 Georgia Pitsava
Georgia Pitsava Andrea G. Maria2
Andrea G. Maria2- 1Division of Intramural Research, Division of Population Health Research, Eunice Kennedy Shriver National Institutes of Child Health and Human Development, National Institutes of Health, Bethesda, MD, United States
- 2Section on Endocrinology and Genetics, Eunice Kennedy Shriver National Institute of Child Health and Human Development, National Institutes of Health, Bethesda MD, United States
- 3Molecular Genomics Core (MGC), Eunice Kennedy Shriver National Institute of Child Health and Human Development, National Institutes of Health, Bethesda MD, United States
Adrenal cortex produces glucocorticoids, mineralocorticoids and adrenal androgens which are essential for life, supporting balance, immune response and sexual maturation. Adrenocortical tumors and hyperplasias are a heterogenous group of adrenal disorders and they can be either sporadic or familial. Adrenocortical cancer is a rare and aggressive malignancy, and it is associated with poor prognosis. With the advance of next-generation sequencing technologies and improvement of genomic data analysis over the past decade, various genetic defects, either from germline or somatic origin, have been unraveled, improving diagnosis and treatment of numerous genetic disorders, including adrenocortical diseases. This review gives an overview of disorders associated with the adrenal cortex, the genetic factors of these disorders and their molecular implications.
1. Introduction
Adrenal glands are the major regulators of body homeostasis and endocrine stress response (1). They are small yellowish structures located on the upper poles of the kidneys, in the retroperitoneal area on the lateral edge of the vertebral column. They are found within perirenal fat and are surrounded by the renal fascia. The left adrenal gland is crescent-shaped, while the right is triangular. The weight of each gland in a healthy adult is 8-10 g and the average dimensions are 5.0x3.0x0.6 cm. They are highly vascular and receive their blood supply from 3 arteries: the superior, middle and inferior adrenal arteries. These arise from the inferior phrenic artery, abdominal aorta and renal arteries, respectively (2). The adrenal arteries form a capsular arteriolar plexus, which supplies the adrenal glands. With respect to venous drainage, the right adrenal has a single vein that drains directly to inferior vena cava, whereas the longer left adrenal vein drains into the renal vein (3).
The adrenal glands are comprised of two distinct parts, the cortex and the medulla. The medulla found in the center of the adrenal gland is composed of chromaffin cells and it is dependent on tissue interactions with the adrenal cortex (4). The cortex forms the outer part and is responsible for up to 90% of the adrenal weight. The adult adrenal cortex plays a vital role in normal physiology, being the site of steroid hormone production (3, 5). It consists of three morphologically and functionally distinct compartments. The outer zona glomerulosa (ZG) makes up about 15% of the cortex and produces aldosterone, a mineralocorticoid that controls blood pressure by regulating intravascular volume through sodium and water retention (6, 7). Beneath the ZG, is zona fasciculata (ZF), that comprises a major part of the adrenal gland and is the site of glucocorticoid synthesis. This is where cortisol is produced, a hormone with important effects on the immune system, metabolism and cardiovascular system. The innermost zone is zona reticularis (ZR), which produces adrenal androgens including androstenedione, dehydroepiandrosterone (DHEA), as well as its sulfate, DHEAS (8).
Adrenal cortex cellular function is finely regulated by complex mechanism that involve paracrine and endocrine responses. Dysregulation of signaling pathways in the adrenal cortex is associated with the development of adrenal tumors, some are benign and most rarely, malignant tumors (9).
The advance of new technologies in the field of genetics made possible to determine variations and structures at a genone-wide level (10). Next generation sequencing (NGS) became available at the beginning of the 21st century lowering the costs of DNA sequencing beyond what is possible with standard dye-terminator methods (11). In the clinical context, NGS has greatly improved the discovery of disease associated variants, facilitating not only faster and precise diagnosis but also risk factor prediction for complex disorders (12). For example, a recent study analyzed gene expressions in cortisol-producing adenomas (CPA) with PRKACA mutation and compared to GNAS and CTNNB1 mutant CPAs. NGS analysis revealed differences between PRKACA mutant and GNAS and CTNNB1 mutant CPAs, such as increased cortisol production in PRKACA mutant CPAs (13). This study allows better understanding of pathways involved in CPA and also may direct a more precise treatment approach for those individuals who harbors CPAs. Another study made use of whole-exome sequencing to determine the proportion of cells exhibiting the disease-causing variant KCNJ5 p.G151R in an individual already diagnosed with bilateral adrenal hyperplasia (BAH). The results indicated a very low-level mosaicism (less than 0.5%) in the germline DNA, while all adrenocortical cells tested from 11 different nodules harbored the disease-causing variant. This finding has implication in patient prognosis and, family risk prediction (14). In this review, we intend to highlight the genomic and molecular aspects of adrenocortical tumors and its implication in patient survival.
2. Hormone secretion
The precursor of all adrenal steroid hormones is cholesterol, which is found in circulating low-density lipoprotein (LDL) particles. Briefly, LDL particles are taken up by adrenal cells via LDL-receptor mediated endocytosis (15, 16). The vesicles formed during this process subsequently fuse with lysozymes, where hydrolysis generates free cholesterol. Alternatively, cholesterol can either be uptaken from circulating HDL cholesterol via the scavenger receptor class B type 1 (SR-B1), or produced de novo from the acetyl coenzyme A (CoA) (17). Cellular cholesterol that is in excess is stored in the form of cholesteryl esters (CEs); the conversion of cholesterol to CEs is catalyzed by the enzyme CoA-acetyltransferase (ACAT) (18). In the adrenal glands, CEs act as the cholesterol ‘storage’ for the production of steroid hormones (18).
2.1 Hypothalamic-pituitary-adrenal (Hpa) axis: Glucocorticoid secretion
The secretion of glucocorticoids is regulated by the HPA axis. Their synthesis is stimulated by ACTH, which is released into the bloodstream by the anterior pituitary as part of a 241-amino acid precursor, POMC. In turn, ACTH production is regulated by corticotropin-releasing hormone (CRH), which is released by the neuroendocrine neurons in the paraventricular nucleus of the hypothalamus. Secretion of CRH is dependent on circadian rhythm, as well as various stressors (fever, hypotension, hypoglycemia) acting on the hypothalamus. The HPA axis is a negative feedback system, in which cortisol acts as a direct inhibitor of the synthesis of both ACTH and CRH.
2.2 Renin-angiotensin-aldosterone system (RAAS): Mineralocorticoid secretion
Secretion of mineralocorticoids is regulated mainly by the RAAS and potassium, while it also responds acutely to ACTH (19, 20). The juxtaglomerular (JG) cells in the afferent arterioles of the kidney contain prorenin, which is inactive. When JG cells are activated (in response to intravascular volume depletion, or decreased sodium in the distal convoluted tubule or β-activation) prorenin is cleaved to renin (18, 21). Once renin is released in the blood it acts on angiotensinogen, which is synthesized in the liver and is converted to angiotensin I (Ang I) in the kidney by renin. Ang I is then converted to angiotensin II (Ang II) by the angiotensin converting enzyme (ACE) in the lungs. Ang II and potassium increase the expression of aldosterone synthase (CYP11B2), while they also stimulate aldosterone production and glomerulosa cell proliferation (22). In turn, aldosterone acts on mineralocorticoid receptors in kidney cells, from the distal convoluted tubule to the cortical collecting tubule. The result of its action is increased sodium reabsorption and excretion of potassium and hydrogen ions.
2.3 Adrenal androgen secretion
ZR cells produce androgens, the most important of which are DHEA and DHEAS (23). These are weak precursors that are converted to testosterone and estrogens (such as estradiol) in the peripheral tissues (24). It is established that steroidogenesis is under the control of ACTH which stimulates the transport of intracellular cholesterol into the adrenal cortex (25).
3. Disorders of growth of the adrenal cortex
3.1 Adrenal hyperplasia
3.1.1 Congenital adrenal hyperplasia (CAH)
CAH is a group of autosomal recessive disorders of the adrenal cortex caused by enzymatic deficiencies in the adrenal steroidogenesis pathway (26, 27). Depending on the degree of residual enzymatic activity, various forms of CAH have been described in the literature, including the most severe form (classic salt-wasting variant), followed by the classic simple virilizing form as well as milder forms (non-classical variant).
3.1.1.1 21OH deficiency
More than 90% of CAH cases are due to deficiency in 21-hydroxylase (CYP21A2) (Online Mendelian Inheritance in Man [OMIM] #201910 (28). The gene encoding 21OH, CYP21A2, is located on chromosome 6p21.3, within the human leukocyte antigen (HLA) major histocompatibility complex locus (29). CYP21A2 and CYP21A1P, a homologous pseudogene, are approximately 30kb apart. Because of the high degree of sequence homology between these duplicate genes, meiotic recombination events are common in this region. Almost 95% of CYP21A2 disease causing mutations are CYP21A1P-derived variants or deletions used due to recombination events (30, 31). Defects in 21OH result in impaired production of aldosterone and cortisol and elevated precursors, most notably 17-hydroxyprogesterone (17OHP), elevated levels of 17OHP are used for the diagnosis of CAH. In addition, excess of androgens occurs due to constitutive adrenal androgen synthesis, and results in virilization.
The most severe form of 21OH deficiency is due to variants that inactivate CYP21A2 completely. Without neonatal screening, the phenotype in these cases manifests within the first 2 weeks of life with a life-threatening adrenal crisis (32). In the non-classic cases, the adrenal crisis is prevented. This is because some enzyme activity is preserved, and as a result aldosterone and cortisol production are not completely abolished (28, 33). The non-classic cases are thus characterized by symptoms attributed to the androgen excess: premature puberty, hirsutism and irregular menses. In some cases, patients may present with few or no symptoms and are identified by family genetic studies for other reasons (34). Females with non-classic CAH usually present with similar symptoms as those with polycystic ovary syndrome (PCOS), including hyperandrogenism (clinical or biochemical), and menstrual abnormalities (33, 35–37), and thus is difficult to differentiate between the two, leading to misdiagnosis of non-classic CAH as PCOS in some cases (38–40). Thus, it is suggested that patients undergo measurement of 17OH- progesterone levels followed by ACTH-stimulation test (41, 42).
3.1.1.2 11βOH deficiency
Approximately 8% of CAH cases are due to 11β-hydroxylase (CYP11B1) deficiency (43). CYP11B1, encoded by CYP11B1, is an enzyme regulated by ACTH, which catalyzes the conversion of 11-deoxycortisol to cortisol in the zona fasciculata. Patients with impaired 11-hydroxylation present with decreased corticosterone and cortisol synthesis, accumulation of the precursor deoxycorticosterone, and overproduction of adrenal androgens. Although deoxycorticosterone is a weak mineralocorticoid, in elevated concentrations it mimics the action of aldosterone, suppressing the renin-angiotensin axis, increasing blood pressure, and sometimes causing hypokalemia (43).
3.1.1.3 17OH deficiency
Deficiency of 17α-hydroxylase (CYP17A1) is rare, and severely damaging variants in CYP17A1 result in absent cortisol as well as androgens, causing puberty failure and sexual infantilism (44). CYP17A1 is expressed in the ZF and the ZR, but not in the ZG. Both 46,XY and 46,XX patients with 17OH deficiency have female external genitalia, and present at puberty as phenotypically female. They have hypergonadotropic hypogonadism without secondary sexual characteristics, and low-renin hypertension.
3.1.1.4 3βHSD2 deficiency
There exist two isoforms of 3β-hydroxysteroid dehydrogenase: 3βHSD1 and 3βHSD2, encoded by HSD3B1 (the homologous type I gene) and HSD3B2, respectively. HSD3B1 is expressed in the placental and peripheral tissues (breast, prostate and skin), while HSD3B2 is expressed exclusively in the adrenals and gonads (45). 3βHSD2 deficiency is characterized by deficiency of both glucocorticoids and mineralocorticoids, as well as by dehydroepiandrosterone (DHEA) overproduction. DHEA is converted to testosterone by extra-adrenal 3βHSD1, and patients present in infancy with underdeveloped 46,XY genitalia and – rarely – 46,XX virilization (46).
3.1.1.5 Lipoid congenital adrenal hyperplasia
The most severe defect of steroidogenesis is lipoid congenital adrenal hyperplasia (LCAH). LCAH is caused by defects in the steroidogenic acute regulatory protein (StAR) and is characterized by deficiency of all steroid hormones. StAR regulates the transfer of cholesterol from the outer to the inner mitochondrial membrane, a vital step in the initiation of steroidogenesis. As a result, cholesterol cannot be mobilized. Adrenal lipid droplets subsequently accumulate and are seen on the autopsy, thus the name of the disorder. In both 46,XY and 46,XX patients, it presents with female external genitalia and an adrenal crisis in the neonatal period (47).
Regarding the current treatment for CAH, there is no consensus yet, therefore, it still remains a challenge. It usually includes glucocorticoid and mineralocorticoid replacement therapy (48).
3.2. Adrenocortical tumors
Adrenocortical tumors (ACTs) can be sporadic or familial, unilateral or bilateral, and non-secreting or secreting. The latter secretes various adrenal steroid hormones; the exact hormone varies depending on the tumor type. Unilateral ACTs are common, and approximately 10% of the general population appears to have an adrenal cortical lesion (49). They are often discovered incidentally when evaluating for another disease and are thus called incidentalomas (50, 51). Once discovered, they are evaluated by abdominal computed tomography (CT). The vast majority of them are benign adrenocortical adenomas (ACAs). Some ACAs are non-secreting, while others can secrete cortisol and cause Cushing syndrome (5-47% of cases), or aldosterone and cause Conn syndrome (1.6-3.3%) (50, 52). The rest of ACTs are adrenocortical carcinomas (ACCs), which are rare (prevalence 4-12 cases per million).
3.2.1 Benign cortisol-producing adrenocortical tumors
Cushing’s syndrome (CS) has an estimated incidence of 39-79 per million people per year in various populations, with a female-to-male ratio of 3:1 (53–56). Data from various studies suggest that there is an increased prevalence in people with early-onset osteoporosis, type 2 diabetes, or hypertension, but the precise estimates vary (57–60).
CS is characterized by cortisol overproduction. The cause of 80% of endogenous CS cases is over-secretion of ACTH by a pituitary corticotroph adenoma or – less frequently – by a neuroendocrine tumor (61–63). In rare cases, neuroendocrine tumors such as pheochromocytoma and medullary thyroid carcinoma produce corticotropin-releasing hormone (CRH), which then results in pituitary ACTH over-secretion (61–63). In 20% of the cases, CS is ACTH-independent, and the cause is the primary overproduction of cortisol by the adrenal glands. In such cases, the most frequent underlying pathology is a cortisol-producing adenoma, while adrenocortical carcinomas and bilateral adrenal hyperplasia are responsible for less than10% of the cases (64). Bilateral adrenal hyperplasia in particular may be either isolated, or part of a syndrome, and can be divided into two entities based on the size of the nodules: primary bilateral macronodular adrenal hyperplasia (PBMAH), which is characterized by several nodules (diameter >10mm) (65), and two micronodular forms. The latter are primary pigmented micronodular adrenal hyperplasia (PPNAD) and isolated micronodular adrenocortical disease (iMAD) (diameter <10mm) (61–63, 66).
The cAMP/PKA pathway is the main regulator of cortisol production (67). PKA (protein kinase A) consists of two regulatory subunits and two catalytic subunits that – under normal conditions – are bound together. In adrenocortical cells, the pathway is activated by the binding of ACTH to MC2R, a G protein-coupled receptor (GPCR). This triggers an increase in cAMP levels, which binds to the PKA regulatory subunits causing the release from the catalytic subunits. The catalytic subunits, then translocate to the nucleus, where they phosphorylate, and thus activate, transcription factors that promote cortisol synthesis (Figure 1) (68).
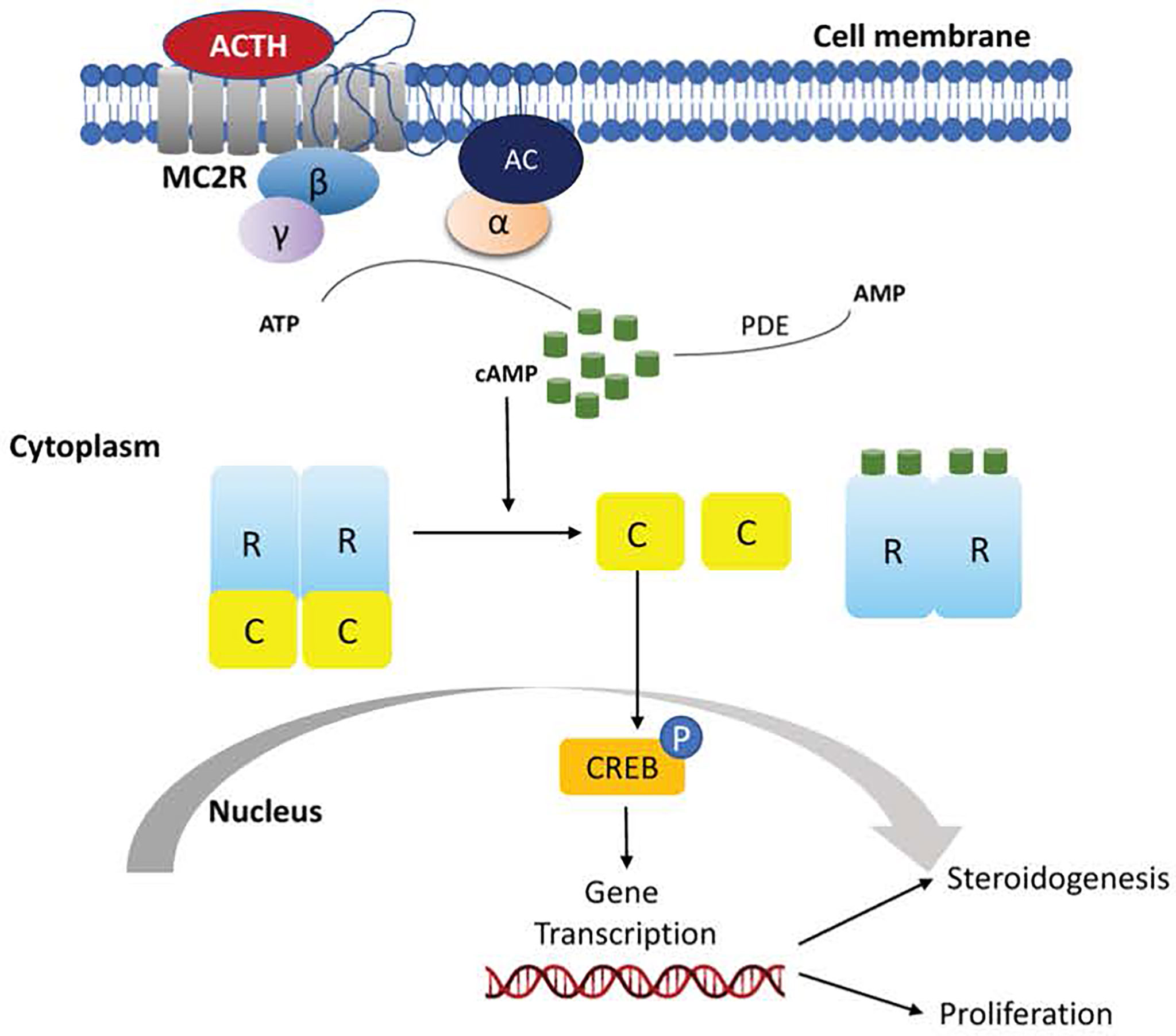
Figure 1 Schematic representation of activation of the cyclic adenosine monophosphate (cAMP) signaling pathway in normal adrenocortical cells.
3.2.2 Primary bilateral macronodular adrenal hyperplasia (PBMAH)
PBMAH is usually diagnosed in patients at 40-65 years old that present CS and low levels of ACTH, or - more recently - when investigating an adrenal incidentaloma. Many terms have been used over the years to describe PBMAH. Such terms include primary macronodular adrenal hyperplasia (PMAH), autonomous macronodular adrenal hyperplasia (AMAH), bilateral macronodular adrenal hyperplasia (BMAH), and ‘huge’ or ‘giant’ macronodular adrenal disease. Another term, ACTH-independent massive bilateral adrenal disease (AIMBAD), has also been used in the past, but in later studies the secretion of cortisol appeared to be regulated by corticotropin and thus, this term is not used anymore (69).
In general, PBMAH presents with bilateral macronodules and enlargement of the adrenal glands. In the majority of cases (77-87%), the macronodules exhibit ectopic or excessive expression of G-protein coupled receptors, including luteinizing hormone/choriogonadotropin (LH/hCG) responsible for Cushing syndrome during pregnancy and after menopause (70), glucose-dependent insulinotropic peptide (GIP) that is responsible for food-dependent Cushing syndrome (71), serotonin 5HT, catecholamines, Ang II, glucagon and vasopressin (65, 72–77). The binding of these receptors to their ligands mimics the result of ACTH binding to MC2R leading to activation of cAMP/PKA pathway and thus excessive cortisol secretion (78). So far, the exact molecular mechanism of the ectopic receptor expression has not been completely elucidated (79).
Genetic variants resulting in increased activity of the cAMP/PKA pathway via a variety of mechanisms have been reported in patients with PBMAH. For example, variants in PDE11A, that encodes phosphodiesterase type 11A, have a prevalence of 24-28%, whereas inactivating germline variants in PDE8B, encoding phosphodiesterase type 8B, and PRKACA copy number gains, have also been encountered (80–83). Another component of the cAMP/PKA pathway associated with PBMAH is the Gα subunit, encoded by GNAS1 (84). Activating variants in GNAS1 cause McCune-Albright syndrome, which is associated with ‘café au lait’ spots, polyostotic fibrous dysplasia, precocious puberty and hyperfunction of multiple endocrine glands (84). These GNAS1 variants are somatic and lead to continuous activation of the cAMP/PKA pathway and thus, cortisol-producing adenomas (85). Finally, in a case report of an isolated case of bilateral adrenal hyperplasia, the synergistic action of two variants (p.C21R and p.S247G) on the same allele of MC2R (encoding the melanocortin 2 receptor/ACTH receptor) resulted in autonomous cortisol secretion via constitutive activation of the cAMP/PKA pathway (86). This case is remarkable in that if those defects had happened in isolation, they would have led to receptor inactivation (86).
Rarely, PBMAH can occur as part of genetic tumor predisposition syndromes such as familial adenomatous polyposis (APC), multiple endocrine neoplasia type 1 (MEN1) and hereditary leiomyomatosis (FH) (83, 87–89). It is important to mention that these genetic alterations are associated with other tumors and are responsible only for a limited number of PBMAH cases. Additionally, PBMAH can be associated with aromatase expression leading to elevated estrogens, independently of sex (79).
More recently, germline variants in the armadillo repeat-containing 5 (ARMC5) have been found to implicate in the pathogenesis of PBMAH, with their prevalence estimated between 21%-26% (90, 91). ARMC5 is a cytosolic protein that had no enzymatic activity, and its function depends on interactions with other proteins (92). ARMC5 is located on chromosome 16p11.2. In mice, as well as in vitro, ARMC5 has been shown to play an important role in regulating steroidogenesis, proliferation, apoptosis, T-cell differentiation and immune responses (92–95). Most of the patients harboring ARMC5 variants had adrenal CS, with the hypersecretion of cortisol being more severe compared to that seen in patients that had ARMC5 variants that were predicted to be benign or did not have ARMC5 variants. In addition, damaging variants or deletions in ARMC5 were identified in several families with PBMAH (96, 97), whereas an association of ARMC5 with primary hyperaldosteronism was also reported in 2015 (98).
In a small number of cases, somatic variants in genes participating in other biological processes have been described. These include two chromatin regulator genes, DOTIL (that encodes a histone H3 lysine methyl-transferase) and HDAC9 (that encodes a histone deacetylase) (99). In addition, a study of two siblings from a family that segregated PBMAH implicated the Endothelin Receptor type A EDNRA, which encodes a G-coupled protein. However, this association remains to be confirmed in follow-up studies (100).
A meaningful update of the 2022 WHO classification of adrenocortical tumors was recently summarized by Mete and co-workers early this year. As a result of the advance on next generation sequencing studies, it is possible to recognize that PBMAH is caused by germline variants in one out of many susceptible genes, with a second hit in the somatic cells. These findings strongly suggest a neoplastic instead of hyperplastic condition. To avoid a misnomer for the disease, the 2022 WHO classification changed the nomenclature of primary bilateral macronodular adrenal hyperplasia to bilateral macronodular adrenocortical disease (101).
3.2.3 Primary pigmented nodular adrenal disease (PPNAD)
PPNAD is most commonly diagnosed in children and young adults and is a rare cause of ACTH-independent hypercortisolism. It is most commonly part of Carney complex (CNC), an autosomal dominant tumor predisposition syndrome. CNC presents with various endocrine tumors including pituitary adenomas, thyroid benign tumors, and testicular Sertoli cell-calcified tumors, as well as non-endocrine tumors, most commonly pigmented skin lesions, skin and cardiac myxomas (102, 103). PPNAD in patients with CNC has a prevalence close to 60% (104, 105).
With respect to the molecular background, CNC is caused by germline inactivating variants in the PRKAR1A gene, located at the 17q24.2-24.3 locus (CNC1 locus). PRKAR1A variants are found in 37% of patients with the sporadic form of the disease and in more than 70% in the typical familial forms, with almost 100% penetrance (105, 106). PRKAR1A encodes the regulatory subunit type 1α (R1α) of PKA. As a result, inactivating PRKAR1A variants result in aberrant activation of the cAMP/PKA pathway. To date, approximately 140 pathogenic variants have been reported (https://PRKAR1A.nichd.nih.gov/hmdb/mutations.html). The majority of them are located in exons 2,3,5,7, and 8 while about 20% are located in intronic sequences and affect splicing (105, 106). Most of the variants are small deletions and insertions, base substitutions or combined rearrangements involving up to 15bp (107). In almost all cases (90%), the genetic alteration leads to a premature stop codon. Subsequently, the transcripts containing the premature stop codon are degraded by nonsense mediated decay (NMD). As a result, the amount of RIα protein produced is half of the normal amount (107–109). Large chromosomal deletions involving the PRKAR1A gene, even though rare, have also been identified (110, 111). Occasionally, the pathogenic variant (missense, short in-frame insertions/deletions and splice variants) may lead to the production of an abnormal protein that is incapable of responding appropriately to cAMP levels or properly bind to the PKA catalytic subunits (108, 112).
In some families without variants in PRKAR1A, the causative gene has not been identified yet; however, genetic linkage analysis of tumors has shown that there is another affected locus on chromosome 2p16 (CNC2 locus) (113, 114). The majority of those cases have been diagnosed with CNC later in life (115). In addition, a single patient with CNC that presented with abnormal skin pigmentation, acromegaly and myxomas, was found to harbor copy number gains of locus containing the PRKACB gene (116). Moreover, a recent study with 353 CNC patients and/or PPNAD, showed that the majority of patients with isolated PPNAD harbored a germline c.709-7del6 variant (105).
During the past years, variants affecting the phosphodiesterase genes PDE11A and PDE8B have emerged as putative causes of PPNAD. The loci harboring these genes had the most significant associations in a genome-wide association study performed on individuals lacking genetic defects in GNAS or PRKAR1A, while the locus harboring PDE11A showed the largest loss-of-heterozygosity in tumor samples (117). In addition, targeted sequencing of PDE11A revealed that patients with CNC that also had PPNAD and/or testicular large cell calcifying Sertoli cell tumors were more likely to have variants in PDE11A, compared to patients without these tumors (118). All of these patients also had germline variants in PRKAR1A, raising the possibility that PDE11A variants act as genetic modifiers that elevate the risk for PPNAD and/or LCCSCT in CNC. With regards to PDE8B, a single nucleotide variant (c.914A>C/p.His305Pro) was detected in a 2 year old female patient diagnosed with PPNAD; the variant was inherited from the patient’s father. This variant was subsequently shown to lead to decreased PDE8B activity in vitro (119).
It is worth noting that variants in PDE11A and PDE8B have been found in other types of adrenocortical tumors as well. In addition to PBMAH tumors (described above), haploinsufficiency of PDE11A has been implicated in ACA and ACC (80), and in vitro studies have demonstrated that putative PBMAH-causing variants compromised the enzymatic activity of PDE11A (81). Furthermore, in a cohort of patients with adrenocortical tumors without variants in PRKAR1A, GNAS, or PDE11A, 7 patients harbored variants in PDE8B (82). Two of these variants were then experimentally shown to decrease protein activity (82).
Recently, genes encoding for the catalytic subunits of PKA have also been implicated in micronodular PBMAH. A study of two patients with familial PBMAH, and of three patients with sporadic iMAD, identified germline copy number gains of the locus that harbors PRKACA, which encodes the Ca catalytic subunit. Tumor samples from these patients revealed elevated basal as well as cAMP-stimulated PKA activity (120, 121). In addition, copy number gains involving the PRKACB locus (which encodes the Cβ catalytic subunit), were reported in a patient with CNC that presented with myxomas, acromegaly, and abnormal skin pigmentation. The patient was found to have increased cAMP-induced kinase activity in lymphocytes, resembling what is seen in CNC patients with PRKAR1A variants. Moreover, increased Cβ levels were found in several cell types as well as in breast myxoma cells (116).
Finally, the wingless-type-(Wnt)-β-catenin pathway has also been suggested to play a role in micronodular BAH. Somatic variants in the β -catenin gene (CTNNB1) were found in two (11%) patients with PPNAD in a previous study, with one of these patients also harboring a PRKAR1A variant. These variants were encountered in larger adrenocortical adenomas that arose within the context of PPNAD, and were absent from the surrounding hyperplastic adrenocortical tissue (122). A different study landed further support to the involvement of the Wnt-β-catenin pathway, by showing accumulation of β-catenin in PPNAD tissues, as well as activating somatic CTNNB1 variants in macronodules, but not in micronodules or the contralateral adrenal gland (123).
3.2.4 Cortisol-producing adrenocortical adenomas (ACA)
Cortisol-producing adenomas exhibit overactivation of the cAMP/PKA signaling pathway. The most prevalent CPA-causing defect is alteration of the catalytic α-subunit of PKA (PRKACA) and the most common variant has been reported to be p.Leu206Arg (124). Other rare variants have been described (120, 125, 126), all localized in a region of PRKACA that affects its interaction with the regulatory subunit 1α. Activating variants in PRKACA lead to continuous activation of PKA by abolishing the interaction between its catalytic and regulatory subunits. In addition, they can lead to hyperphosphorylation of certain substrates, thereby altering substrate specificity (127). An activating somatic variant in PRKACB has also recently been reported in a patient with CPA; in vitro studies showed that this variant confers higher sensitivity to cAMP (128).
Furthermore, somatic inactivating variants in GNAS and PRKAR1A have been found in sporadic adrenocortical tumors (126, 129–133). The genetic alterations in both genes lead to increased signaling via the cAMP/PKA pathway, however they activate different downstream effectors. Adrenal lesions that harbored variants in GNAS or PRKAR1A had overactivation of the p53 and MAPK signaling pathways, respectively. In PRKAR1A-mutant tumors, genes related to Wnt-signaling pathway (CCND1, CTNNB1, LEF1, LRP5, WISP1 and WNT3) were overexpressed, while in GNAS-mutant tumors, there was increased expression of extracellular matrix receptor interaction and focal adhesion pathways (NFKB, NFKBIA and TNFRSF1A) (134).
3.2.5. Aldosterone-producing benign adrenocortical neoplasms
Excess of aldosterone production characterizes a group of adrenal cortical disorders including aldosterone-producing adenomas, adrenal cortical hyperplasia, familial hyperaldosteronism (FH, <1%) and rarely, carcinomas (<1%) (135–137).
3.2.6 Benign adrenocortical tumors producing aldosterone
Primary aldosteronism (PA) is the most frequent secondary form of hypertension, accounting for approximately 10-20% of patients referred with resistant hypertension and 5% of patients in primary care (138, 139). PA is typically due to unilateral adenomas that produce aldosterone (APA) (65%), or BAH (35%) that leads to autonomous aldosterone production (140). The remaining cases include unilateral hyperplasia (2%), familial hyperaldosteronism (FH, <1%) and aldosterone-producing ACC (<1%) (137). The vast majority of PA cases are sporadic and only 6% are familial (141).
3.2.6.1 Inherited forms of PA
Four forms of FH (type I-type IV) have been described so far and they are inherited in an autosomal dominant manner (136).
The underlying cause of FH I (also known as glucocorticoid-remediable aldosteronism-GRA) is the formation of a chimeric gene, resulting from an unequal fusion of the regulatory regions of CYP11B1, which encodes 11β-hydroxylase, and CYP11B2, that encodes aldosterone synthase. Both enzymes are responsible for the last steps of cortisol and aldosterone synthesis, respectively (142, 143). Formation of the chimeric gene leads to aldosterone overproduction under the regulation of ACTH (143). Treatment is based on the use of glucocorticoids (144). Genetic testing for the chimeric gene (CYP11B1/CYP11B2) should be considered for patients who are diagnosed with PA and have a family history of the disease, onset of PA before the age of 20 years, or family history of stroke at a young age (145).
FH II is due to mutations in the CLCN2 gene, which encodes the chloride channel CIC2. Among other tissues, CIC2 is expressed in the adrenal glands. Gain-of-function variants in CLCN2 lead to increased Cl− ions efflux, which causes cell membrane depolarization and opening of voltage-gated calcium channels, triggering aldosterone production (146, 147). FH II is the most common form of FH, with a prevalence of 1.2-6% in patients with PA (141, 148, 149).
FH III is due to coding variants in the G-protein coupled inward rectifying potassium channel 4 (GIRK4), which is encoded by KCNJ5. Genetic defects in this gene cause a lack of ion selectivity and increased sodium influx, which results in cell depolarization triggering calcium entry into the cells. This signals an increase in CYP11B2 expression and increase in aldosterone production. The severity of hyperaldosteronism has been shown to be related to the type of KCNJ5 variant in some patients (150–152), but not in all of them (153). The majority of patients with germline variants in KCNJ5 present with polydipsia, polyuria and refractory hypertension during childhood. In these patients, aldosterone hypersecretion is high enough to require bilateral adrenalectomy (145). There is heterogeneity in the age at which patients present the disease, and in some cases, the symptoms can be controlled with mineralocorticoid-receptor antagonists (MRAs) (150–154).
FH IV is caused by germline variants in CACNA1H, which encodes the pore-forming α1 subunit of the T-type calcium channel Cav3.2. These variants cause alterations in calcium current properties, leading to increased intracellular calcium concentration and production of aldosterone. Germline variants in CACNA1D, which encodes Cav1.3 (the α1D subunit of the voltage-dependent L-type calcium channel) have also been described in patients with PA; these variants occur exclusively de novo. These patients present with a severe early-onset form of hyperaldosteronism associated with a complex neurological disorder, with the phenotype also including seizures and neurological abnormalities (PASNA) (155).
Finally, germline variants in PDE2A, PDE3B and ARMC5 have also been reported in patients with PA (98, 156). The first two genes were associated with PA within the context of BAH; however, these are not considered as genetic causes of FH (156).
3.2.6.2 Aldosterone-producing adrenocortical adenomas (APA)
In the past decade, major advances have been made in unraveling the molecular background of APAs (157, 158). Variants have been identified in genes associated with familial forms of APA, including KCNJ5 and CACNA1D as well as in ATP1A1 and ATP2B3 (which encode two Na+/K+ and Ca2+ ATPases) (155, 159–161). The most frequent defects are recurrent variants in KCNJ5, encountered in more than 40% of APAs in Caucasians, with two particular variants (p.G151R and p.L168R) being responsible for 36% of cases (162). Variants in KCNJ5 appear to be more frequent in Asian cohorts (162–166) (~70% prevalence), and in women compared to men (63% vs 24% prevalence) (163). Variants in CACNA1D are reported in up to 10% of patients with APA, while ATP1A1 and ATP2B3 variants are less frequent (167).
Additionally, the Wnt/β-catenin signaling pathway has been shown to play a vital role in the adrenal cortex development and the biosynthesis of aldosterone (168). It has been shown to be continuously active in approximately 70% of APAs (169). Under normal unstimulated conditions, β-catenin is in the cytosol as part of the axin complex along with Caseine Kinase 1β, glycogen synthase kinase 3β and adenomatous polyposis coli (APC). Binding of the Wnt ligand to its receptor causes β-catenin to dissociate from the axin complex and translocate to the nucleus where it induces the expression of the transcription factors T cell factor (TCF) and lymphocyte enhancer factor (LEF) (168). Somatic variants in CTNNB1 gene, encoding β-catenin, have been identified in 2-5% of patients with sporadic APA (155, 170, 171); similarly, to APAs due to KCNJ5 variants, these APAs are associated with larger adenomas and are more commonly seen in females. Variants in CTNNB1 have also been reported in two pregnant patients with APA that exhibited increased adrenocortical expression of the gonadotropin releasing hormone (GnRH) and LH/hCG receptors (172). Based on this, an association with pregnancy or menopause was suggested, but this was not confirmed in a follow-up study (172). In rare cases, somatic variants in PRKACA and GNAS have been described in patients with cortisol and aldosterone co-secreting adenoma as well (173, 174). However, their role in the development of APA remains unclear, because those variants are similar to the ones found in CPAs and ACC (99, 120, 131, 175–177).
3.3 Adrenocortical carcinoma
ACCs are rare tumors derived from the adrenal cortex. They affect both adults and children with an annual incidence of 0.7-2.0 cases per million per year (178, 179). They are responsible for steroid excess in 60-70% of cases (52, 180, 181) and they represent one of the most aggressive class of endocrine tumors with an overall poor prognosis (5-year survival rate <35%) (129). However, the exact 5-year survival rate varies depending on the tumor stage, from 82% for tumors in stage I to 18% for tumors in stage IV (182). Thus, the stage at the time of the diagnosis is a crucial prognostic factor. Approximately 40-60% of patients present with signs and symptoms related to hormone excess (183–185). Another 30-40% present with non-specific symptoms associated with local tumor growth (30-40%), including early satiety, abdominal fullness and flank or abdominal pain (184, 185). The remaining 20-30% of ACCs are discovered incidentally on imaging studies for unrelated medical conditions (Table 1) (187). With regards to the age distribution, ACC seems relatively more common in children than in adults and is often associated with hereditary tumor syndromes (186, 188). In fact, the elucidation of genetic alterations underlying familial syndromes predisposing to ACC has led to the identification of signaling pathways involved in the development of cancer such as Insulin growth factor 2 (IGF-2), Wnt-beta catenin and p53 pathways (189).

Table 1 Hormone secretion of functional adrenocortical carcinomas.
3.3.1 Molecular basis of ACC: As part of a tumor predisposition syndrome
3.3.1.1 Li-Fraumeni syndrome (LFS)
LFS is an autosomal dominant disorder that predisposes to various types of cancer including brain cancer, leukemia, soft tissue sarcoma and osteosarcoma, premenopausal breast cancer and ACC. LFS accounts for 50-80% of pediatric cases of ACC (190–192). The clinical criteria for the diagnosis of ‘classic’ LFS include a sarcoma diagnosis before the age of 45, with a first-degree relative diagnosed with any type of cancer before 45 years and another first or second-degree relative with any cancer diagnosis before the age of 45 years or a sarcoma diagnosis at any age (193). Germline variants in TP53, the underlying genetic cause of LFS, have been identified in 70% of cases, while de novo variants have been shown to have a prevalence of 7-20% (129, 190, 194). In a cohort of 286 TP53+ patients from 107 families, the cumulative cancer incidence was 50% by 31 years for females and by 46 years for males (190). Of those patients, 67% had their first cancer diagnosis before the age of 17 years, while five patients were diagnosed with ACC before the age of 17 years. Of those patients, 50% had a second cancer diagnosis and ACC was present in one of them (190).
3.3.1.2 Beckwith-Wiedemann syndrome (BWS)
BWS is a systemic overgrowth disorder caused by genetic or epigenetic changes that ultimately result in upregulation of insulin-like growth factor 2 (IGF2) (195). Loss of heterozygosity of the 11p15 locus, which harbors IGF2, is a common finding in childhood ACC (196). BWS is characterized by hemihypertrophy, macrosomia, macroglossia, hyperinsulinism, omphalocele and distinct facial features (197). In addition, in the first 8 years of life, patients with BWS are at increased risk for embryonal tumors including hepatoblastoma, neuroblastoma and Wilms’s tumor (197–201). The risk of developing intra-abdominal tumors is approximately 5-10% and thus patients with BWS need to undergo regular screening for early diagnosis and management. ACC is the next most common type of tumor reported in BWS patients; other common benign adrenal pathologies include adrenal cysts and ACAs (200).
3.3.1.3 Multiple endocrine neoplasia 1 (MEN1)
MEN1 is inherited in an autosomal dominant manner and is caused by germline heterozygous variants in the MEN1 gene on chromosome 11q13. Its main manifestations are hyperparathyroidism (95%), entero-pancreatic neuroendocrine tumors (50%) and pituitary adenomas (40%). Associated adrenal lesions, mostly ACA and hyperplasias, are present in 20-55% of MEN1 cases (202). Most of these adrenal lesions are nonfunctional (87, 203, 204). ACC occurs only in a small fraction of patients with MEN1 (87, 205, 206). In two cohorts of sporadic ACC, somatic variants of MEN1 were shown to have a prevalence of 7% (207, 208).
3.3.1.4 Lynch syndrome
ACC has also been reported in cases of Lynch syndrome (hereditary nonpolyposis colorectal cancer, HNPCC) (209–213). Lynch syndrome is an autosomal dominant disorder caused by germline heterozygous coding variants in DNA-mismatch repair genes (MSH2, MSH6, MLH1 and PMS2). Patients have a significantly increased risk of cancer, especially colorectal and endometrial cancer, and thus screening for Lynch syndrome is recommended in all patients that are diagnosed with colorectal cancer (209, 214). The prevalence of Lynch syndrome in a large cohort of patients with ACC was reported to be approximately 3% (215).
3.3.2 Other
ACC has been reported in patients with neurofibromatosis type 1, familial adenomatous polyposis, and Werner syndrome (186, 216–223). In addition, ACC has been reported in two cases of patients with CNC (224, 225).
In general, the discovery of genetic syndromes that confer an increased risk for ACC has yielded important clues into the molecular basis of ACC development. For example, the association between FAP and adrenal tumors provided the basis for the insights into the role of β-catenin signaling in adrenal tumors, while the link between ACC and BWS combined with gene expression profiling suggested the IGF-1 receptor as a target for ACC therapy. The latter hypothesis has now been tested in clinical trials (226).
4. Treatment approaches and clinical trials
Patients with unilateral adrenal adenomas that are hormonally active receive treatment with unilateral adrenalectomy (25). Individuals diagnosed with CS are evaluated for the need of adrenalectomy based on the degree of cortisol excess, comorbidities, age and preference of the patient; typically, surgical resection of the is the first-line treatment (50). However, in some cases where hypercortisolemia is resistant to surgery or in cases where surgical treatment is contraindicated due to patient co-morbidities pharmacotherapy is indicated (227). This includes ketoconazole along with metyrapone, mitotane, or etomidate (227, 228). Recently, two new drugs were approved by the FDA, levoketoconazole and osilodrostat, which are both inhibitors of steroidogenic enzyme activity. Non-functioning adrenal tumors are evaluated to verify eligibility for partial adrenalectomy based on size and potential to malignancy (229). In general, surgery should be considered in lesions >4cm in size or those that are hormonally indeterminate, even if imaging characteristics resemble those of a benign lesion (50). Based on the guidelines from the Endocrine Society, the diagnosis of PA in patients with hypertension should include screening (elevated aldosterone-renin ratio) followed by confirmatory testing (including salt loading, fludrocortisone or captopril administration, which all fail to sufficiently lower aldosterone levels in patients with PA) (140). Regarding ACCs, complete surgical resection remains the only curative treatment in patients with resectable stages I-III (187, 230), while adjuvant therapies are used to decrease recurrence. In addition, Mitotane, a dichlorodiphenyltrichloroethane analog (DTT), is the only medication that is specifically approved for ACC and can be used either as adjuvant or in advanced stages in combination with classic cytotoxic agents (231). Phase I trial studies using cixutumumab, an insulin growth factor receptor (IGF-1R) antibody have showed promising results in terms of lower toxicity and better disease outcome compared to mitotane (232). Moreover, a trial investigating the combination of cixutumumab with the mTOR inhibitor, temsirolimus, showed that almost half of the patients achieved prolonged stable disease and therefore, current treatment options may be improved (233).
There are at least 15 clinical trials currently recruiting patients diagnosed with ACC. Location includes United States, Europe and China. Over 20 studies, all over the world, are recruiting patients diagnosed with hyperaldosteronism, comprising either observational or interventional studies. There are also about 20 studies currently recruiting patients with CS and many other studies are now recruiting for other adrenal cortex-related diseases. Information about current and past trials, as well as institution and collaborators, can be found at clinicaltrials.gov, a resource provided by the United States National Library of Medicine.
5. Conclusion
Here, we summarized the main genetic and molecular aspects of adrenocortical diseases, which are, in some cases, difficult to diagnose. The rapid progress of next generation sequencing (NGS) techniques has opened new horizons to examine and diagnose adrenocortical diseases with unbiased mechanisms. For example, exome sequencing may be used in patients with a clinical phenotype but no identified variant in one of the known causative genes, in order to perform an unbiased scan and potentially identify causative variants in genes previously not associated with the disorders. This not only yields a diagnosis, but also provides new clues into disease pathophysiology. Despite the progress, diagnosis based on genetic screening is still limited to either large centers and/or patients with financial access to these analyses. Therefore, heath care providers still face limitations in offering access to precise medicine to all patients. Considering that a great number of adrenocortical diseases is due to genetic onset, we believe that increasing access to NGS will greatly improve early and precise diagnosis of adrenocortical disorders. Consequently, more accurate treatments will be delivered to individuals that harbor genetic alterations leading to disorders of the adrenal cortex.
Author contributions
GP: Writing - original draft; AM: Writing - review & editing; FF: Conceptualization; Supervision; Writing - review & editing. All authors contributed to the article and approved the submitted version.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Bechmann N, Berger I, Bornstein SR, Steenblock C. Adrenal medulla development and medullary-cortical interactions. Mol Cell Endocrinol (2021) 528:111258. doi: 10.1016/j.mce.2021.111258
2. Avisse C, Marcus C, Patey M, Ladam-Marcus V, Delattre JF, Flament JB. Surgical anatomy and embryology of the adrenal glands. Surg Clin North Am (2000) 80(1):403–15. doi: 10.1016/s0039-6109(05)70412-6
3. Yates R, Katugampola H, Cavlan D, Cogger K, Meimaridou E, Hughes C, et al. Adrenocortical development, maintenance, and disease. Curr Top Dev Biol (2013) 106:239–312. doi: 10.1016/B978-0-12-416021-7.00007-9
4. Kastriti ME, Kameneva P, Adameyko I. Stem cells, evolutionary aspects and pathology of the adrenal medulla: A new developmental paradigm. Mol Cell Endocrinol (2020) 518:110998. doi: 10.1016/j.mce.2020.110998
5. Gallo-Payet N, Battista MC. Steroidogenesis-adrenal cell signal transduction. Compr Physiol (2014) 4(3):889–964. doi: 10.1002/cphy.c130050
6. Pignatti E, Leng S, Carlone DL, Breault DT. Regulation of zonation and homeostasis in the adrenal cortex. Mol Cell Endocrinol (2017) 441:146–55. doi: 10.1016/j.mce.2016.09.003
7. Magill SB. Pathophysiology, diagnosis, and treatment of mineralocorticoid disorders. Compr Physiol (2014) 4(3):1083–119. doi: 10.1002/cphy.c130042
8. Nishimoto K, Nakagawa K, Li D, Kosaka T, Oya M, Mikami S, et al. Adrenocortical zonation in humans under normal and pathological conditions. J Clin Endocrinol Metab (2010) 95(5):2296–305. doi: 10.1210/jc.2009-2010
9. Lyraki R, Schedl A. Adrenal cortex renewal in health and disease. Nat Rev Endocrinol (2021) 17(7):421–34. doi: 10.1038/s41574-021-00491-4
10. Guerreiro R, Bras J, Hardy J, Singleton A. Next generation sequencing techniques in neurological diseases: redefining clinical and molecular associations . Hum Mol Genet (2014) 23(R1):R47–53. doi: 10.1093/hmg/ddu203
11. Barba M, Czosnek H, Hadidi A. Historical perspective, development and applications of next-generation sequencing in plant virology. Viruses (2014) 6(1):106–36. doi: 10.3390/v6010106
12. Chaitankar V, Karakulah G, Ratnapriya R, Giuste FO, Brooks MJ, Swaroop A. Next generation sequencing technology and genomewide data analysis: Perspectives for retinal research. Prog Retin Eye Res (2016) 55:1–31. doi: 10.1016/j.preteyeres.2016.06.001
13. Baba R, Oki K, Gomez-Sanchez CE, Otagaki Y, Itcho K, Kobuke K, et al. Genotype-specific cortisol production associated with cushing's syndrome adenoma with PRKACA mutations. Mol Cell Endocrinol (2021) 538:111456. doi: 10.1016/j.mce.2021.111456
14. Maria AG, Suzuki M, Berthon A, Kamilaris C, Demidowich A, Lack J, et al. Mosaicism for KCNJ5 causing early-onset primary aldosteronism due to bilateral adrenocortical hyperplasia. Am J Hypertens (2020) 33(2):124–30. doi: 10.1093/ajh/hpz172
15. Gwynne JT, Strauss JF3. The role of lipoproteins in steroidogenesis and cholesterol metabolism in steroidogenic glands. Endocr Rev (1982) 3(3):299–329. doi: 10.1210/edrv-3-3-299
16. Faust JR, Goldstein JL, Brown MS. Receptor-mediated uptake of low density lipoprotein and utilization of its cholesterol for steroid synthesis in cultured mouse adrenal cells. J Biol Chem (1977) 252(14):4861–71.
17. Hoekstra M, Van Berkel TJ, Van Eck M. Scavenger receptor BI: A multi-purpose player in cholesterol and steroid metabolism. World J Gastroenterol (2010) 16(47):5916–24. doi: 10.3748/wjg.v16.i47.5916
18. Chang TY, Li BL, Chang CC, Urano Y. Acyl-coenzyme a:cholesterol acyltransferases . Am J Physiol Endocrinol Metab (2009) 297(1):E1–9. doi: 10.1152/ajpendo.90926.2008
19. Vaidya A, Mulatero P, Baudrand R, Adler GK. The expanding spectrum of primary aldosteronism: Implications for diagnosis, pathogenesis, and treatment. Endocr Rev (2018) 39(6):1057–88. doi: 10.1210/er.2018-00139
20. Byrd JB, Turcu AF, Auchus RJ. Primary aldosteronism: Practical approach to diagnosis and management. Circulation (2018) 138(8):823–35. doi: 10.1161/CIRCULATIONAHA.118.033597
21. Nehme A, Zouein FA, Zayeri ZD, Zibara K. An update on the tissue renin angiotensin system and its role in physiology and pathology. J Cardiovasc Dev Dis (2019) 6(2):14–31. doi: 10.3390/jcdd6020014
22. Rainey WE. Adrenal zonation: clues from 11beta-hydroxylase and aldosterone synthase. Mol Cell Endocrinol (1999) 151(1-2):151–60. doi: 10.1016/s0303-7207(99)00051-9
23. Turcu A, Smith JM, Auchus R, Rainey WE. Adrenal androgens and androgen precursors-definition, synthesis, regulation and physiologic actions. Compr Physiol (2014) 4(4):1369–81. doi: 10.1002/cphy.c140006
24. Labrie F, Luu-The V, Labrie C, Simard J. DHEA and its transformation into androgens and estrogens in peripheral target tissues: Intracrinology. Front Neuroendocrinol (2001) 22(3). doi: 10.1006/frne.2001.0216
25. Sherlock M, Scarsbrook A, Abbas A, Fraser S, Limumpornpetch P, Dineen R, et al. Adrenal incidentaloma. Endocr Rev (2020) 41(6):775–820. doi: 10.1210/endrev/bnaa008
26. Speiser PW, Arlt W, Auchus RJ, Baskin LS, Conway GS, Merke DP, et al. Congenital adrenal hyperplasia due to steroid 21-hydroxylase deficiency: An endocrine society clinical practice guideline. J Clin Endocrinol Metab (2018) 103(11):4043–88. doi: 10.1210/jc.2018-01865
27. Merke DP, Auchus RJ. Congenital adrenal hyperplasia due to 21-hydroxylase deficiency. N Engl J Med (2020) 383(13):1248–61. doi: 10.1056/NEJMra1909786
28. Speiser PW, White PC. Congenital adrenal hyperplasia. N Engl J Med (2003) 349(8):776–88. doi: 10.1056/NEJMra021561
29. Carroll MC, Campbell RD, Porter RR. Mapping of steroid 21-hydroxylase genes adjacent to complement component C4 genes in HLA, the major histocompatibility complex in man. Proc Natl Acad Sci U.S.A. (1985) 82(2):521–5. doi: 10.1073/pnas.82.2.521
30. White PC, Vitek A, Dupont B, New MI. Characterization of frequent deletions causing steroid 21-hydroxylase deficiency. Proc Natl Acad Sci U.S.A. (1988) 85(12):4436–40. doi: 10.1073/pnas.85.12.4436
31. Finkielstain GP, Chen W, Mehta SP, Fujimura FK, Hanna RM, Van Ryzin C, et al. Comprehensive genetic analysis of 182 unrelated families with congenital adrenal hyperplasia due to 21-hydroxylase deficiency. J Clin Endocrinol Metab (2011) 96(1):E161–72. doi: 10.1210/jc.2010-0319
32. Speiser PW, Azziz R, Baskin LS, Ghizzoni L, Hensle TW, Merke DP, et al. Congenital adrenal hyperplasia due to steroid 21-hydroxylase deficiency: An endocrine society clinical practice guideline. J Clin Endocrinol Metab (2010) 95(9):4133–60. doi: 10.1210/jc.2009-2631
33. White PC, Speiser PW. Congenital adrenal hyperplasia due to 21-hydroxylase deficiency. Endocr Rev (2000) 21(3):245–91. doi: 10.1210/edrv.21.3.0398
34. Nandagopal R, Sinaii N, Avila NA, Van Ryzin C, Chen W, Finkielstain GP, et al. Phenotypic profiling of parents with cryptic nonclassic congenital adrenal hyperplasia: Findings in 145 unrelated families. Eur J Endocrinol (2011) 164(6):977–84. doi: 10.1530/EJE-11-0019
35. Sir-Petermann T, Codner E, Perez V, Echiburu B, Maliqueo M, Ladron de Guevara A, et al. Metabolic and reproductive features before and during puberty in daughters of women with polycystic ovary syndrome. J Clin Endocrinol Metab (2009) 94(6):1923–30. doi: 10.1210/jc.2008-2836
36. Bidet M, Bellanne-Chantelot C, Galand-Portier MB, Tardy V, Billaud L, Laborde K, et al. Clinical and molecular characterization of a cohort of 161 unrelated women with nonclassical congenital adrenal hyperplasia due to 21-hydroxylase deficiency and 330 family members. J Clin Endocrinol Metab (2009) 94(5):1570–8. doi: 10.1210/jc.2008-1582
37. Falhammar H, Nordenstrom A. Nonclassic congenital adrenal hyperplasia due to 21-hydroxylase deficiency: Clinical presentation, diagnosis, treatment, and outcome. Endocrine (2015) 50(1):32–50. doi: 10.1007/s12020-015-0656-0
38. Yarman S, Dursun A, Oguz F, Alagol F. The prevalence, molecular analysis and HLA typing of late-onset 21-hydroxylase deficiency in turkish woman with hirsutism and polycystic ovary. Endocr J (2004). doi: 10.1507/endocrj.51.31
39. Kamel N, Tonyukuk V, Emral R, Corapcioglu D, Bastemir M, Gullu S. The prevalence of late onset congenital adrenal hyperplasia in hirsute women from central anatolia. Endocr J (2003) 50(6):815–23. doi: 10.1507/endocrj.50.815
40. Trakakis E, Rizos D, Loghis C, Chryssikopoulos A, Spyropoulou M, Salamalekis E, et al. The prevalence of non-classical congenital adrenal hyperplasia due to 21-hydroxylase deficiency in greek women with hirsutism and polycystic ovary syndrome. Endocr J (2008) 55(1):33–9. doi: 10.1507/endocrj.k07-053
41. Kyritsi EM, Dimitriadis GK, Kyrou I, Kaltsas G, Randeva HS. PCOS remains a diagnosis of exclusion: A concise review of key endocrinopathies to exclude. Clin Endocrinol (2017) 86(1):1–6. doi: 10.1111/cen.13245
42. Moran C, Azziz R. 21-hydroxylase-deficient nonclassic adrenal hyperplasia: The great pretender. Semin Reprod Med (2003) 21(3):295–300. doi: 10.1055/s-2003-43307
43. White PC, Curnow KM, Pascoe L. Disorders of steroid 11 beta-hydroxylase isozymes. Endocr Rev (1994) 15(4):421–38. doi: 10.1210/edrv-15-4-421
44. Auchus RJ. Steroid 17-hydroxylase and 17,20-lyase deficiencies, genetic and pharmacologic. J Steroid Biochem Mol Biol (2017) 165(Pt A):71–8. doi: 10.1016/j.jsbmb.2016.02.002
45. McBride MW, McVie AJ, Burridge SM, Brintnell B, Craig N, Wallace AM, et al. Cloning, expression, and physical mapping of the 3beta-hydroxysteroid dehydrogenase gene cluster (HSD3BP1-HSD3BP5) in human. Genomics (1999) 61(3):277–84. doi: 10.1006/geno.1999.5459
46. Finkielstain GP, Vieites A, Bergada I, Rey RA. Disorders of sex development of adrenal origin. Front Endocrinol (Lausanne) (2021) 12:770782. doi: 10.3389/fendo.2021.770782
47. Miller WL. Disorders in the initial steps of steroid hormone synthesis. J Steroid Biochem Mol Biol (2017) 165(Pt A):18–37. doi: 10.1016/j.jsbmb.2016.03.009
48. Turcu AF, Auchus RJ. The next 150 years of congenital adrenal hyperplasia. J Steroid Biochem Mol Biol (2015) 153:63–71. doi: 10.1016/j.jsbmb.2015.05.013
49. Young WF Jr. Clinical practice. The incidentally discovered adrenal mass. N Engl J Med (2007) 356(6):601–10. doi: 10.1056/NEJMcp065470
50. Fassnacht M, Arlt W, Bancos I, Dralle H, Newell-Price J, Sahdev A, et al. Management of adrenal incidentalomas: European society of endocrinology clinical practice guideline in collaboration with the european network for the study of adrenal tumors. Eur J Endocrinol (2016) 175(2):G1–G34. doi: 10.1530/EJE-16-0467
51. Bertherat J, Mosnier-Pudar H, Bertagna X. Adrenal incidentalomas. Curr Opin Oncol (2002) 14(1):58–63. doi: 10.1097/00001622-200201000-00011
52. Mansmann G, Lau J, Balk E, Rothberg M, Miyachi Y, Bornstein SR. The clinically inapparent adrenal mass: Update in diagnosis and management. Endocr Rev (2004) 25(2):309–40. doi: 10.1210/er.2002-0031
53. Valassi E, Santos A, Yaneva M, Toth M, Strasburger CJ, Chanson P, et al. The european registry on cushing's syndrome: 2-year experience. Baseline demographic and clinical characteristics. Eur J Endocrinol (2011) 165(3):383–92. doi: 10.1530/EJE-11-0272
54. Bolland MJ, Holdaway IM, Berkeley JE, Lim S, Dransfield WJ, Conaglen JV, et al. Mortality and morbidity in cushing's syndrome in new zealand. Clin Endocrinol (Oxf) (2011) 75(4):436–42. doi: 10.1111/j.1365-2265.2011.04124.x
55. Steffensen C, Bak AM, Rubeck KZ, Jorgensen JO. Epidemiology of cushing's syndrome. Neuroendocrinology (2010) 92 Suppl 1:1–5. doi: 10.1159/000314297
56. Lindholm J, Juul S, Jorgensen JO, Astrup J, Bjerre P, Feldt-Rasmussen U, et al. Incidence and late prognosis of cushing's syndrome: A population-based study. J Clin Endocrinol Metab (2001) 86(1):117–23. doi: 10.1210/jcem.86.1.7093
57. Krarup T, Krarup T, Hagen C. Do patients with type 2 diabetes mellitus have an increased prevalence of cushing's syndrome? Diabetes Metab Res Rev (2012) 28(3):219–27. doi: 10.1002/dmrr.2262
58. Terzolo M, Reimondo G, Chiodini I, Castello R, Giordano R, Ciccarelli E, et al. Screening of cushing's syndrome in outpatients with type 2 diabetes: Results of a prospective multicentric study in italy. J Clin Endocrinol Metab (2012) 97(10):3467–75. doi: 10.1210/jc.2012-1323
59. Chiodini I, Mascia ML, Muscarella S, Battista C, Minisola S, Arosio M, et al. Subclinical hypercortisolism among outpatients referred for osteoporosis. Ann Intern Med (2007) 147(8):541–8. doi: 10.7326/0003-4819-147-8-200710160-00006
60. Tabarin A, Perez P. Pros and cons of screening for occult cushing syndrome. Nat Rev Endocrinol (2011) 7(8):445–55. doi: 10.1038/nrendo.2011.51
61. Arnaldi G, Angeli A, Atkinson AB, Bertagna X, Cavagnini F, Chrousos GP, et al. Diagnosis and complications of cushing's syndrome: A consensus statement. J Clin Endocrinol Metab (2003) 88(12):5593–602. doi: 10.1210/jc.2003-030871
62. Biller BM, Grossman AB, Stewart PM, Melmed S, Bertagna X, Bertherat J, et al. Treatment of adrenocorticotropin-dependent cushing's syndrome: A consensus statement. J Clin Endocrinol Metab (2008) 93(7):2454–62. doi: 10.1210/jc.2007-2734
63. Newell-Price J, Bertagna X, Grossman AB, Nieman LK. Cushing's syndrome. Lancet (2006) 367(9522):1605–17. doi: 10.1016/S0140-6736(06)68699-6
64. Lodish M, Stratakis CA. A genetic and molecular update on adrenocortical causes of cushing syndrome. Nat Rev Endocrinol (2016) 12(5):255–62. doi: 10.1038/nrendo.2016.24
65. Lacroix A. ACTH-independent macronodular adrenal hyperplasia. Best Pract Res Clin Endocrinol Metab (2009) 23(2):245–59. doi: 10.1016/j.beem.2008.10.011
66. Stratakis CA. Cushing syndrome caused by adrenocortical tumors and hyperplasias (corticotropin- independent cushing syndrome). Endocr Dev (2008) 13:117–32. doi: 10.1159/000134829
67. Bossis I, Stratakis CA. Minireview: PRKAR1A: normal and abnormal functions. Endocrinology (2004) 145(12):5452–8. doi: 10.1210/en.2004-0900
68. de Joussineau C, Sahut-Barnola I, Levy I, Saloustros E, Val P, Stratakis CA, et al. The cAMP pathway and the control of adrenocortical development and growth. Mol Cell Endocrinol (2012) 351(1):28–36. doi: 10.1016/j.mce.2011.10.006
69. Louiset E, Duparc C, Young J, Renouf S, Tetsi Nomigni M, Boutelet I, et al. Intraadrenal corticotropin in bilateral macronodular adrenal hyperplasia. N Engl J Med (2013) 369(22):2115–25. doi: 10.1056/NEJMoa1215245
70. Lacroix A, Hamet P, Boutin JM. Leuprolide acetate therapy in luteinizing hormone–dependent cushing's syndrome. N Engl J Med (1999) 341(21):1577–81. doi: 10.1056/NEJM199911183412104
71. Reznik Y, Allali-Zerah V, Chayvialle JA, Leroyer R, Leymarie P, Travert G, et al. Food-dependent cushing's syndrome mediated by aberrant adrenal sensitivity to gastric inhibitory polypeptide. N Engl J Med (1992) 327(14):981–6. doi: 10.1056/NEJM199210013271403
72. Vezzosi D, Cartier D, Regnier C, Otal P, Bennet A, Parmentier F, et al. Familial adrenocorticotropin-independent macronodular adrenal hyperplasia with aberrant serotonin and vasopressin adrenal receptors. Eur J Endocrinol (2007) 156(1):21–31. doi: 10.1530/eje.1.02324
73. Libe R, Coste J, Guignat L, Tissier F, Lefebvre H, Barrande G, et al. Aberrant cortisol regulations in bilateral macronodular adrenal hyperplasia: A frequent finding in a prospective study of 32 patients with overt or subclinical cushing's syndrome. Eur J Endocrinol (2010) 163(1):129–38. doi: 10.1530/EJE-10-0195
74. Hofland J, Hofland LJ, van Koetsveld PM, Steenbergen J, de Herder WW, van Eijck CH, et al. ACTH-independent macronodular adrenocortical hyperplasia reveals prevalent aberrant in vivo and in vitro responses to hormonal stimuli and coupling of arginine-vasopressin type 1a receptor to 11beta-hydroxylase. Orphanet J Rare Dis (2013) 8:142. doi: 10.1186/1750-1172-8-142
75. Bourdeau I, Stratakis CA. Cyclic AMP-dependent signaling aberrations in macronodular adrenal disease. Ann N Y Acad Sci (2002) 968:240–55. doi: 10.1111/j.1749-6632.2002.tb04339.x
76. Miyamura N, Taguchi T, Murata Y, Taketa K, Iwashita S, Matsumoto K, et al. Inherited adrenocorticotropin-independent macronodular adrenal hyperplasia with abnormal cortisol secretion by vasopressin and catecholamines: detection of the aberrant hormone receptors on adrenal gland. Endocrine (2002) 19(3):319–26. doi: 10.1385/ENDO:19:3:319
77. Lee S, Hwang R, Lee J, Rhee Y, Kim DJ, Chung UI, et al. Ectopic expression of vasopressin V1b and V2 receptors in the adrenal glands of familial ACTH-independent macronodular adrenal hyperplasia. Clin Endocrinol (Oxf) (2005) 63(6):625–30. doi: 10.1111/j.1365-2265.2005.02387.x
78. Fragoso MC, Alencar GA, Lerario AM, Bourdeau I, Almeida MQ, Mendonca BB, et al. Genetics of primary macronodular adrenal hyperplasia. J Endocrinol (2015) 224(1):R31–43. doi: 10.1530/JOE-14-0568
79. Lampron A, Bourdeau I, Hamet P, Tremblay J, Lacroix A. Whole genome expression profiling of glucose-dependent insulinotropic peptide (GIP)- and adrenocorticotropin-dependent adrenal hyperplasias reveals novel targets for the study of GIP-dependent cushing's syndrome. J Clin Endocrinol Metab (2006) 91(9):3611–8. doi: 10.1210/jc.2006-0221
80. Libe R, Fratticci A, Coste J, Tissier F, Horvath A, Ragazzon B, et al. Phosphodiesterase 11A (PDE11A) and genetic predisposition to adrenocortical tumors. Clin Cancer Res (2008) 14(12):4016–24. doi: 10.1158/1078-0432.CCR-08-0106
81. Vezzosi D, Libe R, Baudry C, Rizk-Rabin M, Horvath A, Levy I, et al. Phosphodiesterase 11A (PDE11A) gene defects in patients with acth-independent macronodular adrenal hyperplasia (AIMAH): Functional variants may contribute to genetic susceptibility of bilateral adrenal tumors. J Clin Endocrinol Metab (2012) 97(11):E2063–9. doi: 10.1210/jc.2012-2275
82. Rothenbuhler A, Horvath A, Libe R, Faucz FR, Fratticci A, Raffin Sanson ML, et al. Identification of novel genetic variants in phosphodiesterase 8B (PDE8B), a cAMP-specific phosphodiesterase highly expressed in the adrenal cortex, in a cohort of patients with adrenal tumours. Clin Endocrinol (Oxf) (2012) 77(2):195–9. doi: 10.1111/j.1365-2265.2012.04366.x
83. Hsiao HP, Kirschner LS, Bourdeau I, Keil MF, Boikos SA, Verma S, et al. Clinical and genetic heterogeneity, overlap with other tumor syndromes, and atypical glucocorticoid hormone secretion in adrenocorticotropin-independent macronodular adrenal hyperplasia compared with other adrenocortical tumors. J Clin Endocrinol Metab (2009) 94(8):2930–7. doi: 10.1210/jc.2009-0516
84. Fragoso MC, Domenice S, Latronico AC, Martin RM, Pereira MA, Zerbini MC, et al. Cushing's syndrome secondary to adrenocorticotropin-independent macronodular adrenocortical hyperplasia due to activating mutations of GNAS1 gene. J Clin Endocrinol Metab (2003) 88(5):2147–51. doi: 10.1210/jc.2002-021362
85. Lumbroso S, Paris F, Sultan C. McCune-albright syndrome: Molecular genetics. J Pediatr Endocrinol Metab (2002) 15 Suppl 3:875–82.
86. Swords FM, Noon LA, King PJ, Clark AJ. Constitutive activation of the human ACTH receptor resulting from a synergistic interaction between two naturally occurring missense mutations in the MC2R gene. Mol Cell Endocrinol (2004) 213(2):149–54. doi: 10.1016/j.mce.2003.10.052
87. Gatta-Cherifi B, Chabre O, Murat A, Niccoli P, Cardot-Bauters C, Rohmer V, et al. Adrenal involvement in MEN1. analysis of 715 cases from the groupe d'etude des tumeurs endocrines database. Eur J Endocrinol (2012) 166(2):269–79. doi: 10.1530/EJE-11-0679
88. Matyakhina L, Freedman RJ, Bourdeau I, Wei MH, Stergiopoulos SG, Chidakel A, et al. Hereditary leiomyomatosis associated with bilateral, massive, macronodular adrenocortical disease and atypical cushing syndrome: A clinical and molecular genetic investigation. J Clin Endocrinol Metab (2005) 90(6):3773–9. doi: 10.1210/jc.2004-2377
89. Shuch B, Ricketts CJ, Vocke CD, Valera VA, Chen CC, Gautam R, et al. Adrenal nodular hyperplasia in hereditary leiomyomatosis and renal cell cancer. J Urol (2013) 189(2):430–5. doi: 10.1016/j.juro.2012.07.139
90. Faucz FR, Zilbermint M, Lodish MB, Szarek E, Trivellin G, Sinaii N, et al. Macronodular adrenal hyperplasia due to mutations in an armadillo repeat containing 5 (ARMC5) gene: A clinical and genetic investigation. J Clin Endocrinol Metab (2014) 99(6):E1113–9. doi: 10.1210/jc.2013-4280
91. Espiard S, Drougat L, Libe R, Assie G, Perlemoine K, Guignat L, et al. ARMC5 mutations in a large cohort of primary macronodular adrenal hyperplasia: Clinical and functional consequences. J Clin Endocrinol Metab (2015) 100(6):E926–35. doi: 10.1210/jc.2014-4204
92. Hu Y, Lao L, Mao J, Jin W, Luo H, Charpentier T, et al. Armc5 deletion causes developmental defects and compromises t-cell immune responses. Nat Commun (2017) 8:13834. doi: 10.1038/ncomms13834
93. Cavalcante IP, Nishi M, Zerbini MCN, Almeida MQ, Brondani VB, Botelho M, et al. The role of ARMC5 in human cell cultures from nodules of primary macronodular adrenocortical hyperplasia (PMAH). Mol Cell Endocrinol (2018) 460:36–46. doi: 10.1016/j.mce.2017.06.027
94. Assie G, Libe R, Espiard S, Rizk-Rabin M, Guimier A, Luscap W, et al. ARMC5 mutations in macronodular adrenal hyperplasia with cushing's syndrome. N Engl J Med (2013) 369(22):2105–14. doi: 10.1056/NEJMoa1304603
95. Berthon A, Faucz FR, Espiard S, Drougat L, Bertherat J, Stratakis CA. Age-dependent effects of Armc5 haploinsufficiency on adrenocortical function. Hum Mol Genet (2017) 26(18):3495–507. doi: 10.1093/hmg/ddx235
96. Suzuki S, Tatsuno I, Oohara E, Nakayama A, Komai E, Shiga A, et al. Germline deletion of Armc5 in familial primary macronodular adrenal hyperplasia. Endocr Pract (2015) 21(10):1152–60. doi: 10.4158/EP15756.OR
97. Bourdeau I, Oble S, Magne F, Levesque I, Caceres-Gorriti KY, Nolet S, et al. ARMC5 mutations in a large french-canadian family with cortisol-secreting beta-adrenergic/vasopressin responsive bilateral macronodular adrenal hyperplasia. Eur J Endocrinol (2016) 174(1):85–96. doi: 10.1530/EJE-15-0642
98. Zilbermint M, Xekouki P, Faucz FR, Berthon A, Gkourogianni A, Schernthaner-Reiter MH, et al. Primary aldosteronism and ARMC5 variants. J Clin Endocrinol Metab (2015) 100(6):E900–9. doi: 10.1210/jc.2014-4167
99. Cao Y, He M, Gao Z, Peng Y, Li Y, Li L, et al. Activating hotspot L205R mutation in PRKACA and adrenal cushing's syndrome. Science (2014) 344(6186):913–7. doi: 10.1126/science.1249480
100. Zhu J, Cui L, Wang W, Hang XY, Xu AX, Yang SX, et al. Whole exome sequencing identifies mutation of EDNRA involved in ACTH-independent macronodular adrenal hyperplasia. Fam Cancer (2013) 12(4):657–67. doi: 10.1007/s10689-013-9642-y
101. Mete O, Erickson LA, Juhlin CC, de Krijger RR, Sasano H, Volante M, et al. Overview of the 2022 WHO classification of adrenal cortical tumors. Endocr Pathol (2022) 33(1):155–96. doi: 10.1007/s12022-022-09710-8
102. Pitsava G, Zhu C, Sundaram R, Mills JL, Stratakis CA. Predicting the risk of cardiac myxoma in carney complex. Genet Med (2021) 23(1):80–5. doi: 10.1038/s41436-020-00956-3
103. Correa R, Salpea P, Stratakis CA. Arney complex: An update. Eur J Endocrinol (2015) 173(4):M85–97. doi: 10.1530/EJE-15-0209
104. Stratakis CA. Genetics of carney complex and related familial lentiginoses, and other multiple tumor syndromes. Front Biosci (2000) 5:D353–66. doi: 10.2741/stratakis
105. Bertherat J, Horvath A, Groussin L, Grabar S, Boikos S, Cazabat L, et al. Mutations in regulatory subunit type 1A of cyclic adenosine 5'-monophosphate-dependent protein kinase (PRKAR1A): Phenotype analysis in 353 patients and 80 different genotypes. J Clin Endocrinol Metab (2009) 94(6):2085–91. doi: 10.1210/jc.2008-2333
106. Cazabat L, Ragazzon B, Groussin L, Bertherat J. PRKAR1A mutations in primary pigmented nodular adrenocortical disease. Pituitary (2006) 9(3):211–9. doi: 10.1007/s11102-006-0266-1
107. Kirschner LS, Carney JA, Pack SD, Taymans SE, Giatzakis C, Cho YS, et al. Mutations of the gene encoding the protein kinase a type i-alpha regulatory subunit in patients with the carney complex. Nat Genet (2000) 26(1):89–92. doi: 10.1038/79238
108. Almeida MQ, Stratakis CA. Carney complex and other conditions associated with micronodular adrenal hyperplasias. Best Pract Res Clin Endocrinol Metab (2010) 24(6):907–14. doi: 10.1016/j.beem.2010.10.006
109. Robinson-White A, Hundley TR, Shiferaw M, Bertherat J, Sandrini F, Stratakis CA. Protein kinase-a activity in PRKAR1A-mutant cells, and regulation of mitogen-activated protein kinases ERK1/2. Hum Mol Genet (2003) 12(13):1475–84. doi: 10.1093/hmg/ddg160
110. Salpea P, Horvath A, London E, Faucz FR, Vetro A, Levy I, et al. Deletions of the PRKAR1A locus at 17q24.2-q24.3 in carney complex: Genotype-phenotype correlations and implications for genetic testing. J Clin Endocrinol Metab (2014) 99(1):E183–8. doi: 10.1210/jc.2013-3159
111. Horvath A, Bossis I, Giatzakis C, Levine E, Weinberg F, Meoli E, et al. Large deletions of the PRKAR1A gene in carney complex. Clin Cancer Res (2008) 14(2):388–95. doi: 10.1158/1078-0432.CCR-07-1155
112. Greene EL, Horvath AD, Nesterova M, Giatzakis C, Bossis I, Stratakis CA. In vitro functional studies of naturally occurring pathogenic PRKAR1A mutations that are not subject to nonsense mRNA decay. Hum Mutat (2008) 29(5):633–9. doi: 10.1002/humu.20688
113. Stratakis CA, Carney JA, Lin JP, Papanicolaou DA, Karl M, Kastner DL, et al. Carney complex, a familial multiple neoplasia and lentiginosis syndrome. analysis of 11 kindreds and linkage to the short arm of chromosome 2. J Clin Invest (1996) 97(3):699–705. doi: 10.1172/JCI118467
114. Matyakhina L, Pack S, Kirschner LS, Pak E, Mannan P, Jaikumar J, et al. Chromosome 2 (2p16) abnormalities in carney complex tumours. J Med Genet (2003) 40(4):268–77. doi: 10.1136/jmg.40.4.268
115. Tirosh A, Valdes N, Stratakis CA. Genetics of micronodular adrenal hyperplasia and carney complex. Presse Med (2018) 47(7-8 Pt 2):e127–37. doi: 10.1016/j.lpm.2018.07.005
116. Forlino A, Vetro A, Garavelli L, Ciccone R, London E, Stratakis CA, et al. PRKACB and carney complex. N Engl J Med (2014) 370(11):1065–7. doi: 10.1056/NEJMc1309730
117. Horvath A, Boikos S, Giatzakis C, Robinson-White A, Groussin L, Griffin KJ, et al. A genome-wide scan identifies mutations in the gene encoding phosphodiesterase 11A4 (PDE11A) in individuals with adrenocortical hyperplasia. Nat Genet (2006) 38(7):794–800. doi: 10.1038/ng1809
118. Libe R, Horvath A, Vezzosi D, Fratticci A, Coste J, Perlemoine K, et al. Frequent phosphodiesterase 11A gene (PDE11A) defects in patients with carney complex (CNC) caused by PRKAR1A mutations: PDE11A may contribute to adrenal and testicular tumors in CNC as a modifier of the phenotype. J Clin Endocrinol Metab (2011) 96(1):E208–14. doi: 10.1210/jc.2010-1704
119. Horvath A, Mericq V, Stratakis CA. Mutation in PDE8B, a cyclic AMP-specific phosphodiesterase in adrenal hyperplasia. N Engl J Med (2008) 358(7):750–2. doi: 10.1056/NEJMc0706182
120. Beuschlein F, Fassnacht M, Assie G, Calebiro D, Stratakis CA, Osswald A, et al. Constitutive activation of PKA catalytic subunit in adrenal cushing's syndrome. N Engl J Med (2014) 370(11):1019–28. doi: 10.1056/NEJMoa1310359
121. Lodish MB, Yuan B, Levy I, Braunstein GD, Lyssikatos C, Salpea P, et al. Germline PRKACA amplification causes variable phenotypes that may depend on the extent of the genomic defect: Molecular mechanisms and clinical presentations. Eur J Endocrinol (2015) 172(6):803–11. doi: 10.1530/EJE-14-1154
122. Tadjine M, Lampron A, Ouadi L, Horvath A, Stratakis CA, Bourdeau I. Detection of somatic beta-catenin mutations in primary pigmented nodular adrenocortical disease (PPNAD). Clin Endocrinol (Oxf) (2008) 69(3):367–73. doi: 10.1111/j.1365-2265.2008.03273.x
123. Gaujoux S, Tissier F, Groussin L, Libe R, Ragazzon B, Launay P, et al. Wnt/beta-catenin and 3',5'-cyclic adenosine 5'-monophosphate/protein kinase a signaling pathways alterations and somatic beta-catenin gene mutations in the progression of adrenocortical tumors. J Clin Endocrinol Metab (2008) 93(10):4135–40. doi: 10.1210/jc.2008-0631
124. Calebiro D, Di Dalmazi G, Bathon K, Ronchi CL, Beuschlein F. cAMP signaling in cortisol-producing adrenal adenoma. Eur J Endocrinol (2015) 173(4):M99–106. doi: 10.1530/EJE-15-0353
125. Di Dalmazi G, Kisker C, Calebiro D, Mannelli M, Canu L, Arnaldi G, et al. Novel somatic mutations in the catalytic subunit of the protein kinase a as a cause of adrenal cushing's syndrome: A european multicentric study. J Clin Endocrinol Metab (2014) 99(10):E2093–100. doi: 10.1210/jc.2014-2152
126. Ronchi CL, Di Dalmazi G, Faillot S, Sbiera S, Assie G, Weigand I, et al. Genetic landscape of sporadic unilateral adrenocortical adenomas without PRKACA p.Leu206Arg mutation. J Clin Endocrinol Metab (2016) 101(9):3526–38. doi: 10.1210/jc.2016-1586
127. Bathon K, Weigand I, Vanselow JT, Ronchi CL, Sbiera S, Schlosser A, et al. Alterations in protein kinase a substrate specificity as a potential cause of cushing syndrome. Endocrinology (2019) 160(2):447–59. doi: 10.1210/en.2018-00775
128. Espiard S, Knape MJ, Bathon K, Assie G, Rizk-Rabin M, Faillot S, et al. Activating PRKACB somatic mutation in cortisol-producing adenomas. JCI Insight (2018) 3(8):e98296. doi: 10.1172/jci.insight.98296
129. Bonnet-Serrano F, Bertherat J. Genetics of tumors of the adrenal cortex. Endocr Relat Cancer (2018) 25(3):R131–52. doi: 10.1530/ERC-17-0361
130. Bertherat J, Groussin L, Sandrini F, Matyakhina L, Bei T, Stergiopoulos S, et al. Molecular and functional analysis of PRKAR1A and its locus (17q22-24) in sporadic adrenocortical tumors: 17q losses, somatic mutations, and protein kinase a expression and activity. Cancer Res (2003) 63(17):5308–19.
131. Goh G, Scholl UI, Healy JM, Choi M, Prasad ML, Nelson-Williams C, et al. Recurrent activating mutation in PRKACA in cortisol-producing adrenal tumors. Nat Genet (2014) 46(6):613–7. doi: 10.1038/ng.2956
132. Libe R, Bertherat J. Molecular genetics of adrenocortical tumours, from familial to sporadic diseases. Eur J Endocrinol (2005) 153(4):477–87. doi: 10.1530/eje.1.02004
133. Kobayashi H, Usui T, Fukata J, Yoshimasa T, Oki Y, Nakao K. Mutation analysis of gsalpha, adrenocorticotropin receptor and p53 genes in japanese patients with adrenocortical neoplasms: including a case of gsalpha mutation. Endocr J (2000) 47(4):461–6. doi: 10.1507/endocrj.47.461
134. Almeida MQ, Azevedo MF, Xekouki P, Bimpaki EI, Horvath A, Collins MT, et al. Activation of cyclic AMP signaling leads to different pathway alterations in lesions of the adrenal cortex caused by germline PRKAR1A defects versus those due to somatic GNAS mutations. J Clin Endocrinol Metab (2012) 97(4):E687–93. doi: 10.1210/jc.2011-3000
135. Duan K, Mete O. Clinicopathologic correlates of primary aldosteronism. Arch Pathol Lab Med (2015) 139(7):948–54. doi: 10.5858/arpa.2014-0156-RS
136. Itcho K, Oki K, Ohno H, Yoneda M. Update on genetics of primary aldosteronism. Biomedicines (2021) 9(4):409–22. doi: 10.3390/biomedicines9040409
137. Young WF. Primary aldosteronism: renaissance of a syndrome. Clin Endocrinol (Oxf) (2007) 66(5):607–18. doi: 10.1111/j.1365-2265.2007.02775.x
138. Brown JM, Siddiqui M, Calhoun DA, Carey RM, Hopkins PN, Williams GH, et al. The unrecognized prevalence of primary aldosteronism: A cross-sectional study. Ann Intern Med (2020) 173(1):10–20. doi: 10.7326/M20-0065
139. Hannemann A, Wallaschofski H. Prevalence of primary aldosteronism in patient's cohorts and in population-based studies–a review of the current literature. Horm Metab Res (2012) 44(3):157–62. doi: 10.1055/s-0031-1295438
140. Funder JW, Carey RM, Mantero F, Murad MH, Reincke M, Shibata H, et al. The management of primary aldosteronism: Case detection, diagnosis, and treatment: An endocrine society clinical practice guideline. J Clin Endocrinol Metab (2016) 101(5):1889–916. doi: 10.1210/jc.2015-4061
141. Mulatero P, Tizzani D, Viola A, Bertello C, Monticone S, Mengozzi G, et al. Prevalence and characteristics of familial hyperaldosteronism: The PATOGEN study (Primary aldosteronism in TOrino-GENetic forms). Hypertension (2011) 58(5):797–803. doi: 10.1161/HYPERTENSIONAHA.111.175083
142. Pascoe L, Curnow KM, Slutsker L, Connell JM, Speiser PW, New MI, et al. Glucocorticoid-suppressible hyperaldosteronism results from hybrid genes created by unequal crossovers between CYP11B1 and CYP11B2. Proc Natl Acad Sci U.S.A. (1992) 89(17):8327–31. doi: 10.1073/pnas.89.17.8327
143. Lifton RP, Dluhy RG, Powers M, Rich GM, Cook S, Ulick S, et al. A chimaeric 11 beta-hydroxylase/aldosterone synthase gene causes glucocorticoid-remediable aldosteronism and human hypertension. Nature (1992) 355(6357):262–5. doi: 10.1038/355262a0
144. Stowasser M, Bachmann AW, Huggard PR, Rossetti TR, Gordon RD. Treatment of familial hyperaldosteronism type i: Only partial suppression of adrenocorticotropin required to correct hypertension. J Clin Endocrinol Metab (2000) 85(9):3313–8. doi: 10.1210/jcem.85.9.6834
145. Young WF Jr. Diagnosis and treatment of primary aldosteronism: Practical clinical perspectives. J Intern Med (2019) 285(2):126–48. doi: 10.1111/joim.12831
146. Scholl UI, Stolting G, Schewe J, Thiel A, Tan H, Nelson-Williams C, et al. CLCN2 chloride channel mutations in familial hyperaldosteronism type II. Nat Genet (2018) 50(3):349–54. doi: 10.1038/s41588-018-0048-5
147. Fernandes-Rosa FL, Daniil G, Orozco IJ, Goppner C, El Zein R, Jain V, et al. A gain-of-function mutation in the CLCN2 chloride channel gene causes primary aldosteronism. Nat Genet (2018) 50(3):355–61. doi: 10.1038/s41588-018-0053-8
148. Pallauf A, Schirpenbach C, Zwermann O, Fischer E, Morak M, Holinski-Feder E, et al. The prevalence of familial hyperaldosteronism in apparently sporadic primary aldosteronism in germany: A single center experience. Horm Metab Res (2012) 44(3):215–20. doi: 10.1055/s-0031-1299730
149. Stowasser M, Gordon RD. Familial hyperaldosteronism. J Steroid Biochem Mol Biol (2001) 78(3):215–29. doi: 10.1016/s0960-0760(01)00097-8
150. Mulatero P, Tauber P, Zennaro MC, Monticone S, Lang K, Beuschlein F, et al. KCNJ5 mutations in european families with nonglucocorticoid remediable familial hyperaldosteronism. Hypertension (2012) 59(2):235–40. doi: 10.1161/HYPERTENSIONAHA.111.183996
151. Scholl UI, Nelson-Williams C, Yue P, Grekin R, Wyatt RJ, Dillon MJ, et al. Hypertension with or without adrenal hyperplasia due to different inherited mutations in the potassium channel KCNJ5. Proc Natl Acad Sci U.S.A. (2012) 109(7):2533–8. doi: 10.1073/pnas.1121407109
152. Monticone S, Hattangady NG, Penton D, Isales CM, Edwards MA, Williams TA, et al. A novel Y152C KCNJ5 mutation responsible for familial hyperaldosteronism type III. J Clin Endocrinol Metab (2013) 98(11):E1861–5. doi: 10.1210/jc.2013-2428
153. Adachi M, Muroya K, Asakura Y, Sugiyama K, Homma K, Hasegawa T. Discordant genotype-phenotype correlation in familial hyperaldosteronism type III with KCNJ5 gene mutation: A patient report and review of the literature. Horm Res Paediatr (2014) 82(2):138–42. doi: 10.1159/000358197
154. Mussa A, Camilla R, Monticone S, Porta F, Tessaris D, Verna F, et al. Polyuric-polydipsic syndrome in a pediatric case of non-glucocorticoid remediable familial hyperaldosteronism. Endocr J (2012) 59(6):497–502. doi: 10.1507/endocrj.ej11-0406
155. Scholl UI, Goh G, Stolting G, de Oliveira RC, Choi M, Overton JD, et al. Somatic and germline CACNA1D calcium channel mutations in aldosterone-producing adenomas and primary aldosteronism. Nat Genet (2013) 45(9):1050–4. doi: 10.1038/ng.2695
156. Rassi-Cruz M, Maria AG, Faucz FR, London E, Vilela LAP, Santana LS, et al. Phosphodiesterase 2A and 3B variants are associated with primary aldosteronism. Endocr Relat Cancer (2021) 28(1):1–13. doi: 10.1530/ERC-20-0384
157. Akerstrom T, Carling T, Beuschlein F, Hellman P. Genetics of adrenocortical tumours. J Intern Med (2016) 280(6):540–50. doi: 10.1111/joim.12452
158. Funder JW. Genetics of primary aldosteronism. Front Horm Res (2014) 43:70–8. doi: 10.1159/000360870
159. Azizan EA, Poulsen H, Tuluc P, Zhou J, Clausen MV, Lieb A, et al. Somatic mutations in ATP1A1 and CACNA1D underlie a common subtype of adrenal hypertension. Nat Genet (2013) 45(9):1055–60. doi: 10.1038/ng.2716
160. Beuschlein F, Boulkroun S, Osswald A, Wieland T, Nielsen HN, Lichtenauer UD, et al. Somatic mutations in ATP1A1 and ATP2B3 lead to aldosterone-producing adenomas and secondary hypertension. Nat Genet (2013) 45(4):440–4, 444e1-2. doi: 10.1038/ng.2550
161. Choi M, Scholl UI, Yue P, Bjorklund P, Zhao B, Nelson-Williams C, et al. K+ channel mutations in adrenal aldosterone-producing adenomas and hereditary hypertension. Science (2011) 331(6018):768–72. doi: 10.1126/science.1198785
162. Fernandes-Rosa FL, Williams TA, Riester A, Steichen O, Beuschlein F, Boulkroun S, et al. Genetic spectrum and clinical correlates of somatic mutations in aldosterone-producing adenoma. Hypertension (2014) 64(2):354–61. doi: 10.1161/HYPERTENSIONAHA.114.03419
163. Akerstrom T, Crona J, Delgado Verdugo A, Starker LF, Cupisti K, Willenberg HS, et al. Comprehensive re-sequencing of adrenal aldosterone producing lesions reveal three somatic mutations near the KCNJ5 potassium channel selectivity filter. PloS One (2012) 7(7):e41926. doi: 10.1371/journal.pone.0041926
164. Lenzini L, Rossitto G, Maiolino G, Letizia C, Funder JW, Rossi GP. A meta-analysis of somatic KCNJ5 k(+) channel mutations in 1636 patients with an aldosterone-producing adenoma. J Clin Endocrinol Metab (2015) 100(8):E1089–95. doi: 10.1210/jc.2015-2149
165. Hong AR, Kim JH, Song YS, Lee KE, Seo SH, Seong MW, et al. Genetics of aldosterone-producing adenoma in korean patients. PloS One (2016) 11(1):e0147590. doi: 10.1371/journal.pone.0147590
166. Wu VC, Huang KH, Peng KY, Tsai YC, Wu CH, Wang SM, et al. Prevalence and clinical correlates of somatic mutation in aldosterone producing adenoma-taiwanese population. Sci Rep (2015) 5:11396. doi: 10.1038/srep11396
167. Zennaro MC, Boulkroun S, Fernandes-Rosa F. Genetic causes of functional adrenocortical adenomas. Endocr Rev (2017) 38(6):516–37. doi: 10.1210/er.2017-00189
168. El Wakil A, Lalli E. The wnt/beta-catenin pathway in adrenocortical development and cancer. Mol Cell Endocrinol (2011) 332(1-2):32–7. doi: 10.1016/j.mce.2010.11.014
169. Berthon A, Drelon C, Ragazzon B, Boulkroun S, Tissier F, Amar L, et al. WNT/beta-catenin signalling is activated in aldosterone-producing adenomas and controls aldosterone production. Hum Mol Genet (2014) 23(4):889–905. doi: 10.1093/hmg/ddt484
170. Akerstrom T, Maharjan R, Sven Willenberg H, Cupisti K, Ip J, Moser A, et al. Activating mutations in CTNNB1 in aldosterone producing adenomas. Sci Rep (2016) 6:19546. doi: 10.1038/srep19546
171. Scholl UI, Healy JM, Thiel A, Fonseca AL, Brown TC, Kunstman JW, et al. Novel somatic mutations in primary hyperaldosteronism are related to the clinical, radiological and pathological phenotype. Clin Endocrinol (Oxf) (2015) 83(6):779–89. doi: 10.1111/cen.12873
172. Teo AE, Garg S, Shaikh LH, Zhou J, Karet Frankl FE, Gurnell M, et al. Pregnancy, primary aldosteronism, and adrenal CTNNB1 mutations. N Engl J Med (2015) 373(15):1429–36. doi: 10.1056/NEJMoa1504869
173. Rhayem Y, Perez-Rivas LG, Dietz A, Bathon K, Gebhard C, Riester A, et al. PRKACA somatic mutations are rare findings in aldosterone-producing adenomas. J Clin Endocrinol Metab (2016) 101(8):3010–7. doi: 10.1210/jc.2016-1700
174. Nakajima Y, Okamura T, Horiguchi K, Gohko T, Miyamoto T, Satoh T, et al. GNAS mutations in adrenal aldosterone-producing adenomas. Endocr J (2016) 63(2):199–204. doi: 10.1507/endocrj.EJ15-0642
175. Sato Y, Maekawa S, Ishii R, Sanada M, Morikawa T, Shiraishi Y, et al. Recurrent somatic mutations underlie corticotropin-independent cushing's syndrome. Science (2014) 344(6186):917–20. doi: 10.1126/science.1252328
176. Tadjine M, Lampron A, Ouadi L, Bourdeau I. Frequent mutations of beta-catenin gene in sporadic secreting adrenocortical adenomas. Clin Endocrinol (Oxf) (2008) 68(2):264–70. doi: 10.1111/j.1365-2265.2007.03033.x
177. Tissier F, Cavard C, Groussin L, Perlemoine K, Fumey G, Hagnere AM, et al. Mutations of beta-catenin in adrenocortical tumors: Activation of the wnt signaling pathway is a frequent event in both benign and malignant adrenocortical tumors. Cancer Res (2005) 65(17):7622–7. doi: 10.1158/0008-5472.CAN-05-0593
178. Fassnacht M, Kroiss M, Allolio B. Update in adrenocortical carcinoma. J Clin Endocrinol Metab (2013) 98(12):4551–64. doi: 10.1210/jc.2013-3020
179. Kerkhofs TM, Verhoeven RH, Bonjer HJ, van Dijkum EJ, Vriens MR, De Vries J, et al. Surgery for adrenocortical carcinoma in the netherlands: Analysis of the national cancer registry data. Eur J Endocrinol (2013) 169(1):83–9. doi: 10.1530/EJE-13-0142
180. Kebebew E, Reiff E, Duh QY, Clark OH, McMillan A. Extent of disease at presentation and outcome for adrenocortical carcinoma: Have we made progress? World J Surg (2006) 30(5):872–8. doi: 10.1007/s00268-005-0329-x
181. Kerkhofs TM, Verhoeven RH, van der Zwan JM, Dieleman J, Kerstens MN, Links TP, et al. Adrenocortical carcinoma: A population-based study on incidence and survival in the netherlands since 1993. Eur J Cancer (2013) 49(11):2579–86. doi: 10.1016/j.ejca.2013.02.034
182. Fassnacht M, Johanssen S, Quinkler M, Bucsky P, Willenberg HS, Beuschlein F, et al. Limited prognostic value of the 2004 international union against cancer staging classification for adrenocortical carcinoma: Proposal for a revised TNM classification. Cancer (2009) 115(2):243–50. doi: 10.1002/cncr.24030
183. Allolio B, Fassnacht M. Clinical review: Adrenocortical carcinoma: clinical update. J Clin Endocrinol Metab (2006) 91(6):2027–37. doi: 10.1210/jc.2005-2639
184. Luton JP, Cerdas S, Billaud L, Thomas G, Guilhaume B, Bertagna X, et al. Clinical features of adrenocortical carcinoma, prognostic factors, and the effect of mitotane therapy. N Engl J Med (1990) 322(17):1195–201. doi: 10.1056/NEJM199004263221705
185. Fassnacht M, Allolio B. Clinical management of adrenocortical carcinoma. Best Pract Res Clin Endocrinol Metab (2009) 23(2):273–89. doi: 10.1016/j.beem.2008.10.008
186. Fassnacht M, Dekkers OM, Else T, Baudin E, Berruti A, de Krijger R, et al. European society of endocrinology clinical practice guidelines on the management of adrenocortical carcinoma in adults, in collaboration with the european network for the study of adrenal tumors. Eur J Endocrinol (2018) 179(4):G1–G46. doi: 10.1530/EJE-18-0608
187. Else T, Kim AC, Sabolch A, Raymond VM, Kandathil A, Caoili EM, et al. Adrenocortical carcinoma. Endocr Rev (2014) 35(2):282–326. doi: 10.1210/er.2013-1029
188. Michalkiewicz E, Sandrini R, Figueiredo B, Miranda EC, Caran E, Oliveira-Filho AG, et al. Clinical and outcome characteristics of children with adrenocortical tumors: A report from the international pediatric adrenocortical tumor registry. J Clin Oncol (2004) 22(5):838–45. doi: 10.1200/JCO.2004.08.085
189. Espiard S, Bertherat J. The genetics of adrenocortical tumors. Endocrinol Metab Clin North Am (2015) 44(2):311–34. doi: 10.1016/j.ecl.2015.02.004
190. Mai PL, Best AF, Peters JA, DeCastro RM, Khincha PP, Loud JT, et al. Risks of first and subsequent cancers among TP53 mutation carriers in the national cancer institute li-fraumeni syndrome cohort. Cancer (2016) 122(23):3673–81. doi: 10.1002/cncr.30248
191. Bougeard G, Sesboue R, Baert-Desurmont S, Vasseur S, Martin C, Tinat J, et al. Molecular basis of the li-fraumeni syndrome: An update from the french LFS families. J Med Genet (2008) 45(8):535–8. doi: 10.1136/jmg.2008.057570
192. Jouinot A, Bertherat J. Diseases predisposing to adrenocortical malignancy (Li-fraumeni syndrome, beckwith-wiedemann syndrome, and carney complex). Exp Suppl (2019) 111:149–69. doi: 10.1007/978-3-030-25905-1_9
193. Schneider K, Zelley K, Nichols KE, Garber J. Li-fraumeni syndrome, in GeneReviews®. Adam MP, editor. Seattle (WA): University of Washington (1993).
194. Wasserman JD, Zambetti GP, Malkin D. Towards an understanding of the role of p53 in adrenocortical carcinogenesis. Mol Cell Endocrinol (2012) 351(1):101–10. doi: 10.1016/j.mce.2011.09.010
195. Weksberg R, Smith AC, Squire J, Sadowski P. Beckwith-wiedemann syndrome demonstrates a role for epigenetic control of normal development. Hum Mol Genet (2003), R61–8. doi: 10.1093/hmg/ddg067
196. Wilkin F, Gagne N, Paquette J, Oligny LL, Deal C. Pediatric adrenocortical tumors: molecular events leading to insulin-like growth factor II gene overexpression. J Clin Endocrinol Metab (2000) 85(5):2048–56. doi: 10.1210/jcem.85.5.6589
197. Shuman C, Beckwith JB, Weksberg R, et al. Beckwith-wiedemann syndrome, in GeneReviews®. Adam MP, editor. Seattle (WA: University of Washington (1993).
198. DeBaun MR, Tucker MA. Risk of cancer during the first four years of life in children from the beckwith-wiedemann syndrome registry. J Pediatr (1998) 132(3 Pt 1):398–400. doi: 10.1016/s0022-3476(98)70008-3
199. Ibrahim A, Kirby G, Hardy C, Dias RP, Tee L, Lim D, et al. Methylation analysis and diagnostics of beckwith-wiedemann syndrome in 1,000 subjects. Clin Epigenet (2014) 6(1):11. doi: 10.1186/1868-7083-6-11
200. Lapunzina P. Risk of tumorigenesis in overgrowth syndromes: a comprehensive review. Am J Med Genet C Semin Med Genet (2005) 137C(1):53–71. doi: 10.1002/ajmg.c.30064
201. Mussa A, Ferrero GB. Screening hepatoblastoma in beckwith-wiedemann syndrome: A complex issue. J Pediatr Hematol Oncol (2015) 37(8):627. doi: 10.1097/MPH.0000000000000408
202. Waldmann J, Bartsch DK, Kann PH, Fendrich V, Rothmund M, Langer P. Adrenal involvement in multiple endocrine neoplasia type 1: Results of 7 years prospective screening. Langenbecks Arch Surg (2007) 392(4):437–43. doi: 10.1007/s00423-006-0124-7
203. Skogseid B, Rastad J, Gobl A, Larsson C, Backlin K, Juhlin C, et al. Adrenal lesion in multiple endocrine neoplasia type 1. Surgery (1995) 118(6):1077–82. doi: 10.1016/s0039-6060(05)80117-5
204. Gibril F, Schumann M, Pace A, Jensen RT. Multiple endocrine neoplasia type 1 and zollinger-ellison syndrome: A prospective study of 107 cases and comparison with 1009 cases from the literature. Med (Baltimore) (2004) 83(1):43–83. doi: 10.1097/01.md.0000112297.72510.32
205. Griniatsos JE, Dimitriou N, Zilos A, Sakellariou S, Evangelou K, Kamakari S, et al. Bilateral adrenocortical carcinoma in a patient with multiple endocrine neoplasia type 1 (MEN1) and a novel mutation in the MEN1 gene. World J Surg Oncol (2011) 9:6. doi: 10.1186/1477-7819-9-6
206. Langer P, Cupisti K, Bartsch DK, Nies C, Goretzki PE, Rothmund M, et al. Adrenal involvement in multiple endocrine neoplasia type 1. World J Surg (2002) 26(8):891–6. doi: 10.1007/s00268-002-6492-4
207. Assie G, Letouze E, Fassnacht M, Jouinot A, Luscap W, Barreau O, et al. Integrated genomic characterization of adrenocortical carcinoma. Nat Genet (2014) 46(6):607–12. doi: 10.1038/ng.2953
208. Zheng S, Cherniack AD, Dewal N, Moffitt RA, Danilova L, Murray BA, et al. Comprehensive pan-genomic characterization of adrenocortical carcinoma. Cancer Cell (2016) 29(5):723–36. doi: 10.1016/j.ccell.2016.04.002
209. Stoffel E, Mukherjee B, Raymond VM, Tayob N, Kastrinos F, Sparr J, et al. Calculation of risk of colorectal and endometrial cancer among patients with lynch syndrome. Gastroenterology (2009) 137(5):1621–7. doi: 10.1053/j.gastro.2009.07.039
210. Medina-Arana V, Delgado L, Gonzalez L, Bravo A, Diaz H, Salido E, et al. Adrenocortical carcinoma, an unusual extracolonic tumor associated with lynch II syndrome. Fam Cancer (2011) 10(2):265–71. doi: 10.1007/s10689-010-9416-8
211. Berends MJ, Cats A, Hollema H, Karrenbeld A, Beentjes JA, Sijmons RH, et al. Adrenocortical adenocarcinoma in an MSH2 carrier: Coincidence or causal relation? Hum Pathol (2000) 31(12):1522–7. doi: 10.1053/hupa.2000.20409
212. Broaddus RR, Lynch PM, Lu KH, Luthra R, Michelson SJ. Unusual tumors associated with the hereditary nonpolyposis colorectal cancer syndrome. Mod Pathol (2004) 17(8):981–9. doi: 10.1038/modpathol.3800150
213. Karamurzin Y, Zeng Z, Stadler ZK, Zhang L, Ouansafi I, Al-Ahmadie HA, et al. Unusual DNA mismatch repair-deficient tumors in lynch syndrome: A report of new cases and review of the literature. Hum Pathol (2012) 43(10):1677–87. doi: 10.1016/j.humpath.2011.12.012
214. Weissman SM, Burt R, Church J, Erdman S, Hampel H, Holter S, et al. Identification of individuals at risk for lynch syndrome using targeted evaluations and genetic testing: National society of genetic counselors and the collaborative group of the americas on inherited colorectal cancer joint practice guideline. J Genet Couns (2012) 21(4):484–93. doi: 10.1007/s10897-011-9465-7
215. Raymond VM, Everett JN, Furtado LV, Gustafson SL, Jungbluth CR, Gruber SB, et al. Adrenocortical carcinoma is a lynch syndrome-associated cancer. J Clin Oncol (2013) 31(24):3012–8. doi: 10.1200/JCO.2012.48.0988
216. Wagner AS, Fleitz JM, Kleinschmidt-Demasters BK. Pediatric adrenal cortical carcinoma: Brain metastases and relationship to NF-1, case reports and review of the literature. J Neurooncol (2005) 75(2):127–33. doi: 10.1007/s11060-005-0376-z
217. Traill Z, Tuson J, Woodham C. Adrenal carcinoma in a patient with gardner's syndrome: Imaging findings. AJR Am J Roentgenol (1995) 165(6):1460–1. doi: 10.2214/ajr.165.6.7484586
218. Marshall WH, Martin FI, Mackay IR. Gardner's syndrome with adrenal carcinoma. Australas Ann Med (1967) 16(3):242–4. doi: 10.1111/imj.1967.16.3.242
219. Fienman NL, Yakovac WC. Neurofibromatosis in childhood. J Pediatr (1970) 76(3):339–46. doi: 10.1016/s0022-3476(70)80472-3
220. Painter TA, Jagelman DG. Adrenal adenomas and adrenal carcinomas in association with hereditary adenomatosis of the colon and rectum. Cancer (1985) 55(9):2001–4. doi: 10.1002/1097-0142(19850501)55:9<2001::aid-cncr2820550929>3.0.co;2-7
221. Wakatsuki S, Sasano H, Matsui T, Nagashima K, Toyota T, Horii A. Adrenocortical tumor in a patient with familial adenomatous polyposis: a case associated with a complete inactivating mutation of the APC gene and unusual histological features. Hum Pathol (1998) 29(3):302–6. doi: 10.1016/s0046-8177(98)90052-1
222. Seki M, Tanaka K, Kikuchi-Yanoshita R, Konishi M, Fukunari H, Iwama T, et al. Loss of normal allele of the APC gene in an adrenocortical carcinoma from a patient with familial adenomatous polyposis. Hum Genet (1992) 89(3):298–300. doi: 10.1007/BF00220544
223. Takazawa R, Ajima J, Yamauchi A, Goto M. Unusual double primary neoplasia: adrenocortical and ureteral carcinomas in werner syndrome. Urol Int (2004) 72(2):168–70. doi: 10.1159/000075974
224. Anselmo J, Medeiros S, Carneiro V, Greene E, Levy I, Nesterova M, et al. A large family with carney complex caused by the S147G PRKAR1A mutation shows a unique spectrum of disease including adrenocortical cancer. J Clin Endocrinol Metab (2012) 97(2):351–9. doi: 10.1210/jc.2011-2244
225. Morin E, Mete O, Wasserman JD, Joshua AM, Asa SL, Ezzat S. Carney complex with adrenal cortical carcinoma. J Clin Endocrinol Metab (2012) 97(2):E202–6. doi: 10.1210/jc.2011-2321
226. Costa R, Carneiro BA, Tavora F, Pai SG, Kaplan JB, Chae YK, et al. The challenge of developmental therapeutics for adrenocortical carcinoma. Oncotarget (2016) 7(29):46734–49. doi: 10.18632/oncotarget.8774
227. Pence A, McGrath M, Lee SL, Raines DE. Pharmacological management of severe cushing's syndrome: The role of etomidate. Ther Adv Endocrinol Metab (2022) 13:20420188211058583. doi: 10.1177/20420188211058583
228. Tatsi C, Maria AG, Malloy C, Lin L, London E, Settas N, et al. Cushing syndrome in a pediatric patient with a KCNJ5 variant and successful treatment with low-dose ketoconazole. J Clin Endocrinol Metab (2021) 106(6):1606–16. doi: 10.1210/clinem/dgab118
229. Alesina PF, Knyazeva P, Hinrichs J, Walz MK. Tailored approach in adrenal surgery: Retroperitoneoscopic partial adrenalectomy. Front Endocrinol (Lausanne) (2022) 13:855326. doi: 10.3389/fendo.2022.855326
230. Fassnacht M, Assie G, Baudin E, Eisenhofer G, de la Fouchardiere C, Haak HR, et al. Adrenocortical carcinomas and malignant phaeochromocytomas: ESMO-EURACAN clinical practice guidelines for diagnosis, treatment and follow-up. Ann Oncol (2020) 31(11):1476–90. doi: 10.1016/j.annonc.2020.08.2099
231. Puglisi S, Calabrese A, Basile V, Pia A, Reimondo G, Perotti P, et al. New perspectives for mitotane treatment of adrenocortical carcinoma. Best Pract Res Clin Endocrinol Metab (2020) 34(3):101415. doi: 10.1016/j.beem.2020.101415
232. Weigel B, Malempati S, Reid JM, Voss SD, Cho SY, Chen HX, et al. Phase 2 trial of cixutumumab in children, adolescents, and young adults with refractory solid tumors: A report from the children's oncology group. Pediatr Blood Cancer (2014) 61(3):452–6. doi: 10.1002/pbc.24605
Keywords: adrenal cortex, genetics, adrenal tumors, aldosterone secretion, cortisol secretion
Citation: Pitsava G, Maria AG and Faucz FR (2022) Disorders of the adrenal cortex: Genetic and molecular aspects. Front. Endocrinol. 13:931389. doi: 10.3389/fendo.2022.931389
Received: 28 April 2022; Accepted: 15 July 2022;
Published: 29 August 2022.
Edited by:
Valentina Morelli, Istituto Auxologico Italiano, ItalyReviewed by:
José Miguel Hinojosa-Amaya, Autonomous University of Nuevo León, MexicoKatherine Araque, Ascendis Pharma, United States
Copyright © 2022 Pitsava, Maria and Faucz. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Fabio R. Faucz, ZmFiaW8uZmF1Y3pAbmloLmdvdg==