 Martina Tetti
Martina Tetti Siyuan Gong
Siyuan Gong Franco Veglio
Franco Veglio Martin Reincke1†
Martin Reincke1† Tracy Ann Williams
Tracy Ann Williams- 1Medizinische Klinik und Poliklinik IV, Klinikum der Universität München, Ludwig-Maximilians-Universität (LMU) München, München, Germany
- 2Division of Internal Medicine and Hypertension, Department of Medical Sciences, University of Turin, Turin, Italy
Primary aldosteronism is the most common surgically curable form of hypertension. The sporadic forms of the disorder are usually caused by aldosterone overproduction from a unilateral adrenocortical aldosterone-producing adenoma or from bilateral adrenocortical hyperplasia. The main knowledge-advances in disease pathophysiology focus on pathogenic germline and somatic variants that drive the excess aldosterone production. Less clear are the molecular and cellular mechanisms that lead to an increased mass of the adrenal cortex. However, the combined application of transcriptomics, metabolomics, and epigenetics has achieved substantial insight into these processes and uncovered the evolving complexity of disrupted cell growth mechanisms in primary aldosteronism. In this review, we summarize and discuss recent progress in our understanding of mechanisms of cell death, and proliferation in the pathophysiology of primary aldosteronism.
Introduction
Primary aldosteronism (PA) is the most common form of secondary hypertension that accounts for 5 to 15% of patients with hypertension (1). The disorder encompasses a group of pathological conditions of the adrenal glands that trigger aldosterone overproduction associated with a higher risk of cardiovascular events (2). Thus, a prompt diagnosis of PA with accurate subtyping of unilateral from bilateral cases is essential to initiate specific treatment strategies (adrenalectomy or pharmacotherapy with a mineralocorticoid receptor antagonist) for optimal patient outcomes (1). Bilateral forms include familial forms of PA, which are rare monogenic forms of hypertension caused by germline pathogenic variants. To date, 4 genetically distinct forms of familial hyperaldosteronism have been identified (FH types 1 to 4), classified according to the affected gene (3, 4). Sporadic aldosterone-producing adenomas (APAs) carry somatic mutations in aldosterone-driver genes and in genes that drive the development of multiple adrenal tumors. In addition to genetic insights, a wealth of data has accumulated from transcriptomics and metabolomics analyses of aldosterone-producing lesions to gain a deeper understanding of PA pathophysiology.
Genetics of primary aldosteronism
An extensive review on the genetics of PA has been published recently (4). In brief, FH type 1 is caused by a chimeric CYP11B1/CYP11B2 gene composed of the adrenocorticotropic hormone (ACTH)-responsive promoter region of CYP11B1 fused to the CYP11B2 coding region (5). As a result, transcription of CYP11B2 (aldosterone synthase) is under the regulatory control of ACTH instead of angiotensin II and potassium as in normal aldosterone physiology. The dysregulated aldosterone production in FH types 2, 3 and 4 is caused by variants in the ion channels encoded by CLCN2 (CIC-2 chloride channel), KCNJ5 (GIRK4 potassium channel), and CACNA1H (Cav3.2 calcium channel), respectively, that function in intracellular ion homeostasis. In addition, germline pathogenic variants in CACNA1D can cause a complex form of PA with seizures and neurologic abnormalities (PASNA) (Table 1).
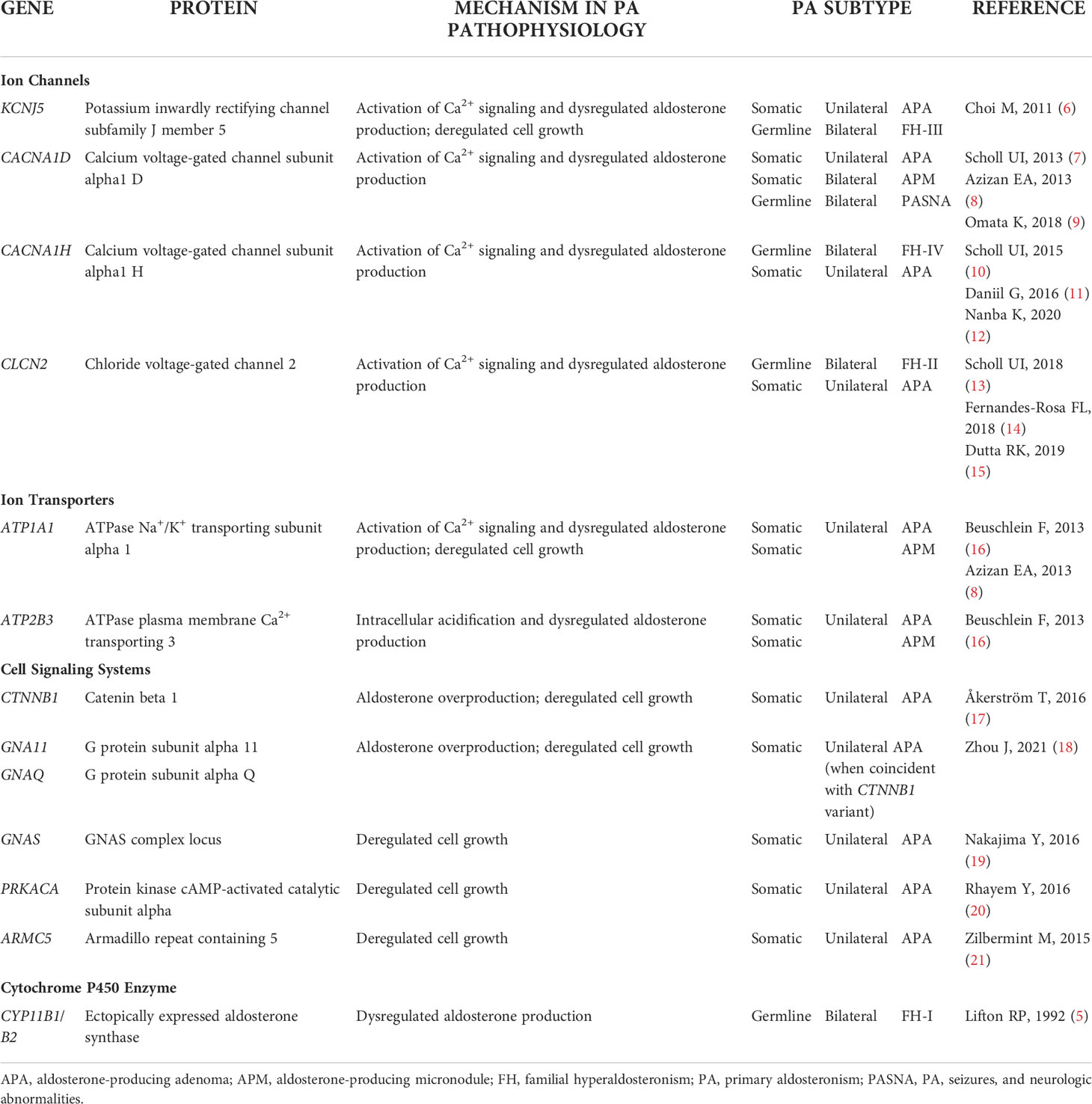
Table 1 Effect Of Gene Variants In Primary Aldosteronism Driver Genes.
The landscape of somatic APA mutations overlaps with the germline pathogenic variants. Thus, KCNJ5, CACNA1D, CACNA1H, and CLCN2 can carry somatic or germline mutations in PA. In APAs, this set of aldosterone-driver genes is complemented by somatic mutations in ATP1A1 and ATP2B3, which encode the ion pumps Na+/K+-ATPase and Ca2+-ATPase, and in genes that are targeted in multiple adrenal tumors such as CTNNB1 (β-catenin), and GNA11, GNAQ, and GNAS, encoding (guanine nucleotide-binding proteins), PRKACA (catalytic subunit of protein kinase A), and ARMC5 (a member of the armadillo/β-catenin-like repeat family) (Table 1). The potential function of PA-related mutations in cell growth mechanisms is discussed below.
Aldosterone-driver mutations and deregulated cell growth
The role of recurrent PA mutations in aldosterone overproduction is clearly demonstrated but less evident is their role in perturbing regulatory mechanisms of adrenal cell growth that lead to an increase of the adrenal cell mass and tumor development.
GIRK4 potassium channel
Choi et al. (6) identified somatic APA mutations and an inherited germline mutation in KCNJ5 (encoding Gly151Arg and Leu168Arg somatic, and Thr158Ala germline variants in GIRK4, G-protein-gated inwardly rectifying K+ channel 4). This triggered a rapid evolution of research and the description of somatic mutations in other genes (7, 8, 12, 16, 17, 22–27) and the identification of additional familial forms of PA (10, 11, 13, 14). The KCNJ5 variants result in single amino acid substitutions in or close to the selectivity filter of the encoded GIRK4 potassium channel. The channel properties are consequently altered causing loss of potassium selectivity and abnormal sodium influx (instead of exclusive K+ efflux) from glomerulosa cells. The resultant depolarization of zona glomerulosa cells causes opening of voltage-gated calcium channels, increased intracellular calcium concentrations and activation of calcium signal transduction and CYP11B2 (aldosterone synthase) gene transcription and increased aldosterone production (6) (Figure 1). KCNJ5 mutations are highly prevalent in APAs (3, 4), associated with larger adenoma size, female sex, and more severe aldosteronism (28). The application of genotype analysis to CYP11B2 (aldosterone synthase) immunopositive regions of paraffin-embedded adrenal sections combined with next generation sequencing (29) has resulted in an increased detection of somatic APA mutations such that prevalence rates are now described in around 90% of cases (22–25, 27, 30).
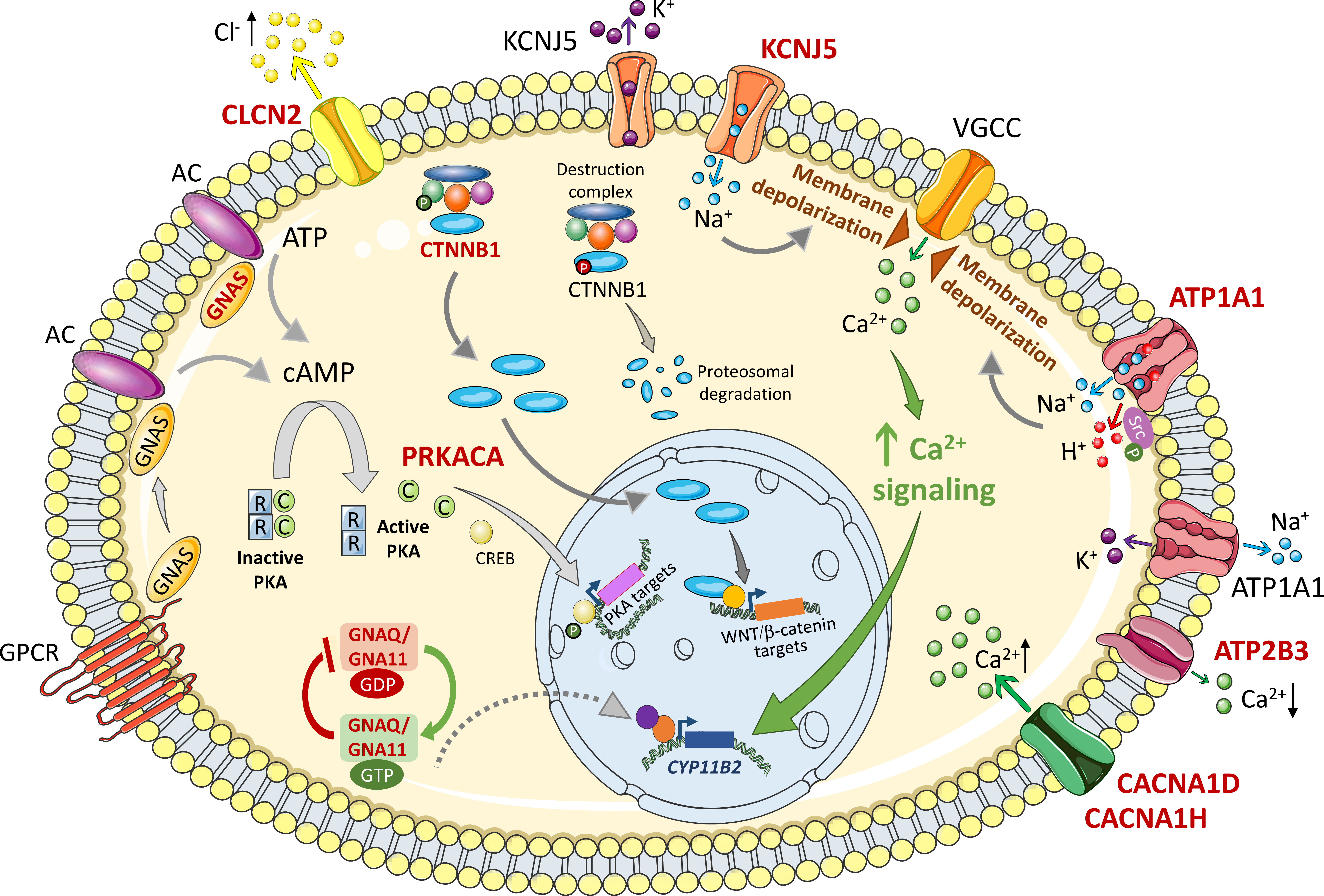
Figure 1 Mechanisms of deregulated cell growth of variants related to primary aldosteronism. The figure shows intracellular signaling pathways activated by APA somatic mutations implicated in cell growth mechanisms. Wild type and mutated forms are labeled in black and red respectively. Membrane depolarization triggered by Na+ or H+ influx causes opening of voltage gated ion channels (VGCC), Ca2+ influx and activation of Ca2+ signaling that activates CYP11B2 gene transcription to drive increased aldosterone biosynthesis and cell proliferation. ATP1A1 is also reported to activate cell proliferation by phosphorylated Src. Intracellular β-catenin concentrations are kept low under basal conditions by continuous proteosomal degradation. Activating mutations in exon 3 of the β-catenin gene bypass this control mechanism resulting in cytoplasmic β-catenin accumulation and nuclear translocation to activate specific gene transcription programs. In many adrenal tumours including some rare APA examples, cAMP/PKA signaling is activated by mutations in GNAS and PRKACA for activation of target genes. GNAQ and GNA11 mutations can cause upregulated aldosterone production in vitro and are associated with hyperplasia of glomerulosa cells adjacent to an APA. They are clinically silent in vivo except when concomitant with CTNNB1 (β-catenin) mutations. See text for details. Somatic mutations in CLCN2, CACNA1D, CACNA1H, and ATP2B3 have been described in APAs which result in increased Cl- efflux (CLCN2), increased Ca2+ influx (CACNA1D, CACNA1H), and decreased Ca2+ efflux (ATP2B3). AC, adenylyl cyclase; ATP, adenosine triphosphate; ATP1A1, ATPase Na+/K+ transporting subunit α1; ATP2B3, ATPase plasma membrane Ca2+ transporting 3; C, catalytic subunit; CACNA1D, calcium voltage-gated channel subunit alpha1 D; CACNA1H, calcium voltage-gated channel subunit alpha1 H; cAMP, cyclic adenosine monophosphate; Ca2+, calcium ions; CLCN2, chloride voltage-gated channel 2; CREB, cAMP response element-binding protein; CYP11B2, cytochrome P450 family 11 subfamily B member 2 (aldosterone synthase); GDP, guanosine diphosphate; GNA11, G protein subunit α11; GNAS, GNAS complex locus; GNAQ, G protein subunit αQ; GTP, guanosine triphosphate; GPCR, G protein coupled receptor; H +, hydrogen ions; K+, potassium ions; KCNJ5, potassium inwardly rectifying channel subfamily J member 5; Na+, sodium ions; PKA, protein kinase A; PRKACA, protein kinase cAMP-activated catalytic subunit α; R, regulatory subunit; VGCC, voltage-gated calcium channel; WNT, wingless-related integration site. Figure produced using Servier Medical Art (https://smart.servier.com/).
Germline KCNJ5 mutations can cause FH type 3 which presents with a variable clinical phenotype that might be explained by the functional properties of the mutated GIRK4 potassium channel. This is supported by the report of 4 families with FH type 3 caused by either of 2 different KCNJ5 mutations (31). Two families with GIRK4-Gly151Arg mutations presented with massive adrenocortical hyperplasia and severe aldosteronism requiring bilateral adrenalectomy. The other 2 families carried a Gly151Glu mutation associated with a milder PA phenotype without evident adrenal hyperplasia on imaging and easily controlled hypertension with pharmacotherapy. The Gly151Glu mutation caused higher sodium influx and greater consequent adrenal cell death than Gly151Arg accounting for the absence of structural changes at adrenal imaging in patients with the GIRK4-Gly151Glu germline variant (31). The cell toxicity of GIRK4 mutants can be circumvented by low levels of gene transcription and expression of the mutated GIRK4 channel (32).
Sodium/potassium-transporting ATPase
The Na+/K+-ATPase pump couples the hydrolysis of an ATP molecule to the transport of 3 Na+ out and 2 K+ into the cell. Azizan et al. (8) and Beuschlein et al. (16), reported somatic gain-of-function mutations in ATP1A1 (Na+/K+-ATPase alpha-1 subunit). ATP1A1 variants are found in 5-17% of APAs (3) and many have been shown to severely impair K+ binding and ATPase activity conferring membrane depolarization in the cells which express them (8, 16, 33). Adrenal cells expressing ATP1A1 mutations show normal intracellular Ca2+ concentrations suggesting that activated Ca2+ export pathways, via a Na+/Ca2+ exchanger or Ca2+-ATPase, or inactivation of Ca2+ channels might function as compensatory mechanisms (34). However, adrenal cells expressing ATP1A1 mutants exhibit abnormal H+ leak currents that cause intracellular acidification that can lead to increased expression of CYP11B2 (aldosterone synthase) (34) (Figure 1).
Na+/K+-ATPase can display a dual function not only as an ion pump but also as a signaling complex relaying extracellular signals to intracellular compartments (35–37). For example, ouabain- the cardiotonic steroid- can bind to Na+/K+-ATPase which in turn interacts, via its alpha-1 subunit, with the Src proto-oncogene tyrosine kinase. Subsequent Src activation by phosphorylation then enhances downstream signaling pathways (37). Human adrenal cells expressing Na+/K+-ATPase-p.Leu104Arg (a recurrent somatic APA mutation) show increased levels of Src phosphorylation and enhanced cell proliferation implicating a potential novel mechanism of the Na+/K+-ATPase-Src functional complex in the progression and development of APAs with an ATP1A1 mutation (38) (Figure 1).
Calcium-transporting ATPase, Cav1.3 and Cav3.2 calcium channels, and the CIC-2 chloride channel
As mentioned above, somatic APA mutations have been identified in other genes involved in intracellular ion homeostasis (ATP2B3, CACNA1D, CACNA1H, and CLCN2). However, there is no experimental evidence to demonstrate a potential role for mutations in these genes in cell proliferation or cell death. ATP2B3 encodes the Ca2+-ATPase which pumps intracellular Ca2+ across the plasma membrane to the extracellular environment. The Ca2+-ATPase mutations in PA interfere with Ca2+ ion binding resulting in decreased export and increased Ca2+ influx.
Heterozygous somatic mutations in the L-type voltage gated Ca2+ channel Cav1.3 (encoded by CACNA1D) are frequently described in aldosterone-producing lesions associated with PA (in APAs and in aldosterone-producing micronodules [APMs]) (7–9) and more rarely in the T-type Ca2+ channel Cav3.2 (encoded by CACNA1H) (12). PA-driver mutations in CACNA1D and CACNA1H confer a gain-of-function and result in increased Ca2+ influx. Somatic APA mutations in CLCN2 encoding the CIC-2 chloride channel have been described in just a few cases (15).
Driver mutations of tumorigenesis in primary aldosteronism
Somatic mutations that constitutively activate WNT/β-catenin (CTNNB1), G-protein (GNAS, GNAQ, GNA11), and cAMP/protein kinase A (PKA) (GNAS, PRKACA) signaling can drive unrestrained cell proliferation and survival in diverse tumors, including adrenal tumors. Activating mutations in β-catenin (encoded by CTNNB1) that cause WNT-independent signaling are widely reported in APAs (17, 39). Constitutive cAMP/PKA signaling due to PRKACA and GNAS activating mutations has been reported in a limited number of APAs (19, 20). Mutations in G protein α subunits, particularly those encoded by GNAS and GNAQ/GNA11, have been reported in many tumours including a wide variety of endocrine tumors (40, 41).
Activation of WNT/β-catenin signaling
The regulated destruction of the transcriptional co-activator β-catenin is central to the WNT-mediated β-catenin signaling cascade. This process involves a multi-subunit destruction complex, comprising AXIN (axis inhibitor protein), APC (tumor suppressor adenomatous polyposis coli), and GSK-3β (glycogen synthase kinase-3), which phosphorylates β-catenin for ubiquitylation and proteasomal degradation. In contrast, WNT-mediated cell stimulation disrupts β-catenin degradation causing its accumulation and subsequent nuclear translocation to co-activate the transcription of WNT target genes (42).
Mutations that drive continuous activation of β-catenin signaling are widely reported in human cancers and in several adrenal tumors (43). Consistent with the proliferative advantage of adrenal cells with activating CTNNB1 mutations, transgenic mice with constitutively active β-catenin display adrenal cell hyperproliferation and adrenal hyperplasia (44) (Figure 1). Many studies implicate disrupted WNT/β-catenin signaling as a key mechanism in APA pathogenesis. This pathway is constitutively activated in 70% APAs, an observation related to the decreased expression of an endogenous inhibitor of the WNT/β-catenin signaling called SFRP2 (secreted frizzled related protein 2) (45). Activating somatic CTNNB1 mutations located in exon 3 have been identified in 1% to 5% of APAs (17, 18, 46). Of note, 16 of 27 (59%) APAs with a CTNNB1 activating mutation carry a concurrent G protein α subunit substitution mutation of a highly conserved glutamine residue (p.Gln209) in GNAQ or GNA11 (18) (discussed in more detail below). Overall, CTNNB1 mutations are found in only a small proportion of APAs with β-catenin cytoplasmic accumulation and nuclear translocation (17, 45, 47) thereby suggesting a role for other factors, other than mutations, in the disruption of WNT/β-catenin signaling in the pathogenesis of APAs (48).
Inactivation of the ARMC5 tumour suppressor protein
Germline and somatic inactivating mutations in the putative tumor-suppressor gene ARMC5 (armadillo repeat containing 5)- a member of the armadillo/β-catenin-like repeat superfamily- can cause primary bilateral macronodular adrenal hyperplasia (a rare form of primary adrenal Cushing syndrome) (49, 50). ARMC5 has multiple binding partners (51) and may function in PKA, and the WNT/β-catenin signaling pathways (52). Armc5 plays a vital role in early mouse embryonic development, and the T-cell immune response. In addition, the development of adrenal hyperplasia with increasing age in Armc5 knock out mice illustrates the key role of Armc5 in the adrenal (51, 52).
Zilbermint et al. (21) identified germline ARMC5 variants in 22 of 56 (39.3%) patients with PA. These included variants predicted to be damaging by in silico analysis in 6 of 56 (10.7%) patients. All patients carrying a damaging ARMC5 variant were African Americans suggesting that ARMC5 pathogenic variants might contribute to the known increased predisposition of this population to low renin hypertension (21). In contrast, ARMC5 mutations in the coding sequence or intron-to-exon boundaries in a cohort of 37 Caucasian patients with PA due to bilateral adrenal lesions were not identified (53).
Activation of G protein signaling
Abnormal expression and activation of G proteins (guanine nucleotide-binding proteins) are frequent features of tumorigenesis (40) with alterations in the G protein α subunits GNAS, GNAQ, and GNA11 commonly detected (41). G protein α subunits are components of the heterotrimeric G protein complex (comprising a Gα, Gβ, and Gγ subunit) which mediates G protein coupled receptor signal transduction. Signal activation is achieved by GDP for GTP exchange on the Gα subunit, hydrolysis of GTP to GDP terminates the signal. Recurrent mutations in GNAS, GNAQ and GNA11 disturb GTPase activity thus driving constitutively active and prolonged signaling (Figure 1).
GNAS is one of the most recurrently mutated G proteins in human cancer (40). GNAS mutations frequently alter either a p.Arg201 or a p.Gln227 residue, which are required for GTPase activity. GNAS mutations are found in a substantial proportion of cortisol-producing adenomas associated with subclinical mild autonomous cortisol excess (5 of 7, 71.4%) (54). Only a few APA with GNAS mutations (GNAS p.Arg201Cys) have been described and these were in cases of PA associated with autonomous cortisol secretion and activation of cAMP/PKA (protein kinase A) signaling (19).
Hotspot somatic mutations in the G protein α subunits GNAQ and GNA11 are found in 295 of 8778 (3.4%) and 155 of 6237 (2.5%) of human tumors, respectively (40, 55). Most are substitution mutations of residues p.Arg183 and p.Gln209 (homologous to p.Arg201 and p.Gln227 in GNAS) that impair GTP hydrolysis. However, mutations of p.Arg183 in GNAQ and GNA11 maintain some sensitivity to regulators of G protein signal termination and are less damaging variants.
As mentioned above, Zhou et al. (18) reported a high prevalence of APA GNA11 or GNAQ mutations coexisting with CTNNB1 mutations. The GNA11 or GNAQ mutations caused substitution of the conserved p.Gln209 residue for either a His, Pro, or Leu amino acid, thereby driving their constitutive activation. Transfection of primary adrenocortical cells with CTNNB1 and GNA11 mutants, individually or combined, caused increased aldosterone production. Solitary GNA11 mutations were also identified in the hyperplastic zona glomerulosa layer of the adrenal cortex adjacent to APAs with GNA11 and CTNNB1 mutations (18). This supports the 2-hit model of tumorigenesis in APA development (56) in which a first event stimulates adrenocortical cell proliferation, and a second event (a somatic aldosterone-driver mutation) drives autonomous aldosterone production (4, 57, 58).
Activation of cAMP/protein kinase A signaling
Cyclic adenosine monophosphate (cAMP) regulates multiple cellular functions in most cell types via the control of target gene transcription mainly mediated by protein kinase A (PKA). Adenylyl cyclase is activated by GNAS binding thus enabling the conversion of adenosine triphosphate (ATP) to cAMP, which in turn activates PKA. The PKA heterotetramer comprises two regulatory subunits (PRKAR1A) and two catalytic subunits (PRKACA). The regulatory subunits maintain the inactivity of the catalytic subunits in the tetrameric complex. cAMP binding to each regulatory subunit causes their dissociation and activation (59). PKA regulates transcription by direct phosphorylation of transcription factors such as CREB (cAMP-response element-binding protein), and CREM (cAMP-responsive modulator) so they can bind to cAMP-response elements in target genes.
Constitutive activation of cAMP/PKA signaling due to GNAS or PRKACA mutations is linked to the formation of many tumors including endocrine tumors (60, 61). The rare examples of APA GNAS mutations are discussed above (under G protein signaling). Somatic PRKACA mutations, encoding a p.Leu206Arg substitution in the catalytic PKA subunit, have been identified in a substantial proportion of cortisol-producing adenomas (54, 60, 62, 63). PRKACA Leu206 directly interacts with the PKA regulatory subunit. The PRKACA p.Leu206Arg mutation disrupts this interaction causing PRKACA constitutive activation with consequent increased phosphorylation of downstream targets (60). Expression of PRKACA p.Leu206Arg in vitro causes increased basal PKA activity and cAMP signaling (60, 62), which can account for the dysregulated cortisol production and cell proliferation of these tumors. In PA, PRKACA p.Leu206Arg mutations are rare (20, 60) and seem to occur in some patients with cortisol co-secretion. In addition, the adenoma with the PRKACA p.Leu206Arg mutation can be negative for CYP11B2 (aldosterone synthase) immunostaining (64, 65) indicating the likely function of this mutation in tumor formation.
Transcriptomics of aldosterone-producing adenomas
APA transcriptome analyses show considerable heterogeneity in methodology (different reference tissues, different gene expression platforms). Despite this, many studies report consistent findings, which have helped define the panorama of transcriptome changes in APA formation and have highlighted genes and signaling pathways that function in dysregulated aldosterone production (Table 2). Below we focus on differentially expressed genes in APAs which are implicated in mechanisms of cell death and proliferation. The application of transcriptomics to identify mechanisms of aldosterone overproduction are described elsewhere (66, 67).
Mechanisms of cell death
Early reports relied on microarray gene expression studies. Transcriptome comparison of 8 APAs with those of 3 normal adrenals identified a range of differentially expressed genes which included TDGF1 (teratocarcinoma-derived growth factor 1)- also reported as upregulated by a SAGE (serial analysis of gene expression) study (68)- and VSNL1 (visinin-like 1) encoding a calcium binding protein (69, 70). Overexpression of TDGF1 and VSNL1 in human adrenocortical cells in vitro suggested that each gene played a role in cell survival by protection against cell death by apoptosis (69, 70). The nuclear transcription factor and WNT/β-catenin target AFF3, also protects adrenal cells from apoptosis and is a positive regulator of adrenal cell proliferation (71). mRNA-seq analysis of 15 APAs suggested a role for AFF3 in APA pathophysiology by demonstrating upregulated AFF3 gene expression in CTNNB1 mutated APAs versus those without a CTNNB1 mutation (72) (Table 2).
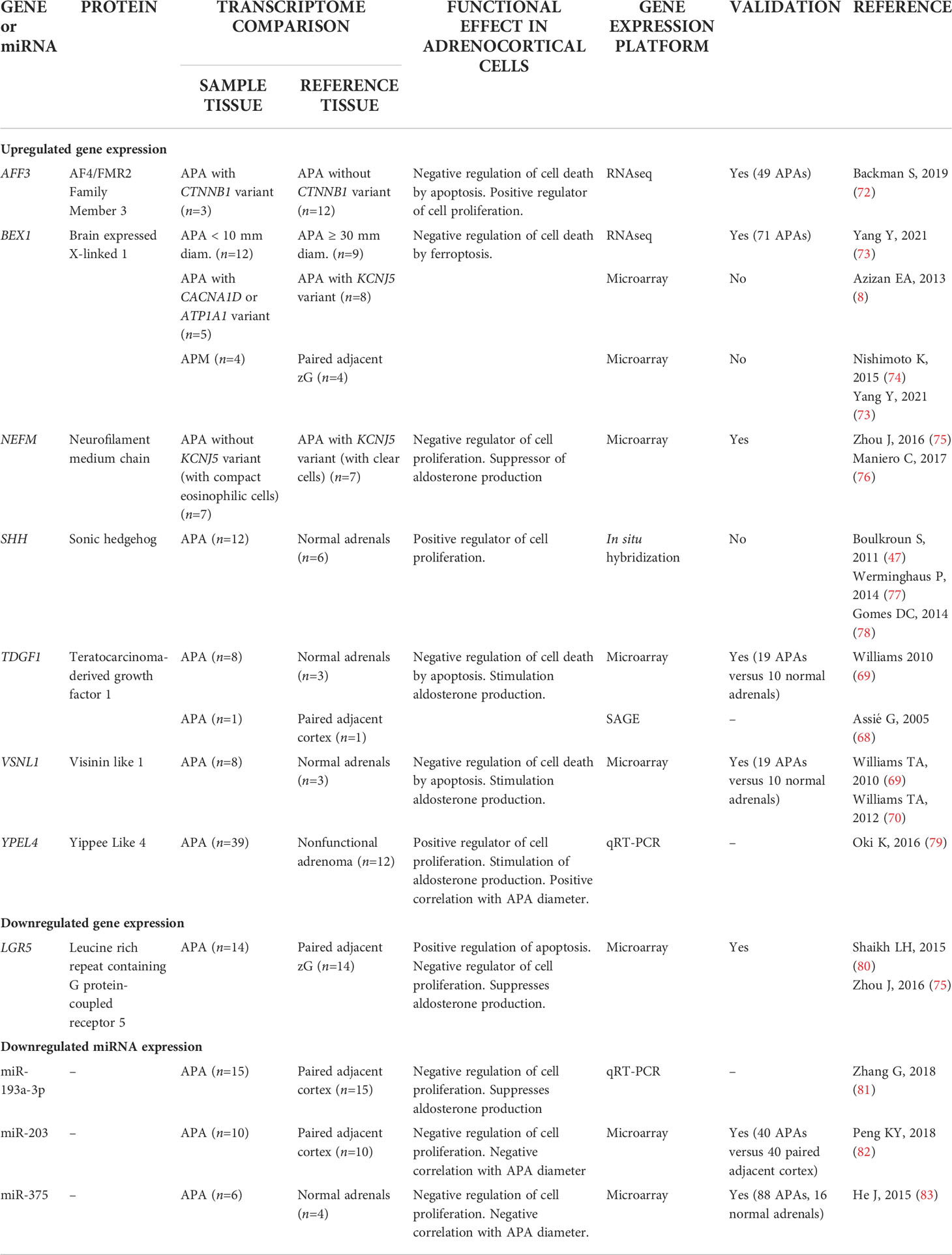
Table 2 Potential function of differentially expressed genes and Mirnas in Aldosterone-Producing Lesions.
Transcriptome analysis of APAs and paired adjacent zona glomerulosa highlighted a potential role for oxidative stress in APA pathogenesis (75). The top canonical biological pathway associated with the differentially expressed genes was NRF2 (nuclear factor erythroid 2–related factor 2)-mediated oxidative stress, which is a critical cellular mechanism to maintain intracellular redox homeostasis and limit oxidative damage (84). An imbalance in redox-based metabolic processes can result in inappropriate production of reactive oxygen species (ROS) and oxidative damage to membrane lipids. If cellular antioxidant systems are unable to inhibit oxidative damage, a chain reaction of lipid peroxidation occurs, and cells commit to a form of regulated cell death called ferroptosis (85). GPX4 (glutathione peroxidase 4) is an essential antioxidant peroxidase which catalyzes the reduction of lipid hydroperoxides to protect cells from ferroptosis. Adrenocortical cells are particularly sensitive to inducers of ferroptosis which act via GPX4 inhibition (86). Functional enrichment analysis of differentially expressed genes in APAs compared with adjacent zona glomerulosa identified a ferroptosis-related gene set (87). The upregulated genes included SCD and GCLC, which encode key enzymes with a ferroptosis protective function in the catalysis of monounsaturated fatty acids and glutathione biosynthesis.
To detect mechanisms of APA tumorigenesis, Yang et al. (73) compared APA transcriptomes of highly diverse sizes (9 macro APAs with adenoma diameter ≥30 mm versus 12 micro APAs ≤10 mm). Over-representation analysis of the transcriptome dataset from Yang et al. (73) illustrates enrichment of cell survival pathways (negative regulation of cell growth, positive regulation of cell death, and positive regulation of apoptotic processes) in the KCNJ5 mutation-negative macro versus micro APAs but not in KCNJ5-mutated APAs (Figures 2A, B). Thus, activation of gene expression programs with a negative impact on cell survival can limit the size of APAs without a KCNJ5 mutation. In contrast, this is not a feature of APAs with a KCNJ5 mutation which are characterized by their larger tumor size (28).
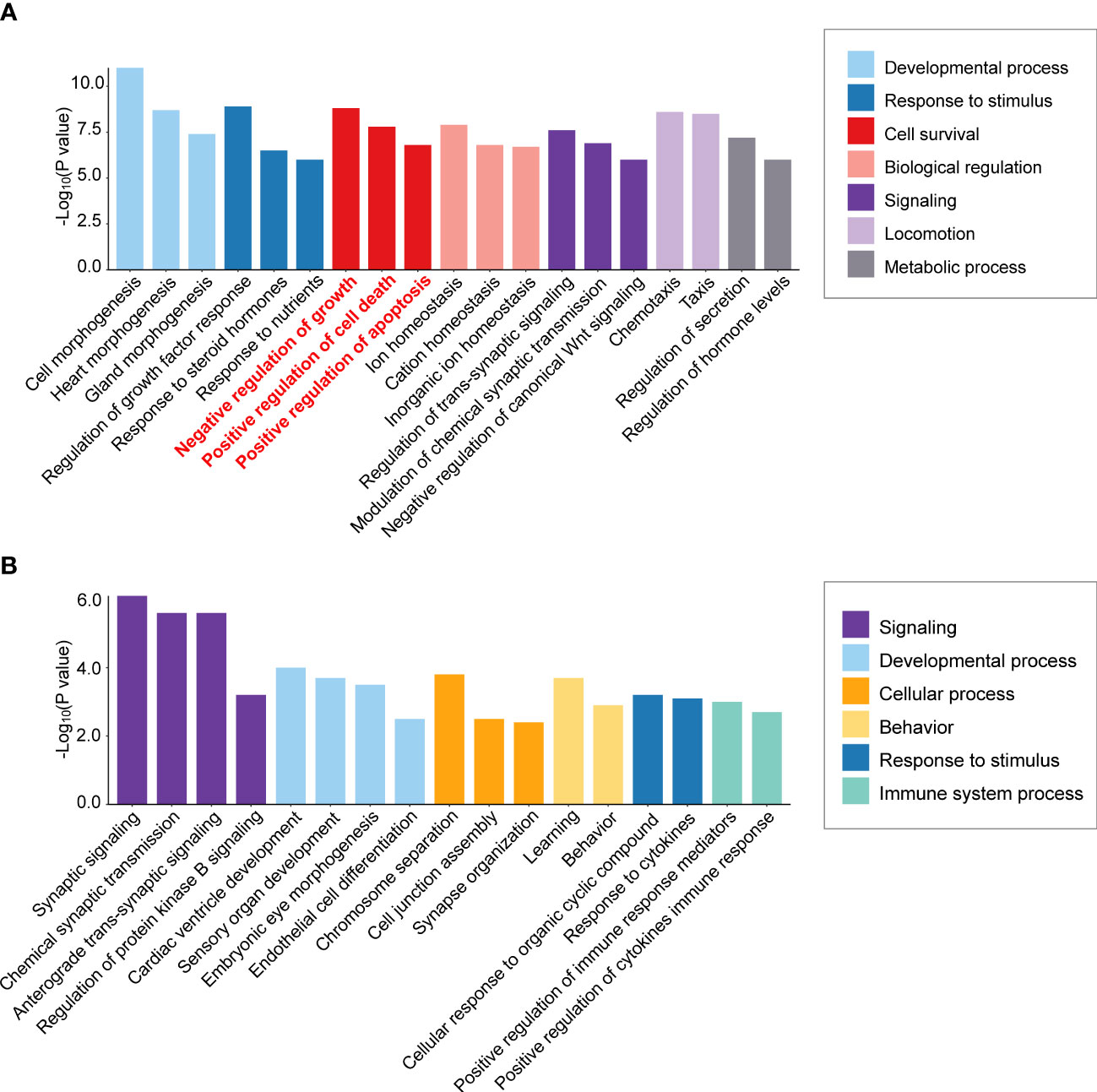
Figure 2 Differential enrichment of cell survival pathways in aldosterone-producing adenomas according to KCNJ5 (GIRK4) mutation status. Biological process enrichment analysis was performed of significantly differentially expressed genes in macro versus micro APAs (≥30 mm versus ≤10 mm adenoma diameter) without a KCNJ5 mutation (A) and with a KCNJ5 mutation (B) using Metascape (http://metascape.org/accessed on 26 February 2022) analysis of publicly available dataset (https://github.com/MedIVLMUMunich/MacroMicroAPA_RNAseq). Enrichment visualization was performed using R package ggplot2 (v3.3.5).
Validation of expression levels of genes associated with processes of cell death and cell proliferation in an expanded sample set of 71 APAs identified a subset of genes with transcription profiles strongly correlated with adenoma diameter in KCNJ5-mutation negative APAs (73). These included BEX1 (brain expressed X-linked 1) which was inversely correlated with APA diameter and was upregulated in APAs relative to paired adjacent cortex. A previous study (8) reported upregulated BEX1 gene expression in APAs with a CACNA1D or ATP1A1 mutation (that tend to be small adenomas) compared with the larger KCNJ5-mutated APAs (8, 28). Flow cytometry analyses of human adrenocortical cells with stable BEX1 overexpression demonstrated its role in protection from cell death by ferroptosis, rather than by apoptosis or by interference with cell cycle progression (73). The relatively high BEX1 gene expression in smaller versus larger APAs and in aldosterone-producing micronodules (APMs) compared with adjacent zona glomerulosa cells (73, 74) implicates a role for BEX1 in the promotion of cell survival in the initiation of adenoma formation.
Mechanisms of cell proliferation
Genes involved in calcium signaling were first highlighted by some of the first APA gene expression studies (88), consistent with the later discovery of activation of the calcium signaling system by aldosterone-driver mutations (89). Signaling cascades initiated by intracellular Ca2+ are ubiquitously employed for the regulation of cell proliferation. This is achieved at several levels including by the promotion of resting G0 cell entry to the cell cycle, activation of the initiation of DNA synthesis at the G1 to S phase transition, and by stimulation of mitosis (90). Thus, in addition to a role in aldosterone production, genes which function in calcium signaling might also mediate cell proliferation, such as those with a calcium binding function such as VSNL1 (visinin-like), CALN1 (calneuron 1) and CLGN (calmegin), which are all significantly upregulated in APAs (70, 91, 92).
The YPEL (yippee-like) gene family encodes proteins with putative zinc-finger-like metal-binding domains (yippee domains). The encoded proteins localize to the centrosome and nucleolus and have been proposed to regulate cell division and proliferation (93). Transcriptome analysis of freshly isolated rat adrenal zona glomerulosa cells treated with angiotensin II (100 nM) or potassium (16 mM KCl) identified a set of genes, comprising YPEL4, that might function in aldosterone production (94). The upregulation of YPEL4 in response to angiotensin II or K+ stimulation in rat adrenal cells was successively observed in human adrenocortical cells which caused increased adrenal cell proliferation (79). A role for YEPL4 in adrenal cell proliferation in APAs was also suggested by the slight but significant positive correlation of YPEL4 gene expression levels with APA diameter (79) (Table 2).
In a microarray study of 14 APAs, LGR5 (leucine rich repeat containing G protein-coupled receptor 5) was downregulated in APA compared with adjacent zona glomerulosa (75, 80). LGR5 overexpression in human adrenal cells caused increased apoptosis, a reduction of proliferation, and decreased aldosterone production (80). Moreover, NEFM (neurofilament medium chain) was upregulated in APAs without a KCNJ5 mutation relative to those with a KCNJ5 mutation (75) and transfection of mutated KCNJ5 into adrenal cells significantly decreased NEFM gene expression levels (60). Consistently, NEFM gene silencing resulted in increased adrenal cell proliferation and amplified aldosterone production (Table 2).
A large transcriptome analysis of APAs identified the potential role of the retinoic acid receptor alpha (RARα) in adrenal cell proliferation. Inactivation of Rarα in transgenic mice caused an inhibition of non-canonical Wnt signaling and increased proliferation in male mice (95). The study suggests that RARα might function in the structural maintenance of the adrenal cortex and that disrupted RARα signaling could be factor which contributes to APA pathogenesis.
In situ metabolomics of aldosterone-producing adenomas and micronodules by mass spectrometry imaging
In situ matrix-assisted laser desorption/ionization mass spectrometry imaging (MALDI-MSI) is a novel technique that can simultaneously measure up to thousands of molecules in a single tissue section. The detected molecules encompass metabolites, lipids, glycans, peptides and proteins, as well as drugs and their metabolites (96). The approach allows co-integration of the detected molecules with conventional hematoxylin and eosin staining or immunohistochemistry and thus enables the spatial visualization of metabolite distribution, and other molecules including hormones, in the context of morphology or protein expression.
According to classical adrenal morphology, the adrenal comprises 3 distinct concentric zones of the cortex (glomerulosa, fasciculata, and reticularis) and the adrenal medulla. Using MALDI-Fourier transform-ion cyclotron resonance-MSI (MALDI-FT-ICR-MSI) applied to fresh frozen normal human adrenal sections, Sun et al. (97) determined a new molecular definition of adrenal gland anatomy. The study established the unexpected complexity of the adrenal by visualization of 10 clearly distinct molecular zones (6 cortical and 4 medullary substructures). The functional and physiological relevance of this multi-layered molecular anatomy remains to be determined but this technique has been extensively applied to extend our knowledge of adrenal pathology (98–100).
In addition to fresh frozen tissue sections, MALDI-FT-ICR MSI can reliably be performed on formalin-fixed paraffin embedded (FFPE) archived tissue samples, including individual biopsy specimens, and tissue microarrays comprising thousands of tissue cores. Comparison of mass spectrometry peaks from tissue specimens that had been fresh frozen or paraffin-embedded, revealed that similar metabolites were detected, with a 72% overlap, of comparable peak intensities, particularly for non-lipid low molecular weight metabolites (101).
MALDI-FT-ICR MSI analysis of a tissue microarray representing 132 APAs with genotype data showed that samples did not cluster according to genotype in the total dataset. However, restricting analysis to adenomas with either a KCNJ5 or a CACNA1D mutation classified these 2 genotype groups with significant differences of 137 metabolites (98). Biological pathway analysis revealed enrichment of purine metabolism with increased purine synthesis in KCNJ5-mutated APAs, which are generally larger, than those with a CACNA1D mutation. Thus, the increased purine synthesis might conceivably result from cell cycle promotion and enhanced cell proliferation (98).
Sugiura et al. (99) used MALDI-MSI with chemical derivatization to visualize specific steroids in fresh frozen adrenal tissue sections. The study generally confirmed the production of aldosterone in areas of CYP11B2 immunostaining in APAs and in aldosterone-producing micronodules (APMs) (99). APMs are small (<10 mm) CYP11B2-positive adrenal lesions located under the adrenal capsule (102, 103). They frequently carry somatic mutations in CACNA1D, ATP2B3 and ATP1A1 corresponding to those found in APAs (104). In contrast, KCNJ5 mutations are largely absent in APMs, despite their high frequency in APAs (74). A small number of adrenal lesions with hybrid immunohistological features composed of an outer APM-like part, characterized by CYP11B2 but not CYP11B1 immunostaining, and an inner APA-like part, with both CYP11B2 and CYP11B1 immunostaining have been described (105). The occurrence of mutations common to both APMs and APAs and the observation of hybrid lesions led to the proposal that APMs might be precursors of APAs in some cases (105, 106) (Figure 3).
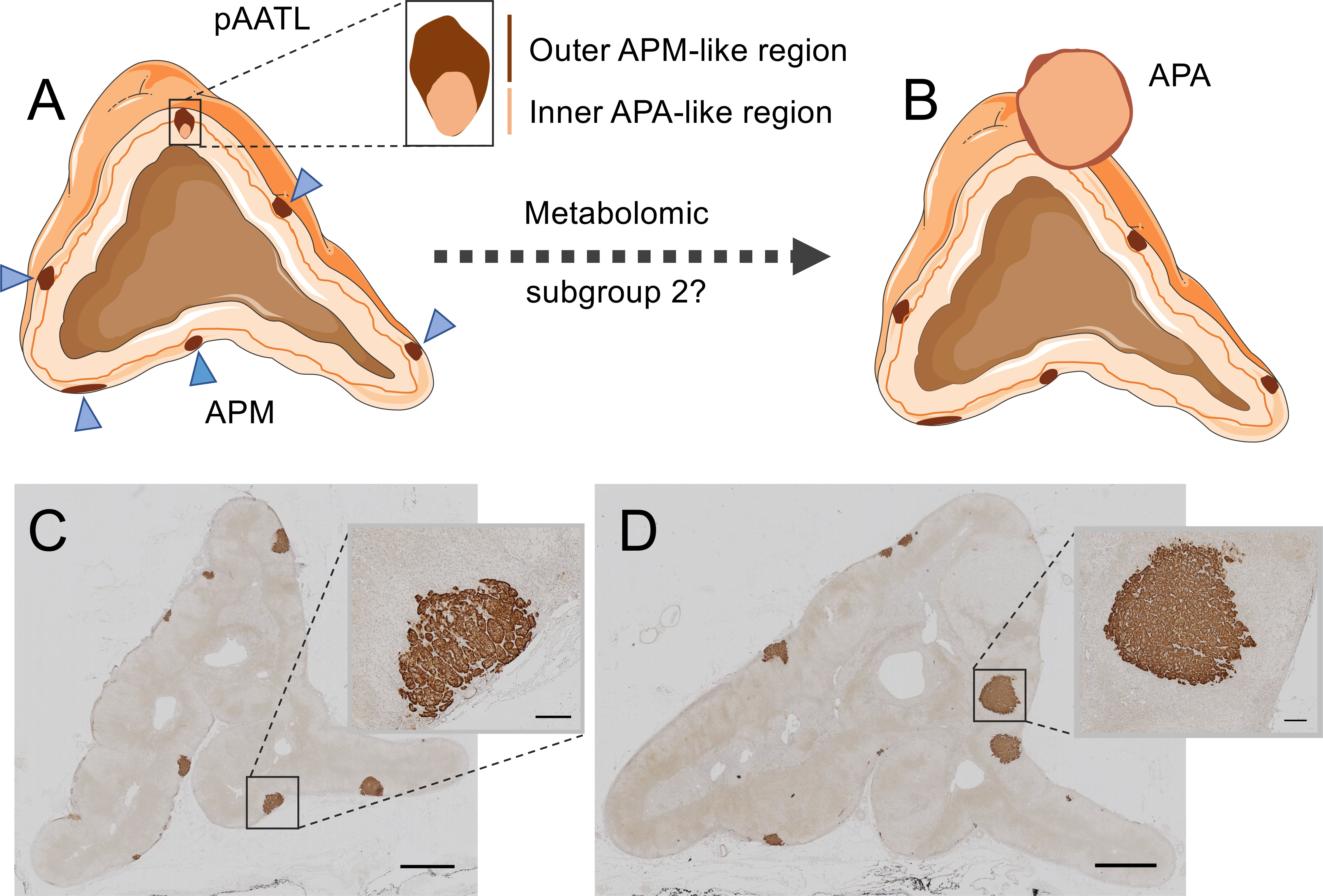
Figure 3 Hybrid lesions in the transition of aldosterone-producing micronodules to adenomas. The pAATL is a lesion composed of an outer APM-like region (with positive immunostaining for CYP11B2 but negative for CYP11B1) and an inner APA-like region (with positive immunostaining for both CYP11B2 and CYP11B1) that can have a different mutation status for PA-driver genes (A). Thus, pAATLs have been proposed as hybrid APM-APA lesions and might represent intermediary lesions in the transit of APMs to APAs. Subgroups of APMs can be differentiated by their highly divergent metabolic profiles. A subset of APMs (metabolic subgroup 2) display a metabolic signature like that of APAs and might suggest progression to APAs (B). CYP11B2 (aldosterone synthase) immunostaining of an adrenal surgically removed from a patient with PA (C, D). The adrenal in Panel C shows multiple APMs (magnified in inset), a different sample block of the same adrenal in Panel D shows formation of aldosterone-producing nodules (magnified in inset). Bars = 2mm (main image) or 200 μm (inset). Blue triangles indicate APMs in Panel (A) APA, aldosterone-producing adenoma; APM, aldosterone-producing micronodule; pAATL, possible APM-to-APA transitional lesion; CYP11B1, 11β-hydroxylase; CYP11B2, aldosterone synthase. Figure produced using Servier Medical Art (https://smart.servier.com/).
MALDI-FT-ICR MSI was used to determine the metabolomic phenotypes of 27 APMs and 6 APAs from surgically resected adrenals from patients with PA (100). The study established 2 subgroups of APMs (subgroups 1 and 2) with distinct metabolic phenotypes. The pattern of metabolites in APM subgroup 1 (20 of 27 APMs) was clearly separated from that of both subgroup 2 (7 of 27 APMs) and APAs. In contrast, the metabolic phenotype of APMs in subgroup 2 closely resembled that of APAs and displayed enrichment of biological pathways supporting cell proliferation and potentially tumour progression with increased purine synthesis characterizing APM subgroup 2 (100). Mutational status did not appear to account for the difference in metabolic signatures between the 2 APM subgroups and they were indistinguishable by immunohistology. Despite the apparent absence of hybrid lesions, these observations support the hypothesis of the “transitional lesion” in which a subgroup of APMs progress to APAs (100, 105) (Figure 3). Such a model in which an APM can transit to form an APA is an alternative to the 2-hit hypothesis of APA development (57) in which an initial event stimulates cell proliferation [such as a genetic variant (107) or circulating factor (108, 109)] and a subsequent somatic mutation in a stimulated cell, drives the aldosterone overproduction.
Emerging role of epigenetics and micro RNAs
Increasing numbers of studies report the disruption of epigenetic and post-transcriptional mechanisms, notably by DNA methylation and microRNAs (miRNA/miR), as mechanisms which might facilitate APA pathogenesis.
DNA methylation in the pathophysiology of aldosterone-producing adenomas
Several studies highlight deregulated DNA methylation as an epigenetic mechanism in APA pathogenesis. Gene expression is typically repressed by cytosine methylation of CpG (5’-cytosine-guanine-3’ dinucleotide) islands in target gene promoter regions. APAs display a distinct pattern of methylation (hypomethylated status) compared with their adjacent adrenal cortex and non-functioning adrenal adenomas (110, 111). CYP11B2 was hypomethylated and upregulated, a process which was bypassed in the presence of either APA KCNJ5 or ATP1A1 mutations (110–112). Murakami et al. (113) integrated a genome-wide methylome analysis with transcriptome data and demonstrated that many genes involved in tumorigenesis (HOX family genes, PRRX1, RAB38, FAP, GCNT2, and ASB4) and steroidogenesis (CYP11B2, MC2R, and HPX) were hypomethylated and upregulated in APAs compared with adjacent cortex.
Micro RNAs in the pathophysiology of aldosterone-producing adenomas
MicroRNAs (miRs, miRNAs) are small molecules, 18-22 nucleotides in length, that regulate post-transcriptional gene expression levels usually via downregulating the expression of specific genes by binding to the 3´untranslated regions of their corresponding mRNAs (114, 115). miRNA expression profiling can characterize diseased states and, as such, might be promising as a diagnostic tool (114). In PA, changes in circulating plasma miRNA expression levels have been reported in patients according to subtype with significant levels of miR-30e-5p, miR-30d-5p, and miR-7-5p overexpression in patients with bilateral adrenal hyperplasia versus those with a unilateral APA (116, 117).
In addition to circulating miRNAs, several studies have reported downregulated miRNAs in APA tissue samples compared with the adjacent adrenal cortex or relative to other resected adrenal specimens (Table 2). Downregulated miR-375 expression was reported in APA relative to both unilateral adrenal hyperplasia and normal adrenals. A role for miR-375 in tumorigenesis is supported by the reduction in adrenal cell viability in response to miR-375 overexpression in vitro. In addition, miR-375 expression levels are negatively correlated with APA diameter and are also downregulated in other tumors (82, 83). The putative tumorigenic function of miR-375 might be mediated by decreased expression of the target gene MTDH (metadherin) and subsequent suppression of Akt signalling (83). Peng et al. identified miRNA-203 as a candidate functional miRNA in APA pathophysiology from microarray-based expression analysis of 10 APA tissues and paired adjacent adrenal cortex (82). Treatment of human adrenal cells with miR-203 inhibitors caused increased aldosterone production and cell proliferation. Moreover, miR-203 mimics resulted in decreased adrenal cell proliferation as well as aldosterone hypersecretion from primary cell cultures derived from APA tissue. WNT5A was identified as a direct target of miR-203 implicating WNT5A/β-catenin signaling in mediating the observed functional effects (82).
Perspectives
Diverse mechanisms alter the balance between adrenal cell death and proliferation to favor APA formation and development. These processes can vary according to genotype. Unrestrained proliferation of adrenal cells carrying a KCNJ5 mutation contrasts with specific gene expression programs elicited in KCNJ5 mutation negative APAs which influence adrenal cell viability and potentially regulate tumor size. The application of advanced omics technologies has tremendous potential to advance our understanding of the underlying biology of APA tumorigenesis. Accordingly, single-cell and spatially resolved transcriptomics will provide a detailed cellular atlas of PA adrenals and define gene expression profiles and genotype in selected cell populations. Integration of spatial transcriptomics with metabolic phenotyping using in situ mass spectrometry imaging will achieve a higher-level definition of biological pathways in cell subpopulations and elucidate the fundamental pathological processes in APA tumor cells.
Author contributions
All authors listed have made a substantial, direct, and intellectual contribution to the work and approved it for publication.
Funding
The Deutsche Forschungsgemeinschaft (DFG, German Research Foundation) supports TW and MR (project number 314061271 - TRR 205, and project number 444776998 [WI 5359/2-1 and RE 752/31-1]). The Else Kröner-Fresenius Stiftung (2012_A103, 2015_A228, and 2019_A104; Else-Kröner Hyperaldosteronismus- German Conn Registry) also supports the work of MR.
Acknowledgments
We gratefully acknowledge the excellent assistance of Isabella-Sabrina Kinker with Figures.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Reincke M, Bancos I, Mulatero P, Scholl UI, Stowasser M, Williams TA. Diagnosis and treatment of primary aldosteronism. Lancet Diabetes Endocrinol (2021) 9:876–92. doi: 10.1016/S2213-8587(21)00210-2
2. Monticone S, D'Ascenzo F, Moretti C, Williams TA, Veglio F, Gaita F, et al. Cardiovascular events and target organ damage in primary aldosteronism compared with essential hypertension: a systematic review and meta-analysis. Lancet Diabetes Endocrinol (2018) 6:41–50. doi: 10.1016/S2213-8587(17)30319-4
3. Williams TA, Reincke M. Pathophysiology and histopathology of primary aldosteronism. Trends Endocrinol Metab (2022) 33:36–49. doi: 10.1016/j.tem.2021.10.002
4. Scholl UI. Genetics of primary aldosteronism. Hypertension (2022) 79(5):887–97. doi: 10.1161/HYPERTENSIONAHA.121.16498
5. Lifton RP, Dluhy RG, Powers M, Rich GM, Cook S, Ulick S, et al. A chimaeric 11 beta-hydroxylase/aldosterone synthase gene causes glucocorticoid-remediable aldosteronism and human hypertension. Nature (1992) 355:262–5. doi: 10.1038/355262a0
6. Choi M, Scholl UI, Yue P, Björklund P, Zhao B, Nelson-Williams C, et al. K+ channel mutations in adrenal aldosterone-producing adenomas and hereditary hypertension. Science (2011) 331:768–72. doi: 10.1126/science.1198785
7. Scholl UI, Goh G, Stölting G, de Oliveira RC, Choi M, Overton JD, et al. Somatic and germline CACNA1D calcium channel mutations in aldosterone-producing adenomas and primary aldosteronism. Nat Genet (2013) 45:1050–4. doi: 10.1038/ng.2695
8. Azizan EA, Poulsen H, Tuluc P, Zhou J, Clausen MV, Lieb A, et al. Somatic mutations in ATP1A1 and CACNA1D underlie a common subtype of adrenal hypertension. Nat Genet (2013) 45:1055–60. doi: 10.1038/ng.2716
9. Omata K, Satoh F, Morimoto R, Ito S, Yamazaki Y, Nakamura Y, et al. Cellular and genetic causes of idiopathic hyperaldosteronism. Hypertension (2018) 72:874–80. doi: 10.1161/HYPERTENSIONAHA.118.11086
10. Scholl UI, Stölting G, Nelson-Williams C, Vichot AA, Choi M, Loring E, et al. Recurrent gain of function mutation in calcium channel CACNA1H causes early-onset hypertension with primary aldosteronism. Elife (2015) 4:e06315. doi: 10.7554/eLife.06315
11. Daniil G, Fernandes-Rosa FL, Chemin J, Blesneac I, Beltrand J, Polak M, et al. CACNA1H mutations are associated with different forms of primary aldosteronism. EBioMedicine (2016) 13:225–36. doi: 10.1016/j.ebiom.2016.10.002
12. Nanba K, Blinder AR, Rege J, Hattangady NG, Else T, Liu CJ, et al. Somatic CACNA1H mutation as a cause of aldosterone-producing adenoma. Hypertension (2020) 75:645–9. doi: 10.1161/HYPERTENSIONAHA.119.14349
13. Scholl UI, Stölting G, Schewe J, Thiel A, Tan H, Nelson-Williams C, et al. CLCN2 chloride channel mutations in familial hyperaldosteronism type II. Nat Genet (2018) 50:349–54. doi: 10.1038/s41588-018-0048-5
14. Fernandes-Rosa FL, Daniil G, Orozco IJ, Göppner C, El Zein R, Jain V, et al. A gain-of-function mutation in the CLCN2 chloride channel gene causes primary aldosteronism. Nat Genet (2018) 50:355–61. doi: 10.1038/s41588-018-0053-8
15. Dutta RK, Arnesen T, Heie A, Walz M, Alesina P, Söderkvist P, et al. A somatic mutation in CLCN2 identified in a sporadic aldosterone-producing adenoma. Eur J Endocrinol (2019) 181:K37–41. doi: 10.1530/EJE-19-0377
16. Beuschlein F, Boulkroun S, Osswald A, Wieland T, Nielsen HN, Lichtenauer UD, et al. Somatic mutations in ATP1A1 and ATP2B3 lead to aldosterone-producing adenomas and secondary hypertension. Nat Genet (2013) 45:440–4, 444e1-2. doi: 10.1038/ng.2550
17. Åkerström T, Maharjan R, Sven Willenberg H, Cupisti K, Ip J, Moser A, et al. Activating mutations in CTNNB1 in aldosterone producing adenomas. Sci Rep (2016) 6:19546. doi: 10.1038/srep19546
18. Zhou J, Azizan EAB, Cabrera CP, Fernandes-Rosa FL, Boulkroun S, Argentesi G, et al. Somatic mutations of GNA11 and GNAQ in CTNNB1-mutant aldosterone-producing adenomas presenting in puberty, pregnancy or menopause. Nat Genet (2021) 53:1360–72. doi: 10.1038/s41588-021-00906-y
19. Nakajima Y, Okamura T, Horiguchi K, Gohko T, Miyamoto T, Satoh T, et al. GNAS mutations in adrenal aldosterone-producing adenomas. Endocr J (2016) 63(2):199–204. doi: 10.1507/endocrj.EJ15-0642
20. Rhayem Y, Perez-Rivas LG, Dietz A, Bathon K, Gebhard C, Riester A, et al. PRKACA somatic mutations are rare findings in aldosterone-producing adenomas. J Clin Endocrinol Metab (2016) 101:3010–7. doi: 10.1210/jc.2016-1700
21. Zilbermint M, Xekouki P, Faucz FR, Berthon A, Gkourogianni A, Schernthaner-Reiter MH, et al. Primary aldosteronism and ARMC5 variants. J Clin Endocrinol Metab (2015) 100:E900–9. doi: 10.1210/jc.2014-4167
22. Nanba K, Omata K, Else T, Beck PCC, Nanba AT, Turcu AF, et al. Targeted molecular characterization of aldosterone-producing adenomas in white americans. J Clin Endocrinol Metab (2018) 103:3869–76. doi: 10.1210/jc.2018-01004
23. Nanba K, Omata K, Gomez-Sanchez CE, Stratakis CA, Demidowich AP, Suzuki M, et al. Genetic characteristics of aldosterone-producing adenomas in blacks. Hypertension (2019) 73:885–92. doi: 10.1161/HYPERTENSIONAHA.118.12070
24. Nanba K, Yamazaki Y, Bick N, Onodera K, Tezuka Y, Omata K, et al. Prevalence of somatic mutations in aldosterone-producing adenomas in Japanese patients. J Clin Endocrinol Metab (2020) 105:e4066–73. doi: 10.1210/clinem/dgaa595
25. De Sousa K, Boulkroun S, Baron S, Nanba K, Wack M, Rainey WE, et al. Genetic, cellular, and molecular heterogeneity in adrenals with aldosterone-producing adenoma. Hypertension (2020) 75:1034–44. doi: 10.1161/HYPERTENSIONAHA.119.14177
26. Rege J, Nanba K, Blinder AR, Plaska S, Udager AM, Vats P, et al. Identification of somatic mutations in CLCN2 in aldosterone-producing adenomas. J Endocr Soc (2020) 4:bvaa123. doi: 10.1210/jendso/bvaa123
27. Meyer LS, Handgriff L, Lim JS, Udager AM, Kinker IS, Ladurner R, et al. Single-center prospective cohort study on the histopathology, genotype, and postsurgical outcomes of patients with primary aldosteronism. Hypertension (2021) 78:738–46. doi: 10.1161/HYPERTENSIONAHA.121.17348
28. Lenzini L, Rossitto G, Maiolino G, Letizia C, Funder JW, Rossi GP. A meta-analysis of somatic KCNJ5 k(+) channel mutations in 1636 patients with an aldosterone-producing adenoma. J Clin Endocrinol Metab (2015) 100:E1089–95. doi: 10.1210/jc.2015-2149
29. Nanba K, Rainey WE, Udager AM. Approaches to gene mutation analysis using formalin-fixed paraffin-embedded adrenal tumor tissue from patients with primary aldosteronism. Front Endocrinol (Lausanne) (2021) 12:683588. doi: 10.3389/fendo.2021.683588
30. Guo Z, Nanba K, Udager A, McWhinney BC, Ungerer JPJ, Wolley M, et al. Biochemical, histopathological, and genetic characterization of posture-responsive and unresponsive APAs. J Clin Endocrinol Metab (2020) 105:e3224–35. doi: 10.1210/clinem/dgaa367
31. Scholl UI, Nelson-Williams C, Yue P, Grekin R, Wyatt RJ, Dillon MJ, et al. Hypertension with or without adrenal hyperplasia due to different inherited mutations in the potassium channel KCNJ5. Proc Natl Acad Sci U.S.A. (2012) 109:2533–8. doi: 10.1073/pnas.1121407109
32. Yang Y, Gomez-Sanchez CE, Jaquin D, Aristizabal Prada ET, Meyer LS, Knösel T, et al. Primary aldosteronism: KCNJ5 mutations and adrenocortical cell growth. Hypertension (2019) 74:809–16. doi: 10.1161/HYPERTENSIONAHA.119.13476
33. Williams TA, Monticone S, Schack VR, Stindl J, Burrello J, Buffolo F, et al. Somatic ATP1A1, ATP2B3, and KCNJ5 mutations in aldosterone-producing adenomas. Hypertension (2014) 63:188–95. doi: 10.1161/HYPERTENSIONAHA.113.01733
34. Stindl J, Tauber P, Sterner C, Tegtmeier I, Warth R, Bandulik S. Pathogenesis of adrenal aldosterone-producing adenomas carrying mutations of the Na(+)/K(+)-ATPase. Endocrinology (2015) 156:4582–91. doi: 10.1210/en.2015-1466
35. Aizman O, Uhlén P, Lal M, Brismar H, Aperia A. Ouabain, a steroid hormone that signals with slow calcium oscillations. Proc Natl Acad Sci U S A (2001) 98:13420–4. doi: 10.1073/pnas.221315298
36. Barwe SP, Anilkumar G, Moon SY, Zheng Y, Whitelegge JP, Rajasekaran SA, et al. Novel role for Na,K-ATPase in phosphatidylinositol 3-kinase signaling and suppression of cell motility. Mol Biol Cell (2005) 16:1082–94. doi: 10.1091/mbc.e04-05-0427
37. Tian J, Cai T, Yuan Z, Wang H, Liu L, Haas M, et al. Binding of src to Na+/K+-ATPase forms a functional signaling complex. Mol Biol Cell (2006) 17:317–26. doi: 10.1091/mbc.e05-08-0735
38. Kobuke K, Oki K, Gomez-Sanchez CE, Gomez-Sanchez EP, Itcho K, Ohno H, et al. ATP1A1 mutant in aldosterone-producing adenoma leads to cell proliferation. Int J Mol Sci (2021) 22:10981. doi: 10.3390/ijms222010981
39. Wu VC, Wang SM, Chueh SJ, Yang SY, Huang KH, Lin YH, et al. The prevalence of CTNNB1 mutations in primary aldosteronism and consequences for clinical outcomes. Sci Rep (2017) 7:39121. doi: 10.1038/srep39121
40. O'Hayre M, Vázquez-Prado J, Kufareva I, Stawiski EW, Handel TM, Seshagiri S, et al. The emerging mutational landscape of G proteins and G-protein-coupled receptors in cancer. Nat Rev Cancer (2013) 13:412–24. doi: 10.1038/nrc3521
41. Parish AJ, Nguyen V, Goodman AM, Murugesan K, Frampton GM, Kurzrock R. GNAS. GNAQ, and GNA11 alterations in patients with diverse cancers. Cancer (2018) 124:4080–9. doi: 10.1002/cncr.31724
42. Lecarpentier Y, Schussler O, Hébert JL, Vallée A. Multiple targets of the canonical WNT/β-catenin signaling in cancers. Front Oncol (2019) 9:1248. doi: 10.3389/fonc.2019.01248
43. Tissier F, Cavard C, Groussin L, Perlemoine K, Fumey G, Hagneré AM, et al. Mutations of beta-catenin in adrenocortical tumors: activation of the wnt signaling pathway is a frequent event in both benign and malignant adrenocortical tumors. Cancer Res (2005) 65:7622–7. doi: 10.1158/0008-5472.CAN-05-0593
44. Berthon A, Sahut-Barnola I, Lambert-Langlais S, de Joussineau C, Damon-Soubeyrand C, Louiset E, et al. Constitutive beta-catenin activation induces adrenal hyperplasia and promotes adrenal cancer development. Hum Mol Genet (2010) 19:1561–76. doi: 10.1093/hmg/ddq029
45. Berthon A, Drelon C, Ragazzon B, Boulkroun S, Tissier F, Amar L, et al. WNT/β-catenin signalling is activated in aldosterone-producing adenomas and controls aldosterone production. Hum Mol Genet (2014) 23:889–905. doi: 10.1093/hmg/ddt484
46. Itcho K, Oki K, Ohno H, Yoneda M. Update on genetics of primary aldosteronism. Biomedicines (2021) 9:409. doi: 10.3390/biomedicines9040409
47. Boulkroun S, Samson-Couterie B, Golib-Dzib JF, Amar L, Plouin PF, Sibony M, et al. Aldosterone-producing adenoma formation in the adrenal cortex involves expression of stem/progenitor cell markers. Endocrinology (2011) 152:4753–63. doi: 10.1210/en.2011-1205
48. Di Dalmazi G, Altieri B, Scholz C, Sbiera S, Luconi M, Waldman J, et al. RNA Sequencing and somatic mutation status of adrenocortical tumors: Novel pathogenetic insights. J Clin Endocrinol Metab (2020) 105:dgaa616. doi: 10.1210/clinem/dgaa616
49. Assié G, Libé R, Espiard S, Rizk-Rabin M, Guimier A, Luscap W, et al. ARMC5 mutations in macronodular adrenal hyperplasia with cushing's syndrome. N Engl J Med (2013) 369:2105–14. doi: 10.1056/NEJMoa1304603
50. Alencar GA, Lerario AM, Nishi MY, Mariani BM, Almeida MQ, Tremblay J, et al. ARMC5 mutations are a frequent cause of primary macronodular adrenal hyperplasia. J Clin Endocrinol Metab (2014) 99:E1501–9. doi: 10.1210/jc.2013-4237
51. Hu Y, Lao L, Mao J, Jin W, Luo H, Charpentier T, et al. Armc5 deletion causes developmental defects and compromises T-cell immune responses. Nat Commun (2017) 8:13834. doi: 10.1038/ncomms13834
52. Berthon A, Faucz FR, Espiard S, Drougat L, Bertherat J, Stratakis CA. Age-dependent effects of Armc5 haploinsufficiency on adrenocortical function. Hum Mol Genet (2017) 26:3495–507. doi: 10.1093/hmg/ddx235
53. Mulatero P, Schiavi F, Williams TA, Monticone S, Barbon G, Opocher G, et al. ARMC5 mutation analysis in patients with primary aldosteronism and bilateral adrenal lesions. J Hum Hypertens (2016) 30:374–8. doi: 10.1038/jhh.2015.98
54. Rege J, Hoxie J, Liu CJ, Cash MN, Luther JM, Gellert L, et al. Targeted mutational analysis of cortisol-producing adenomas. J Clin Endocrinol Metab (2022) 107(2):e594–603. doi: 10.1210/clinem/dgab682
55. Forbes SA, Bindal N, Bamford S, Cole C, Kok CY, Beare D, et al. COSMIC: mining complete cancer genomes in the catalogue of somatic mutations in cancer. Nucleic Acids Res (2011) 39:D945–50. doi: 10.1093/nar/gkq929
56. Knudson AG Jr. Mutation and cancer: statistical study of retinoblastoma. Proc Natl Acad Sci U S A (1971) 68:820–3. doi: 10.1073/pnas.68.4.820
57. Gomez-Sanchez CE, Gomez-Sanchez EP. Mutations of the potassium channel KCNJ5 causing aldosterone-producing adenomas: one or two hits? Hypertension (2012) 59:196–7. doi: 10.1161/HYPERTENSIONAHA.111.186205
58. Zennaro MC, Boulkroun S, Fernandes-Rosa F. Genetic causes of functional adrenocortical adenomas. Endocr Rev (2017) 38:516–37. doi: 10.1210/er.2017-00189
59. Sassone-Corsi P. The cyclic AMP pathway. Cold Spring Harb Perspect Biol (2012) 4:a011148. doi: 10.1101/cshperspect.a011148
60. Goh G, Scholl UI, Healy JM, Choi M, Prasad ML, Nelson-Williams C, et al. Recurrent activating mutation in PRKACA in cortisol-producing adrenal tumors. Nat Genet (2014) 46:613–7. doi: 10.1038/ng.2956
61. Almeida MQ, Stratakis CA. How does cAMP/protein kinase a signaling lead to tumors in the adrenal cortex and other tissues? Mol Cell Endocrinol (2011) 336:162–8. doi: 10.1016/j.mce.2010.11.018
62. Beuschlein F, Fassnacht M, Assié G, Calebiro D, Stratakis CA, Osswald A, et al. Constitutive activation of PKA catalytic subunit in adrenal cushing's syndrome. N Engl J Med (2014) 370:1019–28. doi: 10.1056/NEJMoa1310359
63. Di Dalmazi G, Timmers HJLM, Arnaldi G, Küsters B, Scarpelli M, Bathon K, et al. Somatic PRKACA mutations: Association with transition from pituitary-dependent to adrenal-dependent cushing syndrome. J Clin Endocrinol Metab (2019) 104:5651–7. doi: 10.1210/jc.2018-02209
64. Nanba K, Omata K, Tomlins SA, Giordano TJ, Hammer GD, Rainey WE, et al. Double adrenocortical adenomas harboring independent KCNJ5 and PRKACA somatic mutations. Eur J Endocrinol (2016) 175:K1–6. doi: 10.1530/EJE-16-0262
65. Fallo F, Castellano I, Gomez-Sanchez CE, Rhayem Y, Pilon C, Vicennati V, et al. Histopathological and genetic characterization of aldosterone-producing adenomas with concurrent subclinical cortisol hypersecretion: a case series. Endocrine (2017) 58:503–12. doi: 10.1007/s12020-017-1295-4
66. Aristizabal Prada ET, Castellano I, Sušnik E, Yang Y, Meyer LS, Tetti M, et al. Comparative genomics and transcriptome profiling in primary aldosteronism. Int J Mol Sci (2018) 19:1124. doi: 10.3390/ijms19041124
67. Spyroglou A, Piaditis GP, Kaltsas G, Alexandraki KI. Transcriptomics, epigenetics, and metabolomics of primary aldosteronism. Cancers (Basel) (2021) 13:5582. doi: 10.3390/cancers13215582
68. Assié G, Auzan C, Gasc JM, Baviera E, Balaton A, Elalouf JM, et al. Steroidogenesis in aldosterone-producing adenoma revisited by transcriptome analysis. J Clin Endocrinol Metab (2005) 90:6638–49. doi: 10.1210/jc.2005-1309
69. Williams TA, Monticone S, Morello F, Liew CC, Mengozzi G, Pilon C, et al. Teratocarcinoma-derived growth factor-1 is upregulated in aldosterone-producing adenomas and increases aldosterone secretion and inhibits apoptosis in vitro. Hypertension (2010) 55:1468–75. doi: 10.1161/HYPERTENSIONAHA.110.150318
70. Williams TA, Monticone S, Crudo V, Warth R, Veglio F, Mulatero P. Visinin-like 1 is upregulated in aldosterone-producing adenomas with KCNJ5 mutations and protects from calcium-induced apoptosis. Hypertension (2012) 59:833–9. doi: 10.1161/HYPERTENSIONAHA.111.188532
71. Lefèvre L, Omeiri H, Drougat L, Hantel C, Giraud M, Val P, et al. Combined transcriptome studies identify AFF3 as a mediator of the oncogenic effects of β-catenin in adrenocortical carcinoma. Oncogenesis (2015) 4:e161. doi: 10.1038/oncsis.2015.20
72. Backman S, Åkerström T, Maharjan R, Cupisti K, Willenberg HS, Hellman P, et al. RNA Sequencing provides novel insights into the transcriptome of aldosterone producing adenomas. Sci Rep (2019) 9:6269. doi: 10.1038/s41598-019-41525-2
73. Yang Y, Tetti M, Vohra T, Adolf C, Seissler J, Hristov M, et al. BEX1 is differentially expressed in aldosterone-producing adenomas and protects human adrenocortical cells from ferroptosis. Hypertension (2021) 77:1647–58. doi: 10.1161/HYPERTENSIONAHA.120.16774
74. Nishimoto K, Tomlins SA, Kuick R, Cani AK, Giordano TJ, Hovelson DH, et al. Aldosterone-stimulating somatic gene mutations are common in normal adrenal glands. Proc Natl Acad Sci U S A (2015) 112:E4591–9. doi: 10.1073/pnas.1505529112
75. Zhou J, Lam B, Neogi SG, Yeo GS, Azizan EA, Brown MJ. Transcriptome pathway analysis of pathological and physiological aldosterone-producing human tissues. Hypertension (2016) 68:1424–31. doi: 10.1161/HYPERTENSIONAHA.116.08033
76. Maniero C, Garg S, Zhao W, Johnson TI, Zhou J, Gurnell M, et al. NEFM (Neurofilament medium) polypeptide, a marker for zona glomerulosa cells in human adrenal, inhibits D1R (Dopamine D1 receptor)-mediated secretion of aldosterone. Hypertension (2017) 70:357–64. doi: 10.1161/HYPERTENSIONAHA.117.09231
77. Werminghaus P, Haase M, Hornsby PJ, Schinner S, Schott M, Malendowicz LK, et al. Hedgehog-signaling is upregulated in non-producing human adrenal adenomas and antagonism of hedgehog-signaling inhibits proliferation of NCI-H295R cells and an immortalized primary human adrenal cell line. J Steroid Biochem Mol Biol (2014) 139:7–15. doi: 10.1016/j.jsbmb.2013.09.007
78. Gomes DC, Leal LF, Mermejo LM, Scrideli CA, Martinelli CE Jr, Fragoso MC, et al. Sonic hedgehog signaling is active in human adrenal cortex development and deregulated in adrenocortical tumors. J Clin Endocrinol Metab (2014) 99:E1209–16. doi: 10.1210/jc.2013-4098
79. Oki K, Plonczynski MW, Gomez-Sanchez EP, Gomez-Sanchez CE. YPEL4 modulates HAC15 adrenal cell proliferation and is associated with tumor diameter. Mol Cell Endocrinol (2016) 434:93–8. doi: 10.1016/j.mce.2016.06.022
80. Shaikh LH, Zhou J, Teo AE, Garg S, Neogi SG, Figg N, et al. LGR5 activates noncanonical wnt signaling and inhibits aldosterone production in the human adrenal. J Clin Endocrinol Metab (2015) 100:E836–44. doi: 10.1210/jc.2015-1734
81. Zhang G, Zou X, Liu Q, Xie T, Huang R, Kang H, et al. MiR-193a-3p functions as a tumour suppressor in human aldosterone-producing adrenocortical adenoma by down-regulating CYP11B2. Int J Exp Pathol (2018) 99:77–86. doi: 10.1111/iep.12267
82. Peng KY, Chang HM, Lin YF, Chan CK, Chang CH, Chueh SJ, et al. miRNA-203 modulates aldosterone levels and cell proliferation by targeting Wnt5a in aldosterone-producing adenomas. J Clin Endocrinol Metab (2018) 103:3737–47. doi: 10.1210/jc.2018-00746
83. He J, Cao Y, Su T, Jiang Y, Jiang L, Zhou W, et al. Downregulation of miR-375 in aldosterone-producing adenomas promotes tumour cell growth via MTDH. Clin Endocrinol (Oxf) (2015) 83:581–9. doi: 10.1111/cen.12814
84. Ma Q. Role of nrf2 in oxidative stress and toxicity. Annu Rev Pharmacol Toxicol (2013) 53:401–26. doi: 10.1146/annurev-pharmtox-011112-140320
85. Dixon SJ, Lemberg KM, Lamprecht MR, Skouta R, Zaitsev EM, Gleason CE, et al. Ferroptosis: an iron-dependent form of nonapoptotic cell death. Cell (2012) 149:1060–72. doi: 10.1016/j.cell.2012.03.042
86. Belavgeni A, Bornstein SR, von Mässenhausen A, Tonnus W, Stumpf J, Meyer C, et al. Exquisite sensitivity of adrenocortical carcinomas to induction of ferroptosis. Proc Natl Acad Sci U.S.A. (2019) 116:22269–74. doi: 10.1073/pnas.1912700116
87. Gong S, Tetti M, Reincke M, Williams TA. Primary aldosteronism: Metabolic reprogramming and the pathogenesis of aldosterone-producing adenomas. Cancers (Basel) (2021) 13:3716. doi: 10.3390/cancers13153716
88. Lenzini L, Seccia TM, Aldighieri E, Belloni AS, Bernante P, Giuliani L, et al. Heterogeneity of aldosterone-producing adenomas revealed by a whole transcriptome analysis. Hypertension (2007) 50:1106–13. doi: 10.1161/HYPERTENSIONAHA.107.100438
89. Oki K, Gomez-Sanchez CE. The landscape of molecular mechanism for aldosterone production in aldosterone-producing adenoma. Endocr J (2020) 67:989–95. doi: 10.1507/endocrj.EJ20-0478
90. Berridge MJ. Calcium signalling and cell proliferation. Bioessays (1995) 17:491–500. doi: 10.1002/bies.950170605
91. Kobuke K, Oki K, Gomez-Sanchez CE, Gomez-Sanchez EP, Ohno H, Itcho K, et al. Calneuron 1 increased Ca2+ in the endoplasmic reticulum and aldosterone production in aldosterone-producing adenoma. Hypertension (2018) 71:125–33. doi: 10.1161/HYPERTENSIONAHA.117.10205
92. Itcho K, Oki K, Gomez-Sanchez CE, Gomez-Sanchez EP, Ohno H, Kobuke K, et al. Endoplasmic reticulum chaperone calmegin is upregulated in aldosterone-producing adenoma and associates with aldosterone production. Hypertension (2020) 75:492–9. doi: 10.1161/HYPERTENSIONAHA.119.14062
93. Truong L, Zheng YM, Song T, Tang Y, Wang YX. Potential important roles and signaling mechanisms of YPEL4 in pulmonary diseases. Clin Transl Med (2018) 7:16. doi: 10.1186/s40169-018-0194-5
94. Romero DG, Plonczynski MW, Welsh BL, Gomez-Sanchez CE, Zhou MY, Gomez-Sanchez EP. Gene expression profile in rat adrenal zona glomerulosa cells stimulated with aldosterone secretagogues. Physiol Genomics (2007) 32:117–27. doi: 10.1152/physiolgenomics.00145.2007
95. El Zein RM, Soria AH, Golib Dzib JF, Rickard AJ, Fernandes-Rosa FL, Samson-Couterie B, et al. Retinoic acid receptor α as a novel contributor to adrenal cortex structure and function through interactions with wnt and vegfa signalling. Sci Rep (2019) 9:14677. doi: 10.1038/s41598-019-50988-2
96. Li F, Feuchtinger A, Walch A, Sun N. In situ metabolite mass spectrometry imaging: New insights into the adrenal gland. Horm Metab Res (2020) 52:435–47. doi: 10.1055/a-1129-6947
97. Sun N, Wu Y, Nanba K, Sbiera S, Kircher S, Kunzke T, et al. High-resolution tissue mass spectrometry imaging reveals a refined functional anatomy of the human adult adrenal gland. Endocrinology (2018) 159:1511–24. doi: 10.1210/en.2018-00064
98. Murakami M, Rhayem Y, Kunzke T, Sun N, Feuchtinger A, Ludwig P, et al. In situ metabolomics of aldosterone-producing adenomas. JCI Insight (2019) 4:e130356. doi: 10.1172/jci.insight.130356
99. Sugiura Y, Takeo E, Shimma S, Yokota M, Higashi T, Seki T, et al. Aldosterone and 18-oxocortisol coaccumulation in aldosterone-producing lesions. Hypertension (2018) 72:1345–54. doi: 10.1161/HYPERTENSIONAHA.118.11243
100. Sun N, Meyer LS, Feuchtinger A, Kunzke T, Knösel T, Reincke M, et al. Mass spectrometry imaging establishes 2 distinct metabolic phenotypes of aldosterone-producing cell clusters in primary aldosteronism. Hypertension (2020) 75:634–44. doi: 10.1161/HYPERTENSIONAHA.119.14041
101. Buck A, Ly A, Balluff B, Sun N, Gorzolka K, Feuchtinger A, et al. High-resolution MALDI-FT-ICR MS imaging for the analysis of metabolites from formalin-fixed, paraffin-embedded clinical tissue samples. J Pathol (2015) 237:123–32. doi: 10.1002/path.4560
102. Nishimoto K, Nakagawa K, Li D, Kosaka T, Oya M, Mikami S, et al. Adrenocortical zonation in humans under normal and pathological conditions. J Clin Endocrinol Metab (2010) 95:2296–305. doi: 10.1210/jc.2009-2010
103. Williams TA, Gomez-Sanchez CE, Rainey WE, Giordano TJ, Lam AK, Marker A, et al. International histopathology consensus for unilateral primary aldosteronism. J Clin Endocrinol Metab (2021) 106:42–54. doi: 10.1210/clinem/dgaa484
104. Lim JS, Rainey WE. The potential role of aldosterone-producing cell clusters in adrenal disease. Horm Metab Res (2020) 52:427–34. doi: 10.1055/a-1128-0421
105. Nishimoto K, Seki T, Kurihara I, Yokota K, Omura M, Nishikawa T, et al. Case report: Nodule development from subcapsular aldosterone-producing cell clusters causes hyperaldosteronism. J Clin Endocrinol Metab (2016) 101:6–9. doi: 10.1210/jc.2015-3285
106. Nishimoto K, Koga M, Seki T, Oki K, Gomez-Sanchez EP, Gomez-Sanchez CE, et al. Immunohistochemistry of aldosterone synthase leads the way to the pathogenesis of primary aldosteronism. Mol Cell Endocrinol (2017) 441:124–33. doi: 10.1016/j.mce.2016.10.014
107. Vouillarmet J, Fernandes-Rosa F, Graeppi-Dulac J, Lantelme P, Decaussin-Petrucci M, Thivolet C, et al. Aldosterone-producing adenoma with a somatic KCNJ5 mutation revealing APC-dependent familial adenomatous polyposis. J Clin Endocrinol Metab (2016) 101:3874–8. doi: 10.1210/jc.2016-1874
108. Williams TA, Jaquin D, Burrello J, Philippe A, Yang Y, Rank P, et al. Diverse responses of autoantibodies to the angiotensin II type 1 receptor in primary aldosteronism. Hypertension (2019) 74:784–92. doi: 10.1161/HYPERTENSIONAHA.119.13156
109. Piazza M, Seccia TM, Caroccia B, Rossitto G, Scarpa R, Persichitti P, et al. AT1AA (Angiotensin II type-1 receptor autoantibodies): Cause or consequence of human primary aldosteronism? Hypertension (2019) 74:793–9. doi: 10.1161/HYPERTENSIONAHA.119.13388
110. Howard B, Wang Y, Xekouki P, Faucz FR, Jain M, Zhang L, et al. Integrated analysis of genome-wide methylation and gene expression shows epigenetic regulation of CYP11B2 in aldosteronomas. J Clin Endocrinol Metab (2014) 99:E536–43. doi: 10.1210/jc.2013-3495
111. Di Dalmazi G, Morandi L, Rubin B, Pilon C, Asioli S, Vicennati V, et al. DNA Methylation of steroidogenic enzymes in benign adrenocortical tumors: New insights in aldosterone-producing adenomas. J Clin Endocrinol Metab (2020) 105:dgaa585. doi: 10.1210/clinem/dgaa585
112. Yoshii Y, Oki K, Gomez-Sanchez CE, Ohno H, Itcho K, Kobuke K, et al. Hypomethylation of CYP11B2 in aldosterone-producing adenoma. Hypertension (2016) 68:1432–7. doi: 10.1161/HYPERTENSIONAHA.116.08313
113. Murakami M, Yoshimoto T, Nakabayashi K, Tsuchiya K, Minami I, Bouchi R, et al. Integration of transcriptome and methylome analysis of aldosterone-producing adenomas. Eur J Endocrinol (2015) 173:185–95. doi: 10.1530/EJE-15-0148
114. Gulyaeva LF, Kushlinskiy NE. Regulatory mechanisms of microRNA expression. J Transl Med (2016) 14:143. doi: 10.1186/s12967-016-0893-x
115. Treiber T, Treiber N, Meister G. Regulation of microRNA biogenesis and its crosstalk with other cellular pathways. Nat Rev Mol Cell Biol (2019) 20:5–20. doi: 10.1038/s41580-018-0059-1
116. Decmann A, Nyírö G, Darvasi O, Turai P, Bancos I, Kaur RJ, et al. Circulating miRNA expression profiling in primary aldosteronism. Front Endocrinol (Lausanne) (2019) 10:739. doi: 10.3389/fendo.2019.00739
Keywords: adrenal adenoma, adrenal gland, aldosterone, cell death, endocrine hypertension, ferroptosis, hyperaldosteronism, proliferation
Citation: Tetti M, Gong S, Veglio F, Reincke M and Williams TA (2022) Primary aldosteronism: Pathophysiological mechanisms of cell death and proliferation. Front. Endocrinol. 13:934326. doi: 10.3389/fendo.2022.934326
Received: 02 May 2022; Accepted: 18 July 2022;
Published: 08 August 2022.
Edited by:
Norlela Sukor, Universiti Kebangsaan Malaysia Medical Center (UKMMC), MalaysiaReviewed by:
Zheng-Wei Chen, National Taiwan University, TaiwanSheerazed Boulkroun, Institut National de la Santé et de la Recherche Médicale (INSERM), France
Copyright © 2022 Tetti, Gong, Veglio, Reincke and Williams. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Tracy Ann Williams, VHJhY3kuV2lsbGlhbXNAbWVkLnVuaS1tdWVuY2hlbi5kZQ==
†ORCID: Martin Reincke, orcid.org/0000-0002-9817-9875
Tracy Ann Williams, orcid.org/0000-0002-2388-6444