 Yazhao Mei
Yazhao Mei Hao Zhang
Hao Zhang Zhenlin Zhang
Zhenlin Zhang- Shanghai Clinical Research Center of Bone Disease, Department of Osteoporosis and Bone Diseases, Shanghai Jiao Tong University Affiliated Sixth People’s Hospital, Shanghai, China
Purpose: Nearly 85%-90% of osteogenesis imperfecta (OI) cases are caused by autosome dominant mutations of COL1A1 and COL1A2 genes, of which de novo mutations cover a large proportion, whereas their characteristics remain to be elucidated. This study aims to compare the differences in clinical and genetic characteristics of de novo and inherited COL1A1/COL1A2 mutations of OI, assess the average paternal and maternal age at conception in de novo mutations, and research the rate of nonpenetrance in inherited mutations.
Materials and Methods: A retrospective comparison between de novo and inherited mutations was performed among 135 OI probands with COL1A1/COL1A2 mutations. Mutational analyses of all probands and their family members were completed by Sanger sequencing. A new clinical scoring system was developed to assess the clinical severity of OI quantitatively.
Results: A total of 51 probands (37.78%) with de novo mutations and 84 probands (62.22%) with inherited mutations were grouped by the results of the parental gene verification. The proportion of clinical type III (P<0.001) and clinical scores (P<0.001) were significantly higher in de novo mutations. Missense mutations covered a slightly higher proportion of de novo COL1A1 mutations (46.34%) compared with inherited COL1A1 mutations (33.33%), however, lacking a significant difference (P=0.1923). The mean BMD Z/T-score at the lumbar spine in de novo mutations was -2.3 ± 1.5, lower than inherited mutations (-1.7 ± 1.8), but lacking statistical significance (P=0.0742). There was no significant difference between the two groups in OI-related phenotypes (like fracture frequency, blue sclera, and hearing loss) and biochemical indexes. In de novo mutations, the average paternal and maternal age at conception was 29.2 (P<0.05) and 26.8 (P<0.0001), respectively, which were significantly younger than the average gestational age of the population. Additionally, 98.04% of pedigrees (50/51) with de novo mutations were spontaneous conception. The rate of nonpenetrance of parents with pathogenic variants in the inherited mutation group was 25.64% (20/78).
Conclusions: Our data revealed that the proportion of clinical type III and clinical scores were significantly higher in de novo mutations than in inherited mutations, demonstrating that de novo mutations are more damaging because they have not undergone purifying selection.
Introduction
Osteogenesis imperfecta (OI) is a generalized connective tissue disorder, apart from liability to slight trauma fractures, the phenotypes usually extend to non-skeletal symptoms, like blue sclera, dentinogenesis imperfecta, hearing loss, and ligamentous laxity (1). Based on the clinical and radiographic manifestations, Sillence et al. (2) in 1979 proposed the classic classification of OI: type I-IV: type I is the mildest kind characterized by nondeforming with persistently blue sclerae; type II is the perinatal lethal form; type III is the severest among surviving patients and presents with progressively deforming; type IV is characterized by white sclerae, whose severity is between type I and type III.
Although an increasing number of genes have been identified in OI involving different modes of inheritance (3), about 85%-90% of OI cases are caused by autosome dominant mutations of COL1A1 and COL1A2 genes. They encode the α1(I) and α2(I) chains of type I collagen, which is the main component of the extracellular matrix of bone and skin and is responsible for their elastic properties (4). Mutations in COL1A1 and COL1A2 of collagen-related OI can be divided into two groups: quantitative defect (haploinsufficiency) with synthesizing about half the amount of structurally normal type I collagen, primarily resulting from nonsense, frameshift, and some splice-site variants; another is the structural defect, in which over 80% mutations are the results of glycine substitutions in the helical domain (4). The majority of the type II-IV are the consequences of a structural defect because it has more severe impacts on the extracellular matrix than haploinsufficiency does.
Parental genetic characteristics will be passed on to the next generation. At the same time, de novo mutations, a kind of novel genetic change in a family, will occur during the formation of the gametes or postzygotic in each of us (5, 6). Only mutations present in the germ cells can be passed on to the next generation (7). Distinct from inherited variants, de novo mutations haven’t undergone purifying selection during the reproductive phase, thus making them more harmful (8). Approximately 80% of all de novo germline point mutations occur on the paternal allele because oocytes experience only one additional round of DNA replication during their evolution into a mature ovum, whereas spermatogonia will encounter hundreds of DNA replication and cell fission before becoming sperm cells. What’s more, it has been demonstrated that most types of de novo mutations are much more correlated with increased paternal age at birth (9–11). Rare sporadic diseases in the population are associated with de novo mutations (12). In OI, nearly half of the cases are derived from de novo mutations (13–15).
Previous studies have elucidated the genotype-phenotype correlation and mutation spectrum of collagen-related OI through large cohorts (15–18), which contributes to achieving a more accurate and economical diagnosis of OI. However, in addition to some reports of de novo mutation cases in OI (16, 19), few large cohort studies have compared de novo with inherited mutations in collagen-related OI (14). In this study, we enrolled 135 Chinese probands of OI with de novo or inherited COL1A1/COL1A2 mutations in the department of Osteoporosis and Bone Disease of Shanghai Jiao Tong University Affiliated Sixth People’s Hospital, and then compared the gene mutation spectrum and phenotypic characteristics to investigate differences between de novo and inherited mutations among OI with COL1A1/COL1A2 mutations.
Materials and Methods
Subjects
We carried out a retrospective review of the electronic medical record from January 2005 to January 2022 for all patients at the department of Osteoporosis and Bone Disease of Shanghai Jiao Tong University Affiliated Sixth People’s Hospital who were clinically diagnosed as OI (Figure 1). Our inclusion criteria were (1): probands were confirmed with COL1A1 or COL1A2 mutation; (2) pedigrees had undergone parental gene verification. The Ethics Committee of Shanghai Jiao Tong University Affiliated Sixth People’s Hospital approved the study. Written informed consent was obtained from patients or legal guardians of children younger than 18.
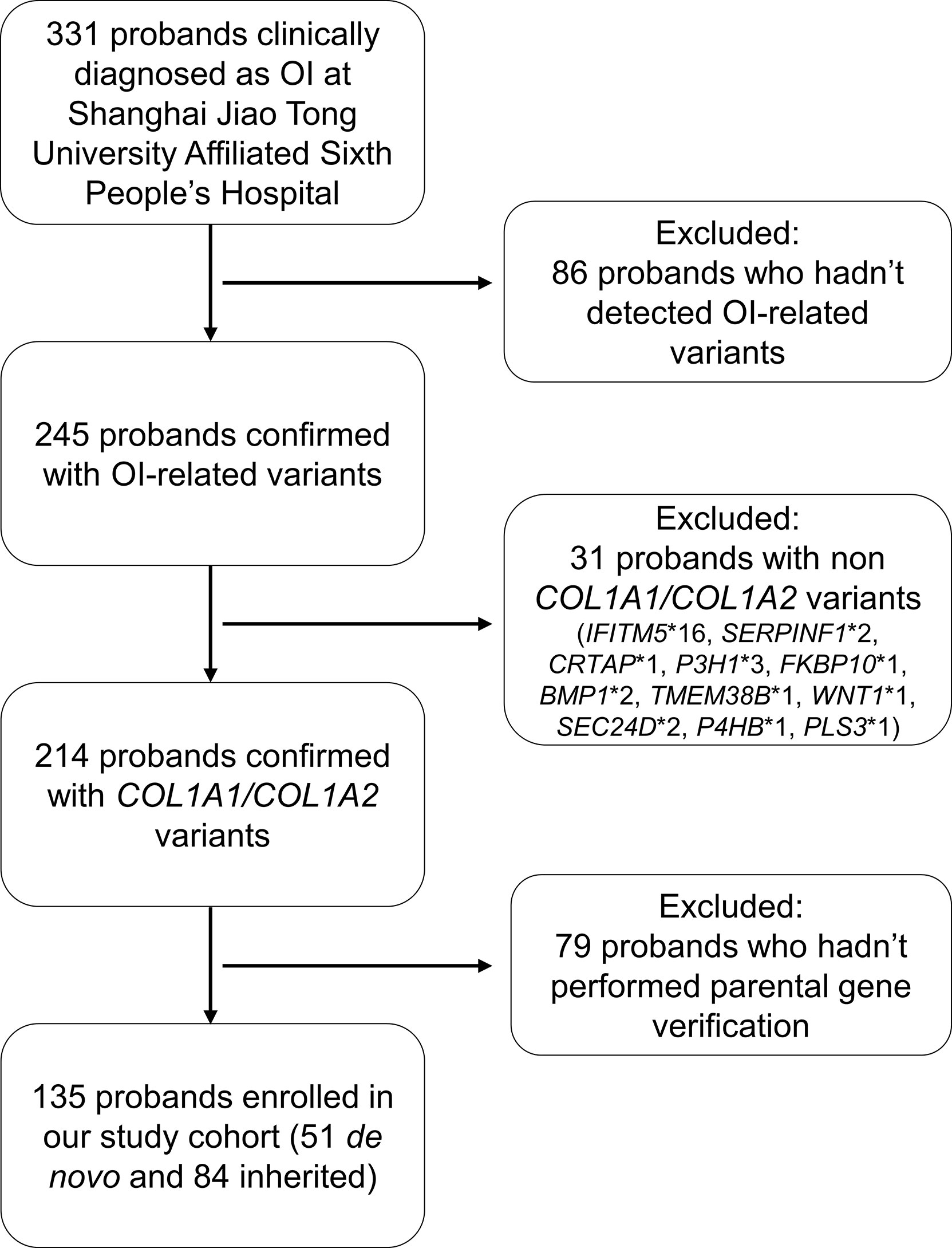
Figure 1 Flowchart of study cohort selection.
Clinical Evaluation
Clinical information, including age at diagnosis, gender, family history, non-skeletal features (like blue sclera, dentinogenesis imperfecta, and hearing loss), and medical history were collected. As for de novo pedigrees, the parental age at conception, methods of conception, and reproductive times were also collected. Information on bone fractures was evaluated, including site, time of initiation, and frequency (per year). The patient’s height was adjusted to standard deviation scores (SDS) based on reference data from the Chinese National Centers for Disease Control and Prevention (20). Baseline bone mineral density (BMD) at the lumbar spine (before bisphosphonate or denosumab treatment) was measured by dual-energy X-ray absorptiometry (DXA) (Lunar, Madison, WI, USA; Hologic, Boston, MA, USA). The coefficient of variability (CV) values of the BMD for the Lunar device at L1-L4 and femoral neck were 1.39% and 2.22%, respectively (21). The CV values of the BMD for the Hologic device at L1-L4 and femoral neck were 0.9% and 0.8%, respectively (21). The results were transformed into age- and sex-specific Z scores (T-scores for males over 50 years old and postmenopausal females) by combing reference data (22–24). In addition, X-ray radiography of the upper and lower extremities and thoracolumbar vertebrae were examined. Then based on the clinical and radiological characteristics, probands were categorized as clinical type I, III, or IV according to the Sillence classification (2). Type II OI was not included in this study as it’s a lethal perinatal form.
Apart from the Sillence classification, we also proposed a new clinical scoring system to assess the severity of OI quantitatively by referring to the scoring criteria of the previous OI researches (18, 25, 26). Clinical score = number of fractures (1-3) + fracture frequency (per year) (1-3) + bowing of lower limbs (0-4) + scoliosis (0-2) + height standard deviation score (SDS) (1-4) + BMD Z/T-score (1-4). The specific contents are displayed in Table 1. The higher the clinical score, the more severe the clinical phenotype.

Table 1 The proposed clinical scoring system for osteogenesis imperfecta patients.
Serum levels of calcium (Ca), phosphate (P), and alkaline phosphatase (ALP) were measured by the Hitachi 7600-020 automatic biochemistry analyzer. Bone formation markers osteocalcin (OC), the bone resorption marker serum beta cross-linked C-terminal telopeptide of type 1 collagen (β-CTX), intact parathyroid hormone (PTH), and 25-hydroxyvitamin D (25OHD) were measured using the Cobas 6000 auto-analyzer (Roche Diagnostics, Mannheim, Switzerland).
COL1A1/COL1A2 Gene Mutation Analysis via Sanger Sequencing
Genomic DNA was extracted from the peripheral blood of all probands and their parents by standard techniques using a DNA extraction kit (Lifefeng Biotech, Shanghai). Exons and exon-intron boundaries of COL1A1 and COL1A2 genes were amplified by polymerase chain reaction (PCR) (GenBank accession NO. NC_000017.11 and NO. NC_000007.14). Primers were designed by the Web-based Primer 3 software (https://bioinfo.ut.ee/primer3-0.4.0/). Direct sequencing was performed using BigDye Terminator Cycle Sequencing Ready Reaction Kit, v. 3.1 (Applied Biosystems, California, USA). The products were analyzed with an ABI 3730 sequencer (Foster, CA, United States). SNPs were identified using Polyphred (https://droog.gs.washington.edu/polyphred/). Then, the parental gene verification was performed in the probands’ parents and siblings by Sanger sequencing. Results absent from the Osteogenesis Imperfecta Variant Database (https://oi.gene.le.ac.uk/) were regarded as novel mutations.
Statistical Analysis
Measurement data (including age at diagnosis, height SDS, BMD Z/T-score, fracture frequency, and clinical score) were presented as mean ± standard deviation, and abnormal distributions were presented as medians (Q1, Q3). The differences between de novo and inherited groups were analyzed as appropriate by Grouped T-Test or Non-parametric Test. Enumeration data (like gender and COL1A1/COL1A2) were expressed as numbers (%) or ratios, and intergroup differences were evaluated with Chi-Square Test or Continuity Adjustment Chi-Square Test as appropriate. The statistical analyses were performed by SAS 8.2 (SAS Institute Inc., Cary, NC, USA). A two-sided P < 0.05 was considered statistically significant.
Results
Altogether, 135 probands from 135 unrelated and non-consanguineous pedigrees, including 118 patients who had been published in our previous studies (15, 27, 28), were enrolled in the study, among which 51 probands (37.78%) belonged to the de novo mutation group, and the remaining 84 probands (62.22%) were from the inherited mutation group. The details of clinical characteristics of each proband, COL1A1 and COL1A2 mutation loci identified in the present study and the parental gene verification are listed in Supplementary Tables 1–4.
Clinical Characteristics of Probands in De novo and Inherited Mutations
The comparisons of clinical characteristics of probands between de novo and inherited mutations were summarized in Table 2. Among 51 de novo mutation cases, twenty-one (41.18%) probands with de novo mutation had at least one unaffected sibling, and one proband (P37, 1.96%) had an affected twin sister. The mean height SDS in de novo mutations was -2.3 ± 3.0, lower than inherited mutations (-1.4 ± 1.9), and the mean BMD Z/T-score at the lumbar spine was -2.3 ± 1.5, also lower than inherited mutations (-1.7 ± 1.8), however, both of them lacking significance (P=0.0767 and 0.0742, respectively). Like inherited mutations, blue sclera covered a substantial proportion of de novo mutations (82.22%), while hearing loss was relatively infrequent (2.22%). There were no significant differences in age at diagnosis, gender, three phenotypes (including the blue sclera, dentinogenesis imperfecta, and hearing loss), and fracture frequency between de novo and inherited mutations. As for biochemical indexes (Ca, P, ALP, β-CTX, OC, PTH, and 25OHD), no statistical difference was identified, either. X-rays radiography of the lower extremities of six de novo and two inherited mutation probands with clinical type III were exhibited in Figure 2. Severe lower limb deformities of these probands could be seen, which resulted in limited movement in their daily life. Although there was no significant difference in the proportion of type I and type IV between de novo and inherited mutations, the proportion of type III in the de novo mutation group was significantly higher than that in the inherited mutation group (P =0.0002).
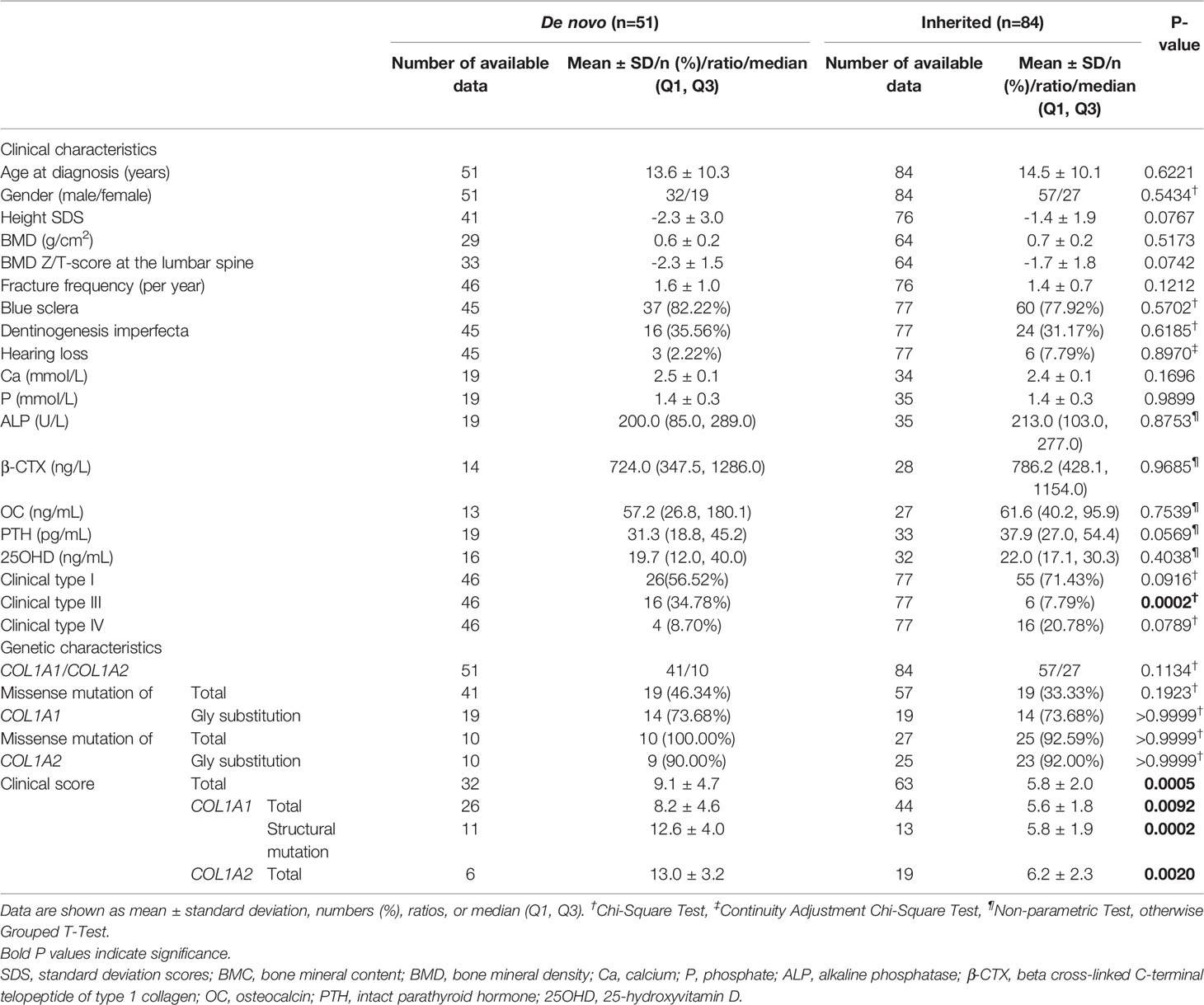
Table 2 Clinical and genetic characteristics of de novo and inherited COL1A1/COL1A2 mutations of OI.
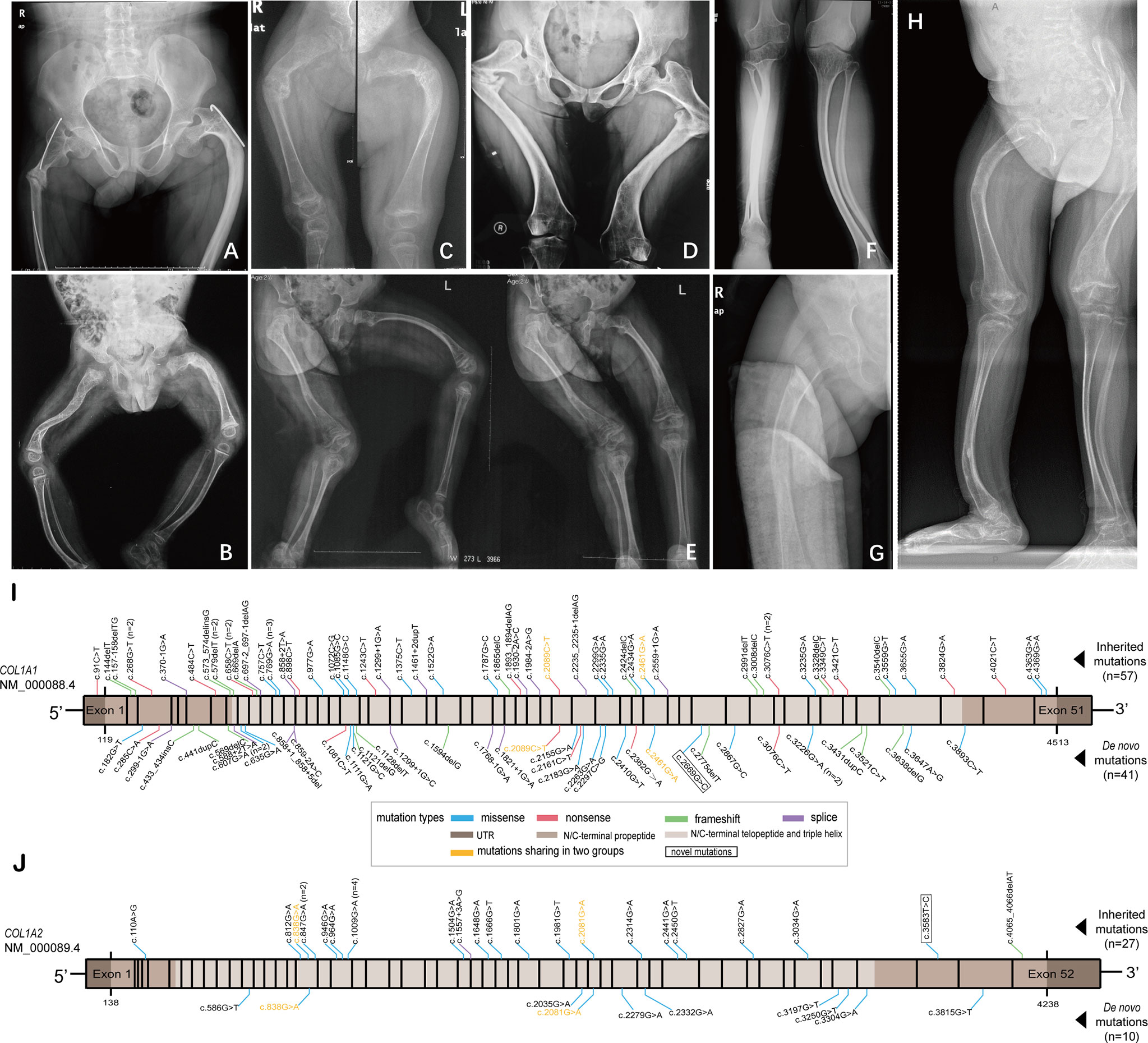
Figure 2 X-ray radiography of the lower extremities of six de novo and two inherited mutation probands with clinical type III, and diagrams showing mutation loci identified in this study. (A, B) X-ray radiography of the femurs of the probands (p68 and p56) with inherited mutations. (C–H) X-ray radiography of the lower extremities of the probands (P9, P3, P49, P43, P13, and P26, respectively) with de novo mutations. (I) Mutation loci in COL1A1 gene identified in two groups. (J) Mutation loci in COL1A2 gene identified in two groups.
A total of 20 probands’ fathers or mothers confirmed with pathogenic variants among 84 hereditary cases exhibited no OI-related phenotype or fracture history, that is, nonpenetrance (listed in Supplementary Table 3). The available data of parents’ medical history was 78. Hence, the rate of nonpentrance in the inherited mutation group was 25.64% (20/78).
Genetic Analysis of COL1A1/COL1A2 in De Novo and Inherited Mutations
All pedigrees in our study had completed parental gene verification to confirm the mutation mode. A total of 123 different collagen type I variants were identified in 135 probands, including 65 (52.85%) missense mutations, 20 (16.26%) nonsense mutations, 21 (17.07%) frameshift mutations, and 17 (13.82%) splice mutations (Figures 2I, J). Of the 123 mutations, two variants (c.2669G>C in COL1A1, and c.3583T>C in COL1A2) absent in the Osteogenesis Imperfecta Variant Database (https://www.le.ac.uk/genetics/collagen/index.html) were novel mutations.
The mutation spectrum of this study comprised four kinds of mutation types, including missense, nonsense, frameshift, and splice mutation. The comparisons between de novo and inherited mutation spectrum were summed up in Table 2. COL1A1 and COL1A2 mutation cases covered a similar proportion in the de novo and inherited groups (P=0.1134). Probands with COL1A1 de novo mutations were 41, containing 19 (46.34%) missense mutations, and probands with COL1A1 inherited mutations were 57, including 19 (33.33%) missense mutations. The missense mutation in the de novo COL1A1 mutation group occupied a larger percentage than in the inherited COL1A1 mutation group, but lacking significance (P=0.1923). All ten probands with COL1A2 de novo mutations were missense mutations, and 92.59% of twenty-seven probands with COL1A2 inherited mutations were missense mutations. No significant intergroup difference in the proportion of missense mutation of COL1A2 was discovered (P>0.9999). Glycine substitutions covered no significantly different proportion of missense mutations in de novo and inherited COL1A1/COL1A2 mutations. Additionally, p.Gly337Ser was seen four times in the COL1A2 inherited mutation group. Intriguingly, mutations of p.Gly821Ser, p.Arg697*, and p.Arg1026* in COL1A1, and p.Gly694Asp in COL1A2 shared in de novo and inherited mutation groups (Table 3). The concrete proportions of each mutation type in COL1A1/COL1A2 for two groups were displayed in Supplementary Figure 1.
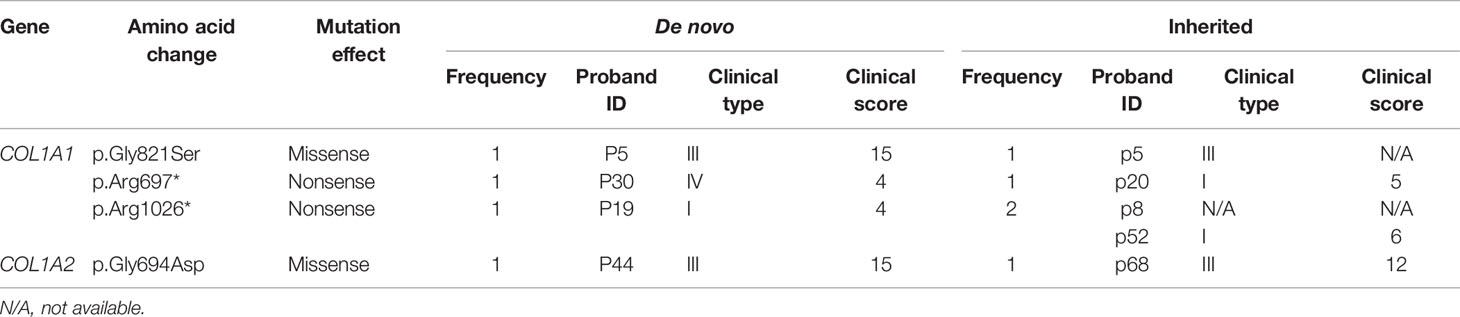
Table 3 Mutations shared in de novo and inherited mutation groups.
Genotype-Phenotype Correlations in De Novo and Inherited Mutations
The frequency of clinical type in each mutation type in de novo and inherited mutations was exhibited in Figure 3. Both in de novo and inherited mutation groups, mutation types of nonsense, frameshift, and splice mainly resulted in clinical type I, and clinical type III and IV (especially type III) were primarily correlated with missense mutations. Compared with inherited mutations in COL1A1/COL1A2, probands with de novo mutations had significantly higher clinical scores (P = 0.0005) (Table 2), which denoted more severe phenotypes. We also compared clinical scores about COL1A1(P = 0.0092), missense mutation of COL1A1(P=0.0002), and COL1A2(P=0.0020) between two groups, respectively, and their significant results were consistent with the outcome mentioned above. At the same time, we presented clinical scores of available probands on different de novo or inherited COL1A1/COL1A2 mutation loci with the help of bubble plots (Figure 3). From four different bubble plots, we could find that: firstly, there was no noticeable difference in the mutation site distribution for the COL1A1/COL1A2 gene between de novo and inherited mutations; secondly, the clinical score of missense mutation was higher than the other three mutation effects in general; lastly, bubbles of the de novo COL1A1/COL1A2 mutations distributed higher in the axis than the inherited did, which means the clinical scores of de novo mutations were higher and phenotypes were more severe. Four mutation loci shared in de novo and inherited mutation groups, mentioned above, had a consistent clinical type and clinical score between the two groups, except for one case with p.Arg1026* mutation (Table 3).
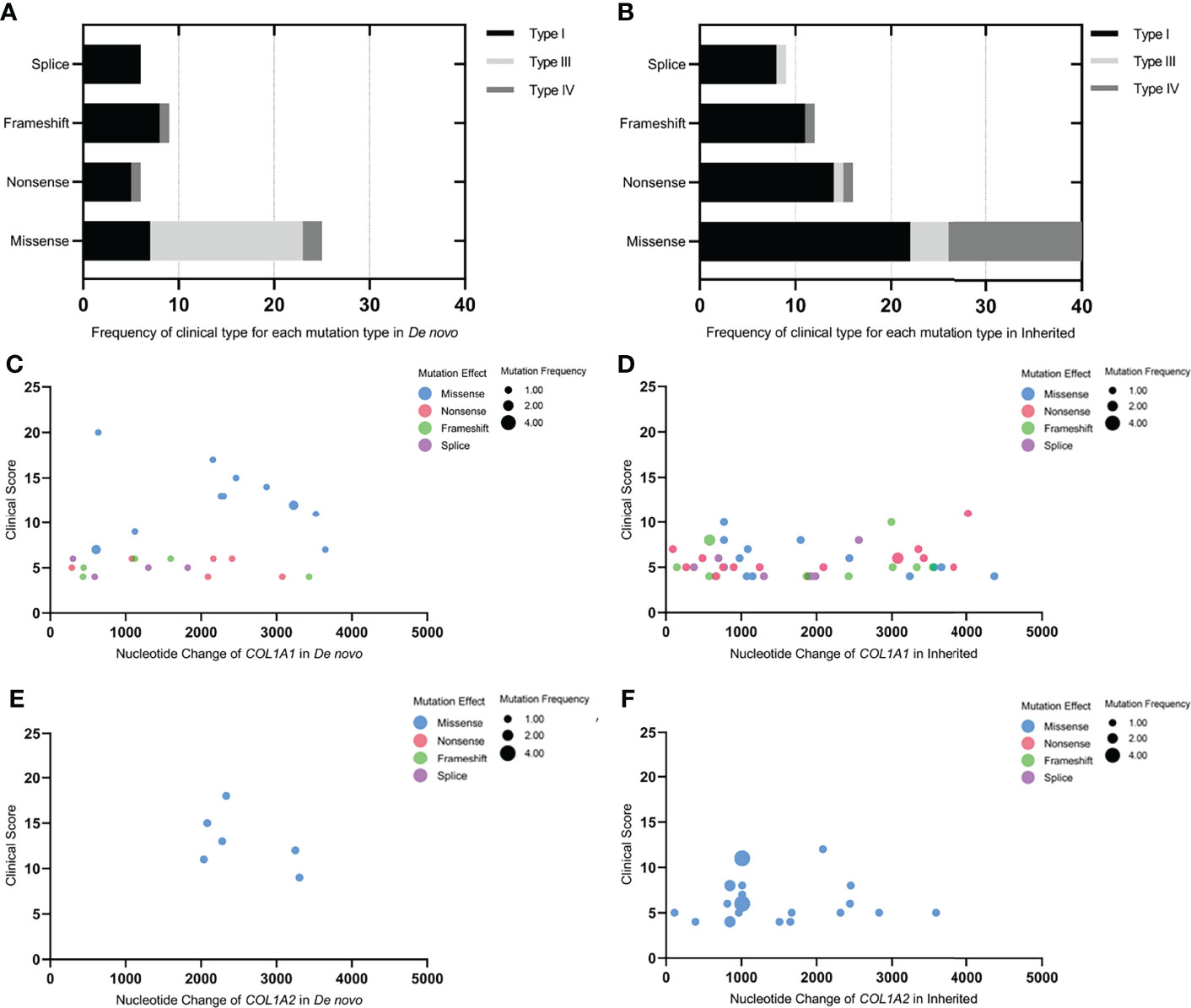
Figure 3 Genotype-phenotype correlations in de novo and inherited mutations. (A, B) Frequency of clinical type for each mutation type in De novo and Inherited. (C–F) The bubble plots of the clinical score of available probands with de novo or inherited COL1A1/COL1A2 mutations. X-axis represented the mutation site on the COL1A1/COL1A2 gene; Y-axis was the clinical score; the colors of the bubble represented different mutation effects (missense, nonsense, frameshift, and splice, respectively); the size of the bubble indicated the mutation frequency of each mutation site in the de novo or inherited mutation group. The bubble was higher and bigger; the phenotype was more severe, and the mutation site was more frequent.
The Parental Age at Conception in De Novo Mutations
In the de novo mutation group, the average paternal and maternal age was 29.2 and 26.8, respectively. In the US, National Vital Statistics System data revealed that the average paternal age at birth was 30.9 in 2015 (29), which was older than the mean paternal age at conception in our study (P = 0.0236). The average maternal age at conception in China in 2017 was 29.7 (30), which was also older than our research data (P <0.0001). Additionally, except for one proband (P36) who was the outcome of medically assisted reproduction, the remaining 50 pedigrees (98.04%) with de novo mutations were spontaneous conception.
Discussion
Previous studies had illustrated the proportion of de novo mutations in OI with COL1A1/COL1A2 mutations: 41.03% in South Korea (13), 56.16% in Estonia, Ukraine, and Vietnam (14), and 54.6% in our previous report (15). The percentage of de novo mutations in our study (37.78%) was significantly lower than our last report (P = 0.0029), which may be due to we excluded cases without performing parental gene verification. OI is a phenotypically heterogeneous disease that familial cases with the same mutation can present remarkably phenotypic diversity, ranging from mild to severe (31, 32). In our study, the rate of nonpenetrance of hereditary probands’ parents was up to 25.64%. Therefore, some affected parents of hereditary probands exhibited very mild or even without symptoms, which may result in missed diagnosis and increasing the false-positive rate of de novo mutations if not performing parental gene verification.
Low bone mass is always presented in patients with OI, and in our study, the de novo mutation group exhibited a lower BMD Z/T-score at the lumbar spine, however, not significantly lower than the inherited group. Fractures of long bones and vertebral compression are the most notable features of OI, while there was no difference in the fracture frequency between de novo and inherited mutation groups in our study. Skeletal deformities like bowing of long bones and scoliosis are usually associated with severe type III. Apart from congenital causes, recurrent fractures of extremities can also result in deformities. As displayed in Figure 2, lower limb deformities would seriously damage the ability to move, and most of them had no alternative but to be in a wheelchair for the rest of their lives. Most of our patients’ severe lower limb deformities were accompanied by severe scoliosis. Activity limitations and participation restrictions are linked to lower quality of life (QOL) (33), and physical QOL is worse in children with more severe OI than in mildly affected children. In an aspect of non-skeletal manifestation, blue sclera was the most prevalent feature of patients with OI (28), and consistent with the previous studies (34, 35), dentinogenesis imperfecta covered a higher proportion in type III and IV OI than type I in our study. Biochemical indexes including bone turnover markers showed no statistical differences in our study. Several papers have revealed that there were no significant differences in levels of the bone turnover markers between different OI clinical types or different OI mutation types (36, 37), therefore, the relationship between severity and bone turnover rates in OI may be more complicated.
Generally, haploinsufficiency of COL1A1 results in the mildest form of OI. In contrast, haploinsufficiency of COL1A2 results in no apparent OI-related phenotype, and homozygous null mutations of COL1A2 bring about phenotypes of varying severity (38). However, in our study, one case (p61, c.1557+3A>G) with haploinsufficiency of COL1A2 mutation showed severe phenotypes and was classified as type III. Most of the structural defects are missense mutations, in which over 80% of mutations are the results of glycine substitutions in the Gly-Xaa-Yaa repeat (4). The most frequent cause of OI with COL1A2 mutation is the structural defect of type I collagen, especially glycine substitutions in the helical domain (16, 39). Consistently, both de novo and inherited COL1A2 mutations in our study were predominantly composed of missense mutations (100% and 92.59%, respectively), especially glycine substitutions. It has been widely demonstrated that structural mutations have more severe clinical consequences than quantitative mutations, and clinical type II-IV are usually caused by mutations altering structures (1, 4). Similarly, clinical type III was mainly driven by missense mutations and type I primarily resulted from quantitative defects in our study, both in de novo and inherited mutation groups. Repeated mutations are often associated with CpG dinucleotides, described as mutational hotspots (40). The mutation of p.Gly337Ser in COL1A2 was detected in four unrelated families in our study, that is, mutational hot spots. Four different mutation loci mentioned above were shared in de novo and inherited mutation groups. Almost all of them had a consistent clinical type and clinical score between the two groups, indicating that the severity of the same mutant locus did not differ between de novo and inherited mutations.
Our results revealed that the proportion of type III and clinical scores were significantly higher in the de novo mutation group. De novo mutations are genetically different from inherited mutations and are more damaging because mutation processes in de novo mutations are happening between generations without undergoing purifying selection (41). Zhytnik et al. (14) revealed that in collagen-related OI, missense mutations and type III occupied a larger proportion in de novo mutations, and type I was lower compared with inherited mutations. In our study, the missense mutations in the de novo COL1A1 mutation group covered a higher proportion than in the inherited COL1A1 mutation group, though lacking significance, which may be a reason why there were more type III cases and higher clinical scores in the de novo mutation group. Although there was no difference in the mutation spectrum between the de novo and inherited COL1A2 mutation groups, type III cases and clinical scores were still higher in de novo mutations than in inherited COL1A2 mutations, probably due to mutation loci in de novo mutations with more serious pathogenicity since they have not undergone purifying selection. Additionally, these significant results confirmed that our clinical scoring system could quantitatively reflect the clinical severity of OI. Errors in DNA replication, exposure to mutagens, or failure to repair DNA damage can result in de novo mutations (12). Nowadays, advanced paternal age at conception has been considered the predominant factor associated with an increasing number of de novo mutations (9, 11, 42, 43). However, in our study, the average paternal age at conception was 29.2 years, significantly younger than the data of the US (29). Thus, paternal age may not be a critical factor contributing to de novo mutations in our research. In addition, Wong et al. (44) reported that medically assisted conception would increase the incidence of de novo mutations compared with natural conception. However, recently, a pilot study by Smits et al. (11) showed no impact of assisted reproductive technologies on the number of de novo mutations in the genome of the offspring. In our study, the majority of the pedigrees (98.04%) with de novo mutations were spontaneous conception, which limited us to investigate the impact of different methods of conception on de novo mutations.
Mosaicism means an individual develops from a single fertilized egg but has two or more genetically distinct cell populations (45). Postzygotic mutations, generated in the first few cell divisions after fertilization, can result in mosaicism and exist in many different tissues (43). It has been estimated that approximately 7% of de novo mutations are early postzygotic events with high-level mosaicism in the blood (46–48). The timing of the postzygotic mutations is of vital importance as it determines the proportion of affected tissues and phenotypes (45). The recurrence risk is very low, the same as the population risk, in parents having a child with a postzygotic mutation (45). Some de novo mutations, but actually because of the parental germline mosaicism, can be shared by more than one sibling. The higher the proportion of mosaicism in parental blood cells, the higher the recurrence risk among siblings (43). Furthermore, some mutations that arise in the germ cell lineage during early embryonic development are undetectable in somatic cells like blood or buccal mucosa, but can also be passed on to the next generation (7). Halvorsen et al. (49) revealed that germline mosaic mutations, which can be transmitted, have not had to undergo purifying selection as de novo mutations. Pyott et al. (50) found that the recurrence of lethal OI usually results from parental mosaicism. Therefore, de novo mutations actually resulting from paternal germline mosaicism have meaningful necessities for genetic counseling about recurrence risks. The detection limit of Sanger sequencing is about 15-20% of somatic mosaic variants. Therefore, deep sequencing, like SNaPshot minisequencing assay or whole-exome sequencing, is recommended to determine the percentage of mosaicism.
Although our comparative study has carried out a meticulous comparison between de novo and inherited COL1A1/COL1A2 mutations of OI, there are still some limitations to the study that must be considered: (1) This study was a retrospective review and suffers from the limitations inherent in such a design. (2) By adhering strictly to the inclusion criteria, we removed over 36% of probands in our institution who were confirmed with COL1A1/COL1A2 pathogenic mutations, which may have resulted in differences in the prevalence of de novo mutations we report when compared with other studies, and influence the clinical and genetical differences in two groups. However, our primary purpose was to compare the difference between two mutational patterns in OI, thus we must be 100% confident with the mode of inheritance of each proband in our study cohort. Therefore, we consider this as less of a limitation and more of a strength of the study. (3) We have not adopted deep sequencing in parental samples to make it clear whether some de novo mutations were actually caused by parental germinal mosaicism. (4) The research about non-skeletal phenotypes is not comprehensive in our study, for instance, cardiovascular abnormalities.
Conclusion
Our study showed that the proportion of clinical type III and clinical scores were significantly higher in de novo mutations compared with inherited COL1A1 mutations, which may demonstrate that the pathogenicity of de novo mutations is, on the whole, more severe than inherited mutations because de novo variants have not undergone purifying selection. These findings highlight the further need to investigate the differences between de novo and inherited mutations in OI and to research the occurrence of germline mosaicism in the parents of de novo mutations, which can provide accurate genetic counseling for both early prenatal and preimplantation diagnosis.
Data Availability Statement
The original contributions presented in the study are included in the article/Supplementary Material. Further inquiries can be directed to the corresponding authors.
Ethics Statement
The studies involving human participants were reviewed and approved by The Ethics Committee of Shanghai Jiao Tong University Affiliated Sixth People’s Hospital. Written informed consent to participate in this study was provided by the participants’ legal guardian/next of kin. Written informed consent was obtained from the individual(s), and minor(s)’ legal guardian/next of kin, for the publication of any potentially identifiable images or data included in this article.
Author Contributions
ZZ, and HZ designed the research, and revised the manuscript. YM summarized the clinical data, analyzed the sequencing data, and drafted the manuscript. HZ contributed to funding acquisition. All authors contributed to the article and approved the submitted version.
Funding
This research was funded by the National Natural Science Foundation of China (81870618).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Acknowledgments
We thank all the patients for their participation in this study and Shanghai Genesky Biotech Co., 102 Ltd. (Shanghai, China) for their technical assistance.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fendo.2022.935905/full#supplementary-material
Supplementary Figure 1 | De novo and inherited mutation spectrum of COL1A1/COL1A2. (A) De novo mutation spectrum of COL1A1. (B) De novo mutation spectrum of COL1A2. (C) Inherited mutation spectrum of COL1A1. (D) Inherited mutation spectrum of COL1A2.
Supplementary Table 1 | Clinical characteristics of De novo mutations.
Supplementary Table 2 | Mutations in COL1A1/COL1A2 of probands with de novo mutations.
Supplementary Table 3 | Clinical characteristics of De novo mutations.
Supplementary Table 4 | Mutations in COL1A1/COL1A2 of probands with inherited mutations.
References
1. Forlino A, Marini JC. Osteogenesis Imperfecta. Lancet (2016) 387(10028):1657–71. doi: 10.1016/S0140-6736(15)00728-X
2. Sillence DO, Senn A, Danks DM. Genetic Heterogeneity in Osteogenesis Imperfecta. J Med Genet (1979) 16(2):101–16. doi: 10.1136/jmg.16.2.101
3. Zhytnik L, Simm K, Salumets A, Peters M, Märtson A, Maasalu K. Reproductive Options for Families at Risk of Osteogenesis Imperfecta: A Review. Orphanet J Rare Dis (2020) 15(1):128. doi: 10.1186/s13023-020-01404-w
4. Marini JC, Forlino A, Cabral WA, Barnes AM, San Antonio JD, Milgrom S, et al. Consortium for Osteogenesis Imperfecta Mutations in the Helical Domain of Type I Collagen: Regions Rich in Lethal Mutations Align With Collagen Binding Sites for Integrins and Proteoglycans. Hum Mutat (2007) 28(3):209–21. doi: 10.1002/humu.20429
5. Roach JC, Glusman G, Smit AFA, Huff CD, Hubley R, Shannon PT, et al. Analysis of Genetic Inheritance in a Family Quartet by Whole-Genome Sequencing. Science (2010) 328(5978):636–9. doi: 10.1126/science.1186802
6. Lynch M. Rate, Molecular Spectrum, and Consequences of Human Mutation. Proc Natl Acad Sci USA (2010) 107(3):961–8. doi: 10.1073/pnas.0912629107
7. Campbell IM, Shaw CA, Stankiewicz P, Lupski JR. Somatic Mosaicism: Implications for Disease and Transmission Genetics. Trends Genet (2015) 31(7):382–92. doi: 10.1016/j.tig.2015.03.013
8. Veltman JA, Brunner HG. De Novo Mutations in Human Genetic Disease. Nat Rev Genet (2012) 13(8):565–75. doi: 10.1038/nrg3241
9. Kong A, Frigge ML, Masson G, Besenbacher S, Sulem P, Magnusson G, et al. Rate of De Novo Mutations and the Importance of Father's Age to Disease Risk. Nature (2012) 488(7412):471–5. doi: 10.1038/nature11396
10. Jónsson H, Sulem P, Kehr B, Kristmundsdottir S, Zink F, Hjartarson E, et al. Parental Influence on Human Germline De Novo Mutations in 1,548 Trios From Iceland. Nature (2017) 549(7673):519–22. doi: 10.1038/nature24018
11. Smits RM, Xavier MJ, Oud MS, Astuti GDN, Meijerink AM, de Vries PF, et al. De Novo Mutations in Children Born After Medical Assisted Reproduction. Hum Reprod (2022) 37(6):1360–9. doi: 10.1093/humrep/deac068
12. Acuna-Hidalgo R, Veltman JA, Hoischen A. New Insights Into the Generation and Role of De Novo Mutations in Health and Disease. Genome Biol (2016) 17(1):241. doi: 10.1186/s13059-016-1110-1
13. Lee K-S, Song H-R, Cho T-J, Kim HJ, Lee T-M, Jin H-S, et al. Mutational Spectrum of Type I Collagen Genes in Korean Patients With Osteogenesis Imperfecta. Hum Mutat (2006) 27(6):599. doi: 10.1002/humu.9423
14. Zhytnik L, Maasalu K, Duy BH, Pashenko A, Khmyzov S, Reimann E, et al. De Novo and Inherited Pathogenic Variants in Collagen-Related Osteogenesis Imperfecta. Mol Genet Genomic Med (2019) 7(3):e559. doi: 10.1002/mgg3.559
15. Xi L, Zhang H, Zhang Z-L. Clinical and Genetic Analysis in 185 Chinese Probands of Osteogenesis Imperfecta. J Bone Miner Metab (2021) 39(3):416–22. doi: 10.1007/s00774-020-01163-5
16. Li L, Mao B, Li S, Xiao J, Wang H, Zhang J, et al. Genotypic and Phenotypic Characterization of Chinese Patients With Osteogenesis Imperfecta. Hum Mutat (2019) 40(5):588–600. doi: 10.1002/humu.23718
17. Bardai G, Moffatt P, Glorieux FH, Rauch F. DNA Sequence Analysis in 598 Individuals With a Clinical Diagnosis of Osteogenesis Imperfecta: Diagnostic Yield and Mutation Spectrum. Osteoporos Int (2016) 27(12):3607–13. doi: 10.1007/s00198-016-3709-1
18. Mrosk J, Bhavani GS, Shah H, Hecht J, Krüger U, Shukla A, et al. Diagnostic Strategies and Genotype-Phenotype Correlation in a Large Indian Cohort of Osteogenesis Imperfecta. Bone (2018) 110:368–77. doi: 10.1016/j.bone.2018.02.029
19. Yin X, Du Y, Zhang H, Wang Z, Wang J, Fu X, et al. Identification of a De Novo Fetal Variant in Osteogenesis Imperfecta by Targeted Sequencing-Based Noninvasive Prenatal Testing. J Hum Genet (2018) 63(11):1129–37. doi: 10.1038/s10038-018-0489-9
20. Li H, Ji C-Y, Zong X-N, Zhang Y-Q. [Height and Weight Standardized Growth Charts for Chinese Children and Adolescents Aged 0 to 18 Years]. Zhonghua Er Ke Za Zhi (2009) 47(7):487–92. doi: 10.3760/cma.j.issn.0578-1310.2009.07.003
21. Zhang H, J-w He, Gao G, Yue H, Yu J-B, Hu W-W, et al. Polymorphisms in the HOXD4 Gene Are Not Associated With Peak Bone Mineral Density in Chinese Nuclear Families. Acta Pharmacol Sin (2010) 31(8):977–83. doi: 10.1038/aps.2010.91
22. Xu H, Zhao Z, Wang H, Ding M, Zhou A, Wang X, et al. Bone Mineral Density of the Spine in 11,898 Chinese Infants and Young Children: A Cross-Sectional Study. PloS One (2013) 8(12):e82098. doi: 10.1371/journal.pone.0082098
23. Khadilkar AV, Sanwalka NJ, Chiplonkar SA, Khadilkar VV, Mughal MZ. Normative Data and Percentile Curves for Dual Energy X-Ray Absorptiometry in Healthy Indian Girls and Boys Aged 5-17 Years. Bone (2011) 48(4):810–9. doi: 10.1016/j.bone.2010.12.013
24. Zhang ZQ, Ho SC, Chen ZQ, Zhang CX, Chen YM. Reference Values of Bone Mineral Density and Prevalence of Osteoporosis in Chinese Adults. Osteoporos Int (2014) 25(2):497–507. doi: 10.1007/s00198-013-2418-2
25. Aglan MS, Hosny L, El-Houssini R, Abdelhadi S, Salem F, Elbanna RAS, et al. A Scoring System for the Assessment of Clinical Severity in Osteogenesis Imperfecta. J Child Orthop (2012) 6(1):29–35. doi: 10.1007/s11832-012-0385-3
26. Li L-J, Lyu F, Song Y-W, Wang O, Jiang Y, Xia W-B, et al. Genotype-Phenotype Relationship in a Large Cohort of Osteogenesis Imperfecta Patients With COL1A1 Mutations Revealed by a New Scoring System. Chin Med J (Engl) (2019) 132(2):145–53. doi: 10.1097/CM9.0000000000000013
27. Zhang Z-L, Zhang H, Y-h Ke, Yue H, Xiao W-J, Yu J-B, et al. The Identification of Novel Mutations in COL1A1, COL1A2, and LEPRE1 Genes in Chinese Patients With Osteogenesis Imperfecta. J Bone Miner Metab (2012) 30(1):69–77. doi: 10.1007/s00774-011-0284-6
28. Zhang H, Yue H, Wang C, Hu W, Gu J, He J, et al. Clinical Characteristics and the Identification of Novel Mutations of COL1A1 and COL1A2 in 61 Chinese Patients With Osteogenesis Imperfecta. Mol Med Rep (2016) 14(5):4918–26. doi: 10.3892/mmr.2016.5835
29. Khandwala YS, Zhang CA, Lu Y, Eisenberg ML. The Age of Fathers in the USA Is Rising: An Analysis of 168 867 480 Births From 1972 to 2015. Hum Reprod (2017) 32(10):2110–6. doi: 10.1093/humrep/dex267
30. Cao J, Xu W, Liu Y, Zhang B, Zhang Y, Yu T, et al. Trends in Maternal Age and the Relationship Between Advanced Age and Adverse Pregnancy Outcomes: A Population-Based Register Study in Wuhan, China, 2010-2017. Public Health (2022) 206:8–14. doi: 10.1016/j.puhe.2022.02.015
31. Liu H-Y, Huang J, Wu D, Li T, Guo L-J, Guo Q-N, et al. Collagen Type I Alpha 1 Mutation Causes Osteogenesis Imperfecta From Mild to Perinatal Death in a Chinese Family. Chin Med J (Engl) (2016) 129(1):88–91. doi: 10.4103/0366-6999.172600
32. Higuchi Y, Hasegawa K, Futagawa N, Yamashita M, Tanaka H, Tsukahara H. Genetic Analysis in Japanese Patients With Osteogenesis Imperfecta: Genotype and Phenotype Spectra in 96 Probands. Mol Genet Genomic Med (2021) 9(6):e1675. doi: 10.1002/mgg3.1675
33. Dahan-Oliel N, Oliel S, Tsimicalis A, Montpetit K, Rauch F, Dogba MJ. Quality of Life in Osteogenesis Imperfecta: A Mixed-Methods Systematic Review. Am J Med Genet A (2016) 170A(1):62–76. doi: 10.1002/ajmg.a.37377
34. Lindahl K, Åström E, Rubin C-J, Grigelioniene G, Malmgren B, Ljunggren Ö, et al. Genetic Epidemiology, Prevalence, and Genotype-Phenotype Correlations in the Swedish Population With Osteogenesis Imperfecta. Eur J Hum Genet (2015) 23(8):1112. doi: 10.1038/ejhg.2015.81
35. Lin H-Y, Chuang C-K, Su Y-N, Chen M-R, Chiu H-C, Niu D-M, et al. Genotype and Phenotype Analysis of Taiwanese Patients With Osteogenesis Imperfecta. Orphanet J Rare Dis (2015) 10:152. doi: 10.1186/s13023-015-0370-2
36. Hald JD, Folkestad L, Harsløf T, Lund AM, Duno M, Jensen JB, et al. Skeletal Phenotypes in Adult Patients With Osteogenesis Imperfecta-Correlations With COL1A1/COL1A2 Genotype and Collagen Structure. Osteoporos Int (2016) 27(11):3331–41. doi: 10.1007/s00198-016-3653-0
37. Wekre LL, Eriksen EF, Falch JA. Bone Mass, Bone Markers and Prevalence of Fractures in Adults With Osteogenesis Imperfecta. Arch Osteoporos (2011) 6:31–8. doi: 10.1007/s11657-011-0054-z
38. Tournis S, Dede AD. Osteogenesis Imperfecta - A Clinical Update. Metabolism (2018) 80:27–37. doi: 10.1016/j.metabol.2017.06.001
39. Liu Y, Asan, Ma D, Lv F, Xu X, Wang J, et al. Gene Mutation Spectrum and Genotype-Phenotype Correlation in a Cohort of Chinese Osteogenesis Imperfecta Patients Revealed by Targeted Next Generation Sequencing. Osteoporos Int (2017) 28(10):2985–95. doi: 10.1007/s00198-017-4143-8
40. Schorderet DF, Gartler SM. Analysis of CpG Suppression in Methylated and Nonmethylated Species. Proc Natl Acad Sci USA (1992) 89(3):957–61. doi: 10.1073/pnas.89.3.957
41. Hurst LD. Fundamental Concepts in Genetics: Genetics and the Understanding of Selection. Nat Rev Genet (2009) 10(2):83–93. doi: 10.1038/nrg2506
42. Goldmann JM, Wong WSW, Pinelli M, Farrah T, Bodian D, Stittrich AB, et al. Parent-Of-Origin-Specific Signatures of De Novo Mutations. Nat Genet (2016) 48(8):935–9. doi: 10.1038/ng.3597
43. Rahbari R, Wuster A, Lindsay SJ, Hardwick RJ, Alexandrov LB, Turki SA, et al. Timing, Rates and Spectra of Human Germline Mutation. Nat Genet (2016) 48(2):126–33. doi: 10.1038/ng.3469
44. Wong WSW, Solomon BD, Bodian DL, Kothiyal P, Eley G, Huddleston KC, et al. New Observations on Maternal Age Effect on Germline De Novo Mutations. Nat Commun (2016) 7:10486. doi: 10.1038/ncomms10486
45. Biesecker LG, Spinner NB. A Genomic View of Mosaicism and Human Disease. Nat Rev Genet (2013) 14(5):307–20. doi: 10.1038/nrg3424
46. Acuna-Hidalgo R, Bo T, Kwint MP, van de Vorst M, Pinelli M, Veltman JA, et al. Post-Zygotic Point Mutations Are an Underrecognized Source of De Novo Genomic Variation. Am J Hum Genet (2015) 97(1):67–74. doi: 10.1016/j.ajhg.2015.05.008
47. Dal GM, Ergüner B, Sağıroğlu MS, Yüksel B, Onat OE, Alkan C, et al. Early Postzygotic Mutations Contribute to De Novo Variation in a Healthy Monozygotic Twin Pair. J Med Genet (2014) 51(7):455–9. doi: 10.1136/jmedgenet-2013-102197
48. Besenbacher S, Liu S, Izarzugaza JMG, Grove J, Belling K, Bork-Jensen J, et al. Novel Variation and De Novo Mutation Rates in Population-Wide De Novo Assembled Danish Trios. Nat Commun (2015) 6:5969. doi: 10.1038/ncomms6969
49. Halvorsen M, Petrovski S, Shellhaas R, Tang Y, Crandall L, Goldstein D, et al. Mosaic Mutations in Early-Onset Genetic Diseases. Genet In Med Off J Am Coll Med Genet (2016) 18(7):746–9. doi: 10.1038/gim.2015.155
50. Pyott SM, Pepin MG, Schwarze U, Yang K, Smith G, Byers PH. Recurrence of Perinatal Lethal Osteogenesis Imperfecta in Sibships: Parsing the Risk Between Parental Mosaicism for Dominant Mutations and Autosomal Recessive Inheritance. Genet In Med Off J Am Coll Med Genet (2011) 13(2):125–30. doi: 10.1097/GIM.0b013e318202e0f6
Keywords: osteogenesis imperfecta, COL1A1, COL1A2, de novo, inherited, clinical scoring system
Citation: Mei Y, Zhang H and Zhang Z (2022) Comparing Clinical and Genetic Characteristics of De Novo and Inherited COL1A1/COL1A2 Variants in a Large Chinese Cohort of Osteogenesis Imperfecta. Front. Endocrinol. 13:935905. doi: 10.3389/fendo.2022.935905
Received: 04 May 2022; Accepted: 17 June 2022;
Published: 14 July 2022.
Edited by:
Giacomina Brunetti, University of Bari Aldo Moro, ItalyReviewed by:
Carla Caffarelli, University of Siena, ItalyVijaya Kumar Pidugu, National Cancer Institute (NIH), United States
Copyright © 2022 Mei, Zhang and Zhang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Zhenlin Zhang, emhhbmd6bEBzanR1LmVkdS5jbg==; Hao Zhang, emhhbmdoYW96Y2xAMTYzLmNvbQ==