022 WHO classification of adrenal cortical tumors. Endocr Pathol (2022) 33(1):155–96. doi: 10.1007/s12022-022-09710-8
6. Özcan HN, Tan AA, Ardıçlı B, Oguz B, Ekinci S, Kutluk T, et al. Imaging findings of primary adrenal tumors in pediatric patients. Diagn Interv Radiol (2021) 27(6):811–5. doi: 10.5152/dir.2021.20701
7. Croteau N, Nuchtern J, LaQuaglia MP. Management of neuroblastoma in pediatric patients. Surg Oncol Clin N Am (2021) 30(2):291–304. doi: 10.1016/j.soc.2020.11.010
8. Pinto EM, Zambetti GP, Rodriguez-Galindo C. Pediatric adrenocortical tumours. Best Pract Res Clin Endocrinol Metab (2020) 34(3):101448. doi: 10.1016/j.beem.2020.101448
9. Erdmann F, Frederiksen LE, Bonaventure A, Mader L, Hasle H, Robison LL, et al. Childhood cancer: Survival, treatment modalities, late effects and improvements over time. Cancer Epidemiol (2021) 71(Pt B). doi: 10.1016/j.canep.2020.101733
10. Winther JF, Kenborg L, Byrne J, Hjorth L, Kaatsch P, Kremer LC, et al. Childhood cancer survivor cohorts in Europe. Acta Oncol (2015) 54(5):655–68. doi: 10.3109/0284186x.2015.1008648
11. Yao W, Dong K, Li K, Zheng S, Xiao X. Comparison of long-term prognosis of laparoscopic and open adrenalectomy for local adrenal neuroblastoma in children. Pediatr Surg Int (2018) 34(8):851–6. doi: 10.1007/s00383-018-4294-5
12. Kelleher CM, Smithson L, Nguyen LL, Casadiego G, Nasr A, Irwin MS, et al. Clinical outcomes in children with adrenal neuroblastoma undergoing open versus laparoscopic adrenalectomy. J Pediatr Surg (2013) 48(8):1727–32. doi: 10.1016/j.jpedsurg.2013.03.056
13. Rodriguez-Galindo C, Krailo MD, Pinto EM, Pashankar F, Weldon CB, Huang L, et al. Treatment of pediatric adrenocortical carcinoma with surgery, retroperitoneal lymph node dissection, and chemotherapy: The children's oncology group ARAR0332 protocol. J Clin Oncol (2021) 39(22):2463–73. doi: 10.1200/jco.20.02871
14. Fascetti-Leon F, Scotton G, Pio L, Beltrà R, Caione P, Esposito C, et al. Minimally invasive resection of adrenal masses in infants and children: results of a European multi-center survey. Surg Endosc (2017) 31(11):4505–12. doi: 10.1007/s00464-017-5506-0
15. Mattone MC, Gil S, Costanzo M, Galluzzo Mutti ML, Casanovas A, Zaidman V, et al. Pediatric adrenocortical tumors cohort characteristics and long-term follow-up at a single Argentinian tertiary center. J Pediatr Endocrinol Metab (2022) 35(1):19–27. doi: 10.1515/jpem-2021-0392
16. Dong R, Yang R, Zhan Y, Lai HD, Ye CJ, Yao XY, et al. Single-cell characterization of malignant phenotypes and developmental trajectories of adrenal neuroblastoma. Cancer Cell (2020) 38(5):716–33.e6. doi: 10.1016/j.ccell.2020.08.014
17. Swift CC, Eklund MJ, Kraveka JM, Alazraki AL. Updates in diagnosis, management, and treatment of neuroblastoma. Radiographics (2018) 38(2):566–80. doi: 10.1148/rg.2018170132
19. Tsubota S, Kadomatsu K. Origin and initiation mechanisms of neuroblastoma. Cell Tissue Res (2018) 372(2):211–21. doi: 10.1007/s00441-018-2796-z
20. Schwab M. Amplification of n-myc as a prognostic marker for patients with neuroblastoma. Semin Cancer Biol (1993) 4(1):13–8.
21. Matthay KK, Maris JM, Schleiermacher G, Nakagawara A, Mackall CL, Diller L, et al. Neuroblastoma. Nat Rev Dis Primers (2016) 2:16078. doi: 10.1038/nrdp.2016.78
22. Lampis S, Raieli S, Montemurro L, Bartolucci D, Amadesi C, Bortolotti S, et al. The MYCN inhibitor BGA002 restores the retinoic acid response leading to differentiation or apoptosis by the mTOR block in MYCN-amplified neuroblastoma. J Exp Clin Cancer Res (2022) 41(1):160. doi: 10.1186/s13046-022-02367-5
23. Peinemann F, van Dalen EC, Enk H, Berthold F. Retinoic acid postconsolidation therapy for high-risk neuroblastoma patients treated with autologous haematopoietic stem cell transplantation. Cochrane Database Syst Rev (2017) 8(8):Cd010685. doi: 10.1002/14651858.CD010685.pub3
24. Pugh TJ, Morozova O, Attiyeh EF, Asgharzadeh S, Wei JS, Auclair D, et al. The genetic landscape of high-risk neuroblastoma. Nat Genet (2013) 45(3):279–84. doi: 10.1038/ng.2529
25. Attiyeh EF, London WB, Mossé YP, Wang Q, Winter C, Khazi D, et al. Chromosome 1p and 11q deletions and outcome in neuroblastoma. N Engl J Med (2005) 353(21):2243–53. doi: 10.1056/NEJMoa052399
26. Gilbert F, Feder M, Balaban G, Brangman D, Lurie DK, Podolsky R, et al. Human neuroblastomas and abnormalities of chromosomes 1 and 17. Cancer Res (1984) 44(11):5444–9.
27. Mlakar V, Jurkovic Mlakar S, Lopez G, Maris JM, Ansari M, Gumy-Pause F. 11q deletion in neuroblastoma: a review of biological and clinical implications. Mol Cancer (2017) 16(1):114. doi: 10.1186/s12943-017-0686-8
28. Spitz R, Hero B, Simon T, Berthold F. Loss in chromosome 11q identifies tumors with increased risk for metastatic relapses in localized and 4S neuroblastoma. Clin Cancer Res (2006) 12(11 Pt 1):3368–73. doi: 10.1158/1078-0432.Ccr-05-2495
29. Brodeur GM, Pritchard J, Berthold F, Carlsen NL, Castel V, Castelberry RP, et al. Revisions of the international criteria for neuroblastoma diagnosis, staging, and response to treatment. J Clin Oncol (1993) 11(8):1466–77. doi: 10.1200/jco.1993.11.8.1466
30. Evans AE, D'Angio GJ, Propert K, Anderson J, Hann HW. Prognostic factor in neuroblastoma. Cancer (1987) 59(11):1853–9. doi: 10.1002/1097-0142(19870601)59:11<1853::aid-cncr2820591102>3.0.co;2-f
31. Li J, Liu X, Chen M, Wang J, Wang X. Values of serum CA125, NSE and 24-hour urine VMA in diagnosis and prediction of treatment of paediatric neuroblastoma. Int J Clin Pract (2021) 75(12):e14932. doi: 10.1111/ijcp.14932
32. Voglino V, Persano G, Crocoli A, Castellano A, Serra A, Giordano U, et al. Hemorrhage during induction chemotherapy in neuroblastoma: Additional risk factors in high-risk patients. Front Pediatr (2021) 9:761896. doi: 10.3389/fped.2021.761896
33. Moroz V, Machin D, Hero B, Ladenstein R, Berthold F, Kao P, et al. The prognostic strength of serum LDH and serum ferritin in children with neuroblastoma: A report from the international neuroblastoma risk group (INRG) project. Pediatr Blood Cancer (2020) 67(8):e28359. doi: 10.1002/pbc.28359
34. Whittle SB, Smith V, Doherty E, Zhao S, McCarty S, Zage PE. Overview and recent advances in the treatment of neuroblastoma. Expert Rev Anticancer Ther (2017) 17(4):369–86. doi: 10.1080/14737140.2017.1285230
35. Laprie A, Michon J, Hartmann O, Munzer C, Leclair MD, Coze C, et al. High-dose chemotherapy followed by locoregional irradiation improves the outcome of patients with international neuroblastoma staging system stage II and III neuroblastoma with MYCN amplification. Cancer (2004) 101(5):1081–9. doi: 10.1002/cncr.20453
36. Park JR, Kreissman SG, London WB, Naranjo A, Cohn SL, Hogarty MD, et al. Effect of tandem autologous stem cell transplant vs single transplant on event-free survival in patients with high-risk neuroblastoma: A randomized clinical trial. Jama (2019) 322(8):746–55. doi: 10.1001/jama.2019.11642
37. Du L, Liu L, Zhang C, Cai W, Wu Y, Wang J, et al. Role of surgery in the treatment of patients with high-risk neuroblastoma who have a poor response to induction chemotherapy. J Pediatr Surg (2014) 49(4):528–33. doi: 10.1016/j.jpedsurg.2013.11.061
38. Ladenstein R, Pötschger U, Pearson ADJ, Brock P, Luksch R, Castel V, et al. Busulfan and melphalan versus carboplatin, etoposide, and melphalan as high-dose chemotherapy for high-risk neuroblastoma (HR-NBL1/SIOPEN): An international, randomised, multi-arm, open-label, phase 3 trial. Lancet Oncol (2017) 18(4):500–14. doi: 10.1016/s1470-2045(17)30070-0
39. Alexander N, Vali R, Ahmadzadehfar H, Shammas A, Baruchel S. Review: The role of radiolabeled DOTA-conjugated peptides for imaging and treatment of childhood neuroblastoma. Curr Radiopharm (2018) 11(1):14–21. doi: 10.2174/1874471011666171215093112
40. Anongpornjossakul Y, Sriwatcharin W, Thamnirat K, Chamroonrat W, Kositwattanarerk A, Utamakul C, et al. Iodine-131 metaiodobenzylguanidine (131I-mIBG) treatment in relapsed/refractory neuroblastoma. Nucl Med Commun (2020) 41(4):336–43. doi: 10.1097/mnm.0000000000001152
41. Yu AL, Gilman AL, Ozkaynak MF, London WB, Kreissman SG, Chen HX, et al. Anti-GD2 antibody with GM-CSF, interleukin-2, and isotretinoin for neuroblastoma. N Engl J Med (2010) 363(14):1324–34. doi: 10.1056/NEJMoa0911123
42. Chudler RM, Kay R. Adrenocortical carcinoma in children. Urol Clin North Am (1989) 16(3):469–79.
43. Pinto EM, Chen X, Easton J, Finkelstein D, Liu Z, Pounds S, et al. Genomic landscape of paediatric adrenocortical tumours. Nat Commun (2015) 6:6302. doi: 10.1038/ncomms7302
44. Zheng S, Cherniack AD, Dewal N, Moffitt RA, Danilova L, Murray BA, et al. Comprehensive pan-genomic characterization of adrenocortical carcinoma. Cancer Cell (2016) 29(5):723–36. doi: 10.1016/j.ccell.2016.04.002
45. Wieneke JA, Thompson LD, Heffess CS. Adrenal cortical neoplasms in the pediatric population: a clinicopathologic and immunophenotypic analysis of 83 patients. Am J Surg Pathol (2003) 27(7):867–81. doi: 10.1097/00000478-200307000-00001
46. Das S, Sengupta M, Islam N, Roy P, Datta C, Mishra PK, et al. Weineke criteria, ki-67 index and p53 status to study pediatric adrenocortical tumors: Is there a correlation? J Pediatr Surg (2016) 51(11):1795–800. doi: 10.1016/j.jpedsurg.2016.07.014
47. Wasserman JD, Zambetti GP, Malkin D. Towards an understanding of the role of p53 in adrenocortical carcinogenesis. Mol Cell Endocrinol (2012) 351(1):101–10. doi: 10.1016/j.mce.2011.09.010
48. Ribeiro RC, Pinto EM, Zambetti GP, Rodriguez-Galindo C. The international pediatric adrenocortical tumor registry initiative: contributions to clinical, biological, and treatment advances in pediatric adrenocortical tumors. Mol Cell Endocrinol (2012) 351(1):37–43. doi: 10.1016/j.mce.2011.10.015
49. Zancanella P, Pianovski MA, Oliveira BH, Ferman S, Piovezan GC, Lichtvan LL, et al. Mitotane associated with cisplatin, etoposide, and doxorubicin in advanced childhood adrenocortical carcinoma: mitotane monitoring and tumor regression. J Pediatr Hematol Oncol (2006) 28(8):513–24. doi: 10.1097/01.mph.0000212965.52759.1c
50. Wiegering V, Riedmeier M, Thompson LDR, Virgone C, Redlich A, Kuhlen M, et al. Radiotherapy for pediatric adrenocortical carcinoma - review of the literature. Clin Transl Radiat Oncol (2022) 35:56–63. doi: 10.1016/j.ctro.2022.05.003
51. Vikner ME, Krogh J, Daugaard G, Andreassen M. Metabolic and hormonal side effects of mitotane treatment for adrenocortical carcinoma: A retrospective study in 50 Danish patients. Clin Endocrinol (Oxf) (2021) 94(2):141–9. doi: 10.1111/cen.14345
52. Oddie PD, Albert BB, Hofman PL, Jefferies C, Laughton S, Carter PJ. Mitotane in the treatment of childhood adrenocortical carcinoma: A potent endocrine disruptor. Endocrinol Diabetes Metab Case Rep (2018) 2018. doi: 10.1530/edm-18-0059
53. Evanoff JD, Patel SG, Hickey KJ, Rensing AJ. Survival characteristics of localized pediatric adrenocortical carcinoma managed with adenectomy: A national cancer center database analysis. J Pediatr Urol (2021) 17(5):735. doi: 10.1016/j.jpurol.2021.06.005
Keywords: child, adrenal malignancy, treatment, survival, prognosis
Citation: Lv Z, Yu Y, Luo Y, Lin S, Xiang X, Mao X and Cheng S (2022) Long-term survival outcomes of pediatric adrenal malignancies: An analysis with the upstaged SEER registry during 2000-2019. Front. Endocrinol. 13:977105. doi: 10.3389/fendo.2022.977105
Received: 24 June 2022; Accepted: 23 August 2022;
Published: 12 September 2022.
Edited by:
David Taïeb, Hôpital de la Timone, FranceReviewed by:
Maria Candida Barisson Villares Fragoso, Institute of Cancer of Sao Paulo, BrazilCopyright © 2022 Lv, Yu, Luo, Lin, Xiang, Mao and Cheng. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Xiaowen Mao, eGlhb3dlbm1hbzA5MjdAMTYzLmNvbQ==; Shigang Cheng, Y2hlbmdnYW5nNTI3QDE2My5jb20=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.