
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Endocrinol., 18 August 2022
Sec. Pediatric Endocrinology
Volume 13 - 2022 | https://doi.org/10.3389/fendo.2022.953707
 Jenifer P. Suntharalingham1
Jenifer P. Suntharalingham1 Miho Ishida1
Miho Ishida1 Ignacio Del Valle1
Ignacio Del Valle1 Susanne E. Stalman2Nita Solanky1Emma Wakeling3Gudrun E. Moore1
Susanne E. Stalman2Nita Solanky1Emma Wakeling3Gudrun E. Moore1 John C. Achermann1*†
John C. Achermann1*† Federica Buonocore1*†
Federica Buonocore1*†Background: Heterozygous de novo variants in SAMD9 cause MIRAGE syndrome, a complex multisystem disorder involving Myelodysplasia, Infection, Restriction of growth, Adrenal hypoplasia, Genital phenotypes, and Enteropathy. The range of additional clinical associations is expanding and includes disrupted placental development, poor post-natal growth and endocrine features. Increasingly, milder phenotypic features such as hypospadias in small for gestational age (SGA) boys and normal adrenal function are reported. Some children present with isolated myelodysplastic syndrome (MDS/monosomy 7) without MIRAGE features.
Objective: We aimed to investigate: 1) the range of reported SAMD9 variants, clinical features, and possible genotype-phenotype correlations; 2) whether SAMD9 disruption affects placental function and leads to pregnancy loss/recurrent miscarriage (RM); 3) and if pathogenic variants are associated with isolated fetal growth restriction (FGR).
Methods: Published data were analyzed, particularly reviewing position/type of variant, pregnancy, growth data, and associated endocrine features. Genetic analysis of SAMD9 was performed in products of conception (POC, n=26), RM couples, (couples n=48; individuals n=96), children with FGR (n=44), SGA (n=20), and clinical Silver-Russell Syndrome (SRS, n=8), (total n=194).
Results: To date, SAMD9 variants are reported in 116 individuals [MDS/monosomy 7, 64 (55.2%); MIRAGE, 52 (44.8%)]. Children with MIRAGE features are increasingly reported without an adrenal phenotype (11/52, 21.2%). Infants without adrenal dysfunction were heavier at birth (median 1515 g versus 1020 g; P < 0.05) and born later (median 34.5 weeks versus 31.0; P < 0.05) compared to those with adrenal insufficiency. In MIRAGE patients, hypospadias is a common feature. Additional endocrinopathies include hypothyroidism, hypo- and hyper-glycemia, short stature and panhypopituitarism. Despite this increasing range of phenotypes, genetic analysis did not reveal any likely pathogenic variants/enrichment of specific variants in SAMD9 in the pregnancy loss/growth restriction cohorts studied.
Conclusion: MIRAGE syndrome is more phenotypically diverse than originally reported and includes growth restriction and multisystem features, but without adrenal insufficiency. Endocrinopathies might be overlooked or develop gradually, and may be underreported. As clinical features including FGR, severe infections, anemia and lung problems can be non-specific and are often seen in neonatal medicine, SAMD9-associated conditions may be underdiagnosed. Reaching a specific diagnosis of MIRAGE syndrome is critical for personalized management.
SAMD9 (sterile α motif domain-containing protein 9) is a single exon gene that encodes a 1,589-amino acid protein and is located on the long arm of chromosome 7 (7q21.2) in humans, next to its paralogous gene SAMD9L (SAMD9-like) in a head-to-toe orientation (1). The molecular role of SAMD9 is not fully elucidated, however it has been shown to be a cytoplasmic protein involved in viral host defense mechanisms, cell proliferation, endosomal fusion and tumor suppression (2–6).
Deleterious loss-of-function variants in SAMD9 were originally reported to be associated with normophosphatemic familial tumoral calcinosis in rare cases (OMIM: 610455) (7, 8). However, it is now well established that heterozygous gain-of-function variants in SAMD9 can be identified in children with a complex multisystem condition named MIRAGE syndrome (myelodysplasia, infection, restriction of growth, adrenal hypoplasia, genital (gonadal) phenotypes, and enteropathy) (OMIM: 617053) (9–11). Although this acronym embraces several common features, the range of additional clinical associations is expanding and includes achalasia/gastroesophageal reflux, recurrent intussusception, renal features (focal segmental glomerulus sclerosis), dysautonomia, autoinflammation, learning difficulties, and hypolacrima/corneal anesthesia, amongst other findings (12–21). Disrupted placental development has also been reported, as well as poor post-natal growth (22).
The missense gain-of-function variants causing MIRAGE syndrome result in decreased cell proliferation and growth restriction in in vitro model systems, highlighting the innate role of SAMD9 as a growth repressor. Most SAMD9 variants occur de novo, however SAMD9 patients can inherit variants from asymptomatic parents either due to germline variants or revertant mosaicism events (20, 23). Indeed, dynamic, somatic revertant mechanisms are now established as frequent genomic events in individuals with pathogenic SAMD9 variants and are often seen in the hematopoietic system/bone marrow. Here, cells with secondary changes that “remove” the primary growth repressive SAMD9 variant have a clonal growth advantage. These changes include the progressive development of monosomy 7 or deletion of 7q (containing SAMD9); secondary loss-of-function variants in SAMD9 (nonsense, frameshift, missense); or acquired uniparental disomy of the “wild-type” allele (9, 10). Secondary events usually occur in cis to ameliorate the effects of the primary gain-of-function variants in the proband and, at least in the hematopoietic lineage, serve to modify disease phenotype. However, when monosomy 7 occurs there is a risk of myelodysplastic syndrome due to loss of SAMD9, SAMD9L, GATA2 and other factors, and an additional risk of leukemia if further somatic changes occur.
To date, more than 50 patients have been reported with severe growth restriction phenotypes due to gain-of-function changes in SAMD9 (9, 10, 12–22, 24–38). Although endocrine features such as primary adrenal insufficiency and gonadal dysgenesis were originally a core part of the syndrome, it is emerging that these features may be more variable. For example, SAMD9 pathogenic variants have now been identified in individuals with hypospadias (46,XY DSD) and born small for gestational age (SGA), and increasingly in children without primary adrenal insufficiency (22). Other endocrine systems may be affected, such as the thyroid gland. Thus, the endocrine phenotype of MIRAGE syndrome is likely to be more variable than originally described and may comprise additional features not always reported.
We hypothesize that: 1) different SAMD9 variants, or variants in different domains of the protein, are associated with milder or diverse endocrine phenotypes, with a focus on adrenal insufficiency; 2) severe gain-of-function of SAMD9 may affect placental function and lead to pregnancy loss and recurrent miscarriage (RM); and that 3) pathogenic variants could be found in children with fetal growth restriction (FGR) as the predominant feature. Determining whether SAMD9 variants cause isolated growth restriction is important as these children could develop monosomy 7, endocrinopathies or immune dysfunction, and could require personalized management such as bone marrow/stem cell transplantation.
A systematic review was undertaken to identify published reports of individuals with SAMD9-associated conditions in the literature. PubMed was searched using the term “SAMD9”, up to May 2022. For this review, a particular focus was put on position and type of variant, pregnancy and growth data, and reported associated endocrine features. All published SAMD9 variant-carrying individuals included in our study and their related publication link are available here http://doi.org/10.17605/OSF.IO/WMVXY (39).
Samples were provided by the Wellbeing of Women Baby Bio Bank (BBB) (http://www.ucl.ac.uk/babybiobank) supported by University College London, Imperial College London and Wellbeing of Women (40) and the Moore Cohort with ethical approval (BBB Research Ethics Committee references: 09/H0405/30 and 09/H0405/30+5; Moore cohort reference: 2001/6029). Five different cohorts were included (Table 1): 1) Products of conception (POC) (n=26), spontaneous loss of pregnancy between 9-11 weeks gestation; 2) Recurrent miscarriage (RM) (n=96; 48 couples), couples who experienced three or more consecutive loss of pregnancies before 20 weeks gestation; 3) FGR (n=44), a fetus with growth restriction of unknown etiology in utero (41) and in this cohort having a weight less than 3rd centile (42); 4) Small for gestational age (SGA) (n=20), a baby with birthweight below the 10th percentile for gestational age (42); 5) Clinical Silver-Russell Syndrome (SRS) (n=8), based on Consensus guidelines for diagnosis (43) and all double negative for H19 hypomethylation and maternal UPD7.

Table 1 Overview of the cohorts included.
DNA was isolated from blood leukocytes and POC as previously described (40, 44, 45). DNA samples were subjected to genetic analysis using the next generation sequencing (NGS) methodologies outlined below.
SGA samples underwent whole exome sequencing (WES) using the Agilent SureSelect Human All Exon V5 kit (Agilent Technologies Inc., Santa Clara, USA) by BGI (BGI Genomics, Hong Kong, China), and were sequenced with the high-throughput sequencing platform of Complete Genomics (Complete Genomics Inc., San Jose, USA) as detailed in Stalman et al. (44).
A HaloPlex DNA targeted gene enrichment panel was designed using SureDesign software (Agilent Technologies Inc., Santa Clara, USA) to capture known and candidate genes involved in fetal growth restriction, including SAMD9. A detailed protocol has been described previously (45). In brief, FGR and POC samples were sequenced on a NextSeq sequencer (Illumina Inc., San Diego, USA) and analyzed with SureCall software (version 4.0.1.46, Agilent Technologies Inc., Santa Clara, USA) using the HaloPlex Default Method.
A Nonacus Cell3TM target NGS panel (Nonacus Ltd., Birmingham, UK) was designed to cover the coding regions of SAMD9. Nine samples from children with FGR and eight with SRS were sequenced on a MiSeq platform (Illumina Inc., San Diego, USA). Binary alignment map (BAM) files were produced by aligning FASTQ files to the GRCh38 reference genome with the Burrows-Wheeler Aligner (v0.7.17). Reads were grouped by unique molecular identifiers (UMIs) performed with fgbio (v0.4.0). Variant calling was performed with Platypus (v 0.8.1). Variant annotation was performed with Ensembl Variant Effect Predictor (VEP) and QIAGEN Clinical Insight (QCI). A detailed description has been previously reported (45, 46).
Individual variant pathogenicity was evaluated using SIFT (http://sift.jcvi.org/) and PolyPhen-2 (http://genetics.bwh.harvard.edu/pph2/), and CADD (Combined Annotation Dependent Depletion) score (https://cadd.gs.washington.edu).
For each cohort, the SAMD9 variant allele frequency was calculated by dividing the number of alleles with any specific variant by the total number of alleles sequenced. Allele frequencies were compared to data in gnomAD v2.1.1 (accessed May 2022, https://gnomad.broadinstitute.org) (47).
RNA expression of SAMD9 in a panel of adult human tissues was obtained from the Human Protein Atlas (https://www.proteinatlas.org) using the consensus RNA-Seq dataset raw counts (https://www.proteinatlas.org/ENSG00000205413-SAMD9/tissue) (48). Relative expression of SAMD9 in a panel of human fetal and adult tissue was adapted from the previous report of Buonocore et al., 2017 (10). Single cell RNA sequencing analysis of placental SAMD9 was evaluated using publicly available data at https://maternal-fetal-interface.cellgeni.sanger.ac.uk (49) and http://placenta.grid.wayne.edu/ (50).
Graphics were generated using GraphPad Prism version 8.4.3 for Windows (GraphPad Software, San Diego, USA; www.graphpad.com). Illustrations of SAMD9 protein domains were created using Domain Graph version 2.0 (51).
Birth SDS values, whenever not reported in the original article, were calculated using the PediTools Web calculator (https://peditools.org) (52).
GraphPad Prism version 8.4.3 for Windows (GraphPad Software, San Diego, USA; www.graphpad.com) was used to perform Chi-squared tests for SAMD9 variants affecting arginine residues, and Mann-Whitney U tests for birth weight, gestational age and birth weight SDS data. Fisher’s exact test was used to test SAMD9 variant allele frequency in the studied cohorts compared to those reported in gnomAD v2.1.1.
Based on a review of the literature, a total of 116 individuals with likely pathogenic variants in SAMD9 were identified. The distribution of all variants associated either with MIRAGE syndrome or MDS is shown in Figure 1A, with secondary somatic changes in SAMD9 indicated in red. Overall, 52 (44.8%) children had features of MIRAGE syndrome (including four individuals in two families), whereas 64 (55.2%) individuals presented with MDS (including five individuals in two families) (Figure 1A).
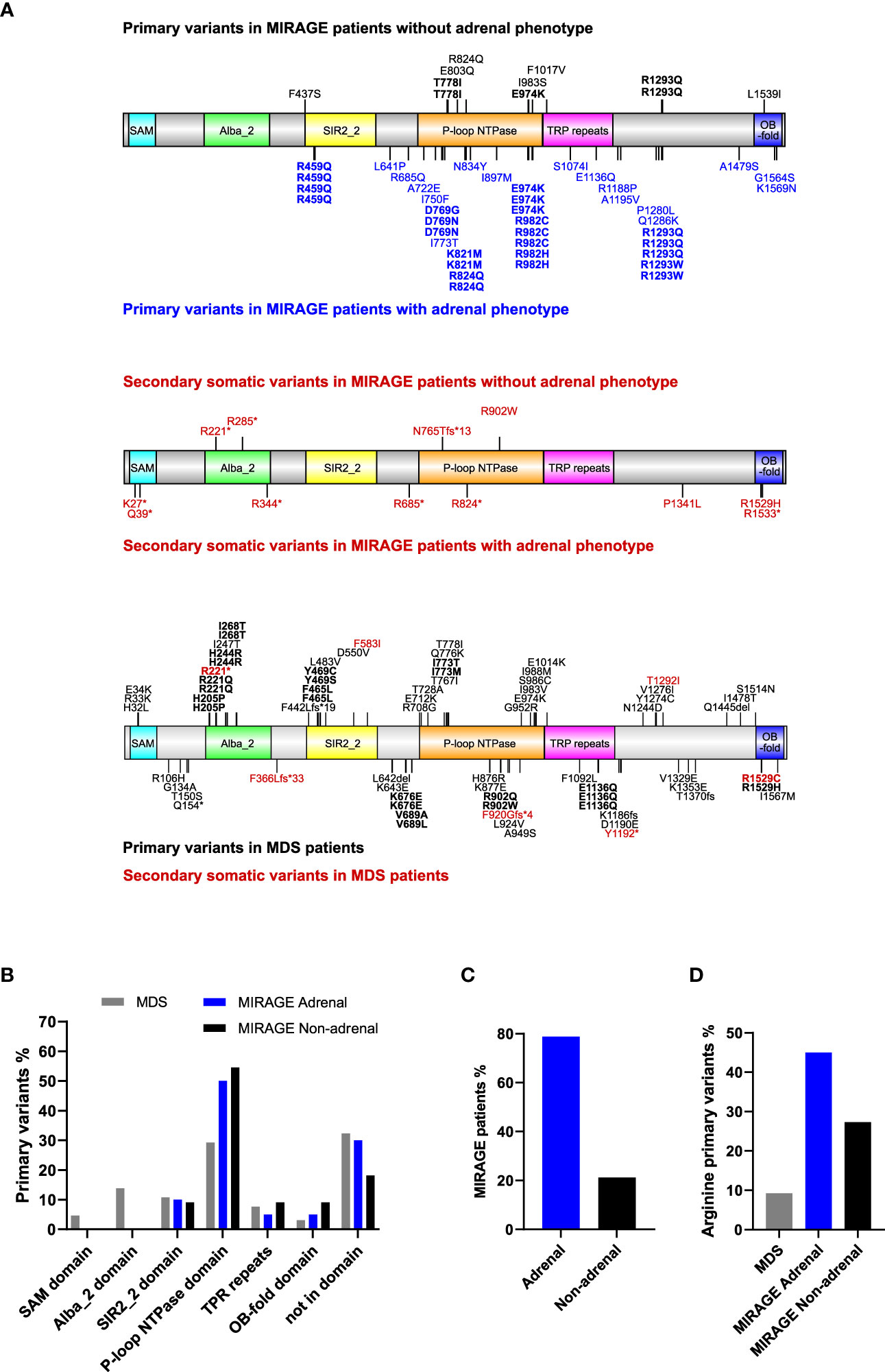
Figure 1 Overview of SAMD9 variants and relation to adrenal insufficiency. (A) Depiction of SAMD9 showing the variants reported in individuals with MIRAGE syndrome (top diagrams) and those with myelodysplastic syndrome (MDS) (bottom diagram). Predicted domains are indicated by different colors. Codons with multiple reported cases are shown in bold. Secondary somatic variants are represented in red. (B) Distribution of SAMD9 primary variants across the specific domains for individuals with MDS, and MIRAGE with or without adrenal insufficiency (Note: secondary somatic variants excluded). (C) MIRAGE patients reported to date with or without adrenal insufficiency. (D) Percentage of gain-of-function variants involving arginine residues in individuals with MDS and MIRAGE syndrome, with or without adrenal insufficiency.
MIRAGE-associated variants tend to cluster within certain regions of the SAMD9 protein, especially the P-loop NTPase domain, whereas MDS-associated variants are distributed across the protein domain structure (Figures 1A, B). Certain “hotspot” variants and codons with recurrent changes were seen in all groups (Figure 1A). Although primary adrenal insufficiency has been considered one of the main features of MIRAGE syndrome since the identification of this multisystem disorder (41/52, 78.8%), a substantial number of children with MIRAGE features are now reported without an adrenal phenotype (11/52, 21.2%) (Figure 1C). Interestingly, changes in arginine residues are more prevalent in MIRAGE patients overall compared to MDS (21/51, 41.2%) MIRAGE; 6/65, 9.2% MDS, χ 2 = 16.33, P < 0.0001), and are potentially enriched in individuals with MIRAGE syndrome who have an adrenal phenotype, but numbers are small (18/40, 45.0% MIRAGE adrenal; 3/11, 27.3% MIRAGE non-adrenal, χ 2 = 1.119, P = 0.2901) (Figure 1D).
To address whether children without an adrenal phenotype have a less severe condition in general, we used birth weight and gestational age as surrogate “markers” of phenotype severity. Infants with MIRAGE syndrome who do not have an adrenal phenotype tend to weigh more at birth (median weight 1515g, range 834 to 2002g) than those with adrenal features (median 1020g, range 464 to 1870g) (P < 0.05). They are delivered later (median age 34.5 weeks, range 30 to 37.3) compared to those having an adrenal phenotype (median 31 weeks, range 25 to 37 weeks) (P < 0.05) (Figures 2A–C). Birth weight SDS was not significantly different (adrenal phenotype, median -2.45, range -4.0 to -0.6; without adrenal phenotype, median -2.05; range -3.3 to -0.9; P = 0.34) (Figure 2D). Taken together, these data suggest that there may be a subtle spectrum of disease phenotypes, and that adrenal features are more common in very small, preterm babies with MIRAGE syndrome.
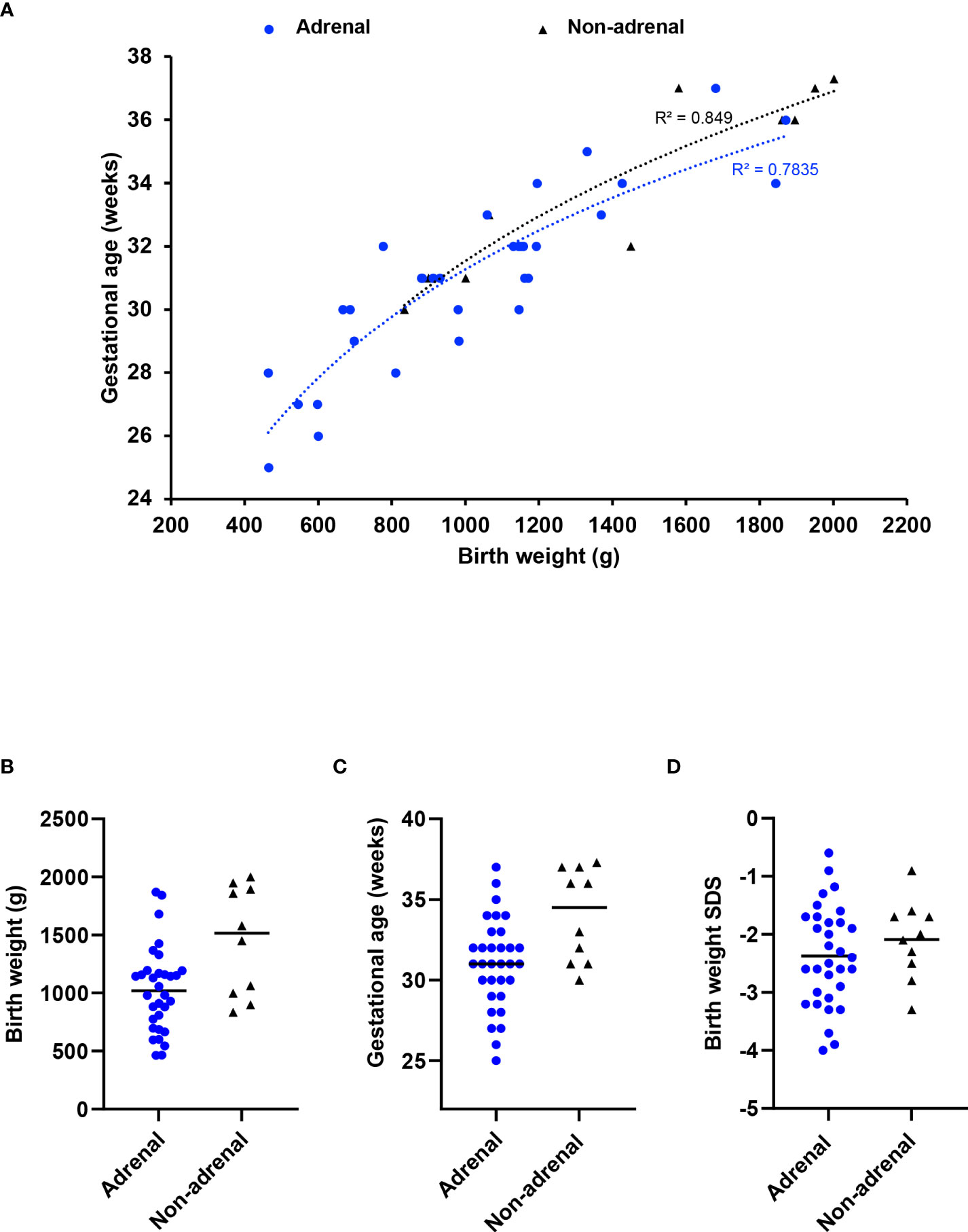
Figure 2 Relation between adrenal insufficiency and gestational age/birth weight. (A) Correlation plot between gestational age and birth weight at delivery for children with MIRAGE syndrome, with or without adrenal insufficiency. A logarithmic trendline and an R squared value for each phenotypic group is shown. Birth weight (B), gestational age in weeks (C) and birth weight standard deviation score (SDS) (D) of children with MIRAGE syndrome reported to date, with and without adrenal insufficiency, with median values shown by the horizontal line.
Since the first description of MIRAGE syndrome in 2016, there has been an increasing number of publications reporting patients with MIRAGE features with variants in SAMD9, and the phenotypic spectrum of SAMD9-related conditions has now expanded. Although adrenal insufficiency was a predominant feature in early reports, the relative proportion of children without adrenal insufficiency has increased with time (Figure 3A). Whilst 46,XY boys with hypospadias are now more common than 46,XY girls with severe gonadal dysfunction, 46,XX girls remain underreported (Figures 3B, C). Several patients have been identified who have additional endocrinopathies including hypothyroidism, hypo- and hyper-glycemia, growth hormone deficiency and panhypopituitarism, although it is often unclear how extensively most children have been investigated for endocrine disorders and some may have died before these were manifest (Figure 3D).
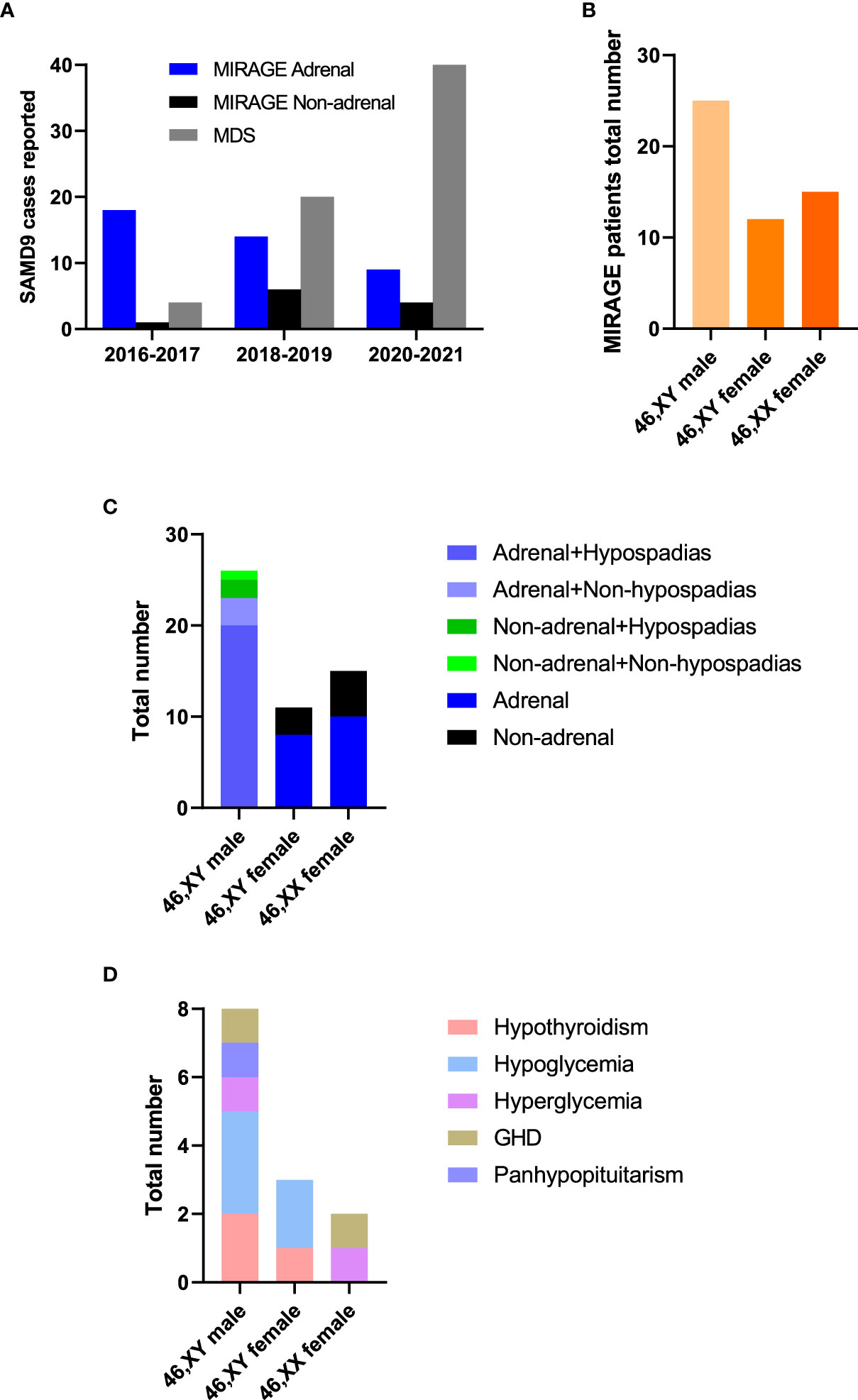
Figure 3 MIRAGE endocrine features. (A) Individuals with SAMD9 pathogenic variants reported from 2016 to 2021. (B) Total numbers of MIRAGE patients based on karyotype and reported sex assignment. “46,XY female” indicates a 46,XY child who was brought up as a girl. (C, D) Endocrine features of children with MIRAGE syndrome, where reported. Data include two reported individuals with MIRAGE syndrome who developed endocrinopathies not originally described in the publication. GHD, growth hormone deficiency.
Analysis of RNA expression in several adult and fetal human tissues shows high levels of SAMD9 in the esophagus, fetal adrenal, colon, bone marrow, thymus, lung and fetal testis (Figures 4A, B), which are tissues particularly affected in MIRAGE syndrome. FGR though, remains a common issue in MIRAGE syndrome and babies are often delivered due to severe FGR. SAMD9 is a growth repressor and could be directly implicated in the pathogenesis of growth restriction in the fetus itself or might affect the fetoplacental unit. To address this further, we investigated the expression of SAMD9 in human placenta using publicly available resources. Data from single cell RNA sequencing indicate that only a small population of annotated cells show SAMD9 expression in first trimester human placenta (Figure 5A). In a study of third trimester placenta (33-40 weeks of gestation), more extensive SAMD9 expression was seen especially in lymphoid tissues (Figure 5B). No enrichment of pathogenic nor polymorphic variants in SAMD9 was identified in DNA from early lost pregnancies (POC), or in couples who had experienced recurrent miscarriage (RM) (Table 2; Figure 6).
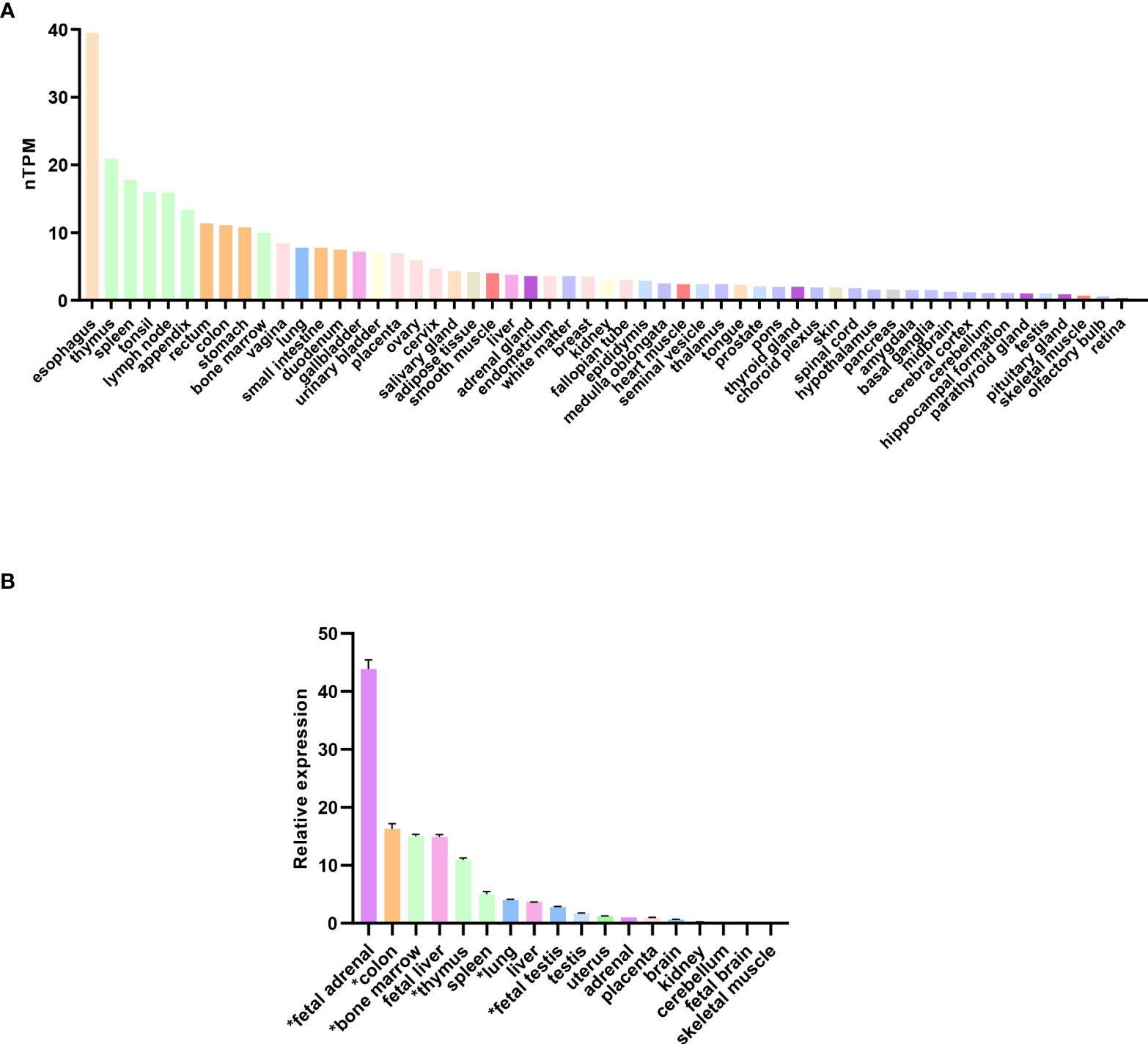
Figure 4 Expression of SAMD9 in human tissues. (A) RNA expression of SAMD9 in several adult human tissues. Data reproduced and modified with permission from the Human Protein Atlas (www.proteinatlas.org) (48). nTPM = normalized Transcripts Per Million. (B) SAMD9 expression in different human fetal and adult tissues showing high expression in fetal adrenal as well as in tissues affected in the clinical phenotype (adapted from Buonocore et al., 2017). Tissues especially relevant to the phenotype are highlighted (*). Data are shown as relative expression compared with GAPDH. Representative data are shown as mean ± SEM for a single study performed with triplicate technical replicates.
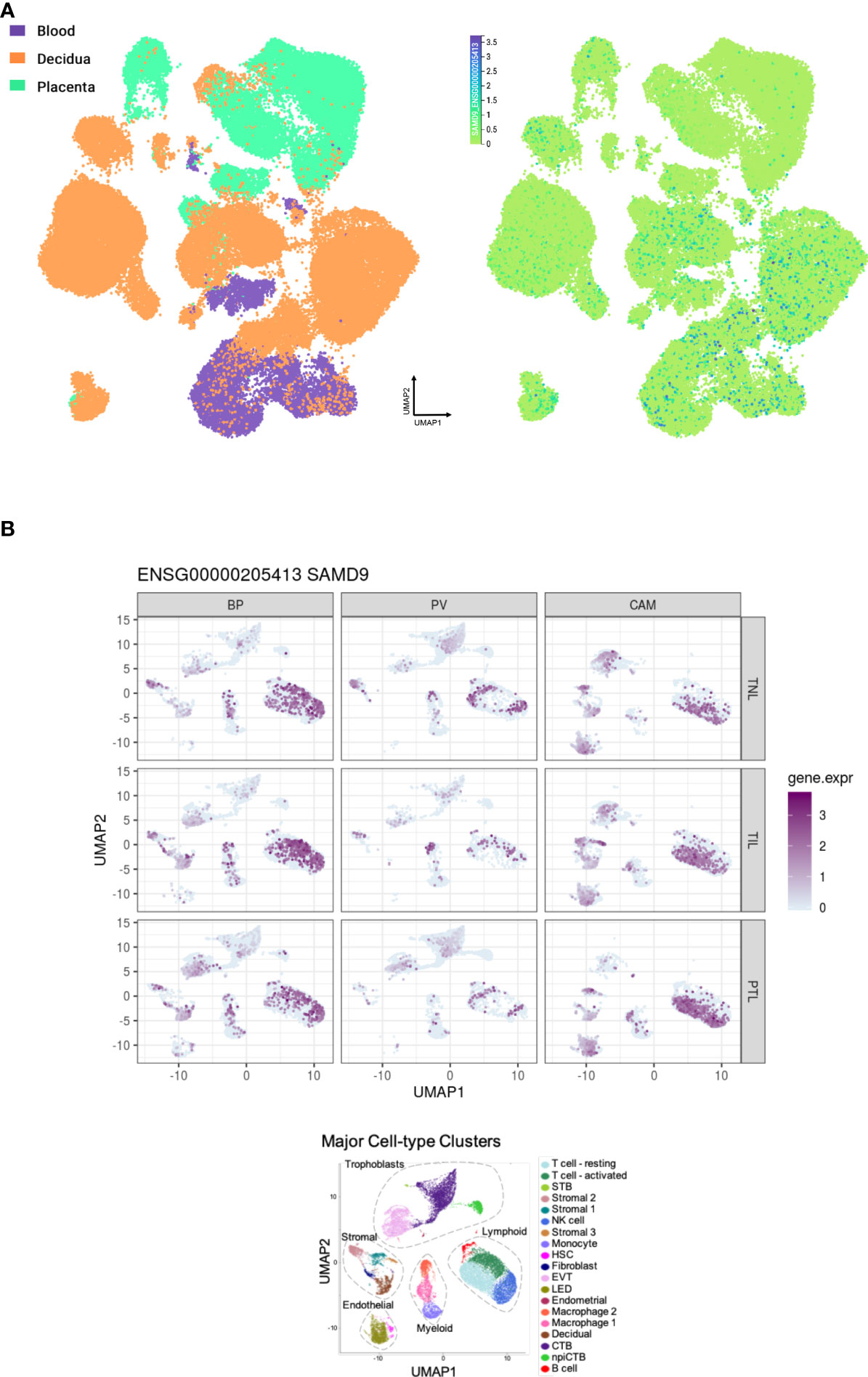
Figure 5 Expression of SAMD9 in human placenta. (A) Single cell RNA sequencing expression in first trimester placenta shown in the left panel in orange. Data accessed from https://maternal-fetal-interface.cellgeni.sanger.ac.uk/ (49). Higher color intensity is directly proportional to high levels of gene expression. (B) Expression of SAMD9 in single cell RNA data from late trimester human placenta. Highest expression of SAMD9 is in purple. Major cell-type clusters are indicated by different colors in the bottom panel. Reproduced from Pique-Regi et al., 2019. BP, Basal Plate; PV, Placental Villi; CAM, Chorioamniotic membranes; TNL, Term No Labor; TIL, Term In Labor; PTL, Preterm Labor; UMAP, Uniform manifold approximation and projection. Data accessed from http://placenta.grid.wayne.edu/ (50).
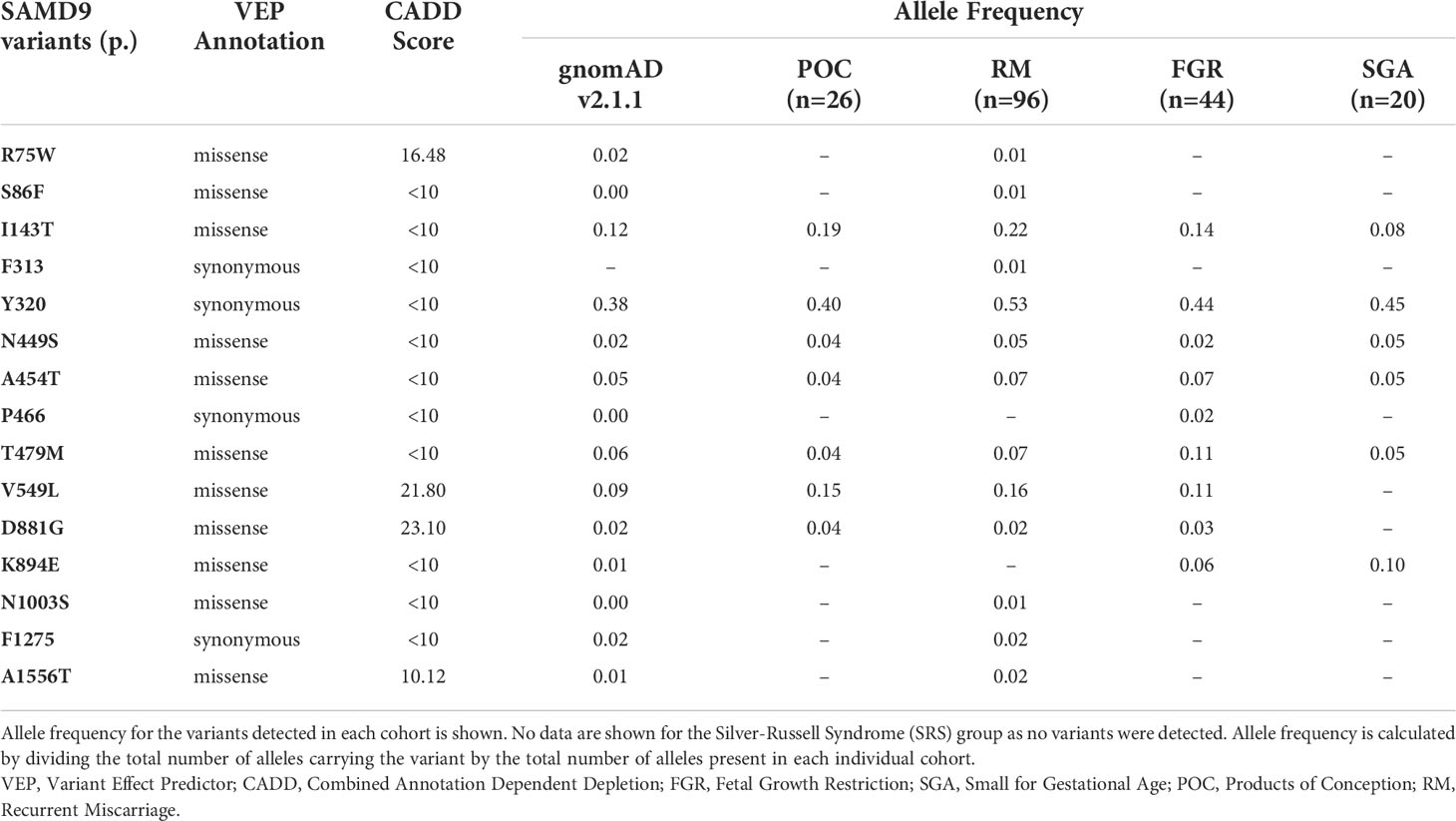
Table 2 SAMD9 allele frequency in the study cohorts.
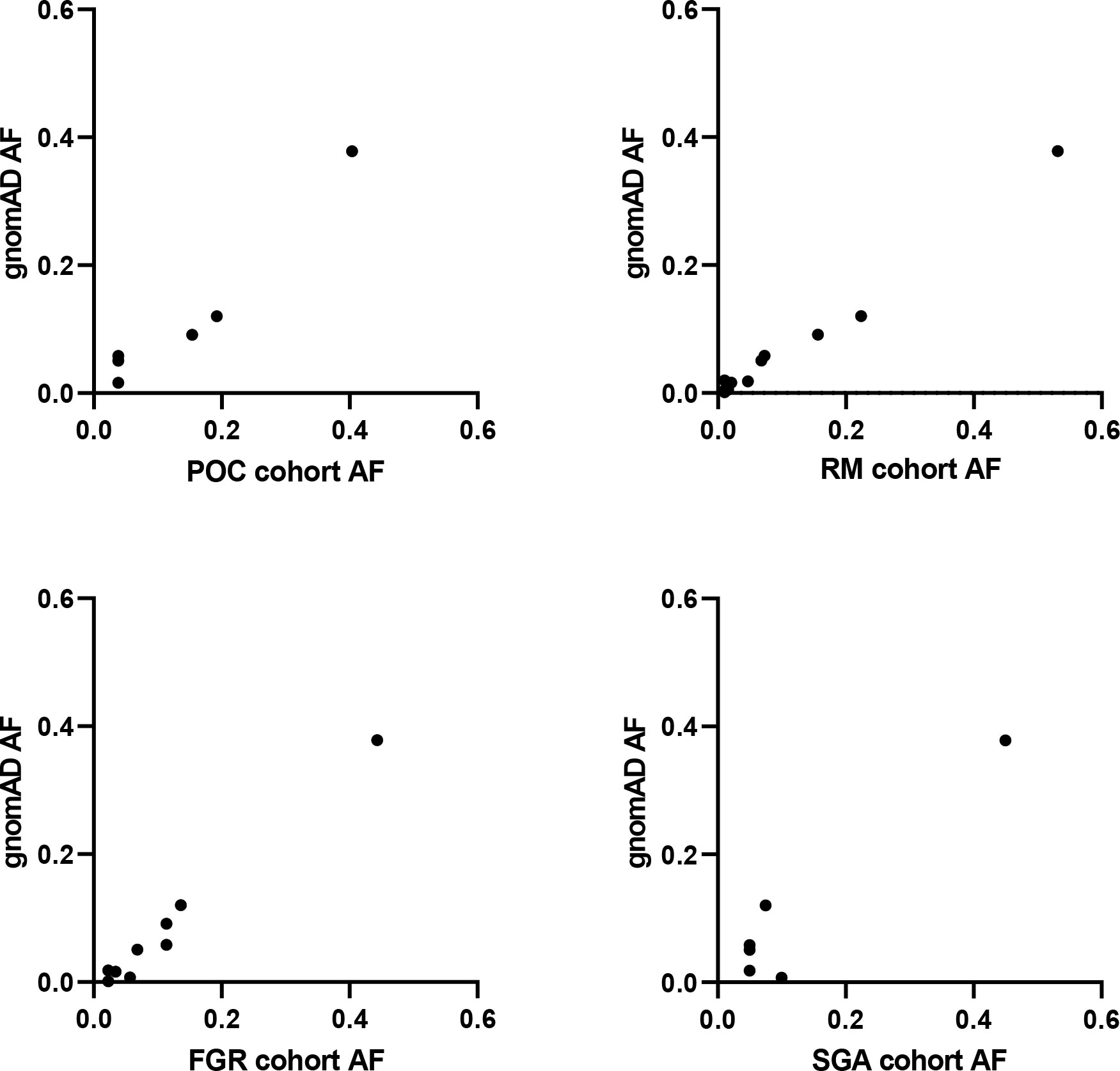
Figure 6 SAMD9 variants in studied cohorts. Correlation plots between allele frequency (AF) of SAMD9 variants detected in our cohorts and that present in gnomAD. No data are shown for Silver-Russell Syndrome (SRS) as no variants in SAMD9 were detected. POC, Product of Conception; RM, Recurrent Miscarriage; FGR, Fetal Growth Restriction; SGA, Small for Gestational Age.
Given the range of phenotypes associated with SAMD9 that have now been reported, we investigated the potential role of SAMD9 variants in a mixed cohort of children with FGR, SGA, and SRS (Table 1). Samples were subjected to targeted NGS and WES, with the aim of identifying any potential pathogenic variants and/or any enrichment of rare variants in SAMD9 in any of the groups. We did not find any predicted pathogenic variants in the cohorts studied, and we did not observe any significant differences in SAMD9 variant allele frequencies in our cohorts compared to those reported in gnomAD (Fisher’s exact test, all analyses P = 1) (Table 2 and Figure 6).
MIRAGE defines a multisystem disorder caused by heterozygous missense variants in SAMD9, which is located on the long arm of chromosome 7. These variants lead to a gain-of-function of the growth repressor SAMD9, causing tissue hypoplasia and growth restriction. The natural history of the condition can be modified in the hematopoietic system at least by revertant somatic rescue mechanisms that can occur in cis to “remove” the affected allele. These events include somatic nonsense or frameshift variants, often only present in a small proportion of sequencing reads, and progressive loss of chromosome 7 or more specifically 7q, where SAMD9 is located. Cells carrying the mutated SAMD9, which then develop the secondary somatic changes, have a clonal advantage allowing partial rescue. However, this can lead to the development of myelodysplasia in the hematopoietic system secondary to the loss of chromosome 7.
As is often the situation when new clinical associations are identified, the first MIRAGE patients reported exhibited the most severe phenotypic features. Many children died in the first few months of life or in utero. More recently additional children with MIRAGE have been described, many of whom have more variable or milder features, and an even larger cohort of children have been identified who present with MDS. Interestingly, MIRAGE phenotypic features have also recently been described in two patients carrying SAMD9L variants, which are associated with MDS and in some cases Ataxia pancytopenia (OMIM:159550) and CANDLE phenotypes (37). Here we show that children with MIRAGE features and growth restriction are more likely to have recurrent or “hotspot” variants compared to children with MDS, and more likely affecting amino acids in the P-loop NTPase domain or arginine residues, suggesting a potential emerging genotype-phenotype correlation. Of note, a SAMD9/9L effector domain (codons 134-385, which include the Alba domain) in association with DNA has recently been crystalized (53), but this structure does not contain most of the key hotspot codons relevant to MIRAGE syndrome.
Adrenal hypoplasia has been one of the key features of MIRAGE syndrome, with most children initially described having an adrenal phenotype. As more cases have been reported, it has emerged that an increasing number of patients do not have an adrenal phenotype, as we have shown here. In general, early adrenal dysfunction is associated with a lower birthweight and lower gestational age, as well as greater degree of gonadal dysfunction as indicated by the genital phenotype. Whether adrenal insufficiency contributes to prenatal fetal distress is unclear. Therefore, a spectrum of phenotypic severity seems to be emerging. This range of features could be linked to the underlying genetic variant; for example, on the limited data available, changes in codon 982 (p.R982C, p.R982H) seem to associate with adrenal dysfunction, whereas two children with changes in the neighboring codon (p.I983S) did not have adrenal features. More data are needed in this regard. Alternatively, some children without adrenal insufficiency could have adrenal rescue mechanisms such as somatic revertant mosaicism in progenitor cells that repopulate the gland under ACTH stimulation, but this theory is difficult to prove given the challenges in accessing adrenal tissue for investigation. On a practical level, adrenal features may be masked if a child receives steroids for another reason (e.g., chronic lung disease or infection), although to our knowledge this was not the situation in the children described here. Finally, whether any of these individuals develop adrenal insufficiency with time is unknown, so clinical vigilance and potentially periodic screening of adrenal function may be warranted.
The association between growth restriction and primary adrenal insufficiency is not limited to MIRAGE syndrome. Similar growth restriction syndromes characterized by adrenal insufficiency, FGR and other features, include IMAGe syndrome (intrauterine growth restriction, metaphyseal dysplasia, adrenal hypoplasia, genitourinary anomalies), caused by gain-of-function variants in the cell-cycle repressor CDKN1C (54, 55), and “IMAGe-like” syndrome with immunodeficiency, caused by biallelic variants in the DNA polymerase POLE1 (56). To date, current UK data suggest that SAMD9-related conditions have not been found in children with primary adrenal insufficiency of unknown etiology without prematurity or growth restriction (57).
In addition to the well-described gonadal defects that can affect both the testis and ovary (15), a range of additional endocrine features have been reported with MIRAGE syndrome and SAMD9 disruption. These include glucose dysregulation, hypothyroidism, and hypothalamo-pituitary issues such as growth hormone insufficiency and panhypopituitarism. Sometimes these features may be influenced by the clinical status of the child (especially in pre-term babies) or by hypocortisolemia. However, it does seem that additional endocrine features can occur, and these may well be underreported if they are not regularly assessed or if children do not survive long enough for them to manifest. More long-term data are needed in this regard, including whether puberty is affected in MIRAGE syndrome, and how best to optimize growth potential throughout childhood and adolescence.
As prenatal and postnatal growth issues are common in MIRAGE patients, and infants are often delivered early due to severe FGR, we investigated a possible link between SAMD9 and pregnancy loss. Data from single cell RNA sequencing show low levels of SAMD9 expression in first trimester human placenta, suggesting that it is unlikely to be a driver of growth restriction via a placental mechanism during that time, but rather via a fetal specific system. However, higher expression was seen later in pregnancy, and histological evaluation of placenta tissues from SAMD9 patients showed poorly developed distal villous trees with widening of the intervillous space, suggesting abnormal development (22). These findings could indicate a role of SAMD9 in both the placenta and fetus, having therefore a dual function in prenatal growth disruption. In our analysis of a limited number of available samples, we did not identify likely pathogenic SAMD9 variants in early POC tissue (spontaneous pregnancy loss), nor in genomic DNA of couples who had early recurrent miscarriages (RM). Mosaic placental mechanisms occurring de novo in the fetus could not be excluded. This contrasts with CDKN1C, which shows marked expression in first trimester placental tissue, highlighting a potential role for fetal growth and survival (45). Taken together, the higher placental expression of SAMD9 in later pregnancy coupled with evidence of abnormal placental development in affected patients, could be contributing factors to poor growth and fetal distress seen in MIRAGE syndrome, and a reason these babies are delivered early.
SAMD9 variants can be found in children born SGA (22) and interestingly, they can be inherited from asymptomatic parents, exhibiting different rescue mechanisms from offspring (20). This led us to analyze SAMD9 in samples from a cohort of children with FGR, SGA and SRS, to investigate whether potential predicted pathogenic variants could be found that could explain the growth restriction phenotype. We did not find any primary or secondary somatic variants, nor did we observe an enrichment of more common variants in SAMD9 in the cohort studied, suggesting that SAMD9 is not a frequent driver of growth disorders unless other features are present.
This study has several important implications. Establishing the range of endocrinopathies in MIRAGE syndrome is essential to reach a quick diagnosis and tailor patient treatment, as adrenal insufficiency is a life-threatening condition and thyroid hormone imbalance can affect brain development and growth. It is also important to define and treat other features, such as dry eyes, feeding difficulties and esophageal reflux, periodic fever, immune dysfunction and gastrointestinal effects, amongst other considerations (11). Establishing a diagnosis of a SAMD9-related condition enables monitoring of monosomy 7, as a secondary somatic event to remove the primary driver, and consequent potential MDS risk. Finally, the features of MIRAGE syndrome can be very non-specific and often seen in sick babies in neonatal medicine delivered prematurely with growth restriction. Raising the awareness of this condition and having greater access to rapid genetic sequencing could be essential to identify children who harbor pathogenic SAMD9 variants, and to identify some of the “missing” cohort of 46,XX girls with this condition. Indeed, more widespread use of whole exome/genome sequencing for children with growth restriction and associated features is likely to identify more children with MIRAGE-associated conditions and provide more information on the range of phenotypic features.
This study has several limitations: 1) Following the literature review, several case reports lacked complete information such as BW and GA, especially for patients carrying SAMD9 variants in the MDS cohort. Furthermore, detailed genetic studies of genotype-phenotype co-segregation were not always described in MDS patients, and functional work is limited suggesting that some false positives may have been reported. 2) As discussed above, endocrine features may also be overlooked and not reported, or they may develop later with time. There is therefore a need for a life-course data analysis to be able to capture the full range of phenotypes associated with SAMD9 variants. 3) Due to a relatively small cohort of miscarriage tissues and RM couples, we might have missed de novo variants, but potentially parental germline mosaicism would be enriched, and this is important as there are several sibling pairs with SAMD9 variants reported. 4) The FGR cohort analyzed in this study was relatively small. Analysis therefore does not exclude SAMD9 as a potential pathogenic gene in this cohort, but suggests it is not a common occurrence.
SAMD9-associated variants comprise a wider spectrum of phenotypes than originally reported, including children with neonatal growth restriction and multisystem features but without adrenal insufficiency. Endocrinopathies are also emerging, though these are possibly overlooked. Indeed, as these patients may present to a diverse range of health care professionals, and with features beyond those in the classic MIRAGE acronym, perhaps referring to “SAMD9-related syndromes” in the future may be more appropriate. We have also shown that SAMD9 variants are not common in fetal growth disorders unless additional features are present. Nevertheless, monitoring severe FGR children remains essential and has important implications in reaching a specific diagnosis for personalized multidisciplinary management.
The original contributions presented in the study are publicly available and can be found here http://doi.org/10.17605/OSF.IO/WMVXY (39). These data include an overview of SAMD9 publications and linked reported variants, and a summary of all SAMD9 variants identified using the HaloPlex, Nonacus and WES platforms.
The studies involving human participants were reviewed and approved by BBB Research Ethics Committee references: 09/H0405/30 and 09/H0405/30+5; Moore cohort reference: 2001/6029. Written informed consent to participate in this study was provided by the participants’ legal guardian/next of kin.
JS and FB designed the targeted NGS panels. JS carried out NGS experiments. MI and ID conducted bioinformatic pipeline analysis. SS provided variants for the SGA samples. NS coordinated samples and clinical data from the BBB. EW contributed to the clinical evaluation of the article. GM is the founder of the BBB and Moore Cohort and provided all the samples and conceptual input. JA and FB undertook literature reviews, conceptualized, and wrote the article. FB conducted the molecular analysis, produced figures and tables. All authors contributed to manuscript revision, read, and approved the submitted version.
This research was funded in whole, or in part, by the Wellcome Trust (grant 209328/Z/17/Z). For the purpose of Open Access, the author has applied a CC BY public copyright license to any Author Accepted Manuscript version arising from this submission. JA also has research support from Great Ormond Street Hospital Children’s Charity (grant V2518) and the National Institute for Health Research, Great Ormond Street Hospital Biomedical Research Centre (grant IS-BRC-1215-20012). The views expressed are those of the authors and not necessarily those of the National Health Service, National Institute for Health Research, or Department of Health.
We would like to thank the Wellbeing of Women Baby Bio Bank (BBB) (University College London, Imperial College London Wellbeing of Women) for providing the POC, RM, FGR, SGA and SRS samples.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Li CF, MacDonald JR, Wei RY, Ray J, Lau K, Kandel C, et al. Human sterile alpha motif domain 9, a novel gene identified as down-regulated in aggressive fibromatosis, is absent in the mouse. BMC Genomics (2007) 8:92. doi: 10.1186/1471-2164-8-92
2. Liu J, McFadden G. SAMD9 is an innate antiviral host factor with stress response properties that can be antagonized by poxviruses. J Virol (2015) 89:1925–31. doi: 10.1128/JVI.02262-14
3. Sivan G, Ormanoglu P, Buehler EC, Martin SE, Moss B. Identification of restriction factors by human genome-wide RNA interference screening of viral host range mutants exemplified by discovery of SAMD9 and WDR6 as inhibitors of the vaccinia virus K1L-C7L-mutant. MBio (2015) 6:e01122–15. doi: 10.1128/mBio.01122-15
4. Meng X, Krumm B, Li Y, Deng J, Xiang Y. Structural basis for antagonizing a host restriction factor by C7 family of poxvirus host-range proteins. Proc Natl Acad Sci (2015) 112:14858–63. doi: 10.1073/pnas.1515354112
5. Nagamachi A, Matsui H, Asou H, Ozaki Y, Aki D, Kanai A, et al. Haploinsufficiency of SAMD9L, an endosome fusion facilitator, causes myeloid malignancies in mice mimicking human diseases with monosomy 7. Cancer Cell (2013) 24:305–17. doi: 10.1016/j.ccr.2013.08.011
6. Ma Q, Yu T, Ren Y-Y, Gong T, Zhong D-S. Overexpression of SAMD9 suppresses tumorigenesis and progression during non small cell lung cancer. Biochem Biophys Res Commun (2014) 454:157–61. doi: 10.1016/J.BBRC.2014.10.054
7. Topaz O, Indelman M, Chefetz I, Geiger D, Metzker A, Altschuler Y, et al. A deleterious mutation in SAMD9 causes normophosphatemic familial tumoral calcinosis. Am J Hum Genet (2006) 79:759–64. doi: 10.1086/508069
8. Chefetz I, Ben Amitai D, Browning S, Skorecki K, Adir N, Thomas MG, et al. Normophosphatemic familial tumoral calcinosis is caused by deleterious mutations in SAMD9, encoding a TNF-α responsive protein. J Invest Dermatol (2008) 128:1423–9. doi: 10.1038/sj.jid.5701203
9. Narumi S, Amano N, Ishii T, Katsumata N, Muroya K, Adachi M, et al. SAMD9 mutations cause a novel multisystem disorder, MIRAGE syndrome, and are associated with loss of chromosome 7. Nat Genet (2016) 48:792–7. doi: 10.1038/ng.3569
10. Buonocore F, Kühnen P, Suntharalingham JP, Del Valle I, Digweed M, Stachelscheid H, et al. Somatic mutations and progressive monosomy modify SAMD9-related phenotypes in humans. J Clin Invest (2017) 127:1700–13. doi: 10.1172/JCI91913
11. Tanase-Nakao K, Olson TS, Narumi S. MIRAGE syndrome. In: Adam MP, Ardinger HH, Pagon RA, editors. GeneReviews®. Seattle (WA: University of Washington (2020). p. 1993–2022.
12. Baquedano-Lobera I, Romero-Salas Y, Ros-Arnal I, Miramar-Gallart MD, López-Pisón J, Corona-Bellostas C, et al. Achalasia as a symptom guide in MIRAGE syndrome: A novel case with p.R1293Q and p.R902W variants in the SAMD9 gene. Clin Genet (2021), 99:740–1. doi: 10.1111/cge.13914
13. Chin X, Sreedharan AV, Tan EC, Wei H, Kuan JL, Ho CWW, et al. MIRAGE syndrome caused by a de novo c.3406G>C (p. Glu1136Gln) mutation in the SAMD9 gene presenting with neonatal adrenal insufficiency and recurrent intussusception: A case report. Front Endocrinol (Lausanne) (2021) 0:742495. doi: 10.3389/FENDO.2021.742495
14. Ishiwa S, Kamei K, Tanase-Nakao K, Shibata S, Matsunami K, Takeuchi I, et al. A girl with MIRAGE syndrome who developed steroid-resistant nephrotic syndrome: A case report. BMC Nephrol (2020) 21:340. doi: 10.1186/s12882-020-02011-4
15. Sarthy J, Zha J, Babushok D, Shenoy A, Fan J-M, Wertheim G, et al. Poor outcome with hematopoietic stem cell transplantation for bone marrow failure and MDS with severe MIRAGE syndrome phenotype. Blood Adv (2018) 2:120–5. doi: 10.1182/bloodadvances.2017012682
16. Rentas S, Pillai V, Wertheim GB, Akgumus GT, Nichols KE, Deardorff MA, et al. Evolution of histomorphologic, cytogenetic, and genetic abnormalities in an untreated patient with MIRAGE syndrome. Cancer Genet (2020) 245:42–8. doi: 10.1016/j.cancergen.2020.06.002
17. Yoshizaki K, Hachiya R, Tomobe Y, Kaku U, Akiba K, Shima H, et al. MIRAGE syndrome with recurrent pneumonia probably associated with gastroesophageal reflux and achalasia: A case report. Clin Pediatr Endocrinol (2019) 28:147–53. doi: 10.1297/cpe.28.147
18. Shima H, Koehler K, Nomura Y, Sugimoto K, Satoh A, Ogata T, et al. Two patients with MIRAGE syndrome lacking haematological features: Role of somatic second-site reversion SAMD9 mutations. J Med Genet (2018) 55:81–5. doi: 10.1136/jmedgenet-2017-105020
19. Okafor D, AlRabadi L, Alper A, Jeffries L, McGrath J, Porto AF. Refractory infantile chronic diarrhea and failure to thrive in a 6-month-old boy with a complex past medical history. Clin Pediatr (Phila) (2019) 58:707–10. doi: 10.1177/0009922819832034
20. Roucher-Boulez F, Mallet D, Chatron N, Dijoud F, Gorduza DB, Bretones P, et al. Reversion SAMD9 mutations modifying phenotypic expression of MIRAGE syndrome and allowing inheritance in a usually de novo disorder. Front Endocrinol (Lausanne) (2019) 10:625. doi: 10.3389/fendo.2019.00625
21. Basilious A, Basilious A, ElJalbout R, Robert M-C. Lacrimal gland hypoplasia and corneal anesthesia in MIRAGE syndrome. Cornea (2021) 00:1–4. doi: 10.1097/ICO.0000000000002900
22. Shima H, Hayashi M, Tachibana T, Oshiro M, Amano N, Ishii T, et al. MIRAGE syndrome is a rare cause of 46,XY DSD born SGA without adrenal insufficiency. PLoS One (2018) 13:e0206184. doi: 10.1371/journal.pone.0206184
23. Schwartz JR, Wang S, Ma J, Lamprecht T, Walsh M, Song G, et al. Germline SAMD9 mutation in siblings with monosomy 7 and myelodysplastic syndrome. Leukemia (2017) 31:1827–30. doi: 10.1038/leu.2017.142
24. Wilson DB, Bessler M, Ferkol TW, Shenoy S, Amano N, Ishii T, et al. Comment on: Acquired monosomy 7 myelodysplastic syndrome in a child with clinical features of dyskeratosis congenita and IMAGe association. Pediatr Blood Cancer (2018) 65:e26747. doi: 10.1002/pbc.26747
25. Jeffries L, Shima H, Ji W, Panisello-Manterola D, McGrath J, Bird LM, et al. A novel SAMD9 mutation causing MIRAGE syndrome: An expansion and review of phenotype, dysmorphology, and natural history. Am J Med Genet Part A (2018) 176:415–20. doi: 10.1002/ajmg.a.38557
26. Kim Y-M, Seo GH, Kim G-H, Ko JM, Choi J-H, Yoo H-W. A case of an infant suspected as IMAGE syndrome who were finally diagnosed with MIRAGE syndrome by targeted mendelian exome sequencing. BMC Med Genet (2018) 19:35. doi: 10.1186/s12881-018-0546-4
27. Csillag B, Ilencikova D, Meissl M, Webersinke G, Laccone F, Narumi S, et al. Somatic mosaic monosomy 7 and UPD7q in a child with MIRAGE syndrome caused by a novel SAMD9 mutation. Pediatr Blood Cancer (2019) 66:e27589. doi: 10.1002/pbc.27589
28. Perisa MP, Rose MJ, Varga E, Kamboj MK, Spencer JD, Bajwa RPS. A novel SAMD9 variant identified in patient with MIRAGE syndrome: Further defining syndromic phenotype and review of previous cases. Pediatr Blood Cancer (2019) 66:e27726. doi: 10.1002/pbc.27726
29. Ahmed IA, Farooqi MS, Vander Lugt MT, Boklan J, Rose M, Friehling ED, et al. Outcomes of hematopoietic cell transplantation in patients with germline SAMD9/SAMD9L mutations. Biol Blood Marrow Transplant (2019) 25:2186–96. doi: 10.1016/J.BBMT.2019.07.007
30. Formankova R, Kanderova V, Rackova M, Svaton M, Brdicka T, Riha P, et al. Novel SAMD9 mutation in a patient with immunodeficiency, neutropenia, impaired anti-CMV response, and severe gastrointestinal involvement. Front Immunol (2019) 10:2194. doi: 10.3389/fimmu.2019.02194
31. Zhang Y, Zhang Y, Zhang VW, Zhang C, Ding H, Yin A. Mutations in both SAMD9 and SLC19A2 genes caused complex phenotypes characterized by recurrent infection, dysphagia and profound deafness – a case report for dual diagnosis. BMC Pediatr (2019) 19:364. doi: 10.1186/s12887-019-1733-y
32. Mengen E, Küçükçongar Yavaş A, Uçaktürk SA. A rare etiology of 46,XY disorder of sex development and adrenal insufficiency: A case of MIRAGE syndrome caused by mutations in the SAMD9 gene. J Clin Res Pediatr Endocrinol (2020) 12:206–11. doi: 10.4274/jcrpe.galenos.2019.2019.0053
33. Viaene AN, Harding BN. The neuropathology of MIRAGE syndrome. J Neuropathol Exp Neurol (2020) 79:458–62. doi: 10.1093/jnen/nlaa009
34. Onuma S, Wada T, Araki R, Wada K, Tanase-Nakao K, Narumi S, et al. MIRAGE syndrome caused by a novel missense variant (p.Ala1479Ser) in the SAMD9 gene. Hum Genome Var (2020) 7:4. doi: 10.1038/s41439-020-0091-5
35. Tanase-Nakao K, Kawai M, Wada K, Kagami M, Narumi S. Acquired uniparental disomy of chromosome 7 in a patient with MIRAGE syndrome that veiled a pathogenic SAMD9 variant. Clin Pediatr Endocrinol (2021) 30:163–9. doi: 10.1297/CPE.30.163
36. Kawashima-Sonoyama Y, Okuno K, Dohmoto T, Tanase-Nakao K, Narumi S, Namba N. The case of a patient with MIRAGE syndrome with familial dysautonomia-like symptoms. Hum Genome Var (2021) 8:27. doi: 10.1038/s41439-021-00158-6
37. Sahoo SS, Pastor VB, Goodings C, Voss RK, Kozyra EJ, Szvetnik A, et al. Clinical evolution, genetic landscape and trajectories of clonal hematopoiesis in SAMD9/SAMD9L syndromes. Nat Med (2021) 27:1806–17. doi: 10.1038/s41591-021-01511-6
38. Mitsui-Sekinaka K, Narumi S, Sekinaka Y, Uematsu K, Yoshida Y, Amano N, et al. Clinical and immunological analyses of ten patients with MIRAGE syndrome. J Clin Immunol (2021) 41:709–11. doi: 10.1007/s10875-020-00964-7
39. Suntharalingham JP, Ishida M, del Valle I, Stalman SE, Solanky N, Wakeling E, et al. Data from: Emerging phenotypes linked to variants in SAMD9 and MIRAGE syndrome. Open Sci Framework (2022). doi: 10.17605/OSF.IO/WMVXY
40. Leon LJ, Solanky N, Stalman SE, Demetriou C, Abu-Amero S, Stanier P, et al. A new biological and clinical resource for research into pregnancy complications: The baby bio bank. Placenta (2016) 46:31–7. doi: 10.1016/j.placenta.2016.08.085
41. Monk D, Moore GE. Intrauterine growth restriction–genetic causes and consequences. Semin Fetal Neonatal Med (2004) 9:371–8. doi: 10.1016/j.siny.2004.03.002
42. ACOG Practice Bulletins. Clinical management guidelines for obstetrician – gynecologists. Obstet Gynecol (2020) 133:168–86. doi: 10.1097/AOG.0000000000003070
43. Wakeling EL, Brioude F, Lokulo-Sodipe O, O’Connell SM, Salem J, Bliek J, et al. Diagnosis and management of silver–Russell syndrome: first international consensus statement. Nat Rev Endocrinol (2017) 13:105–24. doi: 10.1038/nrendo.2016.138
44. Stalman SE, Solanky N, Ishida M, Alemán-Charlet C, Abu-Amero S, Alders M, et al. Genetic analyses in small-for-gestational-age newborns. J Clin Endocrinol Metab (2018) 103:917–25. doi: 10.1210/jc.2017-01843
45. Suntharalingham JP, Ishida M, Buonocore F, del Valle I, Solanky N, Demetriou C, et al. Analysis of CDKN1C in fetal growth restriction and pregnancy loss. F1000Research (2020) 8:90. doi: 10.12688/f1000research.15016.2
46. Guran T, Buonocore F, Saka N, Ozbek MN, Aycan Z, Bereket A, et al. Rare causes of primary adrenal insufficiency: genetic and clinical characterization of a large nationwide cohort. J Clin Endocrinol Metab (2016) 101:284–92. doi: 10.1210/jc.2015-3250
47. Karczewski KJ, Francioli LC, Tiao G, Cummings BB, Alföldi J, Wang Q, et al. The mutational constraint spectrum quantified from variation in 141,456 humans. Nat (2020) 581:434–43. doi: 10.1038/s41586-020-2308-7
48. Uhlén M, Fagerberg L, Hallström BM, Lindskog C, Oksvold P, Mardinoglu A, et al. Proteomics. tissue-based map of the human proteome. Sci (2015) 347:1260419. doi: 10.1126/science.1260419
49. Vento-Tormo R, Efremova M, Botting RA, Turco MY, Vento-Tormo M, Meyer KB, et al. Single-cell reconstruction of the early maternal–fetal interface in humans. Nat (2018) 563:347–53. doi: 10.1038/s41586-018-0698-6
50. Pique-Regi R, Romero R, Tarca AL, Sendler ED, Xu Y, Garcia-Flores V, et al. Single cell transcriptional signatures of the human placenta in term and preterm parturition. Elife (2019) 8:1–22. doi: 10.7554/eLife.52004
51. Ren J, Wen L, Gao X, Jin C, Xue Y, Yao X. DOG 1.0: illustrator of protein domain structures. Cell Res (2009) 19:271–3. doi: 10.1038/cr.2009.6
52. Chou JH, Roumiantsev S, Singh R. PediTools electronic growth chart calculators: Applications in clinical care, research, and quality improvement. J Med Internet Res (2020) 22:e16204. doi: 10.2196/16204
53. Peng S, Meng X, Zhang F, Pathak PK, Chaturvedi J, Coronado J, et al. Structure and function of an effector domain in antiviral factors and tumor suppressors SAMD9 and SAMD9L. Proc Natl Acad Sci (2022) 119:e2116550119. doi: 10.1073/PNAS.2116550119
54. Vilain E, Merrer MLE, Lecointre C, Desangles F, Kay MA, Maroteaux P, et al. IMAGe, a new clinical association of intrauterine growth retardation, metaphyseal dysplasia, adrenal hypoplasia congenita, and genital anomalies. J Clin Endocrinol Metab (1999) 84:4335–40. doi: 10.1210/JCEM.84.12.6186
55. Arboleda VA, Lee H, Parnaik R, Fleming A, Banerjee A, Ferraz-de-Souza B, et al. Mutations in the PCNA-binding domain of CDKN1C cause IMAGe syndrome. Nat Genet (2012) 44:788–92. doi: 10.1038/ng.2275
56. Logan CV, Murray JE, Parry DA, Robertson A, Bellelli R, Tarnauskaitė Ž, et al. DNA Polymerase epsilon deficiency causes IMAGe syndrome with variable immunodeficiency. Am J Hum Genet (2018) 103:1038–44. doi: 10.1016/j.ajhg.2018.10.024
Keywords: SAMD9, FGR, NGS, adrenal insufficiency, endocrinopathies, myelodysplastic syndrome (MDS), MIRAGE syndrome
Citation: Suntharalingham JP, Ishida M, Del Valle I, Stalman SE, Solanky N, Wakeling E, Moore GE, Achermann JC and Buonocore F (2022) Emerging phenotypes linked to variants in SAMD9 and MIRAGE syndrome. Front. Endocrinol. 13:953707. doi: 10.3389/fendo.2022.953707
Received: 26 May 2022; Accepted: 13 July 2022;
Published: 18 August 2022.
Edited by:
Tsutomu Ogata, Hamamatsu University School of Medicine, JapanReviewed by:
Satoshi Narumi, National Center for Child Health and Development (NCCHD), JapanCopyright © 2022 Suntharalingham, Ishida, Del Valle, Stalman, Solanky, Wakeling, Moore, Achermann and Buonocore. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: John C. Achermann, ai5hY2hlcm1hbm5AdWNsLmFjLnVr; Federica Buonocore, Zi5idW9ub2NvcmVAdWNsLmFjLnVr
†These authors have contributed equally to this work and share last authorship
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.