 Georgia Pitsava
Georgia Pitsava Constantine A. Stratakis
Constantine A. Stratakis
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Endocrinol., 03 August 2022
Sec. Pediatric Endocrinology
Volume 13 - 2022 | https://doi.org/10.3389/fendo.2022.937793
This article is part of the Research TopicPediatric Adrenal NeoplasmsView all 12 articles
Pediatric adrenocortical hyperplasias are rare; they usually present with Cushing syndrome (CS); of them, isolated micronodular adrenal disease and its variant, primary pigmented adrenocortical disease are the most commonly encountered. Most cases are due to defects in the cyclic AMP/protein kinase A (cAMP/PKA) pathway, although a few cases remain without an identified genetic defect. Another cause of adrenal hyperplasia in childhood is congenital adrenal hyperplasia, a group of autosomal recessive disorders that affect steroidogenic enzymes in the adrenal cortex. Clinical presentation varies and depends on the extent of the underlying enzymatic defect. The most common form is due to 21-hydroxylase deficiency; it accounts for more than 90% of the cases. In this article, we discuss the genetic etiology of adrenal hyperplasias in childhood.
Adrenocortical tumors (ACTs) are rare in childhood and adolescence (1). The majority of them (approximately 90%) are functional and thus they can present with hirsutism, acne, rapid clitoral or penile enlargement, sexual hair growth as well as increased height velocity (2). Usually, single tumors are benign unilateral adenomas but they can be malignant in rare cases and physicians should be suspicious of that as an early diagnosis impacts overall survival. Less often, patients can present with benign multinodular hyperplastic lesions including primary pigmented nodular adrenocortical disease (PPNAD), non-pigmented isolated micronodular adrenal disease (iMAD), and corticotropin (ACTH)-independent macronodular adrenal hyperplasia (AIMH) (3). These diseases usually present with cortisol excess or Cushing syndrome (CS). On the other hand, deficits of the steroidogenic enzymes of the adrenal cortex, also cause adrenal hyperplasia but they are usually associated with decreased cortisol synthesis. These diseases are known collectively as “congenital adrenal hyperplasia” or CAH, are all autosomal recessive and have a variable clinical presentation. We review in this report both types of adrenal hyperplasias and any updates on their molecular genetics.
The human adrenal gland arises from two different embryological tissues: the cortex develops from the intermediate mesoderm of the urogenital ridge, while the medulla develops from the neural crest-derived chromaffin cells (4). The earliest recognizable form of the adrenal gland is called ‘adrenal primordium’ and appears at 28-30 days post conception (dpc); it is marked by the expression of steroidogenic factor-1 (SF1; also known as Ad4BP or NR5A1) which is essential for the adrenal development and steroidogenesis (5, 6). By the 7th-8th week of fetal development, the developing adrenal cortex delineates two distinct components, the inner fetal zone (FZ) -that comprises 80-90% of the cortical volume- and the outer definitive zone (DZ) (5, 7). By the 9th week of gestation, the developing adrenal gland becomes completely encapsulated (8). The FZ cells express cytochrome P450 17α (CYP17A1), an enzyme that has both 17 hydroxylase and 17,20 lyase activity and converts pregnenolone to dehydroepiandrosterone (DHEA). At this stage, DHEA is the main product produced at ZF; later, it undergoes conversion to estrogens by the placenta for the maintenance of normal pregnancy.
By the end of the second trimester, the transitional zone (between FZ and DZ) expresses HSD3B2, and thus cortisol synthesis is initiated in the fetus (9). By late gestation, the DZ has begun differentiating into the zona glomerulosa (ZG) and zona fasciculata (ZF). Soon after birth, the FZ undergoes regression due to increased apoptotic activity, resulting in decrease in the weight of the adrenal glands by approximately 50% (2, 10). Zona reticularis (ZR) begins to form later in childhood; around the age of 6-8 years in females and 7-9 years in males. This process is known as adrenarche and is characterized by the production of adrenal androgens (11). Medulla is not recognized as a distinct structure with the exception of small clusters of chromaffin cells scattered throughout the cortex, until after birth (12).
Glucocorticoid secretion is regulated by the HPA axis. The most important components of the HPA axis include: 1) the corticotropin-releasing hormone (CRH)- whose actions are mediated by two receptors, CRHR1 and CRHR2- which is released, along with vasopressin, from the paraventricular nucleus of the hypothalamus and it is carried to the anterior pituitary where it stimulates 2) adrenocorticotropic hormone (ACTH) secreted from the anterior lobe of the pituitary gland, and 3) cortisol that is produced by the adrenal cortex in response to the binding of ACTH to the melanocortin type-2 receptor (MC2-R) in the ZF. The HPA axis is subject to the negative feedback cycle from the glucocorticoids; circulating cortisol inhibits primarily CRH and secondarily ACTH (13).
RAAS is a vital regulator of systemic vascular resistance and blood pressure, by increasing vascular tone, water and sodium reabsorption. It has three main components that include renin, angiotensin II and aldosterone. Renin, a protease synthesized in the juxtaglomerular cells of the kidney, cleaves angiotensinogen -synthesized in the liver- to the biologically inactive angiotensin I (AngI). Following that, AngI is converted to AngII by the angiotensin converting enzyme (ACE), which is found primarily in the vascular endothelium of the lungs and kidneys. AngII acts by activating AngII type 1 (AT1) receptors in the adrenal cortex, kidneys and arterioles, resulting in aldosterone release, vasoconstriction, and blood pressure increase. Release of AngII -the major stimuli of RAAS activation- is triggered by intravascular volume depletion and hyperkalemia.
The ZR in the adrenal glands produces the precursor steroids dehydroepiandrosterone (DHEA) and DHEA sulfate (DHEAS) which are later converted, in the peripheral tissues, to potent androgens and estrogens (14, 15). Their secretion is partially regulated by ACTH; however, in multiple instances, the exact mechanism has not been elucidated yet.
Benign adrenocortical tumors producing cortisol.
The vast majority of cortisol-producing adrenal hyperplasias and tumors harbor defects in the cAMP/PKA signaling pathway (16). Under normal circumstances, in the adrenocortical cells, ACTH binds to MC2R (a G protein-coupled receptor) that causes an increase of the cAMP levels and thus activation of the PKA. PKA phosphorylates various transcription factors including cAMP response element binding protein (CREB), steroidogenic factor 1 (SF-1) and activating transcription factor 1 (ATF-1). Following that, these transcription factors bind to the cAMP response element (CRE) in the nucleus and regulate the expression of genes important in steroidogenesis (17–20).
This group of disorders can be categorized into two groups based on the size of the associated nodules: macronodular hyperplasias (BMAH, nodule size ≥1cm), which more commonly affect older adults and micronodular hyperplasias (MiBAH, nodule size <1cm) that occur more frequently in children and young adults. The latter includes primary pigmented adrenocortical nodular disease (PPNAD) and isolated micronodular adrenal disease (iMAD) (21). Two additional characteristics used for the classification of BAHs include the pigment that is present and whether there is atrophy or hyperplasia of the surrounding cortex tissue (22). The pigment most of the times is lipofuscin and macroscopically is seen as light to dark brown or, rarely, black (22).
PPNAD in children and adolescents has been linked to periodic or atypical CS (23–25). In PPNAD the adrenal glands appear to have multiple cortisol-secreting pigmented nodules usually <4-6mm in diameter. PPNAD can present as sporadic or familial; most commonly it presents as familial PPNAD as part of Carney complex (CNC, OMIM#160980). CNC, that was first described in 1985 (26), is a tumor predisposition syndrome that presents with multiple endocrine tumors in addition to lentigines and myxomas (21, 27). In 2000, germline inactivating variants in the PRKAR1A gene which encodes for the regulatory type 1α-subunit of PKA, mapped on the 17q22-24 chromosomal region, was identified as the causative gene in the majority of CNC cases (28–30). These variants lead to increased activity of the PKA. More genetic alterations in the cAMP/PKA signaling pathway have been identified over the last two decades in patients with MiBAH (31–33).
Cushing syndrome (CS) can be exogenous (or ‘iatrogenic’) when glucocorticoids are used in high doses for a prolonged period of time or endogenous due to overproduction of cortisol either by an ACTH-producing pituitary tumor (also known as Cushing disease -CD-) or by an ACTH-independent ACT. Endogenous CS has an incidence of 0.7-2.4 per million people per year (34); in children BAHs as etiologies of CS are much more common than in adults (23).
In most cases, CS onset is slow and gradual (35). The hallmark of CS in childhood is increasing weight with growth velocity deceleration. Other common presenting symptoms include headaches, facial plethora, hypertension, amenorrhea, hypogonadism and hirsutism (Table 1) (35–39). Virilization is not often present except for cases that the tumor produces androgens. Additional manifestations from skin such as abdominal striae, acne and bruising may also occur often.

Table 1 Phenotypic manifestations of pediatric Cushing syndrome.
Diagnostic evaluation should be prompted in patients with high suspicion (Figure 1) (41, 42). Medical history and clinical evaluation, including growth charts in children, are vital in making the initial diagnosis. According to the Endocrine Society guidelines on the diagnostic workup of CS, after excluding exogenous glucocorticoid exposure, cortisol levels should be measured (43). Increased cortisol levels are confirmed with 24-hour urinary free cortisol (UFC) test, late-night salivary cortisol and/or a low-dose dexamethasone suppression test (1mg overnight or 2mg/day over 48 hours). The diagnostic accuracy is not 100% in any those tests and thus, usually multiple tests may be needed in order to establish the diagnosis. In some cases, falsely high UFC (known as pseudo-CS) may be obtained due to severe obesity, depression, pregnancy, alcoholism, chronic exercise, anorexia, anxiety, malnutrition or excessive water intake (>5L/day) (22). Once the presence of endogenous CS has been confirmed, ACTH is used to differentiate between ACTH-dependent and ACTH-independent CS. If ACTH levels are <5pg/mL, then ACTH-independent disease is suggested, while ACTH levels ≥29pg/ml have approximately 70% sensitivity in identifying ACTH-dependent disease in children (35, 43). Following that, high dose dexamethasone suppression test (Liddle test) is used to differentiate between CS due to adrenal causes or CD from ectopic ACTH secretion (44).
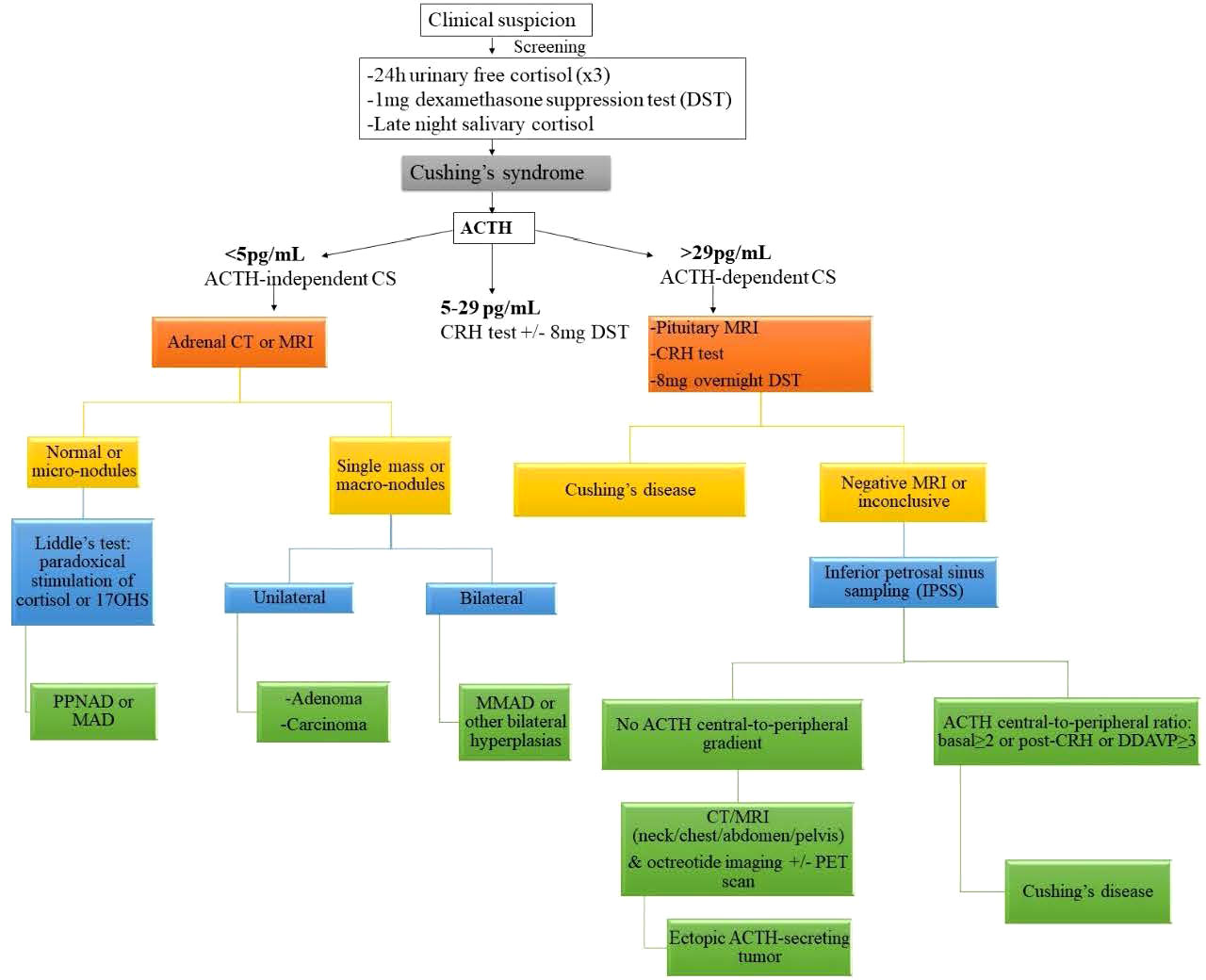
Figure 1 Diagnostic algorithm for suspected Cushing syndrome. ACTH adrenocorticotropic hormone, CS Cushing syndrome, CT computed tomography, DDAVP desmopressin, MAD micronodular adrenal disease, MRI magnetic resonance imaging, PET positron emission tomography, 17OHS 17-hydroxysteroid. Reprinted from Constantine A. Stratakis. Cushing Syndrome in Pediatrics. Endocrinology and Metabolism Clinics of North America. Volume 41, Issue 4, December 2012, Pages 793-803, with permission from Elsevier.
When bilateral adrenocortical hyperplasia is suspected then low-dose dexamethasone test (30ug/kg/dose every 6 hours for 8 doses) followed by high-dose dexamethasone test (120 ug/kg/dose every 6 hours for 8 doses) is performed. However, the diagnosis of CS due to PPNAD can be challenging as often has a cyclical or atypical presentation (e.g. normal or near normal 24-hour UFC). What is very useful though is that these patients appear to have a counterintuitive increase in the production of cortisol in response to high-dose dexamethasone through a PKA-mediated mechanism; this results from the overexpression of glucocorticoids receptors on the adrenocortical cells (45). For distinction from pituitary tumors causing CS, diagnostic imaging may be required, including pituitary magnetic resonance imaging (MRI) (46).
iMAD, like PPNAD, can cause CS usually of mild to moderate severity and it can be cyclical (24, 47). It usually occurs earlier than PPNAD and presents during infancy and childhood (2).
Underlying genetic defects include inactivating variants in phosphodiesterases 11A (PDE11A) and 8B (PDE8) (48–50). PDE11A is located on chromosome 2q31.2, encoding a phosphodiesterase that degrades both cAMP and cGMP. It is expressed in several endocrine tissues, including the adrenal cortex (51). Thus far, five inactivating variants in PDE11A have been identified, three in patients with iMAD and two in patients with PPNAD (31, 32). Out of these variants, three lead to introduction of premature termination codons, whereas the other two disrupt the catalytic domain of PDE11A. Thus, all variants resulted in compromised cAMP degradation, supporting the notion that aberrantly hyperactive cAMP -dependent signaling has a causative role in the development of adrenocortical tumors. Notably, patients harboring such inactivating variants also had decreased PDE11A4 expression (52).
Out of the cAMP-specific PDEs, the one with the highest expression in the adrenal gland is PDE8B (53), and missense variants in PDE8B have been associated with adrenocortical tumors; the mutant forms of PDE8B have compromised ability to degrade cAMP, resulting in abnormally elevated PKA activity. Specifically, a study of 84 patients with adrenal tumors but no identified variants in PRKAR1A, PDE11A or GNAS, discovered a p.H391A substitution in PDE8B (54). In addition, a p.H305P substitution was found in a two year old girl with CS and iMAD (32). The variant was transmitted to the patient from her father. However, the father’s phenotype was characterized as normal/very mild, echoing findings in female patients who inherited PDE11A variants from unaffected fathers.
AIMH/BMAH presenting with CS is more common in older adults and are usually sporadic (47); however, a few familial cases that have been reported appear to be in children (22). It develops progressively over the years with subclinical hypercortisolism (47, 55). In addition, BMAH can rarely occur as part of McCune-Albright syndrome (MAS) where it appears in the infantile period (<6 months old); in a few cases CS spontaneously resolved (24, 56). MAS is due to somatic activating variants in the GNAS gene, that encodes the α-subunit of the Gs protein, causing constant non-ACTH-dependent adrenal cortex stimulation (57).
CAHs is a group of disorders of defective steroidogenesis inherited in an autosomal recessive manner (58). Inactivating variants in genes encoding enzymes that participate in the cortisol synthesis pathway result in a broad spectrum of disease severity; impaired synthesis of cortisol leads to persistently high levels of ACTH via negative feedback, overstimulation of the adrenal cortex and eventually adrenal hyperplasia and oversecretion of the precursors of the enzymatic defect. When the enzymatic defects are complete or almost complete overt enzymatic insufficiency ensues and those defects are known as ‘classic’ CAH. The ‘non classic’ CAH (NCCAH; also termed ‘late onset’) tends to be milder as the enzymatic defects are partial and patients retain some cortisol and aldosterone production. In most cases, patients with NCCAH harbor either two non-classic alleles or one non-classic and one classic, meaning that when two individuals with NCCAH have kids their risk of having a child with classic CAH is approximately 1.5-2.5% (59, 60).
The prevalence of the classic forms is 1:16,000 while for the non-classic forms is approximately 1:2,000 in the Caucasian population in the United States with a higher frequency in Hispanics, Eskimos, people of Mediterranean or Middle-Eastern descent and Ashkenazi Jews (61–65).
21OHD (OMIM#201910, CYP21A2) accounts for approximately 95% of CAH cases (61, 66, 67). The enzyme 21OH catalyzes the conversion of 17-hydroxyprogesterone to 11-deoxycortisol in the ZF and progesterone to 11-deoxycorticosterone (DOC) in ZG. CYP21A2 is located on chromosome 6p21.3, within the human leukocyte antigen (HLA) locus. About 30kb apart, the same locus harbors a non-functional highly homologous pseudogene (CYP21A1P). Because of that, recombination frequently occurs in the region; these recombination events are responsible for most of the CYP21A2 inactivating variants in patients.
21OHD presents with a wide spectrum of phenotypes. Increased ACTH secretion due to inadequate production of cortisol leads to accumulation of 17-hydroxyprogesterone and progesterone. As a result, female infants with classic 21OH deficiency are exposed to excess androgens in utero; at birth, they present with virilization of the genitalia, including rugated or partially fused labia majora, genital hyperpigmentation, enlarged clitoris and a vagina that opens into a common urogenital sinus -like in males- while the ovaries, fallopian tubes and the uterus are normal (61). Males have minimal findings, including genital hyperpigmentation and macrogenitosomia or no physical findings of the disease (61, 68).
Linear growth is also affected as patients are at risk for centrally mediated precocious puberty due to prolonged exposure to high levels of androgens. Data from 18 centers demonstrated that their adult height is on average 1.4SD below the mean compared to the general population (69). Early diagnosis and treatment compliance are vital as both overtreatment and undertreatment increase the risk for short stature, either due to excess glucocorticoid-induced inhibition of the growth axis or due to premature epiphyseal closure (70, 71).
Approximately 75% of classic CAHs represent life-threatening salt-wasting forms and they are a result of large gene conversions, nonsense or frameshift mutations or complete deletions leading to complete absence of 21OH activity. Production of mineralocorticoids and glucocorticoids is ceased resulting in life-threatening adrenal crises in the first two weeks of life (72). When CAH is suspected, then aldosterone, serum electrolytes and plasma renin should be measured. The expected abnormalities are low aldosterone levels, hyperkalemia and elevated renin; however, because renin levels are age-specific appropriate references should be used (73). On the other hand, the NCCAH forms are more often associated with missense variants and they retain some enzymatic activity and thus are able to maintain a relatively normal amount of aldosterone and cortisol at the expense of mild to moderate excess of sex hormone precursors salt balance (72); they present with premature pubarche during childhood; however cases have been described as early as six months old (74). Females most commonly present during adolescence with hirsutism, acne and menstrual abnormalities or infertility, findings similar to polycystic ovarian syndrome (PCOS) while males often remain undiagnosed until they undergo pre-conception genetic screening or after having an affected offspring (74).
Some of the deletions responsible for CAH also lead to Ehlers-Danlos syndrome (hypermobility type). This occurs when the deletion encompasses both CYP21A2 and TNXB (which encodes tenascin-X; a protein of the extracellular matrix). In such cases, the resulting contiguous gene deletion syndrome is called CAH-X syndrome. Overall, CAH-X is estimated to have a prevalence of 9% among CAH patients.
More specifically, there exist 3 subtypes of CAH-X: a) CAH-X CH-1, associated with deletion of exon 35 of TNXB; b) CAH-X CH-2, caused by the c.12174 C>G (p.Cys4058Trp) variant in TNXB; and c) CAH-X CH-3, which is caused by a cluster of 3 variants. The mechanism of two latter subtypes involves dominant negative effects.
17α-hydroxylase (CYP17A1) catalyzes two reactions, the 17-hydroxylase and the 17,20-lyase reaction. Pathogenic variants in CYP17A1 (including splice site alterations, point mutations, small insertions/deletions, and in rare cases, large deletions) result in impairment of both reactions.
Complete 17OHD (OMIM#202110) leads to disrupted steroidogenesis in both the adrenals and the gonads, as it is expressed in both, leading to puberty failure. In addition, both 46,XX and 46,XY will present with female external genitalia due to absent testosterone and dihydrotestosterone; however, 46,XY individuals, due to preservation of anti-Mullerian hormone from the testes do not have an internal Mullerian structure (75). In this form of CAH, in contrast with the others, there is some glucocorticoid production from corticosterone and thus adrenal crisis is very rare. Over the years, accumulation of DOC causes hypokalemia and hypertension. In general, the typical presentation of 17OHD is a girl in adolescent with no secondary sexual characteristics and low-renin hypertension (76, 77). Non-classic forms of 17OHDy have not been well described (75).
About 0.2-8% of all CAH cases are due to 11βOHD (OMIM#202010) (72). 11βOH encoded by CYP11B1, catalyzes the conversion of 11-deoxycortisol and 11-deoxycorticosterone (DOC) to cortisol and corticosterone, respectively. Impaired cortisol and corticosterone production leads to overproduction of the precursors that are later converted to androgens causing virilization in females (78). In addition, patients present with mild to moderate hypertension. Although deoxycorticosterone is not as a potent mineralocorticoid as aldosterone, when it accumulates it leads to retention of salt and hypokalemic hypertension (79).
This form of CAH is very rare accounting for <0.5% of the cases (OMIM#201810) (80). 3β-hydroxysteroid dehydrogenase exists in two isoforms; 3βHSD1, encoded by HSD3B1 (the homologous type I gene) and is expressed in the placenta and peripheral tissue (liver, skin, brain) and 3βHSD2, encoded by HSD3B2 and is found in the adrenals and gonads (72). Inactivating variants in HSD3B2 impair steroidogenesis in both gonads and adrenals, leading to deficiency of glucocorticoids and mineralocorticoids as well as overproduction of dehydroepiandrosterone (DHEA). Subsequently, DHEA is converted to testosterone by 3βHSD1 and thus both 46,XX and 46,XY present with ambiguous genitalia at birth (81). In rare cases, 46,XX can present with virilization, including incomplete fusion of the labia, enlarged clitoris and hyperpigmentation of the genital region due to the increased testosterone (72).
StAR is a mitochondrial protein that regulates cholesterol influx between the outer and inner mitochondrial membrane, a critical and first step of steroid hormone synthesis (82). Inactivating variants in STAR gene cause Lipoid Congenital Adrenal Hyperplasia (LCAH, the most severe form of CAH, OMIM#201710) (83). The adrenals are filled with lipid globules derived from cholesterol and thus the name of the disease. Patients with LCAH have impaired production of mineralocorticoids, glucocorticoids and sex steroids. LCAH appears to be more common in the Korean, Japanese and Palestinian Arab populations (83–92), while certain variants have much higher frequency in specific ethnic groups, including p.Q258X variant in Japanese and Koreans (86, 89) as well as p.R182L variant in Palestinian Arabs (83). Both 46,XX and 46,XY present with female or ambiguous genitalia and adrenal crises during the neonatal period. Hyperpigmentation is also frequent due to the increased levels of ACTH. However, milder forms of the disease have been described in the literature that presented with late-onset adrenal insufficiency and male genitalia (93).
21OHD can be diagnosed via the measurement of 17-hydroxyprogesterone; this measurement does not need necessarily to be performed after ACTH stimulation. Hormonal testing is also used to diagnose the rarer forms of CAH. Because carriers cannot be reliably identified using biochemical methods, genetic testing is necessary to detect heterozygous individuals, who can then be offered genetic counseling. Specifically for the salt-wasting form of CAH, the United States and more than 35 countries around the world have implemented newborn screening programs for early detection and treatment (94).
Adrenocortical hyperplasias are rare in children. Advances over the past two decades have led to a better understanding of their molecular background, including the characterization of the cAMP/PKA signaling pathway as the main component of their pathogenesis. CAH are a group of rare diseases with genetic and clinical heterogeneity and can be life-threatening in their most severe form. The molecular analysis of CAH is useful in confirming the diagnosis and provides a powerful tool in genetic counseling.
GP: Writing-original draft and editing; CAS.: Conseptualization, supervision, writing, review and editing.
This study was supported by the Intramural Research Program, Eunice Kennedy Shriver National Institute of Child Health & Human Development (NICHD), Bethesda, MD, United States. This work was in part supported by the research project Z01-HD008920 (Principal Investigator: Constantine A Stratakis) of the Intramural Research Program of the Eunice Kennedy Shriver National Institute of Child Health & Human Development (NICHD), National Institutes of Health (NIH), Bethesda, MD, USA. Stratakis is currently also funded by IMBB, FORTH intramural funds.
Author CS holds patents on the PRKAR1A, PDE11A and GPR101 genes and/or their function and has received research funding from Pfizer Inc. on the genetics and treatment of abnormalities of growth hormone secretion. CAS is receiving compensation by ELPEN, Inc. Neither Pfizer, Inc nor ELPEN, Inc had any role in the study design, data collection and analysis, decision to publish, or preparation of the manuscript.
The remaining author declares that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Sutter JA, Grimberg A. Adrenocortical tumors and hyperplasias in childhood–etiology, genetics, clinical presentation and therapy. Pediatr Endocrinol Rev (2006) 4(1):32–9.
2. Fuqua JS. Adrenal tumors in childhood. Adv Pediatr (2021) 68:227–44. doi: 10.1016/j.yapd.2021.05.015
3. Bourdeau I, Stratakis CA. Cyclic AMP-dependent signaling aberrations in macronodular adrenal disease. Ann N Y Acad Sci (2002) 968:240–55. doi: 10.1111/j.1749-6632.2002.tb04339.x
4. Xing Y, Lerario AM, Rainey W, Hammer GD. Development of adrenal cortex zonation. Endocrinol Metab Clin North Am (2015) 44(2):243–74. doi: 10.1016/j.ecl.2015.02.001
5. Hanley NA, Rainey WE, Wilson DI, Ball SG, Parker KL. Expression profiles of SF-1, DAX1, and CYP17 in the human fetal adrenal gland: potential interactions in gene regulation. Mol Endocrinol (2001) 15(1):57–68. doi: 10.1210/mend.15.1.0585
6. Hanley NA, Hagan DM, Clement-Jones M, Ball SG, Strachan T, Salas-Cortés L, et al. SRY, SOX9, and DAX1 expression patterns during human sex determination and gonadal development. Mech Dev (2000) 91(1-2):403–7. doi: 10.1016/S0925-4773(99)00307-X
7. Goto M, Hanley KP, Marcos J, Wood PJ, Wright S, Postle AD, et al. In humans, early cortisol biosynthesis provides a mechanism to safeguard female sexual development. J Clin Invest (2006) 116(4):953–60. doi: 10.1172/JCI25091
8. Keegan CE, Hammer GD. Recent insights into organogenesis of the adrenal cortex. Trends Endocrinol Metab (2002) 13(5):200–8. doi: 10.1016/S1043-2760(02)00602-1
9. Pinto EM, Zambetti GP, Rodriguez-Galindo C. Pediatric adrenocortical tumours. Best Pract Res Clin Endocrinol Metab (2020) 34(3):101448. doi: 10.1016/j.beem.2020.101448
10. Ishimoto H, Jaffe RB. Development and function of the human fetal adrenal cortex: a key component in the feto-placental unit. Endocr Rev (2011) 32(3):317–55. doi: 10.1210/er.2010-0001
11. Havelock JC, Auchus RJ, Rainey WE. The rise in adrenal androgen biosynthesis: adrenarche. Semin Reprod Med (2004) 22(4):337–47. doi: 10.1055/s-2004-861550
12. Yon L, Breault L, Contesse V, Bellancourt G, Delarue C, Fournier A, et al. Localization, characterization, and second messenger coupling of pituitary adenylate cyclase-activating polypeptide receptors in the fetal human adrenal gland during the second trimester of gestation. J Clin Endocrinol Metab (1998) 83(4):1299–305. doi: 10.1210/jcem.83.4.4690
13. Smith SM, Vale WW. The role of the hypothalamic-pituitary-adrenal axis in neuroendocrine responses to stress. Dialogues Clin Neurosci (2006) 8(4):383–95. doi: 10.31887/DCNS.2006.8.4/ssmith
14. Rainey WE, Carr BR, Sasano H, Suzuki T, Mason JI. Dissecting human adrenal androgen production. Trends Endocrinol Metab (2002) 13(6):234–9. doi: 10.1016/S1043-2760(02)00609-4
15. Labrie F, Luu-The V, Labrie C, Simard J. DHEA and its transformation into androgens and estrogens in peripheral target tissues: intracrinology. Front Neuroendocrinol (2001) 22(3):185–212. doi: 10.1006/frne.2001.0216
16. Zilbermint M, Stratakis CA. Protein kinase a defects and cortisol-producing adrenal tumors. Curr Opin Endocrinol Diabetes Obes (2015) 22(3):157–62. doi: 10.1097/MED.0000000000000149
17. Bossis I, Stratakis CA. Minireview: PRKAR1A: normal and abnormal functions. Endocrinology (2004) 145(12):5452–8. doi: 10.1210/en.2004-0900
18. Lacroix A, Ndiaye N, Tremblay J, Hamet P. Ectopic and abnormal hormone receptors in adrenal cushing's syndrome. Endocr Rev (2001) 22(1):75–110. doi: 10.1210/edrv.22.1.0420
19. Johannessen M, Delghandi MP, Moens U. What turns CREB on? Cell Signal (2004) 16(11):1211–27. doi: 10.1016/j.cellsig.2004.05.001
20. Rosenberg D, Groussion L, Jullian E, Perlemoine K, Bertagna X, Bertherat J, et al. Role of the PKA-regulated transcription factor CREB in development and tumorigenesis of endocrine tissues. Ann N Y Acad Sci (2002) 968:65–74. doi: 10.1111/j.1749-6632.2002.tb04327.x
21. Almeida MQ, Stratakis CA. Carney Complex and other conditions associated with micronodular adrenal hyperplasias. Best Pract Res Clin Endocrinol Metab (2010) 24(6):907–14. doi: 10.1016/j.beem.2010.10.006
22. Stratakis CA. Cushing syndrome caused by adrenocortical tumors and hyperplasias (corticotropin- independent cushing syndrome). Endocr Dev (2008) 13:117–32. doi: 10.1159/000134829
23. Lodish MB, Keil MF, Stratakis CA. Cushing's syndrome in pediatrics: An update. Endocrinol Metab Clin North Am (2018) 47(2):451–62. doi: 10.1016/j.ecl.2018.02.008
24. Gunther DF, Bourdeau I, Matyakhina L, Cassarino D, Kleiner DE, Griffin K, et al. Cyclical cushing syndrome presenting in infancy: an early form of primary pigmented nodular adrenocortical disease, or a new entity? J Clin Endocrinol Metab (2004) 89(7):3173–82. doi: 10.1210/jc.2003-032247
25. Sarlis NJ, Chrousos GP, Doppman JL, Carney JA, Stratakis CA. Primary pigmented nodular adrenocortical disease: reevaluation of a patient with carney complex 27 years after unilateral adrenalectomy. J Clin Endocrinol Metab (1997) 82(4):1274–8. doi: 10.1210/jcem.82.4.3857
26. Carney JA, Gordon H, Carpenter PC, Shenoy BV, Go VL. The complex of myxomas, spotty pigmentation, and endocrine overactivity. Med (Baltimore) (1985) 64(4):270–83. doi: 10.1097/00005792-198507000-00007
27. Pitsava G, Zhu C, Sundaram R, Mills JL, Stratakis CA. Predicting the risk of cardiac myxoma in Carney complex. Genet Med (2021) 23(1):80–5. doi: 10.1038/s41436-020-00956-3
28. Stratakis CA. Genetics of adrenocortical tumors: Carney complex. Ann Endocrinol (Paris) (2001) 62(2):180–4.
29. Kirschner LS, Carney JA, Pack SD, Taymans SE, Giatzakis C, Cho YS, et al. Mutations of the gene encoding the protein kinase a type I-alpha regulatory subunit in patients with the Carney complex. Nat Genet (2000) 26(1):89–92. doi: 10.1038/79238
30. Kirschner LS, Sandrini F, Monbo J, Lin JP, Carney JA, Stratakis CA. Genetic heterogeneity and spectrum of mutations of the PRKAR1A gene in patients with the carney complex. Hum Mol Genet (2000) 9(20):3037–46. doi: 10.1093/hmg/9.20.3037
31. Horvath A, Boikos S, Giatzakis C, Robinson-White A, Groussin L, Griffin KJ, et al. A genome-wide scan identifies mutations in the gene encoding phosphodiesterase 11A4 (PDE11A) in individuals with adrenocortical hyperplasia. Nat Genet (2006) 38(7):794–800. doi: 10.1038/ng1809
32. Horvath A, Mericq V, Stratakis CA. Mutation in PDE8B, a cyclic AMP-specific phosphodiesterase in adrenal hyperplasia. N Engl J Med (2008) 358(7):750–2. doi: 10.1056/NEJMc0706182
33. Beuschlein F, Fassnacht M, Assié G, Calebiro D, Stratakis CA, Osswald A, et al. Constitutive activation of PKA catalytic subunit in adrenal cushing's syndrome. N Engl J Med (2014) 370(11):1019–28. doi: 10.1056/NEJMoa1310359
34. Sharma ST, Nieman LK, Feelders RA. Cushing's syndrome: epidemiology and developments in disease management. Clin Epidemiol (2015) 7:281–93. doi: 10.2147/CLEP.S44336
35. Batista DL, Riar J, Keil M, Stratakis CA. Diagnostic tests for children who are referred for the investigation of cushing syndrome. Pediatrics (2007) 120(3):e575–86. doi: 10.1542/peds.2006-2402
36. Rahman SH, Papadakis GZ, Keil MF, Faucz FR, Lodish MB, Stratakis CA. Kidney stones as an underrecognized clinical sign in pediatric cushing disease. J Pediatr (2016) 170:273–7.e1. doi: 10.1016/j.jpeds.2015.11.045
37. Lodish MB, Hsiao H-P, Sermbis A, Sinaii N, Rothenbuhler A, Keil MF, et al. Effects of cushing disease on bone mineral density in a pediatric population. J Pediatr (2010) 156(6):1001–5. doi: 10.1016/j.jpeds.2009.12.027
38. Keil MF, Graf J, Gokarn N, Stratakis CA. Anthropometric measures and fasting insulin levels in children before and after cure of cushing syndrome. Clin Nutr (2012) 31(3):359–63. doi: 10.1016/j.clnu.2011.11.007
39. Afshari A, Ardeshirpour Y, Lodish MB, Gourgari E, Sinaii N, Keil M, et al. Facial plethora: Modern technology for quantifying an ancient clinical sign and its use in cushing syndrome. J Clin Endocrinol Metab (2015) 100(10):3928–33. doi: 10.1210/jc.2015-2497
40. Stratakis CA. Cushing syndrome in pediatrics. Endo Metab Clin North America (2012) 41(4):793–803.
41. Lebrethon MC, Grossman AB, Afshar F, Plowman PN, Besser GM, Savage MO. Linear growth and final height after treatment for cushing's disease in childhood. J Clin Endocrinol Metab (2000) 85(9):3262–5. doi: 10.1210/jcem.85.9.6817
42. Magiakou MA, Mastorakos G, Chrousos GP. Final stature in patients with endogenous cushing's syndrome. J Clin Endocrinol Metab (1994) 79(4):1082–5. doi: 10.1210/jcem.79.4.7962277
43. Nieman LK, Biller BMK, Findling JW, Newell-Price J, Savage MO, Stewart PM, et al. The diagnosis of cushing's syndrome: an endocrine society clinical practice guideline. J Clin Endocrinol Metab (2008) 93(5):1526–40. doi: 10.1210/jc.2008-0125
44. Bruno OD, Rossi MA, Contreras LN, Gómez RM, Galparsoro G, Cazado E, et al. Nocturnal high-dose dexamethasone suppression test in the aetiological diagnosis of cushing's syndrome. Acta Endocrinol (Copenh) (1985) 109(2):158–62. doi: 10.1530/acta.0.1090158
45. Louiset E, Stratakis CA, Perraudin V, Griffin KJ, Libé́ R, Cabrol S, et al. The paradoxical increase in cortisol secretion induced by dexamethasone in primary pigmented nodular adrenocortical disease involves a glucocorticoid receptor-mediated effect of dexamethasone on protein kinase a catalytic subunits. J Clin Endocrinol Metab (2009) 94(7):2406–13. doi: 10.1210/jc.2009-0031
46. Oldfield EH MD, Doppman JL MD, Nieman LK MD, Chrousos GP MD, Miller DL MD. Petrosal sinus sampling with and without corticotropin-releasing hormone for the differential diagnosis of cushing's syndrome. N Engl J Med (1991) 325(13):897–905. doi: 10.1056/NEJM199109263251301
47. Bourdeau I, Parisien-La Salle S, Lacroix A. Adrenocortical hyperplasia: A multifaceted disease. Best Pract Res Clin Endocrinol Metab (2020) 34(3):101386. doi: 10.1016/j.beem.2020.101386
48. Vezzosi D, Bertherat J. Phosphodiesterases in endocrine physiology and disease. Eur J Endocrinol (2011) 165(2):177–88. doi: 10.1530/EJE-10-1123
49. Francis SH, Blount MA, Corbin JD. Mammalian cyclic nucleotide phosphodiesterases: molecular mechanisms and physiological functions. Physiol Rev (2011) 91(2):651–90. doi: 10.1152/physrev.00030.2010
50. Levy I, Horvath A, Azevedo M, Bertollo de Alexandre R, Stratakis CA. Phosphodiesterase function and endocrine cells: links to human disease and roles in tumor development and treatment. Curr Opin Pharmacol (2011) 11(6):689–97. doi: 10.1016/j.coph.2011.10.003
51. Boikos SA, Horvath A, Heyerdahl S, Stein E, Robinson-White A, Bossis I, et al. Phosphodiesterase 11A expression in the adrenal cortex, primary pigmented nodular adrenocortical disease, and other corticotropin-independent lesions. Horm Metab Res (2008) 40(5):347–53. doi: 10.1055/s-2008-1076694
52. Horvath A, Giatzakis C, Robinson-White A, Boikos S, Levine E, Griffin K, et al. Adrenal hyperplasia and adenomas are associated with inhibition of phosphodiesterase 11A in carriers of PDE11A sequence variants that are frequent in the population. Cancer Res (2006) 66(24):11571–5. doi: 10.1158/0008-5472.CAN-06-2914
53. Tirosh A, Valdes N, Stratakis CA. Genetics of micronodular adrenal hyperplasia and Carney complex. Presse Med (2018) 47(7-8 Pt 2):e127–37. doi: 10.1016/j.lpm.2018.07.005
54. Rothenbuhler A, Horvath A, Libé R, Faucz FR, Fratticci A, Raffin Sanson ML, et al. Identification of novel genetic variants in phosphodiesterase 8B (PDE8B), a cAMP-specific phosphodiesterase highly expressed in the adrenal cortex, in a cohort of patients with adrenal tumours. Clin Endocrinol (Oxf) (2012) 77(2):195–9. doi: 10.1111/j.1365-2265.2012.04366.x
55. Carney JA, Young WF, Stratakis CA. Primary bimorphic adrenocortical disease: cause of hypercortisolism in McCune-albright syndrome. Am J Surg Pathol (2011) 35(9):1311–26. doi: 10.1097/PAS.0b013e31821ec4ce
56. Stratakis CA, Boikos SA. Genetics of adrenal tumors associated with cushing's syndrome: a new classification for bilateral adrenocortical hyperplasias. Nat Clin Pract Endocrinol Metab (2007) 3(11):748–57. doi: 10.1038/ncpendmet0648
57. Dumitrescu CE, Collins MT. McCune-albright syndrome. Orphanet J Rare Dis (2008) 3:12. doi: 10.1186/1750-1172-3-12
58. Merke DP, Auchus RJ. Congenital adrenal hyperplasia due to 21-hydroxylase deficiency. N Engl J Med (2020) 383(13):1248–61. doi: 10.1056/NEJMra1909786
59. Moran C, Azziz R, Weintrob N, Witchel SF, Rohmer V, Dewailly D, et al. Reproductive outcome of women with 21-hydroxylase-deficient nonclassic adrenal hyperplasia. J Clin Endocrinol Metab (2006) 91(9):3451–6. doi: 10.1210/jc.2006-0062
60. Bidet M, Bellanné́-Chantelot C, Galand-Portier M-B, Golmard J-L, Tardy V, Morel Y, et al. Fertility in women with nonclassical congenital adrenal hyperplasia due to 21-hydroxylase deficiency. J Clin Endocrinol Metab (2010) 95(3):1182–90. doi: 10.1210/jc.2009-1383
61. Speiser PW, White PC. Congenital adrenal hyperplasia. N Engl J Med (2003) 349(8):776–88. doi: 10.1056/NEJMra021561
62. Speiser PW, Arlt W, Auchus RJ, Baskin LS, Conway GS, Merke DP, et al. Congenital adrenal hyperplasia due to steroid 21-hydroxylase deficiency: An endocrine society clinical practice guideline. J Clin Endocrinol Metab (2018) 103(11):4043–88. doi: 10.1210/jc.2018-01865
63. Witchel SF, Azziz R. Nonclassic congenital adrenal hyperplasia. Int J Pediatr Endocrinol (2010) 2010:625105. doi: 10.1186/1687-9856-2010-625105
64. Speiser PW, Dupont B, Rubinstein P, Piazza A, Kastelan A, New MI, et al. High frequency of nonclassical steroid 21-hydroxylase deficiency. Am J Hum Genet (1985) 37(4):650–67.
65. Hannah-Shmouni F, Morissette R, Sinaii N, Elman M, Prezant TR, Chen W, et al. Revisiting the prevalence of nonclassic congenital adrenal hyperplasia in US ashkenazi jews and caucasians. Genet Med (2017) 19(11):1276–9. doi: 10.1038/gim.2017.46
66. Balsamo A, Baronio F, Ortolano R, Menabo S, Baldazzi L, Di Natale V, et al. Congenital adrenal hyperplasias presenting in the newborn and young infant. Front Pediatr (2020) 8:593315. doi: 10.3389/fped.2020.593315
67. Baranowski ES, Arlt W, Idkowiak J. Monogenic disorders of adrenal steroidogenesis. Horm Res Paediatr (2018) 89(5):292–310. doi: 10.1159/000488034
68. Jha S, Turcu AF. Nonclassic congenital adrenal hyperplasia: What do endocrinologists need to know? Endocrinol Metab Clin North Am (2021) 50(1):151–65. doi: 10.1016/j.ecl.2020.10.008
69. Eugster EA, DiMeglio LA, Wright JC, Freidenberg GR, Seshadri R, Pescovitz OH. Height outcome in congenital adrenal hyperplasia caused by 21-hydroxylase deficiency: a meta-analysis. J Pediatr (2001) 138(1):26–32. doi: 10.1067/mpd.2001.110527
70. Jaaskelainen J, Voutilainen R. Growth of patients with 21-hydroxylase deficiency: an analysis of the factors influencing adult height. Pediatr Res (1997) 41(1):30–3. doi: 10.1203/00006450-199701000-00005
71. Yu AC, Grant DB. Adult height in women with early-treated congenital adrenal hyperplasia (21-hydroxylase type): relation to body mass index in earlier childhood. Acta Paediatr (1995) 84(8):899–903. doi: 10.1111/j.1651-2227.1995.tb13789.x
72. Finkielstain GP, Vieites A, Bergadá I, Rey RA. Disorders of sex development of adrenal origin. Front Endocrinol (Lausanne) (2021) 12:770782. doi: 10.3389/fendo.2021.770782
73. Fukushige J, Shimomura K, Ueda K. Influence of upright activity on plasma renin activity and aldosterone concentration in children. Eur J Pediatr (1994) 153(4):284–6. doi: 10.1007/BF01954521
74. Carmina E, Dewailly D, Escobar-Morreale HF, Kelestimur F, Moran C, Oberfield S, Witchel SF, Azziz R. Non-classic congenital adrenal hyperplasia due to 21-hydroxylase deficiency revisited: an update with a special focus on adolescent and adult women. Hum Reprod Update (2017) 23(5):580–99. doi: 10.1093/humupd/dmx014
75. Auchus RJ. Steroid 17-hydroxylase and 17,20-lyase deficiencies, genetic and pharmacologic. J Steroid Biochem Mol Biol (2017) 165(Pt A):71–8. doi: 10.1016/j.jsbmb.2016.02.002
76. Biglieri EG, Kater CE. 17 alpha-hydroxylation deficiency. Endocrinol Metab Clin North Am (1991) 20(2):257–68. doi: 10.1016/S0889-8529(18)30267-6
77. Auchus RJ. The genetics, pathophysiology, and management of human deficiencies of P450c17. Endocrinol Metab Clin North Am (2001) 30(1):101–19. doi: 10.1016/S0889-8529(08)70021-5
78. Mooij CF, Parajes S, Rose IT, Taylor AE, Bayraktaroglu T, Wass JAH, et al. Characterization of the molecular genetic pathology in patients with 11beta-hydroxylase deficiency. Clin Endocrinol (Oxf) (2015) 83(5):629–35. doi: 10.1111/cen.12834
79. Miller WL. MECHANISMS IN ENDOCRINOLOGY: Rare defects in adrenal steroidogenesis. Eur J Endocrinol (2018) 179(3):R125–41. doi: 10.1530/EJE-18-0279
80. Al Alawi AM, Nordenstrom A, Falhammar H. Clinical perspectives in congenital adrenal hyperplasia due to 3beta-hydroxysteroid dehydrogenase type 2 deficiency. Endocrine (2019) 63(3):407–21. doi: 10.1007/s12020-018-01835-3
81. El-Maouche D, Hargreaves CJ, Sinaii N, Mallappa A, Veeraraghavan P, Merke DP. Longitudinal assessment of illnesses, stress dosing, and illness sequelae in patients with congenital adrenal hyperplasia. J Clin Endocrinol Metab (2018) 103(6):2336–45. doi: 10.1210/jc.2018-00208
82. Miller WL. Disorders in the initial steps of steroid hormone synthesis. J Steroid Biochem Mol Biol (2017) 165(Pt A):18–37. doi: 10.1016/j.jsbmb.2016.03.009
83. Bose HS, Sugawara T, Strauss JF, Miller WL. The pathophysiology and genetics of congenital lipoid adrenal hyperplasia. N Engl J Med (1996) 335(25):1870–8. doi: 10.1056/NEJM199612193352503
84. Chen X, Baker BY, Abduljabbar MA, Miller WL. A genetic isolate of congenital lipoid adrenal hyperplasia with atypical clinical findings. J Clin Endocrinol Metab (2005) 90(2):835–40. doi: 10.1210/jc.2004-1323
85. Fujieda K, Tajima T, Nakae J, Sageshima S, Tachibana K, Suwa S, et al. Spontaneous puberty in 46,XX subjects with congenital lipoid adrenal hyperplasia. ovarian steroidogenesis is spared to some extent despite inactivating mutations in the steroidogenic acute regulatory protein (StAR) gene. J Clin Invest (1997) 99(6):1265–71. doi: 10.1172/JCI119284
86. Yoo HW, Kim GH. Molecular and clinical characterization of Korean patients with congenital lipoid adrenal hyperplasia. J Pediatr Endocrinol Metab (1998) 11(6):707–11. doi: 10.1515/JPEM.1998.11.6.707
87. Achermann JC, Meeks JJ, Jeffs B, Das U, Clayton PE, Brook CGD, et al. Molecular and structural analysis of two novel StAR mutations in patients with lipoid congenital adrenal hyperplasia. Mol Genet Metab (2001) 73(4):354–7. doi: 10.1006/mgme.2001.3202
88. Bose HS, Sato S, Aisenberg J, Shalev SA, Matsuo N, Miller WL. Mutations in the steroidogenic acute regulatory protein (StAR) in six patients with congenital lipoid adrenal hyperplasia. J Clin Endocrinol Metab (2000) 85(10):3636–9. doi: 10.1210/jc.85.10.3636
89. Nakae J, Tajima T, Sugawara T, Arakane F, Hanaki K, Hotsubo T, et al. Analysis of the steroidogenic acute regulatory protein (StAR) gene in Japanese patients with congenital lipoid adrenal hyperplasia. Hum Mol Genet (1997) 6(4):571–6. doi: 10.1093/hmg/6.4.571
90. Flück CE, Maret A, Mallet D, Portrat-Doyen S, Achermann JC, Leheup B, et al. A novel mutation L260P of the steroidogenic acute regulatory protein gene in three unrelated patients of Swiss ancestry with congenital lipoid adrenal hyperplasia. J Clin Endocrinol Metab (2005) 90(9):5304–8. doi: 10.1210/jc.2005-0874
91. Lin D, Sugawara T, Strauss JF III, Clark BJ, Stocco DM, Saenger P, et al. Role of steroidogenic acute regulatory protein in adrenal and gonadal steroidogenesis. Science (1995) 267(5205):1828–31. doi: 10.1126/science.7892608
92. Miller WL. Congenital lipoid adrenal hyperplasia: the human gene knockout for the steroidogenic acute regulatory protein. J Mol Endocrinol (1997) 19(3):227–40. doi: 10.1677/jme.0.0190227
93. Kolli V, et al. Characterization of the CYP11A1 nonsynonymous variant p.E314K in children presenting with adrenal insufficiency. J Clin Endocrinol Metab (2019) 104(2):269–76. doi: 10.1210/jc.2018-01661
Keywords: adrenal cortex, Cushing syndrome, adrenal hyperplasia, congenital adrenal hyperplasia, childhood tumors
Citation: Pitsava G and Stratakis CA (2022) Adrenal hyperplasias in childhood: An update. Front. Endocrinol. 13:937793. doi: 10.3389/fendo.2022.937793
Received: 06 May 2022; Accepted: 08 July 2022;
Published: 03 August 2022.
Edited by:
Christina Pamporaki, University Hospital Carl Gustav Carus, GermanyReviewed by:
Angela Huebner, University Hospital Carl Gustav Carus, GermanyCopyright © 2022 Pitsava and Stratakis. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Georgia Pitsava, Zy5waXRzYXZhQGdtYWlsLmNvbQ==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.