
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Endocrinol., 02 September 2022
Sec. Diabetes: Molecular Mechanisms
Volume 13 - 2022 | https://doi.org/10.3389/fendo.2022.947294
This article is part of the Research TopicMolecular Mechanisms and Genetics of Heart Failure Caused by DiabetesView all 5 articles
 Misganaw Asmamaw Mengstie1*†
Misganaw Asmamaw Mengstie1*† Endeshaw Chekol Abebe1
Endeshaw Chekol Abebe1 Awgichew Behaile Teklemariam1
Awgichew Behaile Teklemariam1 Anemut Tilahun Mulu1
Anemut Tilahun Mulu1 Assefa Agegnehu Teshome2
Assefa Agegnehu Teshome2 Edgeit Abebe Zewde3
Edgeit Abebe Zewde3 Zelalem Tilahun Muche3
Zelalem Tilahun Muche3 Muluken Teshome Azezew3
Muluken Teshome Azezew3Diabetes Mellitus (DM) is a worldwide health issue that can lead to a variety of complications. DM is a serious metabolic disorder that causes long-term microvascular and macro-vascular complications, as well as the failure of various organ systems. Diabetes-related cardiovascular diseases (CVD) including heart failure cause significant morbidity and mortality worldwide. Concurrent hypertensive heart disease and/or coronary artery disease have been thought to be the causes of diabetic heart failure in DM patients. However, heart failure is extremely common in DM patients even in the absence of other risk factors such as coronary artery disease and hypertension. The occurrence of diabetes-induced heart failure has recently received a lot of attention. Understanding how diabetes increases the risk of heart failure and how it mediates major cellular and molecular alteration will aid in the development of therapeutics to prevent these changes. Hence, this review aimed to summarize the current knowledge and most recent findings in cellular and molecular mechanisms of diabetes-induced heart failure.
Diabetes Mellitus (DM) is a worldwide health problem characterized by high blood glucose levels caused by a defect in insulin production or insulin resistance, or both (1). DM is a serious metabolic disorder that causes long-term microvascular and macro-vascular complications, as well as the failure of various organ systems (2). Heart Failure (HF) is a long-term condition in which the heart muscles are unable to pump blood effectively enough to meet the body’s needs. It is characterized by cardiomyopathy (cardiac muscle weakness), fibrosis, hypertrophy, cell death, and diastolic dysfunction, followed by systolic dysfunction (3). Uncontrolled DM hurts several systems, including the cardiovascular system. Diabetes-related cardiovascular diseases (CVD) cause significant morbidity and mortality worldwide. Concurrent hypertensive heart disease and/or ischemic coronary artery disease were previously thought to be the cause of HF in diabetic patients. However, even in the absence of predisposing factors for HF, diabetic patients show signs of impaired function and abnormal structure in the heart (4, 5). The occurrence of CVD, including HF in patients with DM, has recently received a lot of attention. According to a plethora of epidemiological evidence, HF is extremely common in DM patients even in the absence of other HF risk factors such as coronary artery disease and hypertension (6–9). HF is four times more prevalent in DM patients than in the general population and is one of the main causes of mortality and morbidity (10). Chronic metabolic imbalance in diabetes can lead to cardiac dysfunction and even HF. Hyperglycemia, insulin resistance, and other factors have been linked to diabetic HF Despite significant advances in our understanding of the pathophysiology of DM-related HF in recent decades, the details of the cellular mechanisms are still not well understood. Understanding how diabetes increases the risk of HF and how it mediates major cellular and molecular alterations will aid in the development of therapeutics to prevent these changes. Hence, this review aimed to summarize the current knowledge and most recent findings in cellular and molecular mechanisms of diabetes-induced HF.
The regulation of cardiac function, both contractility, and relaxation are dependent on energy metabolism. To maintain its contractile function, the heart needs a continuous and high amount of energy in the form of adenosine 5’-triphosphate (ATP) (11). Under normal conditions, 40-60% of cardiac ATP is produced by the oxidation of fatty acids (FA) in the mitochondria. Glucose is the second most important fuel source for the heart, accounting for 20-40% of cardiac ATP through oxidation (12). Free fatty acids, which originate from either serum albumin or lipoprotein triacylglycerol (TAG) enter the cardiac myocyte through sarcolemma membrane fatty acid transporter proteins, cluster of differentiation-36 (CD36) (13). Once in the cytoplasm, fatty acids can then be used to synthesize a variety of lipid intermediates or can be taken up into the mitochondrial matrix to generate ATP via β-oxidation (14). The ability of the heart to switch between available substrates is maintained by tightly regulating fatty acid oxidation at multiple points. The metabolic flexibility of the heart is complex and, it is influenced by several factors such as alteration in contractile work, hormonal change, oxygen supply limitations, and the presence of competing substances (i.e. glucose) (15, 16). Even though fatty acids are the primary fuel source for cardiac cells, they have the highest oxygen requirement to produce ATP and are the least efficient (ATP produced/O2 consumed) myocardial energy substrates. Mitochondrial fatty acid oxidation may also be reduced under stress conditions, resulting in increased glucose utilization (17).
The uptake of glucose into the sarcolemma is mediated by insulin-independent glucose transporter type-1 (GLUT-1), insulin-dependent GLUT-4 transporters as the most abundant isoforms. GLUT-4 is the most common isoform in the adult human heart, accounting for 70% of all glucose transporters, whereas GLUT-1 is highly expressed in the fetal heart (18). Insulin is required for the translocation of GLUT-4 from the intracellular compartment to the sarcolemma membrane. Insulin regulates the expression of the GLUT gene, which affects glucose transport, in addition to GLUT translocation (19). Inside cardiac myocytes, glucose can be converted to glucose-6 phosphate by hexokinase or to sorbitol by polyol pathway. As shown in Figure 1, glucose-6 phosphate can then be utilized in a variety of metabolic pathways including glycolysis, hexose monophosphate pathway (HMP), and hexosamine biosynthetic pathway (HBP) (20).
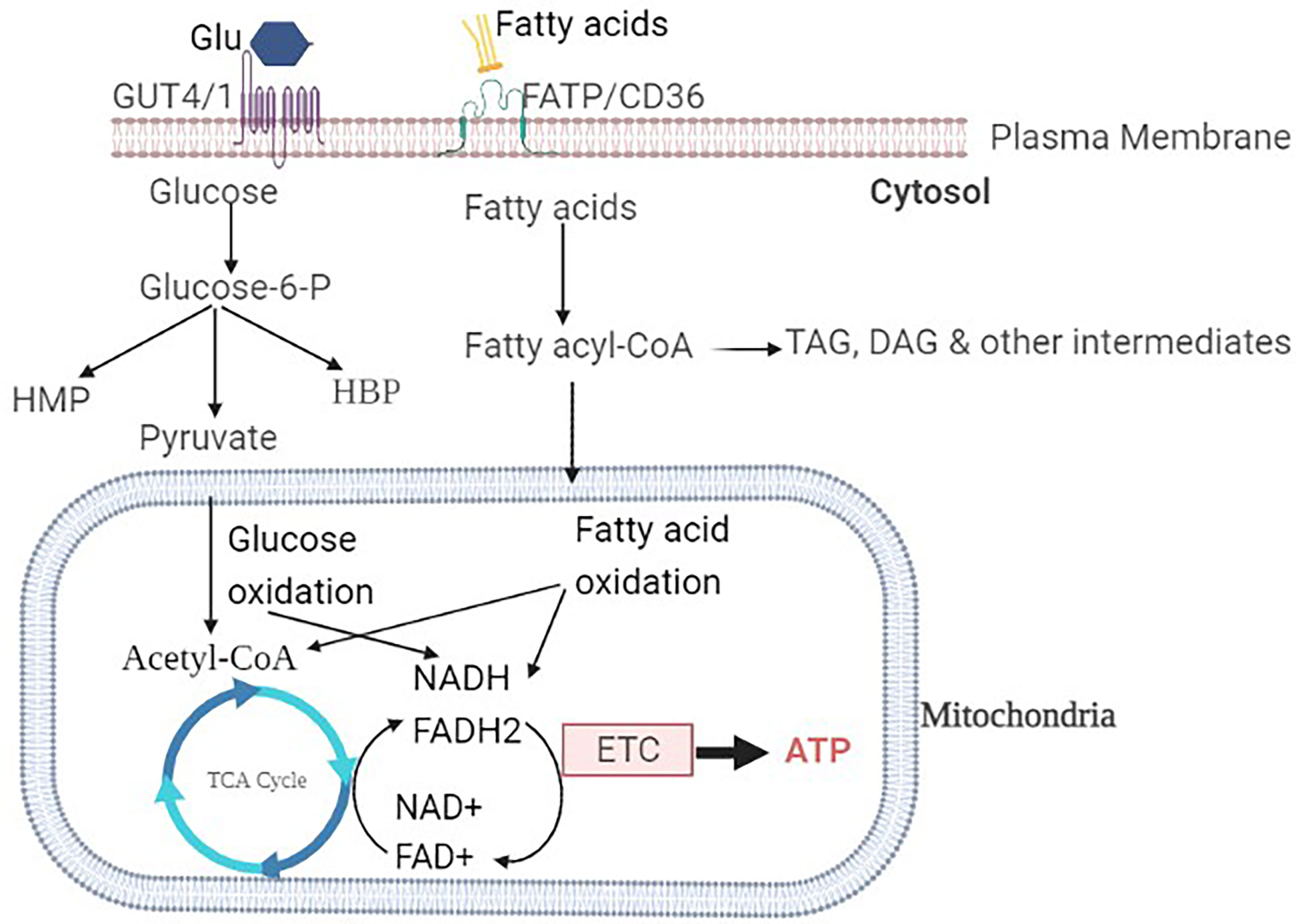
Figure 1 Overview of glucose and fatty acids metabolism in a healthy heart: The glucose transporters GLUT1 and GLUT4 are responsible for glucose uptake into the cardiomyocyte. Once inside the cell, glucose is then phosphorylated to Glucose-6-P. Glucose-6-P is the common intermediate for HMP, HBP, or pyruvate through glycolysis. Pyruvate is then interred to the mitochondria, where it is decarboxylated to acetyl-CoA. Fatty acid transporters (FAT/CD36) transfer non-esterified fatty acids into the cell. Fatty acyl-CoA is formed by intracellular fatty acid activator enzymes and can either be esterified into TAG or reach the mitochondria. Fatty acyl-CoA from mitochondria enters the β-oxidation pathway, where it is converted to acetyl-CoA. Glucose or fatty acid-derived acetyl-CoA enters the TCA cycle, where it undergoes ETC, oxidative phosphorylation, and ATP formation.
Both glucose and fatty acid metabolic abnormalities have been described leading to a better understanding of the complicated process of HF in DM (4). One of the carbohydrate metabolism abnormalities in DM is the reduced expression of glucose transporters. In a diabetic heart, the expression and translocation of these transporters (GLUT-4) are downregulated (21). For example, atrial GLUT-4 trafficking and expression were impaired in an animal model (streptozotocin-induced type-1 diabetic rodents) (22). Concomitantly, there is the reduction of cardiac glucose uptake, glycolysis, and oxidation associated with a shift towards increased concentration of free fatty acids (accelerated cardiac fatty acid oxidation). A marked increase in cardiac fatty acid oxidation rates and the dominance of fatty acids as the major energy source in the heart is the other significant metabolic change seen in DM (23). Elevated levels of circulating FFAs and their increased oxidation are also inhibitors of both glycolysis and glucose oxidation in the heart. Excess fatty acid oxidation increases mitochondrial oxygen consumption (reduces the pool of oxygen). As a result, the cardiac energy exchange efficiency and energetic reserves are reduced because FFA β-oxidation is less efficient in the limited oxygen availability. During times of increased metabolic demands, this may increase the risk of cardiac dysfunction, including HF (24). Furthermore, the fatty acids taken up by cardiac cells are used for the synthesis of triglycerides (TG) and other lipid intermediates in addition to providing energy. Therefore, cardiac cells’ reliance on fatty acids as an energy source results in a dramatic accumulation of lipids within the myocardium leading to myocardial steatosis, which may contribute to the development of HF (25). These changes have been seen in type-1 and type-2 DM patients, as well as in preclinical models (26–28). The combined myocardial alteration of both glycolysis and fatty acid metabolism has been also observed in many forms of heart disease including HF (29). Moreover, free fatty acids and accumulation of intracellular TG and other lipid intermediates can impair insulin signaling and contribute to cardiac insulin resistance (30). Cardiac insulin resistance, the hallmark of type-2 DM is an independent risk factor for the development of HF (31). Cardiac insulin resistance is linked to decreased cardiac insulin metabolic signaling and is caused by a variety of factors including oxidative stress, hyperglycemia, hyperlipidemia, and dysregulated adipokine/cytokine secretion (32).
Mitochondria, the semi-autonomous organelles, and cellular powerhouse play a critical role in ensuring that cells function properly. They are involved in the production of ATP, calcium homeostasis, oxidative stress response, and apoptosis (33). Mitochondrial dysfunction is one of the most principal factors in the development of HF in people with diabetes (34). Cardio myocytes produce about 90% of their ATP through oxidative phosphorylation in the mitochondria (35). As mentioned earlier, cardiac mitochondria in diabetic patients will switch the source of ATP production from glucose to fatty acid oxidation to maintain a constant supply of energy to the cells (36). This process disrupts the oxidative phosphorylation process and causes to produces more reactive oxygen species (ROS) in the electron transport chain. ROS can cause oxidative damage to cellular proteins and lipids. Due to its proximity to the inner membrane, insufficient repair mechanisms, and lack of protective histones, mitochondrial DNA is also extremely vulnerable to ROS (37). ROS also negatively affects myocardial calcium (Ca2+) handling resulting in cytosolic Ca2+ overload (38). Ca2+overload in the cytosol may then cause the opening of the mitochondrial permeability transition pore (mPTP) (39). As illustrated in Figure 2, the opening of mPTP, combined with damage to mitochondrial DNA, causes apoptosis (cell death), which leads to cardiac mitochondrial dysfunction and, eventually, HF (40). Recent evidence showed that increased mPTP also initiates the processes of necroptosis (non-programmed cell death) in the diabetic heart (41). Mitochondrial DNA damage could also trigger the activation of a pro-inflammatory type of programmed cell death (pyroptosis) through the activation of the cGAS-STING pathway (42). The cGAS-STING signaling pathway, consisting of the cyclic GMP-AMP synthase (cGAS) and the cyclic GMP-AMP receptor stimulator of interferon genes (STING), is an innate defense mechanism that detects pathogenic DNA. In addition to sensing microbial DNA, cGAS has been proven to detect endogenous DNA, such as DNA released from mitochondria (43). Activation of cardiac pyroptosis promotes pro-inflammatory responses in cardiomyocytes, thereby exacerbating myocardial hypertrophy during the progression of diabetic cardiomyopathy (44). Generally, these forms of cell death could be attributed to the pathogenesis of diabetic HF involving mitochondrial dysfunction. However, various other forms of cell death including, autophagic cell death, autosis, and ferroptosis have been identified and characterized in diabetic cardiomyopathy (45).
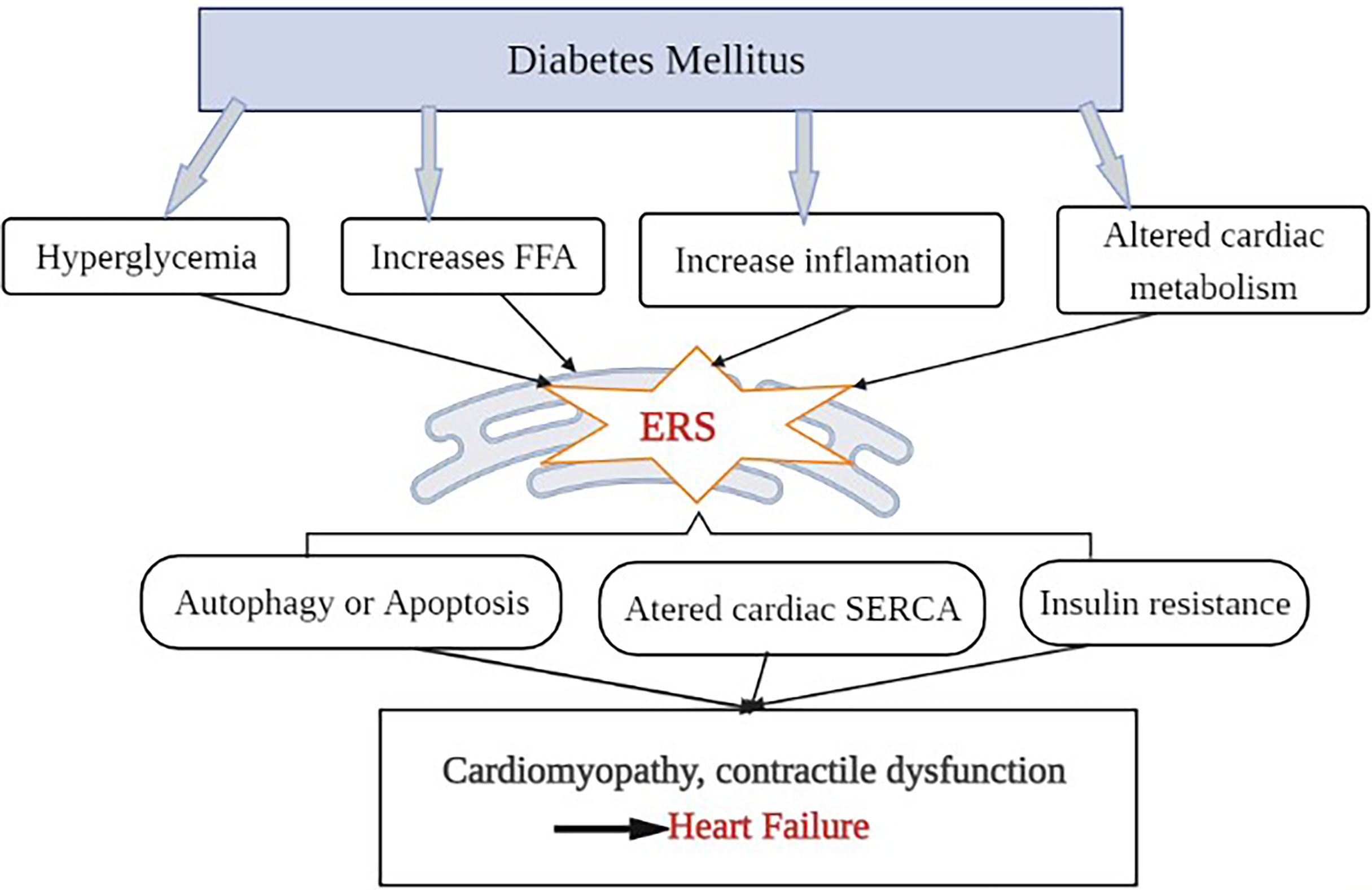
Figure 2 Role of mitochondrial dysfunction in diabetic HF. To maintain ATP production, heart cells in DM adapt to enhanced fatty acid oxidation. Concomitantly, the formation of reactive oxygen species (ROS) is also increased, affecting oxidative phosphorylation, damaging mitochondrial DNA, and disturbing Ca2+ homeostasis. Both mitochondrial DNA damage and mPTP opening (due to Ca2+ overload) promote the cardiac apoptotic (programmed cell death) pathway. Through the activation of the cGAS-STING (cyclic GMP-AMP synthase (cGAS) and the cGAS stimulator of interferon genes) pathway, mitochondrial DNA damage may potentially trigger the activation of a pro-inflammatory type of programmed cell death (pyroptosis). Increased mPTP also starts the necroptosis processes. All of which lead to mitochondrial malfunction and ultimately HF.
The ER is a vital cell organelle that regulates several homeostatic responses, including lipid and steroid hormone synthesis, calcium homeostasis, secretory and transmembrane protein post-translational modifications, and the folding and maturation of newly synthesized proteins (46). ERS is a chronic perturbation of ER homeostasis marked by the accumulation of unfolded/misfolded proteins caused by a variety of factors including altered glucosylation, nutrient deprivation, oxidative stress, and so on. ERS activates a complex signaling pathway referred to as unfolded protein response (UPR) pathway (47). In normal conditions, UPR activates transcriptional and translational pathways in the ER to reduce the rate of general translation and increase the expression of ER-resident protein chaperones, which helps to restore ER homeostasis (48). Three sensor proteins (normally kept inactive by interacting with ER-resident chaperone proteins) initiate the UPR pathway. These sensor proteins are the kinases PERK (protein kinase R-like ER kinase) and IRE1 (inositol requiring enzyme-1), and the transcription factor ATF-6 (activating transcription factor-6) (49). When these sensors are activated, they cause the transcription of UPR target genes, which leads to a decrease in protein synthesis, an increase in ER chaperone expression, and an increase in proteins involved in ER-associated protein degradation (50). Hence, UPR is the mechanism intended to restore cellular function as a compensatory pro-survival response during ERS. However, a prolonged ER stress response can cause cardiac dysfunction by activating the ER-mediated apoptotic pathway and downstream signaling in the cardio-myocytes (51). Chronic ER stress is associated with metabolic disorders like obesity, diabetes, and age-related pathogenesis, according to growing evidence (52).
Cardiac hypertrophy, local inflammation, abnormal intracellular Ca2+ handling, oxidative stress, and endothelial dysfunction are all common symptoms of DM (53). DM raises plasma glucose levels, increases FFA levels, activates inflammation, and alters cardiac metabolism and lipotoxicity, all of which contribute to ERS (54). Cardiac ERS then activates UPR and results in myocardial cell apoptosis. In the DM rat model, upregulation of ERS markers and apoptotic molecules have also confirmed myocardial cell apoptosis (55). The other possible mechanism for the ERS-induced HF in diabetes is the alteration of cardiac sarcoplasmic reticulum Ca 2+ ATPase (SERCA) protein. The alteration of SERCA in diabetes might be due to hyperglycemia-induced non-enzymatic glycation. SERCA is a membrane-bound intracellular enzyme that transports Ca2+ against a concentration gradient by utilizing the free energy of ATP (56). Both human diabetes and experimental animal model studies demonstrated that ERS alters SERCA protein, leading to left ventricular (LV) diastolic dysfunction (57, 58). Reduced cardiac SERCA activity is associated with altered Ca 2+ handling and deficient contractility, which can lead to HF (59, 60). Furthermore, studies have also shown that cardiac ERS alters all aspects of cardiac energy metabolism, from substrate utilization to oxidative phosphorylation and energy transport to myofibrils, potentially contributing to HF (61). Overall changes result in contractile dysfunction, left ventricular (LV) hypertrophy, and cardiomyopathy, which together affect cardiac output and eventually lead to HF (62), as shown in Figure 3. In general, although more study is needed to fully comprehend the mechanisms underlying these processes, appropriate intervention in the ERS process could be a therapeutic strategy for diabetes-related cardiac complications including HF.
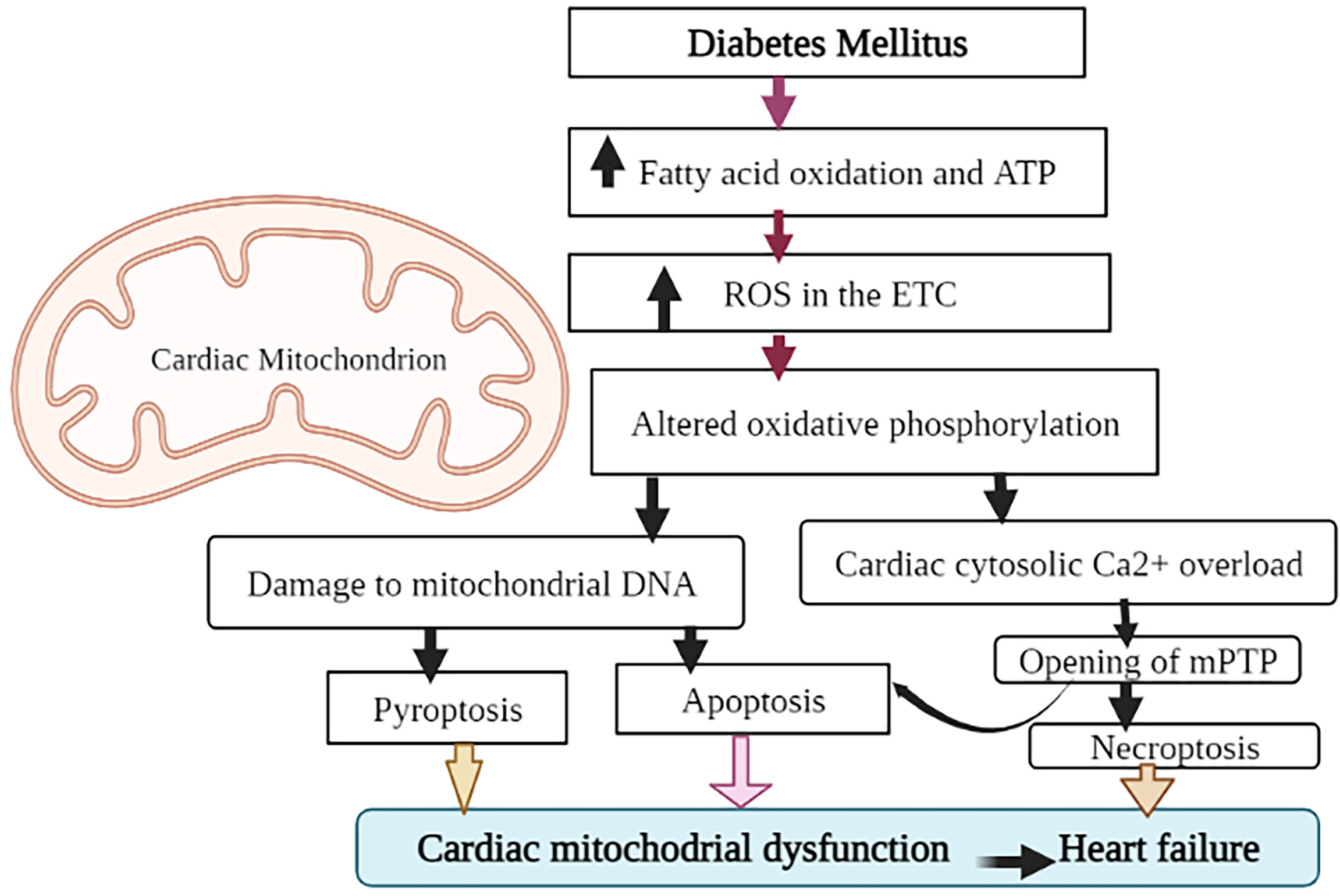
Figure 3 Outlining the role of ER stress in the development of diabetic HF: Diabetes mellitus raises plasma glucose levels, increases the amount of circulating FFAs, alters cardiac metabolism, and triggers an inflammatory response that activates ERS. When ER function is disrupted, it causes cell autophagy or apoptosis, changes in SERCA protein structure and function, and insulin resistance, which leads to diabetic cardiomyopathy, contractile dysfunction, and eventually heart failure.
Systemic chronic inflammation is one of the pathophysiological processes in patients with DM, and it has been a factor in the development of vascular complications (63). Chronic systemic inflammation, caused by hyperglycemia, hyperlipidemia, hyperinsulinemia, and insulin resistance, partially contributes to the development of diabetic HF (64). The accumulation of advanced glycation end products (AGEs), oxidative stress, and hyperlipidemia are followed by activation of the nuclear factor kappa-B (NF-kB) pathway; the renin-angiotensin-aldosterone system (RAAS), and overexpression of inflammatory interleukins are the important molecular mechanisms in the development of diabetic HF (65, 66). Persistent hyperglycemia induces oxidative stress and the accumulation of AGEs, which in turn activates a common signaling pathway involving a transcription factor, known as NF-κB (67). Prolonged hyperglycemia, as previously stated, inhibits glucose oxidation while enhancing fatty acid metabolism in the diabetic heart. Enhanced fatty acid oxidation in the diabetic heart leads to an increase in lipoprotein levels (particularly oxidized LDL is high) (68). It has also been proven that oxidized LDL activates the NF-B signaling pathway (69). NF-κB is a universal transcription factor normally found in an inactive form in the cytoplasm. Once it is activated it trans-locates to the nucleus and is bound with a specific section of DNA that triggers the expression of various cytokines, chemokines, cell adhesion molecules, interleukins, TGFβ, pro-infammatory proteins, and pro-apoptotic genes (70). Finally, the release of those molecules via this mechanism causes ROS stress to the myocardium, fibrosis, hypertrophy, cell death, and eventual myocardial diastolic dysfunction, all of which are the early hallmarks of diabetic HF (71). Hence, NF-κB signaling is one of the promising therapeutic targets for diabetic-related chronic complications including diabetic HF (67).
Diabetes can also promote the RAAS system in the heart through several mechanisms. The first is one is hyperglycemia directly stimulates local Angiotensin-II (Ang-II) production in the cardio-myocytes (72). It was proposed that hyperglycemia-induced intracellular generation of Ang-II in the cardiac cells is by chymase proteins. Chymases are a family of serine proteases found primarily in mast cells, fibroblasts, and vascular endothelial cells (73). It was also demonstrated that intracellular Ang-II is correlated with cardiomyocyte apoptosis, oxidative stress, and cardiac fibrosis in diabetic rats (74). Interestingly inhibition of this enzyme has a significant therapeutic advantage in halting the progression of diabetic-induced cardiac and vascular diseases (75, 76). The second mechanism of RAAS activation in the diabetic heart is that high glucose concentrations can enhance the tissue response to Ang-II. This implies that with hyperglycemia, the tissue response to Ang-II can become more contractile. It has also been demonstrated that high glucose concentration augments Ang-II mediated aortic contraction via an angiotensin-1 receptor (AT1R) in a rat model (77). The third way by which DM promotes RAAS is through metabolic abnormalities associated with hyperglycemia such as AGEs. AGEs are formed after sustained hyperglycemia, oxidative stress, and dyslipidemia in DM. AGEs could mediate intracellular signaling, gene expression, the release of pro-inflammatory molecules and free radicals, and activation of RAAS through receptor-mediated signaling cascade (78, 79). Furthermore, AGEs activate the chymase-dependent production of Ang-II (80). Ang-II plays a significant role in regulating various physiological processes. It acts as a potent vasoconstrictor, promotes growth factors, migration, proliferation, and hypertrophy of vascular smooth muscle cells and cardiac fibroblasts, and increases catecholamine release. Although these mechanisms initially serve to maintain cardiac output, they eventually contribute to the progression of HF (81).
Autophagy is the general term that refers to the process involving the decomposition of intracellular components via lysosomes. It is crucial to all types of cells including cardiovascular cells for maintaining homeostasis and preventing nutritional, metabolic, and infection-related stress (82). Micro-autophagy, macro-autophagy, and chaperone-mediated autophagy (CMA) are the three main types of autophagy in mammalian cells, based on the mode of cargo delivery to the lysosome (83). To capture the cargo in micro-autophagy, direct invaginations of the lysosomal membrane or vacuolar membranes are used. CMA is a type of autophagy that uses chaperones to selectively degrade soluble cytosolic proteins in the lysosomes. Macro-autophagy (hereafter referred to as autophagy) is the most extensively studied type of autophagy characterized by the sequestration of cytoplasmic materials in double-membrane vacuoles called auto-phagosomes, which are then delivered to the lysosome for degradation (84, 85). Several principal factors control this intracellular degradation process. Autophagy-related (ATG) genes and proteins play a crucial role in the process of autophagy such as induction, cargo selection, auto-phagosome formation, fusion, and degradation. To date, more than 30 ATG genes and proteins have been discovered (86). Although there are several regulatory pathways for cellular autophagy, the mammalian target of rapamycin (mTOR) pathway and adenosine 5’monophosphate-activated protein kinase (AMPK) are the most important inhibitors and activators, respectively (87). mTOR is a serine/threonine kinase that is highly conserved from yeast to humans (88). By phosphorylating complex components such as ATG13 and ATG1 (the mammalian ULK1/2 homolog), mTOR inhibits the autophagy-initiating UNC-5 like autophagy activating kinase (ULK) complex. This disrupts the interaction between ATG1 and ATG13, preventing the interaction and phosphorylation of ULK1 by AMPK, and finally inhibiting autophagosome initiation (88, 89). On the other hand, AMPK (a nutrient sensor) is the major activator of autophagy. AMPK promotes autophagy either directly by phosphorylating autophagy-related proteins in the mTOR, such as ULK, or indirectly by regulating autophagy-related gene expression downstream of transcription factors (90).
Although autophagy is important for maintaining cellular homeostasis in healthy cardiac tissues by preventing the accumulation of dysfunctional organelles and cytotoxic protein aggregates, some recent research has found that this process is frequently impaired in diabetic hearts (91). There are contradictory evidences regarding whether autophagy flux is upregulated or downregulated in the hearts of DM patients. Some studies have tried to distinguish autophagy adaptation in the heart of type 1 and type 2 DM. For instance, in animal models, autophagy flux was found to be enhanced in the heart of type 1 diabetics but suppressed in type 2 diabetics (92). Because GLUT-4 and FAT/CD36 cannot translocate to the membrane due to insulin deficiency in type 1 DM, glucose and fatty acid uptake by the membrane is prevented, putting the cell into a state of nutrient deprivation. AMPK is activated by ATP depletion or AMP accumulation. As a result, in type 1 diabetic hearts, autophagy flux is enhanced (93, 94). Diastolic dysfunction and pathogenesis in diabetic cardiomyopathy have been linked to autophagy upregulation in the hearts of type 1 diabetic (95). In contrast, some experimental studies revealed that diminished autophagy in type 1 DM is an adaptive response that limits cardiac dysfunction (96, 97). Unlike type 1 DM, autophagy would be expected to be suppressed in type 2 diabetic hearts (98). Although the exact molecular mechanism is unknown, AMPK must play a major role in the suppression of autophagy in type 2 DM. Overeating and/or obesity, common features of type 2 DM are possible causes that downregulate AMPK activity by increasing intracellular levels of energy substrates like glucose and/or FFA, thereby suppressing autophagy (99). Suppressing AMPK signaling lowers cardiac glucose transport, glycolysis, and fatty acid oxidation, as well as the energy supply required for cardiac function. Indeed, downregulation of AMPK activity is a key factor in the development of diabetic HF in type 2 DM (92). It was also demonstrated that a reduction in cardiac autophagy and the subsequent decrease of cell viability was implicated in cardiac dysfunction in the diabetic heart (78). However, still, there are some contradictory pieces of evidence, which demonstrated that suppression of autophagy in diabetes is protective against cardiac cell dysfunction and HF too (100, 101). Therefore, further study is needed to determine the exact role of autophagy in type 1 or type 2 diabetic HF, as well as whether the response of autophagy in the diabetic heart is protective or not.
Epigenetics is the study of heritable changes in gene expression patterns that are not linked to changes in DNA sequence. DNA methylation and histone modification are the key epigenetic processes that control gene expression (102). Epigenetic dysregulation has a role in the development of many diseases including DM. DNA methylation is a biological event that occurs when DNA methyltransferases add methyl groups to DNA nucleotides (cytosine or adenine). It is usually exerted at the 5′-position of cytosine residues in CpG dinucleotides. DNA methylation regulates gene expression by preventing transcription factors from binding to DNA (103). Recently increasing evidence showed that DNA methylation plays a critical role in the pathogenesis of diabetic complications including HF (104). It was discovered that tumor necrosis factor- α (TNF-α) boosts the SERCA promotor region by enhancing DNA methyltransferase activity in diabetic hyperglycemia. In cardiomyocytes, the expression of the protein is reduced when the SERCA gene is methylated (105). As previously indicated, downregulation of SERCA expression might result in diastolic dysfunction and contractility impairment, which can contribute to diabetic HF (106). Furthermore, a positive correlation was observed between the severity of HF and the expression level of TNF-α (107). This is also supported by the study that aberrant DNA methylation in human cardiomyocytes impairs cardiac contractility, and causes mitochondrial damage, as well as lipid and glucose metabolic problems (108). Taken together, these studies suggest that DNA methylation plays a role in the adverse cardiac remodeling associated with the development of diabetic HF, albeit more research is needed.
Histone modification is another epigenetic process have been also identified as a key contributor to the development of diabetes-related HF (109). Together with DNA, four types of histone proteins (H2A, H2B, H3, and H4) make up the fundamental unit of chromatin known as the nucleosome. Histone modification is an epigenetic process in which histone proteins are methylated, acetylated, phosphorylated, adenylated, ubiquitinated, or ADP ribosylated (110). The transcriptional activity of associated genes is affected by these modifications. Histone modifications also affect the binding ability of other proteins to histones via altering local hydrophobicity, RNA polymerase status, and binding affinity for other transcription coactivators (111). In diabetic myocardium, hyperglycemia promotes the production of mitochondrial ROS, which in turn induces JunD (Jund proto-oncogene subunit) downregulation. Since JunD is one of the oxidative stress gatekeepers, its downregulation causes cardiac dysfunction in both experimental and human DM investigations (112). In vascular cells, ROS also causes long-term epigenetic activation of the NF-B subunit p65. The methylation of histone 3 on lysine 4 has been identified as the epigenetic alteration that enhances p65 gene expression and consequent pro-inflammatory gene expression. These findings show that hyperglycemia-induced histone alteration could be a separate risk factor for diabetes complications including HF (113). Recent evidence from in vitro and in vivo revealed that Histone deacetylation appears to play a role in the etiology of diabetes complications, including HF. So, it has been proposed that the balance between histone acetylation and deacetylation must be tightly regulated (114). Hence, epigenetics in cardiovascular research and therapy is an exciting and promising emerging research field (115).
MicroRNAs (miRNAs) are endogenous, noncoding, single-strand RNAs with an average length of 22 nucleotides that regulate gene expression. miRNAs control gene expression in two ways: by inhibiting translation or by promoting the degradation of target messenger RNA (mRNA) (116). Studies have demonstrated that the expression level of miRNA in the heart of DM patients was found to be different when compared with healthy individuals (117). Changes in miRNA synthesis and levels have been linked to cardiac remodeling and the development of diabetic HF (118). The miRNAs may target mitochondrial function, ROS generation, Ca2+ perturbation, apoptosis, and fibrosis are all thought to be significant pathways for cardiac hypertrophy, remodeling, and HF development (119). One of the distinctive structural characteristics of early HF is cardiomyocyte hypertrophy. Numerous miRNAs were shown to be dysregulated and contributed to the pathophysiology of cardiomyocyte hypertrophy in DM (120). For instance, miR-133 is highly expressed in heart tissue and is known to be involved in diabetic cardiac hypertrophy (121). According to a study done on type 2 DM patients, miR-17 expression was up-regulated, while miR-24, miR-150, miR-199a, miR-214, and miR-320a expression was down-regulated in the diabetic cardiomyopathy patients compared to healthy controls. And it was concluded that dysregulation of circulating miRNA may contribute to the pathogenesis of diabetic induced-cardiomyopathy (122). It has also been demonstrated that upregulation of nuclear miR-320 mediates diabetes-induced cardiac dysfunction by activating the transcription of fatty acid metabolic genes, causing lipotoxicity in the hearts of diabetic mice (123). As a result, it has recently been suggested that measuring circulating miRNAs in combination with other biological markers could substantially improve the diagnostic potential of detecting diabetic cardiomyopathy (124). A significant reduction of miR-21 was also observed in the diastolic dysfunctional heart of diabetic mice. And the delivery of miR-21 efficiently protected against cardiac diastolic dysfunction (one of the early manifestations of HF) by targeting gelsolin protein (125). Furthermore, miRNA can influence other epigenetic changes such as histone H2A mRNA levels, histone deacetylase, and DNA methyltransferases, resulting in gene modification and expression modulation (126). However, the precise involvement of miRNAs in the development of diabetic HF, as well as their cross-talk with other epigenetic processes, is not clearly elucidated.
Metabolic impairment, ERS, autophagy, the inflammatory process, epigenetics, and microRNAs are just a few of the cellular and molecular mechanisms that have been proposed and explored in diabetic HF. As a result, limiting cardiac fatty acid oxidation, boosting glucose oxidation, mitigating ERS, and moderating inflammation could all be promising therapeutic approaches for diabetic HF. However, the role of some key pathways, such as autophagy, epigenetic mechanisms, and microRNAs in the pathogenesis of diabetic HF, are still under-studied and require further investigation.
MM developed the concept of the review. The manuscript of the protocol was drafted by EA, ATek, AM, ATes, EZ, and ZM then critically revised by MM and MA. MM developed and provided feedback for all sections of the review protocol. All authors have approved the final version of the manuscript to be published.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
ATP, adenosine triphosphate; CD36, cluster of differentiation; CVD, cardiovascular diseases; DM, diabetes mellitus; ERS, endoplasmic reticulum stress; ETC, electron transport chain; GLUT, glucose transporters; HBP, hexosamine biosynthetic pathway; HF, heart failure; HMP, hexose monophosphate pathway; miRNA, microRNAs; mPTP, mitochondrial permeability transition pore; mRNA, messenger RNA; ROS, reactive oxygen species; SERCA, sarcoplasmic reticulum calcium ATPase; TAG, triacylglycerol; UPR, unfolded protein response.
1. Al-Lawati JA. Diabetes mellitus: A local and global public health emergency! Oman Med J (2017) 32(3):177–9. doi: 10.5001/omj.2017.34
2. Chawla A, Chawla R, Jaggi S. Microvasular and macrovascular complications in diabetes mellitus: Distinct or continuum? Indian J Endocrinol Metab (2016) 20(4):546–53. doi: 10.4103/2230-8210.183480
3. Jia G, Hill MA, Sowers JR. Diabetic cardiomyopathy: An update of mechanisms contributing to this clinical entity. Circ Res (2018) 122(4):624–38. doi: 10.1161/CIRCRESAHA.117.311586
4. Kenny HC, Abel ED. Heart failure in type 2 diabetes mellitus: Impact of glucose-lowering agents, heart failure therapies, and novel therapeutic strategies. Circ Res (2019) 124(1):121–41. doi: 10.1161/CIRCRESAHA.118.311371
5. Giugliano D, Meier JJ, Esposito K. Heart failure and type 2 diabetes: From cardiovascular outcome trials, with hope. Diabetes Obes Metab (2019) 21(5):1081–7. doi: 10.1111/dom.13629
6. Dunlay SM, Givertz MM, Aguilar D, Allen LA, Chan M, Desai AS, et al. Type 2 diabetes mellitus and heart failure a scientific statement from the American heart association and the heart failure society of America. Circulation (2019) 140:294–324. doi: 10.1161/cir.0000000000000691
7. Thomas MC. Type 2 diabetes and heart failure: Challenges and solutions. Curr Cardiol Rev (2016) 12:249–55. doi: 10.2174/1573403X12666160606120254
8. Schütt KA. Heart failure in patients with diabetes. Diabetologe (2021) 17(5):607–17. doi: 10.1007/s11428-021-00752-w
9. Oo MM, Tan Chung Zhen I, Ng KS, Tan KL, Tan ATB, Vethakkan SR, et al. Observational study investigating the prevalence of asymptomatic stage b heart failure in patients with type 2 diabetes who are not known to have coronary artery disease. BMJ Open (2021) 11(1):1–8. doi: 10.1136/bmjopen-2020-039869
10. Ceriello A, Catrinoiu D, Chandramouli C, Cosentino F, Dombrowsky AC, Itzhak B, et al. Heart failure in type 2 diabetes: Current perspectives on screening, diagnosis and management. Cardiovasc Diabetol (2021) 20(1):1–19. doi: 10.1186/s12933-021-01408-1
11. Martínez MS, García A, Luzardo E, Chávez-Castillo M, Olivar LC, Salazar J, et al. Energetic metabolism in cardiomyocytes: Molecular basis of heart ischemia and arrhythmogenesis. Vessel Plus (2017) 1:230–41. doi: 10.20517/2574-1209.2017.34
12. Hausner EA, Elmore SA, Yang X. Overview of the components of cardiac metabolism. Drug Metab Dispos (2019) 47(6):673–88. doi: 10.1124/dmd.119.086611
13. Glatz JFC, Luiken JJFP, Nabben M. Cd36 (Sr-b2) as a target to treat lipid overload-induced cardiac dysfunction. J Lipid Atheroscler (2020) 9(1):66–78. doi: 10.12997/jla.2020.9.1.66
14. Schulze PC, Konstantinos Drosatos IJG. Lipid use and misuse by the heart. Circ Res (2017) 118(11):1736–51. doi: 10.1161/circresaha.116.306842
15. Angelini A, Pi X, Xie L. Dioxygen and metabolism; dangerous liaisons in cardiac function and disease. Front Physiol (2017) 8(1044):1–20. doi: 10.3389/fphys.2017.01044
16. Karwi QG, Uddin GM, Ho KL, Lopaschuk GD. Loss of metabolic flexibility in the failing heart. Front Cardiovasc Med (2018) 5(68):1–19. doi: 10.3389/fcvm.2018.00068
17. Pascual F, Coleman RA. Fuel availability and fate in cardiac metabolism: A tale of two substrates. Biochim Biophys Acta (2017) 1860(10):1425–33. doi: 10.1016/j.bbalip.2016.03.014
18. Varma U, Koutsifeli P, Benson VL, Mellor KM, Delbridge LMD. Molecular mechanisms of cardiac pathology in diabetes – experimental insights. Biochim Biophys Acta - Mol Basis Dis (2018) 1864(5):1949–59. doi: 10.1016/j.bbadis.2017.10.035
19. Wang T, Wang J, Hu X, Xian-Ju H, Guo-Xun C. Current understanding of glucose transporter 4 expression and functional mechanisms. World J Biol Chem (2020) 11(3):76–118. doi: 10.4331/wjbc.v11.i3.76
20. Tran DH, Wang ZV. Glucose metabolism in cardiac hypertrophy and heart failure. J Am Heart Assoc (2019) 8(12):e012673. doi: 10.1161/JAHA.119.012673
21. Szablewski L. Glucose transporters in healthy heart and in cardiac disease. Int J Cardiol [Internet] (2017) 230:70–5. doi: 10.1016/j.ijcard.2016.12.083
22. Maria Z, Campolo AR, Lacombe VA. Diabetes alters the expression and translocation of the insulin-sensitive glucose transporters 4 and 8 in the atria. PloS One (2015) 10(12):e0146033. doi: 10.1371/journal.pone.0146033
23. Karwi QG, Sun Q, Lopaschuk GD. The contribution of cardiac fatty acid oxidation to diabetic cardiomyopathy severity. Cells (2021) 10(11):3259. doi: 10.3390/cells10113259
24. Kota SK, Kota SK, Jammula S, Panda S, Modi KD. Effect of diabetes on alteration of metabolism in cardiac myocytes: Therapeutic implications. Diabetes Technol Ther (2011) 13(11):1155–60. doi: 10.1089/dia.2011.0120
25. Yamamoto T, Sano M. Deranged myocardial fatty acid metabolism in heart failure. Int J Mol Sci (2022) 23(2):996. doi: 10.3390/ijms23020996
26. Malfitano C, de Souza Junior AL, Carbonaro M, Bolsoni-Lopes A, Figueroa D, de Souza LE, et al. Glucose and fatty acid metabolism in infarcted heart from streptozotocin-induced diabetic rats after 2weeks of tissue remodeling. Cardiovasc Diabetol (2015) 14(1):1–10. doi: 10.1186/s12933-015-0308-y
27. Amaral N, Okonko DO. Metabolic abnormalities of the heart in type II diabetes. Diabetes Vasc Dis Res (2015) 12(4):239–48. doi: 10.1177/1479164115580936
28. Herrero P, Peterson LR, McGill JB, Matthew S, Lesniak D, Dence C, et al. Increased myocardial fatty acid metabolism in patients with type 1 diabetes mellitus. J Am Coll Cardiol (2006) 47(3):598–604. doi: 10.1016/j.jacc.2005.09.030
29. Lopaschuk GD, Karwi QG, Tian R, Wende AR, Abel ED. Cardiac energy metabolism in heart failure. Circ Res (2021) 128(10):1487–513. doi: 10.1161/CIRCRESAHA.121.318241
30. Nakamura M, Sadoshima J. Cardiomyopathy in obesity, insulin resistance and diabetes. J Physiol (2020) 598(14):2977–93. doi: 10.1113/JP276747
31. Zheng L, Li B, Lin S, Chen L, Li H. Role and mechanism of cardiac insulin resistance in occurrence of heart failure caused by myocardial hypertrophy. Aging (Albany NY) (2019) 11(16):6584–90. doi: 10.18632/aging.102212
32. Aroor AR, Chirag H. Mandavia and JRS. insulin resistance and heart failure. Curr Heart Fail Rep (2012) 8(4):609–17. doi: 10.1016/j.hfc.2012.06.005
33. Li A, Gao M, Jiang W, Qin Y, Gong G. Mitochondrial dynamics in adult cardiomyocytes and heart diseases. Front Cell Dev Biol (2020) 8:1555. doi: 10.3389/fcell.2020.584800
34. Jubaidi FF, Zainalabidin S, Mariappan V, Budin SB. Mitochondrial dysfunction in diabetic cardiomyopathy: The possible therapeutic roles of phenolic acids. Int J Mol Sci (2020) 21(17):6043. doi: 10.3390/ijms21176043
35. Dorn GW, Vega RB, Kelly DP. Mitochondrial biogenesis and dynamics in the developing and diseased heart. Genes Dev (2015) 29(19):31–51. doi: 10.1101/gad.269894.115
36. Lopaschuk GD. Fatty acid oxidation and its relation with insulin resistance and associated disorders. Ann Nutr Metab (2016) 68(3):15–20. doi: 10.1159/000448357
37. Gollmer J, Zirlik A, Bugger H. Mitochondrial mechanisms in diabetic cardiomyopathy. Diabetes Metab J (2020) 44(1):33–53. doi: 10.4093/dmj.2019.0185
38. Münzel T, Camici GG, Maack C, Bonetti NR, Fuster V, Kovacic JC. Impact of oxidative stress on the heart and vasculature: Part 2 of a 3-part series. J Am Coll Cardiol (2017) 70(2):212–29. doi: 10.1016/j.jacc.2017.05.035
39. Hurst S, Hoek J, Sheu S-S. Mitochondrial Ca2+ and regulation of the permeability transition pore. J Bioenerg Biomembr (2017) 49:27–47. doi: 10.1007/s10863-016-9672-x
40. Davidson SM, Adameová A, Barile L, Cabrera-Fuentes HA, Lazou A, Pagliaro P, et al. Mitochondrial and mitochondrial-independent pathways of myocardial cell death during ischaemia and reperfusion injury. J Cell Mol Med (2020) 24(7):3795–806. doi: 10.1111/jcmm.15127
41. Bonora M, Patergnani S, Ramaccini D, Morciano G, Pedriali G, Kahsay AE, et al. Physiopathology of the permeability transition pore: Molecular mechanisms in human pathology. Biomolecules (2020) 10(7):1–25. doi: 10.3390/biom10070998
42. Yan M, Li Y, Luo Q, Zeng W, Shao X, Li L, et al. Mitochondrial damage and activation of the cytosolic DNA sensor cGAS–STING pathway lead to cardiac pyroptosis and hypertrophy in diabetic cardiomyopathy mice. Cell Death Discov (2022) 8(1):1–12. doi: 10.1038/s41420-022-01046-w
43. Ou L, Zhang A, Cheng Y, Chen Y. The cGAS-STING pathway: A promising immunotherapy target. Front Immunol (2021) 12:1–15. doi: 10.3389/fimmu.2021.795048
44. Cai Z, Yuan S, Luan X, Feng J, Deng L, Zuo Y, et al. Pyroptosis-related inflammasome pathway: A new therapeutic target for diabetic cardiomyopathy. Front Pharmacol (2022) 13:1–11. doi: 10.3389/fphar.2022.842313
45. Wei J, Zhao Y, Liang H, Du W, Wang L. Preliminary evidence for the presence of multiple forms of cell death in diabetes cardiomyopathy. Acta Pharm Sin B [Internet] (2022) 12(1):1–17. doi: 10.1016/j.apsb.2021.08.026
46. Schwarz DS, Blower MD. The endoplasmic reticulum: Structure, function and response to cellular signaling. Cell Mol Life Sci (2016) 73(1):79–94. doi: 10.1007/s00018-015-2052-6
47. Liu MQ, Chen Z, Chen LX. Endoplasmic reticulum stress: A novel mechanism and therapeutic target for cardiovascular diseases. Acta Pharmacol Sin [Internet] (2016) 37(4):425–43. doi: 10.1038/aps.2015.145
48. Sicari D, Delaunay-Moisan A, Combettes L, Chevet E, Igbaria A. A guide to assessing endoplasmic reticulum homeostasis and stress in mammalian systems. FEBS J (2020) 287(1):27–42. doi: 10.1111/febs.15107
49. Gardner BM, Pincus D, Gotthardt K, Gallagher CM, Walter P. Endoplasmic reticulum stress sensing in the unfolded protein response. Cold Spring Harb Perspect Biol (2013) 5(3):1–15. doi: 10.1101/cshperspect.a013169
50. Adams CJ, Kopp MC, Larburu N, Nowak PR, Ali MMU. Structure and molecular mechanism of ER stress signaling by the unfolded protein response signal activator IRE1. Front Mol Biosci (2019) 6(11):1–12. doi: 10.3389/fmolb.2019.00011
51. Wang S, Binder P, Fang Q, Wang Z, Xiao W, Liu W, et al. Endoplasmic reticulum stress in the heart: Insights into mechanisms and drug targets. Br J Pharmacol (2018) 175(8):1293–304. doi: 10.1111/bph.13888
52. Bhattarai KR, Chaudhary M, Kim HR, Chae HJ. Endoplasmic reticulum (ER) stress response failure in diseases. Trends Cell Biol (2020) 30(9):672–5. doi: 10.1016/j.tcb.2020.05.004
53. Chen Y, Zhao X, Wu H. Metabolic stress and cardiovascular disease in diabetes mellitus: The role of protein O -GlcNAc modification. Arterioscler Thromb Vasc Biol (2019) 39(10):1911–24. doi: 10.1161/ATVBAHA.119.312192
54. Ruan Y, Zeng J, Jin Q, Chu M, Ji K, Wang Z, et al. Endoplasmic reticulum stress serves an important role in cardiac ischemia/reperfusion injury (Review). Exp Ther Med (2020) 20(6):1–1. doi: 10.3892/etm.2020.9398
55. Yang Q, Gao H, Dong R, Wu YQ. Sequential changes of endoplasmic reticulum stress and apoptosis in myocardial fibrosis of diabetes mellitus-induced rats. Mol Med Rep (2016) 13(6):5037–44. doi: 10.3892/mmr.2016.5180
56. Horáková L, Strosova MK, Spickett CM, Blaskovic D. Impairment of calcium ATPases by high glucose and potential pharmacological protection. Free Radic Res (2013) 47(1):81–92. doi: 10.3109/10715762.2013.807923
57. Takada A, Miki T, Kuno A, Kouzu H, Sunaga D, Itoh T, et al. Role of ER stress in ventricular contractile dysfunction in type 2 diabetes. PloS One (2012) 7(6):e39893. doi: 10.1371/journal.pone.0039893
58. Lee Y, Chakraborty S, Muthuchamy M. Roles of sarcoplasmic reticulum Ca2+ ATPase pump in the impairments of lymphatic contractile activity in a metabolic syndrome rat model. Sci Rep [Internet] (2020) 10(1):1–20. doi: 10.1038/s41598-020-69196-4
59. Zhihao L, Jingyu N, Lan L, Michael S, Rui G, Xiyun B, et al. SERCA2a: A key protein in the Ca2+ cycle of the heart failure. Heart Fail Rev (2020) 25(3):523–35. doi: 10.1007/s10741-019-09873-3
60. Wang L, Myles RC, Lee IJ, Bers DM, Ripplinger CM. Role of reduced sarco-endoplasmic reticulum Ca2+-ATPase function on sarcoplasmic reticulum Ca2+ alternans in the intact rabbit heart. Front Physiol (2021) 12:1–11. doi: 10.3389/fphys.2021.656516
61. Prola A, Nichtova Z, Pires Da Silva J, Piquereau J, Monceaux K, Guilbert A, et al. Endoplasmic reticulum stress induces cardiac dysfunction through architectural modifications and alteration of mitochondrial function in cardiomyocytes. Cardiovasc Res (2019) 115(2):328–42. doi: 10.1093/cvr/cvy197
62. Palomer X, Pizarro-Delgado J, Vázquez-Carrera M. Emerging actors in diabetic cardiomyopathy: Heartbreaker biomarkers or therapeutic targets? Trends Pharmacol Sci (2018) 39(5):452–67. doi: 10.1016/j.tips.2018.02.010
63. Tsalamandris S, Antonopoulos AS, Oikonomou E, Papamikroulis GA, Vogiatzi G, Papaioannou S, et al. The role of inflammation in diabetes: Current concepts and future perspectives. Eur Cardiol Rev (2019) 14(1):50–9. doi: 10.15420/ecr.2018.33.1
64. Kaur N, Guan Y, Raja R, Ruiz-Velasco A, Liu W. Mechanisms and therapeutic prospects of diabetic cardiomyopathy through the inflammatory response. Front Physiol (2021) 12:1–11. doi: 10.3389/fphys.2021.694864
65. Senatus L, Maclean M, Arivazhagan L, Egaña-gorroño L, López-díez R, Manigrasso MB, et al. Inflammation meets metabolism: Roles for the receptor for advanced glycation end products axis in cardiovascular disease. Immunometabolism (2021) 3(3):1–23. doi: 10.20900/immunometab20210024
66. Ramesh P, Yeo JL, Brandy EM, McCAnn GP. Role of inflammation in diabetic cardiomyopathy. Ther Adv Endocrinol Metab (2022) 13:1–13. doi: 10.1177/20420188221083530
67. Suryavanshi SV, Kulkarni YA. NF-κβ: A potential target in the management of vascular complications of diabetes. Front Pharmacol (2017) 8(7):1–12.10.3389/fphar.2017.00798
68. Bonilha I, Hajduch E, Luchiari B, Nadruz W, Le GW, Sposito AC. The reciprocal relationship between LDL metabolism and type 2 diabetes mellitus. Metabolites (2021) 11(12):807. doi: 10.3390/metabo11120807
69. Quagliariello V, Bonelli A, Paccone A, Buccolo S, Iovine M, Botti G, et al. Oxidized low-density lipoproteins increases nivolumab-induced cardiotoxicity through TLR4/NF-KB and NLRP3 pathways. Eur Heart J (2021) 42:2837. doi: 10.1093/eurheartj/ehab724.2837
70. Verboom L, Hoste E, van Loo G. OTULIN in NF-κB signaling, cell death, and disease. Trends Immunol (2021) 42(7):590–603. doi: 10.1016/j.it.2021.05.003
71. Yu H, Lin L, Zhang Z, Zhang H, Hu H. Targeting NF-κB pathway for the therapy of diseases: Mechanism and clinical study. Signal Transduct Target Ther (2020) 5(1):1–23. doi: 10.1038/s41392-020-00312-6
72. Skov J, Persson F, Frøkiær J, Christiansen JS. Tissue renin-angiotensin systems: A unifying hypothesis of metabolic disease. Front Endocrinol (Lausanne) (2014) 5:1–7. doi: 10.3389/fendo.2014.00023
73. Froogh G, Kandhi S, Duvvi R, Le Y, Weng Z, Alruwaili N, et al. The contribution of chymase-dependent formation of ANG II to cardiac dysfunction in metabolic syndrome of young rats: Roles of fructose and EETs. Am J Physiol - Hear Circ Physiol (2020) 318(4):985–93. doi: 10.1152/ajpheart.00633.2019
74. Singh VP, Le B, Khode R, Baker KM, Kumar R. Intracellular angiotensin II production in diabetic rats is correlated with cardiomyocyte apoptosis, oxidative stress, and cardiac fibrosis. Diabetes (2008) 57(12):3297–306. doi: 10.2337/db08-0805
75. Ferrario SA, Ahmad S. Chymase inhibitors for the treatment of cardiac diseases: A patent review (2010-2018. Expert Opin Ther Pat (2018) 28(11):755–64. doi: 10.1080/13543776.2018.1531848
76. Maeda Y, Inoguchi T, Takei R, Sawada F, Sasaki S, Fujii M, et al. Inhibition of chymase protects against diabetes-induced oxidative stress and renal dysfunction in hamsters. Am J Physiol - Ren Physiol (2010) 299(6):1328–38. doi: 10.1152/ajprenal.00337.2010
77. Arun KHS, Kaul CL, Ramarao P. High glucose concentration augments angiotensin-II mediated contraction via AT1 receptors in rat thoracic aorta. Pharmacol Res (2004) 50(6):561–8. doi: 10.1016/j.phrs.2004.06.001
78. Wang GY, Bi YG, Liu XD, Zhao Y, Han JF, Wei M, et al. Autophagy was involved in the protective effect of metformin on hyperglycemia-induced cardiomyocyte apoptosis and connexin43 downregulation in H9c2 cells. Int J Med Sci (2017) 14(7):698–704. doi: 10.7150/ijms.19800
79. Yamagishi SI, Matsui T. Advanced glycation end products, oxidative stress and diabetic nephropathy. Oxid Med Cell Longev (2010) 3(2):101–8. doi: 10.4161/oxim.3.2.11148
80. Koka V, Wang W, Xiao RH, Shokei KM, Truong LD, Lan HY. Advanced glycation end products activate a chymase-dependent angiotensin II-generating pathway in diabetic complications. Circulation (2006) 113(10):1353–60. doi: 10.1161/CIRCULATIONAHA.105.575589
81. Sasa V, Kathy G. Angiotensin II, from vasoconstrictor to growth factor: A paradigm shift. Circ Res (2014) 114(5):754–7. doi: 10.1161/circresaha.114.303045
82. Ichimiya T, Yamakawa T, Hirano T, Yokoyama Y, Hayashi Y, Hirayama D, et al. Autophagy and autophagy-related diseases: A review. Int J Mol Sci (2020) 21(23):8974. doi: 10.3390/ijms21238974
83. Parzych KR, Klionsky DJ. An overview of autophagy: Morphology, mechanism, and regulation. Antioxid Redox Signal (2014) 20(3):460–73. doi: 10.1089/ars.2013.5371
84. Wu D, Zhang K, Hu P. The role of autophagy in acute myocardial infarction. Front Pharmacol (2019) 10:551. doi: 10.3389/fphar.2019.00551
85. Yim WWY, Mizushima N. Lysosome biology in autophagy. Cell Discov [Internet] (2020) 6(1):1–12. doi: 10.1038/s41421-020-0141-7
86. Nishimura T, Tooze SA. Emerging roles of ATG proteins and membrane lipids in autophagosome formation. Cell Discov (2020) 6(1):1–18. doi: 10.1038/s41421-020-0161-3
87. Gallagher LE, Williamson LE, Chan EYW. Advances in autophagy regulatory mechanisms. Cells (2016) 5(2):24. doi: 10.3390/cells5020024
88. Zachari M, Ganley IG. The mammalian ULK1 complex and autophagy initiation. Essays Biochem (2017) 61(6):585–96. doi: 10.1042/ebc20170021
89. Al-Bari MAA, Xu P. Molecular regulation of autophagy machinery by mTOR-dependent and -independent pathways. Ann NY Acad Sci (2020) 1467(1):3–20. doi: 10.1111/nyas.14305
90. Tamargo-Gómez I, Mariño G. AMPK: Regulation of metabolic dynamics in the context of autophagy. Int J Mol Sci (2018) 19(12):3812. doi: 10.3390/ijms19123812
91. Dewanjee S, Vallamkondu J, Kalra RS, John A, Reddy PH, Kandimalla R. Autophagy in the diabetic heart: A potential pharmacotherapeutic target in diabetic cardiomyopathy. Ageing Res Rev (2021) 68:101338. doi: 10.1016/j.arr.2021.101338
92. Kanamori H, Takemura G, Goto K, Tsujimoto A, Mikami A, Ogino A, et al. Autophagic adaptations in diabetic cardiomyopathy differ between type 1 and type 2 diabetes. Autophagy (2015) 11(7):1146–60. doi: 10.1080/15548627.2015.1051295
93. Shao D, Tian R. Glucose transporters in cardiac metabolism and hypertrophy. Compr Physiol (2016) 6(1):331–51. doi: 10.1002/cphy.c150016
94. Munasinghe P, Katare R. Maladaptive autophagy in diabetic heart disease. Int J Clin Exp Physiol (2016) 3(4):155–65. doi: 10.4103/2348-8832.196893
95. Kobayashi S, Liang Q. Autophagy and mitophagy in diabetic cardiomyopathy. Biochim Biophys Acta - Mol Basis Dis (2015) 1852(2):252–61. doi: 10.1016/j.bbadis.2014.05.020
96. Li X, Ke X, Li Z, Li B. Vaspin prevents myocardial injury in rats model of diabetic cardiomyopathy by enhancing autophagy and inhibiting inflammation. Biochem Biophys Res Commun (2019) 514(1):1–8. doi: 10.1016/j.bbrc.2019.04.110
97. Xu X, Kobayashi S, Chen K, Timm D, Volden P, Huang Y, et al. Diminished autophagy limits cardiac injury in mouse models of type 1 diabetes. J Biol Chem (2013) 288(25):18077–92. doi: 10.1074/jbc.M113.474650
98. Bhattacharya D, Mukhopadhyay M, Bhattacharyya M, Karmakar P. Is autophagy associated with diabetes mellitus and its complications? A review. EXCLI J (2018) 17:709–20. doi: 10.17179/excli2018-1353
99. Kanamori H, Naruse G, Yoshida A, Minatoguchi S, Watanabe T, Kawaguchi T, et al. Morphological characteristics in diabetic cardiomyopathy associated with autophagy. J Cardiol (2021) 77(1):30–40. doi: 10.1016/j.jjcc.2020.05.009
100. Kobayashi S, Xu X, Chen K, Liang Q. Suppression of autophagy is protective in high glucose-induced cardiomyocyte injury. Autophagy (2012) 8(4):577–92. doi: 10.4161/auto.18980
101. Ouyang C, You J, Xie Z. The interplay between autophagy and apoptosis in the diabetic heart. J Mol Cell Cardiol (2014) 71:71–80. doi: 10.1016/j.yjmcc.2013.10.014
102. Venkatesh I, Makky K. Teaching epigenetic regulation of gene expression is critical in 21st-century science education: Key concepts & teaching strategies. Am Biol Teach (2020) 82(6):372–80. doi: 10.1525/abt.2020.82.6.372
103. Aristizabal MJ, Anreiter I, Halldorsdottir T, Odgers CL, McDade TW, Goldenberg A, et al. Biological embedding of experience: A primer on epigenetics. Proc Natl Acad Sci USA (2020) 117(38):23261–9. doi: 10.1073/pnas.1820838116
104. Zheng J, Cheng J, Zhang Q, Xiao X. Novel insights into DNA methylation and its critical implications in diabetic vascular complications. Biosci Rep (2017) 37(2):1–8. doi: 10.1042/BSR20160611
105. Pepin ME, Wende AR. Epigenetics in the development of diabetic cardiomyopathy. Epigenomics (2019) 11(5):469–72. doi: 10.2217/epi-2019-0027
106. Liu Z, Zhang Y, Qiu C, Zhu H, Pan S, Jia H, et al. Diabetes mellitus exacerbates post-myocardial infarction heart failure by reducing sarcolipin promoter methylation. ESC Hear Fail (2020) 7(4):1935–48. doi: 10.1002/ehf2.12789
107. Tsai CT, Wu CK, Lee JK, Chang SN, Kuo YM, Wang YC, et al. TNF-α down-regulates sarcoplasmic reticulum Ca2+ ATPase expression and leads to left ventricular diastolic dysfunction through binding of NF-κB to promoter response element. Cardiovasc Res (2015) 105(3):318–29. doi: 10.1093/cvr/cvv008
108. Madsen A, Höppner G, Krause J, Hirt MN, Laufer SD, Schweizer M, et al. An important role for DNMT3a-mediated DNA methylation in cardiomyocyte metabolism and contractility. Circulation (2020) 142(16):1562–78. doi: 10.1161/CIRCULATIONAHA.119.044444
109. Yerra VG, Advani A. Histones and heart failure in diabetes. Cell Mol Life Sci [Internet] (2018) 75(17):3193–213. doi: 10.1007/s00018-018-2857-1
110. Qi YK, Ai HS, Li YM, Yan B. Total chemical synthesis of modified histones. Front Chem (2018) 6(19):1–11. doi: 10.3389/fchem.2018.00019
111. Demetriadou C, Koufaris C, Kirmizis A. Histone n-alpha terminal modifications: Genome regulation at the tip of the tail. Epigenet Chromatin (2020) 13(1):1–13. doi: 10.1186/s13072-020-00352-w
112. Hussain S, Khan AW, Akhmedov A, Suades R, Costantino S, Paneni F, et al. Hyperglycemia induces myocardial dysfunction via epigenetic regulation of JunD. Circ Res (2020) 127(10):1261–73. doi: 10.1161/CIRCRESAHA.120.317132
113. Villeneuve LM, Natarajan R. The role of epigenetics in the pathology of diabetic complications. Am J Physiol - Ren Physiol (2010) 299(1):14–25. doi: 10.1152/ajprenal.00200.2010
114. Ke X, Lin Z, Ye Z, Leng M, Chen B, Jiang C, et al. Histone deacetylases in the pathogenesis of diabetic cardiomyopathy. Front Endocrinol (Lausanne) (2021) 12:889. doi: 10.3389/fendo.2021.679655
115. Deng J, Liao Y, Liu J, Liu W, Yan D. Research progress on epigenetics of diabetic cardiomyopathy in type 2 diabetes. Front Cell Dev Biol (2021) 9:1–10. doi: 10.3389/fcell.2021.777258
116. O’Brien J, Hayder H, Zayed Y, Peng C. Overview of microRNA biogenesis, mechanisms of actions, and circulation. Front Endocrinol (Lausanne) (2018) 9:402. doi: 10.3389/fendo.2018.00402
117. Rawal S, Ram TP, Coffey S, Williams MJA, Saxena P, Bunton RW, et al. Differential expression pattern of cardiovascular microRNAs in the human type-2 diabetic heart with normal ejection fraction. Int J Cardiol (2016) 202:40–3. doi: 10.1016/j.ijcard.2015.08.161
118. Mann VD, Mann DL. The emerging role of MicroRNAs in cardiac remodeling and heart failure. Circ Res (2008) 103(10):1072–83. doi: 10.1161/circresaha.108.183087
119. Nair RG, Nair S. Role of MicroRNA in diabetic cardiomyopathy: from mechanism to intervention. Biochim Biophys Acta (2017) 1863(8):2070–7. doi: 10.1016/j.bbadis.2017.03.013
120. Liu X, Liu S. Role of microRNAs in the pathogenesis of diabetic cardiomyopathy. BioMed Rep (2017) 6(2):140–5. doi: 10.3892/br.2017.841
121. Shantikumar S, Caporali A, Emanueli C. Role of microRNAs in diabetes and its cardiovascular complications. Cardiovasc Res (2012) 93(4):583–93. doi: 10.1093/cvr/cvr300
122. Ahmed U, Ashfaq UA, Qasim M, Ahmad I, Ahmad HU, Tariq M, et al. Dysregulation of circulating miRNAs promotes the pathogenesis of diabetes-induced cardiomyopathy. PloS One (2021) 16(4):1–14. doi: 10.1371/journal.pone.0250773
123. Li H, Fan J, Zhao Y, Zhang X, Dai B, Zhan J, et al. Nuclear miR-320 mediates diabetes-induced cardiac dysfunction by activating transcription of fatty acid metabolic genes to cause lipotoxicity in the heart. Circ Res (2019) 125(12):1106–20. doi: 10.1161/CIRCRESAHA.119.314898
124. Moorthy S, Koshy T, Srinivasan V, Silambanan S. Circulating biomarkers and MicroRNAs in the diagnosis, prognosis and treatment of diabetic cardiomyopathy-a review. Cardiol Cardiovasc Med (2020) 04(04):481–97. doi: 10.26502/fccm.92920145
125. Dai B, Li H, Fan J, Zhao Y, Yin Z, Nie X, et al. MiR-21 protected against diabetic cardiomyopathy induced diastolic dysfunction by targeting gelsolin. Cardiovasc Diabetol (2018) 17(1):1–17. doi: 10.1186/s12933-018-0767-z
Keywords: Diabetes Mellitus, heart failure, Diabetic Heart Failure, mechanisms, therapeutic targets
Citation: Mengstie MA, Abebe EC, Teklemariam AB, Mulu AT, Teshome AA, Zewde EA, Muche ZT and Azezew MT (2022) Molecular and cellular mechanisms in diabetic heart failure: Potential therapeutic targets. Front. Endocrinol. 13:947294. doi: 10.3389/fendo.2022.947294
Received: 18 May 2022; Accepted: 12 August 2022;
Published: 02 September 2022.
Edited by:
Ye Ding, Georgia State University, United StatesReviewed by:
Melissa Reichelt, The University of Queensland, AustraliaCopyright © 2022 Mengstie, Abebe, Teklemariam, Mulu, Teshome, Zewde, Muche and Azezew. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Misganaw Asmamaw Mengstie, bWlzZ2FuYXcxMThAZ21haWwuY29t
†ORCID: Misganaw Asmamaw Mengstie, orcid.org/000-0002-9711-8640
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.