 Deng Pan
Deng Pan Lin Xu4†
Lin Xu4† Ming Guo
Ming Guo
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Endocrinol. , 17 August 2022
Sec. Diabetes: Molecular Mechanisms
Volume 13 - 2022 | https://doi.org/10.3389/fendo.2022.973058
Protein kinase C (PKC) is a family of serine/threonine protein kinases, the activation of which plays an important role in the development of diabetic microvascular complications. The activation of PKC under high-glucose conditions stimulates redox reactions and leads to an accumulation of redox stress. As a result, various types of cells in the microvasculature are influenced, leading to changes in blood flow, microvascular permeability, extracellular matrix accumulation, basement thickening and angiogenesis. Structural and functional disorders further exacerbate diabetic microvascular complications. Here, we review the roles of PKC in the development of diabetic microvascular complications, presenting evidence from experiments and clinical trials.
Diabetes mellitus (DM) is widespread globally, and the complications of diabetes are the main causes of morbidity and mortality (1, 2). In patients with DM, hyperglycemia is a major risk factor for the development of microvascular complications (3). Long-term DM affects the function and structure of blood vessels, causing diabetic macrovascular and microvascular complications. Diabetic nephropathy, diabetic retinopathy and diabetic peripheral neuropathy are thought to be the main types of diabetic microvascular complications (4). According to the Outcome Reduction with an Initial Glargine Intervention (ORIGIN) trial, for patients with a baseline HbA1c of more than 6.4%, interventions to normalize normal fasting glucose significantly reduced the risk of diabetic microvascular complications (5). In patients with type 2 diabetes mellitus (T2DM), antihyperglycemic therapy also demonstrated a favorable effect in reducing the risk of microvascular complications (6). However, despite glucose-lowering therapy, a high residual risk of diabetic microvascular complications remains in patients with DM (7). In addition, the interaction of genetic, environmental and metabolic factors is involved in microvascular complications. Thus, there is still an unmet need for a better understanding of the pathogenesis of diabetic microvascular complications (8).
Protein kinase C (PKC) participates in various cellular responses associated with DM (9). Activation of PKC is significantly increased under conditions of elevated glucose levels (22 mM) in vitro (10). Hyperglycemia triggers the glycolysis pathway and further enhances the synthesis of diacylglycerol (DAG), which activates PKC (11). Activation of PKC then facilitates activation of NADPH oxidase, after which the redox reaction starts and redox stress accumulates. As a consequence, reactive oxygen species (ROS) and advanced glycation end products (AGEs) accumulate in cells, leading to cell death and apoptosis (12, 13). The promotion of redox stress is observed in various vascular cells, including pericytes, endothelial cells, smooth muscle cells, podocytes and others (14). Therefore, vascular dysfunction generated by the aforementioned mechanism promotes diabetic microvascular complications, which mainly influence alterations in blood flow (15), extracellular matrix synthesis and basement membrane thickening (16, 17), vascular permeability (18), and angiogenesis (19). In this review, we focus on the roles of PKC and PKC isoforms in the development of diabetic microvascular complications.
PKC is a family of serine/threonine protein kinases, which was discovered over 30 years ago (20). According to the pattern of activation, PKC can be divided into three subgroups, including classical/conventional PKCs (cPKCs): PKC-α, βI, βII, γ; novel PKCs (nPKCs): PKC-δ, ϵ, θ, η, μ; atypical PKCs (aPKCs): PKC-ζ, λ/τ (21, 22) (Figure 1). The activation of cPKCs requires both calcium and DAG or phorbol esters, while nPKCs are activated by DAG or phorbol esters but not by calcium. The aPKCs are not activated by either calcium or DAG/phorbol esters. aPKCs are regulated by protein–protein interactions through the PB1 domain (22, 23). All of the PKC isozymes comprise an N-terminal regulatory region and a C-terminal catalytic region, and the two regions are linked by a V3 hinge (24). In cPKCs and nPKCs, the regulatory region contains the C1 domain, which can be further divided into the tandem C1A and C1B subdomains, which are duplicates and are rich in cysteine sequences (25). In addition, cPKCs contain a C2 domain, which is able to bind to calcium. However, this C2 domain is not present in nPKCs, which have a C2-like domain (cannot bind to calcium) instead. Therefore, nPKCs are not activated by calcium (26). Once the C1 domain binds to DAG, a pseudosubstrate is removed from the catalytic domain, leading to enzyme activation (25, 26).
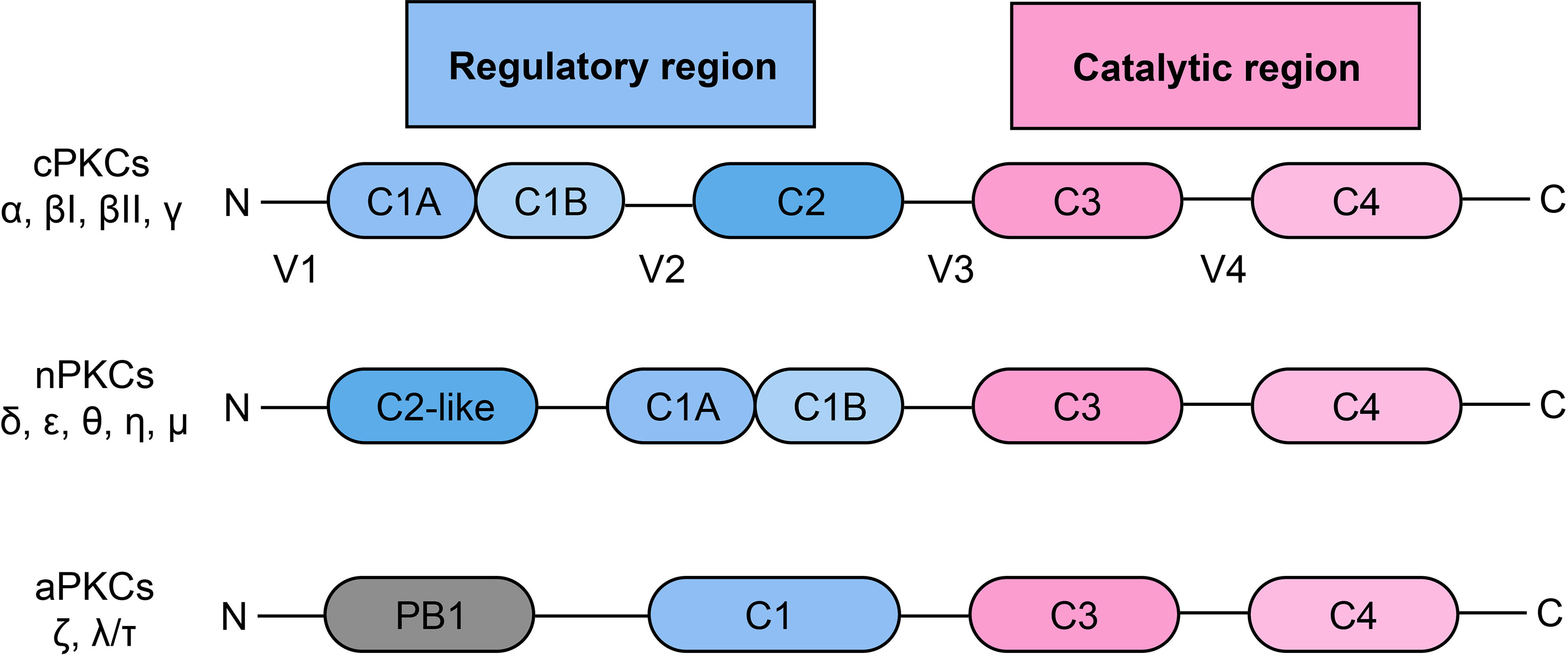
Figure 1 Classification and structural of protein kinase C.PKC: protein kinase C; cPKC: conventional protein kinase C; nPKC: novel protein kinase C; aPKC: atypical protein kinase C.
Diabetic nephropathy (DN) is characterized by proteinuria and a decline in glomerular filtration rate (GFR). If DN is untreated, the resulting end-stage renal failure and uremia are fatal (27). Various kinds of renal cells, including podocytes, endothelial cells, smooth muscle cells, mesangial cells and inflammatory cells, are affected under hyperglycemia (28). Hemodynamic changes play an important role in the pathogenesis of DN, which arises from vasoactive factor release, signal transduction and metabolic changes under hyperglycemic conditions. In the early stage of DN, increased plasma flow and glomerular capillary pressure cause an increase in GFR, termed hyperfiltration (29). Hyperfiltration also precipitates thickening of the glomerular basement membrane (GBM) (hyperplasia) and the detachment, loss, and death of podocytes (30). Moreover, renin and angiotensin-II synthesis in mesangial cells were found to be promoted under high-glucose conditions, which further gave rise to an increase in glomerular capillary pressure, microvascular permeability, and renal cell proliferation (31, 32). As a result, proteinuria and glomerulosclerosis were substantially induced (33). Moreover, with the feedback of hypertrophy and hyperplasia, the proximal tubule length significantly increases, leading to an increased reabsorption of glucose, fatty acids, amino acids, growth factors and cytokines (34). This metabolic production in turn leads to energetic imbalance, such as redox abnormalities and fibrosis. The aforementioned pathological processes ultimately result in extracellular matrix (ECM) deposition in the tubules, which is deemed to be the major determinant in the development of DN (35). Consequently, the GFR is eventually decreased owing to progressive glomerular and tubular injury (36).
The activation of PKC is involved in microvascular contraction within the kidney, affecting the function of glomeruli and contributing to the progression of diabetic nephropathy (Figure 2). The microvascular contraction of glomeruli is enhanced under high-glucose conditions, and blood flow is consequently decreased. This is mainly caused by changes in the levels and sensitivity of contraction factors. Under high-glucose conditions, the levels of vascular contraction factors, such as prostaglandin E2 and prostaglandin F2 alpha, are elevated (15). In addition, the sensitivity of vascular contraction factors is increased in diabetic nephropathy. In diabetic mice, phenylephrine-induced interlobar artery (ILA) contraction was significantly enhanced, which caused ILA dysfunction. ILA dysfunction further reduced blood flow in the glomerulus and induced the progression of diabetic nephropathy. Treatment with rottlerin, a calcium-independent protein kinase C (PKC-δ) inhibitor, alleviated basal overcontraction (37, 38). Similar results were observed in diabetic Zucker rats; the nonselective PKC agonist phorbol-12,13 dibutyrate (PDBU) significantly reduced renal cortical blood flow and increased the mean arterial pressure (39). The results indicate that PKC activation is associated with decreased blood flow and increased renal perfusion pressure, causing glomerulosclerosis and reducing glomerular filtration.
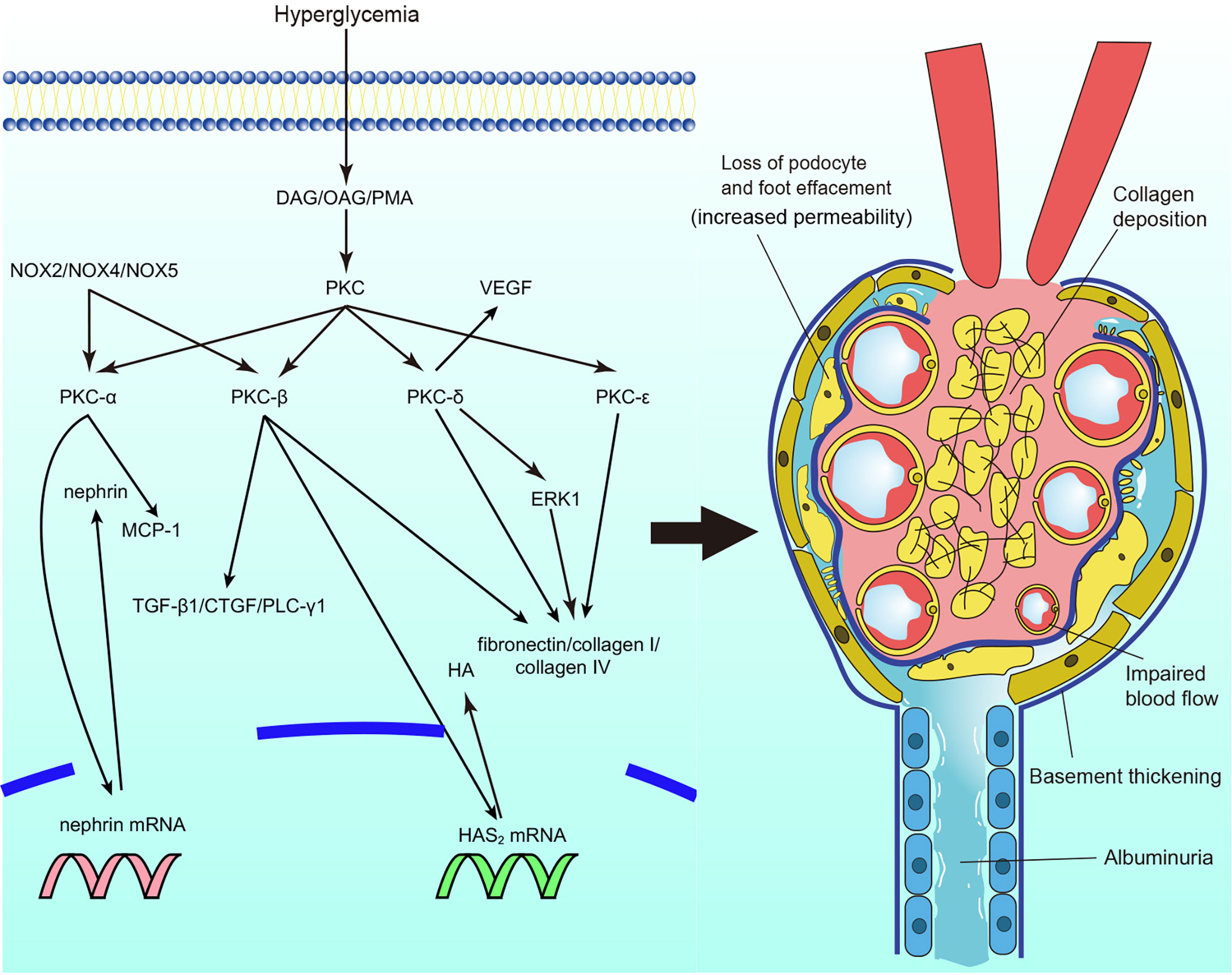
Figure 2 Protein kinase C (PKC) in diabetic nephropathy. PKC activation in diabetic nephropathy impairs glomerular blood flow and filtration, leads to albuminuria, causes extracellular matrix accumulation and collagen deposition particularly. DAG, diacylg lycerol; OAG, oleoyl acetyl glycerol; PKC, protein kinase C; MCP-1, monocyte chemoattractant protein-1; TGF-β1, transforming growth factor β1; CTGF, connective tissue growth factor; PLC-γ1, phospholipase Cγ1; HAS2, Hyaluronan synthase 2; HA, hyaluronan; ERK, extracellular signal-regulated kinase; VEGF, vascu lar endothelial growth factor; NOX, nicotinamide adenine dinucleotide phosphate oxidase.
Impaired barrier function is associated with the deterioration of diabetic nephropathy, during which proteinuria is usually observed. The role of PKC in the vascular permeability of diabetic nephropathy mainly focuses on the glomeruli. Nephrin is one of the most well-studied proteins involved in vascular permeability. Nephrin is essential for podocytes to sustain the integrity of the slit diaphragm to maintain filtration (40, 41). The expression levels of nephrin mRNA and protein were significantly reduced in streptozotocin (STZ)-induced DM mice. However, in global PKC-α knockout mice, nephrin expression was unaffected in the mice with DM (42). Thallas-Bonke et al. also found that PKC-α and PKC-β were overexpressed in DM mice, whereas deletion of NOX4 (nicotinamide adenine dinucleotide phosphate oxidase) reversed the overexpression of PKC-α and PKC-β, with normalized nephrin expression observed (43). In addition, mediation of RhoA downstream of C3aR in endothelial cells by PKC resulted in kidney damage and increased blood vessel permeability (44). Moreover, VEGF plays an important role in increased capillary permeability. Diabetic mice treated with calphostin C showed reduced expression of VEGF in their mesangial cells compared to mice that were not treated with calphostin C (45). Mima et al. explored the interaction of PKC-δ and VEGF in diabetic nephropathy. The expression of VEGF was induced under high-glucose conditions, whereas loss of VEGF signaling was observed, which indicated VEGF resistance. Notably, VEGF signaling glomerular endothelial was not inhibited in global PRKCD knockout mice. In addition, less extracellular matrix and albuminuria were also observed (46). The results suggest that activation of different PKC isoforms is involved in increased vascular permeability and that PKC-α and PKC-β are interesting molecules in the maintenance of glomerular filtration function. PKC-δ may play an important role in the regulation of VEGF expression and prevention of resistance to VEGF in the glomeruli.
Structural abnormalities are the most well-studied pathology in diabetic nephropathy. It has been shown that expansion of the basement membrane and ECM accumulation are induced by high-glucose conditions (47). The synthesis of ECM proteins, such as fibronectin, laminin, and types I, III, and IV collagen, was found to be increased in renal glomerular mesangial cells, accompanied by upregulated activation of PKC (48–50). PMA (PKC activator) and the cell-permeable DAG analog oleoyl acetyl glycerol (OAG) also mimicked these effects (48). Treatment with the PKC inhibitor chelerythrine reduced the expression and synthesis of fibronectin and prevented basement membrane thickening (51). In addition, elements of the ECM regulated by activation of PKC have also been studied. Hyaluronan (HA), a key element of the ECM, was incrementally secreted under hyperglycemia, and inhibition of PKC-β reduced the expression of HAS2 (hyaluronan synthase 2) mRNA and secretion of HA (52). A general PKC inhibitor also reversed the hyperglycemia-induced elevated synthesis of collagenous and total ECM proteins and decreased gelatinase activity in endothelial cells (53). The results suggest that PKC activation is involved in the synthesis of ECM proteins and degradation of the ECM in diabetic nephropathy. Furthermore, studies on the activation of PKC isoforms in DN have also been conducted. PKC-α and PKC-β activation was increased in mesangial cells under high concentrations of glucose, and the synthesis of fibronectin and IV collagen paralleled their activity (54, 55). LY333531 (a specific PKC-β inhibitor) prevented the hyperglycemia-induced expression of ECM components in mesangial cells (55). The same results were observed in global PKC-α/PKC-β double knockout mice, accompanied by alleviation of the development of albuminuria (56). However, despite a reduction in the albumin/creatinine ratio, the ratio was not completely reversed (56). Tokuyama et al. found that a PKC-β inhibitor might enhance reductions in mesangial cell (3) H-thymidine and (3) H-proline incorporation, which reduced the synthesis of collagen by mesangial cell (57). The nPKCs PKC-δ and PKC-ϵ also participate in ECM protein synthesis. The synthesis of ECM proteins in mesangial cells was reduced in PKC-δ-/- and PKC-ϵ-/- STZ-induced DM mice and in mice with exogenous PKC-δ inhibition (46, 58, 59). Interestingly, PKC-ϵ-/- mice did not show a profibrotic phenotype in any organ other than the kidney (58). Of note, Baccora et al. reported that activation of PKC-δ, rather than PKC-α or PKC-β, was increased in mesangial cells under high-glucose conditions (59). The membrane association of PKC-ζ was confirmed to be prevented by rosiglitazone, along with decreased expression of collagen IV in cultured mesangial cells (60). Nevertheless, the role of aPKCs still needs more exploration. Moreover, other fibrotic factors and pathways that interact with PKC are discussed. Transforming growth factor β1 (TGF-β1) is one of the most important factors that causes basement thickening and ECM accumulation in diabetic nephropathy (61). PKC activation and the expression of TGF-β1 in mesangial cells and glomerular endothelial cells were increased under treatment with glycated albumin and advanced oxidation protein products (the level of glycated albumin and advanced oxidation protein products are elevated under high glucose, which mimics the alternations in vivo) (62–64). Inhibition of PKC-α and PKC-β attenuated the expression of TGF-β1 and connective tissue growth factor (CTGF) in glomerular endothelial cells and mesangial cells (54, 55, 62, 64–66). Interactions between other molecules and PKC in terms of ECM protein synthesis and basement thickening have also been explored. Global Akr1b3 knockout mice showed inhibition of PKC activation, with reduced ECM accumulation and glomerular hypertrophy in renal cortical tissue (67). The homologous genes NOX2, NOX4, and NOX5 were also found to inhibit PKC-α and PKC-β activation, followed by decreases in hyperglycemia-induced ROS and expression of TGF-β1 and monocyte chemoattractant protein-1 (MCP-1), which prevented ECM accumulation and basement membrane thickening in mesangial cells and podocyte, and thus decreased albumin excretion (68–70). Extracellular signal-regulated kinase (ERK) is also involved in ECM accumulation. PKC-δ expression was increased in the membranous fraction under high-glucose conditions. Treatment with the PKC-δ inhibitor rottlerin, but not the cPKC inhibitor Gö6976, abrogated ERK expression and decreased hyperglycemia-induced responsiveness to TGF-β1 in mesangial cells, blocking the fibrotic response (71). In contrast, Tuttle et al. discovered that PKC-β inhibition caused decreased expression of ERK1 and ERK2 in mesangial cells (72). Moreover, Wu et al. reported the involvement of phospholipase C γ1 (PLC-γ1) in the development of diabetic nephropathy. PLC-γ1 inhibition suppressed PKC-β-induced protein kinase B (Akt) S473 phosphorylation in mesangial cells under high-glucose conditions, and collagen I upregulation was also prevented by the PLC-γ1 inhibitor U73122 (73). In summary, current studies indicate that overexpression of PKC is involved in the accumulation of the ECM, and mesangial cells account for most of the ECM accumulation; however, regarding PKC isoforms, some studies have shown contradictory results (56, 57, 59). The results may be influenced by the duration of cell culture. The authors suggest that in the development of diabetic nephropathy, specific PKC isoforms may participate in the early process of exacerbation. However, with the development of diabetic nephropathy, more PKC isoforms may be involved in its pathogenesis when high-grade proteinuria occurs. Consequently, it is also vital to explore different PKC isoforms in various types of cells in the kidney and stages of diabetic pathogenesis. In addition, VEGF plays an important role in the development of diabetic nephropathy, the role of VEGF in ECM accumulation may also be a promising topic to be explored in the future.
Oxidative stress is considered an important factor in the pathogenesis of diabetic retinopathy (DR), and accumulated reactive oxygen species ultimately result in the impairment of retinal tissue and vessels. PKC activation is one of the main factors involved in accumulated oxidative stress (74, 75). High glucose concentrations cause damage to the function and structure of the retina, including loss of pericytes, increased vascular permeability, thickening of the retinal capillary basement membrane, tissue ischemia, etc. (Figure 3) (74). The processes mentioned above give rise to the initial stage of DR, termed nonproliferative diabetic retinopathy (NPDR). In the NPDR stage, visual impairment is not always noticed, whereas sight-threating DR and blindness are preventable (76). However, if NPDR is left untreated, vascular endothelial growth factor (VEGF) protein levels are upregulated due to ischemia/hypoxia through hypoxia-inducible factor 1 (HIF-1) activation (77). Upregulation of VEGF causes the formation of new blood vessels, which is called neovascularization. This drives DR into the proliferative diabetic retinopathy (PDR) stage. In this phase, newly formatted permeable vascular tufts and macular edema appear and ultimately cause loss of vision (78, 79).
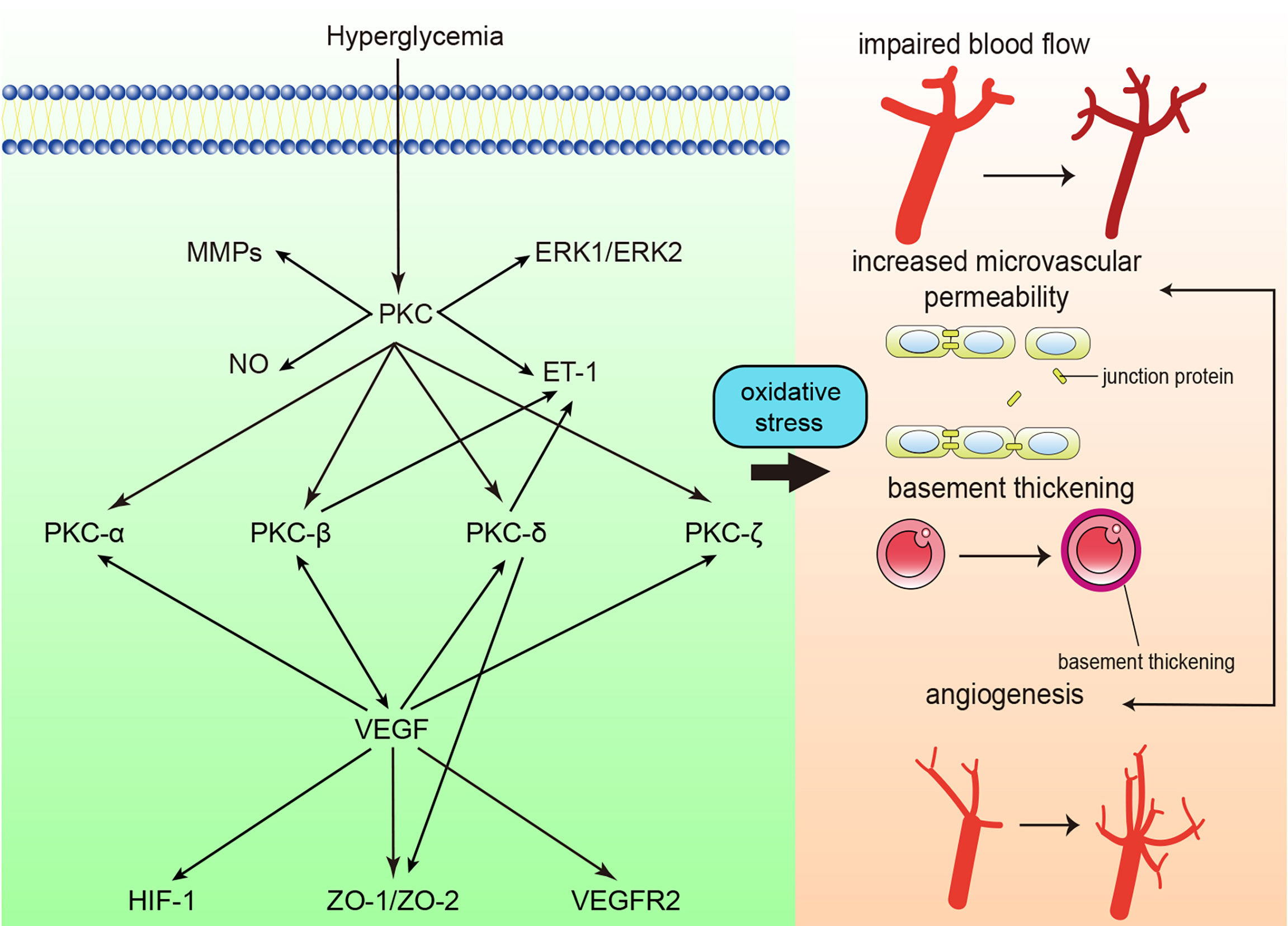
Figure 3 Protein kinase C (PKC) in diabetic retinopathy. PKC activation in diabetic retinopathy influences blood flow, causes retinal microvascular constriction, induces angiogenesis and increases microvascular permeability. ET-1, endothelin-1; NO, nitric oxide; MMP, matrix metalloproteinase; ERK, extracellular signal-regulated kinase ; VEGF, vascular endothelial growth factor; VEGFR2, vascular endothelial growth factor receptor 2; ZO, zonula occludens; HIF-1, Hypoxia-inducible factor 1.
Activation of PKC mediates the progression of decreased retinal blood flow in DM. The mean circulation time (MCT) in diabetic rats was significantly increased. In contrast, when the rats were treated with intravitreal injection of LY333531, the MCT was shortened to half the MCT of those that were not treated (80). PKC-mediated inflammation may be one of the factors of decreased blood flow. In the retinal microcirculation of diabetic rats, an increased number of leukocytes were trapped, leading to blood flow disturbances in the retinal microvasculature. LY333531 significantly reduced the number of trapped leukocytes and increased the retinal blood flow of diabetic rats (81). In addition, the activation of PKC may cause the overexpression of endothelin-1 (ET-1), which can also explain the decrease in retinal blood flow. The expression of ET-1 and activation of PKC were increased in parallel by 2-fold in bovine retinal endothelial cells (BRECs) under high-glucose conditions. The overexpression of ET-1 was inhibited by the general PKC inhibitor GF109203X. It was further discovered that PKC-β and PKC-δ were significantly overexpressed in membranous fractions, which indicated that the activation of PKC-β and PKC-δ mediated ET-1 expression (82). Moreover, nitric oxide (NO) insufficiency participates in the alteration of retinal blood flow. The synthesis of NO from retinal microvascular endothelial cells (RMECs) was downregulated under high-glucose conditions, whereas a PKC inhibitor increased the accumulation of NO and restored the blood flow in the retina (83). PKC-β and PKC-δ are two main isoforms that have been studied regarding blood flow in the retina, and studies on other isoforms are in progress.
It has also been proven that retinal vascular permeability is increased under high-glucose conditions, along with levels of reactive oxygen species (84, 85). The application of a PKC inhibitor reduced high glucose-induced albumin leakage (85). VEGF is mainly studied in terms of retinal vascular permeability. The expression of PKC-β and VEGF were both significantly increased under high-glucose conditions, and the inhibition of PKC-β and PKC-β mutant mice showed attenuated vascular barrier function and less albumin leakage (86–88). PKC-β activation induced occludin Ser490 phosphorylation, which led to the ubiquitination required for VEGF-induced permeability and exacerbated retinal barrier dysfunction (89). In addition to PKC-β, activation of PKC-α, PKC-ζ, PKC-δ and aPKC induced by VEGF is involved in impaired tight junctions and activation of inflammatory pathways and causes hyperpermeability (90–94). PKC-β, PKC-α, and PKC-δ levels were found to be increased in membranous fractions. However, treatment with a PKC-β inhibitor rescued 95% of the VEGF-induced increase in vascular permeability, indicating that PKC-β plays a major role in VEGF-induced hyperpermeability in diabetic retinopathy (90, 91). Moreover, studies focusing on gap junctions were also conducted. Gap function is impaired due to the loss of zonula occludens (ZO)-1, ZO-2, etc. Inhibition of PKC-δ blocked translocation from the cytosol to the membrane and restored the loss of gap junction proteins (95). However, PKC was not found to participate in claudin-1 (another gap junction protein) expression (96). In summary, PKC-β is thought to be the most crucial PKC isoform that participates in increased retinal microvascular permeability, and most related clinical trials have also focused on PKC-β (see below). The role of PKC in specific gap junction proteins still needs further exploration. PKC is also promising for detecting structural impairment in cell junctions visually.
Basement thickening is also one of the most common features in the early stage of diabetic retinopathy (95). As mentioned above, PKC-β overexpression was observed in STZ-induced rats. Treatment with hesperetin (Hsp) suppressed PKC-β expression, abolished DM-induced retinal capillary basement membrane thickness, and prevented retinal microvascular dysfunction (88). Giordo et al. reported an association between PKC activation and endothelial-to-mesenchymal transition (EndMT). In high glucose-exposed human retinal endothelial cells, the general PKC inhibitor chelerythrine significantly reduced ROS levels and EndMT. Moreover, the researchers also observed attenuated fibrotic processes (97). However, due to the correlation between structural and functional changes in diabetic retinopathy (98), more studies on the association between PKC activation and ECM augmentation are expected.
Activation of PKC enhanced angiogenesis in diabetic retinopathy. As mentioned above, PKC activation enhances the expression of VEGF in the retina. Nonetheless, vascular permeability is increased, which exacerbates the development of diabetic retinopathy. The activation of PKC accelerates migration, tube formation, and cellular proliferation in diabetic retinopathy, and the PKC-mediated upregulation of extracellular signal-regulated kinase (ERK) 1/2 and matrix metalloproteinase (MMPs) may account for the enhanced angiogenesis (99, 100). Although enhanced angiogenesis is observed in the retina under high-glucose conditions, the newly formed microvasculature shows impaired function, which accelerates the progression of diabetic retinopathy.
Diabetic neuropathy is a syndrome that affects the peripheral nervous system, and both the somatic and autonomic divisions are impaired. It is mainly thought to be the result of thickening of the capillary basement membrane and endothelial hyperplasia, as mentioned above (101, 102). Sensory axons, autonomic axons and, rarely, motor axons are mainly influenced in diabetic neuropathy. Moreover, the spinal cord is damaged in diabetic neuropathy (31, 103, 104). Damage to sensory axons usually causes symptoms that are easy to notice, such as hyperalgesia, paresthesia, and allodynia. Notably, the loss of nerve conduction velocity and nerve terminals tends to occur in long nerve fibers, which is called a fiber length-dependent pattern. Thus, numbness, nighttime pain, dysesthesia, and sensory loss always appear on the feet and then ascend to the hands, which is called the ‘stocking and glove pattern’ distribution (105). In addition, autonomic axons are also affected. In patients with DM, several autonomic nervous system symptoms can be observed. One of the most common symptoms is orthostatic hypotension, and the autonomic nervous system fails to regulate heart rate and blood flow; consequently, upright position blood pressure cannot be well maintained (106). Other autonomic nervous system symptoms, including gastroparesis, nausea, bloating, and diarrhea, should be considered evidence of the development of diabetic neuropathy. In addition, symptoms such as depression and sleep disturbance should be differentiated and emphasized (107). Motor neurons are located inside the blood–brain barrier, which may explain why damage to motor axons is rarely observed (108). The symptoms of diabetic neuropathy mostly refer to motor dysfunction, including falling, body sway, and dorsiflexion of fingers or toes (109, 110).
Studies concerning activation of PKC in diabetic neuropathy mainly focus on blood flow and conduction velocity. The blood flow in sciatic motor and saphenous nerve sensory were significantly decreased in diabetic rats, along with higher response to noxious mechanical and thermal stimuli. What’s more, NO synthase was also found to be inhibited in diabetic rats. LY333531 treatment attenuated the impaired blood flow and NO synthase activity (111). WAY151003 (general PKC inhibitor) and cremophor (an agent that prevents PKC activation) were also found to be effective in improving motor conduction velocity and correcting blood flow deficits in the sciatic motor and saphenous areas (112, 113). In addition, basement membrane thickening is associated with nerve damage in patients with diabetic neuropathy (114). Nonetheless, as a small number of studies have been conducted on microvascular functional and structural damage in diabetic neuropathy, addressing microvascular problems is thought to be an adjunct therapy in treating diabetic neuropathy.
As mentioned above, in vivo and in vitro experiments have demonstrated that various PKC isoforms participate in the development of diabetic microvascular complications. Since different general and specific PKC inhibitors have been explored, clinical trials have also been conducted to evaluate the efficacy and safety of PKC inhibitors in patients with diabetic microvascular complications (Table 1). One clinical trial showed that the general PKC inhibitor PKC412 reduced macular edema and improved visual acuity. However, gastrointestinal side effects (diarrhea, nausea, and vomiting) were associated with PKC412, along with dose-related liver toxicity, indicating that general PKC inhibitors may not be an appropriate choice for clinical application due to their side effects (115).
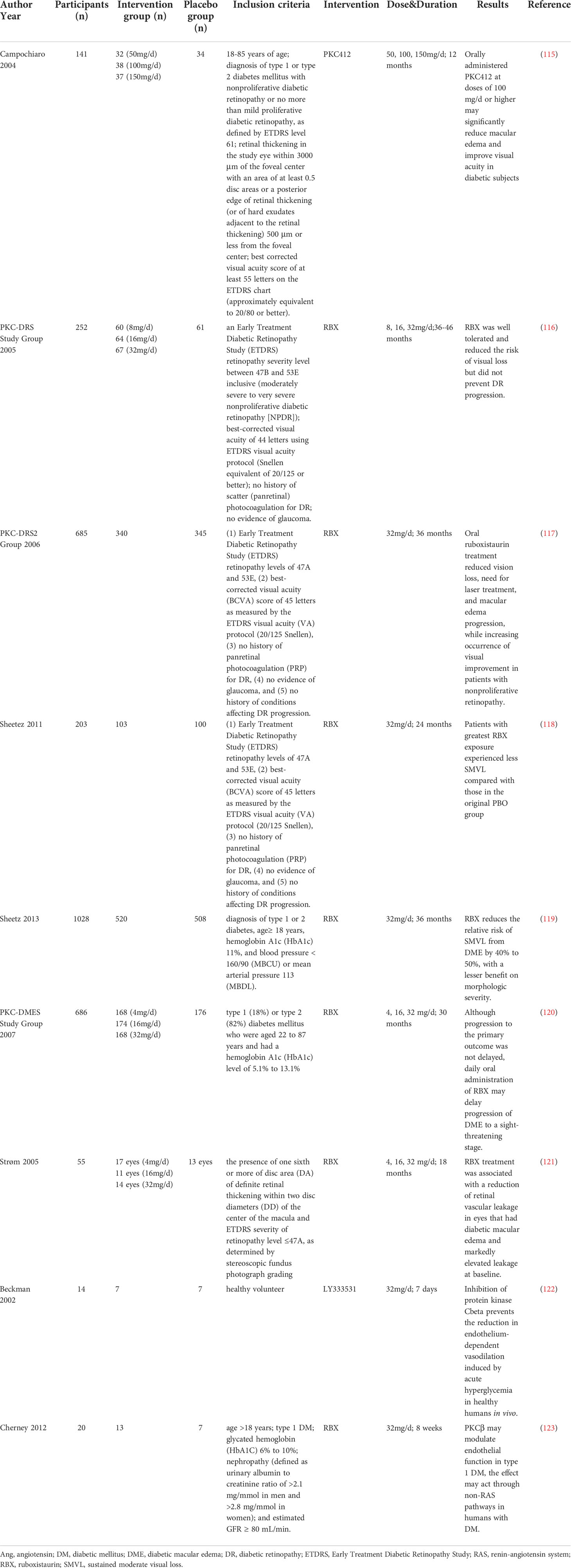
Table 1 Clinical trials of PKC inhibitors on diabetic microvascular complications.
Because of unsatisfactory safety, general PKC inhibitors have not been widely used in clinical trials. Hence, specific PKC inhibitors have been developed. The PKC inhibitors used in trials on diabetic microvascular complications were mainly PKC-β isoform inhibitors, including RBX and LY333531.
The effect of RBX has been evaluated mostly in patients with diabetic retinopathy. The PKC-DES trial evaluated the effect of RBX in patients with severe to very severe nonproliferative diabetic retinopathy. After 36-46 months of treatment with RBX, 32 mg/d RBX significantly reduced the risk of moderate visual loss compared with placebo and was well tolerated. However, the progression of diabetic retinopathy was not deferred (116). Another study (PKC-DRS2) recruited patients with moderate to severe nonproliferative diabetic retinopathy and evaluated the effect of RBX on vision loss. The trial was conducted for 36 months, and retinopathy status was assessed every 6 months. After treatment with RBX for 36 months, significant alleviation of visual loss was observed in the RBX group. In addition, the need for laser treatment and macular edema progression were also improved in the RBX group in patients with nonproliferative retinopathy (117). The PKC-DES2 group also conducted a 2-year open-label extension of PKC-DES2 and found that patients with the most exposure to RBX experienced less sustained moderate vision loss (SMVL) than those in the original placebo group (118). Moreover, two phase 3 trials (MBDL and MBCU) demonstrated similar results: RBX is effective in preventing SMVL by 50% in comparison with standard care; however, a significant difference was not achieved (P=0.06). Moreover, diabetic macular edema (DME) morphology-related measures did not show a trend in favor of or against RBX (119). PKC-DMES recruited patients with diabetic macular edema. Treatment with RBX for 30 months also did not show a satisfactory result in delaying the progression of sight-threatening DME (120). In contrast, Strøm et al. found that for patients with DME, 18 months of RBX treatment significantly prevented retinal vascular leakage compared to placebo, particularly in those with elevated leakage at baseline (121). In summary, the PKC-β inhibitor RBX did not show beneficial results in ameliorating the worsening of diabetic retinopathy. However, comparing PKC-DES with PKC-DES2, we found that RBX might be effective in patients with moderate to severe nonproliferative diabetic retinopathy, in which treatment with 32 mg/d RBX showed a satisfactory effect after 3 years. In addition, RBX may be a promising treatment for DME patients with elevated retinal vascular leakage.
Clinical trials on DM-induced endothelial dysfunction were also conducted. Beckman et al. conducted a randomized controlled trial that included healthy subjects. LY333531 or placebo was administered for 7 days before vascular function testing. Forearm blood flow was significantly reduced in patients treated with placebo, and impaired endothelium-dependent vasodilation was also observed. Pretreatment with LY333531 recovered blood flow and attenuated the blood flow response to methacholine chloride during hyperglycemia (122). Moreover, it was found that 8 weeks of treatment with 32 mg/d RBX blunted the effect of hyperglycemia on flow-mediated vasodilation in patients with type 1 DM (123). However, as most clinical trials have focused on diabetic retinopathy, more clinical trials, especially those on diabetic nephropathy, are expected to translate the results of these experiments to clinical applications.
Given the role of PKC in structural and functional changes in diabetic microvascular complications, it might be a promising target in the treatment of diabetic microvascular complications. However, although many studies have revealed the potential effects of PKC inhibition in alleviating the progression of diabetic microvascular complications, only RBX has shown application potential in diabetic retinopathy, and other PKC inhibitors have not shown satisfactory results in clinical trials. Hence, some challenges and limitations should still be noted in studies at present, as discussed below.
First, many in vivo studies used inappropriate disease models. Diabetic microvascular complications are induced by uncontrolled blood glucose over a long period, which is not well mimicked in current studies. In in vivo studies, DM is mostly induced by STZ intraperitoneal injection, with or without high-sugar and high-fat feed. STZ targets and destroys islet β cells, causing damage to the regulation of blood glucose. Nevertheless, the mechanisms that drive DM in humans are different from the mechanism of STZ-induced DM. Other factors, such as lipids and the genome, affect the development of DM and obvious diabetic microvascular complications. In addition, current experiments focus on certain inbred species, such as Rattus norvegicus and Mus musculus. Therefore, the experiment cannot perfectly explain the role of different PKC isoforms in diabetic microvascular complications in humans due to homogeneity. Therefore, the authors suggest that more species, or at least more animals in a group, should be considered in studies of metabolic diseases such as DM.
Second, there is still a lack of biomarkers to evaluate the effect and pharmacodynamics of PKC inhibitors. In terms of evaluating the effect and safety of PKC inhibitors for diabetic microvascular complications, clinical outcomes are the only evidence. This leads to a heavier economic burden. Thus, there is an urgent need to explore new biomarkers to reflect the role and bioavailability of PKC inhibitors in clinical trials, so that the application prospects of PKC inhibitors can be foreseen to be easier and less expensive.
Third, there are still few clinical trials on specific PKC isoform inhibitors other than RBX. As mentioned before, general PKC inhibitors showed poor bioavailability and tolerability in trials, which makes them unacceptable. In addition, PKC inhibitors may also affect other protein kinases, which cause unintended results (124). Hence, specific PKC isoform inhibitors are vital in clinical application. Despite the satisfactory results of specific PKC isoform inhibitors in animal experiments, PKC-β is the main focus of clinical trials. Inhibitors of PKC-α and PKC-δ may also be promising in terms of the results observed in experiments. However, only a few clinical trials on cancer used PKC inhibitors such as rottlerin (125), and diabetic microvascular complications are not being studied. More studies on the effect and safety of other PKC inhibitors are expected to be conducted to determine their applicability.
Finally, studies of new PKC inhibitors with increased selectivity and sample sizes are anticipated. PKC, a kinase family, influences many types of cells and contributes to different cell responses. As we have reviewed, the activation of PKC influences different downstream molecules, and inhibiting the activation of PKC prevents the exacerbation of diabetic microvascular complications. Nevertheless, unwanted effects of PKC isoform inhibitors should also be noted. For example, inhibition of PKC-β may rescue aggravated angiogenesis in diabetic retinopathy, but delayed wound healing may also be induced. In addition, current clinical trials on diabetic microvascular complications have recruited low numbers of participants. As we mentioned before, due to high heterogeneity among humans, many factors influence the effect of trials. Patients of different ages and races and with different lifestyles are differentially affected by the bioavailability of drugs (e.g., clopidogrel resistance in Asian individuals). However, the number of multicenter clinical trials with large sample sizes on diabetic microvascular complications is still unknown. Thus, it is also vital to conduct trials with more patients and take into account high heterogeneity across human subjects.
In terms of quantities of experiments and clinical trials, PKC can be thought of as a promising and attractive target in the treatment and prevention of diabetic microvascular complications. However, owing to the effects of PKC isoforms in various tissues and organs, it is necessary to develop new chemicals that are tissue- or organ-specific to better prevent adverse effects. The development of new highly selective and well-tolerated drugs that regulate PKC and ultimately translate into clinical medicine is expected to treat and prevent diabetic microvascular complications.
DP and LX formed the reference collection, conducted the reference analysis, and wrote the manuscript. MG contributed to the topic conception and made the decision to submit for publication. All authors contributed to the article and approved the submitted version.
This work was supported by the project of National Natural Science Foundation of China (Grant number: 81904025, 82104907) and Fundamental Research Funds for the Central public welfare research institutes Grant (Grant number: ZZ13-YQ-016 and ZZ13-YQ-016-C1).
We would like to thank American Journal Experts for language editing.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Cho NH, Shaw JE, Karuranga S, Huang Y, da Rocha Fernandes JD, Ohlrogge AW, et al. IDF diabetes atlas: Global estimates of diabetes prevalence for 2017 and projections for 2045. Diabetes Res Clin Pract (2018) 138:271–81. doi: 10.1016/j.diabres.2018.02.023
2. Barr EL, Zimmet PZ, Welborn TA, Jolley D, Magliano DJ, Dunstan DW, et al. Risk of cardiovascular and all-cause mortality in individuals with diabetes mellitus, impaired fasting glucose, and impaired glucose tolerance: The Australian diabetes, obesity, and lifestyle study (AusDiab). Circulation (2007) 116(2):151–7. doi: 10.1161/CIRCULATIONAHA.106.685628
3. Senyigit A, Tabak O, Orhanoglu T, Karadag A, Ugurlu S, Uzun H, et al. Glucagon-like peptide-1 levels and dipeptidyl peptidase-4 activity in type 2 diabetes. Clin Invest Med (2017) 40(5):E188–e199. doi: 10.25011/cim.v40i5.28624
4. Mauricio D, Alonso M N. Gratacòs chronic diabetes complications: The need to move beyond classical concepts. Trends Endocrinol Metab (2020) 31(4):287–95. doi: 10.1016/j.tem.2020.01.007
5. Gilbert RE, Mann JF, Hanefeld M, Spinas G, Bosch J, Yusuf S, et al. Basal insulin glargine and microvascular outcomes in dysglycaemic individuals: Results of the outcome reduction with an initial glargine intervention (ORIGIN) trial. Diabetologia (2014) 57(7):1325–31. doi: 10.1007/s00125-014-3238-4
6. Quality of life in type 2 diabetic patients is affected by complications but not by intensive policies to improve blood glucose or blood pressure control (UKPDS 37). U.K. prospective diabetes study group. Diabetes Care (1999) 22(7):1125–36. doi: 10.2337/diacare.22.7.1125
7. Patel A, MacMahon S, Chalmers J, Neal B, Billot L, Woodward M, et al. Intensive blood glucose control and vascular outcomes in patients with type 2 diabetes. N Engl J Med (2008) 358(24):2560–72. doi: 10.1056/NEJMoa0802987
8. Hiukka A, Maranghi M, Matikainen N, Taskinen MR. PPARalpha: an emerging therapeutic target in diabetic microvascular damage. Nat Rev Endocrinol (2010) 6(8):454–63. doi: 10.1038/nrendo.2010.89
9. Nishizuka Y. Intracellular signaling by hydrolysis of phospholipids and activation of protein kinase c. Science (1992) 258(5082):607–14. doi: 10.1126/science.1411571
10. Xia P, Inoguchi T, Kern TS, Engerman RL, Oates PJ, King GL. Characterization of the mechanism for the chronic activation of diacylglycerol-protein kinase c pathway in diabetes and hypergalactosemia. Diabetes (1994) 43(9):1122–9. doi: 10.2337/diab.43.9.1122
11. Wang QJ. PKD at the crossroads of DAG and PKC signaling. Trends Pharmacol Sci (2006) 27(6):317–23. doi: 10.1016/j.tips.2006.04.003
12. Jang JH, Kim EA, Park HJ, Sung EG, Song IH, Kim JY, et al. Methylglyoxal-induced apoptosis is dependent on the suppression of c-FLIP(L) expression via down-regulation of p65 in endothelial cells. J Cell Mol Med (2017) 21(11):2720–31. doi: 10.1111/jcmm.13188
13. Zhang Y, Peng T, Zhu H, Zheng X, Zhang X, Jiang N, et al. Prevention of hyperglycemia-induced myocardial apoptosis by gene silencing of toll-like receptor-4. J Transl Med (2010) 8133:133. doi: 10.1186/1479-5876-8-133
14. Birkenfeld AL, Shulman GI. Nonalcoholic fatty liver disease, hepatic insulin resistance, and type 2 diabetes. Hepatology (2014) 59(2):713–23. doi: 10.1002/hep.26672
15. Schambelan M, Blake S, Sraer J, Bens M, Nivez MP, Wahbe F. Increased prostaglandin production by glomeruli isolated from rats with streptozotocin-induced diabetes mellitus. J Clin Invest (1985) 75(2):404–12. doi: 10.1172/JCI111714
16. Gutiérrez A, Contreras C, Sánchez A, Prieto D. Role of phosphatidylinositol 3-kinase (PI3K), mitogen-activated protein kinase (MAPK), and protein kinase c (PKC) in calcium signaling pathways linked to the α(1)-adrenoceptor in resistance arteries. Front Physiol (2019) 1055. doi: 10.3389/fphys.2019.00055
17. Zhou SX, Huo DM, He XY, Yu P, Xiao YH, Ou CL, et al. High glucose/lysophosphatidylcholine levels stimulate extracellular matrix deposition in diabetic nephropathy via platelet−activating factor receptor. Mol Med Rep (2018) 17(2):2366–72. doi: 10.3892/mmr.2017.8102
18. Retamal JS, Grace MS, Dill LK, Ramirez-Garcia P, Peng S, Gondin AB, et al. Serotonin-induced vascular permeability is mediated by transient receptor potential vanilloid 4 in the airways and upper gastrointestinal tract of mice. Lab Invest (2021) 101(7):851–64. doi: 10.1038/s41374-021-00593-7
19. Moriya N J. Ferrara inhibition of protein kinase c enhances angiogenesis induced by platelet-derived growth factor c in hyperglycemic endothelial cells. Cardiovasc Diabetol (2015) 1419:19. doi: 10.1186/s12933-015-0180-9
20. Mochly-Rosen D, Das K, Grimes KV. Protein kinase c, an elusive therapeutic target? Nat Rev Drug Discov (2012) 11(12):937–57. doi: 10.1038/nrd3871
21. Carter CA. Protein kinase c as a drug target: implications for drug or diet prevention and treatment of cancer. Curr Drug Targets (2000) 1(2):163–83. doi: 10.2174/1389450003349317
22. P. Geraldes GL. King activation of protein kinase c isoforms and its impact on diabetic complications. Circ Res (2010) 106(8):1319–31. doi: 10.1161/CIRCRESAHA.110.217117
23. Anwer MS. Role of protein kinase c isoforms in bile formation and cholestasis. Hepatology (2014) 60(3):1090–7. doi: 10.1002/hep.27088
24. Newton AC. Protein kinase c: perfectly balanced. Crit Rev Biochem Mol Biol (2018) 53(2):208–30. doi: 10.1080/10409238.2018.1442408
25. Boije af Gennäs G, Talman V, Yli-Kauhaluoma J, Tuominen RK, Ekokoski E. Current status and future prospects of C1 domain ligands as drug candidates. Curr Top Med Chem (2011) 11(11):1370–92. doi: 10.2174/156802611795589584
26. Jornayvaz FR, Shulman GI. Diacylglycerol activation of protein kinase cϵ and hepatic insulin resistance. Cell Metab (2012) 15(5):574–84. doi: 10.1016/j.cmet.2012.03.005
27. Mogensen CE, Christensen E CK. Vittinghus the stages in diabetic renal disease. with emphasis on the stage of incipient diabetic nephropathy. Diabetes (1983) 32:Suppl 264–78. doi: 10.2337/diab.32.2.s64
28. Forbes JM, Cooper ME. Mechanisms of diabetic complications. Physiol Rev (2013) 93(1):137–88. doi: 10.1152/physrev.00045.2011
29. O'Bryan GT, Hostetter TH. The renal hemodynamic basis of diabetic nephropathy. Semin Nephrol (1997) 17(2):93–100.
30. Brosius FC, Coward RJ. Podocytes, signaling pathways, and vascular factors in diabetic kidney disease. Adv Chronic Kidney Dis (2014) 21(3):304–10. doi: 10.1053/j.ackd.2014.03.011
31. Singh R, Singh AK, Alavi N, Leehey DJ. Mechanism of increased angiotensin II levels in glomerular mesangial cells cultured in high glucose. J Am Soc Nephrol (2003) 14(4):873–80. doi: 10.1097/01.ASN.0000060804.40201.6E
32. Gilbert RE, Krum H, Wilkinson-Berka J, Kelly DJ. The renin-angiotensin system and the long-term complications of diabetes: pathophysiological and therapeutic considerations. Diabetes Med (2003) 20(8):607–21. doi: 10.1046/j.1464-5491.2003.00979.x
33. Nakamichi R, Hayashi H K. Itoh effects of high glucose and lipotoxicity on diabetic podocytes. Nutrients (2021) 13(1):241. doi: 10.3390/nu13010241
34. Seyer-Hansen K, J. Hansen HJ. Gundersen renal hypertrophy in experimental diabetes. a morphometric study. Diabetologia (1980) 18(6):501–5. doi: 10.1007/bf00261707
35. Mauer SM, Steffes MW, Ellis EN, Sutherland DE, Brown DM, Goetz FC. Structural-functional relationships in diabetic nephropathy. J Clin Invest (1984) 74(4):1143–55. doi: 10.1172/JCI111523
36. Han Q, Zhu H, Chen X, Liu Z. Non-genetic mechanisms of diabetic nephropathy. Front Med (2017) 11(3). doi: 10.1007/s11684-017-0569-9
37. Nobe K, Takenouchi Y, Kasono K, Hashimoto T, Honda K. Two types of overcontraction are involved in intrarenal artery dysfunction in type II diabetic mouse. J Pharmacol Exp Ther (2014) 351(1):77–86. doi: 10.1124/jpet.114.216747
38. Nobe K, Nezu Y, Tsumita N, Hashimoto T, Honda K. Intra- and extrarenal arteries exhibit different profiles of contractile responses in high glucose conditions. Br J Pharmacol (2008) 155(8):1204–13. doi: 10.1038/bjp.2008.365
39. Ajayi AA, Fidelis P. The effect of flutamide on systemic and renal hemodynamics in zucker diabetic rats: paradoxic renal vasodilator response to endothelin-1 and TXA2 receptor activation in female sex. J Cardiovasc Pharmacol (2006) 48(5):191–8. doi: 10.1097/01.fjc.0000246941.84607.11
40. Youssef N, Noureldein M, Njeim R, Ghadieh HE, Harb F, Azar ST, et al. Reno-Protective effect of GLP-1 receptor agonists in Type1 diabetes: Dual action on TRPC6 and NADPH oxidases. Biomedicines (2021) 9(10):1360. doi: 10.3390/biomedicines9101360
41. Blattner SM, Hodgin JB, Nishio M, Wylie SA, Saha J, Soofi AA, et al. Divergent functions of the rho GTPases Rac1 and Cdc42 in podocyte injury. Kidney Int (2013) 84(5):920–30. doi: 10.1038/ki.2013.175
42. Menne J, Meier M, Park JK, Boehne M, Kirsch T, Lindschau C, et al. Nephrin loss in experimental diabetic nephropathy is prevented by deletion of protein kinase c alpha signaling in-vivo. Kidney Int (2006) 70(8):1456–62. doi: 10.1038/sj.ki.5001830
43. Thallas-Bonke V, Jha JC, Gray SP, Barit D, Haller H, Schmidt HH, et al. Nox-4 deletion reduces oxidative stress and injury by PKC-α-associated mechanisms in diabetic nephropathy. Physiol Rep (2014) 2(11)e12192. doi: 10.14814/phy2.12192
44. Komeno M, Pang X, Shimizu A, Molla MR, Yasuda-Yamahara M, Kume S, et al. Cardio- and reno-protective effects of dipeptidyl peptidase III in diabetic mice. J Biol Chem (2021) 296:296100761. doi: 10.1016/j.jbc.2021.100761
45. Kim NH, Jung HH, Cha DR, Choi DS. Expression of vascular endothelial growth factor in response to high glucose in rat mesangial cells. J Endocrinol (2000) 165(3):617–24. doi: 10.1677/joe.0.1650617
46. Mima A, Kitada M, Geraldes P, Li Q, Matsumoto M, Mizutani K, et al. Glomerular VEGF resistance induced by PKCδ/SHP-1 activation and contribution to diabetic nephropathy. FASEB J (2012) 26(7):2963–74. doi: 10.1096/fj.11-202994
47. Scheinman JI, Fish AJ, Matas AJ, Michael AF. The immunohistopathology of glomerular antigens. II. the glomerular basement membrane, actomyosin, and fibroblast surface antigens in normal, diseased, and transplanted human kidneys. Am J Pathol (1978) 90(1):71–88.
48. Kreisberg JI, Ayo SH. The glomerular mesangium in diabetes mellitus. Kidney Int (1993) 43(1):109–13. doi: 10.1038/ki.1993.18
49. Xia L, Wang H, Goldberg HJ, Munk S, Fantus IG, Whiteside CI. Mesangial cell NADPH oxidase upregulation in high glucose is protein kinase c dependent and required for collagen IV expression. Am J Physiol Renal Physiol (2006) 290(2):F345–56. doi: 10.1152/ajprenal.00119.2005
50. Tahara A, Tsukada J, Tomura Y, Yatsu T, Shibasaki M. Effects of high glucose on AVP-induced hyperplasia, hypertrophy, and type IV collagen synthesis in cultured rat mesangial cells. Endocr Res (2012) 37(4):216–27. doi: 10.3109/07435800.2012.671400
51. Xin X, Khan ZA, Chen S, Chakrabarti S. Extracellular signal-regulated kinase (ERK) in glucose-induced and endothelin-mediated fibronectin synthesis. Lab Invest (2004) 84(11):1451–9. doi: 10.1038/labinvest.3700178
52. Zhuang Q Y. Yin peroxisome proliferator-activated receptor γ agonists attenuate hyperglycaemia-induced hyaluronan secretion in vascular smooth muscle cells by inhibiting PKCβ2. Cell Biochem Biophys (2013) 67(2):583–90. doi: 10.1007/s12013-013-9545-4
53. Grigorova-Borsos AM, Bakillah A, Urios P, Leblond V, Guillot R, Sternberg M. Production of type IV collagen and 72-kDa gelatinase by human endothelial cells cultured in high glucose. effects of a protein kinase c inhibitor, GF 109203X. Biochem Cell Biol (1996) 74(5):659–67. doi: 10.1139/o96-071
54. Soetikno V, Watanabe K, Sari FR, Harima M, Thandavarayan RA, Veeraveedu PT, et al. Curcumin attenuates diabetic nephropathy by inhibiting PKC-α and PKC-β1 activity in streptozotocin-induced type I diabetic rats. Mol Nutr Food Res (2011) 55(11):1655–65. doi: 10.1002/mnfr.201100080
55. Koya D, Jirousek MR, Lin YW, Ishii H, Kuboki K, King GL. Characterization of protein kinase c beta isoform activation on the gene expression of transforming growth factor-beta, extracellular matrix components, and prostanoids in the glomeruli of diabetic rats. J Clin Invest (1997) 100(1):115–26. doi: 10.1172/JCI119503
56. Menne J, Shushakova N, Bartels J, Kiyan Y, Laudeley R, Haller H, et al. Dual inhibition of classical protein kinase c-α and protein kinase c-β isoforms protects against experimental murine diabetic nephropathy. Diabetes (2013) 62(4):1167–74. doi: 10.2337/db12-0534
57. Tokuyama H, Kim S, Zhang Y, Langham RG, Cox AJ, Gow RM, et al. Protein kinase c β inhibition ameliorates experimental mesangial proliferative glomerulonephritis. Nephrol (Carlton) (2011) 16(7):649–55. doi: 10.1111/j.1440-1797.2011.01475.x
58. Meier M, Menne J, Park JK, Holtz M, Gueler F, Kirsch T, et al. Deletion of protein kinase c-epsilon signaling pathway induces glomerulosclerosis and tubulointerstitial fibrosis in vivo. J Am Soc Nephrol (2007) 18(4):1190–8. doi: 10.1681/ASN.2005070694
59. Baccora MH, Cortes P, Hassett C, Taube DW, Yee J. Effects of long-term elevated glucose on collagen formation by mesangial cells. Kidney Int (2007) 72(10):1216–25. doi: 10.1038/sj.ki.5002517
60. Whiteside C, Wang H, Xia L, Munk S, Goldberg HJ, Fantus IG. Rosiglitazone prevents high glucose-induced vascular endothelial growth factor and collagen IV expression in cultured mesangial cells. Exp Diabetes Res (2009) 2009:2009910783. doi: 10.1155/2009/910783
61. K. Sharma, Ziyadeh FN. Hyperglycemia and diabetic kidney disease. The case for transforming growth factor-beta as a key mediator. Diabetes (1995) 44(10):1139–46. doi: 10.2337/diab.44.10.1139
62. Chen S, Cohen MP, Lautenslager GT, Shearman CW, Ziyadeh FN. Glycated albumin stimulates TGF-beta 1 production and protein kinase c activity in glomerular endothelial cells. Kidney Int (2001) 59(2):673–81. doi: 10.1046/j.1523-1755.2001.059002673.x
63. Wei XF, Zhou QG, Hou FF, Liu BY, Liang M. Advanced oxidation protein products induce mesangial cell perturbation through PKC-dependent activation of NADPH oxidase. Am J Physiol Renal Physiol (2009) 296(2):F427–37. doi: 10.1152/ajprenal.90536.2008
64. Ohshiro Y, Ma RC, Yasuda Y, Hiraoka-Yamamoto J, Clermont AC, Isshiki K, et al. Reduction of diabetes-induced oxidative stress, fibrotic cytokine expression, and renal dysfunction in protein kinase cbeta-null mice. Diabetes (2006) 55(11):3112–20. doi: 10.2337/db06-0895
65. Murphy M, Godson C, Cannon S, Kato S, Mackenzie HS, Martin F, et al. Suppression subtractive hybridization identifies high glucose levels as a stimulus for expression of connective tissue growth factor and other genes in human mesangial cells. J Biol Chem (1999) 274(9):5830–4. doi: 10.1074/jbc.274.9.5830
66. Xiao YH, He XY, Han Q, Yang F, Zhou SX. Atorvastatin prevents glomerular extracellular matrix formation by interfering with the PKC signaling pathway. Mol Med Rep (2018) 17(5):6441–8. doi: 10.3892/mmr.2018.8724
67. Liu H, Luo Y, Zhang T, Zhang Y, Wu Q, Yuan L, et al. Genetic deficiency of aldose reductase counteracts the development of diabetic nephropathy in C57BL/6 mice. Diabetologia (2011) 54(5):1242–51. doi: 10.1007/s00125-011-2045-4
68. Jha JC, Thallas-Bonke V, Banal C, Gray SP, Chow BS, Ramm G, et al. Podocyte-specific Nox4 deletion affords renoprotection in a mouse model of diabetic nephropathy. Diabetologia (2016) 59(2):379–89. doi: 10.1007/s00125-015-3796-0
69. Jha JC, Banal C, Okabe J, Gray SP, Hettige T, Chow BSM, et al. NADPH oxidase Nox5 accelerates renal injury in diabetic nephropathy. Diabetes (2017) 66(10):2691–703. doi: 10.2337/db16-1585
70. Jha JC, Dai A, Holterman CE, Cooper ME, Touyz RM, Kennedy CR, et al. Endothelial or vascular smooth muscle cell-specific expression of human NOX5 exacerbates renal inflammation, fibrosis and albuminuria in the akita mouse. Diabetologia (2019) 62(9):1712–26. doi: 10.1007/s00125-019-4924-z
71. Hayashida T, Schnaper HW. High ambient glucose enhances sensitivity to TGF-beta1 via extracellular signal–regulated kinase and protein kinase cdelta activities in human mesangial cells. J Am Soc Nephrol (2004) 15(8):2032–41. doi: 10.1097/01.ASN.0000133198.74973.60
72. Tuttle KR, Johnson EC, Cooney SK, Anderberg RJ, Johnson EK, Clifton GD, et al. Amino acids injure mesangial cells by advanced glycation end products, oxidative stress, and protein kinase c. Kidney Int (2005) 67(3):953–68. doi: 10.1111/j.1523-1755.2005.00159.x
73. Wu D, Peng F, Zhang B, Ingram AJ, Kelly DJ, Gilbert RE, et al. EGFR-PLCgamma1 signaling mediates high glucose-induced PKCbeta1-akt activation and collagen I upregulation in mesangial cells. Am J Physiol Renal Physiol (2009) 297(3):F822–34. doi: 10.1152/ajprenal.00054.2009
74. Kang Q, Yang C. Oxidative stress and diabetic retinopathy: Molecular mechanisms, pathogenetic role and therapeutic implications. Redox Biol (2020) 37:37101799. doi: 10.1016/j.redox.2020.101799
75. Kowluru RA, Chan PS. Oxidative stress and diabetic retinopathy. Exp Diabetes Res (2007) 2007:200743603. doi: 10.1155/2007/43603
76. Sivaprasad S, Pearce E. The unmet need for better risk stratification of non-proliferative diabetic retinopathy. Diabetes Med (2019) 36(4):424–33. doi: 10.1111/dme.13868
77. Wang W, Lo ACY. Diabetic retinopathy: Pathophysiology and treatments. Int J Mol Sci (2018) 19(6):1816. doi: 10.3390/ijms19061816
78. Stitt AW, Curtis TM, Chen M, Medina RJ, McKay GJ, Jenkins A, et al. The progress in understanding and treatment of diabetic retinopathy. Prog Retin Eye Res (2016) 51:51156–86. doi: 10.1016/j.preteyeres.2015.08.001
79. Antonetti DA, Klein R, Gardner TW. Diabetic retinopathy. N Engl J Med (2012) 366(13):1227–39. doi: 10.1056/NEJMra1005073
80. Bursell SE, Takagi C, Clermont AC, Takagi H, Mori F, Ishii H, et al. Specific retinal diacylglycerol and protein kinase c beta isoform modulation mimics abnormal retinal hemodynamics in diabetic rats. Invest Ophthalmol Vis Sci (1997) 38(13):2711–20.
81. Nonaka A, Kiryu J, Tsujikawa A, Yamashiro K, Miyamoto K, Nishiwaki H, et al. PKC-beta inhibitor (LY333531) attenuates leukocyte entrapment in retinal microcirculation of diabetic rats. Invest Ophthalmol Vis Sci (2000) 41(9):2702–6.
82. Park JY, Takahara N, Gabriele A, Chou E, Naruse K, Suzuma K, et al. Induction of endothelin-1 expression by glucose: an effect of protein kinase c activation. Diabetes (2000) 49(7):1239–48. doi: 10.2337/diabetes.49.7.1239
83. Chakravarthy U, Hayes RG, Stitt AW, McAuley E, Archer DB. Constitutive nitric oxide synthase expression in retinal vascular endothelial cells is suppressed by high glucose and advanced glycation end products. Diabetes (1998) 47(6):945–52. doi: 10.2337/diabetes.47.6.945
84. Warboys CM, Toh HB, Fraser PA. Role of NADPH oxidase in retinal microvascular permeability increase by RAGE activation. Invest Ophthalmol Vis Sci (2009) 50(3):1319–28. doi: 10.1167/iovs.08-2730
85. Pricci F, Leto G, Amadio L, Iacobini C, Cordone S, Catalano S, et al. Oxidative stress in diabetes-induced endothelial dysfunction involvement of nitric oxide and protein kinase c. Free Radic Biol Med (2003) 35(6):683–94. doi: 10.1016/S0891-5849(03)00401-5
86. Harhaj NS, Felinski EA, Wolpert EB, Sundstrom JM, Gardner TW, Antonetti DA. VEGF activation of protein kinase c stimulates occludin phosphorylation and contributes to endothelial permeability. Invest Ophthalmol Vis Sci (2006) 47(11):5106–15. doi: 10.1167/iovs.06-0322
87. Ido Y, Chang KC, Lejeune WS, Bjercke RJ, Reiser KM, Williamson JR, et al. Vascular dysfunction induced by AGE is mediated by VEGF via mechanisms involving reactive oxygen species, guanylate cyclase, and protein kinase c. Microcirculation (2001) 8(4):251–63. doi: 10.1111/j.1549-8719.2001.tb00174.x
88. Kumar B, Gupta SK, Srinivasan BP, Nag TC, Srivastava S, Saxena R. Hesperetin ameliorates hyperglycemia induced retinal vasculopathy via anti-angiogenic effects in experimental diabetic rats. Vascul Pharmacol (2012) 57(5-6):201–7. doi: 10.1016/j.vph.2012.02.007
89. Murakami T, Frey T, Lin C, Antonetti DA. Protein kinase cβ phosphorylates occludin regulating tight junction trafficking in vascular endothelial growth factor-induced permeability in vivo. Diabetes (2012) 61(6):1573–83. doi: 10.2337/db11-1367
90. Xu X, Zhu Q, Xia X, Zhang S, Gu Q, Luo D. Blood-retinal barrier breakdown induced by activation of protein kinase c via vascular endothelial growth factor in streptozotocin-induced diabetic rats. Curr Eye Res (2004) 28(4):251–6. doi: 10.1076/ceyr.28.4.251.27834
91. Aiello LP, Bursell SE, Clermont A, Duh E, Ishii H, Takagi C, et al. Vascular endothelial growth factor-induced retinal permeability is mediated by protein kinase c in vivo and suppressed by an orally effective beta-isoform-selective inhibitor. Diabetes (1997) 46(9):1473–80. doi: 10.2337/diab.46.9.1473
92. Song HB, Jun HO, Kim JH, Yu YS, Kim KW, Kim JH. Suppression of protein kinase c-ζ attenuates vascular leakage via prevention of tight junction protein decrease in diabetic retinopathy. Biochem Biophys Res Commun (2014) 444(1):63–8. doi: 10.1016/j.bbrc.2014.01.002
93. Aveleira CA, Lin CM, Abcouwer SF, Ambrósio AF, Antonetti DA. TNF-α signals through PKCζ/NF-κB to alter the tight junction complex and increase retinal endothelial cell permeability. Diabetes (2010) 59(11):2872–82. doi: 10.2337/db09-1606
94. Titchenell PM, Lin CM, Keil JM, Sundstrom JM, Smith CD, Antonetti DA. Novel atypical PKC inhibitors prevent vascular endothelial growth factor-induced blood-retinal barrier dysfunction. Biochem J (2012) 446(3):455–67. doi: 10.1042/BJ20111961
95. Kim JH, Kim JH, Jun HO, Yu YS, Kim KW. Inhibition of protein kinase c delta attenuates blood-retinal barrier breakdown in diabetic retinopathy. Am J Pathol (2010) 176(3):1517–24. doi: 10.2353/ajpath.2010.090398
96. Deissler HL, H. Deissler GE. Lang Inhibition of protein kinase c is not sufficient to prevent or reverse effects of VEGF165 on claudin-1 and permeability in microvascular retinal endothelial cells. Invest Ophthalmol Vis Sci (2010) 51(1):535–42. doi: 10.1167/iovs.09-3917
97. Giordo R, Nasrallah GK, Posadino AM, Galimi F, Capobianco G, Eid AH, et al. Resveratrol-elicited PKC inhibition counteracts NOX-mediated endothelial to mesenchymal transition in human retinal endothelial cells exposed to high glucose. Antioxid (Basel) (2021) 10(2). doi: 10.3390/antiox10020224
98. Cheung N, Mitchell P, Wong TY. Diabetic retinopathy. Lancet (2010) 376(9735):124–36. doi: 10.1016/S0140-6736(09)62124-3
99. Miyata Y, Kase M, Sugita Y, Shimada A, Nagase T, Katsura Y, et al. Protein kinase c-mediated regulation of matrix metalloproteinase and tissue inhibitor of metalloproteinase production in a human retinal müller cells. Curr Eye Res (2012) 37(9):842–9. doi: 10.3109/02713683.2012.682635
100. Ojima T, Takagi H, Suzuma K, Oh H, Suzuma I, Ohashi H, et al. EphrinA1 inhibits vascular endothelial growth factor-induced intracellular signaling and suppresses retinal neovascularization and blood-retinal barrier breakdown. Am J Pathol (2006) 168(1):331–9. doi: 10.2353/ajpath.2006.050435
101. Malik RA, Newrick PG, Sharma AK, Jennings A, Ah-See AK, Mayhew TM, et al. Microangiopathy in human diabetic neuropathy: relationship between capillary abnormalities and the severity of neuropathy. Diabetologia (1989) 32(2):92–102. doi: 10.1007/BF00505180
102. Thrainsdottir S, Malik RA, Dahlin LB, Wiksell P, Eriksson KF, Rosén I, et al. Endoneurial capillary abnormalities presage deterioration of glucose tolerance and accompany peripheral neuropathy in man. Diabetes (2003) 52(10):2615–22. doi: 10.2337/diabetes.52.10.2615
103. Feldman EL, Callaghan BC, Pop-Busui R, Zochodne DW, Wright DE, Bennett DL, et al. Diabetic neuropathy. Nat Rev Dis Primers (2019) 5(1):42. doi: 10.1038/s41572-019-0092-1
104. Selvarajah D, Wilkinson ID, Emery CJ, Harris ND, Shaw PJ, Witte DR, et al. Early involvement of the spinal cord in diabetic peripheral neuropathy. Diabetes Care (2006) 29(12):2664–9. doi: 10.2337/dc06-0650
105. Perkins AT, Morgenlander JC. Endocrinologic causes of peripheral neuropathy. pins and needles in a stocking-and-glove pattern and other symptoms. Postgrad Med (1997) 102(3):81–2, 90-2,102-6. doi: 10.3810/pgm.1997.09.318
106. Spallone V. Update on the impact, diagnosis and management of cardiovascular autonomic neuropathy in diabetes: What is defined, what is new, and what is unmet. Diabetes Metab J (2019) 43(1):3–30. doi: 10.4093/dmj.2018.0259
107. Samakidou G, Eleftheriadou I, Tentolouris A, Papanas N, Tentolouris N. Rare diabetic neuropathies: It is not only distal symmetrical polyneuropathy. Diabetes Res Clin Pract (2021) 177:177108932. doi: 10.1016/j.diabres.2021.108932
108. Feldman EL, Nave KA, Jensen TS, Bennett DLH. New horizons in diabetic neuropathy: Mechanisms, bioenergetics, and pain. Neuron (2017) 93(6):1296–313. doi: 10.1016/j.neuron.2017.02.005
109. Muramatsu K. Diabetes mellitus-related dysfunction of the motor system. Int J Mol Sci (2020) 21(20). doi: 10.3390/ijms21207485
110. Yang Y, Hu X, Zhang Q, Zou R. Diabetes mellitus and risk of falls in older adults: a systematic review and meta-analysis. Age Ageing (2016) 45(6):761–7. doi: 10.1093/ageing/afw140
111. Cotter MA, Jack AM, Cameron NE. Effects of the protein kinase c beta inhibitor LY333531 on neural and vascular function in rats with streptozotocin-induced diabetes. Clin Sci (Lond) (2002) 103(3):311–21. doi: 10.1042/cs1030311
112. Jack AM, Cameron NE, Cotter MA. Effects of the diacylglycerol complexing agent, cremophor, on nerve-conduction velocity and perfusion in diabetic rats. J Diabetes Complications (1999) 13(1):2–9. doi: 10.1016/S1056-8727(98)00014-2
113. Cameron NE, Cotter MA, Jack AM, Basso MD, Hohman TC. Protein kinase c effects on nerve function, perfusion, na(+), k(+)-ATPase activity and glutathione content in diabetic rats. Diabetologia (1999) 42(9):1120–30. doi: 10.1007/s001250051280
114. Coppey LJ, Gellett JS, Davidson EP, Dunlap JA, Lund DD, Yorek MA. Effect of antioxidant treatment of streptozotocin-induced diabetic rats on endoneurial blood flow, motor nerve conduction velocity, and vascular reactivity of epineurial arterioles of the sciatic nerve. Diabetes (2001) 50(8):1927–37. doi: 10.2337/diabetes.50.8.1927
115. Campochiaro PA. Reduction of diabetic macular edema by oral administration of the kinase inhibitor PKC412. Invest Ophthalmol Vis Sci (2004) 45(3):922–31. doi: 10.1167/iovs.03-0955
116. The effect of ruboxistaurin on visual loss in patients with moderately severe to very severe nonproliferative diabetic retinopathy: initial results of the protein kinase c beta inhibitor diabetic retinopathy study (PKC-DRS) multicenter randomized clinical trial. Diabetes (2005) 54(7):2188–97. doi: 10.2337/diabetes.54.7.2188
117. Aiello LP, Davis MD, Girach A, Kles KA, Milton RC, Sheetz MJ, et al. Effect of ruboxistaurin on visual loss in patients with diabetic retinopathy. Ophthalmology (2006) 113(12):2221–30. doi: 10.1016/j.ophtha.2006.07.032
118. Sheetz MJ, Aiello LP, Shahri N, Davis MD, Kles KA, Danis RP. Effect of ruboxistaurin (RBX) on visual acuity decline over a 6-year period with cessation and reinstitution of therapy: results of an open-label extension of the protein kinase c diabetic retinopathy study 2 (PKC-DRS2). Retina (2011) 31(6):1053–9. doi: 10.1097/IAE.0b013e3181fe545f
119. Sheetz MJ, Aiello LP, Davis MD, Danis R, Bek T, Cunha-Vaz J, et al. The effect of the oral PKC β inhibitor ruboxistaurin on vision loss in two phase 3 studies. Invest Ophthalmol Vis Sci (2013) 54(3):1750–7. doi: 10.1167/iovs.12-11055
120. Effect of ruboxistaurin in patients with diabetic macular edema: thirty-month results of the randomized PKC-DMES clinical trial. Arch Ophthalmol (2007) 125(3):318–24. doi: 10.1001/archopht.125.3.318
121. Strøm C, Sander B, Klemp K, Aiello LP, Lund-Andersen H, Larsen M. Effect of ruboxistaurin on blood-retinal barrier permeability in relation to severity of leakage in diabetic macular edema. Invest Ophthalmol Vis Sci (2005) 46(10):3855–8. doi: 10.1167/iovs.05-0096
122. Beckman JA, Goldfine AB, Gordon MB, Garrett LA, Creager MA. Inhibition of protein kinase cbeta prevents impaired endothelium-dependent vasodilation caused by hyperglycemia in humans. Circ Res (2002) 90(1):107–11. doi: 10.1161/hh0102.102359
123. Cherney DZ, Reich HN, Scholey JW, Lai V, Slorach C, Zinman B, et al. Systemic hemodynamic function in humans with type 1 diabetes treated with protein kinase cβ inhibition and renin-angiotensin system blockade: a pilot study. Can J Physiol Pharmacol (2012) 90(1):113–21. doi: 10.1139/y11-106
124. Davis MI, Hunt JP, Herrgard S, Ciceri P, Wodicka LM, Pallares G, et al. Comprehensive analysis of kinase inhibitor selectivity. Nat Biotechnol (2011) 29(11):1046–51. doi: 10.1038/nbt.1990
Keywords: protein kinase C, diabetes mellitus, diabetic microvascular complications, microvascular, review
Citation: Pan D, Xu L and Guo M (2022) The role of protein kinase C in diabetic microvascular complications. Front. Endocrinol. 13:973058. doi: 10.3389/fendo.2022.973058
Received: 19 June 2022; Accepted: 25 July 2022;
Published: 17 August 2022.
Edited by:
Mohd Imtiaz Nawaz, Department of Ophthalmology, College of Medicine, King Saud University, Saudi ArabiaReviewed by:
Ismail Syed, Beth Israel Deaconess Medical Center and Harvard Medical School, United StatesCopyright © 2022 Pan, Xu and Guo. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Ming Guo, NTQ3NDUzMjAyQHFxLmNvbQ==
†These authors have contributed equally to this work and share first authorship
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.