
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Endocrinol., 31 October 2022
Sec. Pediatric Endocrinology
Volume 13 - 2022 | https://doi.org/10.3389/fendo.2022.961650
This article is part of the Research TopicPediatric Adrenal NeoplasmsView all 12 articles
 Maran Ilanchezhian1
Maran Ilanchezhian1 Diana Grace Varghese2
Diana Grace Varghese2 John W. Glod1Karlyne M. Reilly1
John W. Glod1Karlyne M. Reilly1 Brigitte C. Widemann1Yves Pommier2
Brigitte C. Widemann1Yves Pommier2 Rosandra N. Kaplan1
Rosandra N. Kaplan1 Jaydira Del Rivero2*
Jaydira Del Rivero2*Adrenocortical carcinoma (ACC) is a rare endocrine malignancy of the adrenal gland with an unfavorable prognosis. It is rare in the pediatric population, with an incidence of 0.2-0.3 patients per million in patients under 20 years old. It is primarily associated with Li-Fraumeni and Beckwith-Wiedemann tumor predisposition syndromes in children. The incidence of pediatric ACC is 10-15fold higher in southern Brazil due to a higher prevalence of TP53 mutation associated with Li-Fraumeni syndrome in that population. Current treatment protocols are derived from adult ACC and consist of surgery and/or chemotherapy with etoposide, doxorubicin, and cisplatin (EDP) with mitotane. Limited research has been reported on other treatment modalities for pediatric ACC, including mitotane, pembrolizumab, cabozantinib, and chimeric antigen receptor autologous cell (CAR-T) therapy.
Adrenocortical Carcinoma (ACC) is a rare endocrine malignancy with an overall unfavorable prognosis. It is rare amongst children, with an incidence of 0.2-0.3 patients per million, in patients under 20 years of age (1, 2), accounting for less than 0.2% of all pediatric malignancies (3). Pediatric ACC displays a bimodal distribution with peaks under the age of 5 and after 10 years and presents more frequently in females than males (4, 5). Pediatric ACC incidence is 10-15 times higher in southern Brazil, likely due to a higher prevalence of the R337H TP53 mutation (6, 7). Pediatric ACCs are often linked to cancer predisposition syndromes, with most childhood ACCs associated with germline mutations (8, 9).
Pediatric patients with ACC often present differently than adult patients. One key difference from adult ACC is that pediatric ACCs are more often functional, often presenting with excess androgen production (5, 10, 11). Pediatric ACC patients have a 5-year survival rate reported to be between 30% to 70%, depending on disease presentation (11–13). Outcomes in patients with metastatic disease are poor, with a 5-year survival rate estimated to be less than 20% (1, 10, 11, 14, 15). Prognosis of childhood ACC is highly variable and difficult to predict in clinical practice (9).
ACCs that develop in children can either be sporadic or linked to a cancer predisposition syndrome. The genomic landscape of pediatric ACC is characterized by copy-neutral loss of heterozygosity of chromosomes 11 and 17, which is associated with germline TP53 pathogenic variants, insulin-like growth factor-2 overexpression, and somatic mutations in ATRX and CTNNB1 (16).
Pediatric ACCs are most commonly associated with Li-Fraumeni Syndrome, an autosomal dominant familial cancer syndrome associated with a number of malignancies including sarcoma, breast cancer, brain tumors, leukemia, lymphoma, and adrenocortical carcinomas (17–19). Li-Fraumeni Syndrome occurs in the context of germline mutations in the TP53 tumor suppressor gene, which encodes for p53, a transcription factor that helps preserve genomic integrity and activates apoptosis in cells with DNA damage (20). Germline mutations in the gene encoding the p53 tumor suppressor located at 17p13.1 have been found in approximately 50% of children with ACC (21, 22). However, germline p53 mutations are much less common in adults with ACC (23).
In southern Brazil, there is a fifteen-fold increase in the incidence of pediatric ACC compared to other global populations (6). This is due to a unique germline missense mutation in TP53 (R337H). This mutation exists at a high prevalence in this population (0.3%), but with a low penetrance of approximately 2%, which contrasts with the classical presentation of Li-Fraumeni Syndrome that presents with 100% penetrance (24–26). This R337H mutation affects the protein’s oligomerization domain, resulting in pH-dependent instability, predisposing these individuals to adrenocortical tumors (23, 26–29). Additionally, R337H mutation carriers are at increased risk of developing other tumors associated with Li-Fraumeni Syndrome, as well as adult ACC (12, 27). A newborn screening program for R337H mutations in southern Brazil exists and has been successful at early detection of ACCs in this population (24).
Pediatric ACCs have also been associated with Beckwith-Wiedemann syndrome, a systemic overgrowth disorder characterized by macroglossia, macrosomia, organomegaly, and abdominal wall defects (29, 30). This syndrome is a result of genetic defect caused by uniparental disomy in the 11p15 chromosomal region resulting in IGF2 growth factor overexpression (31). Normally, the IGF2 gene is only expressed from the paternal allele due to imprinting (32, 33), but disomy leads to expression from two copies of the gene. In instances of normal functionality, IGF2 activates type 1 IGF receptors, which stimulates cell survival (34). IGF2 plays a major role in fetal adrenal growth and steroidogenesis (35, 36). While IGF2 overexpression alone is not sufficient for adrenal carcinogenesis, it does promote ACCs (36, 37).
Other hereditary tumor syndromes associated with ACC that are less common in the pediatric population include multiple endocrine neoplasia 1 (MEN1), Lynch syndrome, familial adenomatous polyposis (FAP), Neurofibromatosis type 1 (NF1), and Carney complex (Table 1). MEN1 is an autosomal dominant hereditary tumor syndrome characterized by tumors affecting the parathyroid, pituitary, and pancreatic islet (38). Adrenal lesions occur in 20-55% of MEN1 patients, however, these lesions are usually adrenocortical adenomas or hyperplasia; ACC is extremely rare in this population (39). Lynch syndrome is an autosomal dominant inherited cancer predisposition syndrome characterized by an elevated risk of developing colorectal and endometrial cancers (40). The prevalence of Lynch syndrome amongst ACC patients is estimated to be 3.2% (41). FAP is an autosomal dominant hereditary tumor syndrome characterized by the development of adenomas in the rectum and colon followed by colorectal cancer if not treated at an early stage (42). Although rare, ACCs in patients with FAP have been reported in the literature (43–45). NF1 is an autosomal dominant genetic syndrome due to mutations in the NF1 tumor suppressor gene and is characterized by café au lait spots, dermal and plexiform neurofibromas, pheochromocytomas, optic gliomas, and malignant peripheral nerve sheath tumors (46, 47). Rare instances of ACC in patients with NF1 have been reported in the literature (48–51). Carney complex is an autosomal dominant tumor syndrome characterized by skin pigmentary abnormalities, myxomas, endocrine tumors, and schwannomas (52). ACC in the setting of Carney complex is very rare, but has been reported in two cases in the literature (53–55).
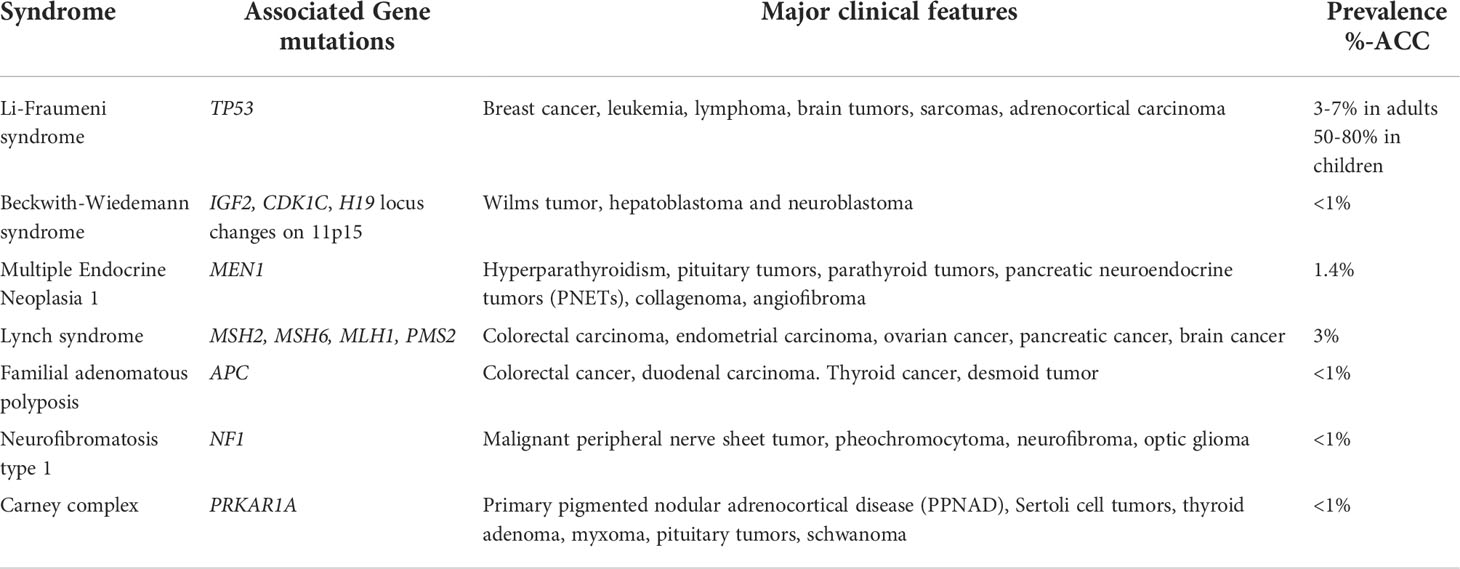
Table 1 Hereditary tumor syndromes associated with ACC.
Pediatric ACC displays a bimodal age distribution with peaks under the age of 5 and after 10 (4), with almost half (46%) of diagnoses occurring in children less than 4 years of age (1). There is a female preponderance for this condition both in childhood and in adulthood. Previous studies have shown the growth-promoting role of estrogen on the ACC cell line NCI-H295 and may explain the basis for this predisposition (56). There is a 2:1 female to male ratio amongst diagnoses of pediatric ACC (1).
Based on international TNM staging, approximately 44% of patients are stage I, 25% stage II, 13% stage III, and 17% stage IV (57). However, differences may exist based on age. Based on data from a SEER study of pediatric ACC patients, 52% of all staged patients presented with localized tumors; however, this was 76% in those aged 4 years and younger compared to 31% in those over the age of 4 (1). Additionally, the same study showed a significantly greater tumor size in older patients. Metastases are found in approximately 25% of pediatric ACC patients (57). The majority of metastases are found in the lungs and liver (14, 57). Other reported sites of metastases include the bone, kidneys, and CNS (57).
Pediatric ACCs are almost always functional, with approximately 95% of tumors exhibiting hormone production, compared to less than 50% of adult ACCs (11, 58, 59). Consequently, diagnoses in children usually follow the presentation of symptoms (12). Amongst all pediatric adrenocortical tumors, the most common presentation is virilization due to excess androgen secretion alone or in combination with hypercortisolism in over 80% of patients (11, 12). Other presentations include Cushing syndrome (15-40%), feminization or gynecomastia due to excess estrogen production (7%), hyperaldosteronism (1-4%), or a mixture of symptoms (60). Cushing’s syndrome without virilization is uncommon (5.5%) (11). Approximately 10% of pediatric patients with ACC have nonfunctional tumors; they are uncommon amongst young children, and are usually only found in adolescents (11, 60).
Michalkiewicz et al. created a registry for pediatric ACCs and provide a descriptive analysis of 254 patients registered on the International Pediatric Adrenocortical Tumor Registry. 254 patients younger than 20 years of age with newly diagnosed or previously treated ACCs were registered. The most common presenting sign (84.2%) was virilization. Cushing’s syndrome without virilization was uncommon (5.5%). Tumors were completely resected in 83% of patients. Patients with disseminated or residual disease received mitotane, cisplatin, etoposide, and/or doxorubicin, and rarely, radiation therapy. At a median follow-up of 2 years and 5 months, 157 patients (61.8%) survived without evidence of disease and 97 patients (38.2%) had died. The 5-year event-free survival estimate was 54.2%. It was concluded that childhood ACCs occur predominantly in females and almost always causes clinical signs. Complete resection is required for cure. Residual or metastatic disease carries a poor prognosis (11). Wieneke’s index has been validated in some studies is a histopathological tool which have shown to predict clinical outcome in pediatric ACC patients (Table 2) (61). Pediatric patients with age <4 y on presentation shows favorable prognosis while metastasis at presentation have guarded prognosis (14).

Table 2A Wieneke’s criteria.

Table 2B Clinical outcomes based on Wieneke’s criteria.
The treatment for pediatric ACC has largely been extrapolated from adult ACC. The European Cooperative Study Group for Pediatric Rare Tumors has published consensus guidelines for the diagnosis and treatment of childhood adrenocortical tumors, derived from the guidelines and data obtained from adult studies (11). Surgery is the primary form of therapy, with an aggressive surgical approach recommended if feasible (62). Results from a recent prospective single-arm risk stratified interventional study with 77 patients showed that surgery has been the mainstay treatment and had shown to have excellent prognosis in stage I ACC (86.2% 5-year event free survival) (63). However, tumor spillage is a frequent concern in these patients with its occurrence in approximately 21% of initial resections and 43% of resections after recurrence (11). Due to this risk of tumor rupture, laparotomy and a curative procedure are recommended, rather than a fine needle aspiration (64). An open adrenalectomy is the standard care for resection of ACC, as laparoscopic resections are associated with a high risk of rupture and peritoneal carcinomatosis (62, 65).
Mitotane is a commonly used single agent in the adjuvant setting following a complete resection of ACC in the adult population and is approved for the treatment of ACC (66). Mitotane inhibits steroidogenesis and has direct adrenolytic functions, inducing permanent atrophy of the fasciculata and reticular zones of the adrenal cortex (67, 68). Mitotane has shown to improve outcomes in the intermediate risk pediatric ACC population if it can be given for more than 6 months and if therapeutic levels (greater than 14mg/L) can be attained (14, 69). In the pediatric ACC population, it has been shown to improve outcomes in stage III and IV, although it is poorly tolerated (63). In a review of 11 children with advanced ACC who were treated with mitotane and a cisplatin based chemotherapeutic regimen, 7 patients showed measurable responses, suggesting that neoadjuvant use can be considered in patients where complete surgical resection is not an option (68). Additionally, a retrospective analysis of 177 patients with adrenocortical carcinomas showed a significant increase in recurrence-free survival amongst patients who received adjuvant mitotane therapy (66). In a interventional open label Phase III trial with pediatric ACC, Stage III ACC patients was shown to have a good clinical outcome with adjuvant chemotherapy with EDP and mitotane but was associated with toxicity. An optimal treatment regimen need to be curated for these patients (63).
Pembrolizumab is a molecular therapy that targets the programed death receptor 1(PD-1) on lymphocytes and has shown to be effective in tumors expressing PD-L1. In a study of Pembrolizumab therapy in pediatric patients with PD-L1 positive tumors, two out of four pediatric ACC patients enrolled showed a partial response (70).
Cabozantinib is a multi-tyrosine kinase inhibitor that has been studied in ACC patients. In a study of 16 patients with advanced ACC treated with cabozantinib, three patients developed a partial response and five patients had stable disease with a median progression free survival of 16 weeks and a median overall survival of 58 weeks (71). Currently, there is an ongoing trial for Cabozantinib-S-Malate for patients with rare tumors including pediatric ACC (Table 3) (72).

Table 3 Clinical trials for pediatric ACC.
B7-H3-specific chimeric antigen receptor autologous T-Cells (CAR-T) have shown success in treating patients with relapsed pediatric acute lymphoblastic leukemia as well as in vivo success in solid tumor types (73). Currently there is a B7-H3-CAR-T cell trial testing the viability of this therapy in pediatric solid tumor types, including pediatric ACC (Table 3).
The use of radiation therapy in pediatric ACC patients has not been widely studied. Since most pediatric ACC cases carry TP53 mutations that predispose them to cancer, radiation can increase the likelihood of developing a secondary tumor. Driver et al., reported that amongst five long term survivors of pediatric ACC in their study, three died of secondary sarcomas that arose in the radiation field (74).
Understanding the oncogenesis and tumor biology of ACC in pediatric patients has been fallen behind compared to adult ACC. The treatment protocols are primarily based on the data from adult patients with adrenocortical cancer. Surgery with lymph node dissection, chemotherapy with EDP (mitotane, cisplatin, etoposide, and/or doxorubicin) and radiation in some cases are the current preferred treatment options in pediatric ACC (Scheme 1).
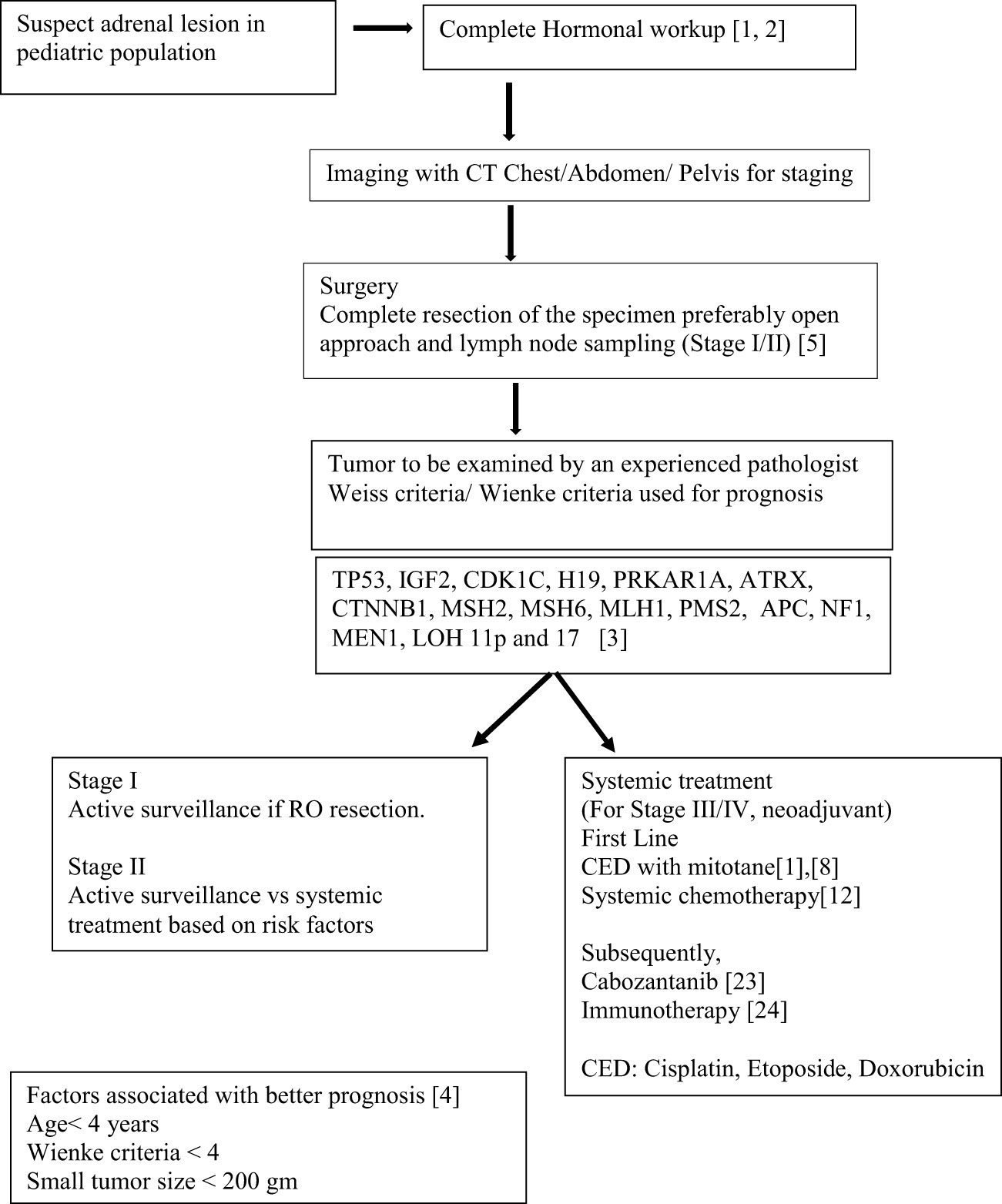
Scheme 1 Schema showing Current guidelines and therapeutics strategies.
It has been shown that the similar histopathology in pediatric and adult populations have different prognosis and the prognostic markers used in adult ACC might not be applicable for pediatric cases (75). Pediatric ACC arises from the fetal zone of adrenal gland and almost always presents with signs of hormone excess (14). As described previously germline mutation (p.R337H) of the TP53 gene and IGF1R overexpression is seen specifically in pediatric population (75). The expression of FATE1 gene has a negative prognostic significance in adult but not in pediatric population. Pediatric ACC is also more frequently associated with cancer predisposition syndromes (76). Ki-67 index and Weiss score are not predictive in pediatric ACC (7). Moreover, even with the differences with adult ACC, the current ongoing trials for adrenocortical cancer may show some future directions for treatment of pediatric ACC. Unfortunately, however, much of the preclinical and clinical trial data we have comes from adult ACC and no pediatric cell line for this tumor exist, limiting our ability to study the pediatric ACC population (77). As such, much of the investigation into treatment modalities for pediatric ACC patients has been extrapolated from adult in-vitro and in-vivo studies.
Targeted molecular therapies have proven to be efficacious in a number of malignancies. Their use in ACC has been studied with limited success. IGF2 overexpression has been found in approximately 90% of ACC cases, making it a potential target for precision molecular therapy (78). Additionally, in pediatric ACC, IGF1R overexpression is associated with a worse prognosis (75, 79). Preclinical studies of IGF2/IGF1R antagonists in ACC xenograft models have been promising, showing a dose dependent growth inhibition (80). Unfortunately, targeted therapies for these targets have not yet been substantiated in vivo. The IGF1R inhibitor lisitinib failed to show improvement in overall or progression free survival in an adult Phase III clinical. However, 5/90 patients showed a stabilization of disease (81).A phase II trial on the IGF1R inhibitor cixutumumab in combination with mitotane was terminated due to slow accrual and limited efficacy (82).
The activation of β-catenin signaling has been found in approximately 40% of ACC tumors (83, 84). Studies done on the NCI-H295R in-vitro model of ACC have shown that inhibition of β-catenin can inhibit tumor proliferation and promote apoptosis (76, 85, 86). However, the clinical utility of Wnt/β-catenin signaling inhibitors has not yet been demonstrated. Wnt/β-catenin signaling is essential for stem/progenitor cell maintenance (83). As such, Wnt/β-catenin signaling inhibition can result in a number of on-target toxicities, which has prevented these agents from advancing to phase III clinical trials (87, 88).
The Ki-67 proliferation index of has demonstrated a major prognostic role in ACC (89). This finding combined with genomic findings in ACC tumors suggest a role for cell cycle activation in advanced ACC (78, 90, 91) The cell cycle inhibitors, gemcitabine, capecitabine, and 5-fluorouracil have shown moderate efficacy and tolerability in patients with advanced ACC. Further investigation of broad and targeted kinase cell cycle inhibitors may be warranted in patients with advanced ACC.
Promising data exists supporting the use of certain modalities of radiotherapy for treating ACC. A study on Iodine-131 Iodometomidate (131I MTO) targeted radionuclide therapy demonstrated one partial response and five patients with stable disease in a cohort of 11 patients with unresectable ACC (92). In ACC patients with somatostatin expressing tumors, Yttrium-90/177Lu-DOTATOC has shown potential as a treatment option. In a study of 19 patients with advanced ACC, eight patients showed radiometabolic uptake of Yttrium-90/177Lu-DOTATOC and two patients showed strong uptake in addition to overall disease control lasting 4 and 12 months (93).
The use of single cell sequencing of tumor specimen from primary and metastasis in larger cohort of patient population to understand of heterogeneity in ACC and use of liquid biopsies will help in more personalized treatment approach for pediatric ACC. The continuation of global consortiums and international collaborations like International Pediatric Adrenocortical Tumor Registry (IPACTR) are necessary to build on the existing knowledge of pediatric ACC (11).
ACCs are a rare tumor in the pediatric population with a poor prognosis. Current treatment algorithms for pediatric ACC patients are based on research in adult populations. Investigation of cisplatin, cabozantinib and CAR-T cell therapy for pediatric ACC patients is ongoing. Research of treatment modalities for childhood ACC is limited and the need for more investigation into the treatment of this unique population exists. Due to the differences in ACC between pediatric and adult populations, we believe additional research in the treatment of childhood ACC is warranted.
MI and DV: prepared the manuscript. JG, KR, RK, BW, YP, JR: reviewed the article and suggested edits. All authors contributed to the article and approved the submitted version.
This project has been funded in whole or in part with federal funds from the National Cancer Institute, National Institutes of Health, under contract number HHSN261200800001E and the MyPART: My Pediatric and Adult Rare Tumor Network - Cures ZIA BC 011852. The content of this publication does not necessarily reflect the views or policies of the Department of Health and Human Services, nor does mention of trade names, commercial products, or organizations imply endorsement by the US Government. Funded by the Division of Intramural Research National Cancer Institute.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. McAteer JP, Huaco JA, Gow KW. Predictors of survival in pediatric adrenocortical carcinoma: a surveillance, epidemiology, and end results (SEER) program study. J Pediatr Surg (2013) 48:1025–31. doi: 10.1016/j.jpedsurg.2013.02.017
2. Miller RW, L Young J Jr, Novakovic B. Childhood cancer. Cancer. (1995) 75:395–405. doi: 10.1002/1097-0142(19950101)75:1+<395::AID-CNCR2820751321>3.0.CO;2-W
3. Rescorla FJ. Malignant adrenal tumors. Semin Pediatr Surg (2006) p:48–56. doi: 10.1053/j.sempedsurg.2005.11.008
4. Ng L, Libertino JM. Adrenocortical carcinoma: diagnosis, evaluation and treatment. J Urol (2003) 169:5–11. doi: 10.1016/S0022-5347(05)64023-2
5. Sabbaga CC, Avilla SG, Schulz C, Garbers JC, Blucher D. Adrenocortical carcinoma in children: clinical aspects and prognosis. J Pediatr Surgery (1993) 28:841–3. doi: 10.1016/0022-3468(93)90341-H
6. Custódio G, Komechen H, Figueiredo FR, Fachin ND, Pianovski MA, Figueiredo BC. Molecular epidemiology of adrenocortical tumors in southern Brazil. Mol Cell endocrinol (2012) 351:44–51. doi: 10.1016/j.mce.2011.10.019
7. Lalli E, Figueiredo BC. Pediatric adrenocortical tumors: what they can tell us on adrenal development and comparison with adult adrenal tumors. Front Endocrinol (2015) 6:23. doi: 10.3389/fendo.2015.00023
8. Jouinot A, Bertherat J. Diseases predisposing to adrenocortical malignancy (Li–fraumeni syndrome, beckwith–wiedemann syndrome, and Carney complex). Genet Endocrine Dis Syndromes (2019) 111:149–69. doi: 10.1007/978-3-030-25905-1_9
9. Miele E, Di Giannatale A, Crocoli A, Cozza R, Serra A, Castellano A, et al. Clinical, genetic, and prognostic features of adrenocortical tumors in children: A 10-year single-center experience. Front Oncol (2020) 10. doi: 10.3389/fonc.2020.554388
10. Ciftci AO, Şenocak ME, Tanyel FC, Büyükpamukçu N. Adrenocortical tumors in children. J Pediatr surgery (2001) 36:549–54. doi: 10.1053/jpsu.2001.22280
11. Michalkiewicz E, Sandrini R, Figueiredo B, Miranda ECM, Caran E, Oliveira-Filho AG, et al. Clinical and outcome characteristics of children with adrenocortical tumors: A report from the international pediatric adrenocortical tumor registry. J Clin Oncol (2004) 22:838–45. doi: 10.1200/JCO.2004.08.085
12. Pinto EM, Zambetti GP, Rodriguez-Galindo C. Pediatric adrenocortical tumours. Best Pract Res Clin Endocrinol Metab (2020) 34:101448. doi: 10.1016/j.beem.2020.101448
13. Sandrini R, Ribeiro RC, DeLacerda L. Childhood adrenocortical tumors. J Clin Endocrinol Metab (1997) 82:2027–31. doi: 10.1210/jc.82.7.2027
14. Gupta N, Rivera M, Novotny P, Rodriguez V, Bancos I, Lteif A. Adrenocortical carcinoma in children: A clinicopathological analysis of 41 patients at the Mayo clinic from 1950 to 2017. Horm Res Paediatr (2018) 90:8–18. doi: 10.1159/000488855
15. Cecchetto G, Ganarin A, Bien E, Vorwerk P, Bisogno G, Godzinski J, et al. Outcome and prognostic factors in high-risk childhood adrenocortical carcinomas: a report from the European cooperative study group on pediatric rare tumors (EXPeRT). Pediatr Blood cancer (2017) 64:e26368. doi: 10.1002/pbc.26368
16. Pinto E, Chen X, Easton J, Finkelstein D, Liu Z, Pounds S, et al. Genomic landscape of paediatric adrenocortical tumours. Nat Commun (2015) 6:6302. doi: 10.1038/ncomms7302
17. Hisada M, Garber JE, Li FP, Fung CY, Fraumeni JF. Multiple primary cancers in families with Li-fraumeni syndrome. JNCI: J Natl Cancer Inst (1998) 90:606–11. doi: 10.1093/jnci/90.8.606
18. Li FP, Fraumeni JFJr. Soft-tissue sarcomas, breast cancer, and other neoplasms: a familial syndrome? Ann Internal Med (1969) 71:747–52. doi: 10.7326/0003-4819-71-4-747
19. Wasserman JD, Zambetti GP, Malkin D. Towards an understanding of the role of p53 in adrenocortical carcinogenesis. Mol Cell Endocrinol (2012) 351:101–10. doi: 10.1016/j.mce.2011.09.010
21. Wagner J, Portwine C, Rabin K, Leclerc JM, Narod SA, Malkin D. High frequency of germline p53 mutations in childhood adrenocortical cancer. J Natl Cancer Inst (1994) 86:1707–10. doi: 10.1093/jnci/86.22.1707
22. Wasserman JD, Novokmet A, Eichler-Jonsson C, Ribeiro RC, Rodriguez-Galindo C, Zambetti GP, et al. Prevalence and functional consequence of TP53 mutations in pediatric adrenocortical carcinoma: a children's oncology group study. J Clin Oncol (2015) 33:602–9. doi: 10.1200/JCO.2013.52.6863
23. Herrmann LJ, Heinze B, Fassnacht M, Willenberg HS, Quinkler M, Reisch N, et al. TP53 germline mutations in adult patients with adrenocortical carcinoma. J Clin Endocrinol Metab (2012) 97:E476–E85. doi: 10.1210/jc.2011-1982
24. Custódio G, Parise GA, Kiesel Filho N, Komechen H, Sabbaga CC, Rosati R, et al. Impact of neonatal screening and surveillance for the TP53 R337H mutation on early detection of childhood adrenocortical tumors. J Clin Oncol (2013) 31:2619. doi: 10.1200/JCO.2012.46.3711
25. Varley JM, McGown G, Thorncroft M, James LA, Margison GP, Forster G, et al. Are there low-penetrance TP53 alleles? evidence from childhood adrenocortical tumors. Am J Hum Genet (1999) 65:995–1006. doi: 10.1086/302575
26. Figueiredo BC, Sandrini R, Zambetti GP, Pereira RM, Cheng C, Liu W, et al. Penetrance of adrenocortical tumours associated with the germline TP53 R337H mutation. J Med Genet (2006) 43:91–6. doi: 10.1136/jmg.2004.030551
27. Latronico AC, Pinto EM, Domenice S, Fragoso MC, Martin RM, Zerbini MC, et al. An inherited mutation outside the highly conserved DNA-binding domain of the p53 tumor suppressor protein in children and adults with sporadic adrenocortical tumors. J Clin Endocrinol Metab (2001) 86:4970–3. doi: 10.1210/jcem.86.10.7957
28. Ribeiro RC, Sandrini F, Figueiredo B, Zambetti GP, Michalkiewicz E, Lafferty AR, et al. An inherited p53 mutation that contributes in a tissue-specific manner to pediatric adrenal cortical carcinoma. Proc Natl Acad Sci U S A. (2001) 98:9330–5. doi: 10.1073/pnas.161479898
29. Sutter JA, Grimberg A. Adrenocortical tumors and hyperplasias in childhood–etiology, genetics, clinical presentation and therapy. Pediatr Endocrinol Rev (2006) 4:32–9.
30. Shuman C, Beckwith JB, Weksberg R. Beckwith-wiedemann syndrome. (2016) Seattle (WA): University of Washington. Available at: https://www.ncbi.nlm.nih.gov/books/NBK1394/
31. Weksberg R, Smith AC, Squire J, Sadowski P. Beckwith–wiedemann syndrome demonstrates a role for epigenetic control of normal development. Hum Mol Genet (2003) 12:R61–R8. doi: 10.1093/hmg/ddg067
32. Koch CA, Pacak K, Chrousos GP. The molecular pathogenesis of hereditary and sporadic adrenocortical and adrenomedullary tumors. J Clin Endocrinol Metab (2002) 87:5367–84. doi: 10.1210/jc.2002-021069
33. Sparago A, Cerrato F, Vernucci M, Ferrero GB, Silengo MC, Riccio A. Microdeletions in the human H19 DMR result in loss of IGF2 imprinting and beckwith-wiedemann syndrome. Nat Genet (2004) 36:958–60. doi: 10.1038/ng1410
34. Grimberg A. Mechanisms by which IGF-I may promote cancer. Cancer Biol Ther (2003) 2:630–5. doi: 10.4161/cbt.2.6.678
35. Coulter CL. Fetal adrenal development: insight gained from adrenal tumors. Trends Endocrinol Metab (2005) 16:235–42. doi: 10.1016/j.tem.2005.05.010
36. MacFarland SP, Mostoufi-Moab S, Zelley K, Mattei PA, States LJ, Bhatti TR, et al. Management of adrenal masses in patients with beckwith-wiedemann syndrome. Pediatr Blood Cancer (2017) 64(8):10.1002/pbc.26432. doi: 10.1002/pbc.26432
37. Guillaud-Bataille M, Ragazzon B, de Reyniès A, Chevalier C, Francillard I, Barreau O, et al. IGF2 promotes growth of adrenocortical carcinoma cells, but its overexpression does not modify phenotypic and molecular features of adrenocortical carcinoma. PLos One (2014) 9:e103744. doi: 10.1371/journal.pone.0103744
38. Skogseid B, Larsson C, Lindgren PG, Kvanta E, Rastad J, Theodorsson E, et al. Clinical and genetic features of adrenocortical lesions in multiple endocrine neoplasia type 1. J Clin Endocrinol Metab (1992) 75:76–81. doi: 10.1210/jcem.75.1.1352309
39. Wang W, Han R, Ye L, Xie J, Tao B, Sun F, et al. Adrenocortical carcinoma in patients with MEN1: a kindred report and review of the literature. Endocr Connect (2019) 8:230–8. doi: 10.1530/EC-18-0526
40. Stoffel E, Mukherjee B, Raymond VM, Tayob N, Kastrinos F, Sparr J, et al. Calculation of risk of colorectal and endometrial cancer among patients with lynch syndrome. Gastroenterology. (2009) 137:1621–7. doi: 10.1053/j.gastro.2009.07.039
41. Raymond VM, Everett JN, Furtado LV, Gustafson SL, Jungbluth CR, Gruber SB, et al. Adrenocortical carcinoma is a lynch syndrome-associated cancer. J Clin Oncol (2013) 31:3012–8. doi: 10.1200/JCO.2012.48.0988
42. Half E, Bercovich D, Rozen P. Familial adenomatous polyposis. Orphanet J Rare Dis (2009) 4:22. doi: 10.1186/1750-1172-4-22
43. Agarwal S, Sharma A, Sharma D, Sankhwar S. Incidentally detected adrenocortical carcinoma in familial adenomatous polyposis: an unusual presentation of a hereditary cancer syndrome. BMJ Case Rep (2018) 2018:bcr–2018-226799. doi: 10.1136/bcr-2018-226799
44. Gaujoux S, Pinson S, Gimenez-Roqueplo A-P, Amar L, Ragazzon B, Launay P, et al. Inactivation of the APC gene is constant in adrenocortical tumors from patients with familial adenomatous polyposis but not frequent in sporadic adrenocortical cancers. Clin Cancer Res (2010) 16:5133–41. doi: 10.1158/1078-0432.CCR-10-1497
45. Shiroky JS, Lerner-Ellis JP, Govindarajan A, Urbach DR, Devon KM. Characteristics of adrenal masses in familial adenomatous polyposis. Dis Colon Rectum (2018) 61:679–85. doi: 10.1097/DCR.0000000000001008
46. Rasmussen SA, Friedman J. NF1 gene and neurofibromatosis 1. Am J Epidemiol (2000) 151:33–40. doi: 10.1093/oxfordjournals.aje.a010118
47. Dare AJ, Gupta AA, Thipphavong S, Miettinen M, Gladdy RA. Abdominal neoplastic manifestations of neurofibromatosis type 1. Neurooncol Adv (2020) 2:i124–i33. doi: 10.1093/noajnl/vdaa032
48. Abrams EM, Gerdts JD, Gruber J, Lemoine-Courcelles C, Simons E, Protudjer JLP. Race/ethnicity, but not income, are associated with increased odds of shellfish allergy. J Allergy Clin Immunol: In Practice (2021) 9:550–2. doi: 10.1016/j.jaip.2020.11.003
49. Menon RK, Ferrau F, Kurzawinski TR, Rumsby G, Freeman A, Amin Z, et al. Adrenal cancer in neurofibromatosis type 1: case report and DNA analysis. Endocrinol Diabetes Metab Case Rep (2014) 2014:140074–. doi: 10.1530/EDM-14-0074
50. Najafi-Semnani M, Rajabi-Moghaddam M, Abbaszadeh H. Adrenocortical carcinoma in a patient with neurofibromatosis type 1: A case report. Caspian J Intern Med (2021) 12:613–7. doi: 10.22088/cjim.12.4.613
51. Sandoval BG, Morales MDMF, Wortham J, Koops M, Granda-Rodriguez R, Bruder JM. Large Invasive adrenal cortical carcinoma in a patient with neurofibromatosis type I. J Endocrine Society (2021) 5:A139–A. doi: 10.1210/jendso/bvab048.281
52. Stratakis CA, Raygada M. Carney Complex. In: GeneReviews® (2018) Seattle (WA): University of Washington. Available at: https://www.ncbi.nlm.nih.gov/books/NBK1286/
53. Morin E, Mete O, Wasserman JD, Joshua AM, Asa SL, Ezzat S. Carney Complex with adrenal cortical carcinoma. J Clin Endocrinol Metab (2012) 97:E202–6. doi: 10.1210/jc.2011-2321
54. Anselmo J, Medeiros S, Carneiro V, Greene E, Levy I, Nesterova M, et al. A large family with Carney complex caused by the S147G PRKAR1A mutation shows a unique spectrum of disease including adrenocortical cancer. J Clin Endocrinol Metab (2012) 97:351–35. doi: 10.1210/jc.2011-2244
55. Bertherat J. Adrenocortical cancer in Carney complex: a paradigm of endocrine tumor progression or an association of genetic predisposing factors? J Clin Endocrinol Metab (2012) 97:387–3. doi: 10.1210/jc.2011-3327
56. Sirianni R, Zolea F, Chimento A, Ruggiero C, Cerquetti L, Fallo F, et al. Targeting estrogen receptor-alpha reduces adrenocortical cancer (ACC) cell growth in vitro and in vivo: potential therapeutic role of selective estrogen receptor modulators (SERMs) for ACC treatment. J Clin Endocrinol Metab (2012) 97:E2238–50. doi: 10.1210/jc.2012-2374
57. Riedmeier M, Decarolis B, Haubitz I, Müller S, Uttinger K, Börner K, et al. Adrenocortical carcinoma in childhood: A systematic review. Cancers (Basel) (2021) 13(21):5266. doi: 10.3390/cancers13215266
58. Allolio B, Fassnacht M. Adrenocortical carcinoma: clinical update. J Clin Endocrinol Metab (2006) 91:2027–37. doi: 10.1210/jc.2005-2639
59. Gundgurthi A, Kharb S, Dutta MK, Garg MK, Khare A, Jacob MJ, et al. Childhood adrenocortical carcinoma: Case report and review. Indian J Endocrinol Metab (2012) 16:431–5. doi: 10.4103/2230-8210.95699
60. Ribeiro R, Figueiredo B. Childhood adrenocortical tumours. Eur J Cancer (2004) 40:1117–26. doi: 10.1016/j.ejca.2004.01.031
61. Wieneke JA, Thompson LD, Heffess CS. Adrenal cortical neoplasms in the pediatric population: a clinicopathologic and immunophenotypic analysis of 83 patients. Am J Surg Pathol (2003) 27:867–81. doi: 10.1097/00000478-200307000-00001
62. Hubertus J, Boxberger N, Redlich A, von Schweinitz D, Vorwerk P. Surgical aspects in the treatment of adrenocortical carcinomas in children: data of the GPOH-MET 97 trial. Klin Padiatr (2012) 224:143–7. doi: 10.1055/s-0032-1304627
63. Rodriguez-Galindo C, Krailo MD, Pinto EM, Pashankar F, Weldon CB, Huang L, et al. Treatment of pediatric adrenocortical carcinoma with surgery, retroperitoneal lymph node dissection, and chemotherapy: The children's oncology group ARAR0332 protocol. J Clin Oncol (2021) 39:2463–73. doi: 10.1200/JCO.20.02871
64. Kardar AH. Rupture of adrenal carcinoma after biopsy. J Urol (2001) 166:984. doi: 10.1016/S0022-5347(05)65881-8
65. Gonzalez RJ, Shapiro S, Sarlis N, Vassilopoulou-Sellin R, Perrier ND, Evans DB, et al. Laparoscopic resection of adrenal cortical carcinoma: a cautionary note. Surgery. (2005) 138:1078–85; discussion 85-6. doi: 10.1016/j.surg.2005.09.012
66. Terzolo M, Angeli A, Fassnacht M, Daffara F, Tauchmanova L, Conton PA, et al. Adjuvant mitotane treatment for adrenocortical carcinoma. N Engl J Med (2007) 356:2372–80. doi: 10.1056/NEJMoa063360
67. Brondani VB, Fragoso MCB. Pediatric adrenocortical tumor–review and management update. Curr Opin Endocrinol Diabetes Obes (2020) 27:177–86. doi: 10.1097/MED.0000000000000540
68. Zancanella P, Pianovski MAD, Oliveira BH, Ferman S, Piovezan GC, Lichtvan LL, et al. Mitotane associated with cisplatin, etoposide, and doxorubicin in advanced childhood adrenocortical carcinoma: Mitotane monitoring and tumor regression. J Pediatr Hematol/Oncol (2006) 28:513–24. doi: 10.1097/01.mph.0000212965.52759.1c
69. Redlich A, Boxberger N, Strugala D, Frühwald MC, Leuschner I, Kropf S, et al. Systemic treatment of adrenocortical carcinoma in children: data from the German GPOH-MET 97 trial. Klin Padiatr (2012) 224:366–71. doi: 10.1055/s-0032-1327579
70. Geoerger B, Kang HJ, Yalon-Oren M, Marshall LV, Vezina C, Pappo A, et al. Pembrolizumab in paediatric patients with advanced melanoma or a PD-L1-positive, advanced, relapsed, or refractory solid tumour or lymphoma (KEYNOTE-051): interim analysis of an open-label, single-arm, phase 1–2 trial. Lancet Oncol (2020) 21:121–33. doi: 10.1016/S1470-2045(19)30671-0
71. Kroiss M, Megerle F, Kurlbaum M, Zimmermann S, Wendler J, Jimenez C, et al. Objective response and prolonged disease control of advanced adrenocortical carcinoma with cabozantinib. J Clin Endocrinol Metab (2020) 105:1461–8. doi: 10.1210/clinem/dgz318
72. Akshintala S, Widemann BC, Barkauskas DA, Hall D, Reid JM, Voss SD, et al. Phase 2 trial of cabozantinib in children and young adults with refractory sarcomas, wilms tumor, and rare tumors: Children's oncology group study (ADVL1622). J Clin Oncol (2021) 39:10010–. doi: 10.1200/JCO.2021.39.15_suppl.10010
73. Majzner RG, Theruvath JL, Nellan A, Heitzeneder S, Cui Y, Mount CW, et al. CAR T cells targeting B7-H3, a pan-cancer antigen, demonstrate potent preclinical activity against pediatric solid tumors and brain tumors. Clin Cancer Res (2019) 25:2560–74. doi: 10.1158/1078-0432.CCR-18-0432
74. Driver CP, Birch J, Gough DC, Bruce J. Adrenal cortical tumors in childhood. Pediatr Hematol Oncol (1998) 15:527–32. doi: 10.3109/08880019809018314
75. Faria AM, Almeida MQ. Differences in the molecular mechanisms of adrenocortical tumorigenesis between children and adults. Mol Cell Endocrinol (2012) 351:52–7. doi: 10.1016/j.mce.2011.09.040
76. Doghman-Bouguerra M, Finetti P, Durand N, Parise IZS, Sbiera S, Cantini G, et al. Cancer-testis antigen FATE1 expression in adrenocortical tumors is associated with a pervasive autoimmune response and is a marker of malignancy in adult, but not children, ACC. Cancers (Basel) (2020) 12(3):689. doi: 10.3390/cancers12030689
77. Pinto EM, Kiseljak-Vassiliades K, Hantel C. Contemporary preclinical human models of adrenocortical carcinoma. Curr Opin Endocr Metab Res (2019) 8:139–44. doi: 10.1016/j.coemr.2019.08.009
78. Giordano TJ, Thomas DG, Kuick R, Lizyness M, Misek DE, Smith AL, et al. Distinct transcriptional profiles of adrenocortical tumors uncovered by DNA microarray analysis. Am J Pathol (2003) 162:521–31. doi: 10.1016/S0002-9440(10)63846-1
79. Ettaieb M, Kerkhofs T, van Engeland M, Haak H. Past, present and future of epigenetics in adrenocortical carcinoma. Cancers (Basel) (2020) 12(5):1218. doi: 10.3390/cancers12051218
80. Barlaskar FM, Spalding AC, Heaton JH, Kuick R, Kim AC, Thomas DG, et al. Preclinical targeting of the type I insulin-like growth factor receptor in adrenocortical carcinoma. J Clin Endocrinol Metab (2009) 94:204–12. doi: 10.1210/jc.2008-1456
81. Fassnacht M, Berruti A, Baudin E, Demeure MJ, Gilbert J, Haak H, et al. Linsitinib (OSI-906) versus placebo for patients with locally advanced or metastatic adrenocortical carcinoma: a double-blind, randomised, phase 3 study. Lancet Oncol (2015) 16:426–35. doi: 10.1016/S1470-2045(15)70081-1
82. Lerario AM, Worden FP, Ramm CA, Hesseltine EA, Stadler WM, Else T, et al. The combination of insulin-like growth factor receptor 1 (IGF1R) antibody cixutumumab and mitotane as a first-line therapy for patients with recurrent/metastatic adrenocortical carcinoma: a multi-institutional NCI-sponsored trial. Horm Cancer (2014) 5:232–9. doi: 10.1007/s12672-014-0182-1
83. Mohan DR, Lerario AM, Hammer GD. Therapeutic targets for adrenocortical carcinoma in the genomics era. J Endocr Soc (2018) 2:1259–74. doi: 10.1210/js.2018-00197
84. Zheng S, Cherniack AD, Dewal N, Moffitt RA, Danilova L, Murray BA, et al. Comprehensive pan-genomic characterization of adrenocortical carcinoma. Cancer Cell (2016) 29:723–36. doi: 10.1016/j.ccell.2016.04.002
85. Gaujoux S, Hantel C, Launay P, Bonnet S, Perlemoine K, Lefèvre L, et al. Silencing mutated β-catenin inhibits cell proliferation and stimulates apoptosis in the adrenocortical cancer cell line H295R. PLos One (2013) 8:e55743. doi: 10.1371/journal.pone.0055743
86. Leal LF, Bueno AC, Gomes DC, Abduch R, de Castro M, Antonini SR. Inhibition of the tcf/beta-catenin complex increases apoptosis and impairs adrenocortical tumor cell proliferation and adrenal steroidogenesis. Oncotarget. (2015) 6:43016–32. doi: 10.18632/oncotarget.5513
87. Chatterjee A, Paul S, Bisht B, Bhattacharya S, Sivasubramaniam S, Paul MK. Advances in targeting the WNT/β-catenin signaling pathway in cancer. Drug Discovery Today (2022) 27:82–101. doi: 10.1016/j.drudis.2021.07.007
88. Krishnamurthy N, Kurzrock R. Targeting the wnt/beta-catenin pathway in cancer: Update on effectors and inhibitors. Cancer Treat Rev (2018) 62:50–60. doi: 10.1016/j.ctrv.2017.11.002
89. Beuschlein F, Weigel J, Saeger W, Kroiss M, Wild V, Daffara F, et al. Major prognostic role of Ki67 in localized adrenocortical carcinoma after complete resection. J Clin Endocrinol Metab (2015) 100:841–9. doi: 10.1210/jc.2014-3182
90. Giordano TJ, Kuick R, Else T, Gauger PG, Vinco M, Bauersfeld J, et al. Molecular classification and prognostication of adrenocortical tumors by transcriptome profiling. Clin Cancer Res (2009) 15:668–76. doi: 10.1158/1078-0432.CCR-08-1067
91. Zsippai A, Szabó DR, Szabó PM, Tömböl Z, Bendes MR, Nagy Z, et al. mRNA and microRNA expression patterns in adrenocortical cancer. Am J Cancer Res (2011) 1:618–28.
92. Hahner S, Kreissl MC, Fassnacht M, Haenscheid H, Knoedler P, Lang K, et al. [131I]iodometomidate for targeted radionuclide therapy of advanced adrenocortical carcinoma. J Clin Endocrinol Metab (2012) 97:914–22. doi: 10.1210/jc.2011-2765
Keywords: pediatric adrenal tumors, adrenocortical cancer (ACC), pediatric adrenocortical carcinoma, endocrine tumors, adrenal tumor
Citation: Ilanchezhian M, Varghese DG, Glod JW, Reilly KM, Widemann BC, Pommier Y, Kaplan RN and Del Rivero J (2022) Pediatric adrenocortical carcinoma. Front. Endocrinol. 13:961650. doi: 10.3389/fendo.2022.961650
Received: 05 June 2022; Accepted: 17 October 2022;
Published: 31 October 2022.
Edited by:
Antje Redlich, University Hospital Magdeburg, GermanyReviewed by:
Verena Wiegering, University Hospital Würzburg, GermanyCopyright © 2022 Ilanchezhian, Varghese, Glod, Reilly, Widemann, Pommier, Kaplan and Del Rivero. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jaydira Del Rivero, SmF5ZGlyYS5kZWxyaXZlcm9AbmloLmdvdg==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.