 Meg Schuurman1,2
Meg Schuurman1,2 Madison Wallace1,3
Madison Wallace1,3 Gurleen Sahi1,2
Gurleen Sahi1,2 Malina Barillaro1,2Siyi Zhang1Mushfiqur Rahman1,2Cynthia Sawyez2
Malina Barillaro1,2Siyi Zhang1Mushfiqur Rahman1,2Cynthia Sawyez2 Nica Borradaile2
Nica Borradaile2 Rennian Wang1,2*
Rennian Wang1,2*- 1Children’s Health Research Institute, London, ON, Canada
- 2Department of Physiology & Pharmacology, University of Western Ontario, London, ON, Canada
- 3Department of Pathology and Laboratory Medicine, University of Western Ontario, London, ON, Canada
Obesity plays a major role in type II diabetes (T2DM) progression because it applies metabolic and oxidative stress resulting in dysfunctional beta-cells and activation of intra-islet pancreatic stellate cells (PaSCs) which cause islet fibrosis. Administration of antioxidant N-acetyl-L-cysteine (NAC) in vivo improves metabolic outcomes in diet-induced obese diabetic mice, and in vitro inhibits PaSCs activation. However, the effects of NAC on diabetic islets in vivo are unknown. This study examined if dosage and length of NAC treatment in HFD-induced diabetic mice effect metabolic outcomes associated with maintaining healthy beta-cells and quiescent PaSCs, in vivo. Male C57BL/6N mice were fed normal chow (ND) or high-fat (HFD) diet up to 30 weeks. NAC was administered in drinking water to HFD mice in preventative treatment (HFDpNAC) for 23 weeks or intervention treatment for 10 (HFDiNAC) or 18 (HFDiNAC+) weeks, respectively. HFDpNAC and HFDiNAC+, but not HFDiNAC, mice showed significantly improved glucose tolerance and insulin sensitivity. Hyperinsulinemia led by beta-cell overcompensation in HFD mice was significantly rescued in NAC treated mice. A reduction of beta-cell nuclear Pdx-1 localization in HFD mice was significantly improved in NAC treated islets along with significantly reduced beta-cell oxidative stress. HFD-induced intra-islet PaSCs activation, labeled by αSMA, was significantly diminished in NAC treated mice along with lesser intra-islet collagen deposition. This study determined that efficiency of NAC treatment is beneficial at maintaining healthy beta-cells and quiescent intra-islet PaSCs in HFD-induced obese T2DM mouse model. These findings highlight an adjuvant therapeutic potential in NAC for controlling T2DM progression in humans.
Introduction
Type 2 diabetes mellitus (T2DM) is a progressive metabolic disease consisting of whole body and pancreatic islet alterations which culminate in dysregulated glycemia (1). T2DM progression, often facilitated by obesity, typically begins with insulin resistance, where peripheral tissue insulin receptors become insensitive and require increased insulin to initiate a response (2, 3). Subclinical inflammation—elevated pro-inflammatory cytokines— is present during obesity which has been associated with metabolic syndrome and peripheral insulin resistance (4). Initially, pancreatic beta-cells accommodate this increase in demand by increasing insulin production as well as their size and number (1, 5). However, this compensatory mechanism is limited and leads to eventual beta-cell failure. The major cause of the clinical progression of T2DM is a decline in beta-cell mass and function within the pancreatic islets (1, 6). Since islet cellular organization is supported by non-islet cells (i.e., pancreatic stellate cells – PaSCs) and the extracellular matrix; this unique microenvironment is crucial for islet function and survival (7–10).
One of the mechanisms facilitating beta-cell failure is believed to be an imbalance between reactive oxygen species (ROS) and antioxidants leading to increased oxidative stress (11). ROS are by-products of metabolism from mitochondria in response to increased glucose, thus in overnutrition settings (i.e. obesity) increased oxidative stress is observed (12). T2DM patients have increased oxidative stress levels compared to non-diabetic controls (11). In addition, diabetic pancreatic islets display lower antioxidant enzyme (i.e. glutathione [GSH], superoxide dismutase [SOD]) expression compared to controls, this further augments beta-cell susceptibility to oxidative stress-induced damage (13, 14). High levels of oxidative stress progresses beta-cell compensation towards dysfunction indicated by reduced nuclear pancreatic and duodenal homeobox 1 (Pdx-1) localization (15). In addition to the beta-cell-specific changes, prolonged high fat diet (HFD)-induced compensation affects the islet microenvironment by increasing intra-islet PaSCs activation and collagen deposition (16). Under conditions of ROS accumulation, mechanical stress and cytokine stimulation, PaSCs are activated and develop a myofibroblast phenotype, gain expression of alpha-smooth muscle actin (aSMA) and are associated with excessive extracellular matrix (ECM) and cytokines production leading to pancreatic inflammation and fibrosis (17, 18). Activated PaSCs are also associated with impaired beta-cell function, in vitro (19). However, whether a definitive relationship exists between activated PaSCs and beta-cell dysfunction has not yet been established. Furthermore, it remains to be questioned whether inhibition of PaSC activation and beta-cell oxidative stress via antioxidants is a viable therapeutic target for T2DM progression.
N-acetyl-L-cysteine (NAC), a biosynthetic precursor to GSH, is a well-known antioxidant that has been studied for its effect on numerous physiological processes and diseases including T2DM (20–22). In addition to acting as a precursor to glutathione, NAC contains a thiol group which enables it to act directly as an antioxidant; it also acts as an anti-inflammatory (23, 24). NAC has been shown to reduce levels of oxidative stress and improve insulin release in isolated Wistar rat islets (25). When administered in drinking water to HFD murine models of T2DM, NAC improved glucose and insulin tolerance tests (22) and provided functional protection in beta-cells of female mice (26). However, NAC has been shown to be ineffective in limited T2DM clinical work (27). NAC efficacy is impacted by timing (22) and dosage (21) of administration. It was observed to be most beneficial for improving metabolic outcomes when administered as a long-term interventional lasting 5 months (21). Although clinical research is limited, animal models show promise for NAC as a potential treatment in T2DM (21, 22, 26, 28). However, the reported literature lacks understanding with the changes in pancreatic islets particularly in beta-cells and PaSCs following NAC treatment in vivo.
In the present study, we aimed to determine the effects of both dosage and timing of NAC administration on pancreatic beta-cells and islet PaSCs in a HFD-induced diabetic mouse model. We hypothesized that the efficiency of NAC treatment will improve metabolic outcomes and beta-cell function by rescuing beta-cell overcompensation while reducing beta-cell stress and PaSC activation induced by HFD. Here, we show that NAC treatment is beneficial for improving metabolic outcomes and beta-cell function, particularly by preserving beta-cell identity, and reducing beta-cell oxidative stress and PaSC activation. However, efficiency of NAC treatment is dependent on both timing and dosage.
Materials and methods
Mouse model of high-fat diet induced obesity T2DM with NAC prevention and intervention treatment
Male C57BL/6N (B6N) mice purchased from Charles River (Charles River Laboratories Quebec, Canada) were housed in our facility under a 12:12 h light/dark cycle with a maximum of 5 mice per cage. At 6-weeks of age, mice maintained a normal chow diet (ND) composed of 22% kcal from fat, 23% kcal protein, and 55% kcal from carbohydrates (Harlan Tekard, Indianapolis, IN, USA) or received a high-fat diet (HFD) composed of 60% kcal from fat, 20% from protein, and 20% from carbohydrates (Research Diets INC, New Brunswick, NJ, USA) ad libitum. Mice received a ND or HFD for 22- or 30-week feeding periods (Supplementary Figure S1).
Antioxidant treatment with N-acetyl-L-cysteine (NAC) (Santa Cruz Biotechnology, Inc., Santa Cruz, CA, USA) was administered in drinking water to HFD fed mice either in 10mM or 50mM dosage (21, 22). Since the effectiveness of 10mM dose did not show improvement of glucose metabolism (Supplementary Figure S2) compared to 50mM dosage, this report was focused on using 50mM NAC for antioxidant groups in a prevention (pNAC) or intervention (iNAC) treatment (Supplementary Figure S1). For pNAC treatment, mice received NAC one week prior to HFD and treated through 22-week HFD feeding (HFDpNAC). For iNAC treatment, mice received NAC at week 12 after HFD and continued NAC intake for the duration of the 22-week HFD (HFDiNAC) and 30-week HFD (HFDiNAC+). NAC intake was assessed in each antioxidant treated group (over a three-day period) by measuring water consumption, expressed as NAC intake (mg) per mouse per day. Control mice were age-matched and consumed either ND or HFD with no NAC treatment.
Body weight was monitored weekly, and food intake was measured at the end of the assigned diet period (29). Food intake is expressed kilocalories per mouse per day. All animal work was conducted based on approved protocols from the University of Western Ontario Animal User Subcommittee (Animal Use Protocol #2021-139) in accordance with the Canadian Council of Animal Care guidelines.
Metabolic studies in experimental mouse models
At the last week of the assigned diet period, fasting blood glucose was examined followed by an intraperitoneal (i.p.) glucose (IPGTT) or insulin (IPITT) tolerance tests. Tests were conducted in the final 2 weeks of diet intervention and at least 5 days apart from one another. For IPGTT, glucose (Dextrose, Sigma, Saint Louis, MO, USA) was administrated at a dose of 2mg/g body weight after an overnight fast for 22 week feeding mice and a 4 hour fast for 30 week feeding mice. For IPITT, insulin (Humulin, Eli Lilly, Toronto, ON, Canada) was administrated at a dose of 1 U/kg body weight after a 4 hour fast. Blood glucose levels were measured before i.p. injection (0 min) and post-injection (15, 30, 60, and 120 mins) and area under the curve (AUC) was used to measure responsiveness. Results of IPITT were normalized to baseline glucose levels (100%) and AUC values were generated to determine changes in insulin sensitivity (30). To determine in vivo glucose-stimulated insulin secretion (GSIS), blood samples were collected following 16 h fasting (0 min) and at 5 and 35 min after i.p. injection of glucose (2mg/g). Plasma insulin levels from GSIS and fed cardiac blood was measured using an ultrasensitive insulin mouse ELISA kit (ALPCO, Salem, NH, USA). Plasma triglycerides and cholesterol were measured using enzymatic, colorimetric assays (Wako Diagnostics, Mountain View, California, USA).
Immunohistological staining and morphometric analyses
At the end of experimental time points, pancreases from mice were collected, weighed, and fixed in 4% paraformaldehyde. Whole pancreatic tissues were embedded in paraffin and sectioned consecutively at 4-µm throughout the length of the pancreas, from head to tail, to avoid any bias due to regional changes in islet distribution and islet cell composition of the pancreas (31). Sections were incubated with the appropriate dilutions of primary antibodies as listed in Supplementary Table S1. As required, 0.2% Triton treatment and/or microwave antigen retrieval with citric acid solution (pH 6.0) were applied to improve detection. Secondary antibodies consisted of fluorescein isothiocyanate (FITC) and tetramethyl rhodamine isothiocyanate (TRITC) (1:50; Jackson Immunoresearch, West Grove, PA, USA). 4’-6’-diamidino-2-phenylindole (DAPI) was used for nuclear counterstaining (Sigma-Aldrich). Images were captured using Nikon Eclipse Ti2 Confocal Microscope (Nikon, Melville, NY, USA) followed by morphometric analysis of islet density, alpha and beta-cell mass and beta-cell size, as previously described (30, 32). Beta-cell proliferation, expression of Pdx-1 transcription factor and oxidative stress were identified by double immunofluorescence staining and quantification from at least 10 random islets per pancreas section. Double positive cells (nuclear marker-positive with insulin-positive) were divided by the total insulin-positive cell population in islets and expressed as a percentage (33). To quantify the percentage of intra-islet PaSCs population, labeled by αSMA and desmin, the positive intra-islet αSMA+ or desmin+ cell area (µm2) was manually traced using Nikon NIS-Element software and divided by the total insulin-positive area (µm2).
Trichrome staining kit (Abcam, Cambridge, MA, USA) was used to measure intra-islet collagen deposition (34). Stained images were acquired at 40x magnification using an Aperio AT2 whole slide scanner (Leica Biosystems Inc, Concord, Ontario, Canada). The percentage of islet collagen deposition was determined using an ImageJ macro, which segments blue areas (collagen) from red areas in the image using colour deconvolution and measures total blue area per image as previously described (35).
Statistical analysis
All data are expressed as means ± SEM. Statistical significance was analyzed using GraphPad Prism (version 6 GraphPad Software, San Diego, CA, USA) with 95% of confidence interval. The difference was analyzed using one-way ANOVA followed by Tukey’s Post-Hoc test. Differences were considered to be statistically significant when p <0.05.
Results
NAC treatment improves glucose metabolism as dose- and time-dependent in HFD-induced diabetic mice
To determine the efficiency of NAC treatment in HFD-induced diabetic mice, both dose- and time-dependent study was established. Dosages of 10mM and 50mM NAC treatment were administered for prevention and intervention groups. HFD mice with 10mM NAC preventive or interventive treatment did not show changes in body weight or fasting blood glucose and had no improvement of glucose metabolism as determined by glucose and insulin tolerance tests (Supplementary Figure S2). Therefore, this report only focused on 50mM dosage of NAC treatment.
Bi-weekly body weights were recorded for the duration of treatment (Figure 1A). HFD mice showed significantly increased body weight compared to ND. There were no body weight significant differences between HFD, HFDpNAC and HFDiNAC groups (Figure 1B). Significant differences in average NAC intake were noted with highest NAC intake observed in the HFDpNAC group (21mg/mouse/day) compared to HFDiNAC (14mg/mouse/day) group and HFDiNAC+ (16mg/mouse/day) group (Supplementary Figure S3A). Only HFDiNAC group showed significantly increased food intake when compared to ND group (Supplementary Figure S3B). A significantly increased fasting blood glucose was determined in the HFD mice when compared to ND (Figure 1C), however, an improved fasting blood glucose level was only observed in HFDpNAC, not HFDiNAC mice (Figure 1C). Fed plasma insulin level was found to be significantly elevated in HFD mice compared to all other groups, and both HFDpNAC and HFDiNAC mice displayed a similar plasma insulin level as ND mice (Figure 1D). Fed circulating triglycerides and cholesterol were assessed and showed no significant differences between ND, HFD, and NAC treated HFD groups (Supplementary Figure S4).
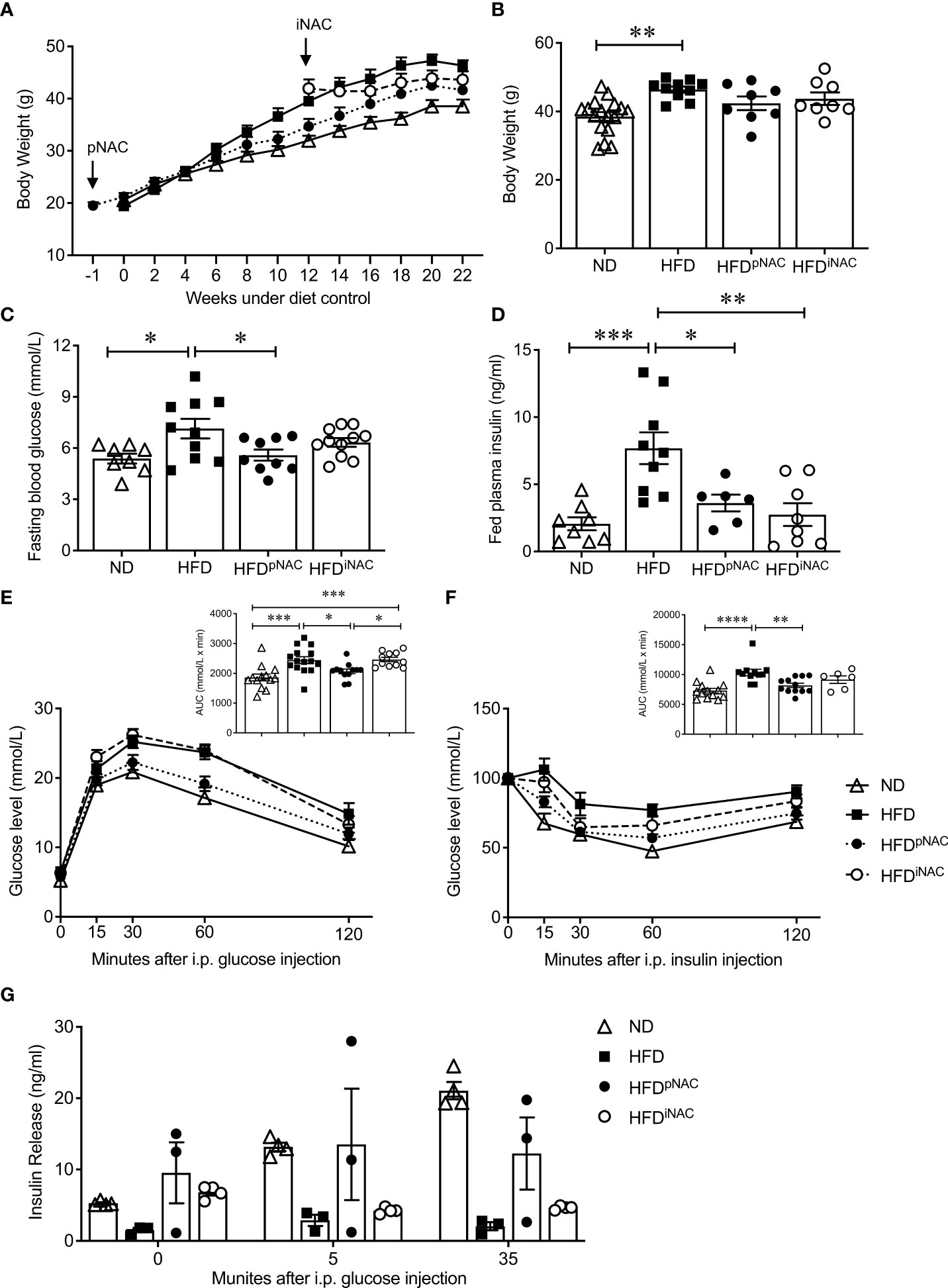
Figure 1 Preventive NAC treatment improves glucose metabolism in HFD-induced diabetic mice. (A) Recording of bi-weekly body weight for the duration of 22 weeks (n=8-17 mice/group). Measurement of (B) body weights, (C) overnight (~16 hours) fasting blood glucose and (D) fed plasma insulin levels (n=6-17 mice/group). (E) IPGTT and area under curve (AUC, n=10-14 mice/group), (F) IPITT and AUC (n=6-12 mice/group) and (G) in vivo GSIS (n=3-5 mice/group) of ND, HFD, HFDpNAC and HFDiNAC mice at 22 weeks. Control diets (ND): open triangle; HFD: closed square; HFDpNAC: closed circle; HFDiNAC: open circle. Data are expressed as means ± SEM. *p<0.05, **p<0.01, ***p<0.001, ****p<0.0001; analyzed using one-way ANOVA followed by Tukey’s Post-Hoc test.
Mice under HFD for 22 weeks demonstrated a progressive T2DM-like phenotype with impaired glucose and insulin tolerance when compared to ND mice (Figures 1E, F). Significantly improved glucose tolerance and insulin sensitivity in HFDpNAC mice was determined, which showed similar response as ND mice, however, this improvement was not found in HFDiNAC mice (Figures 1E, F). Furthermore, in vivo GSIS showed an observable improvement of glucose stimulated insulin secretion in HFDpNAC mice compared to HFD and HFDiNAC groups (Figure 1G). These data indicate that timing efficiency of NAC treatment is critical to normalize glucose metabolism in HFD-induced diabetic mice.
NAC treatment preserves beta-cell mass and identity with reduction of beta-cell oxidative stress in HFD-induced diabetic mice
Double immunofluorescence staining for insulin and glucagon were used for determining islet density, beta- and alpha-cell mass, and beta-cell size (Figure 2A). Morphological analysis of beta-cell mass showed an increase in HFD pancreas, with no discernable changes in alpha cell mass or islet density (Figures 2B, C, F). The increased beta-cell mass in HFD mice was due to an enlargement in beta-cell size with low beta-cell number in the insulin positive area (Figures 2D, E) but was not caused by changes in beta-cell proliferation as determined using Ki67 co-staining (Figure 2G). Interestingly, HFD-induced enlargement of beta-cell mass was not observed in HFDpNAC and HFDiNAC groups (Figure 2B). Beta-cell mass, size, and number in HFDpNAC and HFDiNAC mouse islets displayed similar results as ND mice (Figures 2D, E).
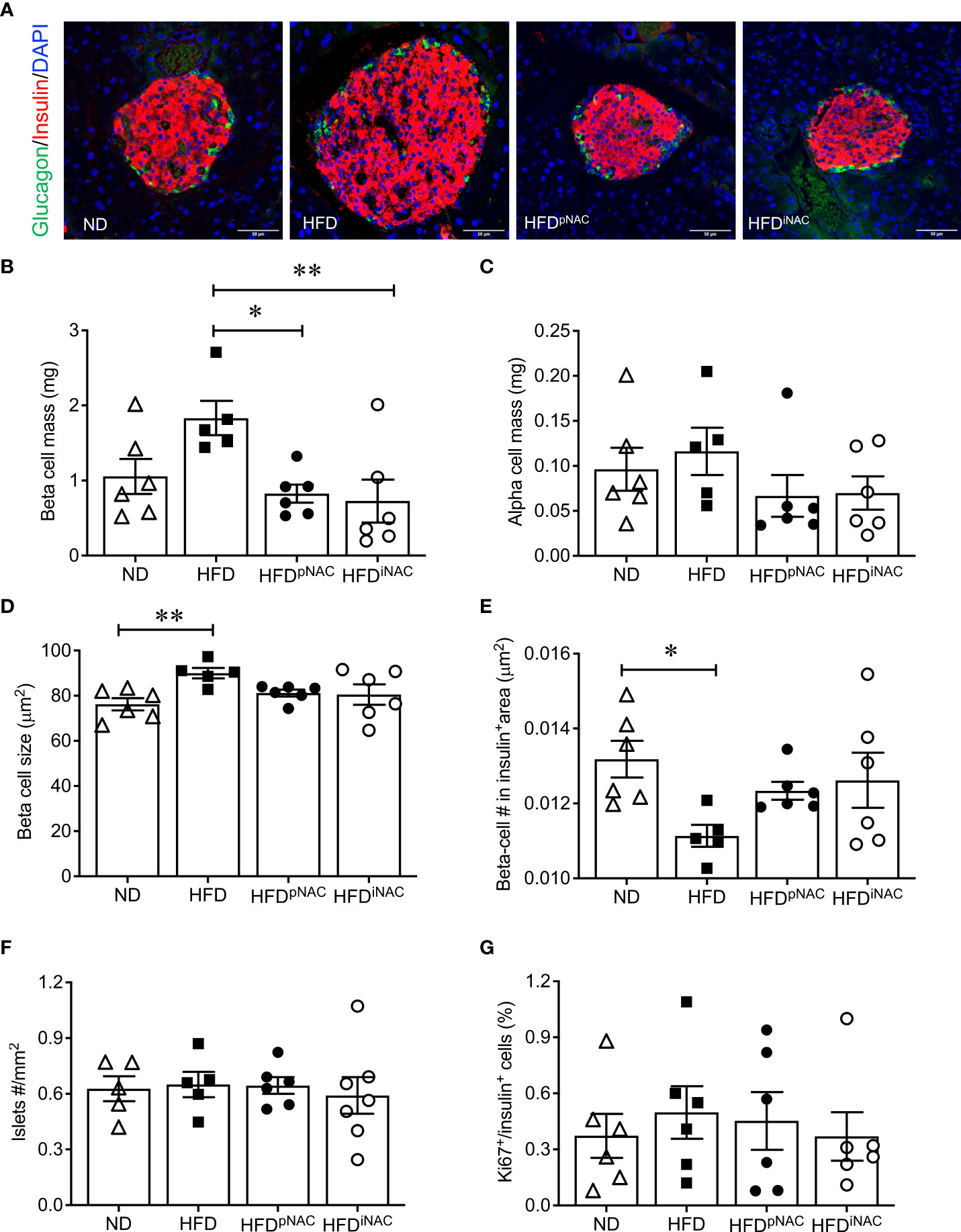
Figure 2 Histological analysis of NAC treated HFD pancreata demonstrated preserved beta-cell mass with no change in cell proliferation. (A) Representative double immunofluorescence images for islet morphology, detected by insulin (red) and glucagon (green) staining, and DAPI labeled nuclei (blue). Scale bars: 50µm. (B) Beta cell mass (C) alpha cell mass (D) beta cell size and (E) number (F) islet density in ND, HFD, HFDpNAC and HFDiNAC mouse pancrea at 22 weeks (n=5-6 pancreata/group). (G) Proliferation of beta cells quantified using Ki67 co-localization with insulin+ cells (n=6 pancreata/group). ND: open triangle; HFD: closed square. HFDpNAC: closed circle; HFDiNAC: open circle. Data are expressed as means ± SEM. *p<0.05, **p<0.01, analyzed using one-way ANOVA followed by Tukey’s Post-Hoc test.
When analyzing transcription factors (Pdx-1, MafA, and FoxO1) involved in beta-cell function and insulin secretion, it was found that nuclear Pdx-1 localization was significantly reduced in HFD mouse beta-cells compared to ND, HFDpNAC and HFDiNAC groups (Figures 3A, B). Both HFDpNAC and HFDiNAC groups showed significantly improved Pdx-1 nuclear localization, similar to ND group (Figures 3A, B). No obvious changes were found between experimental mice for MafA and FoxO1 nuclear localization in beta-cells (data not shown).
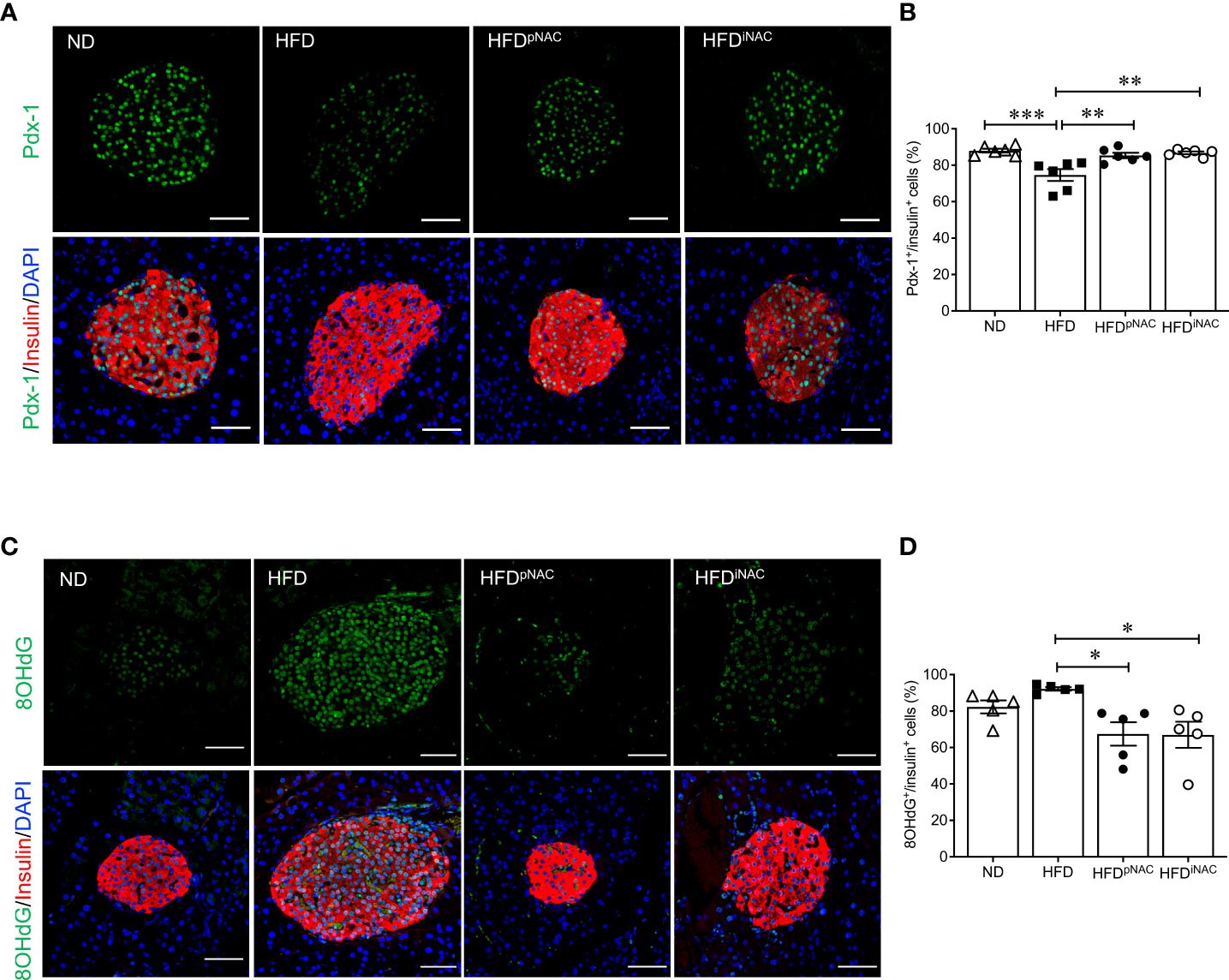
Figure 3 NAC treatment preserves beta-cell identity with reduction of beta-cell oxidative stress in HFD-induced diabetic mice. Representative double immunofluorescence images of (A) Pdx-1 (green) (C) 8OHdG (green) co-stained with insulin (red), nuclei are stained with DAPI (blue). Scale bars: 50µm. Quantification of nuclear (B) Pdx-1 (D) 8OHdG in insulin+ cells of ND, HFD, HFDpNAC and HFDiNAC mouse pancreas at 22 weeks (n=5-6 pancreata/group). ND: open triangle; HFD: closed square; HFDpNAC: closed circle; HFDiNAC: open circle. Data are shown as means ± SEM. *p<0.05, **p<0.01, ***p<0.001, analyzed using one-way ANOVA followed by Tukey’s Post-Hoc test.
To examine NAC’s impact on beta-cell oxidative stress, presence of 8OHdG in beta-cells was quantified using double staining for 8OHdG and insulin. The percentage of nuclear 8OHdG labelling in insulin-positive cells was significantly higher in the HFD mice than that of HFDpNAC and HFDiNAC groups (Figures 3C, D). A significant reduction of beta-cell 8OHdG labelling occurred in both NAC treated HFD mice, but no change observed in the ND mice when compared to HFD mice (Figures 3C, D).
NAC treatment reduces active PaSCs in islets leading to lower intra-islet collagen deposition in HFD-induced diabetic mice
To determine whether NAC influenced intra-islet PaSCs activation during HFD induced oxidative stress, quantification of active PaSCs population was performed. Since marker αSMA labels active PaSCs and desmin marks both quiescent and active PaSCs, double immunofluorescence staining for αSMA and desmin was performed to verify their colocalization (Supplementary Figure S5). Quantification of intra-islet activated PaSCs labeled by αSMA staining revealed a significant increase in αSMA area in HFD mouse islets compared to ND and HFDpNAC mice (Figures 4A, B). Although, αSMA+ staining in HFDiNAC islets showed a slight reduction, there was no statistically significant differences between HFD and HFDiNAC mice (Figures 4A, B). Measuring desmin population in the experimental groups showed a significantly higher percentage of desmin+ staining present in HFD mouse islets compared to HFDpNAC and HFDiNAC mouse islets, but no significance observed in ND group (Figures 4C, D). Both data indicate that HFD induced PaSC activation could be diminished by NAC administration.
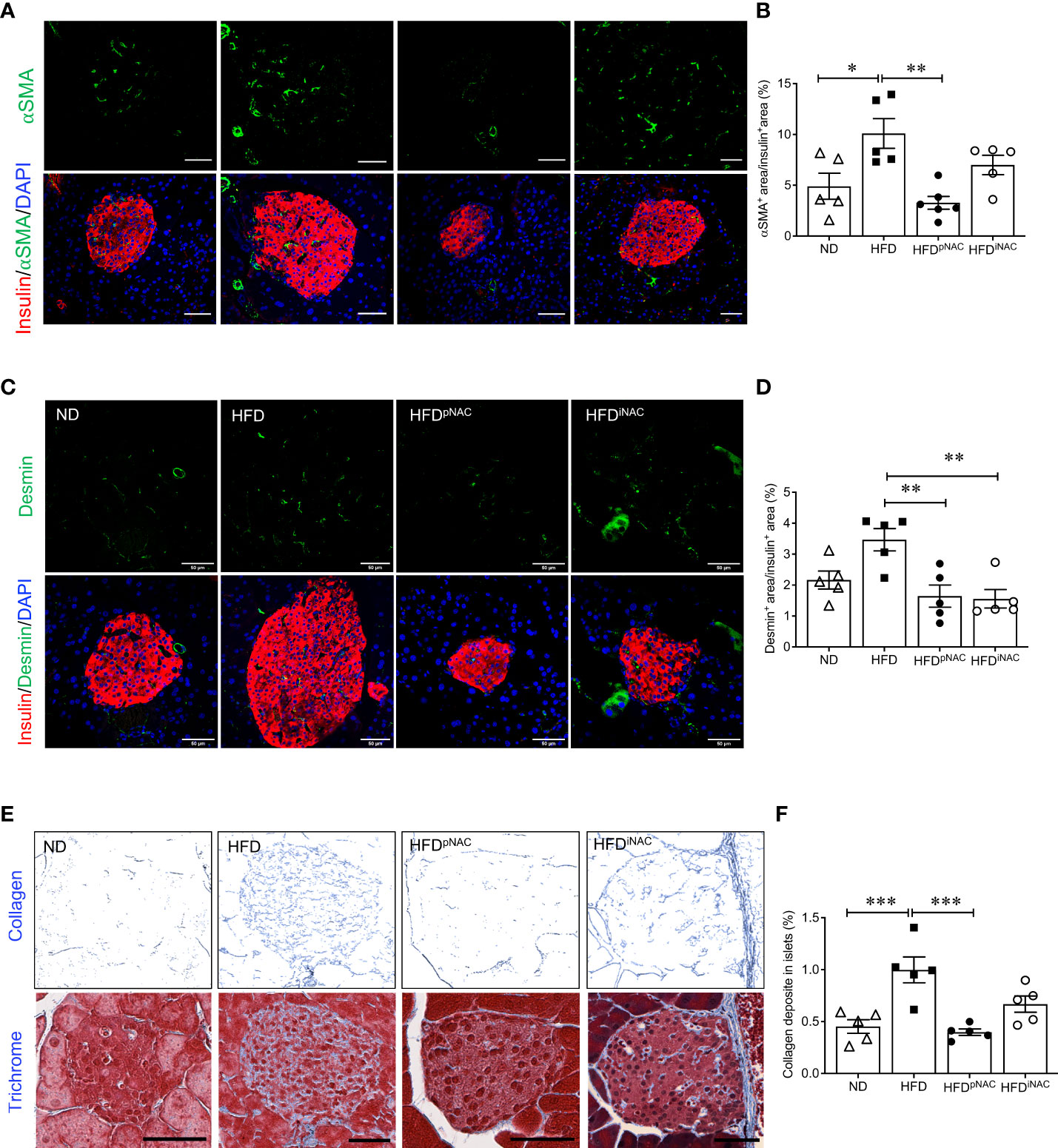
Figure 4 Preventive NAC treatment reduces intra-islet PaSCs activation and collagen deposition in HFD-induced diabetic mice. Representative double immunofluorescence images of (A) α-SMA (green) or (C) desmin (green) co-stained with insulin (red), nuclei are stained with DAPI (blue). Scale bars: 50µm. Quantification of intra-islet α-SMA (B) or desmin (D) area in ND, HFD, HFDpNAC and HFDiNAC mouse islets at 22 weeks (n=5-6 pancreata/group). (E) Representative trichrome staining images, scale bar: 50μm; and (F) quantification of intra-islet collagen deposition (n=5 pancreata/group). ND: open triangle; HFD: closed square; HFDpNAC: closed circle; HFDiNAC: open circle. Data are shown as means ± SEM. *p<0.05, **p<0.01, ***p<0.001, analyzed using one-way ANOVA followed by Tukey’s Post-Hoc test.
To further identify whether NAC treatment could abolish HFD-induced PaSC activation and subsequent intra-islet collagen deposition of islet fibrosis, quantification of intra-islet collagen deposition was performed using trichrome stain (Figure 4E). Intra-islet collagen deposition was significantly increased in HFD mouse islets compared to ND mouse islets (Figure 4F), indicating HFD results in progression of islet fibrosis. A significant reduction of intra-islet collagen deposition in HFDpNAC mouse islets was found (Figures 4E, F), but this observation was not present in HFDiNAC mouse islets (Figures 4E, F).
Prolonged intervention NAC treatment improves glucose metabolism in HFD-induced diabetic mice
Compared to HFDpNAC mice, HFDiNAC mice received NAC treatment 12 weeks after HFD initiation with total duration of 10 weeks NAC treatment. HFDiNAC mice showed no improvement in glucose metabolism, PaSCs activation or collagen deposition, indicating the duration of NAC treatment is critical. Therefore, a prolonged NAC treatment of up to 18 weeks (HFDiNAC+) was established and investigated. Bi-weekly body weights were recorded up to 30 weeks (Figure 5A). HFD-fed mice had significantly increased body weight compared to ND-fed mice and HFDiNAC+ mice (Figure 5B). Reduced body weight was observed in HFDiNAC+ mice compared to HFD mice but remained significantly higher than that of ND mice (Figure 5B). Overnight fasting blood glucose was significantly elevated in both HFD and HFDiNAC+ mice compared to ND mice (Figure 5C). However, hyperinsulinemia observed in HFD mice were significantly improved in HFDiNAC+ mice, but remained significantly higher levels compared to ND mice (Figure 5D). Fed plasma triglyceride levels were significantly lower in HFD mice, but HFDiNAC+ mice showed relatively elevated plasma triglycerides similar to ND mice (Figure 5E). Fed plasma cholesterol showed significant increase in both HFD and HFDiNAC+ mice when compared to ND mice (Figure 5F). A significant impairment of glucose tolerance was present in HFD mice that was not observed in HFDiNAC+ mice. HFDiNAC+ mice which displayed similar glucose tolerance as ND mice (Figures 5G, H). A significant impairment of insulin tolerance was observed in HFD mice when compared to ND mice, however, insulin sensitivity showed a trend toward improvement in HFDiNAC+ mice compared to HFD mice, but statistical significance was not reached (Figures 5I, J).
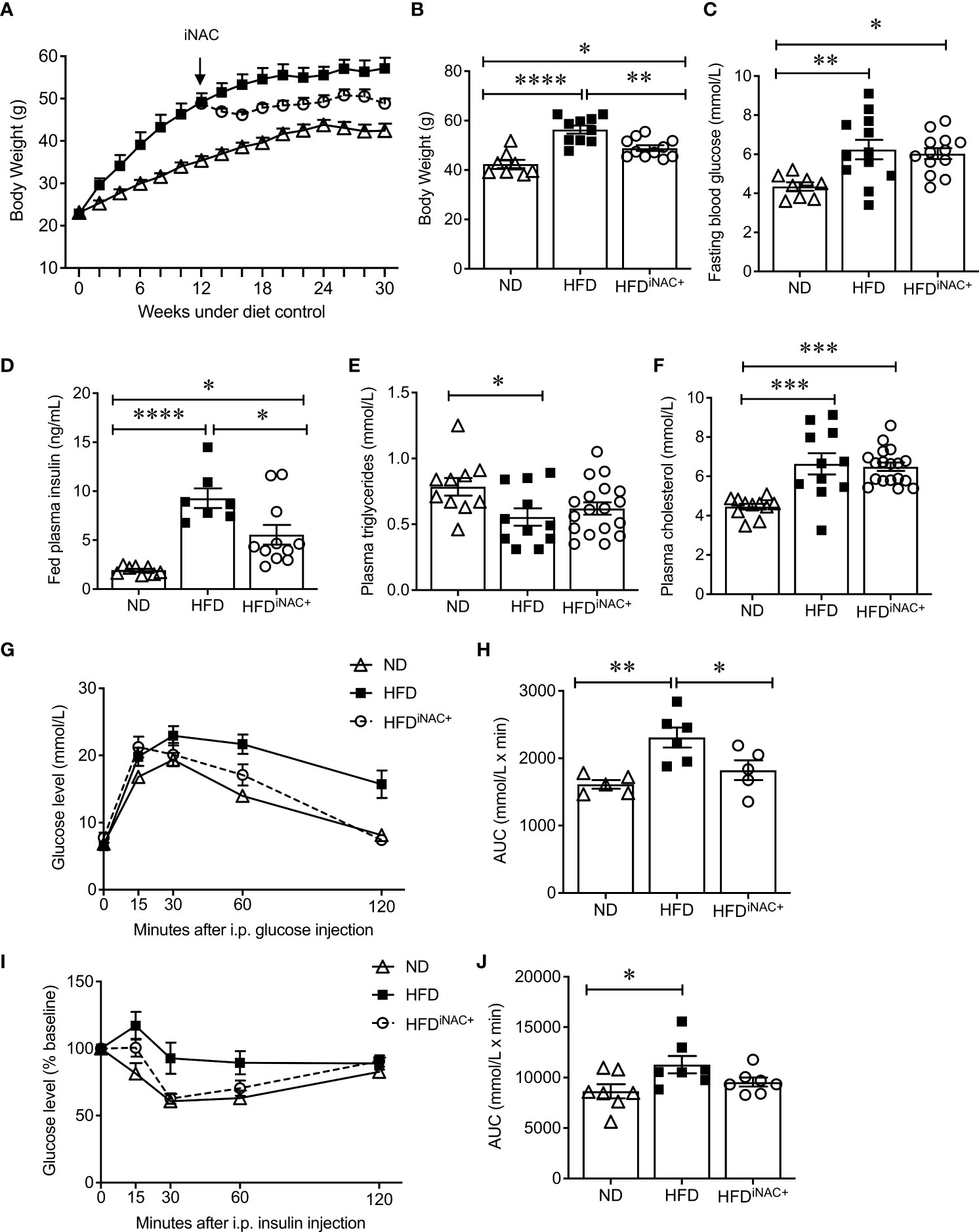
Figure 5 Prolonged intervention NAC treatment improves glucose metabolism in HFD-induced diabetic mice. (A) Recording of biweekly body weight for the duration of 30 weeks (n=8-12 mice/group). Measurement of (B) body weights, (C) overnight (~16 hours) fasting blood glucose, (D) fed plasma insulin, (E) triglycerides and (F) cholesterol levels (n=7-18 mice/group). (G) IPGTT and (H) AUC (n=5-6 mice/group), (I) IPITT and (J) AUC (n=7 mice/group) of ND, HFD and HFDiNAC+ mice at 30 weeks. ND: open triangle; HFD: closed square; HFDiNAC+: open circle. Data are expressed as means ± SEM. *p<0.05, **p<0.01, ***p<0.001, ****p<0.0001, analyzed using one-way ANOVA followed by Tukey’s Post-Hoc test.
Prolonged intervention NAC treatment preserves beta-cell mass and reduces intra-islet PaSCs activation in HFD-induced diabetic mice
Double immunofluorescence staining images for insulin and glucagon showed large islets present in HFD and HFDiNAC+ mouse pancreas compared to ND group (Figure 6A). Morphometric analysis of islet number showed HFD mice have significantly elevated islet numbers compared to ND, however, islet number in HFDiNAC+ is comparable to ND (Figure 6B). Beta-cell mass was significantly increased in HFD and was significantly improved in HFDiNAC+ mice; HFDiNAC+ mice and ND mice showed similar beta-cell masses (Figure 6C). There were no differences observed in alpha-cell mass found between groups (Figure 6D). HFD induced beta-cell dysfunction with loss of nuclear Pdx-1 signals was significantly rescued in HFDiNAC+ islets, which showed similar nuclear Pdx-1 localization as ND islets (Figure 6F). In parallel, HFD induced beta-cell oxidative stress labeled by 8OHdG+ was also significantly improved in HFDiNAC+ islets (Figures 6G, H). Furthermore, HFDiNAC+ mice showed significantly lower intra-islet αSMA labeling (Figures 7A, B) with improved intra-islet collagen deposition (Figure 7C), which was not observed in HFDiNAC islets receiving a shorter duration of NAC treatment. HFD mice maintained higher level of αSMA labeling and collagen deposition in the intra-islets when compared to ND and HFDiNAC+ mice (Figure 7). This data further verified that NAC treatment improves beta-cell identity, oxidative stress and PaSC induced fibrotic in HFD-induced diabetic islets is in a time-dependent manner.
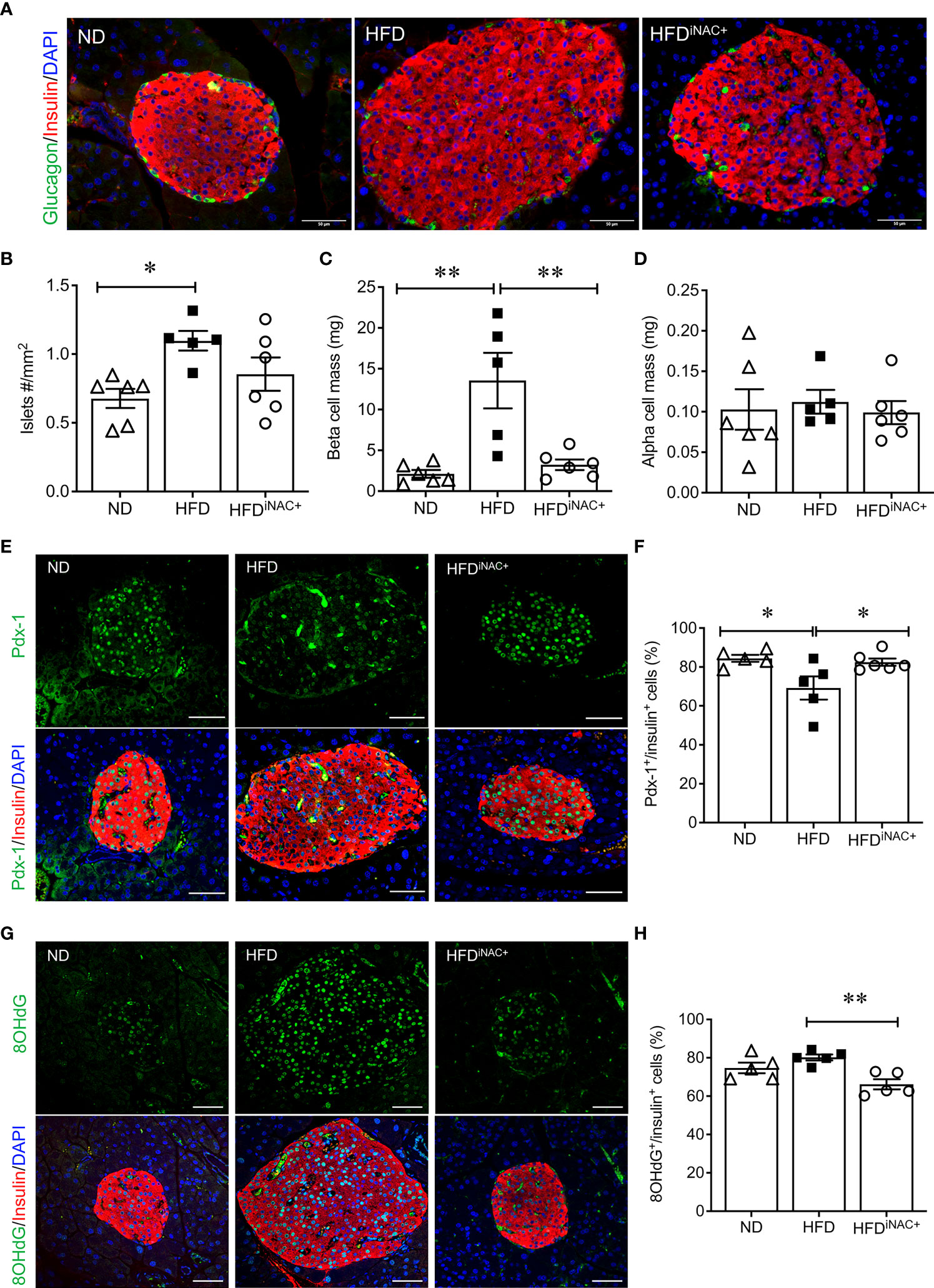
Figure 6 Prolonged intervention NAC treatment preserves beta-cell mass and nuclear Pdx-1 with lower 8OHdG in HFD-induced diabetic mice. (A) Representative double immunofluorescence images for islet morphology, detected by insulin (red) and glucagon (green) staining and DAPI labeled nuclei (blue). Scale bars: 50µm. Morphometric quantification of islet density (B) beta cell mass (C) and alpha cell mass (D) (n=5-6 pancreata/group). Representative double immunofluorescence images for (E) Pdx-1 (green) or (G) 8OHdG (green) and insulin (red), nuclei are stained with DAPI (blue). Scale bars: 50µm. Quantification of nuclear Pdx-1 (F) or 8OHdG (H) in insulin+ cells of ND, HFD and HFDiNAC+ mouse pancreas at 30 weeks (n=5-6 pancreata/group). ND: open triangle; HFD: closed square). HFDiNAC+: open circle. Data are shown as means ± SEM. *p<0.05, **p<0.01, analyzed using one-way ANOVA followed by Tukey’s Post-Hoc test.
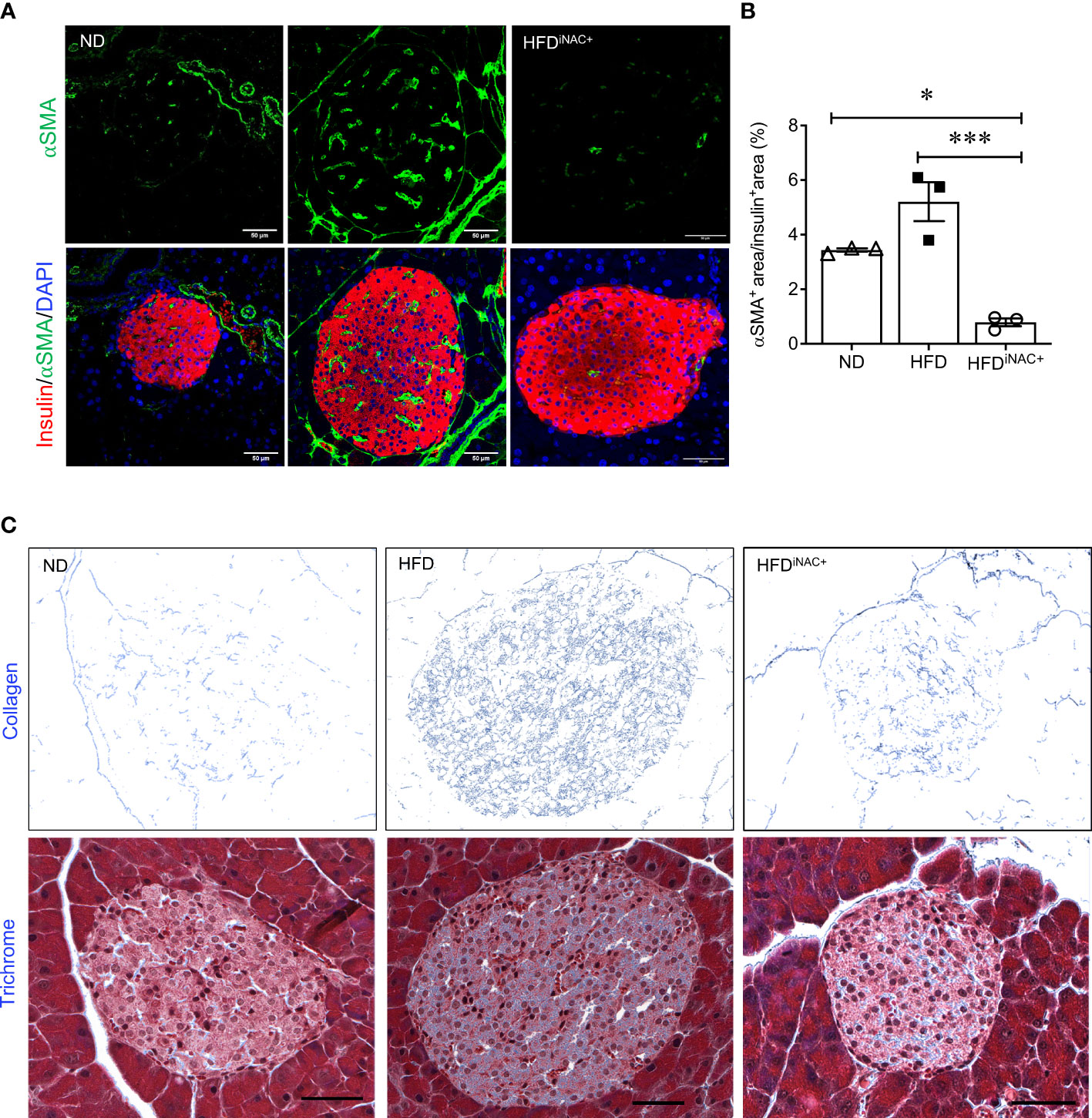
Figure 7 Prolonged intervention NAC treatment reduces intra-islet PaSCs activation and collagen deposition in HFD-induced diabetic mice. (A) Representative double immunofluorescence images of α-SMA (green) co-stained with insulin (red), nuclei are stained with DAPI (blue). Scale bars, 50µm; (B) quantification of intra-islet α-SMA area in ND, HFD and HFDiNAC+ mouse islets at 30 weeks (n=3 pancreata/group). (C) Representative trichrome staining images of intra-islet collagen deposition. Scale bar: 50μm. ND: open triangle; HFD: closed square; HFDiNAC+: open circle) Data are shown as means ± SEM. *p<0.05, ***p<0.001, analyzed using one-way ANOVA followed by Tukey’s Post-Hoc test.
Discussion
This is a first in vivo study investigating the effects of antioxidant NAC on diabetic islets in HFD-induced diabetic mice. It is well documented that mice fed with HFD display dysregulation of glucose metabolism and beta-cell compensation with increased beta-cell oxidative stress and intra-islet PaSC activation, which produces excessive ECM and cytokines resulting in islet fibrosis (Figure 8). When HFD-induced diabetic mice were administered NAC at an effective dose and time, significantly improved glucose tolerance and insulin sensitivity, and rescue from beta-cell overcompensation was determined. NAC treated HFD mice displayed a normalized beta-cell mass and size with significantly improved beta-cell identity, as Pdx-1 nuclear localization, and reduced beta-cell oxidative stress, labeled by 8-OHdG (Figure 8). Importantly, NAC diminished intra-islet PaSCs activation and PaSC-induced collagen deposition preventing fibrosis caused by HFD (Figure 8). This study suggests that antioxidant therapy using NAC is beneficial for maintaining healthy beta-cells and intra-islet quiescent PaSC population in HFD-induced diabetic mice and provides a better understanding of effective dose and time for antioxidant therapy in obesity related diabetes in humans.
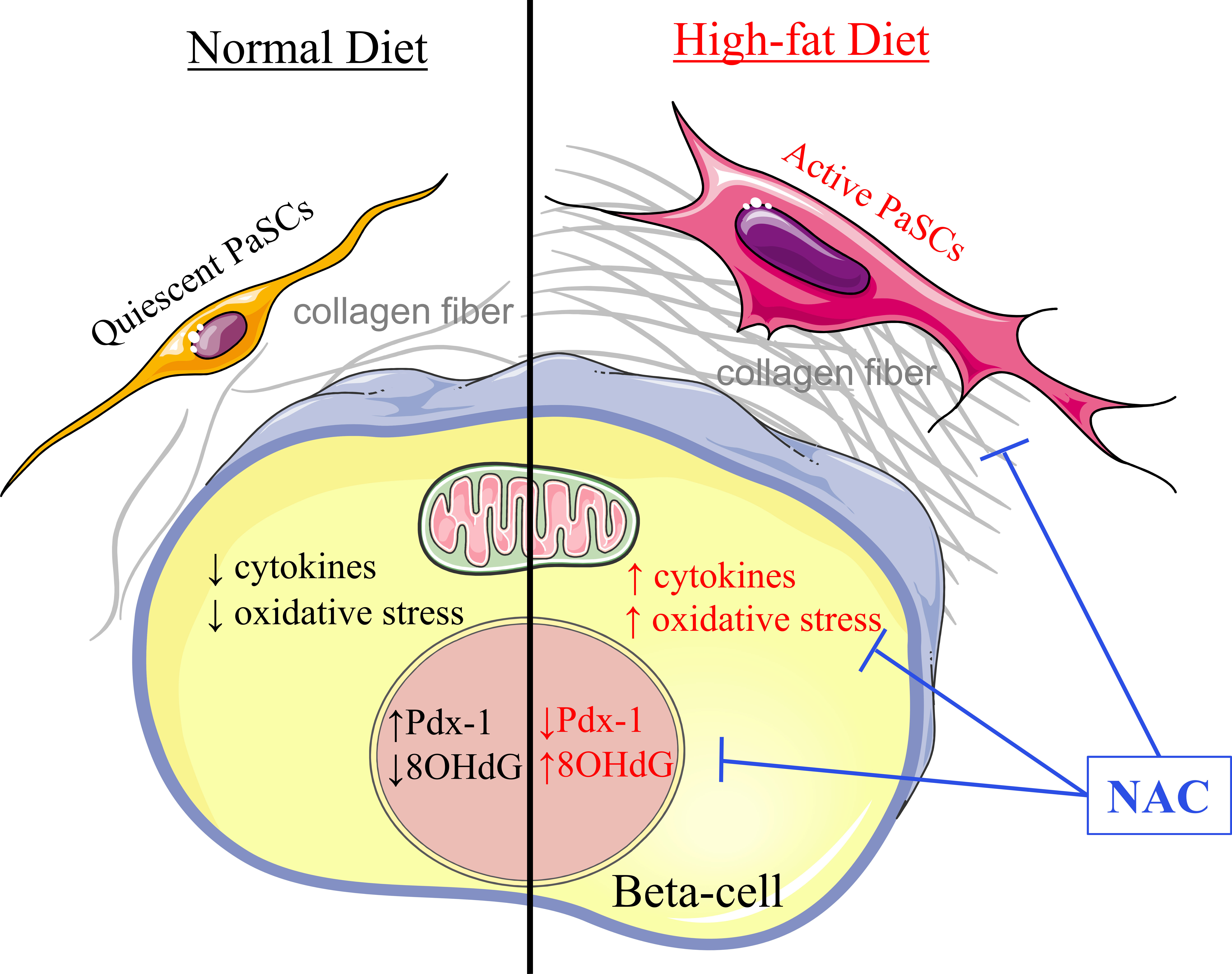
Figure 8 Proposed model of NAC maintains healthy beta-cells and quiescent PaSCs in HFD-induced diabetic mouse islet. In a healthy condition under normal chow diet, intra-islet PaSCs are quiescent; they produce suitable cytokines and ECM for maintaining islet structure and facilitating healthy beta-cell function. Under high-fat diet, increased oxidative stress promoted intra-islet PaSC activation resulting in increased cytokine production and ECM deposition which diminished beta-cell identity and function. Given NAC treatment, HFD islets showed significantly reduced oxidative stress and intra-islet PaSC activation with enhanced beta-cell identity and function.
Identifying the optimal dosage during NAC therapy is critical for designing clinical trials. Use of sub-optimal or excessive antioxidant dosages of NAC could hinder NAC’s positive anti-diabetic effects in T2DM patients (24). Based on previous reports (20, 21), two dosages of NAC (10mM and 50mM) were employed in this study and showed HFD mice that received 10mM NAC in drinking water for up to 22 weeks resulted in no improvements in glucose tolerance and insulin sensitivity. Whereas, administration at 50mM in drinking water showed significant improvements in glucose metabolism, which was consistent with the findings by Falach-Malik et al. (21). It was noted that NAC at 50mM dosage did not cause a significant reduction in body weights, despite increased caloric intake in both HFDpNAC and HFDiNAC mice under 22 weeks HFD feeding. Previous reports suggest that this dosage potentially caused an increase in motor activity and/or metabolic activity which may have been compensated for by increasing food intake resulting in no net loss of weight (22, 36). However, prolonged intervention NAC in HFDiNAC+ mice displayed significantly reduced body weights compared to HFD mice under 30 weeks HFD feeding, suggesting NAC treatment is associated with weight loss and may account for required time to take effect.
Previous studies suggest using NAC at an earlier time point increased effectiveness for improving glucose tolerance and insulin sensitivity related to weight loss in HFD mice (21, 22). HFD mice that received preventive NAC treatment for 23 weeks showed significantly improved glucose and insulin tolerance, but this was not observed at 10 weeks. Prolonged intervention NAC (18 weeks) administration to HFD mice displayed improvement similar to NAC prevention treated mice, indicating that positive anti-diabetic effects of NAC in HFD-induced diabetic mice require up to 18 weeks of prevention or intervention NAC treatment. Although, there was no change in fed plasma lipid level in mice with 22 weeks HFD feeding, HFD-induced hepatic insufficient export of triglycerides was observed in HFD mice with 30 weeks feeding, this impaired liver export of triglycerides was rescued in HFDiNAC+ mice that showed normalised triglycerides level comparable to ND mice. This data indicates that NAC improved glucose metabolism and may be associated with improved liver function to reduce hepatic triglyceride accumulation in order to reduce gluconeogenesis and enhance insulin signalling (37, 38). Taken together, this study suggests that adequate duration of administration is crucial for NAC to be effective as an antioxidative therapy in treating diet-induced obesity diabetes.
Mice fed with HFD for 22 or 30 weeks showed not only impaired metabolic outcomes, but also significant alterations to islet architecture indicating a beta-cell compensation response. HFD mice with NAC prevention or intervention treatment can prevent or rescue this beta-cell overcompensation with significantly normalized beta-cell mass and size. This demonstrates that NAC is effective at maintaining beta-cell function during HFD-induced stress. It is agreed that transcription factor Pdx-1 plays a critical role in regulating beta-cell function by activating genes essential for beta-cell identity; loss of beta-cell identity contributes to the pathogenesis of type 2 diabetes (39). A previous study by Leenders et al. proposed under elevated oxidative stress conditions, beta-cells dedifferentiate into other islet cell types leading to reduced beta-cell identity (40). In the present study, loss of beta-cell identity and increased beta-cell oxidative stress was displayed in HFD mice, however, HFDpNAC and HFDiNAC islets showed preserved or restored beta-cell Pdx-1 nuclear localization, and reduced beta-cell oxidative stress. These findings are supported by previous reports that demonstrated NACs ability to improve Pdx-1 and insulin content in beta-cells in a genetic obesity model of T2DM (26), and to prevent glucose toxicity-induced impaired Pdx-1 binding to the insulin gene promoter in beta-cell line in vitro and reduce plasma 8-OHdG oxidative stress level in Zucker diabetic fatty rats (41). Although the exact mechanism of NACs action on beta-cells remains to be elucidated, it is a well-known that NAC is a biosynthetic precursor of glutathione (23). It also contains a thiol group exerting its antioxidant effects through electron donation (42) and has recently been investigated for its potential intracellular conversion to sulfane sulfur species to scavenge oxidants (43). NAC also produces anti-inflammatory actions by indirectly inhibiting the activation of the redox-sensitive pathway NF-κB in order to prevent inflammatory responses which lead to loss of Pdx-1 expression (42). Since the present study did not evaluate inflammatory markers, it would be interesting for future studies to investigate the role of NAC on inflammatory markers in the setting of diet-induced obesity.
PaSCs in islets play a critical role in controlling ECM, growth factors and cytokine secretion. Over-active PaSCs are involved in beta-cell compensation in the early stages of T2DM progression (44). Furthermore, clinical studies have shown higher levels of αSMA and islet fibrosis in T2DM patients (45), such evidence was also observed in the current study where HFD mice showed significantly elevated intra-islet αSMA+ cells with excessive collagen deposition. Administration of NAC to HFD mice displayed significantly reduced intra-islet oxidative stress and intra-islet αSMA+, which corresponded to significantly reduced intra-islet fibrosis. This observation matches previous in vitro reports that NAC is capable of reducing PaSC activation and reverting cells to a quiescent state (46–48). NAC treatment results in increased vitamin A droplets indicative of quiescent PaSCs (47). It was noted that prevention NAC treated HFDpNAC mice showed similar PaSC activation and islet collagen staining as ND islets, suggesting that initiating NAC therapy is most beneficial to prevent PaSCs activation and intra-islet collagen deposition caused by HFD. However, intervention NAC treated islets did not show a significant improvement until 18 weeks NAC treatment, indicating PaSCs activation begin rapidly after HFD-induced stress, and reverting active PaSCs and collagen accumulation requires sufficient time with antioxidant treatment. A study on diabetic cardiac function in C57/B6 mice supports these results as they determined NAC is beneficial at improving cardiac function and reducing cardiac fibrosis, most noticeably in mice which received earlier and longer treatment (28). However, the potential mechanisms of NAC action on intra-islet PaSCs require further in vivo investigation.
In summary, the present study provides evidence that NAC is effective at protecting beta-cells from overcompensation and loss of beta-cell identity observed during T2DM progression in mice. The efficacy of NAC treatment in HFD-induced diabetic mice was dependent on dosage and duration of treatment. This study supports previous reports showing the ability of NAC to improve metabolic outcomes in mouse models of T2DM. Importantly, it provides first evidence of NAC’s effect on the intra-islet environment where beta-cell identity was protected, and oxidative stress and PaSC activation was diminished. Ultimately, the demonstrated benefits of antioxidant NAC on diet-induced obesity and diabetes indicates that it should be further investigated as an adjuvant therapy in controlling T2DM progression in humans.
Data availability statement
The original contributions presented in the study are included in the article/Supplementary Material. Further inquiries can be directed to the corresponding author.
Ethics statement
The animal study was reviewed and approved by University of Western Ontario Animal User Subcommittee.
Author contributions
MS and MW contributed to an in vivo mouse feeding and glucose metabolic study, data acquisition, interpretation of data, and preparation of the submitted manuscript. MB contributed to the maintenance of animals, data acquisition and preparation of the submitted manuscript. GS, MR, SZ and CS contributed to data acquisition. NB contributed interpretation of data and manuscript discussion. RW contributed to the design of experiments within this project, interpretation of data and preparation of the submitted manuscript. All authors contributed to the article and approved the submitted version.
Funding
This study was funded by the Canadian Institutes of Health Research (grant# 152944).
Acknowledgments
This work was supported by grants from the Canadian Institutes of Health Research.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fendo.2022.938680/full#supplementary-material
Supplementary Figure 1 | Experimental timeline to determine the time-dependent of NAC administration on high fat diet-induced diabetic mice. Both normal diet (ND) and high fat diet (HFD) experimental groups were established at age of 6 weeks. pNAC: preventative NAC was started at 5 weeks of age, one week prior to HFD diet start and had a total of 23 weeks of NAC treatment; iNAC: intervention NAC was given at 18 weeks of age, 12 weeks after HFD diet start and had a total of 10 weeks of NAC treatment; iNAC+: intervention NAC was given at 18 weeks of age, 12 weeks after HFD diet start and had a total of 18 weeks of NAC treatment.
Supplementary Figure 2 | 10mM NAC treatment did not improve glucose metabolism in HFD-induced diabetic mice. (A) Body weight, (B) fasted blood glucose levels, (C) IPGTT, (D) area under curve (AUC) analysis of IPGTT, (E) IPITT and (F) AUC analysis of IPITT results from ND, HFD, HFDpNAC and HFDiNAC mice during 22-week experiment. Data are expressed means ± SEM (n=3-17 mice per experimental group). *p<0.05, **p<0.01, ***p<0.001 vs. ND, determined by one-way ANOVA followed by Tukey’s multiple comparisons test.
Supplementary Figure 3 | NAC and food intake in HFD-induced diabetic mice. (A) Average NAC intake for mice undergoing prevention (pNAC), intervention (iNAC) and extend intervention (iNAC+) treatment. (B) Food intake in ND, HFD, HFDpNAC and HFDiNAC mice and (C) Food intake in ND, HFD and HFDiNAC+ mice. Data are expressed means ± SEM (n=5-11 mice per experimental group). *p<0.05, **p<0.01, ***p<0.001 vs. ND or as indicated, determined by one-way ANOVA followed by Tukey’s multiple comparisons test.
Supplementary Figure 4 | Plasma lipid levels in ND, HFD, HFDpNAC and HFDiNAC mice. Fed plasma triglycerides (A) and cholesterol (B). Data are expressed means ± SEM (n=8-11 mice per experimental group).
Supplementary Figure 5 | Correlation of desmin (green) and αSMA (red) in the islet as dash-line outlined. Lower panel displays enlarged images, arrows indicate co-staining, arrowheads label single staining. Scale bar: 50μm.
References
1. Prentki M, Nolan CJ. Islet β cell failure in type 2 diabetes. J Clin Invest (2006) 116:1802–12. doi: 10.1172/JCI29103
2. Keane KN, Cruzat VF, Carlessi R, de Bittencourt PIH, Newsholme P. Molecular events linking oxidative stress and inflammation to insulin resistance and β-cell dysfunction. Oxid Med Cell Longev (2015) 2015:181643. doi: 10.1155/2015/181643
3. McAdam-Marx C, Mukherjee J, Bellows BK, Unni S, Ye X, Iloeje U, et al. Evaluation of the relationship between weight change and glycemic control after initiation of antidiabetic therapy in patients with type 2 diabetes using electronic medical record data. Diabetes Res Clin Pract (2014) 103:402–11. doi: 10.1016/j.diabres.2013.12.038
4. Temelkova-Kurktschiev T, Siegert G, Bergmann S, Henkel E, Koehler C, Jaroß W, et al. Subclinical inflammation is strongly related to insulin resistance but not to impaired insulin secretion in a high risk population for diabetes. Metabolism (2002) 51:743–9. doi: 10.1053/meta.2002.32804
5. Butler AE, Janson J, Bonner-Weir S, Ritzel R, Rizza RA, Butler PC. β-cell deficit and increased β-cell apoptosis in humans with type 2 diabetes. Diabetes (2003) 52:102–10. doi: 10.2337/diabetes.52.1.102
6. Swisa A, Glaser B, Dor Y. Metabolic stress and compromised identity of pancreatic beta cells. Front Genet (2017) 8:21. doi: 10.3389/fgene.2017.00021
7. Omary MB, Lugea A, Lowe AW, Pandol SJ. The pancreatic stellate cell: a star on the rise in pancreatic diseases. J Clin Invest (2007) 117:50–9. doi: 10.1172/JCI30082
8. Apte MV, Pirola RC, Wilson JS. Pancreatic stellate cells: a starring role in normal and diseased pancreas. Front Physiol (2012) 3:344. doi: 10.3389/FPHYS.2012.00344
9. Riopel M, Li J, Fellows GF, Goodyer CG, Wang R. Ultrastructural and immunohistochemical analysis of the 8-20 week human fetal pancreas. Islets (2014) 6(4):e982949. doi: 10.4161/19382014.2014.982949
10. Li J, Chen B, Fellows GF, Goodyer CG, Wang R. Activation of pancreatic stellate cells is beneficial for exocrine but not endocrine cell differentiation in the developing human pancreas. Front Cell Dev Biol (2021) 9:694276. doi: 10.3389/fcell.2021.694276
11. Sekhar RV, Mckay SV, Patel SG, Guthikonda AP, Reddy VT, Balasubramanyam A, et al. Glutathione synthesis is diminished in patients with uncontrolled diabetes and restored by dietary supplementation with cysteine and glycine. Diabetes Care (2011) 34:162–7. doi: 10.2337/DC10-1006
12. Rehman K, Akash MSH. Mechanism of generation of oxidative stress and pathophysiology of type 2 diabetes mellitus: How are they interlinked? J Cell Biochem (2017) 118:3577–85. doi: 10.1002/JCB.26097
13. Sakuraba H, Mizukami H, Yagihashi N, Wada R, Hanyu C, Yagihashi S. Reduced beta-cell mass and expression of oxidative stress-related DNA damage in the islet of Japanese type II diabetic patients. Diabetologia (2002) 45:85–96. doi: 10.1007/S125-002-8248-Z
14. Miki A, Ricordi C, Sakuma Y, Yamamoto T, Misawa R, Mita A, et al. Divergent antioxidant capacity of human islet cell subsets: A potential cause of beta-cell vulnerability in diabetes and islet transplantation. PloS One (2018) 13:e0196570. doi: 10.1371/JOURNAL.PONE.0196570
15. Kawamori D, Kajimoto Y, Kaneto H, Umayahara Y, Fujitani Y, Miyatsuka T, et al. Oxidative stress induces nucleo-cytoplasmic translocation of pancreatic transcription factor PDX-1 through activation of c-jun NH2-terminal kinase. Diabetes (2003) 52:2896–904. doi: 10.2337/diabetes.52.12.2896
16. Lee E, Ryu GR, Ko SH, Ahn YB, Song KH. A role of pancreatic stellate cells in islet fibrosis and β-cell dysfunction in type 2 diabetes mellitus. Biochem Biophys Res Commun (2017) 485:328–34. doi: 10.1016/j.bbrc.2017.02.082
17. Hong O-K, Lee S-H, Rhee M, Ko S-H, Cho J-H, Choi Y-H, et al. Hyperglycemia and hyperinsulinemia have additive effects on activation and proliferation of pancreatic stellate cells: possible explanation of islet-specific fibrosis in type 2 diabetes mellitus. J Cell Biochem (2007) 101:665–75. doi: 10.1002/jcb.21222
18. Hayden MR, Habibi J, Joginpally T, Karuparthi PR, Sowers JR. Ultrastructure study of transgenic Ren2 rat aorta – part 1: Endothelium and intima. Cardiorenal Med (2012) 2:66–82. doi: 10.1159/000335565
19. Kikuta K, Masamune A, Hamada S, Takikawa T, Nakano E, Shimosegawa T. Pancreatic stellate cells reduce insulin expression and induce apoptosis in pancreatic β-cells. Biochem Biophys Res Commun (2013) 433:292–7. doi: 10.1016/J.BBRC.2013.02.095
20. Ma Y, Gao M, Liu D. N-acetylcysteine protects mice from high fat diet-induced metabolic disorders. Pharm Res (2016) 33:2033–42. doi: 10.1007/s11095-016-1941-1
21. Falach-Malik A, Rozenfeld H, Chetboun M, Rozenberg K, Elyasiyan U, Sampson SR, et al. N-Acetyl-L-Cysteine inhibits the development of glucose intolerance and hepatic steatosis in diabetes-prone mice. Am J Transl Res (2016) 8:3744–56.
22. Shen F-C, Weng S-W, Tsao C-F, Lin H-Y, Chang C-S, Lin C-Y, et al. Early intervention of n-acetylcysteine better improves insulin resistance in diet-induced obesity mice. Free Radic Res (2018) 52:1296–310. doi: 10.1080/10715762.2018.1447670
23. Zhitkovich A. N -acetylcysteine: Antioxidant, aldehyde scavenger, and more. Chem Res Toxicol (2019) 32:1318–9. doi: 10.1021/acs.chemrestox.9b00152
24. Uraz S, Tahan G, Aytekin H, Tahan V. N-acetylcysteine expresses powerful anti-inflammatory and antioxidant activities resulting in complete improvement of acetic acid-induced colitis in rats. Scand J Clin Lab Invest (2013) 73:61–6. doi: 10.3109/00365513.2012.734859
25. Roma LP, Oliveira CAM, Carneiro EM, Albuquerque GG, Boschero AC, Souza KLA. N-acetylcysteine protects pancreatic islet against glucocorticoid toxicity. Redox Rep (2011) 16:173–80. doi: 10.1179/1351000211Y.0000000006
26. Kaneto H, Kajimoto Y, Miyagawa J, Matsuoka T, Fujitani Y, Umayahara Y, et al. Beneficial effects of antioxidants in diabetes: possible protection of pancreatic beta-cells against glucose toxicity. Diabetes (1999) 48:2398–406. doi: 10.2337/DIABETES.48.12.2398
27. Szkudlinska MA, Von Frankenberg AD, Utzschneider KM. The antioxidant n-acetylcysteine does not improve glucose tolerance or β-cell function in type 2 diabetes. J Diabetes Complications (2016) 30:618. doi: 10.1016/J.JDIACOMP.2016.02.003
28. Liu C, Lu XZ, Shen MZ, Xing CY, Ma J, Duan YY, et al. N-acetyl cysteine improves the diabetic cardiac function: possible role of fibrosis inhibition. BMC Cardiovasc Disord (2015) 15:84. doi: 10.1186/S12872-015-0076-3
29. Oakie A, Feng ZC, Li J, Silverstein J, Yee SP, Wang R. Long-term c-kit overexpression in beta cells compromises their function in ageing mice. Diabetologia (2019) 62:1430–44. doi: 10.1007/S00125-019-4890-5
30. Feng ZC, Riopel M, Popell A, Wang R. A survival kit for pancreatic beta cells: stem cell factor and c-kit receptor tyrosine kinase. Diabetologia (2015) 58:654–65. doi: 10.1007/S00125-015-3504-0
31. Yashpal NK, Li J, Wang R. Characterization of c-kit and nestin expression during islet cell development in the prenatal and postnatal rat pancreas. Dev Dyn (2004) 229:813–25. doi: 10.1002/DVDY.10496
32. Feng Z-C, Riopel M, Li J, Donnelly L, Wang R. Downregulation of fas activity rescues early onset of diabetes in c-kit wv/+ mice. Am J Physiol Metab (2013) 304:E557–65. doi: 10.1152/ajpendo.00453.2012
33. Feng ZC, Donnelly L, Li J, Krishnamurthy M, Riopel M, Wang R. Inhibition of Gsk3β activity improves β-cell function in c-KitWv/+ male mice. Lab Invest (2012) 92:543–55. doi: 10.1038/LABINVEST.2011.200
34. Riopel M, Krishnamurthy M, Li J, Liu S, Leask A, Wang R. Conditional β1-integrin-deficient mice display impaired pancreatic β cell function. J Pathol (2011) 224:45–55. doi: 10.1002/PATH.2849
35. Peters KM, Zhang R, Park C, Nong Z, Yin H, Wilson RB, et al. Vitamin d intervention does not improve vascular regeneration in diet-induced obese male mice with peripheral ischemia. J Nutr Biochem (2019) 70:65–74. doi: 10.1016/J.JNUTBIO.2019.04.010
36. Novelli ELB, Santos PP, Assalin HB, Souza G, Rocha K, Ebaid GX, et al. N-acetylcysteine in high-sucrose diet-induced obesity: Energy expenditure and metabolic shifting for cardiac health. Pharmacol Res (2009) 59:74–9. doi: 10.1016/J.PHRS.2008.10.004
37. Ress C, Kaser S. Mechanisms of intrahepatic triglyceride accumulation. World J Gastroenterol (2016) 22:1664–73. doi: 10.3748/WJG.V22.I4.1664
38. Samuel VT, Liu ZX, Qu X, Elder BD, Bilz S, Befroy D, et al. Mechanism of hepatic insulin resistance in non-alcoholic fatty liver disease. J Biol Chem (2004) 279:32345–53. doi: 10.1074/JBC.M313478200
39. Gao T, McKenna B, Li C, Reichert M, Nguyen J, Singh T, et al. Pdx1 maintains β cell identity and function by repressing an α cell program. Cell Metab (2014) 19:259–71. doi: 10.1016/J.CMET.2013.12.002
40. Leenders F, Groen N, de Graaf N, Engelse MA, Rabelink TJ, de Koning EJP, et al. Oxidative stress leads to β-cell dysfunction through loss of β-cell identity. Front Immunol (2021) 12:690379. doi: 10.3389/fimmu.2021.690379
41. Tanaka Y, Gleason CE, Tran POT, Harmon JS, Robertson RP. Prevention of glucose toxicity in HIT-T15 cells and zucker diabetic fatty rats by antioxidants. Proc Natl Acad Sci U.S.A. (1999) 96:10857–62. doi: 10.1073/PNAS.96.19.10857
42. Lasram MM, Dhouib IB, Annabi A, El Fazaa S, Gharbi N. A review on the possible molecular mechanism of action of n-acetylcysteine against insulin resistance and type-2 diabetes development. Clin Biochem (2015) 48:1200–8. doi: 10.1016/J.CLINBIOCHEM.2015.04.017
43. Pedre B, Barayeu U, Ezeriņa D, Dick TP. The mechanism of action of n-acetylcysteine (NAC): The emerging role of H2S and sulfane sulfur species. Pharmacol Ther (2021) 228:107916. doi: 10.1016/J.PHARMTHERA.2021.107916
44. Ryu GR, Lee E, Chun H-J, Yoon K-H, Ko S-H, Ahn Y-B, et al. Oxidative stress plays a role in high glucose-induced activation of pancreatic stellate cells. Biochem Biophys Res Commun (2013) 439:258–63. doi: 10.1016/j.bbrc.2013.08.046
45. Kim JW, Ko SH, Cho JH, Sun C, Hong OK, Lee SH, et al. Loss of beta-cells with fibrotic islet destruction in type 2 diabetes mellitus. Front Biosci (2008) 13:6022–33. doi: 10.2741/3133
46. Jesnowski R, Fürst D, Ringel J, Chen Y, Schrödel A, Kleeff J, et al. Immortalization of pancreatic stellate cells as an in vitro model of pancreatic fibrosis: deactivation is induced by matrigel and n-acetylcysteine. Lab Invest (2005) 85:1276–91. doi: 10.1038/labinvest.3700329
47. Feng H, Moriyama T, Ohuchida K, Sheng N, Iwamoto C, Shindo K, et al. N-acetyl cysteine induces quiescent-like pancreatic stellate cells from an active state and attenuates cancer-stroma interactions. J Exp Clin Cancer Res (2021) 40:133. doi: 10.1186/S13046-021-01939-1
Keywords: N-acetyl-L-cysteine (NAC), HFD-induced diabetes, beta-cell oxidative stress, pancreatic stellate cells (PaSCs), collagen fiber
Citation: Schuurman M, Wallace M, Sahi G, Barillaro M, Zhang S, Rahman M, Sawyez C, Borradaile N and Wang R (2022) N-acetyl-L-cysteine treatment reduces beta-cell oxidative stress and pancreatic stellate cell activity in a high fat diet-induced diabetic mouse model. Front. Endocrinol. 13:938680. doi: 10.3389/fendo.2022.938680
Received: 07 May 2022; Accepted: 08 August 2022;
Published: 25 August 2022.
Edited by:
Alexandre Gabarra Oliveira, São Paulo State University, BrazilCopyright © 2022 Schuurman, Wallace, Sahi, Barillaro, Zhang, Rahman, Sawyez, Borradaile and Wang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Rennian Wang, cndhbmdAdXdvLmNh