 Rosario Le Moli*
Rosario Le Moli* Veronica Vella
Veronica Vella Dario Tumino
Dario Tumino Tommaso Piticchio
Tommaso Piticchio Adriano Naselli
Adriano Naselli Antonino Belfiore
Antonino Belfiore Francesco Frasca
Francesco Frasca- Endocrinology Unit, Department of Clinical and Experimental Medicine, University of Catania, Garibaldi-Nesima Hospital, Catania, Italy
Obesity is strongly associated with chronic low-grade inflammation. Obese patients have an increased risk to develop thyroid autoimmunity and to became hypothyroid, suggesting a pathogenetic link between obesity, inflammation and autoimmunity. Moreover, type 2 diabetes and dyslipidemia, also characterized by low-grade inflammation, were recently associated with more aggressive forms of Graves’ ophthalmopathy. The association between obesity and autoimmune thyroid disorders may also go in the opposite direction, as treating autoimmune hyper and hypothyroidism can lead to weight gain. In addition, restoration of euthyroidism by L-T4 replacement therapy is more challenging in obese athyreotic patients, as it is difficult to maintain thyrotropin stimulation hormone (TSH) values within the normal range. Intriguingly, pro-inflammatory cytokines decrease in obese patients after bariatric surgery along with TSH levels. Moreover, the risk of thyroid cancer is increased in patients with thyroid autoimmune disorders, and is also related to the degree of obesity and inflammation. Molecular studies have shown a relationship between the low-grade inflammation of obesity and the activity of intracellular multiprotein complexes typical of immune cells (inflammasomes). We will now highlight some clinical implications of inflammasome activation in the relationship between obesity and thyroid disease.
Introduction
Obesity is a public health concern with a prevalence rapidly increasing in the last ten years from 3.2% to 10.8% in adult men and from 6.4% to 14.9% in adult women (1). Adipose tissue should be considered an endocrine organ comprising various cell types such as adipocytes, preadipocytes and immune cells (2). Adipocytes are prone to secrete pro-inflammatory adipokines, such as tumor necrosis factor-α (TNF-α), monocyte chemoattractant protein-1 (MCP-1), and interleukin-6 (IL-6), which recruit immune cells and amplify the inflammation process (3, 4). These mechanisms may play a major role in obesity-related disorders, including atherosclerosis, diabetes, neurodegenerative diseases and cancer (5–7). New emerging data reveal a relationship between the low-grade inflammatory state of obesity and inflammasome activation level (8, 9). Inflammasomes are intracellular multiprotein complexes typical of immune cells, such as monocytes and macrophages, which mediate the first line of defense in response to both sterile (absence of microbial and virus particles) and non-sterile (microbial and/or virus infection) injuries, leading to the processing and release of pro-inflammatory cytokines (10–16). The inflammasome oligomeric structure includes three elements: I) NOD-like receptor (NLR), that is a stress signal receptor (17–19). NLRs can contain a Pirin Domain 3 (NLRPD3) or a caspase recruitment domain (NLRC4) and are present in several cells of different tissues; II) adaptor protein, that is an apoptosis associated speck-like protein containing caspase activation recruitment domain (ASC); III) pro-caspase-1(pro-CASP1), pro-IL-β and pro-IL-18 (Figure 1).
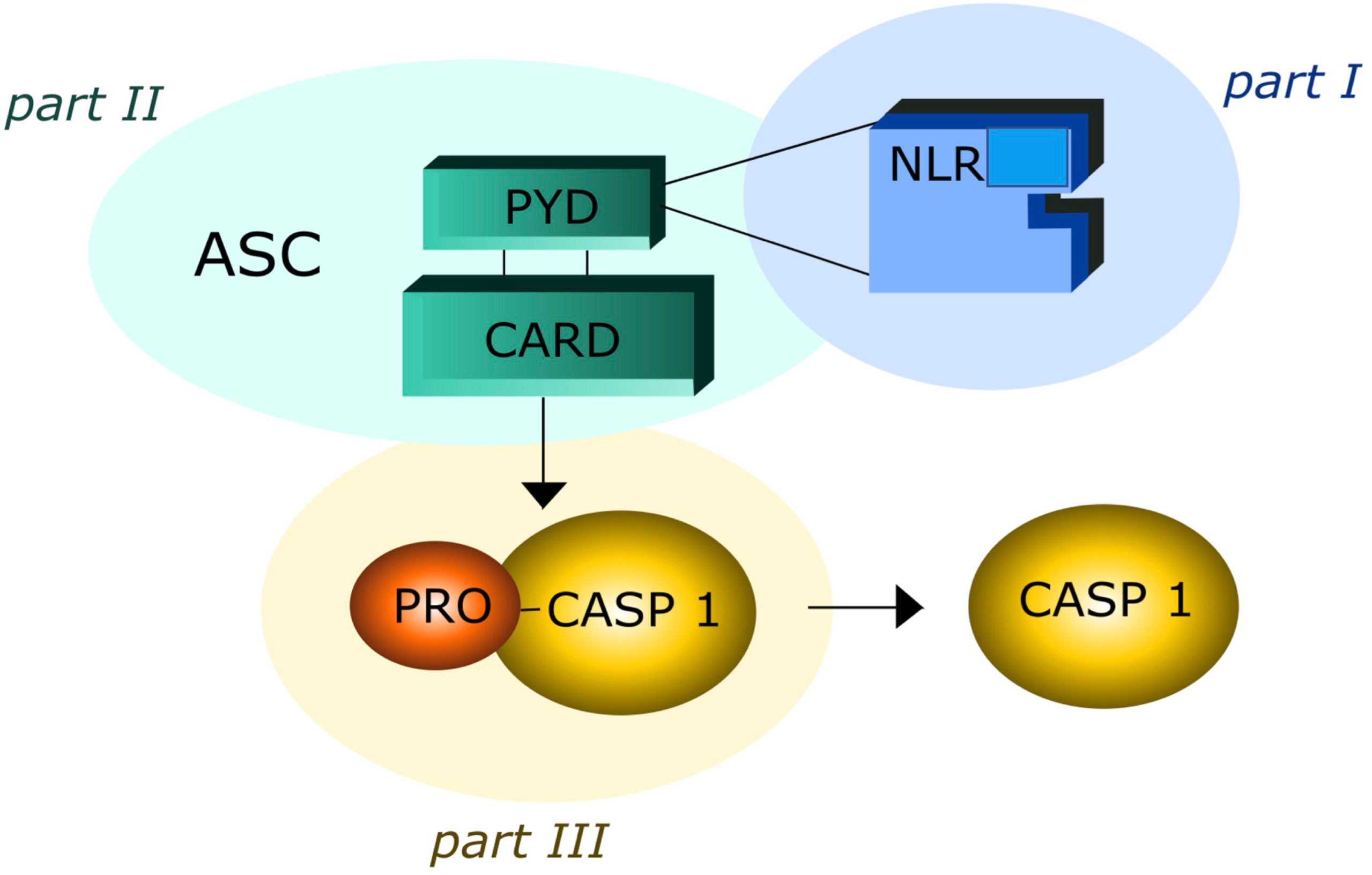
Figure 1 The three elements of inflammsome oligomeric structure: part I contain Nod-Like Receptor (NLR); part II contain Adaptor Protein Apoptosis-Associated Speck-Like Protein (ASC), Piriin Domain (PYD), Caspase Recruitment Domain (CARD); part III contain Pro Caspase I (PRO –CASP-1).
A further mechanism that contributes to the background activation of inflammasome in obese patients is the reduced efficiency of innate immunity caused by the inhibition of autophagic processes (20, 21). This mechanism contributes to the activation of the NLRPD3 inflammasome, which in turn (22) enhances chronic low-grade inflammation in adipose tissue (23, 24), thereby resulting in systemic insulin resistance and metabolic impairment (25). Here we highlight some emerging clinically relevant implications of inflammasome activation and chronic low-grade inflammation associated with obesity for patients with thyroid disorders.
Obesity, inflammasome and immune related thyroid diseases
Recent epidemiological and experimental data provided evidence of a bidirectional interaction between obesity and thyroid autoimmunity (26–28). In particular, obesity is associated with an enhanced risk of hypothyroidism and an increased risk of developing anti-thyroperoxidase antibodies (TPOAbs) (27). One prospective study of 783 obese patients referred to bariatric surgery reported a prevalence of 17.1% and 12.3% of autoimmune thyroiditis and autoimmune hypothyroidism, respectively (29). This association was confirmed by the National Health and Nutrition Examination Survey (NHANES III) which described the presence of TPOAbs in 11.3% and hypothyroidism in 4.6% of 17,353 patients (30). Autoimmune thyroid disease (AITD), including Graves’ disease (GD) and Hashimoto’s disease (HT), include thyroid disorders characterized by an immune reaction against thyroid autoantigens that occur in individuals with a distinct genetic background after exposure to certain environmental factors (31–36). GD and HT share similar pathological features characterized by lymphocyte infiltration of the thyroid tissue and antibodies production (37). The relationship between obesity and thyroid autoimmunity is not only based on epidemiological aspects but also on common pathogenetic mechanisms. A major player of this association is insulin resistance (IR), that is present in 15.5 - 46% of the obese patients (38). IR attenuates the insulin-stimulated phosphoinositide 3-kinase (PI3K) signaling pathway, causing hyperinsulinemia and a variety of adverse effects, including increased oxidative stress due to the production of reactive oxygen radical species (ROS) which activate the inflammasome complex in different tissues (25, 39) and reduces the vasoactive/anti-inflammatory effect of nitric oxide (NO) (39, 40). A variable degree of leptin resistance (LR) is usually associated with IR and obesity and can be considered a second mechanism in the interplay between obesity and thyroid disorders. Indeed, chronic exposure of endothelial cells to increased circulating levels of leptin also leads to a decrease in NO production (41). In addition, ROS induce pro-inflammatory MCP-1 expression which further enhances leukocyte infiltration into vascular cells. Chronic activation of inflammatory response contributes to the impairment of a correct immune response to exogenous antigens and to the persistence of autoimmune processes in predisposed subjects (42–44). This concept is supported by the observation that increased NLR inflammasome activity in both obese patients and experimental animals can play a major role in macrophage recruitment and immune activation (45). The third mechanism is the increased oxidation of low-density lipoproteins (LDL) occurring in obese patients with IR. Several lines of evidence indicate that increased oxidized low-density lipoproteins (oxLDLc) and cholesterol crystals can trigger the activation of NLR inflammasome, that, in turn, leads to a tonic activation of innate immune system (45, 46) (Figure 2). Hence, insulin/leptin system dysregulation along with oxLDLc increase and inflammasome activation are associated with impaired immunological tolerance and metabolic changes favoring thyroid autoimmunity (41, 45–48). The complete NLR-mediated inflammasome activation in thyroid cells is a complex event that involves the engagement of the tool - like receptors (TLRs) and the recruitment of the myddosome complex, which triggers downstream nuclear factor - KB (NF-kB). cell signaling, resulting in the increased levels of NLR and pro-ILs (49, 50) (Figure 3).
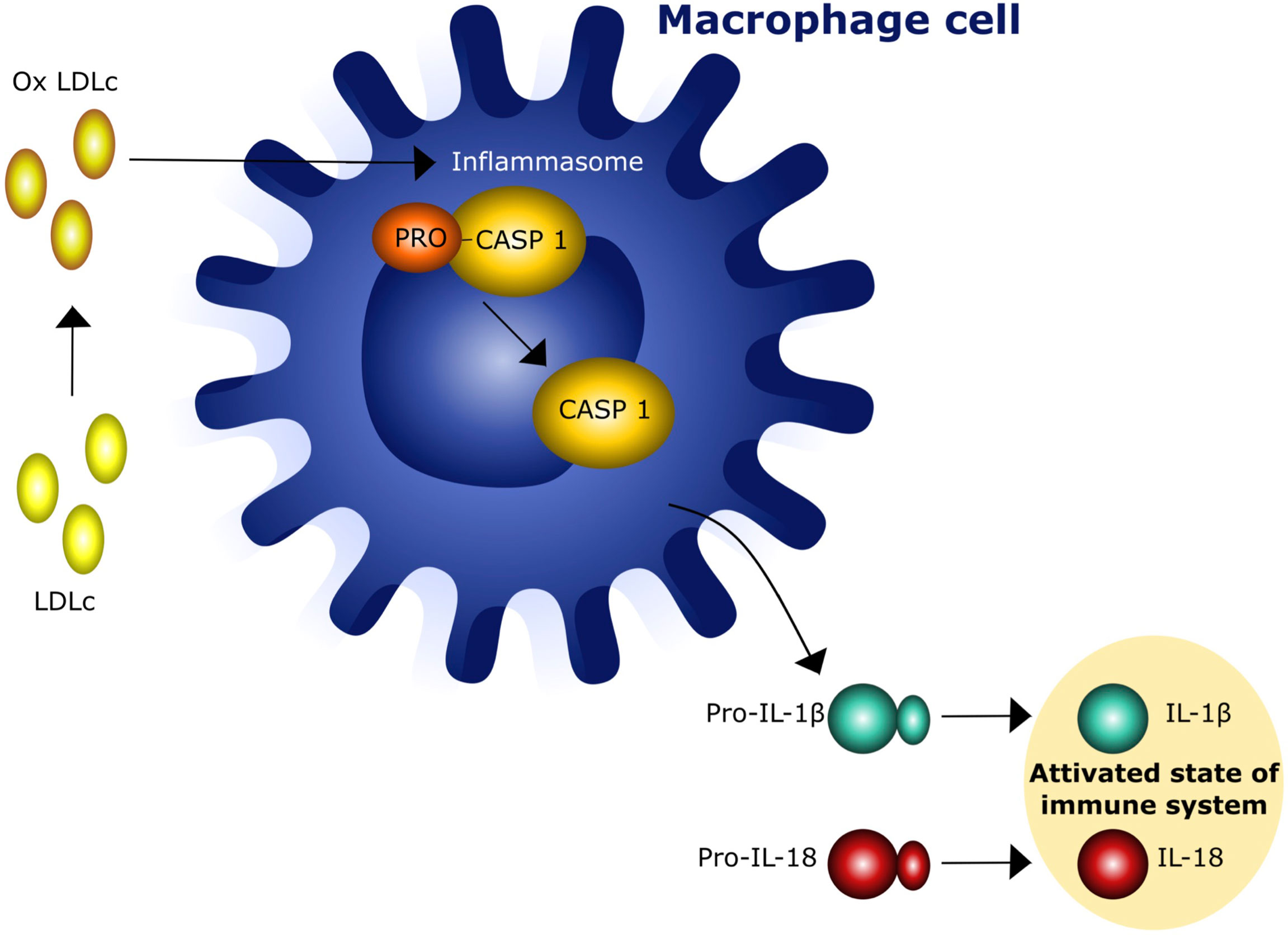
Figure 2 Interaction of oxidized low density lipoprotein cholesterol (oxLDLc) with cells of innate immunity. IL - 1β = Interleukin 1β, IL-15 = interleukin 18.
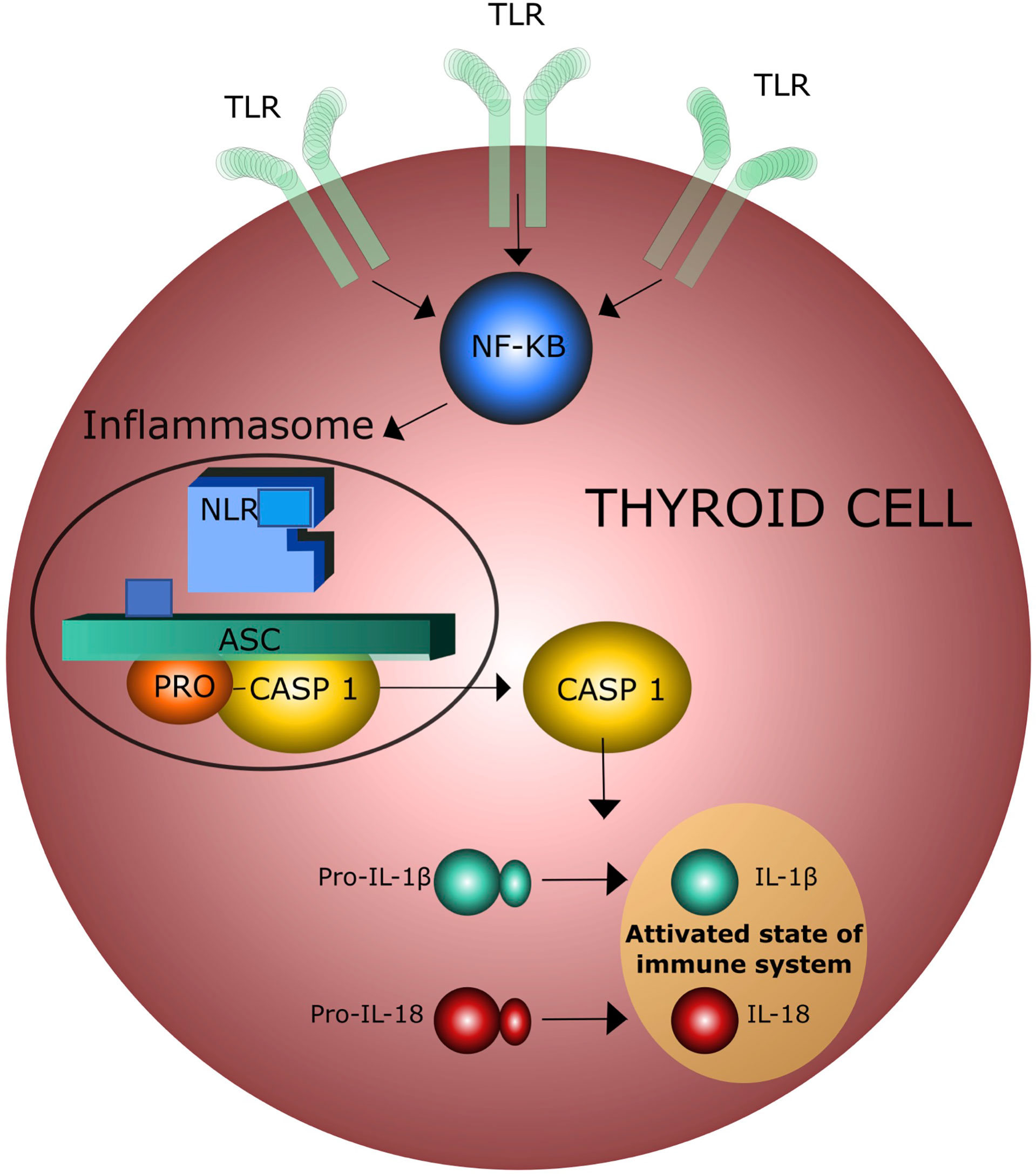
Figure 3 Activation of inflammasome by toll-like receptors (TLR) in thyroid cells; NF-KB = Nuclear Factor – KB.
More specifically, low-grade inflammation causes a predominance of CD4 + T helper cells over the proinflammatory Th17 subsets, increasing the IL-1β production which plays a major role in the pathogenesis of Hashimoto’s thyroiditis (HT) (40, 51, 52). It is interesting to note that both human and rat thyroid cells express functional TLRs on their surface and these receptors may directly bind both endogenous and exogenous ‘danger molecules’, thereby promptly activating the innate immune system. Several experimental evidences support the idea that the innate immune response is linked to thyroid tissue damage. Thyroid cell damage leads to the release of intracellular proteins that can be recognized by antigen-presenting cells (APCc), thereby activating acquired immune response, which, in turn, amplifies thyroid cell damage and ultimately causes thyroid tissue destruction (35, 53–55) (Figure 4).
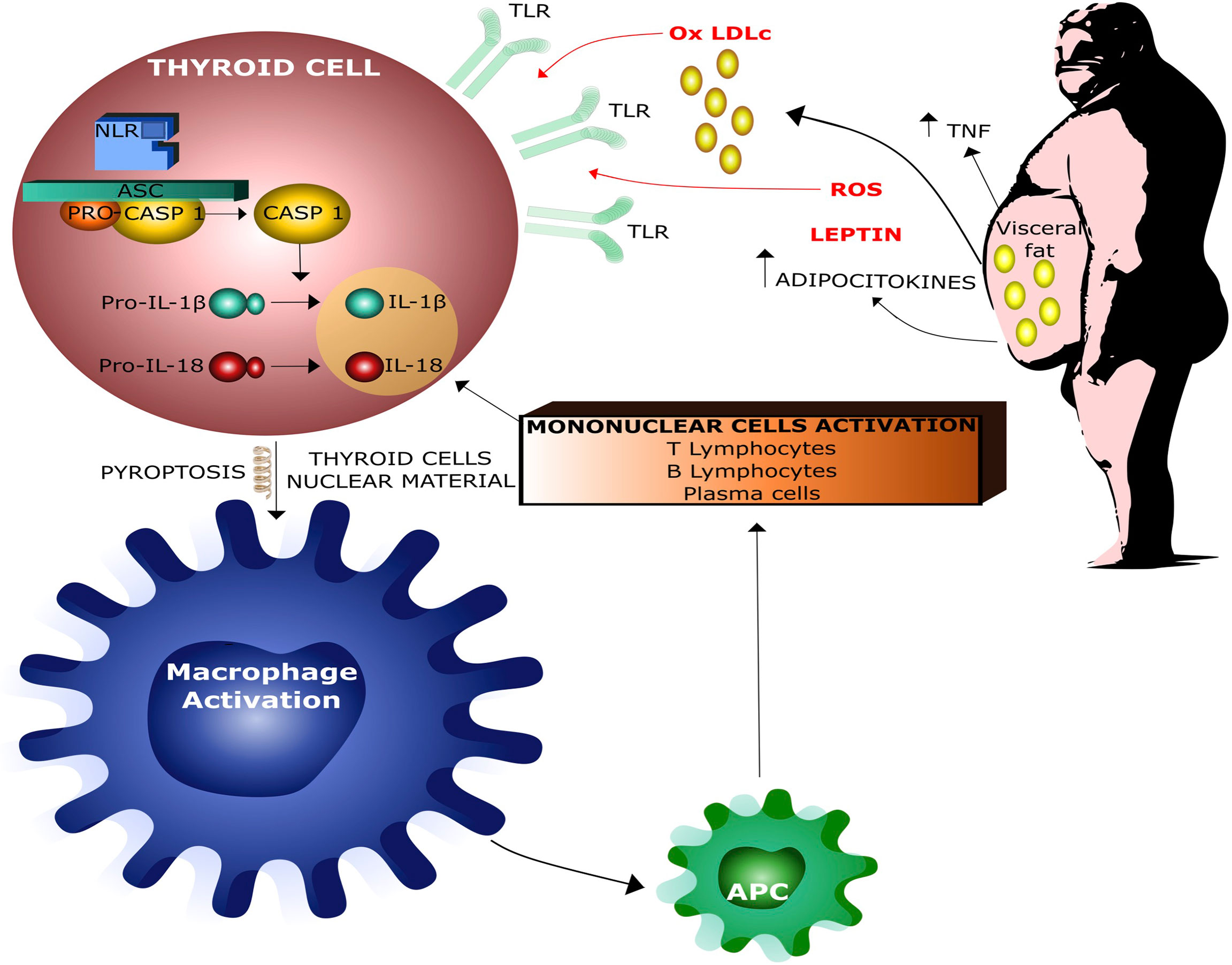
Figure 4 Interaction between obesity and thyroid damage by infammasome activation. Ox LDLc = Oxidized Low Density Lipoproteins Cholesterol, ROS = Radical Oxigen Species, APC = Antigen Presenting Cells.
Thyroid hormone are another important component of the putative bidirectional cross-talk between obesity and thyroid immunity process. All clinical conditions causing altered thyroid hormone levels may contribute to the extent of activation of inflammation and immunity response. On the other hand, alterations in thyroid hormone levels may favor obesity, atherosclerosis, and inflammation. Recent studies indicate that triiodothyronine (T3) negatively modulates NLR inflammasome and macrophage function. Conversely, low levels of thyroxine (T4) may contribute to inflammation and immune response activation in hypothyroid patients (56–59). This effect of T4 may also occur through an extra-genomic pathway by binding the cell membrane integrin receptor avβ3, which, in turn, may modulate the immune response via the activation of signaling pathways including mitogen-activated protein kinase (MAPK), cyclooxygenase 2 (COX-2) and hypoxia factor 1 alpha (HIF-1a), all converging into inflammasome activation (60). Taken together, these studies suggest that thyroid hormone effect on immune response is indirect and can be mediated by the combination of both genomic and nongenomic actions in specific cellular/clinical contexts (61) and these mechanisms are of primary importance in obese patients who frequently display reduced T4 levels (56).
Key points
Available evidence suggests a two-way interaction between obesity and inflammation and autoimmunity. In obese, insulin-resistant patients, careful attention to thyroid hormone homeostasis can prevent worsening of metabolic conditions and prevent thyroid dysfunction.
Obesity, inflammasome and thyroid eyes disease
Graves’ ophthalmopathy (GO) is an autoimmune thyroid related eyes disease (TED) occurring in about 25% of patients with Graves’ disease (GD). Elevated levels of thyroid stimulating receptor antibodies (TRAbs) are considered a risk factor for severe GO. Gender, age, smoking, and duration of hyperthyroidism are additional risk factors for the severity of GO (62–64). Recently, diabetes mellitus (DM) has been related to GO severity. Moreover, the prevalence of Type I diabetes is higher in patients with GO than in the normal population and dysthyroid optic neuropathy (DON) occurs more frequently in patients with both GO and DM and have a worse prognosis than in those with GO alone (65). Moreover, one important study showed evidence that DM and cigarette smoking are significant predictors of severe GO. In particular, DM was a stronger determinant of GO severity (p=0.001) and significant predictor of diplopia (p= 0.03). Additionally, in Graves’ patients with Type II diabetes (T2DM), GO preceded the onset of hyperthyroidism more frequently than in control patients (p < 0.05). GO severity was significantly associated to T2DM with a duration longer than five years (Odds Ratio = 4.9; p = 0.045) and the presence of micro and macro vascular complications (Odds Ratio = 4.8; p = 0.048). GO severity was also related to patient overweight (BMI > 26) (66). These clinical evidences suggest that GO may be related to a peculiar pathophysiologic background linked to the metabolic syndrome and obesity. Indeed, insulin-resistance and compensatory hyperinsulinemia in obese patients may exert proliferative effects on human orbital preadipocytes/fibroblasts via the phosphatidylinositol 3-kinase (PI3-K) and the mammalian target of rapamycin (mTOR) signaling pathways (67). In addition, elevated levels of insulin increase IGF-1 bioavailability, induce overexpression of the IGF-I receptors (IGF-IR) in orbital preadipocytes/fibroblasts and enhance IGF-IR/TSHR cross-talk thereby contributing to the severity of GO (68–72). Systemic low-grade chronic inflammation causes NLR inflammasome activation in fat tissues and production of neurotoxic factors such as IL-1β and TNF-α, that are determinants of GO pathogenesis (73). Moreover, low-density lipoproteins cholesterol (LDLc) and their oxidized forms are increased in obese and diabetic patients leading to reduced or incomplete autophagic processes (74) and to the activation of NLR inflammasome in macrophage cells. In GO of obese patients, activated macrophage population within the orbital microenvironment may increase the secretion of IGF-1 by fibroblasts thereby promoting adipogenesis, hyaluronan synthesis and the expansion of soft orbital tissues (50, 75). Stein et al. reported that statins are able to reduce the onset of GO in patients with GD. A recent cross-sectional study indicated that total and LDL cholesterol is associated with GO activity, suggesting that high serum cholesterol level is a risk factor for GO development (64, 76). Considering that GO is an inflammatory and autoimmune response against thyroid and orbital autoantigens, statins may protect from GO presentation and activity owing to their anti-inflammatory and hypolipemic action. Hypercholesterolemia can cause chronic inflammation per se through oxLDL by modulating inflammasome activity via TLRs and increasing the expression of dipeptidyl dipeptidase IV (DPP4) in macrophages. Raised DPP4 activity leads to an increase of CD36+ cells, which is a hallmark of the atherosclerotic inflammatory process in obese and insulin resistant patients (50). In particular, LDLc may attenuate corticosteroid transcriptional and translational activities thereby reducing their anti-inflammatory efficacy in GO (77). Taken together, these observations support the notion that chronic low-grade inflammation and inflammasome activation in dysmetabolic subjects may sustain the autoimmune process underlying TED pathogenesis and their clinical and phenotypic expression.
Key points
Obesity, diabetes and hypercholesterolemia contribute to GO disease. The use of statins and strict cholesterol control should be considered as part of the GO treatment plan.
Therapy of hyperthyroidism, cytokines, and weight gain
It is a common observation that hyperthyroidism is associated with weight loss in most cases, while approximately 10% of hyperthyroid patients experience weight increase. Therefore, restoration of euthyroidism usually results into weight regain that is sometimes excessive. The mechanism underlying this phenomenon is not entirely clear and it is commonly believed that the transition from hyper to euthyroidism can promote obesity in predisposed patients (78). On the other hand, some studies tried to identify specific risk factors for such weight increase that may include the severity of the autoimmune process in Graves’ disease (78, 79). Data regarding post-therapy body composition (lean and fat body mass) in terms of preferential accumulation and timing are still conflicting (79, 80). However, recent studies focus on the effect of dietary programs in the prevention of weight gain compared to standard care (81). Taken together, these studies strongly suggest that weight regain after treatment of hyperthyroidism is not only caused by a decreased catabolic effect of thyrotoxicosis, but rather linked to additional, independent mechanisms (82). This hypothesis is supported by the observation of a relationship between treatment modalities for hyperthyroidism and weight regain, with a greater effect for radioiodine and surgery, compared to anti-thyroid drugs. Indeed, several studies indicate a statistically and clinically significant excess of weight gain after thyroidectomy for hyperthyroidism (83). However, this observation is mitigated by studies showing body weight gain in all thyroidectomized patients regardless of underlying thyroid disorder (83, 84). Accordingly, excessive body weight gain after thyroidectomy has been attributed to deficient replacement therapy with L-T4 and to an imbalance of thyroid hormone homeostasis due to poor tissue conversion of T4 to T3 (85). However, several studies indicate that radioiodine (RAI) therapy is also associated with significant weight gain (by 5-6 kg on average over 1year post-treatment). This weight gain can last up to 5 years and lead to excess body weight compared to the premorbid condition similarly to what is observed in Graves’ patients after total thyroidectomy. Treatment of hyperthyroidism with anti-thyroid drugs (ATDs) is also associated with 2-4 kg weight gain which starts after the start of the initial treatment and may last one year after the end of the treatment (86, 87). This effect occurs with both treatment with ATDs alone and block-and-replace therapy regimen (ATDs plus L-T4) although studies directly comparing these two regimens are lacking (88). However, there is no clear evidence that ATD treatment leads to lasting weight gain. Conversely, several observations indicate that body weight remains unchanged after completion of ATD therapy, suggesting that the impact of these drugs on weight and energy balance differs from surgery and radioactive iodine and depends on residual thyroid function (84). These considerations were reinforced by a comparative study between different treatment modalities for hyperthyroidism. Indeed, a retrospective study including 133 patients with Graves’ disease who received ATDs, surgery or RAI indicated that treatment of hypothyroidism post-thyroidectomy or post-RAI was associated with significantly greater weight gain compared to patients who achieved euthyroidism with ATDs or hemi-thyroidectomy (78). Interestingly, this study found that these Graves’ patients experienced more weight gain compared to patients with thyroid cancer subjected to total thyroidectomy. Weight increments for both the surgical- or RAI- induced hypothyroidism groups were 10 vs 4 kg in the ATD or hemithyroidectomy-treated euthyroid groups vs only 0.6 kg in the surgically treated thyroid cancer cohort (78). Hence, these results suggested that thyroid autoimmune disease is associated with weight gain per se. The mechanisms underlying the direct effect of Graves’ disease on weight increase is still unclear. One hypothesis is that brown adipose tissue (BAT) expresses TSH receptors which are activated by anti-TSH-R stimulating antibodies of Graves’ patients. In fact, TSH may stimulate thermogenesis by binding to the TSH receptor expressed in adipocytes, a function involved in maintaining thermal status during hypothyroidism. Moreover, in vivo acute administration of recombinant human TSH at supraphysiological doses induced the release of small but significant amounts of leptin which was proportional to the adipose mass. Hence, it is reasonable to suppose that TSH stimulating activity may have per se an important effect on adipogenesis, mainly on white adipose tissue. On the other hand, this effect may be enhanced by inflammatory cytokines present in Graves’ patients (IL-1, IL-6 and TNFα) which are able to activate the hypothalamic, pituitary, adrenal axis, and cortisol secretion. In conclusion, several lines of evidence suggest that Graves’ disease is associated with excessive weight regain during therapy and may represent an important risk factor for the development of obesity. It is also possible, that the different effect of AITs on residual overweight may be due to their immunosuppressive effect (along with the use of corticocosteroids).
Key points
Excessive weight regain may occur after treatment of hyperthyroidism, especially after radioiodine or surgery. Thyroid autoimmunity should be considered an independent mechanism that leads to inappropriate body weight increase.
Therapy of hypothyroidism in obese patients: role of inflammasome, ubiquitination, glucurono- coniugation and activity of deiodinases
Replacement therapy with Levothyroxine (LT4) is currently the standard treatment for hypothyroid patients. Its therapeutic goal is a TSH value within the age-related range, which is associated with an improvement in symptoms, quality of life and cardiovascular risk. However, the most appropriate treatment of hypothyroid patients is still under debate, especially in some particular conditions including old-age and obesity. In particular, daily calorie intake and nutritional status may affect leptin levels, which, in turn, may upregulate hypothalamic TRH and deiodinase 1 (D1) activity (38, 41, 89–91). Therefore, obese patients show a higher average TSH level than non-obese subjects (61). This positive correlation between BMI and TSH may reflect a compensatory pituitary-thyroid axis activation by leptin and insulin (48) to increase energy expenditure. However, it may also suggest a certain degree of thyroid hormone resistance in obese patients (92) related to the chronic low-grade inflammation and inflammasome activation but also to the regulation of 5’ adenosine monophosphate-activated protein kinase (AMPK), a central target not only for the modulation of insulin sensitivity but also for the effect of thyroid hormones on appetite and energy expenditure (93). Indeed, in vivo studies performed in obese mice indicate that obesity-induced inflammasome activation up-regulates deiodinase activity by down-regulating ubiquitin-mediated protein degradation, thereby contributing to thyroid hormone (TH) resistance at pituitary level and body mass gain (94, 95). Moreover, impaired transport of TH through cell membrane by low-grade inflammation may represent an additional mechanism of thyroid hormone resistance in obese patients (96). Pituitary resistance and impaired cell membrane transport of thyroid hormones may explain the positive correlation between FT3 levels and body weight. Taken together, these studies suggest that TSH levels in obese hypothyroid patients may not recapitulate the adequacy of L-T4 replacement dose (61). Indeed, some studies suggest that optimization of L-T4 replacement dose should be performed according to lean body mass (LBM), rather than whole body weight (RBW). In particular, LBM is related to body mass cells (BMC) that corresponds to lean mass of body organs (liver, kidney and heart) and correlates to D1 activity, L-T4 glucurono-conjugation and basal metabolic rate (BMR). In contrast, fat mass, which is one of the major determinants of low-grade inflammation, may exert a confounding effect on L-T4 requirement (97, 98). Hence, further studies are needed to evaluate the role of different LT-4 dose and formulations or L-T4/L-T3 combination in the treatment of hypothyroidism in obese patients.
Key points
The most appropriate treatment of hypothyroidism in obese patients is still debated. These patients may have some degree of resistance to thyroid hormone related to chronic low-grade inflammation and activation of the inflammasome.
Bariatric surgery: impact on thyroid inflammasome, autoimmunity and thyroid function
Bariatric surgery has become a safe and efficacious treatment for patients with severe and complex obesity, who do not respond adequately to caloric restriction (99). Several studies reported a significant effect of bariatric surgery on many inflammatory markers, including thyroid autoantibodies, and a significant effect on thyroid hormone levels (100, 101). As discussed above, obesity is a condition associated with a low-grade systemic inflammation, leading to altered levels of various inflammatory markers (102, 103) and imbalanced adipokine secretion (104). For instance, adiponectin and orexin-A, two adipokines related to insulin sensitivity and calorie intake, are markedly decreased in obese patients along with a significant increase in inflammatory cytokines (105). Interestingly, there is evidence that patients with morbid obesity, subjected to bariatric surgery, display reduced levels of pro-inflammatory cytokines along with increased adiponectin levels that play an anti-inflammatory action (106). The effect of bariatric surgery in reverting the dysregulation of the inflammasome system has been extensively studied by Vicente et al. (107), who evaluated changes in inflammasome components in 22 patients with morbid-obesity before and after six months of bariatric-surgery (sleeve-gastrectomy and roux-en-Y gastric bypass). In this study, the most important changes were found in NOD-like-receptors, cell-cycle and DNA-damage regulators, while, at baseline, several components of the inflammasome were significantly related to metabolic alterations, including T2DM (CCL2/CXCR1/SIRT1), hypertension (AIM2/ASC/P2RX7) and dyslipidemia (CXCL3/NLRP7). It was interesting to note that the levels of all these markers significantly changed six-months after bariatric-surgery. Gene-expression levels of NLRC4/NLRP12/CXCL3/CCL8/TLR4 were related to pre and post-operative peripheral-blood mononuclear-cells. In vitro experiments, performed to validate the mechanistic insights of these findings indicated that NLRC4/NLRP12 silencing in the hepatoma cell line HEPG2 resulted in increased cell-viability and lipid-accumulation along with a reduction in the apoptosis rate. In respect to the relationship between inflammasome activation in morbid obesity subclinical hypothyroidism (108), a report by Zhu et al. indicates that obese patients subjected to laparoscopic sleeve gastrectomy display a significant decrease in inflammation markers, such as IL-6, TNF-α, and CRP (109) that is concomitant with a significant TSH reduction. In agreement with these results, some reports indicate a reduction/normalization in TSH levels in obese patients after weight loss induced by either bariatric surgery or a low-calorie diet (110). A recent meta-analysis confirmed that bariatric surgery is able to revert subclinical hypothyroidism in obese patients and to reduce L-T4 replacement dose. It was interesting to note that, while TSH, FT3 and T3 levels decreased significantly after bariatric surgery, FT4, T4 and rT3 levels remained unchanged (100). Although the association between bariatric surgery and thyroid function seems clear and mainly based upon thyroid autoimmunity, the mechanism of this relationship may be complex and bidirectional: some studies indicate a role for autoimmune subclinical hypothyroidism in the pathogenesis of obesity (111),, while others support the hypothesis that the excess of dysfunctional adipose tissue may be responsible for changes in serum thyroid hormone levels (90, 112). In line with this notion, Xia et al. reported a significant reduction in serum TPOAbs and thyroglobulin antibodies (TgAbs) in 101 patients with morbid obesity (BMI ≥ 32 kg/m2 or BMI ≥ 27.5 kg/m2 with one or more comorbidities) treated with bariatric surgery (101). The reduction in TPOAbs and TgAbs was from 79.3 and 177.1 IU/mL to 57.8 and 66.0 IU/mL, respectively (P < 0.05). In accordance with these results, Kyrou et al, albeit in a small population study, evaluated ultrasound thyroid morphology in obese patients treated with bariatric surgery and found a significant relationship between weight loss and an increase in thyroid echogenic pattern (113). In conclusion, these data indicate that bariatric surgery is able to attenuate inflammasome activity and may exert a beneficial effect on thyroid autoimmunity and function.
Key points
Bariatric surgery can attenuate the activity of the inflammasome and can exert a beneficial effect on autoimmunity and thyroid function.
Obesity, inflammasome and thyroid cancer
A functional relationship between chronic inflammation and cancer was first proposed by Virchow in 1863 and is supported by clinical and epidemiological evidence. As mentioned above, obesity is characterized by chronic low-grade chronic inflammation and insulin resistance. It is now widely accepted that the consequent deranged immunity, compensatory hyperinsulinemia, hyperlipidemia, and enhanced oxidative stress are risk factors for cancer development and/or progression (114–116). Several data indicate that increased inflammasome activity in adipose tissue is an important mediator of obesity-induced inflammation and insulin resistance. Inflammasome-induced cytokines are mainly produced from the hematopoietic compartment and act in autocrine and paracrine manners to form an inflammatory microenvironment (117), recruit immune cells and interfere with insulin signaling processes thus promoting the development of diabetes (118). Obesity increases oxidative stress that in turn upregulates inflammatory cytokines (119). Indeed, chronic inflammation activates transcription of factors such as nuclear factor kappa-light-chain-enhancer of activated B (NF-kB), STAT3, and activator protein 1 (AP1) in pre-malignant cells. All these pathways enhance cell proliferation and survival promoting angiogenesis in conjunction with hypoxia (120). Various infiltrating cells have been identified in tumors, namely, tumor-associated lymphocytes, tumor-associated macrophages (TAM), immature dendritic cells, mast cells and myeloid-derived suppressor cells. The presence of specific inflammatory-immune cells such as macrophages and mast cells in tumor sites has been associated with a poor prognosis of thyroid cancer (121). Thyroid cancer cells and the tumoral stroma may secrete several cytokines and chemokines, thus sustaining the survival of cancer cells promoting the selection of clones that acquire additional genetic lesions becoming resistant to oncogene-induced apoptosis. Proinflammatory cytokines produced in tumor sites by inflammatory and epithelial cancer cells depend mainly on the NF-kB transcription factor and are pivotal in cancer progression (121). Consistently, several cancer histotypes are more common and more aggressive in obese patients than in the normal weight population. Several lines of evidence suggest that obesity is also associated with thyroid cancer. Various epidemiologic studies and metanalysis have found a statistically significant association between BMI and thyroid cancer, especially in males (122). The risk of thyroid cancer appears to be positively related with the degree of obesity (123). Moreover, obesity is related to more aggressive histotypes (123). Animal models support these studies and explain the complex mechanisms between obesity and thyroid tumors demonstrating that a high-fat diet (HFD) induces thyroid cancer cell proliferation by enhancing cyclin D1 protein levels as well as the phosphorylation of RB protein, serum leptin levels and STAT3 expression (124). In addition, an inhibitor of STAT3 was able to inhibit the proliferation of HFD-induced thyroid cancer cells through the reduction of cyclins D1 and B1, CDK4, CDK6 and pRB (125). A role of inflammation and immune response in the onset of thyroid cancer and autoimmune thyroid diseases (such as Hashimoto’s thyroiditis and Graves’ disease) has already been demonstrated (126, 127). Moreover, increased TAM infiltration in poorly differentiated thyroid cancers (PDTCs) was found to be positively correlated with capsule invasion, extrathyroidal extension and poor prognosis (128). It has been also reported that the expression of BRAF V600E mutation and the RET/PTC gene rearrangement promote the activity of NF-kB, the expression of inflammatory mediators, and lymph node metastases in patients with papillary thyroid cancer (PTC) (129). Inflammation is intimately linked to the metabolic reprogramming of cancer cells. Indeed, compared with healthy tissues or benign lesions thyroid cancer is characterized by increased lactic acid production, regardless of its histotype. Moreover, PKM2, a key enzyme of glycolysis, has been found overexpressed in advanced and poorly differentiated thyroid cancer and associated with tumor aggressiveness and negative prognosis of PTC (130). In other cell models it has been shown that PKM2-dependent glycolysis promotes NLP3 inflammasome activation (131). Indeed, pharmacological inhibition of PKM2 attenuates the release of IL-1b, IL-18 and HMGB1 due to NLP3 activation (131). Although a direct link between thyroid cancer and obesity-induced inflammasome activation has not been clearly demonstrated, several lines of evidence support the hypothesis that a relationship may be also present in thyroid cancer. Recent studies indicate that the inflammasomes can be activated by fatty acids and high glucose levels linking metabolic danger signals to the activation of inflammation and cancer development. These data suggest that activation of the inflammasomes may represent a crucial step in the obesity-associated thyroid cancer development.
Key point
Inflammasomes can be activated by fatty acids and high glucose levels linking metabolic danger signals to the activation of inflammation and cancer development.
A possible role of vitamin D in autoimmune thyroid diseases and obesity
Several studies have shown that the risk and the clinical evolution of autoimmune thyroid disease and other chronic inflammatory diseases are associated with low plasma levels of vitamin D (132). This is not surprising as vitamin D modulates the synthesis of IL-1, IL-6, and other cytokines, suppresses dendritic cell differentiation, and influences regulatory T cell activity. Vitamin D inhibits the activity of TLRs (Toll-like receptors) by inhibiting the identification of pathogenic or non-pathogenic molecular patterns (e.g. LDLc binding with TLR in thyroid cells) by TLRs. In addition, vitamin D via its receptor (VDR), inhibits the deubiquitination and activation of NLRP3 inflammasome (133, 134). Notably, hyperthyroidism is associated with reduced levels of vitamin D (135). Moreover, vitamin D negatively modulate lipogenesis controlling calcium influx into adipocytes (136). These data are in agreement with studies showing that vitamin D deficiency is associated with overweight, obesity and insulin resistance (137, 138). Accordingly, vitamin D supplementation may decrease the incident risk of autoimmune disease (139) and increases insulin sensitivity (140, 141).
Key points
Vitamin D attenuates the activation of the inflammasome and its immune response, modulates lipogenesis and increases insulin sensitivity. Vitamin D deficiency is related to the risk and severity of thyroid autoimmunity and obesity.
Conclusions and perspectives
Recent epidemiological and experimental data provided evidence of a bidirectional interaction between obesity and thyroid autoimmunity. In particular, obesity is associated with an enhanced risk of hypothyroidism and of developing anti-thyroperoxidase antibodies. This is also true for thyroid autoimmunity related disorders like GO: low-grade inflammation and inflammasome activation in dysmetabolic subjects may enhance the autoimmune process underlying GO pathogenesis and its clinical and phenotypic expression. On the other hand, thyroid autoimmunity may favor overweight and obesity. One empirical evidence supporting this hypothesis is Graves’ disease. Indeed, a marked weight gain during GD therapy may represent an important risk factor for the development of obesity. Moreover, the lower extent of weight gain with immunosuppressive therapies unravels the possibility that thyroid autoimmunity per se is able to favor obesity, although the exact mechanisms underlying this phenomenon are not fully understood. In addition to the positive effect of obesity in thyroid autoimmunity and hypothyroidism, several lines of evidence suggest that obesity related inflammasome activation may induce a certain degree of TH resistance both in peripheral tissues and hypothalamus leading to a significant increase of TSH, thereby exerting confounding effects on L-T4 requirement in obese people. Inflammasome activation in obesity may also increase the risk of thyroid cancer and is related with a more aggressive thyroid cancer phenotype. Several in vitro data support and explain this observation. A further observation confirms the role of inflammasome related obesity in thyroid disorders: bariatric surgery is able to attenuate inflammasome activity and may exert a beneficial effect on thyroid autoimmunity and function. Taken together these results indicate that thyroid function should be always screened in obese people and that thyroid disorders should be carefully addressed in obese and dysmetabolic people. Further studies are needed to unravel the mechanisms underlying the bidirectional relationship between obesity and thyroid disorders as well as to evaluate the role of different LT-4 doses and formulations or L-T4/L-T3 combination in the treatment of hypothyroidism in obese patients.
Ethics statement
Written informed consent for participation was not required for this study in accordance with the national legislation and the institutional requirements.
Author contributions
Conceptualization, RM, AB, VV, FF; methodology, RM, VV; software, RM, AN, TP; validation, AB, FF, RM,VV; investigation, RM, TP, VV, FF, AB; resources, RM, FF, AB; writing—original draft preparation, RM, FF, AB, VV; writing—review and editing, RM, FF, AB, VV; supervision, RM, FF, AB, VV; project administration, AB. funding acquisition, AB. All authors have read and agreed to the published version of the manuscript.
Funding
This research was supported in part by Fondazione AIRC: IG n. 23369 to AB and by Ministero della Salute, Italy, grant RF-2019-12368937 to AB.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Di Cesare M, Bentham J, Stevens GA, Zhou B, Danaei G, Lu Y, et al. Trends in adult body-mass index in 200 countries from 1975 to 2014: A pooled analysis of 1698 population-based measurement studies with 19.2 million participants. Lancet (2016) 387(10026):1377–96. doi: 10.1016/S0140-6736(16)30054-X
2. Blüher M. Obesity: global epidemiology and pathogenesis. Nat Rev Endocrinol (2019) 15(5):288–98. doi: 10.1038/s41574-019-0176-8
3. Coppack SW. Pro-inflammatory cytokines and adipose tissue. Proc Nutr Soc (2001) 60(3):349–56. doi: 10.1079/PNS2001110
4. Wang C, Ha X, Li W, Xu P, Gu Y, Wang T, et al. Correlation of TLR4 and KLF7 in inflammation induced by obesity. Inflammation (2017) 40(1):42–51. doi: 10.1007/s10753-016-0450-z
5. Wu KKL, Cheung SWM, Cheng KKY. NLRP3 inflammasome activation in adipose tissues and its implications on metabolic diseases. Int J Mol Sci (2020) 21(11):1–22. doi: 10.3390/ijms21114184
6. He Y, Hara H, Núñez G. Mechanism and regulation of NLRP3 inflammasome activation. Trends Biochem Sci (2016) 41(12):1012–21. doi: 10.1016/j.tibs.2016.09.002
7. Mangan MSJ, Olhava EJ, Roush WR, Seidel HM, Glick GD, Latz E. Targeting the NLRP3 inflammasome in inflammatory diseases. Nat Rev Drug Discov (2018) 17(8):588–606. doi: 10.1038/nrd.2018.97
8. Schroder K, Tschopp J. The inflammasomes. Cell (2010) 140(6):821–32. doi: 10.1016/j.cell.2010.01.040
9. Lumeng CN, Bodzin JL, Saltiel AR. Obesity induces a phenotypic switch in adipose tissue macrophage polarization. J Clin Invest (2007) 117(1):175–84. doi: 10.1172/JCI29881
10. Benham V, Chakraborty D, Bullard B, Bernard JJ. A role for FGF2 in visceral adiposity-associated mammary epithelial transformation. Adipocyte (2018) 7(2):113–20. doi: 10.1080/21623945.2018.1445889
11. Chakraborty D, Benham V, Bullard B, Kearney T, Hsia HC, Gibbon D, et al. Fibroblast growth factor receptor is a mechanistic link between visceral adiposity and cancer. Oncogene (2017) 36(48):6668–79. doi: 10.1038/onc.2017.278
12. Addobbati C, da Cruz HLA, Adelino JE, Melo Tavares Ramos AL, Fragoso TS, Domingues A, et al. Polymorphisms and expression of inflammasome genes are associated with the development and severity of rheumatoid arthritis in Brazilian patients. Inflammation Res (2018) 67(3):255–64. doi: 10.1007/s00011-017-1119-2
13. Leavy O. Inflammasome: NAIPs: Pathogen-sensing proteins. Nat Rev Immunol (2011) 11(10):644. doi: 10.1038/nri3069
14. Malhotra S, Rio J, Urcelay E, Nurtdinov R, Bustamante MF, Fernandez O, et al. NLRP3 inflammasome is associated with the response to IFN-b in patients with multiple sclerosis. Brain (2015) 138(3):644–52. doi: 10.1093/brain/awu388
15. Wynn TA, Chawla A, Pollard JW. Macrophage biology in development, homeostasis and disease. Nature (2013) 496(7446):445–55. doi: 10.1038/nature12034
16. Dombrowski Y, Peric M, Koglin S, Kammerbauer C, Göß C, Anz D, et al. Cytosolic DNA triggers inflammasome activation in keratinocytes in psoriatic lesions. Sci Transl Med (2011) 3(82):82ra38. doi: 10.1126/scitranslmed.3002001
17. Bergsbaken T, Fink SL, Cookson BT. Pyroptosis: Host cell death and inflammation. Nat Rev Microbiol (2009) 7(2):99–109. doi: 10.1038/nrmicro2070
18. He Y, Zeng MY, Yang D, Motro B, Núñez G. NEK7 is an essential mediator of NLRP3 activation downstream of potassium efflux. Nature (2016) 530(7590):354–7. doi: 10.1038/nature16959
19. Okondo MC, Johnson DC, Sridharan R, Go EB, Chui AJ, Wang MS, et al. DPP8 and DPP9 inhibition induces pro-caspase-1-dependent monocyte and macrophage pyroptosis. Nat Chem Biol (2017) 13(1):46–53. doi: 10.1038/nchembio.2229
20. Mariño G, Pietrocola F, Eisenberg T, Kong Y, Malik SA, Andryushkova A, et al. Regulation of autophagy by cytosolic acetyl-coenzyme a. Mol Cell (2014) 53(5):710–25. doi: 10.1016/j.molcel.2014.01.016
21. Pietrocola F, Galluzzi L, Bravo-San Pedro JM, Madeo F, Kroemer G. Acetyl coenzyme a: A central metabolite and second messenger. Cell Metab (2015) 21(6):805–21. doi: 10.1016/j.cmet.2015.05.014
22. Kelley N, Jeltema D, Duan Y, He Y. Nlrp3炎症小体的激活和调控机制综述. Int J Mol Sci (2019) 20(13):1–24. doi: 10.3390/ijms20133328
23. Rocha VZ, Libby P. Obesity, inflammation, and atherosclerosis. Nat Rev Cardiol (2009) 6(6):399–409. doi: 10.1038/nrcardio.2009.55
24. Camell CD, Günther P, Lee A, Goldberg EL, Youm Y, Bartke A, et al. Aging induces an Nlrp3 inflammasome-dependent expansion of adipose B cells that impairs metabolic homeostasis. Cell Metab (2019) 30(6):1024–39. doi: 10.1016/j.cmet.2019.10.006
25. Abais JM, Xia M, Zhang Y, Boini KM, Li PL. Redox regulation of NLRP3 inflammasomes: ROS as trigger or effector? Antioxid Redox Signal (2015) 22(13):1111–29. doi: 10.1089/ars.2014.5994
26. Weetman AP. An update on the pathogenesis of hashimoto’s thyroiditis. J Endocrinol Invest (2021) 44(5):883–90. doi: 10.1007/s40618-020-01477-1
27. Song RH, Wang B, Yao QM, Li Q, Jia X, Zhang JA. The impact of obesity on thyroid autoimmunity and dysfunction: A systematic review and meta-analysis. Front Immunol (2019) 10:2349. doi: 10.3389/fimmu.2019.02349
28. Biondi B. Thyroid and obesity: An intriguing relationship. J Clin Endocrinol Metab (2010) 95(8):3614–7. doi: 10.1210/jc.2010-1245
29. Fierabracci P, Pinchera A, Martinelli S, Scartabelli G, Salvetti G, Giannetti M, et al. Prevalence of endocrine diseases in morbidly obese patients scheduled for bariatric surgery: Beyond diabetes. Obes Surg (2011) 21(1):54–60. doi: 10.1007/s11695-010-0297-6
30. Hollowell JG, Staehling NW, Dana Flanders W, Harry Hannon W, Gunter EW, Spencer CA, et al. Serum TSH, T4, and thyroid antibodies in the united states population (1988 to 1994): National health and nutrition examination survey (NHANES III). J Clin Endocrinol Metab (2002) 87(2):489–99. doi: 10.1210/jcem.87.2.8182
31. Wiersinga WM. Clinical relevance of environmental factors in the pathogenesis of autoimmune thyroid disease. Endocrinol Metab (2016) 31(2):213–22. doi: 10.3803/EnM.2016.31.2.213
32. Liu J, Mao C, Dong L, Kang P, Ding C, Zheng T, et al. Excessive iodine promotes pyroptosis of thyroid follicular epithelial cells in hashimoto’s thyroiditis through the ROS-NF-κB-NLRP3 pathway. Front Endocrinol (Lausanne) (2019) 10:778. doi: 10.3389/fendo.2019.00778
33. Klecha AJ, Barreiro Arcos ML, Frick L, Genaro AM, Cremaschi G. Immune-endocrine interactions in autoimmune thyroid diseases. Neuroimmunomodulation (2008) 15(1):68–75. doi: 10.1159/000135626
34. Klecha AJ, Genaro AM, Lysionek AE, Caro RA, Coluccia AG, Cremaschi GA. Experimental evidence pointing to the bidirectional interaction between the immune system and the thyroid axis. Int J Immunopharmacol (2000) 22(7):491–500. doi: 10.1016/S0192-0561(00)00012-6 doi: 10.3389/fcell.2020.614030
35. De Luca R, Davis PJ, Lin HY, Gionfra F, Percario ZA, Affabris E, et al. Thyroid hormones interaction with immune response, inflammation and non-thyroidal illness syndrome. Front Cell Dev Biol (2021) 8:1–9.
36. Lombardi A, Menconi F, Greenberg D, Concepcion E, Leo M, Rocchi R, et al. Dissecting the genetic susceptibility to graves’ disease in a cohort of patients of Italian origin. Front Endocrinol (Lausanne) (2016) 7:21. doi: 10.3389/fendo.2016.00021
37. Smith TJ, Hegedus L. Graves’ disease. N Engl J Med (2016) 375(16):1552–65. doi: 10.1056/NEJMra1510030
38. Fahed M, Abou Jaoudeh MG, Merhi S, Mosleh JMB, Ghadieh R, Ghadieh R, et al. Evaluation of risk factors for insulin resistance: A cross sectional study among employees at a private university in Lebanon. BMC Endocr Disord (2020) 20(1):1–14. doi: 10.1186/s12902-020-00558-9
39. Kwon H, Pessin JE. Adipokines mediate inflammation and insulin resistance. Front Endocrinol (Lausanne) (2013) 4:71. doi: 10.3389/fendo.2013.00071
40. Mollaei M, Abbasi A, Hassan ZM, Pakravan N. The intrinsic and extrinsic elements regulating inflammation. Life Sci (2020) 260:118258. doi: 10.1016/j.lfs.2020.118258
41. Duntas LH, Biondi B. The interconnections between obesity, thyroid function, and autoimmunity: The multifold role of leptin. Thyroid (2013) 23(6):646–53. doi: 10.1089/thy.2011.0499
42. Kawashima A, Tanigawa K, Akama T, Yoshihara A, Ishii N, Suzuki K. Innate immune activation and thyroid autoimmunity. J Clin Endocrinol Metab (2011) 96(12):3661–71. doi: 10.1210/jc.2011-1568
43. Kawashima A, Yamazaki K, Hara T, Akama T, Yoshihara A, Sue M, et al. Demonstration of innate immune responses in the thyroid gland: Potential to sense danger and a possible trigger for autoimmune reactions. Thyroid (2013) 23(4):477–87. doi: 10.1089/thy.2011.0480
44. Kanda H, Tateya S, Tamori Y, Kotani K, Hiasa K, Kitazawa R, et al. MCP-1 contributes to macrophage infiltration into adipose tissue, insulin resistance, and hepatic steatosis in obesity. J Clin Invest (2006) 116(6):1494. doi: 10.1172/JCI26498
45. Yang CA, Chiang BL. Inflammasomes and human autoimmunity: A comprehensive review. J Autoimmun (2015) 61:1–8. doi: 10.1016/j.jaut.2015.05.001
46. Duewell P, Kono H, Rayner KJ, Sirois CM, Bauernfeind FG, Abela GS, et al. NLRP3 inflamasomes are required for atherogenesis and activated by cholesterol crystals that form early in disease. Nature (2010) 464(7293):1357–61. doi: 10.1038/nature08938
47. McLachlan SM, Rapoport B. Breaking tolerance to thyroid antigens: Changing concepts in thyroid autoimmunity. Endocr Rev (2014) 35(1):59–105. doi: 10.1210/er.2013-1055
48. Santini F, Marzullo P, Rotondi M, Ceccarini G, Pagano L, Ippolito S, et al. Mechanisms in endocrinology: The crosstalk between thyroid gland and adipose tissue: Signal integration in health and disease. Eur J Endocrinol (2014) 171(4):R137–52. doi: 10.1530/EJE-14-0067
49. Harii N, Lewis CJ, Vasko V, McCall K, Benavides-Peralta U, Sun X, et al. Thyrocytes express a functional toll-like receptor 3: Overexpression can be induced by viral infection and reversed by phenylmethimazole and is associated with hashimoto’s autoimmune thyroiditis. Mol Endocrinol (2005) 19(5):1231–50. doi: 10.1210/me.2004-0100
50. Rao X, Zhao S, Braunstein Z, Mao H, Razavi M, Duan L, et al. Oxidized LDL upregulates macrophage DPP4 expression via TLR4/TRIF/CD36 pathways. EBioMedicine (2019) 41:50–61. doi: 10.1016/j.ebiom.2019.01.065
51. Guo Q, Wu Y, Hou Y, Liu Y, Liu T, Zhang H, et al. Cytokine secretion and pyroptosis of thyroid follicular cells mediated by enhanced NLRP3, NLRP1, NLRC4, and AIM2 inflammasomes are associated with autoimmune thyroiditis. Front Immunol (2018) 9. doi: 10.3389/fimmu.2018.01197
52. Hepp M, Werion A, De Greef A, de Ville de Goyet C, de Bournonville M, Behets C, et al. Oxidative stress-induced sirtuin1 downregulation correlates to hif-1α, glut-1, and vegf-a upregulation in th1 autoimmune hashimoto’s thyroiditis. Int J Mol Sci (2021) 22(8):3806. doi: 10.3390/ijms22083806
53. Ishii KJ, Suzuki K, Coban C, Takeshita F, Itoh Y, Matoba H, et al. Genomic DNA released by dying cells induces the maturation of APCs. J Immunol (2001) 167(5):2602–7. doi: 10.4049/jimmunol.167.5.2602
54. Kawashima A, Tanigawa K, Akama T, Wu H, Sue M, Yoshihara A, et al. Fragments of cenomic DNA released by injured cells activate innate immunity and suppress endocrine function in the thyroid. Endocrinology (2011) 152(4):1702–12. doi: 10.1210/en.2010-1132
55. Evavold CL, Ruan J, Tan Y, Xia S, Wu H, Kagan JC. The pore-forming protein gasdermin d regulates interleukin-1 secretion from living macrophages. Immunity (2018) 48(1):35–44.e6. doi: 10.1016/j.immuni.2017.11.013
56. De Vito P, Incerpi S, Pedersen JZ, Luly P, Davis FB, Davis PJ. Thyroid hormones as modulators of immune activities at the cellular level. Thyroid (2011) 21(8):879–90. doi: 10.1089/thy.2010.0429
57. Panveloski-Costa AC, Silva Teixeira S, Ribeiro IMR, Serrano-Nascimento C, das Neves RX, Favaro RR, et al. Thyroid hormone reduces inflammatory cytokines improving glycaemia control in alloxan-induced diabetic wistar rats. Acta Physiol (2016) 217(2):130–40. doi: 10.1111/apha.12647
58. Andersen CJ, Murphy KE, Fernandez ML. Impact of obesity and metabolic syndrome on immunity. Adv Nutr (2016) 7(1):66–75. doi: 10.3945/an.115.010207
59. Kwakkel J, Surovtseva OV, De Vries EM, Stap J, Fliers E, Boelen A. A novel role for the thyroid hormone-activating enzyme type 2 deiodinase in the inflammatory response of macrophages. Endocrinology (2014) 155(7):2725–34. doi: 10.1210/en.2013-2066
60. De Vito P, Balducci V, Leone S, Percario Z, Mangino G, Davis PJ, et al. Nongenomic effects of thyroid hormones on the immune system cells: New targets, old players. Steroids (2012) 77(10):988–95. doi: 10.1016/j.steroids.2012.02.018
61. Rotondi M, Leporati P, La Manna A, Pirali B, Mondello T, Fonter R, et al. Raised serum TSH levels in patients with morbid obesity: Is it enough to diagnose subclinical hypothyroidism? Eur J Endocrinol (2009) 160(3):403–8. doi: 10.1530/EJE-08-0734
62. Bahn RS. Current insights into the pathogenesis of graves’ ophthalmopathy. Horm Metab Res (2015) 47(10):773–8.
64. Stein JD, Childers D, Gupta S, Talwar N, Nan B, Lee BJ, et al. Risk factors for developing thyroid-associated ophthalmopathy among individuals with graves disease. JAMA Ophthalmol (2015) 133(3):290–6. doi: 10.1001/jamaophthalmol.2014.5103
65. Kalmann R, Mourits MP. Diabetes mellitus: A risk factor in patients with graves’ orbitopathy. Br J Ophthalmol (1999) 83(4):463–5. doi: 10.1055/s-0035-1555762
66. Le Moli R, Muscia V, Tumminia A, Frittitta L, Buscema M, Palermo F, et al. Type 2 diabetic patients with graves’ disease have more frequent and severe graves’ orbitopathy. Nutr Metab Cardiovasc Dis (2015) 25(5):452–7. doi: 10.1016/j.numecd.2015.01.003
67. Zhang L, Grennan-Jones F, Draman MS, Lane C, Morris D, Dayan CM, et al. Possible targets for nonimmunosuppressive therapy of graves’ orbitopathy. J Clin Endocrinol Metab (2014) 99(7):1183–90. doi: 10.1210/jc.2013-4182
68. Sciacca L, Vigneri R, Tumminia A, Frasca F, Squatrito S, Frittitta L, et al. Clinical and molecular mechanisms favoring cancer initiation and progression in diabetic patients. Nutr Metab Cardiovasc Dis (2013) 23(9):808–15. doi: 10.1016/j.numecd.2013.05.006
69. Smith TJ, Huetwell FG, Hegedus L, Douglas RS. Role of IGF-1 pathway in the pathogenesis of graves’ orbitopathy. Best Pract Res Clin Endocrinol Metab (2012) 26(734):291–302. doi: 10.1016/j.beem.2011.10.002
70. Wiersinga WM. Autoimmunity in graves’ ophthalmopathy: The result of an unfortunate marriage between TSH receptors and IGF-1 receptors? J Clin Endocrinol Metab (2011) 96(8):2386–94. doi: 10.1210/jc.2011-0307
71. Zhang L, Grennan-Jones F, Lane C, Rees DA, Dayan CM, Ludgate M. Adipose tissue depot-specific differences in the regulation of hyaluronan production of relevance to graves’ orbitopathy. J Clin Endocrinol Metab (2012) 97(2):653–62. doi: 10.1210/jc.2011-1299
72. Valyasevi RW, Erickson DZ, Harteneck DA, Dutton CM, Heufelder AE, Jyonouchi SC, et al. Differentiation of human orbital preadipocyte fibroblasts induces expression of functional thyrotropin receptor. J Clin Endocrinol Metab (1999) 84(7):2257–562. doi: 10.1210/jc.84.7.2557
73. Matheis F, Muller PA, Graves CL, Gabanyi I, Zachary J, Costa-borges D, et al. Adrenergic signaling in muscularis macrophages limits infection-induced neuronal loss. Cell (2021) 180(1):64–78. doi: 10.1016/j.cell.2019.12.002
74. Tall AR, Westerterp M. Inflammasomes, neutrophil extracellular traps, and cholesterol. J Lipid Res (2019) 60(4):721–7. doi: 10.1194/jlr.S091280
75. Janssen JAMJL, Smith TJ. Lessons learned from targeting IGF-I receptor in thyroid-associated ophthalmopathy. Cells (2021) 10(2):1–18. doi: 10.3390/cells10020383
76. Sabini E, Mazzi B, Profilo MA, Mautone T, Casini G, Rocchi R, et al. High serum cholesterol is a novel risk factor for graves’ orbitopathy: Results of a cross-sectional study. Thyroid (2018) 28(3):386–94. doi: 10.1089/thy.2017.0430
77. Naselli A, Moretti D, Regalbuto C, Arpi ML, Lo Giudice F, Frasca F, et al. Evidence that baseline levels of low-density lipoproteins cholesterol affect the clinical response of graves’ ophthalmopathy to parenteral corticosteroids. Front Endocrinol (Lausanne) (2020) 11:609895. doi: 10.3389/fendo.2020.609895
78. Kyriacou A, Kyriacou A, Makris KC, Syed AA, Perros P. Weight gain following treatment of hyperthyroidism–a forgotten tale. Clin Obes (2019) 9(5):1–12. doi: 10.1111/cob.12328
79. Karmisholt J, Carlé A, Andersen S. Body weight changes in hyperthyroidism: Timing and possible explanations during a one year repeated measurement study. Eur Thyroid J (2021) 10(3):208–14. doi: 10.1159/000512078
80. Sun L, Goh HJ, Verma S, Govindharajulu P, Sadananthan SA, Michael N, et al. Metabolic effects of brown fat in transitioning from hyperthyroidism to euthyroidism. Eur J Endocrinol (2021) 185(4):553–63. doi: 10.1530/EJE-21-0366
81. Liu G, Liang L, Bray GA, Qi L, Hu FB, Rood J, et al. Thyroid hormones and changes in body weight and metabolic parameters in response to weight loss diets: The POUNDS LOST trial. Int J Obes (2017) 41(6):878–86. doi: 10.1038/ijo.2017.28
82. Baranowska-Bik A, Bik W. The association of obesity with autoimmune thyroiditis and thyroid function-possible mechanisms of bilateral interaction. Int J Endocrinol (2020) 2020:8894792. doi: 10.1155/2020/8894792
83. Huynh CN, Pearce JV, Kang L, Celi FS. Weight gain after thyroidectomy: A systematic review and meta-analysis. J Clin Endocrinol Metab (2021) 106(1):282–91. doi: 10.1210/clinem/dgaa754
84. Tigas S, Idiculla J, Beckett G, Toft A. Is excessive weight gain after ablative treatment of hyperthyroidism due to inadequate thyroid hormone therapy? Thyroid (2000) 10(12):1107–11. doi: 10.1089/thy.2000.10.1107
85. Torlinska B, Nichols L, Mohammed MA, McCabe C, Boelaert K. Patients treated for hyperthyroidism are at increased risk of becoming obese: Findings from a Large prospective secondary care cohort. Thyroid (2019) 29(10):1380–9. doi: 10.1089/thy.2018.0731
86. Yotsapon T, Waralee C, Hussamon P, Panita S, Siriwan B, Soontaree N, et al. Duration of antithyroid drug treatment may predict weight gain after radioactive iodine therapy in patients with graves’ disease. Heliyon (2022) 8(5):e09471. doi: 10.1016/j.heliyon.2022.e09471
87. Dale J, Daykin J, Holder R, Sheppard MC, Franklyn JA. Weight gain following treatment of hyperthyroidism. Clin Endocrinol (Oxf) (2001) 55(2):233–9. doi: 10.1046/j.1365-2265.2001.01329.x
88. Klieverik LP, Kalsbeek A, Ackermans MT, Sauerwein HP, Wiersinga WM, Fliers E. Energy homeostasis and body weight before and after cessation of block and replacement therapy in euthyroid patients with graves’ disease. Int J Endocrinol (2011) 2011:1–8. doi: 10.1155/2011/715370
89. Kozłowska L, Rosołowska-Huszcz D. Leptin, thyrotropin, and thyroid hormones in obese/overweight women before and after two levels of energy deficit. Endocrine (2004) 24(2):147–53. doi: 10.1385/ENDO:24:2:147
90. Marzullo P, Minocci A, Tagliaferri MA, Guzzaloni G, Di Blasio A, De Medici C, et al. Investigations of thyroid hormones and antibodies in obesity: Leptin levels are associated with thyroid autoimmunity independent of bioanthropometric, hormonal, and weight-related determinants. J Clin Endocrinol Metab (2010) 95(8):3965–72. doi: 10.1210/jc.2009-2798
91. Zhang Y, Scarpace PJ. The role of leptin in leptin resistance and obesity. Physiol Behav (2006) 88(3):249–56. doi: 10.1016/j.physbeh.2006.05.038
92. Laclaustra M, Moreno-Franco B, Lou-Bonafonte JM, Mateo-Gallego R, Casasnovas JA, Guallar-Castillon P, et al. Impaired sensitivity to thyroid hormones is associated with diabetes and metabolic syndrome. Diabetes Care (2019) 42(2):303–10. doi: 10.2337/dc18-1410
93. Duntas LH, Orgiazzi J, Brabant G. The interface between thyroid and diabetes mellitus. Clin Endocrinol (Oxf) (2011) 75(1):1–9. doi: 10.1111/j.1365-2265.2011.04029.x
94. Ignacio-Souza LM, Bombassaro B, Pascoal LB, Portovedo MA, Razolli DS, Coope A, et al. Defective regulation of the ubiquitin/proteasome system in the hypothalamus of obese male mice. Endocrinology (2014) 155(8):2831–44. doi: 10.1210/en.2014-1090
95. Van Der Spek AH, Fliers E, Boelen A. Thyroid hormone and deiodination in innate immune cells. Endocrinol (2021) 162(1):1–15. doi: 10.1210/endocr/bqaa200
96. de Hoogd S, Välitalo PAJ, Dahan A, van Kralingen S, Coughtrie MMW, van Dongen EPA, et al. Influence of morbid obesity on the pharmacokinetics of morphine, morphine-3-Glucuronide, and morphine-6-Glucuronide. Clin Pharmacokinet (2017) 56(12):1577–87. doi: 10.1007/s40262-017-0544-2
97. Santini F, Pinchera A, Marsili A, Ceccarini G, Castagna MG, Valeriano R, et al. Lean body mass is a major determinant of levothyroxine dosage in the treatment of thyroid diseases. J Clin Endocrinol Metab (2005) 90(1):124–7. doi: 10.1210/jc.2004-1306
98. Juiz-valiña P, Cordido M, Outeiriño-blanco E, Pértega S, Urones P, García-brao MJ, et al. Evaluation of thyroid hormone replacement dosing in morbidly obese hypothyroid patients after bariatric surgery-induced weight loss. J Clin Med (2021) 10(16):3685. doi: 10.3390/jcm10163685
99. Dixon JB, Straznicky NE, Lambert EA, Schlaich MP, Lambert GW. Surgical approaches to the treatment of obesity. Nat Rev Gastroenterol Hepatol (2011) 8(8):429–37. doi: 10.1038/nrgastro.2011.112
100. Guan B, Chen YY, Yang J, Yang W, Cunchuan W. Effect of bariatric surgery on thyroid function in obese patients: a systematic review and meta-analysis. Obes Surg (2017) 27(12):3292–305. doi: 10.1007/s11695-017-2965-2
101. Xia MF, Chang XX, Zhu XP, Yan HM, Shi CY, Wu W, et al. Preoperative thyroid autoimmune status and changes in thyroid function and body weight after bariatric surgery. Obes Surg (2019) 29(9):2904–11. doi: 10.1007/s11695-019-03910-8
102. Pradhan AD, Manson JE, Rifai N, Buring JE, Ridker PM. C-reactive protein, interleukin 6, and risk of developing type 2 diabetes mellitus. J Am Med Assoc (2001) 286(3):327–34. doi: 10.1001/jama.286.3.327
103. Spranger J, Kroke A, Mo M, Hoffmann K, Bergmann MM. Inflammatory cytokines and the risk to develop type 2 diabetes. Diabetes (2003) 52:812–7. doi: 10.2337/diabetes.52.3.812
104. Su X, Peng D. Adipokines as novel biomarkers of cardio-metabolic disorders. Clin Chim Acta (2020) 507:31–8. doi: 10.1016/j.cca.2020.04.009
105. Valenzano A, Tartaglia N, Ambrosi A, Tafuri D, Monda M, Messina A, et al. The metabolic rearrangements of bariatric surgery: Focus on orexin-a and the adiponectin system. J Clin Med (2020) 9(10):1–11. doi: 10.3390/jcm9103327
106. Zhang C, Zhang J, Liu Z, Zhou Z. More than an anti-diabetic bariatric surgery, metabolic surgery alleviates systemic and local inflammation in obesity. Obes Surg (2018) 28(11):3658–68. doi: 10.1007/s11695-018-3400-z
107. Herrero-Aguayo V, Sáez-Martínez P, Cánovas JL, Prados-Carmona J, Alcántara-Laguna M, López F, et al. Dysregulation of components of the inflammasome machinery after bariatric surgery: Novel targets for a chronic disease. J Clin Endocrinol Metab (2021) 106(12):4917–34. doi: 10.1210/clinem/dgab586
108. Gupta G, Sharma P, Kumar P, Itagappa M. Study on subclinical hypothyroidism and its association with various inflammatory markers. J Clin Diagn Res (2015) 9(11):BC04–6. doi: 10.7860/JCDR/2015/14640.6806
109. Zhu C, Gao J, Mei F, Lu L, Zhou D, Qu S. Reduction in thyroid-stimulating hormone correlated with improved inflammation markers in Chinese patients with morbid obesity undergoing laparoscopic sleeve gastrectomy. Obes Surg (2019) 29(12):3954–65. doi: 10.1007/s11695-019-04063-4
110. Reinehr T, De Sousa G, Andler W. Hyperthyrotropinemia in obese children is reversible after weight loss and is not related to lipids. J Clin Endocrinol Metab (2006) 91(8):3088–91. doi: 10.1210/jc.2006-0095
111. Hu Y, Zheng J, Ye X, Song Y, Wu X. Association between elevated thyroid peroxidase antibody and abdominal fat distribution in patients with type 2 diabetes mellitus. Diabetes Metab Syndr Obes Targets Ther (2022) 15:863–71. doi: 10.2147/DMSO.S345507
112. Rotondi M, Magri F, Chiovato L. Thyroid and obesity: Not a one-way interaction. J Clin Endocrinol Metab (2011) 96(2):344–6. doi: 10.1210/jc.2010-2515
113. Kyrou I, Adesanya O, Hedley N, Wayte S, Grammatopoulos D, Thomas CL, et al. Improved thyroid hypoechogenicity following bariatric-induced weight loss in euthyroid adults with severe obesity-a pilot study. Front Endocrinol (Lausanne) (2018) 9:488. doi: 10.3389/fendo.2018.00488
114. Olefsky JM, Glass CK. Macrophages, inflammation, and insulin resistance. Annu Rev Physiol (2009) 72:219–46. doi: 10.1146/annurev-physiol-021909-135846
115. Deng T, Lyon CJ, Bergin S, Caligiuri MA, Hsueh WA. Obesity, inflammation, and cancer. Annu Rev Pathol Mech Dis (2016) 11:421–49. doi: 10.1146/annurev-pathol-012615-044359
116. Avgerinos KI, Spyrou N, Mantzoros CS, Dalamaga M. Obesity and cancer risk: Emerging biological mechanisms and perspectives. Metabol: Clin Exp (2019) 92:121–35. doi: 10.1016/j.metabol.2018.11.001
117. Guo B, Fu S, Zhang J, Liu B, Li Z. Targeting inflammasome/IL-1 pathways for cancer immunotherapy. Sci Rep (2016) 6:1–12. doi: 10.1038/srep36107
118. Wen H, Gris D, Lei Y, Jha S, Zhang L, Huang MTH, et al. Fatty acid-induced NLRP3-ASC inflammasome activation interferes with insulin signaling. Nat Immunol (2011) 12(5):408–15. doi: 10.1038/ni.2022
119. Cnop M, Foufelle F, Velloso LA. Endoplasmic reticulum stress, obesity and diabetes. Trends Mol Med (2012) 18(1):59–68. doi: 10.1016/j.molmed.2011.07.010
120. Grivennikov SI, Greten FR, Karin M. Immunity, inflammation, and cancer. Cell (2010) 140(6):883–99. doi: 10.1016/j.cell.2010.01.025
121. Ferrari SM, Fallahi P, Galdiero MR, Ruffilli I, Elia G, Ragusa F, et al. Immune and inflammatory cells in thyroid cancer microenvironment. Int J Mol Sci (2019) 20(18):1–22. doi: 10.3390/ijms20184413
122. Zhao ZG, Guo XG, Ba CX, Wang W, Yang YY, Wang J, et al. Overweight, obesity and thyroid cancer risk: A meta-analysis of cohort studies. J Int Med Res (2012) 40(6):2041–50. doi: 10.1177/030006051204000601
123. Schmid D, Ricci C, Behrens G, Leitzmann MF. Adiposity and risk of thyroid cancer: A systematic review and meta-analysis. Obes Rev (2015) 16(12):1042–54. doi: 10.1111/obr.12321
124. Kim WG, Park JW, Willingham MC, Cheng SY. Diet-induced obesity increases tumor growth and promotes anaplastic change in thyroid cancer in a mouse model. Endocrinology (2013) 154(8):2936–47. doi: 10.1210/en.2013-1128
125. Park JW, Han CR, Zhao L, Willingham MC, Cheng SY. Inhibition of stat3 activity delays obesity-induced thyroid carcinogenesis in a mouse model. Endocr Relat Cancer (2016) 23(1):53–63. doi: 10.1530/ERC-15-0417
126. Dias Lopes NM, Mendonça Lens HH, Armani A, Marinello PC, Cecchini AL. Thyroid cancer and thyroid autoimmune disease: A review of molecular aspects and clinical outcomes. Pathol Res Pract (2020) 216(9)153098. doi: 10.1016/j.prp.2020.153098
127. Feldt-Rasmussen U. Hashimoto’s thyroiditis as a risk factor for thyroid cancer. Curr Opin Endocrinol Diabetes Obes (2020) 27(5):364–71. doi: 10.1097/MED.0000000000000570
128. Ryder M, Ghossein RA, Ricarte-Filho JCM, Knauf JA, Fagin JA. Increased density of tumor-associated macrophages is associated with decreased survival in advanced thyroid cancer. Endocr Relat Cancer (2008) 15(4):1069–74. doi: 10.1677/ERC-08-0036
129. Zhou D, Li Z, Bai X. Braf v600e and RET/PTC promote the activity of nuclear factor-κb, inflammatory mediators, and lymph node metastasis in papillary thyroid carcinoma: A study of 50 patients in inner mongolia. Med Sci Monit (2018) 24:6795–808. doi: 10.12659/MSM.909205
130. Feng C, Gao Y, Wang C, Yu X, Zhang W, Guan H, et al. Aberrant overexpression of pyruvate kinase m2 is associated with aggressive tumor features and the BRAF mutation in papillary thyroid cancer. J Clin Endocrinol Metab (2013) 98(9):1–10. doi: 10.1210/jc.2012-4258
131. Xie M, Yu Y, Kang R, Zhu S, Yang L, Zeng L, et al. PKM2-dependent glycolysis promotes NLRP3 and AIM2 inflammasome activation. Nat Commun (2016) 7:1–13. doi: 10.1038/ncomms13280
132. Altieri B, Muscogiuri G, Barrea L, Mathieu C, Vallone CV, Mascitelli L, et al. Does vitamin d play a role in autoimmune endocrine disorders? a proof of concept. Rev Endocr Metab Disord (2017) 18(3):335–46. doi: 10.1007/s11154-016-9405-9
133. Rao Z, Chen X, Wu J, Xiao M, Zhang J, Wang B, et al. Vitamin d receptor inhibits NLRP3 activation by impeding its BRCC3-mediated deubiquitination. Front Immunol (2019) 10. doi: 10.3389/fimmu.2019.02783
134. Sadeghi K, Wessner B, Laggner U, Ploder M, Tamandl D, Friedl J, et al. Vitamin D3 down-regulates monocyte TLR expression and triggers hyporesponsiveness to pathogen-associated molecular patterns. Eur J Immunol (2006) 36(2):361–70. doi: 10.1002/eji.200425995
135. Chao G, Zhu Y, Fang L. Correlation between hashimoto’s thyroiditis–related thyroid hormone levels and 25-hydroxyvitamin d. Front Endocrinol (Lausanne) (2020) 11:4. doi: 10.3389/fendo.2020.00004
136. Martini LA, Wood RJ. Vitamin d status and the metabolic syndrome. Nutr Rev (2006) 64(11):479–86. doi: 10.1111/j.1753-4887.2006.tb00180.x
137. Green TJ, Skeaff CM, Rockell JEP, Venn BJ, Lambert A, Todd J, et al. Vitamin d status and its association with parathyroid hormone concentrations in women of child-bearing age living in Jakarta and Kuala Lumpur. Eur J Clin Nutr (2008) 62(3):373–8. doi: 10.1038/sj.ejcn.1602696
138. Alkharfy KM, Al-Daghri NM, Yakout SM, Hussain T, Mohammed AK, Krishnaswamy S. Influence of vitamin d treatment on transcriptional regulation of insulin-sensitive genes. Metab Syndr Relat Disord (2013) 11(4):283–8. doi: 10.1089/met.2012.0068
139. Tsugawa Y, Jena AB, Orav EJ, Blumenthal DM, Tsai TC, Mehtsun WT, et al. Age and sex of surgeons and mortality of older surgical patients: observational study. BMJ (2018) 361:k1343. doi: 10.1136/bmj.k1343
140. Talaei A, Mohamadi M, Adgi Z. The effect of vitamin d on insulin resistance in patients with type 2 diabetes. Diabetol Metab Syndr (2013) 5(8). doi: 10.1186/1758-5996-5-8
141. Galușca D, Popoviciu MS, Babeș EE, Vidican M, Zaha AA, Babeș VV, et al. Vitamin d implications and effect of supplementation in endocrine disorders: Autoimmune thyroid disorders (Hashimoto’s disease and grave’s disease), diabetes mellitus and obesity. Med (2022) 58(2):194. doi: 10.3390/medicina58020194
Keywords: obesity, inflammasome, thyroid autoimmunity, weight regain, thyroid cancer, Lt-4 therapy
Citation: Le Moli R, Vella V, Tumino D, Piticchio T, Naselli A, Belfiore A and Frasca F (2022) Inflammasome activation as a link between obesity and thyroid disorders: Implications for an integrated clinical management. Front. Endocrinol. 13:959276. doi: 10.3389/fendo.2022.959276
Received: 01 June 2022; Accepted: 03 August 2022;
Published: 19 August 2022.
Edited by:
Marian Elizabeth Ludgate, Cardiff University, United KingdomReviewed by:
Sandra Incerpi, Roma Tre University, ItalyLeonidas H Duntas, National University Of Athens, Greece
Copyright © 2022 Le Moli, Vella, Tumino, Piticchio, Naselli, Belfiore and Frasca. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Rosario Le Moli, cmxlbW9saUB1bmljdC5pdA==