 Amber Lockridge
Amber Lockridge John A. Hanover
John A. Hanover- Laboratory of Cell and Molecular Biology, National Institute for Diabetes and Digestive and Kidney Diseases, National Institutes of Health, Bethesda, MD, United States
Although traditionally considered a glucose metabolism-associated modification, the O-linked β-N-Acetylglucosamine (O-GlcNAc) regulatory system interacts extensively with lipids and is required to maintain lipid homeostasis. The enzymes of O-GlcNAc cycling have molecular properties consistent with those expected of broad-spectrum environmental sensors. By direct protein-protein interactions and catalytic modification, O-GlcNAc cycling enzymes may provide both acute and long-term adaptation to stress and other environmental stimuli such as nutrient availability. Depending on the cell type, hyperlipidemia potentiates or depresses O-GlcNAc levels, sometimes biphasically, through a diversity of unique mechanisms that target UDP-GlcNAc synthesis and the availability, activity and substrate selectivity of the glycosylation enzymes, O-GlcNAc Transferase (OGT) and O-GlcNAcase (OGA). At the same time, OGT activity in multiple tissues has been implicated in the homeostatic regulation of systemic lipid uptake, storage and release. Hyperlipidemic patterns of O-GlcNAcylation in these cells are consistent with both transient physiological adaptation and feedback uninhibited obesogenic and metabolic dysregulation. In this review, we summarize the numerous interconnections between lipid and O-GlcNAc metabolism. These links provide insights into how the O-GlcNAc regulatory system may contribute to lipid-associated diseases including obesity and metabolic syndrome.
Introduction
Lipid homeostasis is crucial to normal physiological function. Lipids provide substrates for anabolic synthesis, act as signaling molecules or membrane property modifiers and they constitute an energy dense form of nutrient storage that can supply regulated access to cellular fuel for high demand tissues or when glucose availability is low. In the context of human evolution, the relative scarcity of high fat foods placed a premium on maximal uptake and storage of dietary lipids to ensure against future periods of nutrient scarcity. Thus, the tongue and the intestine respond to elevated lipid content by signaling to the hedonic reward system of the brain to override homeostatic control of eating (i.e. hunger and satiety) and stimulate increased consumption. Intestinal enterocytes adapt to extract more dietary fatty acids (FAs) and secrete them into the bloodstream in the form of triacylglycerol (TAG) and peripheral tissues, namely the adipose and liver, are activated to expand their cellular uptake and lipid storage capacity. On the other hand, prolonged or excessive lipid accumulation is a source of considerable cellular toxicity and stress. To mitigate these concerns, dietary lipid exposure also provides negative feedback to normalize eating patterns after a transient period of hyperlipidemia. Many cell types mitigate the risk of lipotoxicity by sequestering TAG inside phospholipid coated lipoproteins called lipid droplets (LDs). Particularly in adipocytes and hepatocytes, these can then be released back into the bloodstream during the post-absorptive state. In the state of obesity, nutrient regulation of these carefully balanced controls is diminished leading to hyperphagia irrelevant to satiety, tonically elevated systemic lipid uptake and a loss of metabolic flexibility in the liver and adipose that fuels dysregulations of lipid and glucose homeostasis, inflammation and insulin resistance that are the hallmarks of metabolic syndrome.
Metabolic flexibility is key to maintaining physiological homeostasis in response to hyperlipidemia. To make decisions about hunger vs. hedonic eating, lipid opportunity vs. toxicity and FA storage vs. release, cells need input about acute nutrient conditions but also historic nutrient availability, energy demand, storage occupancy and reserve capacity as well as cellular and tissue level stress. This information is integrated and encoded by nutrient sensitive molecules like insulin, which alters cellular behavior through receptor induced signaling, and intracellular enzymes such as AMPK and O-GlcNAc Transferase (OGT) that directly modify target proteins through phosphorylation and O-linked β-N-acetylglucosamine (O-GlcNAc) modification, respectively. Protein O-GlcNAcylation has traditionally been framed in the context of glucose metabolism, related to the derivation of its substrate, UDP-GlcNAc, from a glycolytic branchpoint. However, there is a growing appreciation of this post-translational modification (PTM) as responsive to and a regulator of all classes of nutrient metabolism, including lipids. Not only are lipids also required for the material synthesis of UDP-GlcNAc but they influence the availability and activity of all the major O-GlcNAc regulatory enzymes. Lipid responsive O-GlcNAcylation patterns show remarkable diversity in terms of tissue and context specificity, substrate targeting and temporal dynamism and are implicated in both vital physiological functions and hyperlipidemic pathology. As such, dissecting the relationship between lipids and the O-GlcNAc regulatory system has become increasingly relevant to understanding the physiology and pathophysiology of lipid homeostasis, with particular relevance to the onset and progression of obesity.
Lipid influence on the O-GlcNAc regulatory system
Much of the classical depiction of O-GlcNAcylation as a nutrient-sensing modification comes from the incorporation of nutrient-derived metabolites through the various steps of the Hexosamine Biosynthetic Pathway (HBP), which produces UDP-GlcNAc. When used for post-translational O-GlcNAcylation, OGT catalyzes the transfer of the GlcNAc portion of this molecule onto the hydroxyl side chain of one or more serine and threonine residues inside a target protein. The O-GlcNAc regulatory system, as referenced in this review, is thus comprised of the enzymes that permit and facilitate HBP flux as well as the glycosylation enzymes - OGT and its GlcNAc-removing counterpart, O-GlcNAcase (OGA). Importantly, OGT, OGA and the HBP metabolites contribute to other molecular reactions as well (e.g. complex scaffolding, other glycosylation types) and so the activity and regulation of this system cannot be considered strictly synonymous with protein O-GlcNAcylation in all cases. Canonically, lipid influence over the O-GlcNAc regulatory system arises from the use of acetyl-CoA, derived from fatty acid oxidation (FAO) in many cell types, in the commitment step of UDP-GlcNAc synthesis. However, lipids also interact directly and indirectly with the expression, localization, protein binding and/or activity of all the major enzymes in this regulatory system. The partitioning of fructose-6-phosphate (F6P) between glycolysis and the HBP appears particularly relevant while the fat-sensitive targeting of OGT and OGA has been documented through a surprising diversity of mechanisms, possibly as a means towards context-specific substrate specificity. In addition, factors such as cell type, energy status, insulin signaling, lipid species and duration appear important in shaping unique relationships between hyperlipidemia and the O-GlcNAc regulatory system. A more precise delineation of these specific interactions is necessary, therefore, to understand the complexity and dynamism of both homeostatic and obesogenic lipid responses.
Protein O-GlcNAcylation
As a nutrient sensing post-translational modification (PTM), protein O-GlcNAcylation is utilized to varying degrees in all cell types but is particularly active in tissues and on proteins with high relevance to metabolism (1, 2). To date, thousands of O-GlcNAcylated proteins have been identified (3, 4), primarily in the cytoplasm and nucleus but also in various organelles (e.g. mitochondria, LDs) (5). Proteins can have one or many OGT target sites and the modification of those sites can be acutely transient, long-lasting or semi-permanent depending on the context. The effect of O-GlcNAcylation can alter a protein’s stability, transcriptional, enzymatic or protein-binding activity, sub-cellular localization and/or the patterning of other PTMs (e.g. phosphorylation) (6, 7) and these changes can be inhibitory or activating, depending on the target, cell type and cellular conditions. Since its discovery, a substantial literature has developed connecting increased O-GlcNAcylation with elevated glucose levels (8, 9) but this response is also seen in other conditions such as cellular stress (10) and hyperlipidemia.
Both high fat-diet (HFD) and genetic obesity have been corelated to increased O-GlcNAcylation in multiple tissues, with some notable exceptions. Based on models of varying dietary fat (40-65%) and duration (1-5 months), elevated O-GlcNAcylation was observed in mouse liver (11, 12), aorta (13), retina (14), kidney (15), white adipose tissue (16), intestine (17) and skeletal muscle (18) and in the rat heart (19) and cerebral arteries (20). A specific increase in nuclear O-GlcNAcylation was noted in the heart and liver but not kidneys of HFD rats (21). Young male swine also had higher cardiac O-GlcNAcylation after 3 months on an obesogenic hypercholesterolemic diet (22). Additionally, two mouse models of non-HFD obesity (db/db and ob/ob) showed higher kidney and liver O-GlcNAcylation, respectively (15, 23). By contrast, hippocampal O-GlcNAcylation was reduced by ~30% in 6 week (wk) HFD mice compared to normal chow controls (12). This was similar to findings in male human islets in which obesity (BMI≥30) was correlated to hypo-O-GlcNAcylation compared to lean controls (BMI ≤ 25) (24). In some tissues, the impact of hyperlipidemia was not unidirectional but temporally dynamic and biphasic. Mouse pancreatic islets and macrophages both showed higher O-GlcNAcylation during the early stages of diet-induced obesity (4-6 wks) but levels fell below standard chow fed controls after 3 months in both cases (24, 25). Therefore, the effects of in vivo hyperlipidemia on protein O-GlcNAcylation, while primarily potentiating in most tissues, were depressive in the hippocampus or in islets and macrophages after prolonged exposure (Table 1).
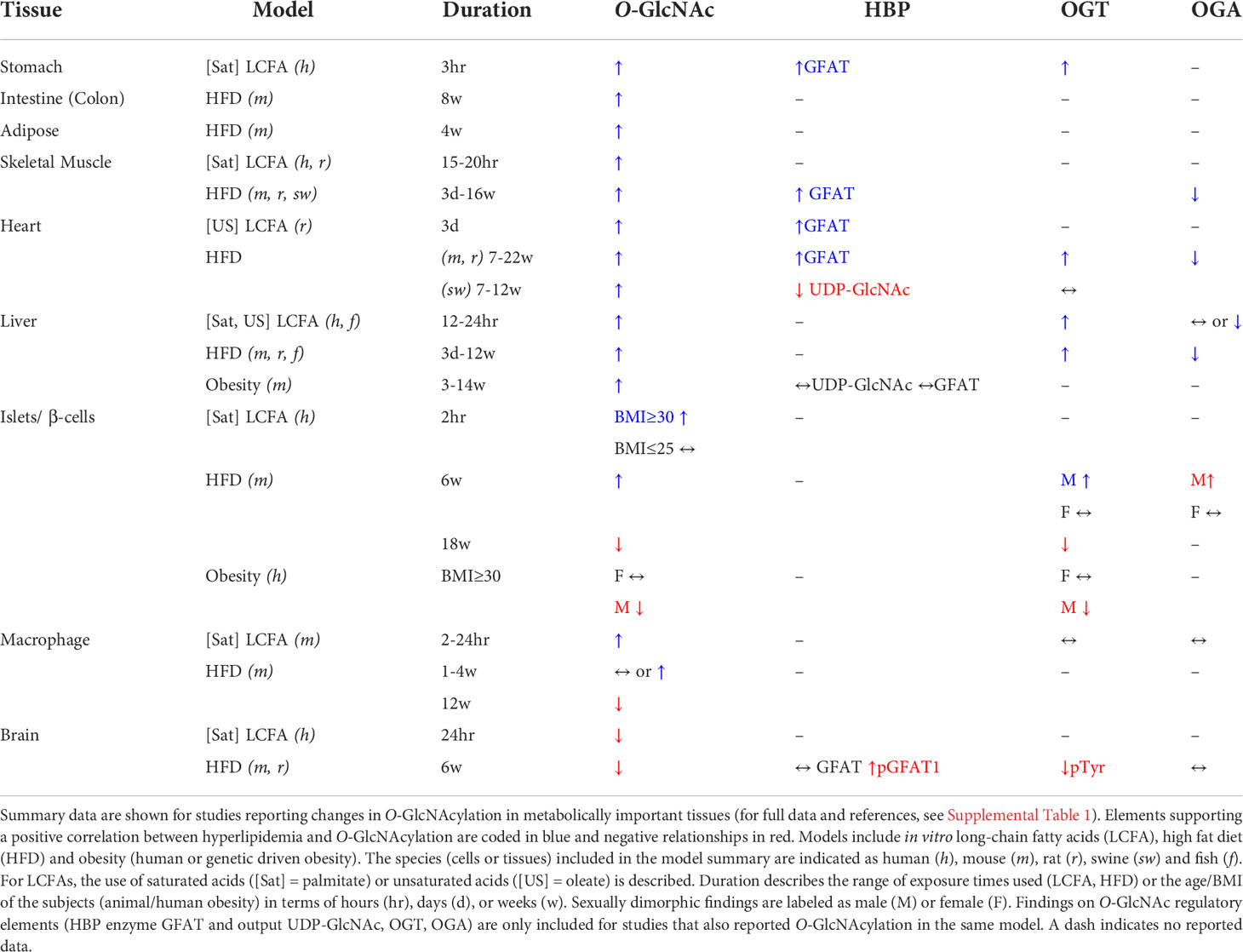
Table 1 Effects of hyperlipidemia on protein O-GlcNAcylation in different tissues.
Similar to the obesity models, in vitro exposure to lipids, especially long-chain fatty acids (LCFA), triggered a rise in O-GlcNAcylation in all but a select few cell types. Palmitate (PA), which is the most abundant saturated LCFA in most HFDs, increased total protein O-GlcNAcylation in immortalized cell lines from mouse and human liver (26, 27), rat retina (14) and two human-derived gastric cancer cell lines (28). PA also stimulated O-GlcNAcylation in primary isolated cells including rat L6 myotubes (29), kidney mesangial cells (30), mouse bone-marrow-derived macrophages (25) and human obese donor male islets (24). Notably, this latter observation was not seen for the lean donor cells, suggesting that pre-existing obesity may have sensitized the islets to PA’s acute effects on the O-GlcNAc regulatory system (24). By contrast, PA led to a 50% decrease in the O-GlcNAcylation of SHSY-5Y human neuroblastoma cells, mimicking the similar effect of HFD on the hippocampus (12). Oleate (OA), an endogenously abundant unsaturated LCFA, was reported to drive protein O-GlcNAcylation in primary hepatocytes from fish (31) and rat neonatal cardiomyocytes (32). Importantly, LCFA concentrations in these experiments, 100-600 uM PA or 400-800 uM OA, were within the physiological range for human plasma (33) although the durations of exposure varied widely by experiment, from 2 to 48 hours (see Supplementary Table S1 for study details). Beyond LCFAs, increased cellular O-GlcNAcylation was documented in response to multi-lipid enriched media for mouse oocytes (34), a biologically active ceramide analog (Cer6) in rat retinal cells (14) and short chain fatty acids in intestinal epithelial cells (17). Both the in vivo and in vitro outcomes are largely aligned with the perception of O-GlcNAcylation as a general marker of nutrient excess in most tissues. It is worth noting, however, that relatively few studies reported multiple timepoints, which is likely important in distinguishing the physiological and pathophysiological relevance of outcome.
A notable exception to the predominant pattern of hyperlipidemic hyper-O-GlcNAcylation was the lipid-stimulated depression of O-GlcNAcylation in the mouse hippocampus and human neuroblastoma cells. The rationale for this contrary effect is not definitive but several interesting hypotheses arise. Most brain cells, including neurons, have a low capacity for FAO, relying almost exclusively on glucose oxidation or sometimes ketones, to fuel cellular activity [see (35)]. At the same time, glucose-driven O-GlcNAcylation is neuroprotective against multiple cognitive disorders [for review (36)]. Lipotoxic insulin resistance in the brain disrupts glucose metabolism and has been proposed as a causal link between obesity and Alzheimer’s disease (12). Therefore, the rationale and/or mechanisms for lipid sensing and lipid modulation of protein O-GlcNAcylation may be different in these cells compared to others. For example, in hyper-O-GlcNAcylated cell types, lipids appear to partition F6P towards the HBP at the expense of glycolysis (see “UDP-GlcNAc Synthesis” for details). In cells that cannot alternately utilize fatty acids as a fuel source, this would come with considerable energetic costs, especially in electrically active neurons. To that point, neural hypo-O-GlcNAcylation was dependent on the low energy sensor AMPK, both in this model (12) and in glucose-deprived neuroblastoma cells (37). In addition to preserving glycolytic substrate, reducing neural O-GlcNAcylation may stimulate LD growth as a method to sequester toxic lipid excess. It was recently shown that decreased O-GlcNAcylation of the TATA box binding protein (TBP) shifts its influence over the transcription pre-initiation complex proteins, leading to broadly increased lipogenic gene expression associated with rat hippocampal LD accumulation (38). Hyperlipidemic O-GlcNAcylation patterns have not yet been characterized in areas of the brain more directly involved in lipid sensing and nutrient metabolism (e.g. the hypothalamus). However, fasting represents an endogenous state of low energy with high circulating free FAs (FFAs) and has been shown to increase O-GlcNAcylation in FAO-capable AgRP neurons of the hypothalamus while the opposite occurs in glucose-utilizing POMC neurons (discussed in section OGT activity and dietary lipid uptake). Therefore, the metabolic flexibility and lipotoxic vulnerability of a given cell type, even more than its anatomical location, may be a significant contributor to its O-GlcNAc lipid response.
Both in vivo and in vitro hyperlipidemia models demonstrate that lipids increase protein O-GlcNAcylation in most cell types. Macrophages and islet cells, however, showed a biphasic relationship between the duration of lipid exposure and O-GlcNAcylation, first potentiating and then inhibitory. This is consistent with the dynamism of their functional adaptation to progressive obesity, discussed later in this review [but see also (39, 40)]. Furthermore, hyperlipidemia suppressed O-GlcNAcylation in some brain cells, possibly related to the unique neuroprotective role of this modification and/or an enhanced sensitivity to lipotoxicity in the absence of lipid oxidation as a viable fuel source.
UDP-GlcNAc synthesis
One method for lipids to impact cellular O-GlcNAcylation levels is through the enzymes that regulate UDP-GlcNAc synthesis. The production of UDP-GlcNAc through the HBP starts with F6P, primarily derived from the early steps of glycolysis. If phosphorylated by phosphofructokinase 1 (PFK-1), F6P can continue down the glycolytic path as fructose-1,6-bisphosphate (FBP) or it can be diverted towards the HBP by the rate-limiting enzyme glucosamine-F6P-aminotransferase (GFAT). Conversely, F6P can be pulled back from either pathway through the reversal of these reactions, mediated by the enzymes FBPase-1 and glucosamine-6-phosphate deaminase (GNPDA), respectively. Thus, PFK/FBPase and GFAT/GNPDA represent critical choice points over the metabolic fate of F6P, which ultimately dictates the proportion of glucose dedicated to UDP-GlcNAc synthesis [see (41–43)]. Importantly, GFAT is expressed in two paralogous forms as Gfpt1/GFAT1 and Gfpt2/GFAT2. Despite having a similar role in the HBP and ~75% sequence conservation, these two proteins exhibit independent regulatory, developmental and tissue expression patterns (14, 44–47) suggesting unique but as yet poorly distinguished physiological roles. GFAT transfers an amide group from the amino acid glutamine (Gln) to convert F6P to glucosamine-6-phosphate (GlcN-6-P). Subsequently, GlcN-6-P acetyltransferase (GNAT) uses acetyl-CoA as a substrate to form N-acetylglucosamine-6-phosphate (GlcNAc-6-P), committing pathway flux in the forward direction. In the final steps of the HBP, UTP hydrolysis fuels the formation of uridine diphosphate-N-acetylglucosamine (UDP-GlcNAc). UDP-GlcNAc, and its epimers UDP-GalNAc and ManNAc, contribute to multiple pathways (e.g. mucin-like O-glycosylation, glycosaminoglycan synthesis), but UDP-GlcNAc availability is most sensitively linked to OGT-mediated O-GlcNAcylation (48). This pathway, and its putative regulation by lipids, are presented graphically as a component of Figure 1.
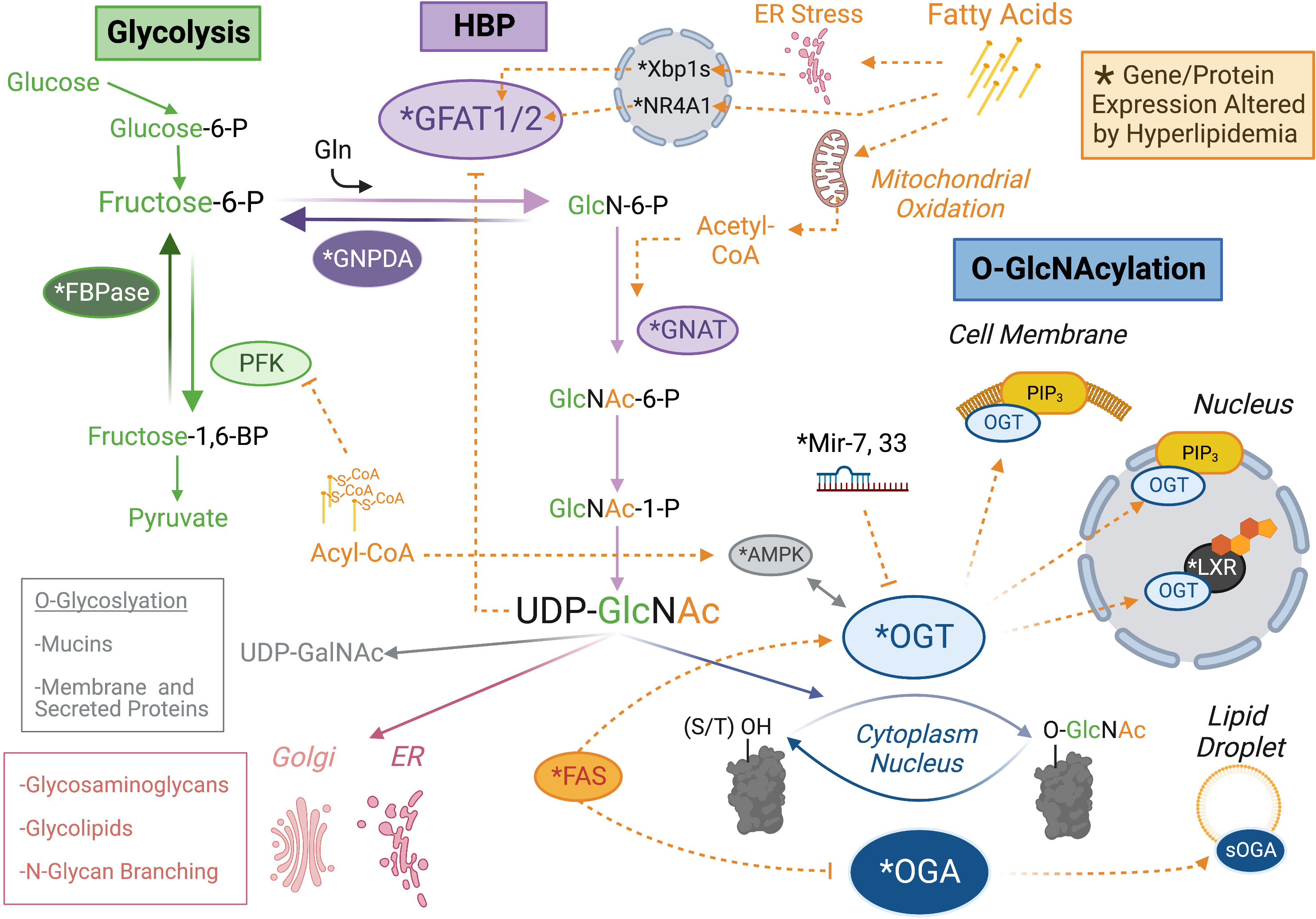
Figure 1 Lipid Control Over the O-GlcNAc Regulatory System. Protein O-GlcNAcylation (blue) is regulated by the activity or localization of the glycoslylation enzymes, O-GlcNAc Transferase (OGT) and O-GlcNAcase (OGA) through direct binding to lipid species (e.g. PIP3) or downstream of lipid sensitive proteins and miRNAs. The substrate for OGT is UDP-GlcNAc, which is synthesized through an offshoot of glycolysis (green) called the hexosamine biosynthetic pathway (HBP, purple). Lipid regulation of the HBP seems particularly focused on expression of the rate limiting enzyme GFAT and control over the metabolic fate of fructose-6-phosphate (P), which represents the divergence point between glycolysis and the HBP. Proteins (or microRNA) with an altered expression profile in obesity are indicated by asterisk (see main text for details). Orange dashed lines indicate agonistic (➔) or inhibitory (–|) relationships between lipid metabolites (e.g. Acyl-CoA) or lipid regulated species (e.g. Fatty Acid Synthase, FAS). It is important to note that UDP-GlcNAc has multiple utilizations in the cell, which might also be influenced by the regulatory relationship indicated. Post-translational modifications are not included for the sake of visual clarity. Created with BioRender.com.
The reported outcomes of hyperlipidemia on HBP activity have been varied, dependent on the experimental approach. UDP-GlcNAc was elevated in the skeletal muscle of ob/ob and KKAy genetically obese mice (49, 50) and in WT mice after 3 days HFD (51) or following in vivo lipid infusion in rats (52). Despite this, a later study found no effect of intralipid infusion on muscle HBP products (53) and UDP-GlcNAc levels were unchanged in the skeletal muscle of Zucker Diabetic Fatty rats compared to lean controls (54). Furthermore, UDP-GlcNAc was normal in the livers of the same ob/ob and KKAy mice with elevated muscle levels (49, 50). This last finding may be attributable, in part, to the higher Km of GFAT for F6P in rodent skeletal muscle compared to other tissue types (~2.4 vs. <0.5 mM), meaning GFAT could have been saturated in the liver (49). However, UDP-GlcNAc was decreased in the HFD swine heart simultaneous with a measure of increased O-GlcNAcylation (22), illustrating the fact that the static pool of UDP-GlcNAc is an imperfect measure of either HBP flux or HBP-driven O-GlcNAcylation. UDP-GlcNAc is rapidly depleted as a substrate and feeds back to inhibit GFAT, such that the impact of transient elevations on OGT activity may be missed (33, 53). In fact, a general limitation of the field is that HBP flux, defined as the rate of molecular turnover through this pathway, is not well understood.
As an alternate approach, several studies have focused on lipid regulation of the HBP enzymes themselves. Gfat mRNA was increased in HFD mouse retina (14) and porcine muscle (55) while protein levels were up in the HFD mouse aorta (56) and higher activity was directly measured in ob/ob mouse muscle and fat tissue (49). Similarly, human BMI was positively correlated to GFAT activity in non-diabetic human muscle cells (57). GFAT’s counter-regulatory enzyme partner, GNPDA, has also been implicated in human obesity (58) and was transcriptionally downregulated in the rat hypothalamus after 6 wks of HFD (59). The exception to these findings was an increase in inhibitory phosphorylation of GFAT1 in the mouse hippocampus, but this was consistent with the decrease in protein O-GlcNAcylation observed in that tissue after HFD (12). In vitro, human-derived myocytes showed increased GFAT protein and/or mRNA after a 20-hour exposure to saturated LCFAs (PA, stearate) while unsaturated LCFAs (OA, linoleate, palmitoylate) had little or no effect in the same study (33). However, a longer 3-day OA incubation did elevate GFAT protein in a separate investigation on rat neonatal cardiomyocytes (32). Alltogether, the evidence suggests that GFAT activity is closely correlated to the outcome of hyperlipidemia on O-GlcNAcylation, presumably through its effects on the availability of UDP-GlcNAc.
However, several studies suggest that GFAT1 and GFAT2 are differentially impacted by lipid exposure. Dai et al. reported that PA, Cer6 and HFD all increased rodent retinal Gfpt2 transcription, as well as protein O-GlcNAcylation, but Gfpt1 mRNA was increased, decreased or unchanged by each respective condition (14). In mouse oocytes, multi-lipid media supplementation triggered an increase in Gfpt1 after 8 hours, falling to baseline by 16 hours whereas Gfpt2 was only observed to decrease and at the latter timepoint (34). Some increases in GFAT transcription might be explained by lipotoxicity. ER stress, a well-characterized downstream effect of lipid accumulation (60), triggers the splicing activation of Xbp1, which can then act in the nucleus as a Gfpt1 transcription factor (61), driving up UDP-GlcNAc synthesis (61, 62). In addition, both HFD and in vitro lipids (PA, Cer6) increased the expression of lipotoxicity sensor and nuclear receptor protein NR4A1 leading to the specific upregulation of GFAT2, but not GFAT1, in the rodent retina (14). Differential nutrient regulation of the GFAT paralogs was also observed in Drosophila wherein dietary GlcN-6-P, the product of GFAT HBP activity, rescues lethality in gfat2 but not gfat1 knockout (KO) flies (63). Nevertheless, ectopic overexpression of either protein is sufficient to rescue loss of the other (47), suggesting that the nutrient access and/or responsivity of gfat1 and gfat2 expressing cells may be meaningfully distinct. Indeed, a recent study in WT flies confirms that of the two genes, only gfat2 expression is correlated with circadian eating patterns (64). By contrast, gfat1 may be more sensitive to lipid-induced stress, which is consistent with its proposed role in mediating the depressive effects of hyperlipidemia on neural O-GlcNAcylation. Unfortunately, much of the available data on lipids/obesity and GFAT does not differentiate between the paralogous forms, which may be a key detail underlying the diversity of hyperlipidemic outcomes in different cell types or conditions.
The availability of F6P for GFAT, which is rate-limiting in mammalian cells (49), is another target for lipid regulation of HBP flux. The enzyme PFK, which moves F6P towards glycolysis, is inhibited by LCFA-CoAs, an activated form of LCFA important in cellular lipid metabolism. LCFA-CoA binding to purified rabbit muscle PFK-1 in vitro exposed a tryptic cleavage site for the enzyme (65). Palmitoyl-CoA also triggered the inhibitory acylation of PFK-1 cysteine residues near the enzyme’s nucleotide-binding site. Furthermore, cytoplasmic citrate, which accumulates in response to high FAO activity, is also a well-known allosteric inhibitor of PFK [for review (66)]. Since PFK inhibition blocks glycolysis, this could increase F6P availability for GFAT-mediated HBP flux. Towards a similar outcome, LCFAs potentiate FBPase, which would pull F6P back from glycolysis by dephosphorylating FBP. PA or OA-supplemented media increased gene expression of both the liver and muscle isoforms of FBPase, as well as the HBP enzyme GNAT, in pancreatic β-cell line Min6 (67). These findings are consistent with a hypothesis of competitive glycolytic and HBP flux in response to normoglycemic but hyperlipidemic conditions.
In sum, HBP throughput appears to be a common target of lipid regulation with particular emphasis on GFAT and enzymes that direct the metabolic fate of F6P. In most cases, lipid exposure potentiated HBP-promoting proteins (GFAT, GNAT, FBPase-1) and inhibited HBP-detracting proteins (GNPDA, PFK-1), consistent with models of hyper-O-GlcNAcylation. However, the GFAT1 and GFAT2 paralogs appear to be independently regulated by lipids, dependent in part on cell type, lipid species and duration of exposure, energy status and cell stress. In particular, AMPK-dependent inhibition of GFAT1 was associated with the lipid depression of O-GlcNAcylation in some brain cells.
O-GlcNAc enzymes
In addition to UDP-GlcNAc synthesis, lipids may impact protein O-GlcNAcylation through the GlcNAc attachment and detachment enzymes, OGT and OGA. Although this reaction is catalyzed by only a single pair of enzymes, the capacity for multiple regulatory levels (i.e. expression, function, protein binding and/or localization) provides opportunities for substrate or context-specificity [for review (68)]. The ordered bi-bi enzyme mechanism utilized by OGT requires that UDP-GlcNAc bind prior to peptide binding (69). Both OGT and OGA act in a distributive rather than processive fashion suggesting that multiple cycles of binding and dissociation reactions are required (70). Moreover, both enzymes express multiple splice isoforms, including a shortened version of OGA (sOGA), which lacks the histone acetyltransferase domain of the longer and predominantly active protein and shows selective localization to LDs (71) and mitochondria (72) where lipid metabolic processes are highly active. The three OGT isoforms differ in localization but also in the length of their tetratricopeptide repeat (TPR) domains, which mediate non-catalytic protein binding, interactions that facilitate glycosyltransferase substrate selectivity as well as OGT’s non-enzymatic structural functions (e.g (73).). Furthermore, both OGT and OGA are subject to post-translational modifications, including O-GlcNAcylation and phosphorylation, that impact their function and protein interactions (74). Lipids appear to target OGT and OGA at multiple levels within this mechanistic diversity. It is important to acknowledge, however, that many counter-regulatory mechanisms exist to keep O-GlcNAcylation levels within a physiologically optimal range [see (75, 76)], such that effects on any particular element of the system may indirectly influence other elements.
In obesity models, OGT and/or OGA protein were not always altered but when they were, it was generally consistent with the change in protein O-GlcNAcylation. Among tissues with HFD hyper-O-GlcNAcylation – OGT was elevated in the mouse liver and heart (11, 27, 56) while OGA was suppressed in skeletal muscle and heart (18, 22, 56). In islets, depressed O-GlcNAcylation was accompanied by reduced OGT levels (24). Interestingly, higher islet O-GlcNAcylation after a moderate HFD period, was associated with either no change in OGT and OGA (females) or simultaneous unidirectional change (males), indicating an increased O-GlcNAc cycling rate. Contrary to islets, O-GlcNAcylation loss in the mouse HFD hippocampus was correlated with normal OGT protein and OGA activity but OGT PTMs were altered (decreased O-GlcNAcylation and tyrosine phosphorylation, increased Ser/Thr phosphorylation) (12). Recently, LCFA-CoAs were found to allosterically activate AMPK (77), a Ser/Thr kinase with high expression in hippocampal neurons (78) and increased activation in the HFD hippocampus (12). OGT phosphorylation by AMPK shifts its subcellular localization and substrate specificity in myotubes (79) and it would be interesting to see whether it has a similar role in the brain. Only one study reported on GFAT, OGT and OGA together, finding that HFD increased the first two and decreased the latter in the mouse aorta (56), working collectively to elevate O-GlcNAcylation by increasing substrate and transferase activity while decreasing O-GlcNAc removal.
Data from in vitro experiments is more preliminary but suggests that lipid regulation of OGT and OGA is less direct than for F6P and HBP enzymes. Two studies found increased OGT/ogt mRNA following LCFA exposures – with PA in human gastric cancer cells (28) or OA in primary hepatocytes from yellow croaker fish (31). However, PA in mouse liver and macrophage cells had no effect on transcription of Ogt or Oga (25, 26). A hyperlipidemia responsive Ogt transcription factor has not been definitively identified. Nevertheless, two microRNAs that are highly involved in lipid metabolism target OGT post-transcriptionally – Mir-7 [confirmed (80, 81)] and Mir-33 [putative (82)]. Mir-33 is co-expressed with the lipid transcription factor, SREBP2, in response to low cholesterol levels (83). Mir7 expression is bidirectionally regulated by in vitro LCFAs (84) and HFD feeding (85), possibly downstream of another lipid transcription factor, PPARα (86). In islets, Mir-7a expression was reduced after 5 wks HFD but increasingly expressed beyond that timepoint (85), consistent with the timeframe of elevated and then repressed OGT expression found in a separate HFD islet study (24). On the other hand, fatty acid synthase (FAS), which is the rate limiting enzyme of de novo lipogenesis, and also binds to and influences both OGT (87) and OGA (23) in an O-GlcNAc elevating direction. Pharmacological FAS inhibition dose-dependently suppressed HepG2 OGT protein levels (23). By contrast, FAS-bound OGA showed an ~85% decrease in activity compared to unbound OGA in a human-derived bone cancer cell line (88). Murine obesity increases the interaction between FAS and OGT in the liver, correlated to higher protein O-GlcNAcylation and lipogenic activity in that tissue (87). These connections provide plausible paths by which hyperlipidemia may influence the protein expression or activity of OGT and OGA.
As a means to guide substrate targeting, lipids have also been shown to influence the subcellular localization of the O-GlcNAc enzymes. OGT binds to phosphatidylinositol phosphate (PIP) and its derivatives, showing high specificity for PIP3 in vitro (89, 90), although a definitive binding domain has yet to be identified (91). PIP3 binding does not alter OGT enzymatic activity but it was reported to recruit OGT to the plasma membrane in response to serum or insulin in Cos7 kidney-derived cells (89) and HepG2 human liver cells (92). Following high-glucose culture, OGT moved to the nuclear membrane of MIN6 β-cells and co-immunoprecipitated with PIP3 in nuclear extracts (90). In all cases, the triggering stimulus was pharmacologically identified to be PI3 kinase activation, which converts membrane bound PIP2 to PIP3. Accordingly, loss of PTEN, the enzyme that dephosphorylates PIP3, was correlated to decreased OGT protein in the mouse liver (23). In addition to PIP3, OGT binds to oxysterol-activated liver-X receptor (LXR) and co-localizes with it in the nucleus of transfected Huh7 human hepatoma cells (93). LXR loss in mice does not alter OGA mRNA or OGT protein level but does selectively reduce nuclear O-GlcNAcylation (93). Protein expression of the LXRα paralog is enhanced by HFD in the rat liver (94–96) and in HepG2 cells by 24 hours in vitro PA (96). Less data is available on OGA localization patterns but acute OA potentiates the activity of the sOGA isoform and stimulates its accumulation on LD membranes in HeLa cells and 3T3-L1 pre-adipocytes (71) through an unknown mechanism. The extent and physiological relevance of these localization shifts in the endogenous cellular environment, whether enzymatic or non-catalytic, remain largely unknown but suggest an intriguing complexity to lipid regulation of OGT and OGA that may not be apparent in common endpoint measures of total protein level or activity.
Changes in OGT and OGA were sometimes but not always apparent in models of lipid altered O-GlcNAcylation. Plausible mechanisms linking lipids to OGT/OGA activity include lipid sensitive micoRNA transcription and interactions with nutrient/energy-sensing proteins that impact protein stability, enzymatic activity, PTM patterns and subcellular localization. As a general conclusion, lipids not only impact overall O-GlcNAc tone but are capable of diverse mechanistic influences that tune elements of the O-GlcNAc regulatory system in different ways. Simultaneously, hyperlipidemia can be expected to modify the ambient cellular environment, incorporating tissue and context dependent cues such as nutrient history, energy status and stress to further direct O-GlcNAcylation patterns towards specific outcomes.
OGT activity and dietary lipid uptake
As described in section Lipid influence on the O-GlcNAc regulatory system, protein O-GlcNAcylation is regulated by hyperlipidemia and obesity. However, this relationship is bidirectional. Many of the factors which contribute to circulatory and tissue hyperlipidemia are controlled, directly or indirectly, by O-GlcNAcylated proteins. This includes influence over total food consumption and specific lipid uptake, at the level of fat preference and intestinal absorption. The homeostatic drive to eat is controlled primarily in the hypothalamus by the balance between orexigenic (hunger) and anorectic (satiety) signaling. However, eating can also be stimulated by appetitive drives towards palatable foods (e.g. high fat, high sugar) that are regulated by the brain’s mesolimbic reward circuitry. Activity in these regions is responsive to the local cellular environment but also to secreted signals from the body about historic and acute conditions of nutrient availability, energy demand and dietary content. Beyond food quantity and content, actual lipid uptake depends on the rate and capacity of intestinal absorption. Ingested lipids are primarily broken down into FFAs to be packaged by enterocytes into TAG or other storage lipids, at the core of chylomicron particles that are secreted into the bloodstream. Obesity is associated with a loss of acute nutrient signaling in the brain, intestine and peripheral metabolic tissues combined with tonic physiological shifts that stimulate overconsumption, dietary lipid preference and enhanced uptake. OGT appears necessary for the nutrient-sensitive regulation of normal eating patterns but O-GlcNAcylation is also implicated in mechanisms underlying these obesogenic phenotypes. Figure 2 summarizes the known and suspected roles of the O-GlcNAc regulatory system in dietary lipid uptake under physiological and obese conditions, as detailed below.
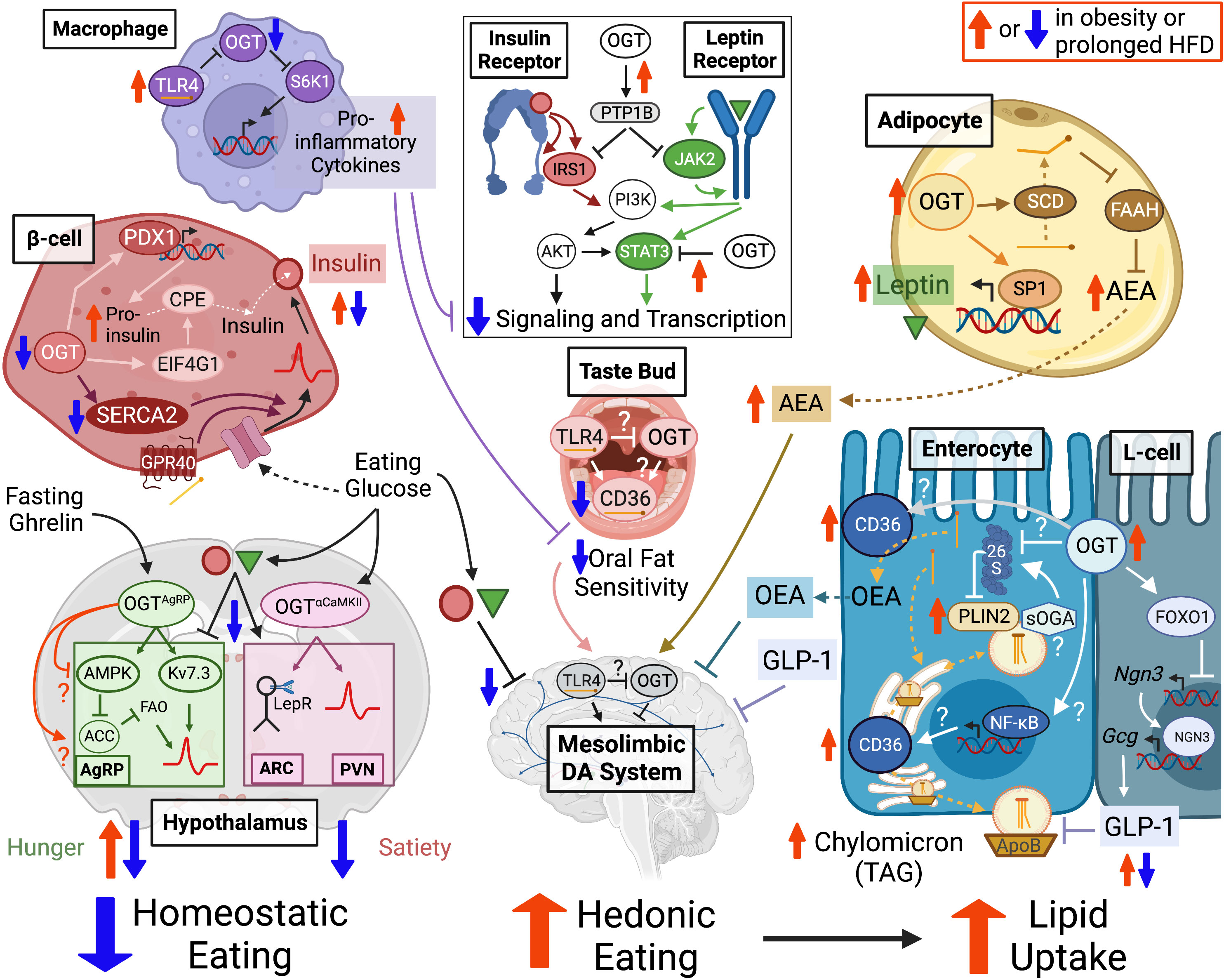
Figure 2 OGT supports neural activity in hypothalamic nuclei that regulate homeostatic eating drives including hunger-stimulating AgRP neurons and satiety-promoting neurons in the ARC and PVN. OGT in β-cells and adipocytes is important for the production of insulin and leptin hormones, which promote satiety and inhibit mesolimbic dopamine circuits that control hedonic eating motivation towards high fat foods. OGT activity facilitates transient lipid hyperphagia through the stabilization of adipocyte endocannabinoid AEA but may also contribute to counter-balancing feedback through oral fat sensitivity and orexigenic intestinal hormone secretion (i.e. OEA, GLP-1). The O-GlcNAc potentiated and lipid binding proteins CD36 and PLIN2 are promising candidates for regulating systemic lipid uptake through lingual fat detection and/or enterocyte extraction of dietary fatty acids into circulatory particles of chylomicron TAG. These mechanisms support a physiological eating pattern driven by homeostatic motivations punctuated by transient hedonic overconsumption of high palatability foods. However, OGT is also implicated in mechanisms underlying obesogenic pathophysiology. Prolonged hyperlipidemia decreases macrophage O-GlcNAcylation to disinhibit TL4-mediated pro-inflammatory cytokine secretion that can blunt insulin and leptin receptor sensitivity as could OGT targeting of receptor signaling modulators PTP1B and STAT3 in multiple cell types. Persistent high fat exposure decreases O-GlcNAcylation in β-cells but potentiates it in adipocytes and intestinal epithelial cells, potentially contributing to the high basal but impaired nutrient regulation of mature insulin and leptin secretion, elevated AEA levels and increased dietary lipid extraction that characterize a shift in obesity away from homeostasis and towards the chronic overconsumption of fat. OGT in this diagram represents the output of OGT activity (e.g. O-GlcNAcylation or protein interactions), whether driven by UDP-GlcNAc synthesis, OGA or OGT itself. “?” indicates a regulatory mechanism that has been demonstrated in other cell types but only hypothesized in the current setting. Dashed arrows show movement and solid arrows show effect, whether potentiating (–>) or inhibitory (–|). A double arrow combination (1 up, 1 down) indicates an increase in tonic tone with a depression in nutrient-responsive potentiation. Created with BioRender.com. Abbreviations: Agouti-related protein (AgRP) expressing, Arcuate Nucleus (ARC), Paraventricular Nucleus (PVN), O-GlcNAc Transferase (OGT), Dopamine (DA), N-arachidonoylethanolamine (AEA), Oleoylethanolamide (OEA), Cluster of Differentiation 36 (CD36), Triacylglycerol (TAG), Protein Tyrosine Phosphatase 1B (PTP1B), short-isoform O-GlcNAcase (sOGA), Perilipin 2 (PLIN2), Toll-Like Receptor 4 (TLR4).
Hunger
Hunger is stimulated by the activity of orexigenic NPY/AgRP neurons in the hypothalamic arcuate (ARC) nucleus in response to fasting conditions. Progressively during the pre-prandial period, gastric ghrelin (97) and adipocyte-secreted uridine and FFAs (98) are released into the bloodstream where they can be taken up by cells in the hypothalamus. Ghrelin and purine receptor activation, the latter from uridine-driven UDP synthesis, potentiate NPY/AgRP neural activity (99, 100). AgRP activity subsequently inhibits anorectic signaling in the brain’s satiety centers, namely ARC POMC neurons and the paraventricular nucleus (PVN), as well as influencing peripheral glucose metabolism [for review (101)]. ARC neurons are also capable of direct nutrient sensing of glucose and fatty acids [see (102)]. Fasting increases circulatory FFAs in the absence of elevated glucose/insulin (see section O-GlcNAcylation in lipid storage and release ). Moderate LCFA uptake under these conditions stimulates AgRP neural activity by fueling FAO-dependent ATP production. This is facilitated by ghrelin receptor signaling, which mitigates negative feedback on the FAO rate from lipogenesis and oxidative stress (103). At the same time, basal glucose levels in the fasting state limit energetic support for anorectic POMC neurons, which derive ATP through glucose oxidation (104). Thus, a suppression in satiety signaling works with increasing hunger signals to provide a building trigger to eat.
Obesity decreases fasting-stimulated hunger while maintaining anti-satiety signaling during all nutrient states. In mice, obesity abrogates the fasting-induced rise in serum uridine (105). By contrast, post-prandial uridine is elevated in obese humans (106) and hypothalamic UDP is increased in obese mice (100). Similarly, fasting levels of ghrelin are reduced in human obesity but so is the magnitude of meal-induced ghrelin suppression (101, 107). Ghrelin resistance develops in AgRP neurons, depressing their stimulated activation, but inhibition of the PVN is maintained (101). While short-term high fat feeding in rats enhances AgRP FAO capacity (108), contributing to HFD hyperphagia (see “Hedonic Eating”) long-term HFD desensitizes AgRP neurons to both fasting and dietary fat exposure (109) and abrogates the glucose sensitivity of POMC neurons (110) in mice. These changes demonstrate the importance of AgRP activity in lipid-sensitive physiological responses and show how chronic hyperlipidemia uncouples orexigenic signaling from its appropriate nutrient context.
Protein O-GlcNAcylation has been implicated in AgRP excitability and ghrelin sensitivity. Both fasting and ghrelin increase OGT and O-GlcNAcylation levels in ARC AgRP neurons, specifically including the Kv7.3 potassium channel (111). Kv7.3 potentiates neural firing rate by helping to re-polarize the cell after an action potential, which shortens the inhibitory period before the next depolarization. Mutagenic loss of the primary Kv7.3 O-GlcNAc site decreased its activity at depolarized potentials, correlating to a reduction in AgRP excitation rate in AgRP-Ogt KO mice. Furthermore, these mice were insensitive to ghrelin or fasting induced suppression of thermogenesis in retroperitoneal white adipose tissue, which the authors show is driven by AgRP activation. Despite these changes, AgRP-Ogt KO mice had no food intake phenotype. However, this may be due to the genetic loss occurring early in development in this model. AgRP neuronal ablation in neonates also has little effect on feeding but when induced in adults leads to rapid starvation (112). It’s possible, therefore, that acute and post-developmental changes in AgRP OGT activity in response to ghrelin or fasting would support hunger signaling.
OGT activity may also contribute to AgRP ghrelin sensitivity through its influence on FAO capacity. In a fed or glucose abundant state, lipogenesis typically suppresses FAO through the inhibition of its gatekeeper enzyme, CPT1, by the metabolic product of the acetyl-coA carboxylase (ACC) enzyme (103). In the pre-prandial period, not only are lipogenic stimulants low but AgRP ghrelin receptor signaling potentiates FAO flux by activating AMPK to inhibit ACC through a pair of sequential phosphorylation reactions (pAMPK activated, pACC inhibited). A similar mechanism for the upregulation of FAO has been observed in peripheral tissues and is subject to regulation by O-GlcNAcylation. Oral O-GlcNAc and adipose (aP2-cre) GFAT overexpression (OE) increase pAMPK/pACC levels in murine liver (113) and fat (114), respectively. Importantly, these observations were made in tissues from fasted animals and in the former study, no differences were found in the livers of fed mice (113). In perfused rat hearts, GFAT inhibition (azaserine) depresses pAMPK/pACC and reverses GlcN’s potentiation of FAO (115). AMPK activity may be further driven by a potentiating O-GlcNAcylation of the AMPK α1 catalytic subunit, which was increased in the adipocyte GFAT OE model (114). Like the Kv7.3 results, these data suggest that O-GlcNAcylation in AgRP neurons may maintain their physiological functions, including orexigenesis and lipid sensing.
Nevertheless, some studies have characterized a negative relationship between OGT activity and the regulatory mechanisms of FAO. Multiple models targeting OGT or OGA have correlated increased O-GlcNAc tone with a systemic shift away from FAO and towards glucose oxidation (16, 116–118). A similar respiratory shift is believed to underlie the suppression of AgRP neural activity in response to exogenously induced hypothalamic hyperlipidemia [see (119)]. In addition, the OGT/AMPK relationship is complex and contextual (120). O-GlcNAcylation has been shown to potentiate AMPK, as referenced above, or inhibit it, dependent on the pre-existence (or not) of activating conditions such as low glucose. Furthermore, O-GlcNAc promoting conditions have led to increased ACC transcription (121–125), including of the specific mitochondrial ACC2 isoform associated with CPT1/FAO inhibition (121), while direct O-GlcNAcylation of the lipogenically predominant ACC1 inhibits its activity in CD4+ T-cells (126). Notably, many of these ACC upregulations were exclusive to the fed state (123, 124), independent of AMPK activity (126), and/or associated with O-GlcNAc potentiated lipogenic transcription factors (e.g. ChREBP (124), SREBP1 [125)]. It is reasonable to hypothesize that these relationships are less relevant in the fasting environment of elevated AgRP OGT activity, except perhaps in the case of obesity and its effects on the fasting milieu (e.g. systemically elevated glucose/insulin/lipids and ghrelin receptor desensitization). In this case, a suppressive effect of O-GlcNAcylation on AgRP FAO could contribute to the nutrient insensitivity and loss of fasting-stimulated hunger associated with the obese state.
AgRP/NPY neural activity and ghrelin response underlie the pre-prandial hunger drive and are important for hypothalamic lipid sensing. Fasting and ghrelin-stimulated OGT activity in these neurons is linked to increased firing rate and ghrelin sensitivity but a direct evidential connection to consumptive outcomes has not yet been made. Nevertheless, the lipogenesis-FAO counter-regulatory relationship, which is essential for diet-related AgRP functions, is also well-documented to be under context-dependent O-GlcNAc control in peripheral cell types. This raises intriguing questions about the orexigenic consequences of the nutrient-uncoupled obese OGT interactome.
Satiety
Food consumption is associated with an inhibition of orexigenic signaling and increased satiety factors. Following a meal, stomach distension and gastrointestinal nutrient signaling inhibit ghrelin and stimulate the release of satiety hormones [e.g. gastric leptin (127) and intestinal CCK, PYY, GIP (128), GLP-1 (129)]. Most of these activate local vagal nerves that run to the brainstem and regulate gastric emptying, suppress hunger signaling and promote fullness. Incretins (GIP, GLP-1) and post-prandial glucose trigger the pancreatic secretion of insulin, which is a multi-potent anorectic hormone. Insulin and incretins inhibit AgRP neurons and upregulate POMC and PVN neural activity to suppress feeding and promote systemic energy storage. Adipocyte-secreted leptin has similar effects in the brain but also acts as an insulin sensitizer and suppresses circulatory uridine (98) and orexigenic endocannabinoid level in the hypothalamus (130). Leptin does not increase acutely following a meal (131) but as a response to basal adiposity/insulin levels and influences overall perception of satiety.
As in AgRP neurons, dietary hyperlipidemia leads to a complex and multiphasic response in satiety-regulating cells. High fat consumption initially triggers a short-term drop in leptin levels that contribute to transient HFD hyperphagia (131–133) and potentiates glucose-stimulated insulin secretion to shore up nutrient uptake and storage (40, 134). Established obesity, on the other hand, is characterized by constitutive basal hyperleptinemia and hyperinsulinemia but with impaired nutrient regulation of secretion. In addition, insulin and leptin resistance develops in numerous cell types, including ARC neurons [see (101)]. Receptor resistance and numerous other cellular pathologies are exacerbated by a chronic state of systemic inflammation. Similar to orexigenic factors, then, satiety signaling in obese individuals becomes less coupled to the nutrient environment while shifts in hypothalamic insulin and leptin sensitivity contribute to an overactivation of post-prandial AgRP neurons but a suppression of POMC and PVN activity that promotes increased food intake.
Conditional OGT loss studies in the brain tie protein O-GlcNAcylation to hypothalamic satiety processing. Adult tamoxifen induced Ogt KO in the mouse forebrain (αCaMKII-CreERT2), including the hypothalamus, leads to transient obesity due to increased meal size and duration (135, 136). Lagerlof et al. attributed this to a decrease in the excitability of a subpopulation of CamKIIα+/OGT-deficient neurons in the PVN, re-iterating the in vivo phenotype through stereotactically-targeted OGT deletion in these cells (135). Additionally, they show that PVN CamKIIα+ (but not adjacent CamKIIα-) neural O-GlcNAcylation is bidirectionally regulated by glucose (increased) and fasting (decreased). In vivo, αCaMKII-CreERT2 Ogt KO prevented activation of these cells following food intake. Interestingly, hypothalamic O-GlcNAcylation was recently suggested to function as a type of satiation memory whereby prior nutrient intake can influence subsequent consumption and energy expenditure (137). In this model, a larger meal size induces more O-GlcNAcylation in brain satiety centers such as the PVN. As protein O-GlcNAcylation can be sustained longer than its acute nutrient stimulation, this then serves as a form of long-term potentiation, lowering the excitation threshold for subsequent satiety signaling. It would be interesting to see whether O-GlcNAcylation could serve as a nutrient context memory in AgRP neurons as well, for example contributing to the pro-thermogenic effect of intermittent fasting (138). Regardless, this data further supports the notion that hypothalamic OGT activity is an important regulator of homeostatic eating in both the pre- and post-prandial state.
In addition to changes in PVN O-GlcNAcylation and excitation, αCaMKII-CreERT2 Ogt KO mice exhibit significant neuronal cell loss in multiple brain regions. Dai et al. reported a 60% decrease in leptin receptor (LepR) expressing hypothalamic neurons, primarily in the ARC nucleus, at a timepoint two weeks later than used in the Lagerlof study (136). A loss of hypothalamic LepR activity is the driving cause of hyperphagic obesity in the db/db mouse model and believed to contribute to both decreased satiety and increased leptin secretion in the state of obesity. Shortly following induction, αCaMKII-CreERT2 Ogt KO mice develop systemic hyperleptinemia, prior to significant weight gain, which is consistent with a loss of LepR-mediated hypothalamic feedback inhibition over peripheral leptin production. Consequently, neuronal OGT may contribute to central satiety signaling through both leptin-dependent and independent mechanisms.
In addition to its central nervous system (CNS) effects, O-GlcNAcylation in the periphery directly promotes the secretion of many anorectic hormones (e.g. leptin, insulin) and is required for their upregulation by high fat feeding (24, 139). Leptin gene expression and serum levels are increased in rodent models of adipocyte hyper-O-GlcNAcylation including GlcN infusion (140), GFAT OE [Glut4-cre (141), ap2-cre (142)] and Glut4-cre OGT OE (143). By contrast, adipocyte-specific OGT loss depressed leptin transcripts and circulatory protein in HFD mice while having no effect on leptin levels in standard chow fed mice (139). These expression changes likely involve O-GlcNAcylation of the Sp1 transcription factor (144). Deletion of the Sp1 binding site in the leptin promoter depresses both basal and GlcN-stimulated transcription in vitro in 3T3-L1 adipocytes (145). Based on these studies, hyperlipidemia stimulated O-GlcNAcylation of adipose tissue, as described in section Lipid influence on the O-GlcNAc regulatory system, is a plausible contributor to progressive hyperleptinemia in obese individuals.
In the last decade, a considerable literature has developed to support the requirement of β-cell OGT activity for the efficient and lipid-responsive secretion of insulin. β-cell Ogt KO mice (Rip-cre) are severely hypoinsulinemic due to depressed β-cell mass and insulin transcription as well as proinsulin maturation deficits (146). This has been attributed, in part, to O-GlcNAc regulation of the transcription factor Pdx1, a master regulator of both pancreatic and β-cell development and function [see (147–149)]. Pdx1 O-GlcNAcylation increases its DNA binding and potentiates insulin secretion in Min6 insulinoma cells (150). Pdx1 protein levels were also depressed in OGT depleted β-cells (146, 151). In addition, O-GlcNAcylation of the translation initiation protein eIF4G1 has been implicated in the proinsulin processing deficits of β-cell Ogt KO mice, upstream of the protein stability of prohormone convertase enzyme CPE (152). More specific to the hyperlipidemic context, induced adult deletion of β-cell OGT prevents adaptive hyperinsulinemia during early high fat feeding and abolishes LCFA potentiation of glucose stimulated insulin secretion (FASIS) (24). This latter impairment was rescued by pharmacological activation of the ER calcium importer and OGT targeted protein SERCA2. Pdx1 O-GlcNAcylation has also been connected to FASIS through its transcriptional upregulation of the LCFA receptor GPR40 (90). Interestingly, the timing of the rise and fall of mouse islet (~80% β-cells) O-GlcNAcylated proteins in response to high fat feeding (24) mirrors the transient potentiation of nutrient-stimulated insulin secretion under the same conditions (24, 134). Consequently, the decrease in islet OGT activity observed in mouse and human models of progressed obesity (see Table 1 summary) is consistent with the high proinsulin levels and acute nutrient desensitization also seen in those models.
Central and peripheral leptin and insulin resistance are also key obesity pathologies that have been definitively linked to O-GlcNAcylation in the latter system. Leptin and insulin receptor signaling depend on many of the same OGT targeted proteins (e.g. IRS-1, PI3K, STAT3, PTP1B). The insulin receptor is an autophosphorylating tyrosine kinase that initiates the sequential phosphorylation and activation of IRS, PI3K and especially Akt, which has a multitude of transcription and protein regulatory roles [see (153)]. LepR activation relies on ligand-stimulated JAK2 kinase, which similarly stimulates the phosphorylation of IRS/PI3K/Akt but also STAT3 and its downstream transcriptional programs [for review (154)]. Hypothalamic loss of either insulin or leptin receptors leads to hyperphagic obesity in rodents (51, 155), as does the specific disruption of leptin stimulated STAT3 phosphorylation (156). Protein tyrosine phosphatase 1B (PTP1B) is a negative regulator of insulin and leptin receptor sensitivity in multiple tissues through the dephosphorylation of IRS1 and JAK2, respectively, and is a major contributor to obesity precipitated resistance to these hormones in the brain (157, 158). PTP1B and STAT3 have been characterized as OGT targets in the liver in association with diminished insulin and leptin signaling, respectively (27, 159). In HepG2 cells, PA increased PTP1B O-GlcNAcylation and potentiated its activity. By contrast, mutagenic loss of PTP1B’s OGT target sites rescued hyperlipidemia induced insulin resistance. The consequences of STAT3 O-GlcNAcylation are more complicated. Two activating phosphorylation sites have been characterized for STAT3, pY705 and pS727, which appear differentially regulated by STAT3 O-GlcNAcylation under hyperglycemic conditions (159, 160). Nevertheless, Zimmerman et al. showed that HepG2 GFAT inhibition (azaserine) interferes with the specific leptin activation of both residues (159). Although these mechanistic investigations were conducted in liver cells, they are suggestive that excess O-GlcNAcylation in the hypothalamus, as much as its absence, could contribute to a loss of sensitivity to nutrient satiety signals. To that end, GlcN infusion in rats prevents leptin’s suppression of food intake (161) and a pan-neuronal loss of OGA (Nestin-cre) results in hyperleptinemic obese mice (162), consistent with a phenotype of central leptin resistance. Interestingly, food intake is not altered in the Oga KO model, which manifests during early gestation. But like the AgRP neuronal ablation models, this may speak more to the high plasticity of food regulatory mechanisms during early development. Models of adult conditional O-GlcNAc modification in specific populations of hypothalamic neurons under varying conditions of lipid exposure would be helpful in clarifying the role of the O-GlcNAc regulatory system in mediating CNS leptin and insulin sensitivity as well as homeostatic eating.
In sum, OGT appears to be required for normal satiety signaling in the brain by supporting hypothalamic excitability in the PVN and the viability of leptin sensitive neurons in the ARC. In addition, protein O-GlcNAcylation in adipose and pancreatic tissue supports the basal and lipid-mediated secretion of leptin and insulin as both satiety hormones and master regulators of nutrient metabolism. On the other hand, OGT activity in liver cells has been linked to insulin and leptin resistance through STAT3 and PTP1B dependent pathways that are known to drive receptor desensitization in the hypothalamus, specifically in response to obesity. The potential for O-GlcNAc-driven mechanisms to contribute to physiological homeostasis as well as obesogenic pathology was also noted in the previous sub-section on AgRP-mediated hunger.
Hedonic eating
The hypothalamus is not the only brain region that contributes to food intake. Mesolimbic dopamine circuitry controls hedonic eating, associated with food cravings and motivation towards high palatability, high calorie foods. In addition, inputs from regions related to self-regulation, emotion and memory play a role, particularly in humans (163). In lean individuals, acute exposure to high fat foods triggers HFD hyperphagia due to the aforementioned changes in homeostatic signaling as well as the FA stimulated activation of hedonic circuitry. This is followed by various repressive mechanisms that work to limit prolonged or excessive caloric intake. This positive-negative feedback loop is exemplified by oral fat detection. During a meal, FAs derived from food bind to CD36 receptors on the tongue (164, 165) to stimulate mesolimbic neural activity (166). However, lingual CD36 mRNA and protein is also rapidly depressed by lipid exposure, to two-fold within an hour of re-feeding in mice (167). On a slightly longer timescale, high fat consumption stimulates the adipocytic synthesis and secretion of lipid-derived endocannabinoids (e.g. anandamide/AEA, 2-AG) that promote both orexigenic and hedonic eating drives (168, 169). In terms of negative feedback, dietary fat absorption also potentiates the secretion of the anorectic hormones insulin, GLP-1 and oleoylethanolamide (OEA), which suppress mesolimbic activation [for review (170)]. This balance of pro- and anti-consumptive factors allows for an organism to take advantage of the possibly transient availability of high caloric value foods but also to limit against the lipotoxic consequences of prolonged hyperlipidemia.
In a common theme, chronic obesity disrupts the nutrient-sensitive balance of these counter-regulatory relationships leading to a tonic overactivation of lipid-stimulated orexigenic and reward circuitry. Endocannabinoid levels increase progressively over the course of 20 wks HFD in mice (168) and, in humans, are positively correlated to markers of obesity [reviewed in (171)]. Moreover, while lean humans show a post-prandial decrease in plasma AEA and leptin levels, this does not occur in individuals with obesity (131, 172). Similarly, basal levels of GLP-1 are elevated in obese mice (173) but nutrient stimulated secretion is impaired in both mice (173) and humans (174). By contrast, circulatory ghrelin, which is typically suppressed by eating, rises in response to palatable food consumption in obese humans (174). Despite a sensitized hedonic perception of high fat foods, oral fat sensitivity and lingual CD36 expression are depressed during chronic obesity as is their dynamic regulation by dietary lipids [(175) and for review (176)]. Furthermore, central insulin and leptin resistance disinhibits dopaminergic excitability and contributes to mesolimbic overactivation (163). Thus, obesity abrogates acute nutrient feedback to the brain and shifts the motivation for eating away from homeostatic drives and towards hedonic ones that favor overconsumption of high calorie or high fat foods.
Loss of OGT in the brain and peripheral tissues lead to specific changes in HFD hyperphagia, implicating O-GlcNAcylation in hedonic eating. αCaMKII-CreERT2 Ogt KO mice were hyperphagic under standard chow conditions but HFD feeding failed to further potentiate this in either of the studies described previously. In fact, absolute food intake decreased in these mice a few days after switching to HFD, in contrast to the chow-fed Ogt KO or WT HFD control mice in the same timeframe (136). Although the underlying mechanisms have not been explicitly elucidated, the death of hypothalamic leptin-sensing neurons could have rendered them insensitive to changes in circulatory leptin levels and/or interrupted the CNS-mediated feedback loop of leptin-stimulated leptin inhibition. Alternately, OGT loss was also observed in the nucleus of the solitary tract (NTS) (135), which is the primary source of CNS GLP-1 and a critical integration site for nutrient-sensitive peripheral signals (e.g. taste, vagal afferents) and their effects on both homeostatic and hedonic neural circuits [for review (177)]. Several studies have re-iterated the importance of OGT for neuronal viability or excitability [e.g. (178, 179)] so it would be interesting to see whether O-GlcNAcylation in the NTS or regions of the mesolimbic system associate with dietary fat preference.
OGT loss in adipose and macrophage cells was also associated with lipid preference but in opposite directions. Fat specific (Adipoq-cre) Ogt KO mice displayed normal standard chow eating patterns but lacked HFD hyperphagia (139). The expected pattern of overconsumption was restored in these mice through dietary supplementation with monounsaturated fatty acids (MUFA). The authors show that MUFA inhibit the degradation of adipocyte AEA by fatty acid amide hydrolase (FAAH) leading to increased circulatory AEA and food intake. By contrast, adipocyte OGT loss depressed MUFA synthesis through hypoexpression of the lipid desaturase SCD, which would otherwise couple a rise in adipocyte FFA uptake to pro-consumptive signaling through suppression of FAAH. A decrease in FAAH expression or activity occurs in multiple models of obesity and HFD eating [described in (168, 180)] which, based on the pathway above, is also a predictable outcome of increased OGT activity in hyperlipidemic adipocytes. Opposite to adipocytes, OGT loss in macrophages (LysM-cre) stimulates HFD food intake (25). This may be related to the pro-inflammatory profile of these cells. The LysM-cre driver targets macrophages in the brain (181) and in the periphery, where increased circulatory cytokines can cross the blood brain barrier (182). Neural inflammation, including in response to HFD, alters dopaminergic neurotransmission and has been suggested to trigger insulin resistance in the nucleus accumbens, associated with the overconsumption of palatable foods [for review (183)]. HFD induced inflammation has also been linked to taste bud remodeling believed to underlie the gustatory desensitization of obese individuals (184). Therefore, increased macrophage OGT activity during a transient period of hyperlipidemia is likely protective against excessive inflammation and may stimulate negative feedback on overeating. However, prolonged hyperlipidemia depresses macrophage O-GlcNAcylation, which in combination with hyper-O-GlcNAcylated adipocyte secretion of AEA, would support persistently enhanced lipid uptake.
Although OGT can be linked to mesolimbic and lingual fat responses upstream of macrophage inflammatory signaling, the mechanistic pathway implicated in the LysM-cre Ogt KO cells suggests additional, more speculative, hypotheses. The potentiation of pro-inflammatory signaling in these cells was specifically observed downstream of the TLR4 activating drug LPS (25), in agreement with previous findings associating a TLR4 triggered suppression of macrophage O-GlcNAcylation with exacerbated inflammation (185). Both TLR4 receptors and OGT are expressed in mesolimbic dopaminergic neurons where they have been implicated, individually, in the perception of hedonic food reward (186, 187). More specifically, dopaminergic TLR4 activity, which is endogenously activated by saturated LCFAs, is required for dietary sugar and lipid preference in mice (186). By contrast, OGT hyperactivity in this cell type is linked to the loss of sweet taste perception in sugar fed flies (187, 188). Consequently, the mesolimbic O-GlcNAc regulatory system appears well positioned to mediate lipid-responsive hedonic circuitry in the brain, whether independently or downstream of TLR4-dependent signaling. In addition, TLR4 and OGT have been found in taste-responsive mammalian and/or fly tongue cells (188, 189). Both proteins are positively associated with CD36 expression and function in a variety of cell types (28, 190, 191). This specifically includes a reduction of lingual CD36 mRNA in lipid insensitive TLR4 KO mice (192). While nothing is known about lipid-driven O-GlcNAcylation patterns in the tongue and mesolimbic neurons or the role of OGT in mediating lipid responses in those areas, these circumstantial associations warrant further investigation.
OGT activity in adipose tissue and the brain is required for transient HFD hyperphagia in rodents by supporting FA-stimulated endocannabinoid secretion and, likely, dynamic leptin signaling. Macrophage O-GlcNAcylation resists dietary lipid overconsumption, possibly related to anti-inflammatory effects in hedonic CNS circuitry or the tongue epithelium. However, direct OGT activity and OGT interactions with TLR4-mediated signaling in these tissues should be considered for future experimentation. The implications of persistent O-GlcNAcylation in adipocytes or its eventual suppression in macrophages, however, are consistent with obesogenic endophenotypes that facilitate the transition from homeostatic to hedonically dominated eating patterns that favor lipid overconsumption.
Intestinal absorption
Once dietary lipids have reached the small intestine, the capacity of intestinal enterocytes to absorb and store them, as well as to synthesize and release TAG-rich chylomicrons, determines practical lipid uptake. Diet-derived monoacylglycerol and FFAs primarily diffuse passively across the enterocyte membrane. Inside the cell, however, they are escorted by proteins to the ER membrane for re-esterification into TAG [for review (193)]. This neutral lipid is then packaged into ER luminal LDs, used for chylomicron biogenesis, or into cytoplasmic LDs for storage. Lipids stored from prior ingestion are believed to fuel the first wave of post-prandial chylomicron secretion as well as between meal release (194). Chylomicron synthesis is closely dependent on two ER proteins - apolipoprotein B48 (ApoB48) and microsomal triglyceride transfer protein (MTTP), which facilitate formation of the pre-chylomicron phospholipid membrane and the transfer of neutral lipids from luminal LDs into its core (195). These particles mature in the Golgi, receiving additional TAG and membrane modifications, before release into the lymphatic system where they can acquire plasma-derived modifications including apoC-ii (196), which is critical for cellular uptake at target tissues. Uptake-competent chylomicrons are then secreted into the bloodstream where they can deliver diet-derived lipids to peripheral tissues.
Obesity is associated with hypertriglyceridemia due in part to increased enterocyte lipid uptake. Diet induced obesity depresses fecal lipid content in mice as a consequence of the enhanced extraction of dietary fat (197). One reason for this is an expansion of the intestinal surface area, downstream of stem cell proliferation and factors related to increased food intake (197). In addition, individual enterocytes adapted to obesity show an enhanced capacity for lipid uptake, TAG storage and secretion. Prolonged HFD, or in vitro PA, induces expression of enterocytic periliplin 2 (PLIN2) (198), an LD membrane protein important for long-term cytosolic LD storage [reviewed in (194)]. Two mouse models with intestinal PLIN2 depletion showed reduced cytosolic LD expansion and suppressed dietary lipid extraction under HFD conditions (197, 198). The multi-potent FA binding and transport protein CD36 has also been implicated. CD36 deficient mice and humans produce fewer and smaller chylomicrons (199–201) but the mechanisms behind this shift are not precisely delineated [for review (202)]. CD36 appears to be dispensable for net intestinal FFA uptake (199, 203). However, LCFA binding and activation of the receptor leads to the rapid induction of ApoB48 and MTTP protein (204), as well as a slower transcriptional regulation (205), but also to pre-chylomicron assembly and transport (199, 206). In lean subjects, enterocyte CD36 activation is transient due to lipid-stimulated proteasomal degradation within ~1 hour of high fat ingestion (204). However, this downregulation is absent in obese mice, leading to prolonged secretion of large particle chylomicrons (205), which both hold more TAG and are more rapidly depleted by target tissue uptake (207, 208). In sum, obesity related changes in enterocyte number, TAG storage and chylomicron secretion proteins exacerbate systemic hyperlipidemia by enhancing the capacity for dietary lipid extraction.
Although not specific to the gut, O-GlcNAcylation is associated with the increased stability of both PLIN2 and CD36 in vitro. As mentioned in “O-GlcNAc Enzymes”, OA potentiates sOGA activity and promotes its accumulation on LD membranes where it co-localizes with PLIN2 in ad-sOGA OE HeLa cells (71). RNAi knockdown of sOGA in that model stabilized PLIN2 and PLIN3 protein level through reduced proteasomal activation. OGT activity regulates the ubiquitin-dependent degradation of multiple intracellular proteins (209), including through inhibitory O-GlcNAcylation of the S26 proteasome itself (210–213). In HeLa cells, sOGA inhibition increased general ubiquitinylated protein abundance while its overexpression accelerated proteasomal degradation of PLIN2/3 at the LD membrane (71). It would be interesting to see whether the lipid-stimulated proteolysis of CD36 might be similarly regulated. Nevertheless, CD36 is also a direct and transcriptional OGT target. OGA inhibition (Thiamet-G) of gastric cancer cells increased CD36 mRNA and protein levels while Ogt KO or siRNA diminished both basal and PA-stimulated CD36 abundance (28). The transcriptional upregulation was attributed to NF-KB O-GlcNAcylation, which increases its activity at the CD36 promoter (28). In addition, CD36 contains at least two O-GlcNAcylated residues (28, 190) that, when mutated, reduce the rate of cellular FFA uptake and PA-stimulated LD accumulation (28). With the exception of sOGA, all of these proteins have demonstrated expression in enterocytes where analogous regulatory relationships, if true, would link hyperlipidemia-driven O-GlcNAcylation to enhanced intestinal uptake and secretion of dietary fat as well as to fat-stimulated satiety through CD36-dependent enterocyte synthesis of OEA (214, 215).
OGT has been directly knocked out in the intestinal epithelium (Villin-cre), which includes enterocytes, but the outcome on lipid uptake is unclear. Villin-cre Ogt KO mice demonstrate intestinal hypertrophy and hyperplasia (216), similar to the effects of obesity. However, in studies from the same lab, these mice also have lower body weight and improved glucose tolerance (17), inconsistent with a higher rate of nutrient absorption. Furthermore, HFD or in vitro short chain fatty acids (SCFAs) consistently increased intestinal O-GlcNAcylation (17), which argues against a potentiating role for OGT activity in expansion of the obese gut. Instead, the predominant Ogt KO phenotype characterized by the authors was an increase in serum GLP-1, apparently due to hyperplastic expansion of intestinal L-cells (17). This data was supported by a knockin model of rat OGT OE with transcriptional deregulation and impaired L-cell and GLP-1 levels, reflecting obesity conditions related to L-cell dysfunction [e.g. (173)]. OGT’s suppressive effects were tied in part to FOXO1 O-GlcNAcylation leading to the inhibition of Ngn3-mediated gene expression (17). In the CNS and periphery, GLP-1 dampens palatable food intake and potentiates glucose-stimulated insulin secretion [for review (217)]. The systemic role of intestinal GLP-1 (vs. NTS or intra-islet GLP-1) is somewhat in question, however, due to its rapid degradation in the gut and liver [see (218)]. Moreover, fecal transplantation from Villin-Ogt KOs to WT mice transferred the body weight and L-cell hyperplasia phenotype, implicating causative changes in the intestinal microbiome (17). The impact of any of these changes under HFD conditions is unknown. However, both gut microbiota and intestinal GLP-1 have regulatory roles in local lipid metabolism [for review (219)]. According to these studies, increased GLP-1 signaling depresses dietary fat absorption and chylomicron TAG secretion, with similar results in germ-free or antibiotic-treated animals. Therefore, HFD stimulated O-GlcNAcylation of the intestinal epithelium, and its subsequent effects on microbiome activity and L-cell GLP-1 hyposecretion, could conceivably contribute to increased dietary lipid extraction.
Dietary lipid absorption increases during obesity as does the secretion of TAG-rich chylomicron particles from intestinal enterocytes. Protein O-GlcNAcylation has not been directly linked to enterocyte physiology but OGT and sOGA activity in other cell types regulate the stability and activity of CD36 and PLIN2, which are central to hyperlipidemia adaptations in the intestine. Furthermore, O-GlcNAc’s transcriptional regulation of L-cell GLP-1 secretion has implications for both local lipid extraction and meal related satiety. On the basis of these mechanisms, intestinal hyper-O-GlcNAcylation in response to dietary or even microbiome-derived hyperlipidemia would positively contribute to the enhanced lipid uptake.
O-GlcNAcylation in lipid storage and release
Section OGT activity and dietary lipid uptake described the complex and temporally dynamic relationships between dietary fat and lipid uptake, at both the motivational and absorptive level. OGT activity supports both hunger and satiety regulating mechanisms as well as lipid-potentiated hyperphagia and its feedback inhibition through dynamic, cell type-specific responses to both low and high nutrient conditions. By contrast, persistent and excessive lipid exposure overwhelms the homeostatic forces of both the lipid response and O-GlcNAc regulatory systems, further driving their reciprocal dysfunction. A similar pattern also emerges for the role of O-GlcNAcylation in peripheral lipid uptake, storage and release.
Lipids in the bloodstream are taken up by numerous cell types, some primarily for utilization (e.g. heart, skeletal muscle) and others for the purpose of storage and regulated release (i.e. white adipose tissue, liver). The source of these lipids can be diet-derived chylomicron TAG but also adipocyte-released FFAs and hepatic lipoprotein TAG, with varying contributions to fasting and post-prandial circulation. FFAs at the cell surface, either albumin-bound or lipolytically released from TAG, move across the plasma membrane with the aid of FFA binding proteins under tissue specific nutrient and hormone regulation. White adipose tissue (WAT) is the primary site for dietary lipid uptake and long-term fat storage. Adipocytes possess a specialized regulatory system that facilitates switching between meal stimulated FFA uptake and lipogenic LD expansion or fasting dominant LD TAG lipolysis and FFA secretion. Furthermore, WAT storage capacity can expand to accommodate a higher lipid load through adipogenesis and hypertrophy. The liver is also a major source for nutrient uptake and short-term storage as well as producing energy-generating substrates when exogenous sources are low. This includes a careful regulation of interconnected lipid and glucose metabolic pathways. Due to these dual roles, WAT and the liver are particularly sensitive to intra- and extracellular cues that differentiate the post-prandial and fasting state. Under physiological conditions, O-GlcNAcylation facilitates adaptive nutrient uptake, production and storage but differentially between tissues according to their specific roles throughout the day or over a developmental time course. In obesity, the signals that guide dynamic O-GlcNAc activity, substrate targeting and functional outcome become less context-specific such that static and tone-shifted O-GlcNAcylation appears to exacerbate the negative consequences of persistent hyperlipidemia.
Cellular lipid uptake
While cellular FFA uptake can occur passively, membrane-bound and adjacent proteins facilitate transport efficiency in both generic and tissue-specific ways. Circulatory TAG is first be hydrolyzed by endothelial lipoprotein lipase (LPL), which is activated by ApoC-ii proteins in the lipoprotein membrane. The LPL protein is synthesized in capillary-adjacent cells then transported to the epithelial wall, allowing differences in local secretion and activation to drive tissue-specific lipid uptake profiles independent of circulatory abundance [for review (220)]. LPL expression and activity is highest in white adipose and cardiac tissue (221–223). However feeding, glucose and insulin potentiate adipocyte LPL hydrolysis while these conditions are repressive in the heart (221, 224). LPL is also active in skeletal muscle lipid uptake (221, 225) but largely in response to fasting and exercise (226, 227) with little insulin sensitivity (228–230) and only a mild and transient response to eating (226, 231). Cell membrane proteins that facilitate FFA uptake (i.e. FATP1-6, FABPpm, CD36) also demonstrate context-dependent regulation. Insulin sensitive FATP1 and CD36 are particularly important in adipose, heart and skeletal muscle. Interestingly, myocyte CD36 acts as a direct FFA translocase while adipocyte CD36 facilitates insulin/lipolysis stimulated endocytosis (232, 233). FATP1 and LPL expression are low in the liver, although some evidence suggests hepatic LPL contributes to circulatory LPL levels (234). Hepatic CD36, on the other hand, is elevated during fasting (235), when FFA uptake by the liver is highest. These unique regulatory profiles help direct circulatory lipids towards vitally active cells (heart, exercising muscle) and energy production (hepatic gluconeogenesis, ketogenesis) under high demand or low nutrient conditions but also funnel the majority of post-prandial dietary fat towards WAT adipocytes that have a higher capacity for non-toxic lipogenic storage.
The machinery and rate of lipid uptake is altered by obesity in a similarly tissue-specific manner. LCFA uptake is elevated in adipose, cardiac and hepatic tissues from obese individuals (236–239) but lower in skeletal muscle (240, 241). This is consistent with the up-or-downregulation of CD36 and, excepting the liver, FATP1 in those cell types (238, 242–248). Obesity also upregulates LPL expression and activity in fat and liver while levels in cardiac and skeletal muscle are unchanged or diminished [for review (220, 239)]. However, the outcome of obesity on adipocyte lipid uptake is dependent on insulin signaling. Overweight insulin resistant individuals have reduced adipose LPL expression/activity and slower post-prandial TAG clearance compared to weight-matched but more insulin sensitive controls (249). This is likely exacerbated by obesity precipitated changes in β-cell insulin secretion (24, 40, 134) and intestinal ApoC-ii expression (204, 205, 250), namely a basal elevation with a loss of nutrient stimulation in both. During early hyperlipidemia, when insulin signaling is potentiated, these adaptations boost cardiac utilization and hepatic buffering while further directing excess dietary lipid towards adipocytes. In progressed obesity, however, the capacity for both adipose and muscle FFA uptake is reduced, exacerbating circulatory hyperlipidemia and pushing a steatogenic burden onto the heart and liver.
While investigation in this area is not extensive, changes in cellular lipid uptake proteins have been observed in O-GlcNAc modified models. Oga KO in mouse embryonic fibroblasts (MEFs) depresses transcription of Apoc2, Slc27a1 (FATP1) and Cd36 (116). These results implicate O-GlcNAcylation in the regulation of relevant genes but the lack of tissue specificity limits broader interpretation based on this model. By contrast, multiple investigations have demonstrated the effects of HBP-driven O-GlcNAcylation on CD36 in the rodent heart. As described in section OGT activity and dietary lipid uptake, CD36 is a direct and indirect OGT target and the dominant source of FFA influx in the heart. Cardiac perfusion with GlcN (190) or Gln (115) increases membrane-specific expression of cardiomyocyte CD36. Similarly, in vitro GlcNAc rescued a depression in CD36 O-GlcNAcylation and membrane expression in a model of cardiomyocyte stress (191). This data is in addition to the study referenced earlier showing that a reduction in CD36 O-GlcNAcylation suppresses FFA uptake rate in gastric cancer cells (28). Besides CD36, LPL has been implicated in a small number of studies. Ogt KO β-cells (RIP-cre) have fewer Lpl transcripts (24) while OGA inhibition increases LPL secretion in primary rat adipocytes (251). LPL in the liver has not been investigated under O-GlcNAc modified conditions but the O-GlcNAc potentiated transcription factor LXR (252), is a major driver of hepatic Lpl expression (253), including in response to dietary hyperlipidemia (254). Oral O-GlcNAc also increases hepatic FFA content, predominantly in fed mice (113). These results are largely consistent with the conclusion that OGT activity supports FFA uptake through the upregulation of LPL and CD36, notably in tissues with a hyper-O-GlcNAcylation response to hyperlipidemia.
It is not clear to what extent O-GlcNAc facilitated FFA uptake might underlie constructive hyperlipidemia adaptations, such as enhanced cardiac utilization or protective lipid sequestration by adipose and liver, as opposed to further driving lipotoxic overwhelm. However, in addition to cellular uptake proteins, HBP and/or OGT activity has been extensively linked to insulin resistance, particularly in adipose and skeletal muscle (16, 53, 142, 143, 255). OGT depletion rescues HFD insulin sensitivity in these same tissues (139, 256, 257). Therefore, elevated O-GlcNAc levels are expected to diminish the post-prandial and insulin-dependent stimulation of CD36 membrane expression in muscle and both CD36 internalization and LPL activation in adipose tissue in contribution to the reduced TAG clearance rate of the obese state.
The available literature supports the hypothesis that O-GlcNAcylation can regulate tissue specific cellular lipid uptake through the expression or activity of proteins required for lipoprotein TAG hydrolysis (i.e. LPL) and efficient FFA membrane transport (i.e. FATP1, CD36). OGT activity in adipose tissue, the liver and heart upregulate these mechanisms. Hyperlipidemia potentiates O-GlcNAcylation in these tissues, which may accelerate lipid clearance from the blood as an adaptive strategy to fat overconsumption or desensitize insulin stimulated uptake while potentiating basal and ectopic uptake, further driving serum hyperlipidemia and pathological lipid accumulation.
Adipocyte lipolysis
Adipose tissue is a dominant contributor to post-prandial lipid uptake and fasting FFA secretion, making its capacity for lipid storage and metabolic flexibility critical for systemic nutrient homeostasis. As in most cell types, WAT FAs are packaged into cytosolic LD TAG, which under physiological conditions constitute ~ 80% of adipocyte weight (258). TAG content is dynamically controlled by LD membrane proteins including lipogenic acyltransferases, lipolytic lipases and PLINs. There are multiple PLIN isoforms associated with WAT LDs that take on structural, signaling and regulatory roles [see (259)]. PLIN1, which is predominantly expressed in adipocytes, is important for the regulation of lipolysis. TAG lipolysis occurs through the sequential action of three lipases (ATGL, HSL, MGL), which progressively liberate one FFA from each TAG fatty acyl chain [for review (260)]. In the basal lipolytic state, low ATGL activity on the LD membrane provides constitutive FFA flux. Fasting and exercise enhance the lipolytic rate, however, by triggering sympathetic catecholamine release to stimulate adipocyte β-adrenergic receptors, leading to the cAMP/PKA-mediated phosphorylation of PLIN1 and HSL. Phosphorylated PLIN1 dissociates from the ATGL co-activator protein CGI-58, freeing it to potentiate ATGL’s catalytic activity up to 20-fold (261). Between rising levels of diacylglycerol and its own phosphorylation, cytoplasmic HSL translocates to the LD surface to further promote lipolysis [for recent review (262)]. By contrast, post-prandial insulin signaling suppresses lipolysis by triggering HSL dephosphorylation and cAMP degradation and by interfering with the PKA phosphorylation of PLIN1 (263) as well as by blocking CD36 dependent FFA exocytosis (233). Moreover, insulin drives LD expansion by amplifying FFA uptake, acyltransferase activity, lipogenic gene expression and glucose-derived lipogenic substrate availability [for review (264)]. These regulatory relationships ensure that the balance between adipocyte FFA storage and release is matched to the acute nutrient conditions of the body.
Obesity is associated with an increase in basal lipolysis and a decrease in catecholamine-stimulated FFA secretion (260). Increased leptin levels, described in the previous section, and a reduced LD membrane density of PLIN1 contribute to higher constitutive FFA release (265, 266). The hypoexpression and protein degradation of PLIN1 may be downstream of pro-inflammatory cytokine TNFα (266, 267), which has a higher adipose tissue concentration in obese individuals (268). At the same time, changes in adrenergic receptor expression/composition, associated with a reduction in cAMP generation, as well as decreased HSL transcription have been blamed for the blunting of stimulated lipolysis (260). Insulin receptor resistance, also associated with inflammatory cytokines in obesity (269), is expected to potentiate both these lipolytic dysregulations, exacerbating both adipose and hepatic lipid accumulation while diminishing the validity of serum FFA level as a marker of the fasting state.
The adipocyte O-GlcNAc regulatory system influences LD FFA storage and release through effects on PLIN1. Yang et al. have shown that OGT and PKA compete for two of the same PLIN1 target sites, S517 and S492, with opposing effects on the stimulated activation of lipolysis (16). Accordingly, adipocyte OGT loss (Adipoq-CreER) increases the interaction between CGI-58 and ATGL on the LD surface and enhances both fasting and β-adrenergic stimulated lipolysis. Consistently, OGA inhibition protects WT adipocytes from forskolin (i.e. cAMP) induced LD depletion. Furthermore, overexpression of rat OGT in mouse adipose tissue exaggerates HFD WAT expansion and diminishes in vitro stimulated lipolysis but only in adipocytes derived from HFD fed animals. Surprisingly, O-GlcNAc modifying conditions have similar effects on the LD dynamics of HeLa cells, which do not express PLIN1. Oleic acid loading followed by serum depletion (i.e. cellular fasting) triggers lipolytic LD depletion in WT but not OGT OE HeLa cells while LD loss is exaggerated in OGA OE cells (16). This could be attributable to sOGA mediated proteolytic degradation of PLIN2 and PLIN3 (see “Intestinal Absorption”). Like HeLa cells, sOGA in 3T3-L1 adipocytes co-localizes with PLIN2/3 but only on smaller, nascent LDs while sOGA is absent on larger PLIN1-coated LDs (71). The switch from statically lipogenic PLIN2 to lipolytically dynamic PLIN1 is a physiologically important component of LD maturation [for review (270)]. Some evidence suggests that PLIN2 and PLIN1 compete for occupancy in the LD membrane (271), in which case adipocyte sOGA might facilitate PLIN1 expression and LD maturation by stimulating PLIN2 degradation. To that point, OGA inhibition (NButGT) during 3T3L1 differentiation decreases PLIN1 levels and LD biogenesis in vitro (272). Physiologically, PLIN1 also plays a role in suppressing constitutive lipolysis but the consequences of OGT/OGA activity on this have not been directly studied. Basal lipolysis is not significantly altered in the models described above but was found to be elevated by GFAT inhibition in cultured human adipocytes (273). Nevertheless, sOGA and OGT activity in adipocytes appears to support adaptive functioning by adjusting lipolytic sensitivity while hyper-O-GlcNAcylation in obesity may limit metabolic flexibility by suppressing LD maturation and PLIN1-mediated lipolysis.
In addition to direct effects, OGT-driven changes in adipose insulin sensitivity and macrophage cytokine secretion may further fuel lipolytic dysfunction in obesity. Adipocyte insulin resistance in models of elevated HBP flux (142, 255, 274, 275) was among the first pieces of evidence linking O-GlcNAcylation to insulin sensitivity. In vivo, this may be restricted to the context of hyperlipidemia as numerous adipocyte OGT models fail to impact insulin sensitivity in standard chow fed mice (16, 139, 276). By contrast, the same studies show that under HFD conditions, adipose OGT OE (rat OGT knockin) worsens insulin resistance (16) and Ogt KO (adiponectin-cre) prevents it altogether (139). These models are also associated with changes in lipolysis or circulating FFA levels consistent with an exacerbating role for OGT in obesity related lipolytic dysfunction. Another pathway towards this conclusion is through O-GlcNAc regulation of macrophage cytokine transcription. As previously described, prolonged HFD depresses macrophage O-GlcNAcylation, correlated to pro-inflammatory polarization and cytokine secretion in adipose tissue (25). Obesity substantially increases adipose macrophage content, from ~10% up to 40% (277), accelerating the local (and systemic) inflammation that also contributes to insulin desensitization (269). Consistent with cytokine induced insulin resistance, WAT from LysM-cre macrophage Ogt KO mice show increased β-adrenergic stimulated lipolysis (25). Similarly, SV-differentiated WT adipocytes co-cultured with Ogt KO macrophages exhibit stronger lipolytic activation to acute inflammatory stimulation (25). Thus, adipocyte and macrophage O-GlcNAc responses to prolonged hyperlipidemia, despite their opposite directions, likely both contribute to adipose tissue insulin resistance and the dysregulation of lipolysis in obese individuals.
Adipogenesis
Adipose tissue adapts to persistent nutrient excess by first expanding individual cell size (hypertrophy) and subsequently the number of mature adipocytes (hyperplasia) [for review (278)]. Moderate hypertrophy, which is driven by LD expansion, preserves systemic insulin sensitivity and protects more vulnerable tissues against lipotoxicity. However, very large adipocytes develop significant stress (mechanical, hypoxic, ER, oxidative, etc.) that leads to intracellular insulin resistance and necrotic features [reviewed in (279)]. This reduces adipose tissue lipogenic and lipolytic capacity and stimulates inflammatory cytokine secretion. As a mitigating strategy against excessive hypertrophy, hyperplasia generates new adipocytes from a pool of precursor stem cells.
Adipogenesis begins with the irreversible commitment of stem cells to a pre-adipocyte form (279). When triggered to differentiate, pre-adipocytes undergo clonal mitotic expansion followed by terminal differentiation, the latter characterized by growth arrest, morphological changes, LD formation/maturation and adipokine synthesis. Terminal differentiation begins with the transient expression and activation of the beta and delta isoforms of CAAT/enhancer-binding proteins (C/EBP), which initiate expression of Pparɣ and C/ebpα during a first transcriptional wave (278). During the second wave, C/ebpα and PPARɣ (+co-activator RXR) act in a positive feedback loop to rapidly promote their mutual expression. Subsequently, they work cooperatively to promote the gene expression of lipogenic and adipokinic proteins. Adipogenic success, therefore, influences both the size of the adipocyte pool and the capacity of newly differentiated adipocytes to take up, store and release FFAs through their expression of relevant enzymes and regulatory proteins. By contrast, decreased adipogenesis and increased hypertrophy contribute significantly to metabolic dysfunction in progressed obesity.
Dynamic O-GlcNAcylation is a hallmark and requisite for adipocyte differentiation. Ishihara et al. were the first to demonstrate that O-GlcNAcylation increases progressively over the course of an 8-day 3T3-L1 differentiation. In that study, OGT protein expression mirrored O-GlcNAcylation while OGA was upregulated on day 1 then remained at that level throughout (280). This O-GlcNAcylation pattern was recapitulated by Hsieh et al., who also showed that Gfat mRNA and protein were increased but in a cyclical pattern, with transient protein peaks on days 1 and 4 (281). Multiple studies have noted that different target proteins appear to be O-GlcNAcylated at different timepoints (71, 273, 280, 281), with several heavier weight molecular bands only appearing in western blots of differentiated cells. Among the identified OGT targets in these studies were proteins involved in nutrient metabolism, cytoskeletal structure, nuclear transport and transcription regulation (280), including PPARɣ and C/EBPβ (281). Inhibition of GFAT (DON, azaserine, siGFAT) prevented the differentiation-induced increase in O-GlcNAcylation and forestalled adipogenesis based on the absence of phenotypic markers (morphology, LD accumulation), transcriptional markers (C/ebpβ, Pparɣ, Srebf1) and functional markers (Ir, Glut4, Fas, Adiponectin, Plin1, Plin2) (280, 281). Nevertheless, OGA inhibition has also been linked to depressed differentiation, although not consistently across studies. Applied during 3T3-L1 differentiation, pharmacological OGA inhibition by NAG-thiazoline delayed Pparɣ and ap2 expression in one study (282) but NButGT did not in another (272). However, the latter investigation did note that 3T3-L1 lipid accumulation and PPARɣ transcriptional activity were decreased. On the other hand, gene trap Oga KO WAT cells, generated from differentiated MEFs, showed no differences compared to WT controls (118). Given the importance of transient, timed protein activity for differentiation to progress, it is not surprising that OGT targeting shifts over time and that disruption of that timing, by elevating or depressing global O-GlcNAcylation, would impair adipogenesis.
PPARɣ and C/EBPβ are strongly O-GlcNAcylated during normal 3T3-L1differentiation. Activation of either of these transcription factors is sufficient to independently trigger adipocyte differentiation but only PPARɣ is essential for it to occur ( (283) ISBN 9780128146491). PPARɣ is expressed in two splice isoforms. The adipocyte-exclusive PPARɣ2, which is O-GlcNAcylated at Thr84, is the isoform required for adipogenesis (139, 284). Mutagenic loss of T84 O-GlcNAcylation decreases PPARɣ2 protein and activity by 45% (ligand-stimulated) to 74% (basal) in transfected HEK293 cells (139). Similarly, HFD adipose tissue from Adipoq-cre Ogt KO mice show depressed PPARɣ2 levels with increased inhibitory phosphorylation at Ser82. The PPARɣ1 isoform is more broadly expressed across tissues, including in adipocytes, but its functional role is not well understood (284). Thr54 is an OGT target in PPARɣ1 but increasing its O-GlcNAcylation (NButGT) reduces enzymatic activity (272), making its contribution to adipogenesis unclear. It is worth noting that elevated glucose during 3T3-L1 differentiation increased the O-GlcNAcylation of PPARɣ1 but not PPARɣ2 in the same study, suggesting independent regulation of the two isoforms.
C/EBPβ is O-GlcNAcylated at two adjacent serine residues (Ser180, Ser181) (282). When occupied, these sites prevent the sequential activating phosphorylations of C/EBPβ, first by MAPK/CDK2 (Thr188) and then by GSK3β (Ser184, Thr179). These modifications are required for C/EBPβ to bind to DNA during the early stage of terminal differentiation. Consequently, OGA inhibition delays the expression of C/EBPβ differentiation targets, Pparɣ and ap2, while mutagenesis of the S180/181 residues strongly potentiate C/EBPβ transactivation of the C/ebpα promoter. Therefore, it is likely that rising O-GlcNAcylation plays a constructive role in terminating the transient elevation of C/EBPβ activity during the first transcriptional wave and enhancing PPARɣ2 function in the second wave of adipocyte differentiation. Interestingly, the Ogt promoter contains a binding site for C/EBPβ which is required for its transcriptional potentiation by OGA and p300 (an acetyltransferase C/EBPβ co-activator) in transfected HEK293T and pancreatic cancer cells (285). OGA, p300 and C/EBPβ are all elevated during early adipocyte differentiation (280, 286) whereupon they could mediate the transition from the first to the second transcriptional wave by facilitating early C/EBPβ driven transcription, including Ogt expression, that then provides negative feedback on further C/EBPβ activity. A simplified model of this regulatory hypothesis is summarized in Figure 3. Unfortunately, the effect of obesity on O-GlcNAcylation patterns in differentiating adipocytes is not known but the oppositional effects of O-GlcNAcylation on C/EBPβ and PPARɣ in relation to the timing of their adipogenic activities suggests that any shift in the ratio of OGT/OGA activity could delay or inhibit hyperplastic expansion.
Figure 3 Model for O-GlcNAc Regulation of Adipogenesis. Adipocyte terminal differentiation is characterized by a progressive increase in O-GlcNAcylation but the precise rationale for this is not fully elucidated. One possibility is that a high OGA/OGT ratio at the start of differentiation (1) permits C/EBPβ kinase activation by keeping O-GlcNAcylation off of nearby inhibitory residues (2). Activated C/EBPβ can then act at the Pparɣ and Ogt promoters (3). Both C/EBPβ and PPARɣ stimulate expression of C/EBPα (4), which marks the transition from the 1st to the 2nd transcriptional wave (—–) on ~ day 3 of a standard 8-day 3T3L1 differentiation protocol [see (272, 280, 282)]. Building OGT levels, with static OGA expression, could then provide negative feedback on C/EBPβ activity and simultaneously potentiate PPARɣ through their respective O-GlcNAcylations (5). In the final phase of differentiation, C/EBPα and PPARɣ act as transcription factors for each other and for adipogenic and adipokinic genes that characterize a mature adipocyte (6). The left side graph shows the approximate relative activity of each protein (x-axis) over an 8-day time course (y-axis). On the right, solid arrows represent positive (green, –>) and negative (red, –|) effects. Dashed arrows indicate translocation. Phosphorylation is shown as an encircled pink P and O-GlcNAcylation as a blue square with a G. Created with BioRender.com
Dynamic activity of the O-GlcNAc regulatory system in adipose tissue contributes to constructive adaptations to transient hyperlipidemia. These include modulating the activation threshold for PLIN1 mediated lipolysis (see previous sub-section) and controlling the activity of adipogenic transcription factors that permit expansion of WAT storage through hypertrophy and hyperplasia. At the same time, persistent elevations in adipocyte O-GlcNAc tone as well as macrophage OGT depletion in the face of prolonged overnutrition/obesity are implicated in adipose tissue dysfunctions including a loss of nutrient sensitive lipolysis, increased inflammation, excessive lipid retention, impaired adipocyte differentiation and both local and systemic insulin resistance.
Liver
The liver is an important tissue for overflow nutrient uptake and in the maintenance of fasting nutrient availability. Hepatocytes take up TAG-depleted chylomicron remnants and FFAs from the circulation but also generate de novo FFAs when glucose is abundant. FFAs that are not oxidized in the liver are sequestered into TAG and either stored in cytoplasmic LDs or packaged for secretion into very low density lipoproteins (VLDL). The capacity for hepatic TAG storage under physiological conditions is limited and the majority is released back into the bloodstream in response to elevated FFA uptake (287, 288) or VLDL-TAG accumulation [for review (289, 290)]. Consequently, VLDL synthesis, which largely mirrors chylomicron biogenesis and its dependency on MTTP and ApoB availability (see “Intestinal Absorption”), is closely tied to hepatic TAG release. Unlike in enterocytes, however, insulin limits hepatic TAG secretion by suppressing ApoB100 translation and stability [reviewed in (291)] and by excluding MTTP transcription factor FOXO1 from the nucleus (290). Even so, hepatic cytosolic TAG storage is highest during physiological fasting [e.g. (124)] and VLDL secretion contributes significantly to circulatory TAG in the late post-prandial period (292). It should be noted that the liver plays a central role in the synthesis, storage and post-absorptive release of glucose as well, which is heavily interconnected with lipid metabolism [recently reviewed in (293)]. Like adipose, then, the liver depends on insulin and nutrient sensitivity, lipogenic capacity and metabolic flexibility to maintain control over systemic nutrient homeostasis.
Both hepatic fat content and circulatory VLDL-TAG increase in response to obesity. Lipid uptake to the liver is potentiated, as previously discussed, but de novo lipogenesis also increases. This is partly because insulin’s potentiation of lipogenic gene expression is selectively preserved (294) even as VLDL secretion and then gluconeogenesis become disinhibited (290). Synthesized FFAs typically contribute to <5% of circulatory TAG in metabolically healthy fasted individuals (295). However, that proportion increases under hyperlipidemic insulin resistant conditions (295) and in response to dietary lipogenic stimulation (296). Moreover, TAG incorporation of saturated long-chain FAs, which are abundant in high fat foods, favors higher VLDL release [reviewed in (297)]. Dietary hyperlipidemia, then, initially elevates both fasting and post-prandial TAG secretion, sparing the liver from excessive accumulation. Prolonged exposure of hepatocytes to high FFA concentrations, however, triggers ER stress and ApoB100 degradation leading to enhanced lipid retention (287). Furthermore, disturbances in glucose and lipid metabolism positively feedback on each other, with hyperglycemia driving lipogenesis and lipid accumulation disrupting both hepatic and whole-body glucose metabolism [for review (293)]. The liver thus becomes both a victim of and a source for pathological hyperlipidemia and systemic metabolic dysregulation in progressed obesity.
Lipid handling by the liver incorporates many elements of FFA uptake, hormone receptor sensitivity and lipoprotein synthesis already discussed, including their susceptibility to regulation by O-GlcNAcylation. sOGA stimulates the proteolysis of PLIN2, which is highly abundant on liver cell LD membranes (298, 299). PLIN2 OE in vitro suppresses VLDL secretion by recruiting FFAs into cytosolic storage (300), a pathway implicated in hepatic lipid accumulation following diet-induced obesity (301). O-GlcNAc stabilization of PLIN2, though not yet demonstrated in the liver, could contribute to cytosolic lipid hoarding, as could an OGT-driven increase in CD36 activity and expression. The facilitative role of OGT in PTP1B-driven insulin resistance has been directly demonstrated in the liver. As described in “Satiety”, PTP1B is a multi-tissue negative regulator of insulin and leptin signaling and its expression is positively correlated to VLDL secretion in the rodent liver (302). Reduced O-GlcNAcylation of PTP1B improves HepG2 insulin sensitivity and decreases lipid accumulation following PA enrichment of the media (27). However, PTP1B O-GlcNAcylation is specifically increased in the liver of HFD mice (27). Hepatic insulin resistance in the context of obesity increases the nuclear localization of FOXO1 and the expression of its gluconeogenic and lipogenic gene targets (303), driving up VLDL-TAG secretion (304). High glucose also potentiates hepatic FOXO1 transcription through O-GlcNAcylation of nuclear-permitted protein (305, 306). These conditions are potentially additive in progressed obesity, although the effects of OGT activity on non-gluconeogenic FOXO1 gene expression (e.g. MTTP) have not been tested. Consistent with these mechanisms in principle, however, both liver Ad-OGT OE (89, 124) and human PEPCK-cre GFAT OE (307) mice are insulin resistant with disturbed hepatic and systemic glucose metabolism as well as fasting or random-fed plasma hypertriglyceridemia. By contrast, Ad-OGA OE, targeted to the liver, rescues plasma/hepatic TAG levels and glucose tolerance in db/db obese mice (124), implicating static hyper-O-GlcNAcylation of the liver as a contributing factor to local and circulatory hyperlipidemia in obesity.
In addition to modulating the response to extrahepatic nutrients and hormones, OGT activity drives de novo lipogenesis in the liver through posttranslational enzyme targeting. Among the most important lipogenic enzymes are FAS and ACC, whose regulation by OGT was referenced in earlier sections. FAS has an intriguingly reciprocal relationship to OGT and OGA, as it binds both enzymes to influence their activity as well as its own (see “O-GlcNAc Enzymes”). Hepatic FAS is directly O-GlcNAcylated, more so in re-fed vs. fasted liver or in fasted livers from obese vs. lean mice (87). OGA inhibition upregulates FAS protein levels in human hepatocyte cells by increasing its interaction with the deubiquitinase USP2A (87). Moreover, an increase in FAS-dependent PA accumulation in non-alcoholic fatty liver disease (NAFLD) liver cancer cells was attributed to higher OGT levels (308). Directly upstream of FAS in the lipogenesis pathway is ACC, whose AMPK-dependent inhibition by OGT was discussed in “Hunger”. However, these studies were reported in the context of FAO activation, where the mitochondria bound ACC2 paralog (309) is the more likely target. By contrast, cytoplasmic ACC1 (309), contains multiple OGT target sites that potentiate its activity, independent of AMPK, in CD4+ T cells (126). As an alternate route to the same end, O-GlcNAcylation of inactivated AMPK is inhibitory in vitro and associated with ACC disinhibition (79). This mechanism was blamed for the correlation between OGA inhibition and reduced pAMPK/pACC in fish liver (31). These post-translational relationships provide a mechanism for rapidly amplifying lipogenesis in response to the specific combination of elevated nutrient levels and OGT activity in the liver.
Nutrient-sensitive gene expression is another major regulatory pathway for hepatic lipogenesis [for review (290)]. Nearly all of the principal transcription factors for this are O-GlcNAc regulated including ChREBP, LXRα, SREBP-1c, and FXR. Liver ad-OGA OE in db/db obese mice diminishes total and O-GlcNAcylated ChREBP levels with a concomitant decrease in transcriptional targets, FAS and ACC (124). Ad-OGT OE in WT mice has the opposite effect, potentiating ChREBP O-GlcNAcylation and lipogenic enzyme transcription (i.e. Fas/Acc/Scd1), as well as hepatic lipid accumulation (124). However this phenotype was only observed in tissues from fed mice, emphasizing the importance of insulin and nutrient availability in mediating OGT’s pro-lipogenic effects. Interestingly, the feeding-stimulated O-GlcNAcylation of nuclear ChREBP in the liver is dependent on LXR (93), either due to its recruitment of OGT to the nucleus (see “O-GlcNAc Enzymes”) or its role as an upstream transcription factor. LXR is itself O-GlcNAcylated in response to elevated glucose (93, 252), which also potentiates its transactivation of SREBP-1c (252). Although not directly O-GlcNAcylated (93, 124, 125), hepatic SREBP-1c is further supported by OGT-dependent protection against proteasomal degradation (125). The nuclear receptor FXR regulates bile acid metabolism but also targets proteins involved in lipogenesis and VLDL synthesis and acts as a transrepressor of ChREBP at some promoters [for review (310, 311)]. FXR O-GlcNAcylation and transcriptional activity is increased in glucose-treated HepG2 cells or re-fed mouse liver (312). All of these glucose and/or insulin stimulated proteins contribute to normal glucose-derived lipid storage in the liver, which is likely facilitated by post-prandial OGT activity. In obesity, however, persistent O-GlcNAcylation may exacerbate both hyperlipidemia by facilitating hepatic insulin resistance to VLDL repression and amplifying de novo FFA and VLDL production.
While post-prandial conditions potentiate the O-GlcNAcylation of most of the hepatic proteins described above, prolonged fasting is the driver in other cases. Both p53 and PGC1-α are more O-GlcNAcylated in fasted (24 hrs) vs. re-fed liver (11, 313). These proteins share a common regulatory target with fasting glycosylated AgRP neurons (see “Hunger”). Namely, their enhanced activities in this context all potentiate gluconeogenic gene expression, dependent on their association with OGT (11, 313). In the p53 and AgRP studies, starvation is associated with an increase in OGT protein levels (111, 313). This has also been seen in glucose deprived cells in vitro (37, 314, 315) and variously attributed to AMPK-dependent (37) or GlcNAc-mediated (314) Ogt transcription. Ruan et al., however, showed that HCF-1 helps recruit OGT specifically to PGC1-α, with peak in vitro effects near average basal blood glucose levels (i.e. ~5 mM) (11). This is consistent with the expected range for physiological gluconeogenesis regulation. Shifts in hepatic OGT targeting have also been seen in response to fasting elevations of glucagon (316). In that study, glucagon receptor activation led to CaMKII phosphorylation of OGT and its increased pro-activating association with autophagic protein ULK1 (316). Moreover, starvation (24-48 hrs) of liver-specific ad-Ogt KO mice resulted in fewer autophagy markers and reduced hepatic glucose and FFA content (316), the latter presumably due to reduced LD catabolism. Hepatic lipophagy has been observed as early as 6 hrs after fasting, believed to contribute additional substrate for FAO (317). p53 activity also upregulates multiple proteins that promote FAO and negatively regulate FAS and SREBP-1c [for review (318)]. Ad-PGC1-α OE in the rat liver potentiates FAO and diminishes hepatic and plasma TAG levels (319). Although gluconeogenesis has been the primary endpoint assessed in models of low nutrient hyper-O-GlcNAcylation, it would be interesting to see whether the fasting-appropriate shifts in lipid metabolism associated with the activation of these proteins is similarly upregulated.
Like the ARC hypothalamus and adipose tissue, the liver has dual roles, both reigning in post-prandial nutrient excess and ensuring a systemic supply of energy substrates (i.e. glucose, FFAs, ketones) during the post-absorptive and fasting periods. Feeding-stimulated O-GlcNAcylation of glucose and insulin sensitive targets facilitates nutrient storage by potentiating lipogenic transcription factors and enzymes. Fasting also induces O-GlcNAcylation of select targets involved in glucose and energy production, suggesting the importance of dynamic OGT activity for metabolic flexibility in this tissue. Consequently, static hyper-O-GlcNAcylation in obesity is likely to contribute to hepatic steatosis, circulatory hyperlipidemia and hyperglycemia by over-driving lipogenesis and glucose production while accelerating insulin resistance that disinhibits VLDL-TAG secretion.
Postprandial vs. fasting state
An important theme throughout this review is that the regulation and consequences of OGT activity differ in fasting and fed tissues as well as between physiological and obese conditions. For example, OGT OE in the mouse liver only manifests as hepatic steatosis in the fed state (124) while the lipolytic phenotypes of OGT KO in adipose (16) or kidney proximal tube epithelial cells (15) are stronger in fasted mice. Moreover, OGT loss facilitates fasting lipolysis in the first tissue but inhibits it in the second. This type of tissue specific outcome may help explain why whole body null mutations of either ogt or oga produce similar lipid storage deficiencies in C. elegans (320, 321). Cell type discrepancies are also apparent in the O-GlcNAc regulatory system’s response to systemic nutrient status. Fasting decreases the overall O-GlcNAcylation of inguinal WAT in mice without effecting epididymal WAT (16). In channel catfish, fasting induces Ogt mRNA in the liver, with no significant change in muscle or brain, while overfeeding selectively potentiates ogt expression in the brain (322). And as discussed in the previous section, even the OGT targeting of different proteins within the same cell can be reciprocally regulated by fasting and feeding to drive diverse metabolic adaptation strategies. These findings illustrate the complexity of the O-GlcNAc regulatory system as a dynamic mediator of nutrient context homeostasis that depends on unique interactions with extracellular signaling and the intracellular environment to achieve the right outcome at the right time for different tissues. By contrast, the metabolic dysfunctions that arise from persistent hyperlipidemia dull the distinction between fasting and post-prandial conditions, setting the stage for O-GlcNAc confusion to exacerbate obesity pathology.
The post-prandial state is marked by high levels of circulatory glucose and insulin as well as increased ER stress that coincide with an overall elevation in meal-related glycosylation. Following nutrient consumption, elevations in O-GlcNAcylation are driven by the mass action of enhanced glycolytic flux, which potentiates not just HBP activity but all glucose derived metabolic branches. Among these, NADPH (from the pentose phosphate pathway) and acetyl-coA (from glucose oxidation) act as substrates for FAS in the initial steps of de novo lipogenesis. FAS activation stimulates OGT protein expression and sets up a positive feedback loop between the two, enhancing lipogenic flux and suppressing FAO and changing the relative abundance of lipid metabolites. Evidence for the direct and indirect effects of different lipid species (e.g. palmitate, LCFA-CoAs) on the O-GlcNAc regulatory system and its modifiers was reviewed in section Lipid influence on the O-GlcNAc regulatory system. Insulin signaling alters the OGT substrate landscape through numerous effects on transcription (e.g. ChREBP), protein stability (e.g. CD36) and localization (e.g. FOXO1). However, it also potentiating OGT activity through tyrosine phosphorylation (323) and directs its translocation to cell membranes, particularly lipid rafts (92) that play a significant role in protein complex assembly and signal transduction [for review (324)]. In a detailed characterization of feeding-related O-GlcNAc regulatory patterns in the fly body, Liu et al. demonstrate how both eating and circadian rhythmicity drive transient elevations in pre-prandial Ogt and fasting Oga (64). These cycles guide global O-GlcNAcylation levels to rise and fall in tandem with eating (64), evidencing the important role of this modification in the ingestion and post-prandial response to food.
Moreover, insulin signaling has a particular impact on nutrient-GFAT coupling. As mentioned in section Lipid influence on the O-GlcNAc regulatory system, hyperphysiological levels of insulin appeared to be required for lipid infusion to potentiate HBP product synthesis in skeletal muscle (52, 53) and obesity was associated with higher skeletal muscle UDP-GlcNAc in two hyperinsulinemic mouse models (49, 50) but not in insulin deficient Zucker Diabetic Fatty rats (54). In humans, the positive correlation between obese BMI and muscle GFAT activity is lost in insulin resistant diabetic individuals (57). At the cellular level, insulin appears to suppress the feeding associated potentiation of cytosolic Gfat2 mRNA and protein in the liver of normal weight, standard chow fed mice (93). However, a similar elevation in ubiquitously expressed Gfat1 was insulin insensitive. This, in conjunction with an increase in OGT-binding nuclear receptor LXRα, may have contributed to the selective elevation in nuclear O-GlcNAcylation seen between fasted and re-fed mice in this tissue (93). These adaptations likely facilitate OGT activation of transcription factors such as the lipogenic transactivators described in the previous sub-section. It should be noted, however, that the durations of fasting (24 hrs) and re-feeding (12 hrs) in this study were not equivalent to physiological timepoints (325). On the other hand, XBP1 splicing, and ER stress response that potentiates Gfat1 transcription in cancer cells (61), does increase in mouse liver within a post-prandial timeframe [≤ 4 hrs post-meal (62)]. Importantly, induced XBP1s was also associated with a substantial increase in hepatic UDP-GlcNAc (62). In the fly body, Gfat2 was found to be the more nutrient sensitive paralog. Feeding induced the transcription of Gfat2, with some concurrent inhibition of activity, but had little effect on Gfat1 (64). Nevertheless, these relationships elucidate how post-prandial conditions potentiate and guide O-GlcNAcylation to promote the transcription and activity of proteins involved in the response to nutrient abundance.
By contrast, fasting is characterized by high FFA and low glucose availability with unique regulatory influences over HBP flux. Most models of nutrient deprivation are associated with a decrease in UDP-GlcNAc levels, even in models with simultaneous protein hyper-O-GlcNAcylation (37, 314, 315). These studies suggest that changes in OGT level, activity and substrate affinity are responsible, despite a loss of glucose driven HBP activity. Surprisingly, then, glucose and amino acid starvation stimulate XBP1s and GFAT1 levels in vitro (326) while nuclear receptor NR4A1, a known transcriptional enhancer of Gfat2 (14, 327), is increased by fasting in rodent WAT (328) and liver (329). Higher GFAT availability could be a means to boost a localized and/or rapidly utilized supply of OGT substrate, even as the detection of overall UDP-GlcNAc levels remains low. Given that one endpoint of fasting stimulated O-GlcNAcylation is hepatic gluconeogenesis, which cannot run concurrently with glycolysis, it is feasible that lipid-directed F6P partitioning towards the HBP (see “UDP-GlcNAc Synthesis”) also facilitates targeted O-GlcNAcylation when extracellular glucose levels are low.
Outside the post-prandial period, the substrate specificity and outcome of O-GlcNAcylation is influenced by AMPK and p38 activation as well low insulin and dysregulated glucagon signaling. Fasting stimulated NR4A1 is downstream of CaMKII activation in the heart (327) and sensitive to hepatic glucagon signaling (329). These are interrelated processes in the liver wherein glucagon receptor activation induces a rise in intracellular Ca2+ that activates CaMKII [for review (330)]. This pathway was identified as the driving mechanism behind fasting O-GlcNAcylation of ULK1, which relied on CaMKII phosphorylation of OGT (316) but could also have turned on NR4A1 mediated Gfat2 to enhance substrate supply. Furthermore, the outcome of ULK1 O-GlcNAcylation was to enhance its interaction with and activation by AMPK (316). This is conceptually in line with the observation that the O-GlcNAcylation of AMPK itself is stimulatory under pre-existing low glucose conditions. AMPK-dependent stimulation of OGT mRNA/protein has also been observed in glucose-deprived neuroblastoma cells (37). In that study, OGT substrate selection was also guided by increased interaction with p38 MAPK, which is activated in liver cells by both post-absorptive and prolonged fasting and by in vitro glucagon and LCFAs (331, 332). In addition to these OGT targeting mechanisms, low insulin signaling unmasks some O-GlcNAc regulatory relationships such as the potentiation of hepatic FoxO1 or the inhibition of PKA-dependent adipocyte PLIN1 activation, as previously discussed. While some of these mechanisms may be exclusive to starvation [e.g. XBP1s/GFAT1 (326)], others appear to be an early response to nutrient depression [e.g. p38 activation (37)] suggesting a role in physiological fasting as well. Regardless, these examples are merely a representative sampling of how feeding related shifts in intracellular conditions can alter the substrate-specific status and effect of O-GlcNAcylation.
Obesity is associated with systemic and intracellular metabolic changes that flatten the fed to fasting transition and its capacity to direct distinct O-GlcNAcylation patterns. The efficacy of insulin as a post-prandial signal is blunted due to high constitutive levels with depressed glucose-stimulated secretion plus multi-tissue insulin resistance. Hepatic de novo lipogenesis remains insulin sensitive, however, elevating its level of activation during the fasting period. Liver FFA content and a hyperglucagonemia (333) overdrive fasting gluconeogenesis while reduced insulin stimulation of post-prandial cellular uptake also diminishes glucose as a demarcation of recent consumption. Persistent hyperlipidemia, related to the disinhibition of adipocyte lipolysis/FFA leak (fed state) plus high VLDL production rate with delayed and less effective TAG clearance (fasting), drives ER stress and shifts the circulatory lipid signature throughout the day. This may be exacerbated inside cells by potentiated FFA but not LPL-dependent uptake as well as a reduced utilization of FAO and upregulated lipid storage mechanisms. Persistent nutrient excess, including HFD, depresses AMPK expression and activation [see (334)]. Consequently, nutrient and hormone responsive OGT targeting or interaction-based outcomes are likely to become less dynamic and coupled to eating in obese individuals. In addition, however, this scenario introduces mixed state regulatory forces on O-GlcNAcylation such as glucose-driven HBP activity with lipid F6P partitioning, glucagon stimulated OGT targeting without AMPK activation and the unmasking of insulin-inhibited substrates in a high UDP-GlcNAc/OGT environment. These combinations create the potential for novel and dysfunction-oriented O-GlcNAcylation patterns that are not seen in a physiological state.
Furthermore, proteins that target OGT and HBP flux are specifically dysregulated in response to obesity or HFD. Most of the proteins described above are elevated in the liver of obese humans and/or rodents including the protein level and activity of FAS (335–337) and LXRα (94–96). An increase in XBP1 splicing (338) and p38 activation (21, 339, 340) have also been reported. Contrastingly in adipose tissue, prolonged hyperlipidemia depresses FAS (337, 341, 342) and p38 transcription and activation [reviewed in (343)] while reports on LXRα have been mixed (338, 344, 345). In some cases, these tissue specific differences have been linked to a shift in lipid storage from adipose to liver in progressed obesity (337, 345). To that end, FAS and LXRα stimulate de novo lipogenesis in both tissues (346) while p38 alters hepatic FGF21 secretion to stimulate adipocyte FFA lipolysis (347). In the liver, it seems likely that the upregulation of these proteins contributes to obesity associated hepatic hyper-O-GlcNAcylation, particularly in the nucleus (21). However their role in shaping HFD WAT O-GlcNAcylation patterns is less clear. It is worth noting that NR4A1 gene expression is higher in obese human and rodent WAT (348, 349) while nutrient driven UDP-GlcNAc synthesis is expected to contribute in both tissues as well. One precaution to interpretation is that these proteins and transcripts were investigated exclusively in random-fed animals. Nevertheless, the sensitivity of their expression to in vitro hyperlipidemia and/or cellular stress, which are tonically elevated in obese tissues, suggests that the changes are likely constitutive and contribute to inappropriate regulation of O-GlcNAc-mediated cellular programming in both states. Table 2 summarizes qualitative differences between the fasting and post-prandial milieu under physiological and obese conditions.
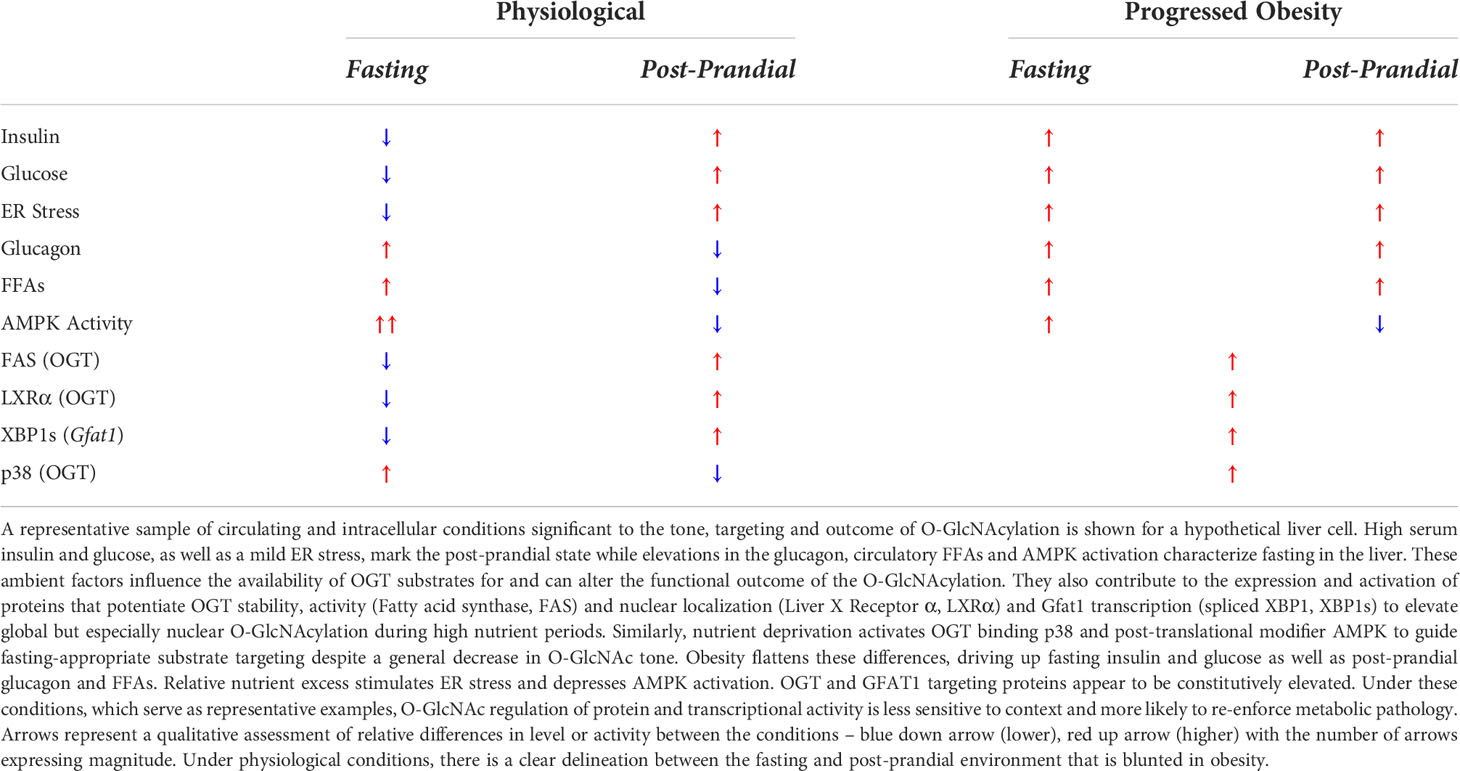
Table 2 Hepatic O-GlcNAc Interactome in Physiology and Obesity.
Summary and future directions
Despite its traditional classification as a glucose metabolism associated modification, the O-GlcNAc regulatory system interacts extensively with lipids and appears central to support lipid homeostasis. Hyperlipidemia (e.g. in vitro fatty acid exposure, diet induced obesity) potentiates protein O-GlcNAcylation in many cell types including gastrointestinal tissue, adipose, muscle, heart and liver. A biphasic response was noted in pancreatic β-cells and macrophages with an early rise in O-GlcNAcylation followed by a drop below baseline in models of more progressed obesity. In some neural cells (i.e. hippocampus, neuroblastoma cells), elevated lipid exposure depressed O-GlcNAcylation under all tested conditions. It is not clear whether these examples of lipid stimulated hypo-O-GlcNAcylation are exclusive to the noted tissue types or if the available data is restricted by experimental variability. More consistent standards in the use of subject sex and species, lipid type and duration, endpoint measurements and the use of multi-timepoint assessment would significantly aid in understanding this complex physiology.
Cellular O-GlcNAcylation patterns are commonly associated with changes in the enzymes that direct HBP UDP-GlcNAc synthesis and with the glycosylation enzymes OGT and OGA. However the relative contributions of each were context dependent. Mechanisms linking lipids to the HBP rate-limiting enzyme GFAT include transcriptional potentiation by ER stress protein Xbp1s and lipotoxicity sensor NR4A1 as well as inhibitory AMPK phosphorylation. Despite serving the same HBP function, the paralogous GFAT1 and GFAT2 proteins, which are not always experimentally distinguished, appear to be independently regulated and show distinct responses to ingested nutrients and different lipid species. Moreover, most of the enzymes that regulate glycolysis and the HBP are fat sensitive, particularly around the nexus of F6P metabolism. The effects of LCFA or LCFA-CoAs on GFAT/GNPDA and PFK/FBPase all appear to direct glucose towards the HBP at the expense of glycolysis under hyper-O-GlcNAcylating conditions. Numerous lipid-sensitive OGT modification strategies have been documented including transcription changes, post-transcriptional suppression by micro-RNAs, FAS-potentiated protein stabilization and activity, AMPK-dependent substrate preference and shifts in subcellular localization to bind nuclear receptor LXR or membrane-bound PIP3. OGA regulation has not been deeply characterized in this context but FAS depresses its activity and a mitochondria/lipid droplet associated splice isoform, sOGA, has been identified. Thus, the O-GlcNAc regulatory system evinces a surprising flexibility in lipid response strategies despite its limited diversity of direct enzyme regulators.
In addition to being lipid responsive, the O-GlcNAc regulatory system plays a significant role in lipid physiology, including dietary fat intake. Systemic lipid uptake is determined by the overall amount of food consumed and the preference and intestinal absorption of its fat content. Under physiological conditions, hypothalamic O-GlcNAcylation is increased by fasting in orexigenic AgRP neurons and by feeding in anorectic ARC and PVN neurons. In both cases, OGT activity is required to maintain normal excitatory activity and/or neuronal viability, as well as AgRP ghrelin sensitivity and ARC leptin sensitivity. In addition, OGT acts outside the CNS to support the synthesis and HFD upregulation of satiety hormones, including adipocyte-secreted leptin and β-cell insulin. These findings implicate O-GlcNAcylation in the homeostatic regulation of eating but OGT also influences dietary fat preference specifically. Adipocyte OGT is required for transient HFD hyperphagia by coupling lipid level to the secretion of hunger-promoting endocannabinoid hormone AEA. Macrophage OGT depletion also potentiates HFD overconsumption, possibly downstream of inflammation-related taste bud remodeling or insulin resistance in mesolimbic dopamine neurons that control the hedonic motivation to eat. In macrophages, OGT is a downstream inhibitor of TLR4 signaling, which is also necessary for dietary sugar and fat preference suggesting that its interactions with OGT in mesolimbic cells and on the tongue merit further investigation. O-GlcNAcylation in the intestine may support dietary lipid extraction by limiting local GLP-1 secretion, which otherwise suppresses enterocyte chylomicron-TAG output. Furthermore, O-GlcNAcylation in vitro upregulates two important lipid sensing proteins - PLIN2 and CD36. LD membrane protein PLIN2 facilitates cytosolic LD storage in multiple cells, including as a means to potentiate enterocyte lipid extraction. CD36 is similarly involved in multi-cellular FFA uptake and signal transduction that contributes to lingual fat sensitivity and enterocyte synthesis of chylomicrons and anorectic hormone OEA. Importantly, O-GlcNAcylation has not been phenotypically characterized in fasting-stimulated hunger or intestinal lipid absorption but has been directly tied to meal-related satiety and fat preference.
Context-sensitive OGT activity supports both pro-consumptive and negative feedback mechanisms on eating and lipid uptake emphasizing its role in homeostatic balancing. By contrast, hyperlipidemia-driven O-GlcNAcylation patterns show the potential to exacerbate obesity endophenotypes that drive pathological overeating. These include the uncoupling of orexigenic and anorectic signals from their nutrient context, an overall decrease in satiety and tonic elevations in the hedonic value and lipid extraction of food. Towards that end, adipocyte hyper-O-GlcNAcylation in obesity is consistent with the progressive increase in plasma AEA that supports food craving and reward. The opposite condition in macrophages, hypo-O-GlcNAcylation, is linked to proinflammatory signaling known to facilitate ghrelin, leptin and insulin resistance that numbs homoeostatic circuitry to nutrient status signals and feedback triggers. This is likely accelerated by O-GlcNAc stabilization of PTP1B and STAT3, proteins with a primary role in insulin and leptin resistance in the brain and peripheral tissues. Shifts in oxidative capacity or fuel preference, seen systemically in both OGA and OGT depressed animals, are likely to disrupt direct nutrient sensing in AgRP (FAO) and/or POMC (glucose oxidizing) neurons, consistent with the nutrient insensitivity of these regions in obese individuals. Unfortunately, O-GlcNAc responses to hyperlipidemia and obesity in most areas of the brain are not known so the relevant mechanisms in this state remain speculative.
Beyond ingestion, dynamic O-GlcNAcylation in peripheral cell types has been implicated in lipid uptake and utilization as well as the balance between nutrient storage and release. OGT activity stimulates CD36 dependent FFA uptake in heart cells and likely in adipocytes by potentiating LPL secretion, which is required for circulatory TAG hydrolysis. While these proteins have similar roles in other tissues (e.g. liver, skeletal muscle), their O-GlcNAc regulation has not been tested in that context. In addition to LPL, however, adipocyte O-GlcNAcylation tunes lipolytic capacity by inhibiting the PKA (i.e. fasting) phosphorylation of LD protein and lipolysis gatekeeper PLIN1 and possibly by mediating its membrane expression during LD maturation. In addition, dynamic OGT/OGA activity are required for the terminal differentiation phase of adipogenesis. This is likely related to O-GlcNAc’s inhibition of C/EBPβ and its potentiation of PPARɣ2, the latter of which drives lipogenic and adipokinic gene expression. Thus, the O-GlcNAc regulatory system contributes to the maximal lipid storage capacity of WAT and its metabolic flexibility to switch between post-prandial FFA uptake and fasting FFA release. The liver also takes on dual roles in lipid storage and release as well as fasting glucose production. Hepatic OGT potentiates the meal related activation of lipogenic enzymes (FAS, ACC) and transcription factors (ChREBP, LXR, SREBP-1c, FXR). However during fasting, hepatic AMPK and glucagon signaling steer O-GlcNAcylation onto select protein targets that activate gluconeogenesis, autophagy and FAO. Proper WAT and liver function, therefore, rely on differential detection of and regulation by the post-prandial and fasting states including relative glucose/FFA abundance, insulin/glucagon signaling and ER stress/AMPK activation. The O-GlcNAc regulatory system is similarly sensitive to these conditions, leading to unique variations in HBP flux, OGT behavior and substrate interactions that guide its context-dependent support for nutrient homeostasis in these tissues.
During early exposure, lipid-responsive O-GlcNAcylation in peripheral cells helps to accommodate dietary fat overconsumption by upregulating the mechanisms behind circulatory TAG clearance, cardiac FAO supply, hepatic nutrient buffering and WAT lipid storage. In the face of persistent hyperlipidemia, however, these same changes are counter-productive to cellular health due to excessive lipid accumulation, exacerbated by overactive hepatic lipogenesis and impaired adipocyte lipolysis and differentiation. Insulin resistance, facilitated by OGT depleted pro-inflammatory macrophages and adipose/liver hyper-O-GlcNAcylation, further impairs adipocyte FFA uptake and disinhibits FFA/VLDL secretion, adding back to circulatory hyperlipidemia. The imbalance of nutrient and hormone signaling blunts the fed to fasting transition, which feeds back onto the O-GlcNAc regulatory system itself. An oversupply of OGT, GFAT and HBP nutrient input together with context-insensitive OGT targeting and substrate access could fundamentally change the functional impact of O-GlcNAcylation and divorce it from sources of negative feedback. While not proven, this hypothesis suggests how the O-GlcNAc regulatory system, with its sole pair of enzyme controllers and a nutrient-promiscuous source of effector, can nevertheless diversely contribute to complex homeostatic relationships. Furthermore, it adds nuance to the “goldilocks zone” theory for how the same directional change in O-GlcNAc tone can be both beneficial and harmful under different circumstances.
In conclusion, O-GlcNAcylation is best understood as a multi-nutrient sensitive modification that has distinct glucose and lipid sensitive response profiles as well as a flexible array of outcomes dependent on cellular context. Under physiological conditions, cell type specific O-GlcNAcylation patterns contribute to low and high nutrient-cued homeostasis to influence the balance between hunger vs. satiety, fat preference vs. overconsumption, lipid utilization vs. storage vs. release. It may also serve as a form of intermediate length metabolic memory to tie recent or expected nutrient availability with markers of energy supply, demand and cell stress in guiding cellular adaptation strategies to acute nutrient conditions. Prolonged hyperlipidemia, however, appears to disrupt this carefully balanced system and shift the net outcome of O-GlcNAc regulatory activity towards obesogenic and metabolic dysfunction. Future investigations into the source of this shift - whether differences in O-GlcNAc levels, OGT/OGA targeting, or the larger O-GlcNAc interactome – may prove fruitful in delineating and abrogating the self-sustaining pathways between progressive obesity and metabolic disease.
Author contributions
AL conceived the initial review topic and created the first draft of the manuscript and figures. AL and JH contributed together to the ongoing conceptual development and revision of the final manuscript and figures.
Funding
Funding was provided by NIDDK intramural research program grant ZIADK060103.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fendo.2022.943576/full#supplementary-material
References
1. Hanover JA, Krause MW, Love DC. The hexosamine signaling pathway: O-GlcNAc cycling in feast or famine. Biochim Biophys Acta (2010) 1800(2):80–95. doi: 10.1016/j.bbagen.2009.07.017
2. Chatham JC, Zhang J, Wende AR. Role of O-linked n-acetylglucosamine protein modification in cellular (Patho)Physiology. Physiol Rev (2021) 101(2):427–93. doi: 10.1152/physrev.00043.2019
3. Wulff-Fuentes E, Berendt RR, Massman L, Danner L, Malard F, Vora J, et al. The human O-GlcNAcome database and meta-analysis. Sci Data (2021) 8(1):25. doi: 10.1038/s41597-021-00810-4
4. Ma J, Li Y, Hou Wu, Wu C. O-GlcNAcAtlas: A database of experimentally identified O-GlcNAc sites and proteins. Glycobiology (2021) 31(7):719–23. doi: 10.1093/glycob/cwab003
5. Zachara N, Akimoto Y, Hart GW. The O-GlcNAc modification. In: Essentials of glycobiology Cold spring harbor (NY): Cold Spring Harbor Laboratory Press (2015). p. 239–51. rd.
6. Hart GW, Slawson C, Ramirez-Correa G, Lagerlof O. Cross talk between O-GlcNAcylation and phosphorylation: roles in signaling, transcription, and chronic disease. Annu Rev Biochem (2011) 80:825–58. doi: 10.1146/annurev-biochem-060608-102511
7. Yang X, Qian K. Protein O-GlcNAcylation: emerging mechanisms and functions. Nat Rev Mol Cell Biol (2017) 18(7):452–65. doi: 10.1038/nrm.2017.22
8. Peterson SB, Hart GW. New insights: A role for O-GlcNAcylation in diabetic complications. Crit Rev Biochem Mol Biol (2016) 51(3):150–61. doi: 10.3109/10409238.2015.1135102
9. Ning J, Yang H. O-GlcNAcylation in hyperglycemic pregnancies: Impact on placental function. Front Endocrinol (Lausanne) (2021) 12:659733. doi: 10.3389/fendo.2021.659733
10. Groves JA, Lee A, Yildirir G, Zachara NE. Dynamic O-GlcNAcylation and its roles in the cellular stress response and homeostasis. Cell Stress Chaperones (2013) 18(5):535–58. doi: 10.1007/s12192-013-0426-y
11. Ruan HB, Han X, Li MD, Singh JP, Qian K, Azarhoush S, et al. O-GlcNAc transferase/host cell factor C1 complex regulates gluconeogenesis by modulating PGC-1alpha stability. Cell Metab (2012) 16(2):226–37. doi: 10.1016/j.cmet.2012.07.006
12. Zuliani I, Lanzillotta C, Tramutola A, Barone E, Perluigi M, Rinaldo S, et al. High-fat diet leads to reduced protein O-GlcNAcylation and mitochondrial defects promoting the development of alzheimer's disease signatures. Int J Mol Sci (2021) 22(7). doi: 10.3390/ijms22073746
13. Luo B, Soesanto Y, McClain DA. Protein modification by O-linked GlcNAc reduces angiogenesis by inhibiting akt activity in endothelial cells. Arterioscler Thromb Vasc Biol (2008) 28(4):651–7. doi: 10.1161/ATVBAHA.107.159533
14. Dai W, Dierschke SK, Toro AL, Dennis MD. Consumption of a high fat diet promotes protein O-GlcNAcylation in mouse retina via NR4A1-dependent GFAT2 expression. Biochim Biophys Acta Mol Basis Dis (2018) 1864(12):3568–76. doi: 10.1016/j.bbadis.2018.09.006
15. Sugahara S, Kume S, Chin-Kanasaki M, Yasuda-Yamahara M, Yamahara K, et al. Protein O-GlcNAcylation is essential for the maintenance of renal energy homeostasis and function via lipolysis during fasting and diabetes. J Am Soc Nephrol (2019) 30(6):962–78. doi: 10.1681/ASN.2018090950
16. Yang Y, Fu M, Li MD, Zhang K, Zhang B, Wang S, et al. O-GlcNAc transferase inhibits visceral fat lipolysis and promotes diet-induced obesity. Nat Commun (2020) 11(1):181. doi: 10.1038/s41467-019-13914-8
17. Zhao M, Ren K, Xiong X, Cheng M, Zhang Z, Huang Z, et al. Protein O-GlcNAc modification links dietary and gut microbial cues to the differentiation of enteroendocrine l cells. Cell Rep (2020) 32(6):108013. doi: 10.1016/j.celrep.2020.108013
18. Wang X, Feng Z, Wang X, Yang L, Han S, Cao K, et al. O-GlcNAcase deficiency suppresses skeletal myogenesis and insulin sensitivity in mice through the modulation of mitochondrial homeostasis. Diabetologia (2016) 59(6):1287–96. doi: 10.1007/s00125-016-3919-2
19. Rajamani U, Joseph D, Roux S, Essop MF, Rajamani U, et al. The hexosamine biosynthetic pathway can mediate myocardial apoptosis in a rat model of diet-induced insulin resistance. Acta Physiol (Oxf) (2011) 202(2):151–7. doi: 10.1111/j.1748-1716.2011.02275.x
20. Lima VV, Giachini FR, Matsumoto T, Li W, Bressan AF, Chawla D, et al. High-fat diet increases O-GlcNAc levels in cerebral arteries: a link to vascular dysfunction associated with hyperlipidaemia/obesity? Clin Sci (Lond) (2016) 130(11):871–80. doi: 10.1042/CS20150777
21. Li SY, Liu Y, Sigmon VK, McCort A, Ren J, et al. High-fat diet enhances visceral advanced glycation end products, nuclear O-Glc-Nac modification, p38 mitogen-activated protein kinase activation and apoptosis. Diabetes Obes Metab (2005) 7(4):448–54. doi: 10.1111/j.1463-1326.2004.00387.x
22. Karimi M, Pavlov VI, Ziegler O, Sriram N, Yoon SY, Agbortoko V, et al. Robust effect of metabolic syndrome on major metabolic pathways in the myocardium. PloS One (2019) 14(12):e0225857. doi: 10.1371/journal.pone.0225857
23. Raab S, Gadault A, Very N, Decourcelle A, Baldini S, Schulz C, et al. Dual regulation of fatty acid synthase (FASN) expression by O-GlcNAc transferase (OGT) and mTOR pathway in proliferating liver cancer cells. Cell Mol Life Sci (2021) 78(13):5397–413. doi: 10.1007/s00018-021-03857-z
24. Lockridge A, Jo S, Gustafson E, Damberg N, Mohan R, Olson M, et al. Islet O-GlcNAcylation is required for lipid potentiation of insulin secretion through SERCA2. Cell Rep (2020) 31(5):107609. doi: 10.1016/j.celrep.2020.107609
25. Yang Y, Li X, Luan HH, Zhang B, Zhang K, Nam JH, et al. OGT suppresses S6K1-mediated macrophage inflammation and metabolic disturbance. Proc Natl Acad Sci U.S.A. (2020) 117(28):16616–25. doi: 10.1073/pnas.1916121117
26. Lee DE, Lee SJ, Kim SJ, Lee SH, Kwon OS. Curcumin ameliorates nonalcoholic fatty liver disease through inhibition of O-GlcNAcylation. Nutrients (2019) 11(11). doi: 10.3390/nu11112702
27. Zhao Y, Tang Z, Shen A, Tao C, Wan C, Zhu X, et al. The role of PTP1B O-GlcNAcylation in hepatic insulin resistance. Int J Mol Sci (2015) 16(9):22856–69. doi: 10.3390/ijms160922856
28. Jiang M, Wu N, Xu B, Chu X, Li X, Su S, et al. Fatty acid-induced CD36 expression via O-GlcNAcylation drives gastric cancer metastasis. Theranostics (2019) 9(18):5359–73. doi: 10.7150/thno.34024
29. Habegger KM, Penque BA, Sealls W, Tackett L, Bell LN, Blue EK, et al. Fat-induced membrane cholesterol accrual provokes cortical filamentous actin destabilisation and glucose transport dysfunction in skeletal muscle. Diabetologia (2012) 55(2):457–67. doi: 10.1007/s00125-011-2334-y
30. Park MJ, Kim DI, Lim SK, Choi JH, Han HJ, Yoon KC, et al. High glucose-induced O-GlcNAcylated carbohydrate response element-binding protein (ChREBP) mediates mesangial cell lipogenesis and fibrosis: the possible role in the development of diabetic nephropathy. J Biol Chem (2014) 289(19):13519–30. doi: 10.1074/jbc.M113.530139
31. Pang Y, Xu X, Xiang X, Li Y, Zhao Z, Li J, et al. High fat activates O-GlcNAcylation and affects AMPK/ACC pathway to regulate lipid metabolism. Nutrients (2021) 13(6). doi: 10.3390/nu13061740
32. Fricovsky ES, Suarez J, Ihm SH, Scott BT, Suarez-Ramirez JA, Banerjee I, et al. Excess protein O-GlcNAcylation and the progression of diabetic cardiomyopathy. Am J Physiol Regul Integr Comp Physiol (2012) 303(7):R689–99. doi: 10.1152/ajpregu.00548.2011
33. Weigert C, Klopfer K, Kausch C, Brodbeck K, Stumvoll M, Haring HU, et al. Palmitate-induced activation of the hexosamine pathway in human myotubes: increased expression of glutamine:fructose-6-phosphate aminotransferase. Diabetes (2003) 52(3):650–6. doi: 10.2337/diabetes.52.3.650
34. Wong SL, Wu LL, Robker RL, Thompson JG, McDowall ML. Hyperglycaemia and lipid differentially impair mouse oocyte developmental competence. Reprod Fertil Dev (2015) 27(4):583–92. doi: 10.1071/RD14328
35. Schonfeld P, Reiser G. Why does brain metabolism not favor burning of fatty acids to provide energy? reflections on disadvantages of the use of free fatty acids as fuel for brain. J Cereb Blood Flow Metab (2013) 33(10):1493–9. doi: 10.1038/jcbfm.2013.128
36. Park J, Lai MKP, Arumugam TV, Jo DG, et al. O-GlcNAcylation as a therapeutic target for alzheimer's disease. Neuromolecular Med (2020) 22(2):171–93. doi: 10.1007/s12017-019-08584-0
37. Cheung WD, Hart GW. AMP-activated protein kinase and p38 MAPK activate O-GlcNAcylation of neuronal proteins during glucose deprivation. J Biol Chem (2008) 283(19):13009–20. doi: 10.1074/jbc.M801222200
38. Hardiville S, Banerjee PS, Selen Alpergin ES, Smith DM, Han G, Ma J, et al. TATA-box binding protein O-GlcNAcylation at T114 regulates formation of the b-TFIID complex and is critical for metabolic gene regulation. Mol Cell (2020) 77(5):1143–1152.e7. doi: 10.1016/j.molcel.2019.11.022
39. Kraakman MJ, Murphy AJ, Jandeleit-Dahm K, Kammoun HL. Macrophage polarization in obesity and type 2 diabetes: weighing down our understanding of macrophage function? Front Immunol (2014) 5:470. doi: 10.3389/fimmu.2014.00470
40. Alejandro EU, Gregg B, Blandino-Rosano M, Cras-Meneur C, Bernal-Mizrachi E. Natural history of beta-cell adaptation and failure in type 2 diabetes. Mol Aspects Med (2015) 42:19–41. doi: 10.1016/j.mam.2014.12.002
41. Visinoni S, Khalid NF, Joannides CN, Shulkes A, Yim M, Whitehead J, et al. The role of liver fructose-1,6-bisphosphatase in regulating appetite and adiposity. Diabetes (2012) 61(5):1122–32. doi: 10.2337/db11-1511
42. Oikari S, Makkonen K, Deen AJ, Tyni I, Karna R, Tammi RH, et al. Hexosamine biosynthesis in keratinocytes: roles of GFAT and GNPDA enzymes in the maintenance of UDP-GlcNAc content and hyaluronan synthesis. Glycobiology (2016) 26(7):710–22. doi: 10.1093/glycob/cww019
43. Araujo L, Khim P, Mkhikian H, Mortales CL, Demetriou M, et al. Glycolysis and glutaminolysis cooperatively control T cell function by limiting metabolite supply to n-glycosylation. Elife (2017) 6. doi: 10.7554/eLife.21330
44. Kroef V, Ruegenberg S, Horn M, Allmeroth K, Ebert L, Bozkus S, et al. GFPT2/GFAT2 and AMDHD2 act in tandem to control the hexosamine pathway. Elife (2022) 11. doi: 10.7554/eLife.69223.sa2
45. Hu Y, Riesland L, Paterson AJ, Kudlow JE. Phosphorylation of mouse glutamine-fructose-6-phosphate amidotransferase 2 (GFAT2) by cAMP-dependent protein kinase increases the enzyme activity. J Biol Chem (2004) 279(29):29988–93. doi: 10.1074/jbc.M401547200
46. Oki T, Yamazaki K, Kuromitsu J, Okada M, Tanaka I. cDNA cloning and mapping of a novel subtype of glutamine:fructose-6-phosphate amidotransferase (GFAT2) in human and mouse. Genomics (1999) 57(2):227–34. doi: 10.1006/geno.1999.5785
47. Cotsworth S, Jackson CJ, Hallson G, Fitzpatrick KA, Syrzycka M, Coulthard AB, et al. Characterization of Gfat1 (zeppelin) and Gfat2, essential paralogous genes which encode the enzymes that catalyze the rate-limiting step in the hexosamine biosynthetic pathway in drosophila melanogaster. Cells (2022) 11(3). doi: 10.3390/cells11030448
48. Biwi J, Biot C, Guerardel Y, Vercoutter-Edouart AS, Lefebvre T. The many ways by which O-GlcNAcylation may orchestrate the diversity of complex glycosylations. Molecules (2018) 23(11). doi: 10.3390/molecules23112858
49. Buse MG, Robinson KA, Gettys TW, McMahon EG, Gulve EA. Increased activity of the hexosamine synthesis pathway in muscles of insulin-resistant ob/ob mice. Am J Physiol (1997) 272(6 Pt 1):E1080–8. doi: 10.1152/ajpendo.1997.272.6.E1080
50. Fujita T, Furukawa S, Morita K, Ishihara T, Fujita M, Shiotani T, Matsushita Y, et al. Glucosamine induces lipid accumulation and adipogenic change in C2C12 myoblasts. Biochem Biophys Res Commun (2005) 328(2):369–74. doi: 10.1016/j.bbrc.2004.12.185
51. Obici S, Feng Z, Karkanias G, Baskin DG, Rossetti L, et al. Decreasing hypothalamic insulin receptors causes hyperphagia and insulin resistance in rats. Nat Neurosci (2002) 5(6):566–72. doi: 10.1038/nn0602-861
52. Hawkins M, Barzilai N, Liu R, Hu M, Rossetti L, et al. Role of the glucosamine pathway in fat-induced insulin resistance. J Clin Invest (1997) 99(9):2173–82. doi: 10.1172/JCI119390
53. Choi CS, Lee FN, Youn JH. Free fatty acids induce peripheral insulin resistance without increasing muscle hexosamine pathway product levels in rats. Diabetes (2001) 50(2):418–24. doi: 10.2337/diabetes.50.2.418
54. Bosch RR, Janssen SW, Span PN, Olthaar A, van Emst-de Vries SE, Willems PH, et al. Exploring levels of hexosamine biosynthesis pathway intermediates and protein kinase c isoforms in muscle and fat tissue of zucker diabetic fatty rats. Endocrine (2003) 20(3):247–52. doi: 10.1385/ENDO:20:3:247
55. Meadus WJ, MacInnis R, Dugan ME. Prolonged dietary treatment with conjugated linoleic acid stimulates porcine muscle peroxisome proliferator activated receptor gamma and glutamine-fructose aminotransferase gene expression in vivo. J Mol Endocrinol (2002) 28(2):79–86. doi: 10.1677/jme.0.0280079
56. Cui YL, Xue RQ, Xi H, Ming Z, Yu XJ, Lui LZ, et al. Cholinergic drugs ameliorate endothelial dysfunction by decreasing O-GlcNAcylation via M3 AChR-AMPK-ER stress signaling. Life Sci (2019) 222:1–12. doi: 10.1016/j.lfs.2019.02.036
57. Daniels MC, Ciaraldi TP, Nikoulina S, Henry RR, McClain DA. Glutamine:fructose-6-phosphate amidotransferase activity in cultured human skeletal muscle cells: relationship to glucose disposal rate in control and non-insulin-dependent diabetes mellitus subjects and regulation by glucose and insulin. J Clin Invest (1996) 97(5):1235–41. doi: 10.1172/JCI118538
58. Willer CJ, Speliotes EK, Loos RJ, Li S, Lindgren CM, Heid IM, et al. Six new loci associated with body mass index highlight a neuronal influence on body weight regulation. Nat Genet (2009) 41(1):25–34. doi: 10.1038/ng.287
59. Gutierrez-Aguilar R, Kim DH, Woods SC, Seeley RJ. Expression of new loci associated with obesity in diet-induced obese rats: from genetics to physiology. Obes (Silver Spring) (2012) 20(2):306–12. doi: 10.1038/oby.2011.236
60. Han J, Kaufman RJ. The role of ER stress in lipid metabolism and lipotoxicity. J Lipid Res (2016) 57(8):1329–38. doi: 10.1194/jlr.R067595
61. Liu Y, Cao Y, Pan X, Shi M, Wu Q, Huang T, et al. O-GlcNAc elevation through activation of the hexosamine biosynthetic pathway enhances cancer cell chemoresistance. Cell Death Dis (2018) 9(5):485. doi: 10.1038/s41419-018-0522-0
62. Deng Y, Wang ZV, Tao C, Gao N, Holland WL, Ferdous A, et al. The Xbp1s/GalE axis links ER stress to postprandial hepatic metabolism. J Clin Invest (2013) 123(1):455–68. doi: 10.1172/JCI62819
63. Chen P, Visokay S, Abrams JM. Drosophila GFAT1 and GFAT2 enzymes encode obligate developmental functions. Fly (Austin) (2020) 14(1-4):3–9. doi: 10.1080/19336934.2020.1784674
64. Liu X, Blazenovic I, Contreras AJ, Pham TM, Tabuloc CA, Li YH, et al. Hexosamine biosynthetic pathway and O-GlcNAc-processing enzymes regulate daily rhythms in protein O-GlcNAcylation. Nat Commun (2021) 12(1):4173. doi: 10.1038/s41467-021-24301-7
65. Jenkins CM, Yang J, Sims HF, Gross RW. Reversible high affinity inhibition of phosphofructokinase-1 by acyl-CoA: a mechanism integrating glycolytic flux with lipid metabolism. J Biol Chem (2011) 286(14):11937–50. doi: 10.1074/jbc.M110.203661
66. Hue L, Taegtmeyer H. The randle cycle revisited: a new head for an old hat. Am J Physiol Endocrinol Metab (2009) 297(3):E578–91. doi: 10.1152/ajpendo.00093.2009
67. Busch AK, Cordery D, Denyer GS, Biden TJ, et al. Expression profiling of palmitate- and oleate-regulated genes provides novel insights into the effects of chronic lipid exposure on pancreatic beta-cell function. Diabetes (2002) 51(4):977–87. doi: 10.2337/diabetes.51.4.977
68. Ong Q, Han W, Yang X. O-GlcNAc as an integrator of signaling pathways. Front Endocrinol (Lausanne) (2018) 9:599. doi: 10.3389/fendo.2018.00599
69. Lazarus MB, Jiang J, Gloster TM, Zandberg WF, Whitworth GE, Vocadlo DJ, et al. Structural snapshots of the reaction coordinate for O-GlcNAc transferase. Nat Chem Biol (2012) 8(12):966–8. doi: 10.1038/nchembio.1109
70. Lu L, Fan D, Hu CW, Worth M, Lu L, Ma ZX, Jiang J, et al. Distributive O-GlcNAcylation on the highly repetitive c-terminal domain of RNA polymerase II. Biochemistry (2016) 55(7):1149–58. doi: 10.1021/acs.biochem.5b01280
71. Keembiyehetty CN, Krzeslak A, Love DC, Hanover JA. A lipid-droplet-targeted O-GlcNAcase isoform is a key regulator of the proteasome. J Cell Sci (2011) 124(Pt 16):2851–60. doi: 10.1242/jcs.083287
72. Pagesy P, Bouaboud A, Feng Z, Hulin P, Issad T. Short O-GlcNAcase is targeted to the mitochondria and regulates mitochondrial reactive oxygen species level. Cells (2022) 11(11). doi: 10.3390/cells11111827
73. Urso SJ, Comly M, Hanover JA, Lamitina T. The O-GlcNAc transferase OGT is a conserved and essential regulator of the cellular and organismal response to hypertonic stress. PloS Genet (2020) 16(10):e1008821. doi: 10.1371/journal.pgen.1008821
74. Ma J, Wu C, Hart GW. Analytical and biochemical perspectives of protein O-GlcNAcylation. Chem Rev (2021) 121(3):1513–81. doi: 10.1021/acs.chemrev.0c00884
75. Tan ZW, Fei G, Paolo JA, Bellaousov S, Martin SES, Duveau DY, et al. O-GlcNAc regulates gene expression by controlling detained intron splicing. Nucleic Acids Res (2020) 48(10):5656–69. doi: 10.1093/nar/gkaa263
76. Lin CH, Liao CC, Chen MY, Chou TY. Feedback regulation of O-GlcNAc transferase through translation control to maintain intracellular O-GlcNAc homeostasis. Int J Mol Sci (2021) 22(7). doi: 10.3390/ijms22073463
77. Pinkosky SL, Scott JW, Desjardins EM, Smith BK, Day EA, Ford RJ, et al. Long-chain fatty acyl-CoA esters regulate metabolism via allosteric control of AMPK beta1 isoforms. Nat Metab (2020) 2(9):873–81. doi: 10.1038/s42255-020-0245-2
78. Han Y, Luo Y, Sun J, Ding Z, Liu J, Yan W, et al. AMPK signaling in the dorsal hippocampus negatively regulates contextual fear memory formation. Neuropsychopharmacology (2016) 41(7):1849–64. doi: 10.1038/npp.2015.355
79. Bullen JW, Balsbaugh JL, Chanda D, Shabanowitz J, Hunt DF, Neumann D, et al. Cross-talk between two essential nutrient-sensitive enzymes: O-GlcNAc transferase (OGT) and AMP-activated protein kinase (AMPK). J Biol Chem (2014) 289(15):10592–606. doi: 10.1074/jbc.M113.523068
80. Babae N, Bourajjaj M, Liu Y, Van Beijnum JR, Cerisoli F, Scaria PJ, et al. Systemic miRNA-7 delivery inhibits tumor angiogenesis and growth in murine xenograft glioblastoma. Oncotarget (2014) 5(16):6687–700. doi: 10.18632/oncotarget.2235
81. Woo SY, Lee SY, Yu SL, Park SJ, Kang D, Kim JS, et al. MicroRNA-7-5p's role in the O-GlcNAcylation and cancer metabolism. Noncoding RNA Res (2020) 5(4):201–7. doi: 10.1016/j.ncrna.2020.11.003
82. Afonso MS, Verma N, van Solingen C, Cyr Y, Sharma M, Perie L, et al. MicroRNA-33 inhibits adaptive thermogenesis and adipose tissue beiging. Arterioscler Thromb Vasc Biol (2021) 41(4):1360–73. doi: 10.1161/ATVBAHA.120.315798
83. Marquart TJ, Allen RM, Ory DS, Baldan A. miR-33 links SREBP-2 induction to repression of sterol transporters. Proc Natl Acad Sci U.S.A. (2010) 107(27):12228–32. doi: 10.1073/pnas.1005191107
84. Li ZY, Na HM, Peng G, Pu J, Liu P. Alteration of microRNA expression correlates to fatty acid-mediated insulin resistance in mouse myoblasts. Mol Biosyst (2011) 7(3):871–7. doi: 10.1039/C0MB00230E
85. Latreille M, Hausser J, Stutzer I, Zhang Q, Hastoy B, Gargani S, et al. MicroRNA-7a regulates pancreatic beta cell function. J Clin Invest (2014) 124(6):2722–35. doi: 10.1172/JCI73066
86. Singaravelu R, Quan C, Powdrill MH, Shaw TA, Srinivasan P, Lyn RK, et al. MicroRNA-7 mediates cross-talk between metabolic signaling pathways in the liver. Sci Rep (2018) 8(1):361. doi: 10.1038/s41598-017-18529-x
87. Baldini SF, Wavelet C, Hainault I, Guinez C, Lefebvre T, et al. The nutrient-dependent O-GlcNAc modification controls the expression of liver fatty acid synthase. J Mol Biol (2016) 428(16):3295–304. doi: 10.1016/j.jmb.2016.04.035
88. Groves JA, Maduka AO, O'Meally RN, Cole RN, Zachara NE. Fatty acid synthase inhibits the O-GlcNAcase during oxidative stress. J Biol Chem (2017) 292(16):6493–511. doi: 10.1074/jbc.M116.760785
89. Yang X, Ongusaha PP, Miles PD, Havstad JC, Zhang F, So WV, et al. Phosphoinositide signalling links O-GlcNAc transferase to insulin resistance. Nature (2008) 451(7181):964–9. doi: 10.1038/nature06668
90. Kebede M, Ferdaoussi M, Mancini A, Alquier T, Kulkarni RN, Walker MD, et al. Glucose activates free fatty acid receptor 1 gene transcription via phosphatidylinositol-3-kinase-dependent O-GlcNAcylation of pancreas-duodenum homeobox-1. Proc Natl Acad Sci U.S.A. (2012) 109(7):2376–81. doi: 10.1073/pnas.1114350109
91. Lazarus MB, Nam Y, Jiang J, Sliz P, Walker S. Structure of human O-GlcNAc transferase and its complex with a peptide substrate. Nature (2011) 469(7331):564–7.
92. Perez-Cervera Y, Dehennaut YV, Aquino Gil M, Guedri K, Solorzano Mata CJ, Olivier-Van Stichelen S, et al. Insulin signaling controls the expression of O-GlcNAc transferase and its interaction with lipid microdomains. FASEB J (2013) 27(9):3478–86. doi: 10.1096/fj.12-217984
93. Bindesboll C, Fan Q, Norgaard RC, MacPherson L, Ruan HB, Wu J, et al. Liver X receptor regulates hepatic nuclear O-GlcNAc signaling and carbohydrate responsive element-binding protein activity. J Lipid Res (2015) 56(4):771–85. doi: 10.1194/jlr.M049130
94. Rey M, Kruse MS, Magrini-Huaman RN, Gomez J, Simirgiotis MJ, Tapia A, et al. Tessaria absinthioides (Hook. & arn.) DC. (Asteraceae) decoction improves the hypercholesterolemia and alters the expression of LXRs in rat liver and hypothalamus. Metabolites (2021) 11(9). doi: 10.3390/metabo11090579
95. Ghoneim RH, Ngo Sock ET, Lavoie JM, Piquette-Miller M. Effect of a high-fat diet on the hepatic expression of nuclear receptors and their target genes: relevance to drug disposition. Br J Nutr (2015) 113(3):507–16. doi: 10.1017/S0007114514003717
96. Kim JH, Jung DY, Kim HR, Jung MH. Histone H3K9 demethylase JMJD2B plays a role in LXRalpha-dependent lipogenesis. Int J Mol Sci (2020) 21(21). doi: 10.3390/ijms21218313
97. Cummings DE, Purnell JQ, Frayo RS, Schmidova K, Wisse BE, Weigle DS. A preprandial rise in plasma ghrelin levels suggests a role in meal initiation in humans. Diabetes (2001) 50(8):1714–9. doi: 10.2337/diabetes.50.8.1714
98. Deng Y, Wang ZV, Gordillo R, An Y, Zhang C, Liang Q, et al. An adipo-biliary-uridine axis that regulates energy homeostasis. Science (2017) 355(6330). doi: 10.1126/science.aaf5375
99. Chen HY, Trumbauer ME, Chen AS, Weingarth DT, Adams JR, Frazier EG, et al. Orexigenic action of peripheral ghrelin is mediated by neuropeptide y and agouti-related protein. Endocrinology (2004) 145(6):2607–12. doi: 10.1210/en.2003-1596
100. Steculorum SM, Paeger L, Bremser S, Evers N, Hinze Y, Idzko M, et al. Hypothalamic UDP increases in obesity and promotes feeding via P2Y6-dependent activation of AgRP neurons. Cell (2015) 162(6):1404–17. doi: 10.1016/j.cell.2015.08.032
101. Timper K, Bruning JC. Hypothalamic circuits regulating appetite and energy homeostasis: pathways to obesity. Dis Model Mech (2017) 10(6):679–89. doi: 10.1242/dmm.026609
102. Cota D, Proulx K, Seeley RJ. The role of CNS fuel sensing in energy and glucose regulation. Gastroenterology (2007) 132(6):2158–68. doi: 10.1053/j.gastro.2007.03.049
103. Andrews ZB, Liu ZW, Walllingford N, Erion DM, Borok E, Friedman JM, et al. UCP2 mediates ghrelin's action on NPY/AgRP neurons by lowering free radicals. Nature (2008) 454(7206):846–51. doi: 10.1038/nature07181
104. Drougard A, Fournel A, Valet P, Knauf C, et al. Impact of hypothalamic reactive oxygen species in the regulation of energy metabolism and food intake. Front Neurosci (2015) 9:56. doi: 10.3389/fnins.2015.00056
105. Deng Y, Wang ZV, Gordillo R, Zhu Y, Ali A, Zhang C, et al. Adipocyte Xbp1s overexpression drives uridine production and reduces obesity. Mol Metab (2018) 11:1–17. doi: 10.1016/j.molmet.2018.02.013
106. Kohli R, Bhattacharjee J, Inge TH. Postprandial uridine physiology is altered by obesity. Gastroenterology (2018) 155(5):1645–6. doi: 10.1053/j.gastro.2018.07.043
107. Alvarez-Castro P, Pena L, Cordido F. Ghrelin in obesity, physiological and pharmacological considerations. Mini Rev Med Chem (2013) 13(4):541–52. doi: 10.2174/1389557511313040007
108. Pocai A, Lam TK, Obici S, Gutierrez-Juarez R, Muse ED, Arduini A, et al. Restoration of hypothalamic lipid sensing normalizes energy and glucose homeostasis in overfed rats. J Clin Invest (2006) 116(4):1081–91. doi: 10.1172/JCI26640
109. Beutler LR, Corpuz TV, Ahn JS, Kosar S, Song W, Chen Y, et al. Obesity causes selective and long-lasting desensitization of AgRP neurons to dietary fat. Elife (2020) 9. doi: 10.7554/eLife.55909
110. Parton LE, Ye LE, Coppari R, Enriori PJ, Choi B, Zhang CY, et al. Glucose sensing by POMC neurons regulates glucose homeostasis and is impaired in obesity. Nature (2007) 449(7159):228–32. doi: 10.1038/nature06098
111. Ruan HB, Dietrich MO, Liu ZW, Zimmer MR, Li MD, Singh JP, et al. O-GlcNAc transferase enables AgRP neurons to suppress browning of white fat. Cell (2014) 159(2):306–17. doi: 10.1016/j.cell.2014.09.010
112. Luquet S, Perez FA, Hnasko TS, Palmiter RD. NPY/AgRP neurons are essential for feeding in adult mice but can be ablated in neonates. Science (2005) 310(5748):683–5. doi: 10.1126/science.1115524
113. Ryczko MC, Pawling J, Chen R, Abdel Rahman AM, Yau K, Copeland JK, et al. Metabolic reprogramming by hexosamine biosynthetic and golgi n-glycan branching pathways. Sci Rep (2016) 6:23043. doi: 10.1038/srep23043
114. Luo B, Parker GJ, Cooksey RC, Soesanto Y, Evans M, Jones D, et al. Chronic hexosamine flux stimulates fatty acid oxidation by activating AMP-activated protein kinase in adipocytes. J Biol Chem (2007) 282(10):7172–80. doi: 10.1074/jbc.M607362200
115. Lauzier B, Vaillant F, Merlen C, Gelinas R, Bouchard B, Rivard ME, et al. Metabolic effects of glutamine on the heart: anaplerosis versus the hexosamine biosynthetic pathway. J Mol Cell Cardiol (2013) 55:92–100. doi: 10.1016/j.yjmcc.2012.11.008
116. Keembiyehetty C, Love DC, Harwood KR, Gavrilova O, Comly ME, Hanover JA, et al. Conditional knock-out reveals a requirement for O-linked n-acetylglucosaminase (O-GlcNAcase) in metabolic homeostasis. J Biol Chem (2015) 290(11):7097–113. doi: 10.1074/jbc.M114.617779
117. Tan EP, McGreal SR, Graw S, Tessman R, Koppel SJ, Dhakal P, et al. Sustained O-GlcNAcylation reprograms mitochondrial function to regulate energy metabolism. J Biol Chem (2017) 292(36):14940–62. doi: 10.1074/jbc.M117.797944
118. Yang YR, Jang HJ, Choi SS, Lee YH, Lee GH, Seo YK, et al. Obesity resistance and increased energy expenditure by white adipose tissue browning in oga(+/-) mice. Diabetologia (2015) 58(12):2867–76. doi: 10.1007/s00125-015-3736-z
119. Lam TK, Schwartz GJ, Rossetti L. Hypothalamic sensing of fatty acids. Nat Neurosci (2005) 8(5):579–84. doi: 10.1038/nn1456
120. Gelinas R, Dontaine J, Horman S, Beauloye C, Bultot L, Bertrand L, et al. AMP-activated protein kinase and O-GlcNAcylation, two partners tightly connected to regulate key cellular processes. Front Endocrinol (Lausanne) (2018) 9:519. doi: 10.3389/fendo.2018.00519
121. Imbriolo J, Mapanga RF, Essop MF. The hexosamine biosynthetic pathway induces gene promoter activity of acetyl-CoA carboxylase beta. Biochem Biophys Res Commun (2014) 452(3):734–9. doi: 10.1016/j.bbrc.2014.08.142
122. Rumberger JM, Wu T, Hering MA, Marshall S. Role of hexosamine biosynthesis in glucose-mediated up-regulation of lipogenic enzyme mRNA levels: effects of glucose, glutamine, and glucosamine on glycerophosphate dehydrogenase, fatty acid synthase, and acetyl-CoA carboxylase mRNA levels. J Biol Chem (2003) 278(31):28547–52. doi: 10.1074/jbc.M302793200
123. Sekine O, Love DC, Rubenstein DS, Hanover JA. Blocking O-linked GlcNAc cycling in drosophila insulin-producing cells perturbs glucose-insulin homeostasis. J Biol Chem (2010) 285(49):38684–91. doi: 10.1074/jbc.M110.155192
124. Guinez C, Filhoulaud G, Rayah-Benhamed F, Marmier S, Dubuquoy C, Dentin R, et al. O-GlcNAcylation increases ChREBP protein content and transcriptional activity in the liver. Diabetes (2011) 60(5):1399–413. doi: 10.2337/db10-0452
125. Sodi VL, Bacigalupa ZA, Ferrer CM, Lee JV, Gocal WA, Mukhopadhyay D, et al. Nutrient sensor O-GlcNAc transferase controls cancer lipid metabolism via SREBP-1 regulation. Oncogene (2018) 37(7):924–34. doi: 10.1038/onc.2017.395
126. Machacek M, Saunders H, Zhang Z, Tan EP, Li J, Li T, et al. Elevated O-GlcNAcylation enhances pro-inflammatory Th17 function by altering the intracellular lipid microenvironment. J Biol Chem (2019) 294(22):8973–90. doi: 10.1074/jbc.RA119.008373
127. Kim MH, Kim H. Role of leptin in the digestive system. Front Pharmacol (2021) 12:660040. doi: 10.3389/fphar.2021.660040
128. Fukuda M. The role of GIP receptor in the CNS for the pathogenesis of obesity. Diabetes (2021) 70(9):1929–37. doi: 10.2337/dbi21-0001
129. van Bloemendaal L, Ten Kulve JS, Fleur SE, Ijzerman RG, Diamant M. Effects of glucagon-like peptide 1 on appetite and body weight: focus on the CNS. J Endocrinol (2014) 221(1):T1–16. doi: 10.1530/JOE-13-0414
130. Balsevich G, Sticht M, Bowles NP, Singh A, Lee TTY, Li Z, et al. Role for fatty acid amide hydrolase (FAAH) in the leptin-mediated effects on feeding and energy balance. Proc Natl Acad Sci U.S.A. (2018) 115(29):7605–10. doi: 10.1073/pnas.1802251115
131. Larsen MA, Isaksen VT, Paulssen EJ, Goll R, Florholmen JR. Postprandial leptin and adiponectin in response to sugar and fat in obese and normal weight individuals. Endocrine (2019) 66(3):517–25. doi: 10.1007/s12020-019-02102-9
132. Ainslie DA, Proietto J, Fam BC, Thorburn AW. Short-term, high-fat diets lower circulating leptin concentrations in rats. Am J Clin Nutr (2000) 71(2):438–42. doi: 10.1093/ajcn/71.2.438
133. Havel PJ, Townsend R, Chaump L, Teff K. High-fat meals reduce 24-h circulating leptin concentrations in women. Diabetes (1999) 48(2):334–41. doi: 10.2337/diabetes.48.2.334
134. Gupta D, Jetton TL, LaRock K, Monga N, Satish B, Lausier J, et al. Temporal characterization of beta cell-adaptive and -maladaptive mechanisms during chronic high-fat feeding in C57BL/6NTac mice. J Biol Chem (2017) 292(30):12449–59. doi: 10.1074/jbc.M117.781047
135. Lagerlof O, Slocomb JE, Hong I, Aponte Y, Blackshaw S, Hart GW, et al. The nutrient sensor OGT in PVN neurons regulates feeding. Science (2016) 351(6279):1293–6. doi: 10.1126/science.aad5494
136. Dai CL, Gu JH, Liu F, Iqbal K, Gong CX. Neuronal O-GlcNAc transferase regulates appetite, body weight, and peripheral insulin resistance. Neurobiol Aging (2018) 70:40–50. doi: 10.1016/j.neurobiolaging.2018.05.036
137. Andersson B, Tan EP, McGreal SR, Apte U, Hanover JA, Slawson C, et al. O-GlcNAc cycling mediates energy balance by regulating caloric memory. Appetite (2021) 165:105320. doi: 10.1016/j.appet.2021.105320
138. Kim KH, Kim YH, Son JE, Lee JH, Kim S, Choe MS, et al. Intermittent fasting promotes adipose thermogenesis and metabolic homeostasis via VEGF-mediated alternative activation of macrophage. Cell Res (2017) 27(11):1309–26. doi: 10.1038/cr.2017.126
139. Li MD, Vera NB, Yang Y, Zhang B, Ni W, Ziso-Qejvanaj E, et al. Adipocyte OGT governs diet-induced hyperphagia and obesity. Nat Commun (2018) 9(1):5103. doi: 10.1038/s41467-018-07461-x
140. Einstein FH, Atzmon G, Yang XM, Ma XH, Rincon M, Rudin E, et al. Differential responses of visceral and subcutaneous fat depots to nutrients. Diabetes (2005) 54(3):672–8. doi: 10.2337/diabetes.54.3.672
141. McClain DA, Alexander T, Cooksey RC, Considine RV. Hexosamines stimulate leptin production in transgenic mice. Endocrinology (2000) 141(6):1999–2002. doi: 10.1210/endo.141.6.7532
142. Hazel M, Cooksey RC, Jones D, Parker G, Neidigh JL, Witherbee B, et al. Activation of the hexosamine signaling pathway in adipose tissue results in decreased serum adiponectin and skeletal muscle insulin resistance. Endocrinology (2004) 145(5):2118–28. doi: 10.1210/en.2003-0812
143. McClain DA, Lubas WA, Cooksey RC, Hazel M, Parker GJ, Love DC, et al. Altered glycan-dependent signaling induces insulin resistance and hyperleptinemia. Proc Natl Acad Sci U.S.A. (2002) 99(16):10695–9. doi: 10.1073/pnas.152346899
144. Jackson SP, Tjian R. O-Glycosylation of eukaryotic transcription factors: implications for mechanisms of transcriptional regulation. Cell (1988) 55(1):125–33. doi: 10.1016/0092-8674(88)90015-3
145. Zhang P, Klenk ES, Lazzaro MA, Williams LB, Considine RV. Hexosamines regulate leptin production in 3T3-L1 adipocytes through transcriptional mechanisms. Endocrinology (2002) 143(1):99–106. doi: 10.1210/endo.143.1.8568
146. Alejandro EU, Bozadjieva N, Kumusoglu D, Abdulhamid S, Levine H, Haataja L, et al. Disruption of O-linked n-acetylglucosamine signaling induces ER stress and beta cell failure. Cell Rep (2015) 13(11):2527–38. doi: 10.1016/j.celrep.2015.11.020
147. Jara MA, Werneck-De-Castro JP, Lubaczeuski C, Johnson JD, Bernal-Mizrachi E. Pancreatic and duodenal homeobox-1 (PDX1) contributes to beta-cell mass expansion and proliferation induced by Akt/PKB pathway. Islets (2020) 12(2):32–40. doi: 10.1080/19382014.2020.1762471
148. Zhu Y, Liu Q, Zhou Z, Ikeda Y. PDX1, neurogenin-3, and MAFA: critical transcription regulators for beta cell development and regeneration. Stem Cell Res Ther (2017) 8(1):240. doi: 10.1186/s13287-017-0694-z
149. Gao T, McKenna B, Li C, Reichert M, Nguyen J, Singh T, et al. Pdx1 maintains beta cell identity and function by repressing an alpha cell program. Cell Metab (2014) 19(2):259–71. doi: 10.1016/j.cmet.2013.12.002
150. Gao Y, Miyazaki J, Hart GW. The transcription factor PDX-1 is post-translationally modified by O-linked n-acetylglucosamine and this modification is correlated with its DNA binding activity and insulin secretion in min6 beta-cells. Arch Biochem Biophys (2003) 415(2):155–63. doi: 10.1016/S0003-9861(03)00234-0
151. Mohan R, Jo S, Lockridge A, Ferrington DA, Murray K, Eschenlauer A, et al. OGT regulates mitochondrial biogenesis and function via diabetes susceptibility gene Pdx1. Diabetes (2021) 70(11):2608–25. doi: 10.2337/db21-0468
152. Jo S, Lockridge A, Alejandro EU. eIF4G1 and carboxypeptidase e axis dysregulation in O-GlcNAc transferase-deficient pancreatic beta-cells contributes to hyperproinsulinemia in mice. J Biol Chem (2019) 294(35):13040–50. doi: 10.1074/jbc.RA119.008670
153. De Meyts P. The insulin receptor and its signal transduction network. In: Feingold KR, et al, editors. Endotext. MDText.com, Inc. South Dartmouth (MA) (2000).
154. Wauman J, Zabeau L, Tavernier J. The leptin receptor complex: Heavier than expected? Front Endocrinol (Lausanne) (2017) 8:30. doi: 10.3389/fendo.2017.00030
155. Ring LE, Zeltser LM. Disruption of hypothalamic leptin signaling in mice leads to early-onset obesity, but physiological adaptations in mature animals stabilize adiposity levels. J Clin Invest (2010) 120(8):2931–41. doi: 10.1172/JCI41985
156. Bates SH, Stearns WH, Dundon TA, Schubert M, Tso AW, Wang Y, et al. STAT3 signalling is required for leptin regulation of energy balance but not reproduction. Nature (2003) 421(6925):856–9. doi: 10.1038/nature01388
157. White CL, Whittington A, Barnes MJ, Wang Z, Bray GA, Morrison CD. HF Diets increase hypothalamic PTP1B and induce leptin resistance through both leptin-dependent and -independent mechanisms. Am J Physiol Endocrinol Metab (2009) 296(2):E291–9. doi: 10.1152/ajpendo.90513.2008
158. Picardi PK, Calegari VC, Prada PO, Moraes JC, Araujo E, Marcondes MC, et al. Reduction of hypothalamic protein tyrosine phosphatase improves insulin and leptin resistance in diet-induced obese rats. Endocrinology (2008) 149(8):3870–80. doi: 10.1210/en.2007-1506
159. Zimmerman AD, Harris RB. In vivo and in vitro evidence that chronic activation of the hexosamine biosynthetic pathway interferes with leptin-dependent STAT3 phosphorylation. Am J Physiol Regul Integr Comp Physiol (2015) 308(6):R543–55. doi: 10.1152/ajpregu.00347.2014
160. Xu C, Liu GD, Feng L, Zhang CH, Wang F. Identification of O-GlcNAcylation modification in diabetic retinopathy and crosstalk with phosphorylation of STAT3 in retina vascular endothelium cells. Cell Physiol Biochem (2018) 49(4):1389–402. doi: 10.1159/000493444
161. Vasselli JR, Scarpace PJ, Harris RB, Banks WA. Dietary components in the development of leptin resistance. Adv Nutr (2013) 4(2):164–75. doi: 10.3945/an.112.003152
162. Olivier-Van Stichelen S, Wang P, Comly M, Love DC, Hanover JA. Nutrient-driven O-linked n-acetylglucosamine (O-GlcNAc) cycling impacts neurodevelopmental timing and metabolism. J Biol Chem (2017) 292(15):6076–85. doi: 10.1074/jbc.M116.774042
163. Belfort-DeAguiar R, Seo D. Food cues and obesity: Overpowering hormones and energy balance regulation. Curr Obes Rep (2018) 7(2):122–9. doi: 10.1007/s13679-018-0303-1
164. Laugerette F, Passilly-Degrace P, Patris B, Niot I, Febbraio M, Montmayeur JP, et al. CD36 involvement in orosensory detection of dietary lipids, spontaneous fat preference, and digestive secretions. J Clin Invest (2005) 115(11):3177–84. doi: 10.1172/JCI25299
165. Pepino MY, Love-Gregory L, Klein S, Abumrad NA. The fatty acid translocase gene CD36 and lingual lipase influence oral sensitivity to fat in obese subjects. J Lipid Res (2012) 53(3):561–6. doi: 10.1194/jlr.M021873
166. Peterschmitt Y, Abdoul-Azize S, Murtaza B, Barbier M, Khan AS, Millot JL, et al. Fatty acid lingual application activates gustatory and reward brain circuits in the mouse. Nutrients (2018) 10(9). doi: 10.3390/nu10091246
167. Martin C, Passilly-Degrace P, Gaillard D, Merlin JF, Chevrot M, Besnard P. The lipid-sensor candidates CD36 and GPR120 are differentially regulated by dietary lipids in mouse taste buds: impact on spontaneous fat preference. PloS One (2011) 6(8):e24014. doi: 10.1371/journal.pone.0024014
168. Kuipers EN, Kantae V, Maarse BCE, van den Berg SM, van Eenige R, Nahon KJ, et al. High fat diet increases circulating endocannabinoids accompanied by increased synthesis enzymes in adipose tissue. Front Physiol (2018) 9:1913. doi: 10.3389/fphys.2018.01913
169. Di Marzo V, Matias I. Endocannabinoid control of food intake and energy balance. Nat Neurosci (2005) 8(5):585–9. doi: 10.1038/nn1457
170. Coccurello R, Maccarrone M. Hedonic eating and the "Delicious circle": From lipid-derived mediators to brain dopamine and back. Front Neurosci (2018) 12:271. doi: 10.3389/fnins.2018.00271
171. Mazier W, Saucisse N, Gatta-Cherifi B, Cota D. The endocannabinoid system: Pivotal orchestrator of obesity and metabolic disease. Trends Endocrinol Metab (2015) 26(10):524–37. doi: 10.1016/j.tem.2015.07.007
172. Gatta-Cherifi B, Matias I, Vallee M, Tabarin A, Marsicano G, Piazza PV, et al. Simultaneous postprandial deregulation of the orexigenic endocannabinoid anandamide and the anorexigenic peptide YY in obesity. Int J Obes (Lond) (2012) 36(6):880–5. doi: 10.1038/ijo.2011.165
173. Richards P, Pais R, Habib AM, Brighton CA, Yeo GS, Reimann F, et al. High fat diet impairs the function of glucagon-like peptide-1 producing l-cells. Peptides (2016) 77:21–7. doi: 10.1016/j.peptides.2015.06.006
174. Rigamonti AE, Piscitelli F, Aveta T, Agosti F, Col De A, Bini S, et al. Anticipatory and consummatory effects of (hedonic) chocolate intake are associated with increased circulating levels of the orexigenic peptide ghrelin and endocannabinoids in obese adults. Food Nutr Res (2015) 59:29678. doi: 10.3402/fnr.v59.29678
175. Chevrot M, Bernard A, Ancel D, Buttet M, Martin C, Abdoul-Azize S, et al. Obesity alters the gustatory perception of lipids in the mouse: plausible involvement of lingual CD36. J Lipid Res (2013) 54(9):2485–94. doi: 10.1194/jlr.M039446
176. Brondel L, Quilliot D, Mouillot T, Khan NA, Bastable P, Boggio V, et al. Taste of fat and obesity: Different hypotheses and our point of view. Nutrients (2022) 14(3). doi: 10.3390/nu14030555
177. Browning KN, Carson KE. Central neurocircuits regulating food intake in response to gut inputs-preclinical evidence. Nutrients (2021) 13(3). doi: 10.3390/nu13030908
178. Wang AC, Jensen EH, Rexach JE, Vinters HV, Hsieh-Wilson LC. Loss of O-GlcNAc glycosylation in forebrain excitatory neurons induces neurodegeneration. Proc Natl Acad Sci U.S.A. (2016) 113(52):15120–5. doi: 10.1073/pnas.1606899113
179. Su C, Schwarz TL. O-GlcNAc transferase is essential for sensory neuron survival and maintenance. J Neurosci (2017) 37(8):2125–36. doi: 10.1523/JNEUROSCI.3384-16.2017
180. Engeli S. Dysregulation of the endocannabinoid system in obesity. J Neuroendocrinol (2008) 20 Suppl 1:110–5. doi: 10.1111/j.1365-2826.2008.01683.x
181. Kotoda M, Furukawa H, Miyamoto T, Korai M, Shikata F, Kuwabara A, et al. Role of myeloid lineage cell autophagy in ischemic brain injury. Stroke (2018) 49(6):1488–95. doi: 10.1161/STROKEAHA.117.018637
182. Banks WA, Kastin AJ, Broadwell RD. Passage of cytokines across the blood-brain barrier. Neuroimmunomodulation (1995) 2(4):241–8. doi: 10.1159/000097202
183. Wallace CW, Fordahl SC. Obesity and dietary fat influence dopamine neurotransmission: exploring the convergence of metabolic state, physiological stress, and inflammation on dopaminergic control of food intake. Nutr Res Rev (2021), 1–16. doi: 10.1017/S0954422421000196
184. Kaufman A, Choo E, Koh A, Dando R. Inflammation arising from obesity reduces taste bud abundance and inhibits renewal. PloS Biol (2018) 16(3):e2001959. doi: 10.1371/journal.pbio.2001959
185. Li X, Gong W, Wang H, Li T, Attri KS, Lewis RE, et al. O-GlcNAc transferase suppresses inflammation and necroptosis by targeting receptor-interacting Serine/Threonine-protein kinase 3. Immunity (2019) 50(3):576–590.e6. doi: 10.1016/j.immuni.2019.01.007
186. Li Y, Chen L, Zhao W, Sun L, Zhang R, Zhu S, et al. Food reward depends on TLR4 activation in dopaminergic neurons. Pharmacol Res (2021) 169:105659. doi: 10.1016/j.phrs.2021.105659
187. May CE, Rosander J, Gottfried J, Dennis E, Dus M. Dietary sugar inhibits satiation by decreasing the central processing of sweet taste. Elife (2020) 9. doi: 10.7554/eLife.54530
188. May CE, Vaziri A, Lin YQ, Grushko O, Khabiri M, Wang QP, et al. High dietary sugar reshapes sweet taste to promote feeding behavior in drosophila melanogaster. Cell Rep (2019) 27(6):1675–1685.e7. doi: 10.1016/j.celrep.2019.04.027
189. Wang H, Zhou M, Brand J, Huang L. Inflammation and taste disorders: mechanisms in taste buds. Ann N Y Acad Sci (2009) 1170:596–603. doi: 10.1111/j.1749-6632.2009.04480.x
190. Laczy B, Fulop N, Onay-Besikci A, Rosiers Des C, Chatham JC. Acute regulation of cardiac metabolism by the hexosamine biosynthesis pathway and protein O-GlcNAcylation. PloS One (2011) 6(4):e18417. doi: 10.1371/journal.pone.0018417
191. Nabeebaccus AA, Zoccarato A, Hafstad AD, Santos CX, Aasum E, Brewer AC, et al. Nox4 reprograms cardiac substrate metabolism via protein O-GlcNAcylation to enhance stress adaptation. JCI Insight (2017) 2(24). doi: 10.1172/jci.insight.96184
192. Camandola S, Mattson MP. Toll-like receptor 4 mediates fat, sugar, and umami taste preference and food intake and body weight regulation. Obes (Silver Spring) (2017) 25(7):1237–45. doi: 10.1002/oby.21871
193. D'Aquila T, Hung YH, Carreiro A, Buhman KK. Recent discoveries on absorption of dietary fat: Presence, synthesis, and metabolism of cytoplasmic lipid droplets within enterocytes. Biochim Biophys Acta (2016) 1861(8 Pt A):730–47. doi: 10.1016/j.bbalip.2016.04.012
194. Beilstein F, Carriere V, Leturque A, Demignot S. Characteristics and functions of lipid droplets and associated proteins in enterocytes. Exp Cell Res (2016) 340(2):172–9. doi: 10.1016/j.yexcr.2015.09.018
195. van Greevenbroek MM, de Bruin TW. Chylomicron synthesis by intestinal cells in vitro and in vivo. Atherosclerosis (1998) 141 Suppl 1:S9–16. doi: 10.1016/S0021-9150(98)00212-3
196. Wolska A, Dunbar RL, Freeman LA, Ueda M, Amar MJ, Sviridov DO, et al. Apolipoprotein c-II: New findings related to genetics, biochemistry, and role in triglyceride metabolism. Atherosclerosis (2017) 267:49–60. doi: 10.1016/j.atherosclerosis.2017.10.025
197. Stojanovic O, Altirriba J, Rigo D, Spiljar M, Evrard E, Roska B, et al. Dietary excess regulates absorption and surface of gut epithelium through intestinal PPARalpha. Nat Commun (2021) 12(1):7031. doi: 10.1038/s41467-021-27133-7
198. Frank DN, Bales ES, Monks J, Jackman MJ, MacLean PS, Ir D, et al. Perilipin-2 modulates lipid absorption and microbiome responses in the mouse intestine. PloS One (2015) 10(7):e0131944. doi: 10.1371/journal.pone.0131944
199. Drover VA, Ajmal M, Nassir F, Davidson NO, Nauli AM, Sahoo D, et al. CD36 deficiency impairs intestinal lipid secretion and clearance of chylomicrons from the blood. J Clin Invest (2005) 115(5):1290–7. doi: 10.1172/JCI21514
200. Nauli AM, Nassir F, Zheng S, Yang Q, Lo CM, Vonlehmden SB, et al. CD36 is important for chylomicron formation and secretion and may mediate cholesterol uptake in the proximal intestine. Gastroenterology (2006) 131(4):1197–207. doi: 10.1053/j.gastro.2006.08.012
201. Masuda D, Hirano K, Oku H, Sandoval JC, Kawase R, Yuasa-Kawase M, et al. Chylomicron remnants are increased in the postprandial state in CD36 deficiency. J Lipid Res (2009) 50(5):999–1011. doi: 10.1194/jlr.P700032-JLR200
202. Zhao L, Li Y, Ding Q, Li Y, Chen Y, Ruan XZ. CD36 senses dietary lipids and regulates lipids homeostasis in the intestine. Front Physiol (2021) 12:669279. doi: 10.3389/fphys.2021.669279
203. Cifarelli V, Abumrad NA. Intestinal CD36 and other key proteins of lipid utilization: Role in absorption and gut homeostasis. Compr Physiol (2018) 8(2):493–507. doi: 10.1002/cphy.c170026
204. Tran TT, Poirier H, Clement L, Nassir F, Pelsers MM, Petit V, et al. Luminal lipid regulates CD36 levels and downstream signaling to stimulate chylomicron synthesis. J Biol Chem (2011) 286(28):25201–10. doi: 10.1074/jbc.M111.233551
205. Buttet M, Poirier H, Traynard V, Gaire K, Tran TT, Sundaresan S, et al. Deregulated lipid sensing by intestinal CD36 in diet-induced hyperinsulinemic obese mouse model. PloS One (2016) 11(1):e0145626. doi: 10.1371/journal.pone.0145626
206. Siddiqi S, Saleem U, Abumrad NA, Davidson NO, Storch J, Siddiqi SA, et al. A novel multiprotein complex is required to generate the prechylomicron transport vesicle from intestinal ER. J Lipid Res (2010) 51(7):1918–28. doi: 10.1194/jlr.M005611
207. Quarfordt SH, Goodman DS. Heterogeneity in the rate of plasma clearance of chylomicrons of different size. Biochim Biophys Acta (1966) 116(2):382–5. doi: 10.1016/0005-2760(66)90019-1
208. Martins IJ, Mortimer BC, Miller J, Redgrave TG. Effects of particle size and number on the plasma clearance of chylomicrons and remnants. J Lipid Res (1996) 37(12):2696–705. doi: 10.1016/S0022-2275(20)37472-1
209. Ruan HB, Nie Y, Yang X. Regulation of protein degradation by O-GlcNAcylation: crosstalk with ubiquitination. Mol Cell Proteomics (2013) 12(12):3489–97. doi: 10.1074/mcp.R113.029751
210. Zhang F, Su K, Yang X, Bowe DB, Paterson AJ, Kudlow JE, et al. O-GlcNAc modification is an endogenous inhibitor of the proteasome. Cell (2003) 115(6):715–25. doi: 10.1016/S0092-8674(03)00974-7
211. Liu K, Paterson AJ, Zhang F, McAndrew J, Fukuchi K, Wyss JM, et al. Accumulation of protein O-GlcNAc modification inhibits proteasomes in the brain and coincides with neuronal apoptosis in brain areas with high O-GlcNAc metabolism. J Neurochem (2004) 89(4):1044–55. doi: 10.1111/j.1471-4159.2004.02389.x
212. Zhang F, Hu Y, Huang P, Toleman CA, Paterson AJ, Kudlow JE. Proteasome function is regulated by cyclic AMP-dependent protein kinase through phosphorylation of Rpt6. J Biol Chem (2007) 282(31):22460–71. doi: 10.1074/jbc.M702439200
213. Xu J, Wang S, Viollet B, Zou MH. Regulation of the proteasome by AMPK in endothelial cells: the role of O-GlcNAc transferase (OGT). PloS One (2012) 7(5):e36717. doi: 10.1371/journal.pone.0036717
214. Schwartz GJ, Fu J, Astarita G, Li X, Gaetani S, Campolongo P, et al. The lipid messenger OEA links dietary fat intake to satiety. Cell Metab (2008) 8(4):281–8. doi: 10.1016/j.cmet.2008.08.005
215. Guijarro A, Fu J, Astarita G, Piomelli D. CD36 gene deletion decreases oleoylethanolamide levels in small intestine of free-feeding mice. Pharmacol Res (2010) 61(1):27–33. doi: 10.1016/j.phrs.2009.09.003
216. Zhao M, Xiong X, Ren K, Xu B, Cheng M, Sahu C, et al. Deficiency in intestinal epithelial O-GlcNAcylation predisposes to gut inflammation. EMBO Mol Med (2018) 10(8). doi: 10.15252/emmm.201708736
217. Eren-Yazicioglu CY, Yigit A, Dogruoz RE, Yapici-Eser H. Can GLP-1 be a target for reward system related disorders? a qualitative synthesis and systematic review analysis of studies on palatable food, drugs of abuse, and alcohol. Front Behav Neurosci (2020) 14:614884. doi: 10.3389/fnbeh.2020.614884
218. Drucker DJ. GLP-1 physiology informs the pharmacotherapy of obesity. Mol Metab (2022) 57:101351. doi: 10.1016/j.molmet.2021.101351
219. Yu Y, Raka F, Adeli K. The role of the gut microbiota in lipid and lipoprotein metabolism. J Clin Med (2019) 8(12). doi: 10.3390/jcm8122227
220. Wang H, Eckel RH. Lipoprotein lipase: from gene to obesity. Am J Physiol Endocrinol Metab (2009) 297(2):E271–88. doi: 10.1152/ajpendo.90920.2008
221. Fu Z, Abou-Samra AB, Zhang R. A lipasin/Angptl8 monoclonal antibody lowers mouse serum triglycerides involving increased postprandial activity of the cardiac lipoprotein lipase. Sci Rep (2015) 5:18502. doi: 10.1038/srep18502
222. Symposium: Cancer of the colon and rectum: discussion. Dis Colon Rectum (1976) 19(1):20–4. doi: 10.1007/BF02590846
223. Zhao WS, Hu SL, Yu K, Wang H, Wang W, Loor J, et al. Lipoprotein lipase, tissue expression and effects on genes related to fatty acid synthesis in goat mammary epithelial cells. Int J Mol Sci (2014) 15(12):22757–71. doi: 10.3390/ijms151222757
224. Shang R, Rodrigues B. Lipoprotein lipase and its delivery of fatty acids to the heart. Biomolecules (2021) 11(7). doi: 10.3390/biom11071016
225. Savonen R, Hiden M, Hultin M, Zechner R, Levak-Frank S, Olivecrona G, et al. The tissue distribution of lipoprotein lipase determines where chylomicrons bind. J Lipid Res (2015) 56(3):588–98. doi: 10.1194/jlr.M056028
226. Sugden MC, Holness MJ, Howard RM. Changes in lipoprotein lipase activities in adipose tissue, heart and skeletal muscle during continuous or interrupted feeding. Biochem J (1993) 292 (Pt 1):113–9. doi: 10.1042/bj2920113
227. Taskinen MR, Nikkila EA. Effect of acute vigorous exercise on lipoprotein lipase activity of adipose tissue and skeletal muscle in physically active men. Artery (1980) 6(6):471–83.
228. Ruge T, Sukonina V, Kroupa O, Makoveichuk E, Lundgren M, Svensson MK, et al. Effects of hyperinsulinemia on lipoprotein lipase, angiopoietin-like protein 4, and glycosylphosphatidylinositol-anchored high-density lipoprotein binding protein 1 in subjects with and without type 2 diabetes mellitus. Metabolism (2012) 61(5):652–60. doi: 10.1016/j.metabol.2011.09.014
229. Ryan AS, Ortmeyer HK. Insulin suppression of fatty acid skeletal muscle enzyme activity in postmenopausal women, and improvements in metabolic flexibility and lipoprotein lipase with aerobic exercise and weight loss. Int J Obes (Lond) (2019) 43(2):276–84. doi: 10.1038/s41366-018-0068-3
230. Taskinen MR, Nikkila EA, Nousiainen R, Gordin A. Lipoprotein lipase activity in adipose tissue and skeletal muscle of human diabetics during insulin deprivation and restoration. Scand J Clin Lab Invest (1981) 41(3):263–8. doi: 10.3109/00365518109092043
231. Picard F, Boivin A, Lalonde J, Deshaies Y. Resistance of adipose tissue lipoprotein lipase to insulin action in rats fed an obesity-promoting diet. Am J Physiol Endocrinol Metab (2002) 282(2):E412–8. doi: 10.1152/ajpendo.00307.2001
232. Hao JW, Wang J, Guo H, Zhao YY, Sun HH, Li YF, et al. CD36 facilitates fatty acid uptake by dynamic palmitoylation-regulated endocytosis. Nat Commun (2020) 11(1):4765. doi: 10.1038/s41467-020-18565-8
233. Daquinag AC, Gao Z, Fussell C, Immaraj L, Pasqualini R, Arap W, et al. Fatty acid mobilization from adipose tissue is mediated by CD36 posttranslational modifications and intracellular trafficking. JCI Insight (2021) 6(17). doi: 10.1172/jci.insight.147057
234. Liu G, Xu JN, Liu D, Ding Q, Liu MN, Chen R, et al. Regulation of plasma lipid homeostasis by hepatic lipoprotein lipase in adult mice. J Lipid Res (2016) 57(7):1155–61. doi: 10.1194/jlr.M065011
235. Xu J, Donepudi AC, Moscovitz JE, Slitt AL. Keap1-knockdown decreases fasting-induced fatty liver via altered lipid metabolism and decreased fatty acid mobilization from adipose tissue. PloS One (2013) 8(11):e79841. doi: 10.1371/journal.pone.0079841
236. Berk PD, Zhou SL, Kiang CL, Stump D, Bradbury M, Isola LM. Uptake of long chain free fatty acids is selectively up-regulated in adipocytes of zucker rats with genetic obesity and non-insulin-dependent diabetes mellitus. J Biol Chem (1997) 272(13):8830–5. doi: 10.1074/jbc.272.13.8830
237. Lopaschuk GD, Folmes CD, Stanley WC. Cardiac energy metabolism in obesity. Circ Res (2007) 101(4):335–47. doi: 10.1161/CIRCRESAHA.107.150417
238. Ge F, Hu C, Hyodo E, Arai K, Zhou S, Lobdell HT, et al. Cardiomyocyte triglyceride accumulation and reduced ventricular function in mice with obesity reflect increased long chain fatty acid uptake and de novo fatty acid synthesis. J Obes (2012) 2012:205648. doi: 10.1155/2012/205648
239. Geisler CE, Renquist BJ. Hepatic lipid accumulation: cause and consequence of dysregulated glucoregulatory hormones. J Endocrinol (2017) 234(1):R1–R21. doi: 10.1530/JOE-16-0513
240. Colberg SR, Simoneau JA, Thaete FL, Kelley DE. Skeletal muscle utilization of free fatty acids in women with visceral obesity. J Clin Invest (1995) 95(4):1846–53. doi: 10.1172/JCI117864
241. Wu Q, Ortegon AM, Tsang B, Doege H, Feingold KR, Stahl A. FATP1 is an insulin-sensitive fatty acid transporter involved in diet-induced obesity. Mol Cell Biol (2006) 26(9):3455–67. doi: 10.1128/MCB.26.9.3455-3467.2006
242. Bonen A, Tandon NN, Glatz JF, Luiken JJ, Heigenhauser GJ. The fatty acid transporter FAT/CD36 is upregulated in subcutaneous and visceral adipose tissues in human obesity and type 2 diabetes. Int J Obes (Lond) (2006) 30(6):877–83. doi: 10.1038/sj.ijo.0803212
243. Luo X, Li Y, Yang P, Chen Y, Wei L, Yu T, et al. Obesity induces preadipocyte CD36 expression promoting inflammation via the disruption of lysosomal calcium homeostasis and lysosome function. EBioMedicine (2020) 56:102797. doi: 10.1016/j.ebiom.2020.102797
244. Cameron-Smith D, Burke LM, Angus DJ, Tunstall RJ, Cox GR, Bonen A, et al. A short-term, high-fat diet up-regulates lipid metabolism and gene expression in human skeletal muscle. Am J Clin Nutr (2003) 77(2):313–8. doi: 10.1093/ajcn/77.2.313
245. Koonen DP, Jacobs RL, Febbraio M, Young ME, Soltys CL, Ong H, et al. Increased hepatic CD36 expression contributes to dyslipidemia associated with diet-induced obesity. Diabetes (2007) 56(12):2863–71. doi: 10.2337/db07-0907
246. Binnert C, Koistinen HA, Martin G, Andreelli F, Ebeling P, Koivisto VA, et al. Fatty acid transport protein-1 mRNA expression in skeletal muscle and in adipose tissue in humans. Am J Physiol Endocrinol Metab (2000) 279(5):E1072–9. doi: 10.1152/ajpendo.2000.279.5.E1072
247. Miquilena-Colina ME, Lima-Cabello E, Sanchez-Campos S, Garcia-Mediavilla MV, Fernandez-Bermejo M, Lozano-Rodriguez T, et al. Hepatic fatty acid translocase CD36 upregulation is associated with insulin resistance, hyperinsulinaemia and increased steatosis in non-alcoholic steatohepatitis and chronic hepatitis c. Gut (2011) 60(10):1394–402. doi: 10.1136/gut.2010.222844
248. Tsai SF, Hung HC, Shih MM, Chang FC, Chung BC, Wang CY, et al. High-fat diet-induced increases in glucocorticoids contribute to the development of non-alcoholic fatty liver disease in mice. FASEB J (2022) 36(1):e22130. doi: 10.1096/fj.202101570R
249. Panarotto D, Remillard P, Bouffard L, Maheux P. Insulin resistance affects the regulation of lipoprotein lipase in the postprandial period and in an adipose tissue-specific manner. Eur J Clin Invest (2002) 32(2):84–92. doi: 10.1046/j.1365-2362.2002.00945.x
250. D'Aquila T, Zembroski AS, Buhman KK. Diet induced obesity alters intestinal cytoplasmic lipid droplet morphology and proteome in the postprandial response to dietary fat. Front Physiol (2019) 10:180. doi: 10.3389/fphys.2019.00180
251. Lim JM, Sherling D, Teo CF, Hausman DB, Lin D, Well L, et al. Defining the regulated secreted proteome of rodent adipocytes upon the induction of insulin resistance. J Proteome Res (2008) 7(3):1251–63. doi: 10.1021/pr7006945
252. Anthonisen EH, Berven L, Holm S, Nygard M, Nebb HI, Gronning-Wang LM, et al. Nuclear receptor liver X receptor is O-GlcNAc-modified in response to glucose. J Biol Chem (2010) 285(3):1607–15. doi: 10.1074/jbc.M109.082685
253. Peng D, Hiipakka RA, Xie JT, Reardon CA, Getz GS, Liao S. Differential effects of activation of liver X receptor on plasma lipid homeostasis in wild-type and lipoprotein clearance-deficient mice. Atherosclerosis (2010) 208(1):126–33. doi: 10.1016/j.atherosclerosis.2009.07.016
254. Zhang Y, Repa JJ, Gauthier K, Mangelsdorf DJ. Regulation of lipoprotein lipase by the oxysterol receptors, LXRalpha and LXRbeta. J Biol Chem (2001) 276(46):43018–24. doi: 10.1074/jbc.M107823200
255. Hebert LF Jr., Daniels MC, Zhou J, Crook ED, Turner RL, Simmons ST, et al. Overexpression of glutamine:fructose-6-phosphate amidotransferase in transgenic mice leads to insulin resistance. J Clin Invest (1996) 98(4):930–6. doi: 10.1172/JCI118876
256. Shi H, Munk A, Nielsen TS, Daughtry MR, Larsson L, Li S, et al. Skeletal muscle O-GlcNAc transferase is important for muscle energy homeostasis and whole-body insulin sensitivity. Mol Metab (2018) 11:160–77. doi: 10.1016/j.molmet.2018.02.010
257. Murata K, Morino K, Ida S, Ohashi N, Lemecha M, Park SY, et al. Lack of O-GlcNAcylation enhances exercise-dependent glucose utilization potentially through AMP-activated protein kinase activation in skeletal muscle. Biochem Biophys Res Commun (2018) 495(2):2098–104. doi: 10.1016/j.bbrc.2017.12.081
258. Stahl A, Evans JG, Pattel S, Hirsch D, Lodish HF. Insulin causes fatty acid transport protein translocation and enhanced fatty acid uptake in adipocytes. Dev Cell (2002) 2(4):477–88. doi: 10.1016/S1534-5807(02)00143-0
259. Kimmel AR, Sztalryd C. The perilipins: Major cytosolic lipid droplet-associated proteins and their roles in cellular lipid storage, mobilization, and systemic homeostasis. Annu Rev Nutr (2016) 36:471–509. doi: 10.1146/annurev-nutr-071813-105410
260. Duncan RE, Ahmadian M, Jaworski K, Sarkadi-Nagy E, Sul HS. Regulation of lipolysis in adipocytes. Annu Rev Nutr (2007) 27:79–101. doi: 10.1146/annurev.nutr.27.061406.093734
261. Miyoshi H, Perfield JW 2nd, Obin MS, Greenberg AS. Adipose triglyceride lipase regulates basal lipolysis and lipid droplet size in adipocytes. J Cell Biochem (2008) 105(6):1430–6. doi: 10.1002/jcb.21964
262. Yang A, Mottillo EP. Adipocyte lipolysis: from molecular mechanisms of regulation to disease and therapeutics. Biochem J (2020) 477(5):985–1008. doi: 10.1042/BCJ20190468
263. Choi SM, Tucker DF, Gross DN, Easton RM, DiPilato LM, Dean AS, et al. Insulin regulates adipocyte lipolysis via an akt-independent signaling pathway. Mol Cell Biol (2010) 30(21):5009–20. doi: 10.1128/MCB.00797-10
264. Carpentier AC. 100(th) anniversary of the discovery of insulin perspective: insulin and adipose tissue fatty acid metabolism. Am J Physiol Endocrinol Metab (2021) 320(4):E653–70. doi: 10.1152/ajpendo.00620.2020
265. Wang Y, Sullivan S, Trujillo M, Lee MJ, Schneider SH, Brolin RE, et al. Perilipin expression in human adipose tissues: effects of severe obesity, gender, and depot. Obes Res (2003) 11(8):930–6. doi: 10.1038/oby.2003.128
266. Ju L, Han J, Zhang X, Deng Y, Yan H, Wang C, et al. Obesity-associated inflammation triggers an autophagy-lysosomal response in adipocytes and causes degradation of perilipin 1. Cell Death Dis (2019) 10(2):121. doi: 10.1038/s41419-019-1393-8
267. Ryden M, Arvidsson E, Blomqvist L, Perbeck L, Dicker A, Arner P. Targets for TNF-alpha-induced lipolysis in human adipocytes. Biochem Biophys Res Commun (2004) 318(1):168–75. doi: 10.1016/j.bbrc.2004.04.010
268. Prins JB, Niesler CU, Winterford CM, Bright NA, Siddle K, O'Rahilly S, et al. Tumor necrosis factor-alpha induces apoptosis of human adipose cells. Diabetes (1997) 46(12):1939–44. doi: 10.2337/diab.46.12.1939
269. Zatterale F, Longo M, Naderi J, Raciti GA, Desiderio A, Miele C, et al. Chronic adipose tissue inflammation linking obesity to insulin resistance and type 2 diabetes. Front Physiol (2019) 10:1607. doi: 10.3389/fphys.2019.01607
270. Itabe H, Yamaguchi T, Nimura S, Sasabe N. Perilipins: a diversity of intracellular lipid droplet proteins. Lipids Health Dis (2017) 16(1):83. doi: 10.1186/s12944-017-0473-y
271. Bersuker K, Olzmann JA. Establishing the lipid droplet proteome: Mechanisms of lipid droplet protein targeting and degradation. Biochim Biophys Acta Mol Cell Biol Lipids (2017) 1862(10 Pt B):1166–77. doi: 10.1016/j.bbalip.2017.06.006
272. Ji S, Park SY, Roth J, Kim HS, Cho JW. O-GlcNAc modification of PPARgamma reduces its transcriptional activity. Biochem Biophys Res Commun (2012) 417(4):1158–63. doi: 10.1016/j.bbrc.2011.12.086
273. O'Rourke RW, Meyer KA, Gaston G, White AE, Lumeng CN, Marks DL. Hexosamine biosynthesis is a possible mechanism underlying hypoxia's effects on lipid metabolism in human adipocytes. PloS One (2013) 8(8):e71165. doi: 10.1371/journal.pone.0071165
274. Traxinger RR, Marshall S. Role of amino acids in modulating glucose-induced desensitization of the glucose transport system. J Biol Chem (1989) 264(35):20910–6. doi: 10.1016/S0021-9258(19)30022-5
275. Marshall S, Bacote V, Traxinger RR. Discovery of a metabolic pathway mediating glucose-induced desensitization of the glucose transport system. role of hexosamine biosynthesis in the induction of insulin resistance. J Biol Chem (1991) 266(8):4706–12. doi: 10.1016/S0021-9258(19)67706-9
276. Ohashi N, Morino K, Ida S, Sekine O, Lemecha M, Kume S, et al. Pivotal role of O-GlcNAc modification in cold-induced thermogenesis by brown adipose tissue through mitochondrial biogenesis. Diabetes (2017) 66(9):2351–62. doi: 10.2337/db16-1427
277. Weisberg SP, McCann D, Desai M, Rosenbaum M, and Leibel RL, Ferrante AW Jr. Obesity is associated with macrophage accumulation in adipose tissue. J Clin Invest (2003) 112(12):1796–808. doi: 10.1172/JCI200319246
278. Longo M, Zatterale F, Naderi J, Parrillo L, Formisano P, Raciti GA, et al. Adipose tissue dysfunction as determinant of obesity-associated metabolic complications. Int J Mol Sci (2019) 20(9). doi: 10.3390/ijms20092358
279. Ali AT, Hochfeld WE, Myburgh R, Pepper MS. Adipocyte and adipogenesis. Eur J Cell Biol (2013) 92(6-7):229–36. doi: 10.1016/j.ejcb.2013.06.001
280. Ishihara K, Takahashi I, Tsuchiya Y, Hasegawa M, Kamemura K. Characteristic increase in nucleocytoplasmic protein glycosylation by O-GlcNAc in 3T3-L1 adipocyte differentiation. Biochem Biophys Res Commun (2010) 398(3):489–94. doi: 10.1016/j.bbrc.2010.06.105
281. Hsieh TJ, Lin T, Hsieh PC, Liao MC, Shin SJ. Suppression of glutamine:fructose-6-phosphate amidotransferase-1 inhibits adipogenesis in 3T3-L1 adipocytes. J Cell Physiol (2012) 227(1):108–15. doi: 10.1002/jcp.22707
282. Li X, Molina H, Huang H, Zhang YY, Liu M, Qian SW, et al. O-Linked n-acetylglucosamine modification on CCAAT enhancer-binding protein beta: role during adipocyte differentiation. J Biol Chem (2009) 284(29):19248–54. doi: 10.1074/jbc.M109.005678
283. Tungland B. Human microbiota in health and disease : pathogenesis to therapy. London, United Kingdom: Elsevier/Academic Press (2018). 655 pages.
284. Audano M, Pedretti S, Caruso D, Crestani M, De Fabiani E, Mitro N. Regulatory mechanisms of the early phase of white adipocyte differentiation: an overview. Cell Mol Life Sci (2022) 79(3):139. doi: 10.1007/s00018-022-04169-6
285. Qian K, Wang S, Fu M, Zhou J, Singh JP, Li MD, et al. Transcriptional regulation of O-GlcNAc homeostasis is disrupted in pancreatic cancer. J Biol Chem (2018) 293(36):13989–4000. doi: 10.1074/jbc.RA118.004709
286. Zhao Y, Zhang YD, Zhang YY, Qian SW, Zhang ZC, Li SF, et al. p300-dependent acetylation of activating transcription factor 5 enhances C/EBPbeta transactivation of C/EBPalpha during 3T3-L1 differentiation. Mol Cell Biol (2014) 34(3):315–24. doi: 10.1128/MCB.00956-13
287. Ota T, Gayet C, Ginsberg HN. Inhibition of apolipoprotein B100 secretion by lipid-induced hepatic endoplasmic reticulum stress in rodents. J Clin Invest (2008) 118(1):316–32. doi: 10.1172/JCI32752
288. Zhang YL, Hernandez-Ono A, Ko C, Yasunaga K, Huang LS, Ginsberg HN. Regulation of hepatic apolipoprotein b-lipoprotein assembly and secretion by the availability of fatty acids. i. differential response to the delivery of fatty acids via albumin or remnant-like emulsion particles. J Biol Chem (2004) 279(18):19362–74. doi: 10.1074/jbc.M400220200
289. Alves-Bezerra M, Cohen DE. Triglyceride metabolism in the liver. Compr Physiol (2017) 8(1):1–8. doi: 10.1002/cphy.c170012
290. Choi SH, Ginsberg HN. Increased very low density lipoprotein (VLDL) secretion, hepatic steatosis, and insulin resistance. Trends Endocrinol Metab (2011) 22(9):353–63. doi: 10.1016/j.tem.2011.04.007
291. Sparks JD, Sparks CE, Adeli K. Selective hepatic insulin resistance, VLDL overproduction, and hypertriglyceridemia. Arterioscler Thromb Vasc Biol (2012) 32(9):2104–12. doi: 10.1161/ATVBAHA.111.241463
292. Cohn JS, Johnson EJ, Millar JS, Cohn SD, Milne RW, Marcel YL, et al. Contribution of apoB-48 and apoB-100 triglyceride-rich lipoproteins (TRL) to postprandial increases in the plasma concentration of TRL triglycerides and retinyl esters. J Lipid Res (1993) 34(12):2033–40. doi: 10.1016/S0022-2275(20)35345-1
293. Jones JG. Hepatic glucose and lipid metabolism. Diabetologia (2016) 59(6):1098–103. doi: 10.1007/s00125-016-3940-5
294. Brown MS, Goldstein JL. Selective versus total insulin resistance: a pathogenic paradox. Cell Metab (2008) 7(2):95–6. doi: 10.1016/j.cmet.2007.12.009
295. Diraison F, Moulin P, Beylot M. Contribution of hepatic de novo lipogenesis and reesterification of plasma non esterified fatty acids to plasma triglyceride synthesis during non-alcoholic fatty liver disease. Diabetes Metab (2003) 29(5):478–85. doi: 10.1016/S1262-3636(07)70061-7
296. Aarsland A, Wolfe RR. Hepatic secretion of VLDL fatty acids during stimulated lipogenesis in men. J Lipid Res (1998) 39(6):1280–6. doi: 10.1016/S0022-2275(20)32553-0
297. Sundaram M, Yao Z. Recent progress in understanding protein and lipid factors affecting hepatic VLDL assembly and secretion. Nutr Metab (Lond) (2010) 7:35. doi: 10.1186/1743-7075-7-35
298. Fujimoto Y, Itabe H, Sakai J, Makita M, Noda J, Mori M, et al. Identification of major proteins in the lipid droplet-enriched fraction isolated from the human hepatocyte cell line HuH7. Biochim Biophys Acta (2004) 1644(1):47–59. doi: 10.1016/j.bbamcr.2003.10.018
299. Najt CP, Senthivinayagam S, Aljazi MB, Fader KA, Olenic SD, Brock JR, et al. Liver-specific loss of perilipin 2 alleviates diet-induced hepatic steatosis, inflammation, and fibrosis. Am J Physiol Gastrointest Liver Physiol (2016) 310(9):G726–38. doi: 10.1152/ajpgi.00436.2015
300. Magnusson B, Asp L, Bostrom P, Ruiz M, Stillemark-Billton P, Linden D, et al. Adipocyte differentiation-related protein promotes fatty acid storage in cytosolic triglycerides and inhibits secretion of very low-density lipoproteins. Arterioscler Thromb Vasc Biol (2006) 26(7):1566–71. doi: 10.1161/01.ATV.0000223345.11820.da
301. Libby AE, Bales E, Orlicky DJ, McManaman JL. Perilipin-2 deletion impairs hepatic lipid accumulation by interfering with sterol regulatory element-binding protein (SREBP) activation and altering the hepatic lipidome. J Biol Chem (2016) 291(46):24231–46. doi: 10.1074/jbc.M116.759795
302. Qiu W, Avramoglu RK, Dube N, Chong TM, Naples M, Au C, et al. Hepatic PTP-1B expression regulates the assembly and secretion of apolipoprotein b-containing lipoproteins: evidence from protein tyrosine phosphatase-1B overexpression, knockout, and RNAi studies. Diabetes (2004) 53(12):3057–66. doi: 10.2337/diabetes.53.12.3057
303. Qu S, Altomonte J, Perdomo G, He J, Fan Y, Kamagate A, et al. Aberrant forkhead box O1 function is associated with impaired hepatic metabolism. Endocrinology (2006) 147(12):5641–52. doi: 10.1210/en.2006-0541
304. Kamagate A, Qu S, Perdomo G, Su D, Kim DH, Slusher S, et al. FoxO1 mediates insulin-dependent regulation of hepatic VLDL production in mice. J Clin Invest (2008) 118(6):2347–64. doi: 10.1172/JCI32914
305. Housley MP, Rodgers JT, Udeshi ND, Kelly TJ, Shabanowitz J, Hunt DF, et al. O-GlcNAc regulates FoxO activation in response to glucose. J Biol Chem (2008) 283(24):16283–92. doi: 10.1074/jbc.M802240200
306. Housley MP, Udeshi ND, Rodgers JT, Shabanowitz J, Puigserver P, Hunt DF, et al. A PGC-1alpha-O-GlcNAc transferase complex regulates FoxO transcription factor activity in response to glucose. J Biol Chem (2009) 284(8):5148–57. doi: 10.1074/jbc.M808890200
307. Veerababu G, Tang J, Hoffman RT, Daniels MC, Jr Hebert LF, Crook ED, et al. Overexpression of glutamine: fructose-6-phosphate amidotransferase in the liver of transgenic mice results in enhanced glycogen storage, hyperlipidemia, obesity, and impaired glucose tolerance. Diabetes (2000) 49(12):2070–8. doi: 10.2337/diabetes.49.12.2070
308. Xu W, Zhang X, Wu JL, Fu L, Liu K, Liu D, et al. O-GlcNAc transferase promotes fatty liver-associated liver cancer through inducing palmitic acid and activating endoplasmic reticulum stress. J Hepatol (2017) 67(2):310–20. doi: 10.1016/j.jhep.2017.03.017
309. Abu-Elheiga L, Brinkley WR, Zhong L, Chirala SS, Woldegiorgis G, Wakil SJ. The subcellular localization of acetyl-CoA carboxylase 2. Proc Natl Acad Sci U.S.A. (2000) 97(4):1444–9. doi: 10.1073/pnas.97.4.1444
310. Jiao Y, Lu Y, Li XY. Farnesoid X receptor: a master regulator of hepatic triglyceride and glucose homeostasis. Acta Pharmacol Sin (2015) 36(1):44–50. doi: 10.1038/aps.2014.116
311. Benhamed F, Filhoulaud G, Caron S, Lefebvre P, Staels B, Postic C, et al. O-GlcNAcylation links ChREBP and FXR to glucose-sensing. Front Endocrinol (Lausanne) (2014) 5:230. doi: 10.3389/fendo.2014.00230
312. Berrabah W, Aumercier P, Gheeraert C, Dehondt H, Bouchaert E, Alexandre J, et al. Glucose sensing O-GlcNAcylation pathway regulates the nuclear bile acid receptor farnesoid X receptor (FXR). Hepatology (2014) 59(5):2022–33. doi: 10.1002/hep.26710
313. Gonzalez-Rellan MJ, Fondevila MF, Fernandez U, Rodriguez A, Varela-Rey M, Veyrat-Durebex C, et al. O-GlcNAcylated p53 in the liver modulates hepatic glucose production. Nat Commun (2021) 12(1):5068. doi: 10.1038/s41467-021-25390-0
314. Taylor RP, Geisler TS, Chambers JH, McClain DA. Up-regulation of O-GlcNAc transferase with glucose deprivation in HepG2 cells is mediated by decreased hexosamine pathway flux. J Biol Chem (2009) 284(6):3425–32. doi: 10.1074/jbc.M803198200
315. Taylor RP, Parker GJ, Hazel MW, Soesanto Y, Fuller W, Yazzie MJ, et al. Glucose deprivation stimulates O-GlcNAc modification of proteins through up-regulation of O-linked n-acetylglucosaminyltransferase. J Biol Chem (2008) 283(10):6050–7. doi: 10.1074/jbc.M707328200
316. Ruan HB, Ma Y, Torres S, Zhang B, Feriod C, Heck RM, et al. Calcium-dependent O-GlcNAc signaling drives liver autophagy in adaptation to starvation. Genes Dev (2017) 31(16):1655–65. doi: 10.1101/gad.305441.117
317. Schulze RJ, Drizyte K, Casey CA, McNiven MA. Hepatic lipophagy: New insights into autophagic catabolism of lipid droplets in the liver. Hepatol Commun (2017) 1(5):359–69. doi: 10.1002/hep4.1056
318. Laubach K, Zhang J, Chen X. The p53 family: A role in lipid and iron metabolism. Front Cell Dev Biol (2021) 9:715974. doi: 10.3389/fcell.2021.715974
319. Morris EM, Meers GM, Booth FW, Fritsche KL, Hardin CD, Thyfault JP, et al. PGC-1alpha overexpression results in increased hepatic fatty acid oxidation with reduced triacylglycerol accumulation and secretion. Am J Physiol Gastrointest Liver Physiol (2012) 303(8):G979–92. doi: 10.1152/ajpgi.00169.2012
320. Hanover JA, Forsythe ME, Hennessey PT, Brodigan TM, Love DC, Ashwell G, et al. A caenorhabditis elegans model of insulin resistance: altered macronutrient storage and dauer formation in an OGT-1 knockout. Proc Natl Acad Sci U.S.A. (2005) 102(32):11266–71. doi: 10.1073/pnas.0408771102
321. Forsythe ME, Love DC, Lazarus BD, Kim EJ, Prinz WA, Ashwell G, et al. Caenorhabditis elegans ortholog of a diabetes susceptibility locus: oga-1 (O-GlcNAcase) knockout impacts O-GlcNAc cycling, metabolism, and dauer. Proc Natl Acad Sci U.S.A. (2006) 103(32):11952–7. doi: 10.1073/pnas.0601931103
322. Abernathy O, Kostner D, Buer P, Dougherty M, Schmidtberger A, Spainhour R, et al. Expression of messenger RNA encoding two cellular metabolic regulators, AMP-activated protein kinase (AMPK) and O-GlcNAc transferase (OGT), in channel catfish: Their tissue distribution and relationship with changes in food intake. Comp Biochem Physiol A Mol Integr Physiol (2019) 235:12–21. doi: 10.1016/j.cbpa.2019.04.023
323. Whelan SA, Lane MD, Hart GW. Regulation of the O-linked beta-n-acetylglucosamine transferase by insulin signaling. J Biol Chem (2008) 283(31):21411–7. doi: 10.1074/jbc.M800677200
324. Sviridov D, Miller YI. Biology of lipid rafts: Introduction to the thematic review series. J Lipid Res (2020) 61(5):598–600. doi: 10.1194/jlr.IN119000330
325. Monnier L, Colette C. Target for glycemic control: concentrating on glucose. Diabetes Care (2009) 32 Suppl 2:S199–204. doi: 10.2337/dc09-S310
326. Chaveroux C, Sarcinelli C, Barbet V, Belfeki S, Barthelaix A, Ferraro-Peyret C, et al. Nutrient shortage triggers the hexosamine biosynthetic pathway via the GCN2-ATF4 signalling pathway. Sci Rep (2016) 6:27278. doi: 10.1038/srep27278
327. Lehmann LH, Jebessa ZH, Kreusser MM, Horsch A, He T, Kronlage M, et al. A proteolytic fragment of histone deacetylase 4 protects the heart from failure by regulating the hexosamine biosynthetic pathway. Nat Med (2018) 24(1):62–72. doi: 10.1038/nm.4452
328. Duszka K, Bogner-Strauss JG, Hackl H, Rieder D, Neuhold C, Prokesch A, et al. Nr4a1 is required for fasting-induced down-regulation of Ppargamma2 in white adipose tissue. Mol Endocrinol (2013) 27(1):135–49. doi: 10.1210/me.2012-1248
329. Min AK, Bae KH, Jung YA, Choi YK, Kim MJ, Kim JH, et al. Orphan nuclear receptor Nur77 mediates fasting-induced hepatic fibroblast growth factor 21 expression. Endocrinology (2014) 155(8):2924–31. doi: 10.1210/en.2013-1758
330. Ozcan L, Wong CC, Li G, Xu T, Pajvani U, Park SK, et al. Calcium signaling through CaMKII regulates hepatic glucose production in fasting and obesity. Cell Metab (2012) 15(5):739–51. doi: 10.1016/j.cmet.2012.03.002
331. Collins QF, Xiong Y, Lupo EG Jr, Liu HY, Cao W. p38 mitogen-activated protein kinase mediates free fatty acid-induced gluconeogenesis in hepatocytes. J Biol Chem (2006) 281(34):24336–44. doi: 10.1074/jbc.M602177200
332. Cao W, Collins QF, Becker TC, Robidoux J, Lupo EG Jr, Xiong Y, et al. p38 mitogen-activated protein kinase plays a stimulatory role in hepatic gluconeogenesis. J Biol Chem (2005) 280(52):42731–7. doi: 10.1074/jbc.M506223200
333. Stern JH, Smith GI, Chen S, Unger RH, Klein S, Scherer PE, et al. Obesity dysregulates fasting-induced changes in glucagon secretion. J Endocrinol (2019) 243(2):149–60. doi: 10.1530/JOE-19-0201
334. Zhao P, Saltiel AR. From overnutrition to liver injury: AMP-activated protein kinase in nonalcoholic fatty liver diseases. J Biol Chem (2020) 295(34):12279–89. doi: 10.1074/jbc.REV120.011356
335. Choi BH, Jin Z, Yi CO, Oh J, Jeong EA, Lee JY, et al. Effects of lobeglitazone on insulin resistance and hepatic steatosis in high-fat diet-fed mice. PloS One (2018) 13(7):e0200336. doi: 10.1371/journal.pone.0200336
336. Kim YR, Lee EJ, Shin KO, Kim MH, Pewzner-Jung Y, Lee YM, et al. Hepatic triglyceride accumulation via endoplasmic reticulum stress-induced SREBP-1 activation is regulated by ceramide synthases. Exp Mol Med (2019) 51(11):1–16. doi: 10.1038/s12276-019-0340-1
337. Eissing L, Scherer T, Todter K, Knippschild U, Greve JW, Buurman WA, et al. De novo lipogenesis in human fat and liver is linked to ChREBP-beta and metabolic health. Nat Commun (2013) 4:1528. doi: 10.1038/ncomms2537
338. Dahlman I, Nilsson M, Jiao H, Hoffstedt J, Lindgren CM, Humphreys K, et al. Liver X receptor gene polymorphisms and adipose tissue expression levels in obesity. Pharmacogenet Genomics (2006) 16(12):881–9. doi: 10.1097/01.fpc.0000236334.49422.48
339. Gum RJ, Gaede LL, Heindel MA, Waring JF, Trevillyan JM, Zinker BA, et al. Antisense protein tyrosine phosphatase 1B reverses activation of p38 mitogen-activated protein kinase in liver of ob/ob mice. Mol Endocrinol (2003) 17(6):1131–43. doi: 10.1210/me.2002-0288
340. Liu Z, Patil IY, Jiang T, Sancheti H, Walsh JP, Stiles BL, et al. High-fat diet induces hepatic insulin resistance and impairment of synaptic plasticity. PloS One (2015) 10(5):e0128274. doi: 10.1371/journal.pone.0128274
341. Kern M, Kloting N, Niessen HG, Thomas L, Stiller D, Mark M, et al. Linagliptin improves insulin sensitivity and hepatic steatosis in diet-induced obesity. PloS One (2012) 7(6):e38744. doi: 10.1371/journal.pone.0038744
342. Wang Q, Dong Z, Liu X, Song X, Song Q, Shang Q, et al. Programmed cell death-4 deficiency prevents diet-induced obesity, adipose tissue inflammation, and insulin resistance. Diabetes (2013) 62(12):4132–43. doi: 10.2337/db13-0097
343. Leiva M, Matesanz N, Pulgarin-Alfaro M, Nikolic I, Sabio G. Uncovering the role of p38 family members in adipose tissue physiology. Front Endocrinol (Lausanne) (2020) 11:572089. doi: 10.3389/fendo.2020.572089
344. Juvet LK, Andresen SM, Schuster GU, Dalen KT, Tobin KA, Hollung K, et al. On the role of liver X receptors in lipid accumulation in adipocytes. Mol Endocrinol (2003) 17(2):172–82. doi: 10.1210/me.2001-0210
345. Beaven SW, Matveyenko A, Wroblewski K, Chao L, Wilpitz D, Hsu TW, et al. Reciprocal regulation of hepatic and adipose lipogenesis by liver X receptors in obesity and insulin resistance. Cell Metab (2013) 18(1):106–17. doi: 10.1016/j.cmet.2013.04.021
346. Nguyen P, Leray V, Diez M, Serisier S, Le Bloc'h J, Siliart B, et al. Liver lipid metabolism. J Anim Physiol Anim Nutr (Berl) (2008) 92(3):272–83. doi: 10.1111/j.1439-0396.2007.00752.x
347. Liu W, Sun C, Yan Y, Cao H, Niu Z, Shen S, et al. Hepatic P38 activation modulates systemic metabolism through Fgf21-mediated interorgan communication. Diabetes (2021) 71(1):60–72. doi: 10.2337/figshare.16811332
348. Veum VL, Dankel SN, Gjerde J, Nielsen HJ, Solsvik MH, Haugen C, et al. The nuclear receptors NUR77, NURR1 and NOR1 in obesity and during fat loss. Int J Obes (Lond) (2012) 36(9):1195–202. doi: 10.1038/ijo.2011.240
Keywords: O-GlcNAc, glycosylation, hexosamine biosynthetic pathway, lipid, fatty acid, metabolism, obesity, homeostasis
Citation: Lockridge A and Hanover JA (2022) A nexus of lipid and O-Glcnac metabolism in physiology and disease. Front. Endocrinol. 13:943576. doi: 10.3389/fendo.2022.943576
Received: 13 May 2022; Accepted: 27 July 2022;
Published: 30 August 2022.
Edited by:
Pierre De Meyts, de Duve Institute, Université Catholique de Louvain, BelgiumReviewed by:
Tony Lefebvre, Lille University of Science and Technology, FranceJohn C. Chatham, University of Alabama at Birmingham, United States
Chad Slawson, University of Kansas Medical Center Research Institute, United States
Copyright © 2022 Lockridge and Hanover. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Amber Lockridge, YW1iZXIubG9ja3JpZGdlQG5paC5nb3Y=; John A. Hanover, amFoQGhlbGl4Lm5paC5nb3Y=