 Yunbo Jia
Yunbo Jia Yang Liu1
Yang Liu1 Siyu Sun
Siyu Sun Guangwei Sun
Guangwei Sun
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
MINI REVIEW article
Front. Endocrinol., 16 June 2022
Sec. Diabetes: Molecular Mechanisms
Volume 13 - 2022 | https://doi.org/10.3389/fendo.2022.928016
This article is part of the Research TopicInsights in Diabetes: Molecular Mechanisms 2021View all 10 articles
Various theories for the hormonal basis of diabetes have been proposed and debated over the past few decades. Insulin insufficiency was previously regarded as the only hormone deficiency directly leading to metabolic disorders associated with diabetes. Although glucagon and its receptor are ignored in this framework, an increasing number of studies have shown that they play essential roles in the development and progression of diabetes. However, the molecular mechanisms underlying the effects of glucagon are still not clear. In this review, recent research on the mechanisms by which glucagon and its receptor contribute to the pathogenesis of diabetes as well as correlations between GCGR mutation rates in populations and the occurrence of diabetes are summarized. Furthermore, we summarize how recent research clearly establishes glucagon as a potential therapeutic target for diabetes.
Diabetes is a metabolic disorder characterized by hyperglycemia resulting from an absolute deficiency of insulin secretion (type 1 diabetes, T1D), or a combination of insulin resistance and an inadequate compensatory insulin secretion (type 2 diabetes, T2D) (1). However, each type of diabetes in animals and humans is accompanied by hyperglucagonemia (2–4), so glucagon excess is more critical to the development of diabetes than insulin deficiency (4, 5). Increasing evidence indicates that blocking glucagon and glucagon receptor (GCGR) can relieve hyperglycemia in animals and humans, clearly establishing the important roles of glucagon and GCGR in the pathogenesis of diabetes (6, 7).
Glucagon is a linear peptide containing 29 amino acids. It is secreted by islet α cells and mainly targets the liver cells (8). GCGR is a G-protein-coupled receptor (GPCR) mainly detected in islet β cells and liver cells (9). After glucagon specifically binds to GCGR, it promotes liver glycogen breakdown and increases blood glucose levels to stimulate insulin release (10, 11). Glucagon-like peptide 1 (GLP-1), mainly expressed in intestinal L cells, activates glucagon-like peptide-1 receptor (GLP-1R) to adjust metabolism (12, 13). Glucagon and GLP-1 are derived from the same biosynthetic precursor proglucagon and are involved in the regulation of lipid and cholic acid metabolism, thereby playing pivotal roles in glucose metabolism and the pathogenesis of diabetes (7, 12, 13).
In this review, we explore the controversial relationships between glucagon and metabolic disorders associated with diabetes based on recent research with an emphasis on recent evidence supporting the important role of glucagon. We also elucidate the correlation between GCGR mutations in populations and the occurrence of diabetes. Furthermore, we summarize drug strategies to provide a new basis for the treatment of diabetes.
The debate over the relative roles of hormones in the regulation of diabetes-related metabolic disorders has spanned decades. In 1921, the discovery of insulin was regarded as one of the greatest breakthroughs in the history of medicine. This led to the establishment of the insulinocentric view, which proposes that all diabetes-related metabolic disorders are directly caused by a lack of insulin secretion (14). Glucagon was not yet characterized and accordingly was not associated with these metabolic disorders. The insulinocentric theory was accepted for over half a century until Unger et al. proposed the bihormone theory at a conference in 1975 (15, 16).
According to the theory of bihormonal regulation, diabetes results from the abnormal secretion of both insulin and glucagon (15, 16). Some metabolic disorders associated with diabetes are directly caused by insulin deficiency, such as elevated lipolysis, increased proteolysis, and decreased glucose utilization. Others, such as decreased glycogen synthesis, increased ketogenesis, elevated hepatic glycogenolysis, and gluconeogenesis, are direct effects of excess glucagon (15–18) (Figure 1). Glucagon has glucogenic, ketogenic, and gluconeogenic functions and mediates severe endogenous hyperglycemia and hyperketonemia under a state of insulin deficiency; thus, it is a direct cause of the substantial increases in the levels of glucose and ketone in severe presentations of diabetes (19). In patients with diabetes with relatively steady levels of insulin, a rise in glucagon causes hyperglycemia and glycosuria (17). Glucagon suppression may be an effective adjunct to routine antihyperglycemic therapy in patients with diabetes (20–22).
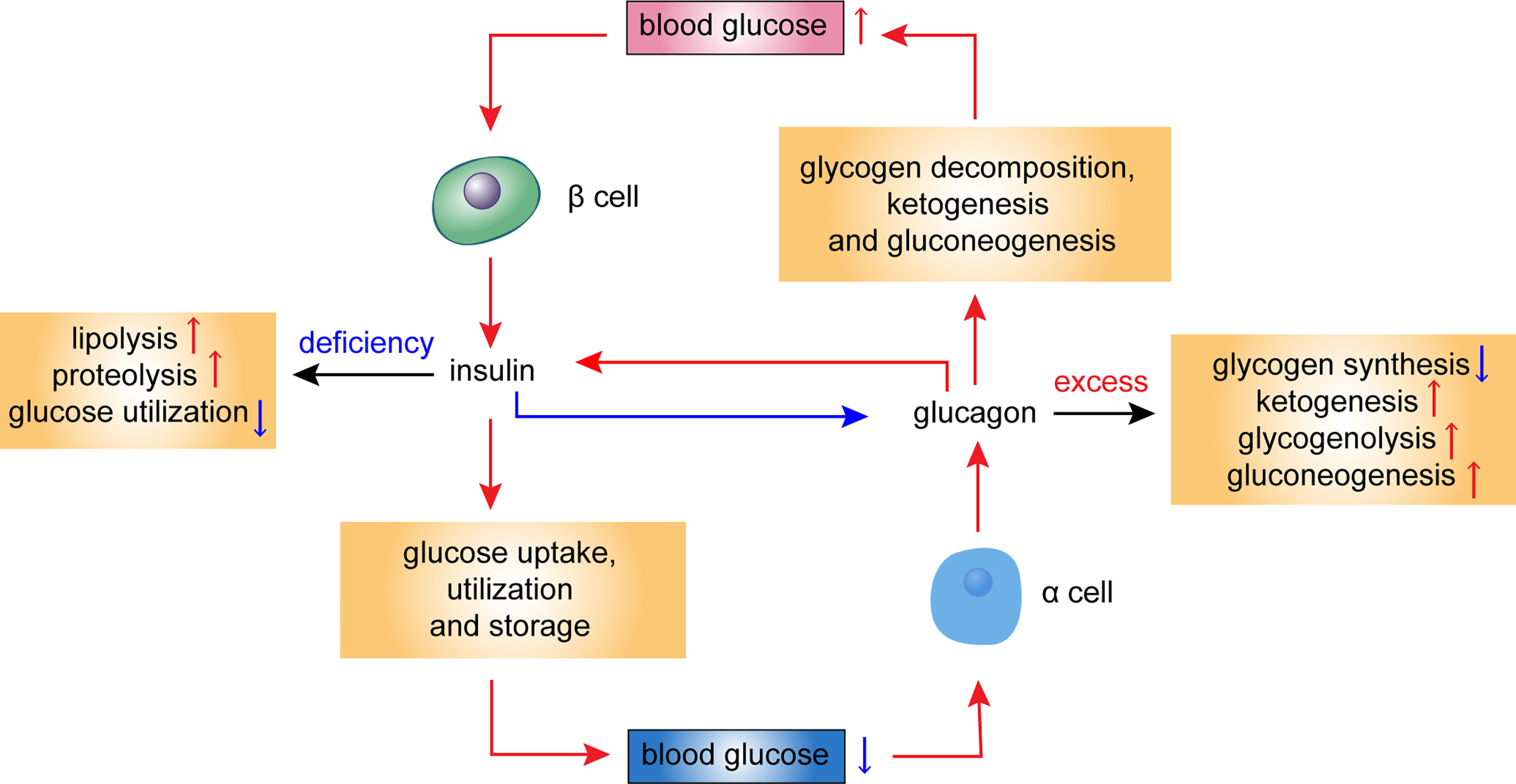
Figure 1 Hormonal regulation of glucose homeostasis in the islet cells. This diagram illustrates the metabolic effects of glucagon and insulin. Blood glucose levels influence secretion of insulin and glucagon. Insulin deficiency leads to elevated lipolysis, increased proteolysis, and decreased glucose utilization, while excess glucagon leads to decreased glycogen synthesis, increased ketogenesis, elevated glycogenolysis, and gluconeogenesis. Red arrows refer to a stimulatory effect, while blue arrows refer to an inhibitory effect.
Glucagonocentric hypothesis was proposed by Unger et al. based on the following evidence: (a) hyperglucagonemia is present in all forms of diabetes; (b) marked hyperglucagonemia is caused by perfusing anti-insulin serum to the normal pancreas; (c) during a total insulin deficiency, all metabolic manifestations of diabetes can be suppressed by glucagon suppressors, like somatostatin, and in global Gcgr knockout (Gcgr-/-) mice, demonstrating that β cell destruction does not cause diabetes (4). Thus, compared with insulin deficiency, glucagon excess plays a more essential role in the development of diabetes.
Gcgr -/- mice were designed to further understand the role of GCGR in the development of diabetes; these mice do not respond to glucagon at any concentration, and their fasting blood glucose levels are lower than those of wild-type mice. These knockout mice exhibit enhanced glucose tolerance and elevated insulin sensitivity during insulin tolerance testing (23). When β cells of Gcgr -/- mice were destroyed by streptozotocin (STZ) and insulin secretion was inhibited, animals did not develop hyperglycemia, suggesting that Gcgr -/- mice do not develop T1D, even under a state of insulin deficiency (24). After the transient repair of defective Gcgr with an adenovirus vector, the blood glucose levels of the mice increased after β cell destruction (25). When Gcgr was inactivated again, blood glucose levels returned to normal, suggesting that in the absence of glucagon, insulin deficiency does not result in abnormal blood glucose levels, and that the abnormal blood glucose concentration caused by insulin deficiency can be restored by eliminating the effect of glucagon (25). Hence, blocking Gcgr can restore hyperglycemia in rodent models with insufficient insulin secretion; however, this effect requires a certain number of β cells (26). Active GLP-1 was identified in pancreatic perfusate from Gcgr -/- but not wild-type mice (27), and FGF21 acts additively with GLP-1 to prevent insulinopenic diabetes in mice lacking glucagon action (28), which further reduces the risk of Gcgr -/- mice developing diabetes. On the contrary, Gcgr knockout implies that glucagon cannot function normally, which can cause a series of metabolic problems, such as hyperglucagonemia and compensatory hyperplasia of α cells (23, 29, 30). Therefore, the above phenomena should be monitored in the development of GCGR antagonists. The therapeutic potential of GCGR is not fully recognized and should be a basis of further studies; however, the established animal models provide an effective means for the development of strategies to reduce the incidence of diabetes.
In healthy people, high blood glucose stimulates β-cell insulin secretion, and glucagon secretion is suppressed; low blood glucose inhibits β-cell insulin secretion, and glucagon secretion is stimulated (Figure 1). Nevertheless, hyperglucagonemia was present in patients with diabetes, including T1D (31) and T2D (32). No significant difference of plasma glucagon level was found between T1D and T2D (31, 32). Absolute deficiency or relative deficiency of insulin secretion weakened the inhibition of insulin on glucagon (4).
Glucagon’s role in intra-islet paracrine regulation is essential. Svendsen et al. (27) used isolated perfused pancreas from wild-type, Glp-1r knockout, diphtheria toxin-induced proglucagon knockdown, β cell-specific Gcgr knockout, and Gcgr−/− mice to examine glucagon-induced insulin secretion. They found that paracrine glucagon actions are required for maintenance of normal insulin secretion, and intra-islet glucagon signaling involves the activation of both GCGR and GLP-1R. Loss of either GCGR or GLP-1R does not change insulin responses, whereas combined blockage of both receptors significantly reduces insulin secretion (27). Additionally, Gcgr -/-mice show normal blood glucose levels and increased glucagon levels in glucose-stimulated insulin secretion (GSIS) tests after treatment with 10 mM (33) or 12 mM (27) glucose. This is similar to levels observed in control mice, suggesting that the insulin-promoting effect of glucagon is achieved mainly via GLP-1R. However, as the cognate downstream receptor of glucagon, the physiological significance of β-cell GCGR remains subtle. Zhang et al. (34) states that glucagon potentiates insulin secretion via β-cell GCGR at physiological but not high concentrations of glucose, and β-cell GCGR activation promotes GSIS more than GLP-1R in high fat diet. These findings indicate that GCGR contributes to glucose homeostasis maintenance during nutrient overload. These studies emphasized the indispensable roles of GCGR on β cells in mediating both the glucose balance and catabolic state and implied that GCGR is closely related to the pathogenesis of diabetes. Accordingly, studies of the mechanisms by which GCGR regulates insulin secretion are of great significance.
In pancreatic β cells, GLUT2, a glucose transporter protein, is required for GSIS (35). Glucose binding to GLUT2 is a key pathway leading to increased ATP levels, deionization, increased intracellular calcium concentration, and enhanced insulin exocytosis. GLUT1 expression decreased in Gcgr–/– mice but increased in wild-type mice after glucose stimulation (36). As a paracrine hormone, glucagon binds to GCGR with high affinity, while also exerting a “spillover” effect by binding to GLP-1R with low affinity (37). After glucagon binds to GCGR and GLP-1R on β cells, the activated receptors engage the G protein Gαs, which stimulate the generation of cyclic adenosine monophosphate (cAMP) (34, 38–40). The response of glucagon to glucose mainly depends on cAMP signaling in islet β cells and the increased cAMP level promotes insulin release (39, 41) (Figure 2).
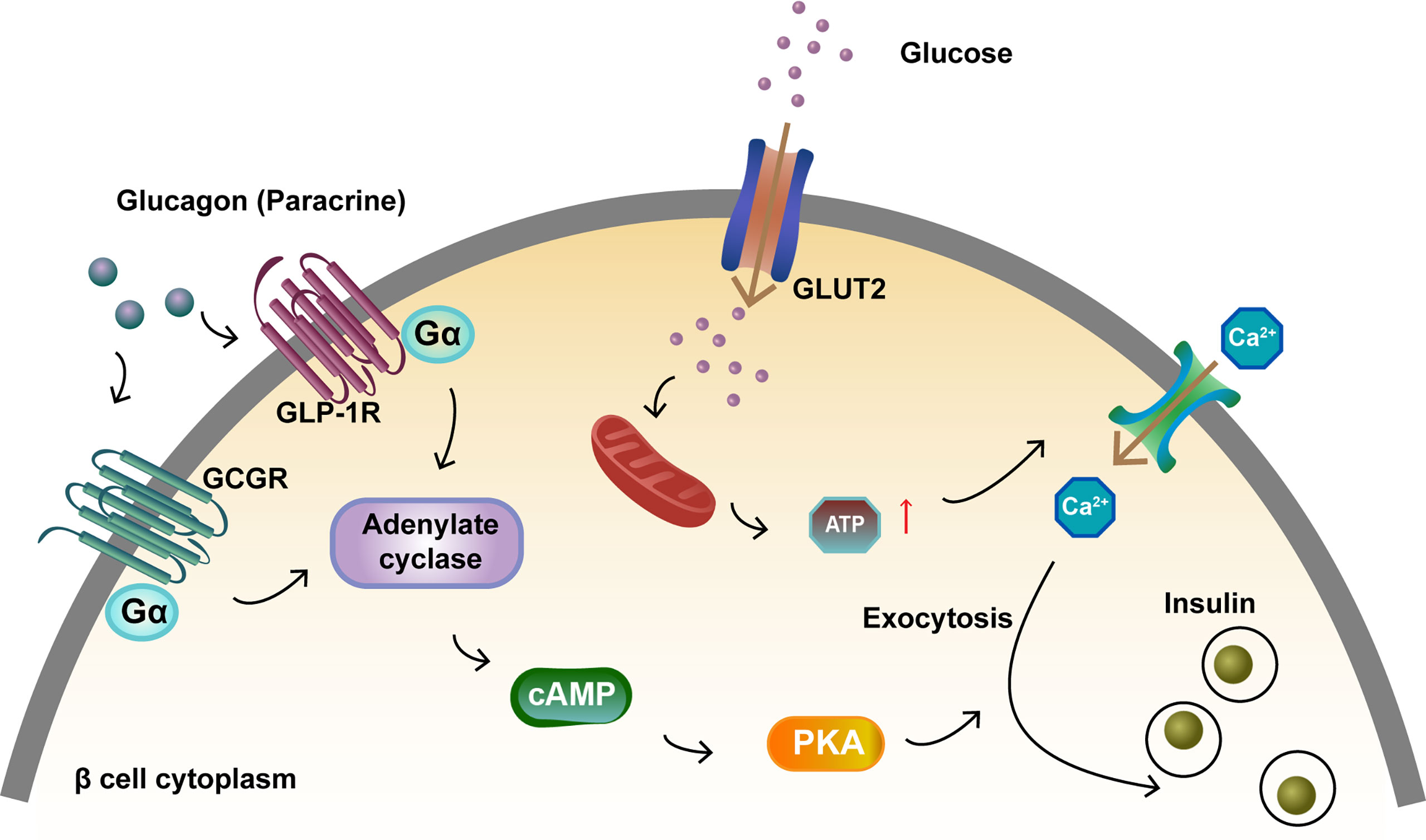
Figure 2 Activation of GCGR and GLP-1R to promote insulin secretion in islet β cells. Glucagon binds to GCGR and GLP-1R on β cells and the activated receptors engage the G protein Gαs. This results in adenylate cyclase activation and cAMP formation. Glucose binds to GLUT2, which increases ATP levels and intracellular calcium concentration, and enhances insulin exocytosis. The increase in intracellular cAMP levels activates PKA, which also promotes insulin exocytosis.
T2D, also called non-insulin dependent diabetes mellitus, is a common disorder with complex traits. Multiple genomic scans have identified different loci associated with T2D, including a locus on chromosome 17q24-25 (42, 43) and GCGR on chromosome 17q25, which might be explained by linkage identified in the same region (44). GCGR mediates glucose homeostasis by binding to glucagon and may contribute to the pathogenesis of T2D and the development of β-cell dysfunction, resulting in a deficient insulin response in some patients with T2D. Further studies are needed to determine the effect of hepatic glucagon resistance on metabolic disorders and its association with the occurrence of diabetes. Chronic hyperglycemia increases the protein expression of GCGR in the liver and decreases downstream glucagon signaling, leading to liver glucagon resistance (45, 46). GCGR mutations may be related to hyperglucagonemia via the impairment of endogenous glucagon autofeedback, to high hepatic glucose output in T2D via elevated glycogenolysis and/or gluconeogenesis, and to abnormal insulin secretion via the glucagon resistance of β cells in T2D.
GCGR is regarded as a candidate gene for the pathogenesis of T2D and GCGR mutations with similar frequencies have been found associated with T2D (47). Polymorphisms in the GCGR gene are associated with T2D in Caucasians (48). The Gly40Ser variant of GCGR (c.118G >A) causes a change from glycine (at the 40th amino acid residue) to serine. In French and Sardinian familial T2D groups, 5% and 8% of randomly selected patients with diabetes, respectively, showed Gly40Ser mutations. These percentages are substantially higher than the frequencies of any other candidate gene mutations reported previously (47). Gough et al. examined patients from three geographically distinct regions in the United Kingdom and the Gly40Ser mutation was present in 15/691 patients with T2D and 1/425 geographically matched controls (48), suggesting that individuals with the Gly40Ser mutation may be predisposed to T2D. GCGR mutation frequencies have been examined in other populations and regions. However, the Gly40Ser mutation was not detected in studies involving subjects of Japanese (49–52), Finnish (53), Dutch (54), Utahans (55), German (54, 56), Russian (57), Indian Tamil (58), Han Chinese (59), Taiwanese (60), Brazilian (61), and Italian (44) descents. Another study (62) conducted in different areas of Sardinia did not find low insulin secretion in the population carrying this mutation in contrast to the earlier 1995 study (47). It showed that the Gly40Ser variation was not related to T2D in the Sardinian population and that its frequency varied among regions in Sardinia. Although no such association was found in Brazil, reduced insulin secretion was observed in Gly40Ser carriers (61). Based on a genetic analysis of 64 children with diabetes, the Gly40Ser mutation may be associated with T2D susceptibility in China (63). It reduces the binding of GCGR and glucagon and insulin secretion; this observation led Hansen to hypothesize that the Gly40Ser mutation in GCGR can lead to the abnormal functioning of islet β cells and may predispose carriers to diabetes, possibly by impairing glucagon-mediated signaling and decreasing the sensitivity of the target tissues to glucagon (64).
In addition to the relationship between the Gly40Ser mutation and T2D, an elevated frequency of GCGR mutations has been found in probands from multiple (affected sibling pair) families with T1D, also known as insulin-dependent diabetes; however, the lack of preferential transmission from heterozygous parents to affected siblings with T1D suggests population stratification (48). Overall, this Gly40Ser mutation may promote islet β-cell dysfunction, resulting in deficient insulin responses in patients with diabetes.
Together, these findings suggest that the contribution of GCGR to diabetes may vary and mutations in this gene play only a small role in determining the susceptibility of an individual to diabetes and the observed genetic heterogeneity of diabetes. Given the heterogeneity of the disease, the importance of GCGR for diabetes susceptibility may vary among ethnicities owing to the differences in genetic and environmental factors. GCGR is a polymorphic gene. The absence of a GCGR polymorphism (Gly40Ser) at one site does not rule out mutations associated with susceptibility to diabetes in other regions. For example, in addition to Gly40Ser, homozygous missense mutations (P86S) have been found in GCGR; these mutations contribute to the formation of an ineffective GCGR, resulting in hyperglycemia and extreme α-cell proliferation (65). Recent studies have reported 250 missense variants in human GCGR (66, 67). GCGR shows lower allelic diversity and fewer missense variants and variants with trait associations than the other class B1 GPCRs. These observations support the crucial role of the glucagon system in metabolism and indicate that the predominant signaling pathway mediating the physiological effects of GCGR is the one mediated by Gαs. These findings provide a clear link between molecular mechanisms and clinical phenotypes. The metabolic phenotypes related to several missense variants of GCGR have been investigated in case studies and in studies of genetically engineered animals, including V368M and V369M (68, 69). Further research is needed to explore the relationship between GCGR variants and diabetes.
Several emerging glucagon-based therapies are under pre-clinical and clinical development.
GCGR antagonism has been proposed as a pharmacological approach to treat T1D or T2D, including the use of small molecule antagonists, monoclonal antibodies (mAb) against GCGR, and antisense oligonucleotides that reduce expression of the receptor (70–73). Relevant clinical trials have shown that they can reduce blood glucose levels through inhibition of glucagon action (74–76); however, several adverse effects, such as increased LDL-cholesterol (LDL-c), ALT level, and bodyweight, have been observed (74, 77).
Several GCGR antagonists have been developed to improve glucose tolerance, insulin secretion, and glucose control in animals (78, 79), and have shown remarkable efficacy in patients with T2D, such as MK-0893, MK-3577, LY2409021 and LGD-6972 (76, 80–82). They upregulate circulating GLP-1 level by promoting intestinal L-cell proliferation and GLP-1 production in T2D (82). MK-0893 and MK-3577, which were advanced to phase II clinical trials, led to robust glucose lowering in patients with T2D; however, their adverse effects, such as increased LDL-c and ALT level, have hindered their clinical development (83–86). LY2409021 significantly reduced blood glucose and HbA1c levels with a lower risk of hypoglycemia (80, 81), but it increased liver fat (87). LGD-6972 is an allosteric GCGR antagonist, structurally different from other small molecule GCGR antagonists. It was well tolerated at all tested doses and did not cause hypoglycemia (88, 89), but additional details on biochemical differentiation are lacking and this compound does not appear to be under active clinical development (71).
With the cessation of clinical trials of GCGR antagonists and better understanding of the protein structure of GCGR, antibodies against GCGR have been developed. GCGR mAbs have good specificity, strong targeting, and are relatively easy to source. They can not only return blood glucose and HbA1c to normal levels when administered to mice with T1D not treated with insulin (73), as well as patients with T1D (90), but also show a strong hypoglycemic effect in mice and monkeys with T2D (91, 92). They can even induce β cell regeneration by the transdifferentiation of a portion of pancreatic α cells or δ cells into β cells (93). REMD 477 is a fully competitive mAb against GCGR. A single dose of REMD-477 significantly reduces insulin requirement in patients with T1D, which improves glycemic control in patients without serious adverse reactions (90). Another GCGR mAb, REGN1193, has good safety and tolerability, but transient elevation of transaminases was also observed (94). Overall, GCGR mAbs are promising for improving glycemic control and have great research promise.
GR-ASO inhibits the effect of glucagon mainly by decreasing the expression of GCGR mRNA (95). The intraperitoneal administration of GR-ASO to db/db mice and Zucker diabetic fatty (ZDF) rats decreases (nearly normalizes) non-fasting blood glucose levels (95). GR-ASO improves β-cell function (i.e., improves the insulin response to intraperitoneal glucose stimulation) and substantially improves glucose tolerance in normal and ZDF rats. However, Gcgr-/- mice and other animals treated with GR-ASO show extensive islet α-cell proliferation and significantly elevated circulating proglucagon-related peptide levels (96). Recently, ISIS-GCGRRx (76), IONIS-GCGRRxN (97), and ISIS 325568 (98) have been shown to attenuate glucagon-stimulated hepatic glucose production and glucose fluctuations. They support the treatment of GR-ASO in patients with T2D.
The most well-characterized biological function of GLP-1 is to potentiate glucose-dependent insulin secretion, which makes the GLP-1R an attractive target in the treatment of T2D (99). Thus, GLP-1R agonists are clinically used as anti-diabetic drugs (100). Glucagon not only acts to antagonize insulin in the fasting state but also functions in the fed state and promotes insulin secretion to maintain normal blood glucose levels (34). The insulin-promoting properties of glucagon are mediated by GCGR and GLP-1R in β cells (27, 33, 101); however, GLP-1R is the main receptor to exert an insulin-stimulating effect (101). It is reasonable to assume that even with GCGR mutations in β cells, glucagon binding to GLP-1R exerts an insulin-promoting effect that can reduce blood glucose concentrations in patients with diabetes. Although GLP-1R agonists have been used for the treatment of diabetes, their efficacy is limited by target receptor desensitization and downregulation via the recruitment of β-arrestins (102, 103). GLP-1R agonists with decreased β-arrestin-2 recruitment have shown promising effects in recent preclinical and clinical studies (104). Understanding the mechanisms of action may resolve these issues with the application of GLP-1R agonists.
Owing to the traditional view that the main effect of glucagon is to increase blood glucose levels, the idea of increasing glucagon concentration as a means of reducing glucose levels initially met resistance. Nevertheless, the action of glucagon on GCGR and GLP-1R (regulators of insulin secretion and energy metabolism) has a significant effect on systemic glucose homeostasis (105). On the one hand, GCGR and GLP-1R co-agonists can activate GLP-1R to promote insulin secretion and then reduce blood glucose. On the other hand, they can activate GCGR, promote lipid metabolism and reduce body weight (106–108). Since human islets have more mixed α-β cell interfaces, the ratio of GCGR to GLP-1R may be particularly vital to human islet function (8, 109). SAR425899 is a novel polypeptide with a co-excitatory effect on GCGR and GLP-1R, which can reduce blood glucose and HbA1c levels and reduce body weight in patients with T2D; however, it has an adverse effect on the gastrointestinal tract (110). It also improves postprandial blood glucose control by significantly enhancing β cell function and slowing glucose absorption rate (111). These findings highlight the possible clinical relevance of dual agonist peptides that simultaneously stimulate the synthesis of GCGR and GLP-1R and may drive the development of novel antidiabetic drugs.
In this review, we provide a clear overview of various theories of hormonal regulation of diabetes, with a focus on the essential roles of glucagon and its specific receptor in the pathogenesis of diabetes. Although GCGR and glucagon play important roles in diabetes, the mechanisms and role of mutations still needs to be explored. We summarized the pleiotropic effects of glucagon, future research prospects, and the development of novel therapeutic strategies. This area of research remains challenging but exciting. Further research on islet α cells, glucagon, and GCGR signaling pathways is expected to provide a basis for developing new strategies for diabetes prevention.
YJ wrote the manuscript. GS designed and critically reviewed the manuscript. SS critically revised the manuscript. YL and LF supervised the writing of the manuscript. All authors contributed to the article and approved the submitted version.
Funding was received from 345 talent project of Shengjing hospital of China Medical University and the National Natural Science Foundation of China, grant # 82070683.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. American Diabetes A. Diagnosis and Classification of Diabetes Mellitus. Diabetes Care (2013) 36 (Suppl 1):S67–74. doi: 10.2337/dc13-S067
2. Muller WA, Faloona GR, Unger RH. Hyperglucagonemia in Diabetic Ketoacidosis. Its Prevalence and Significance. Am J Med (1973) 54:52–7. doi: 10.1016/0002-9343(73)90083-1
3. Muller WA, Girardier L, Seydoux J, Berger M, Renold AE, Vranic M. Extrapancreatic Glucagon and Glucagonlike Immunoreactivity in Depancreatized Dogs. A Quantitative Assessment of Secretion Rates and Anatomical Delineation of Sources. J Clin Invest (1978) 62:124–32. doi: 10.1172/JCI109096
4. Unger RH, Cherrington AD. Glucagonocentric Restructuring of Diabetes: A Pathophysiologic and Therapeutic Makeover. J Clin Invest (2012) 122:4–12. doi: 10.1172/JCI60016
5. Lee YH, Wang MY, Yu XX, Unger RH. Glucagon Is the Key Factor in the Development of Diabetes. Diabetologia (2016) 59:1372–5. doi: 10.1007/s00125-016-3965-9
6. Conarello SL, Jiang G, Mu J, Li Z, Woods J, Zycband E, et al. Glucagon Receptor Knockout Mice Are Resistant to Diet-Induced Obesity and Streptozotocin-Mediated Beta Cell Loss and Hyperglycaemia. Diabetologia (2007) 50:142–50. doi: 10.1007/s00125-006-0481-3
7. Habegger KM, Heppner KM, Geary N, Bartness TJ, DiMarchi R, Tschop MH. The Metabolic Actions of Glucagon Revisited. Nat Rev Endocrinol (2010) 6:689–97. doi: 10.1038/nrendo.2010.187
8. Muller TD, Finan B, Clemmensen C, DiMarchi RD, Tschop MH. The New Biology and Pharmacology of Glucagon. Physiol Rev (2017) 97:721–66. doi: 10.1152/physrev.00025.2016
9. Zhang H, Qiao A, Yang D, Yang L, Dai A, de Graaf C, et al. Structure of the Full-Length Glucagon Class B G-Protein-Coupled Receptor. Nature (2017) 546:259–64. doi: 10.1038/nature22363
11. Huypens P, Ling Z, Pipeleers D, Schuit F. Glucagon Receptors on Human Islet Cells Contribute to Glucose Competence of Insulin Release. Diabetologia (2000) 43:1012–9. doi: 10.1007/s001250051484
12. Nauck MA, Meier JJ. The Incretin Effect in Healthy Individuals and Those With Type 2 Diabetes: Physiology, Pathophysiology, and Response to Therapeutic Interventions. Lancet Diabetes Endocrinol (2016) 4:525–36. doi: 10.1016/S2213-8587(15)00482-9
13. Campbell JE, Drucker DJ. Pharmacology, Physiology, and Mechanisms of Incretin Hormone Action. Cell Metab (2013) 17:819–37. doi: 10.1016/j.cmet.2013.04.008
14. Banting FG, Best CH, Collip JB, Campbell WR, Fletcher AA. Pancreatic Extracts in the Treatment of Diabetes Mellitus. Can Med Assoc J (1922) 12:141–6.
15. Unger RH, Orci L. The Essential Role of Glucagon in the Pathogenesis of Diabetes Mellitus. Lancet (1975) 1:14–6. doi: 10.1016/S0140-6736(75)92375-2
16. Unger RH. The Banting Memorial Lecture 1975. Diabetes and the Alpha Cell. Diabetes (1976) 25:136–51. doi: 10.2337/diab.25.2.136
17. Unger RH. Role of Glucagon in the Pathogenesis of Diabetes: The Status of the Controversy. Metabolism (1978) 27:1691–709. doi: 10.1016/0026-0495(78)90291-3
18. Unger RH, Orci L. Glucagon and the A Cell: Physiology and Pathophysiology (Second of Two Parts). N Engl J Med (1981) 304:1575–80. doi: 10.1056/NEJM198106253042604
19. Muller WA, Faloona GR, Unger RH. The Effect of Experimental Insulin Deficiency on Glucagon Secretion. J Clin Invest (1971) 50:1992–9. doi: 10.1172/JCI106691
20. Dobbs R, Sakurai H, Sasaki H, Faloona G, Valverde I, Baetens D, et al. Glucagon: Role in the Hyperglycemia of Diabetes Mellitus. Science (1975) 187:544–7. doi: 10.1126/science.1089999
21. Gerich JE, Lorenzi M, Bier DM, Schneider V, Tsalikian E, Karam JH, et al. Prevention of Human Diabetic Ketoacidosis by Somatostatin. Evidence for an Essential Role of Glucagon. N Engl J Med (1975) 292:985–9. doi: 10.1056/NEJM197505082921901
22. Raskin P, Unger RH. Hyperglucagonemia and its Suppression. Importance in the Metabolic Control of Diabetes. N Engl J Med (1978) 299:433–6. doi: 10.1056/NEJM197808312990901
23. Gelling RW, Du XQ, Dichmann DS, Romer J, Huang H, Cui L, et al. Lower Blood Glucose, Hyperglucagonemia, and Pancreatic Alpha Cell Hyperplasia in Glucagon Receptor Knockout Mice. Proc Natl Acad Sci USA (2003) 100:1438–43. doi: 10.1073/pnas.0237106100
24. Lee Y, Wang MY, Du XQ, Charron MJ, Unger RH. Glucagon Receptor Knockout Prevents Insulin-Deficient Type 1 Diabetes in Mice. Diabetes (2011) 60:391–7. doi: 10.2337/db10-0426
25. Lee Y, Berglund ED, Wang MY, Fu X, Yu X, Charron MJ, et al. Metabolic Manifestations of Insulin Deficiency do Not Occur Without Glucagon Action. Proc Natl Acad Sci USA (2012) 109:14972–6. doi: 10.1073/pnas.1205983109
26. Damond N, Thorel F, Moyers JS, Charron MJ, Vuguin PM, Powers AC, et al. Blockade of Glucagon Signaling Prevents or Reverses Diabetes Onset Only If Residual Beta-Cells Persist. Elife (2016) 5:e13828. doi: 10.7554/eLife.13828
27. Svendsen B, Larsen O, Gabe MBN, Christiansen CB, Rosenkilde MM, Drucker DJ, et al. Insulin Secretion Depends on Intra-Islet Glucagon Signaling. Cell Rep (2018) 25:1127–34.e1122. doi: 10.1016/j.celrep.2018.10.018
28. Omar BA, Andersen B, Hald J, Raun K, Nishimura E, Ahren B. Fibroblast Growth Factor 21 (FGF21) and Glucagon-Like Peptide 1 Contribute to Diabetes Resistance in Glucagon Receptor-Deficient Mice. Diabetes (2014) 63:101–10. doi: 10.2337/db13-0710
29. Longuet C, Robledo AM, Dean ED, Dai C, Ali S, McGuinness I, et al. Liver-Specific Disruption of the Murine Glucagon Receptor Produces Alpha-Cell Hyperplasia: Evidence for a Circulating Alpha-Cell Growth Factor. Diabetes (2013) 62:1196–205. doi: 10.2337/db11-1605
30. Wei R, Gu L, Yang J, Yang K, Liu J, Le Y, et al. Antagonistic Glucagon Receptor Antibody Promotes Alpha-Cell Proliferation and Increases Beta-Cell Mass in Diabetic Mice. iScience (2019) 16:326–39. doi: 10.1016/j.isci.2019.05.030
31. Kawamori D, Katakami N, Takahara M, Miyashita K, Sakamoto F, Yasuda T, et al. Dysregulated Plasma Glucagon Levels in Japanese Young Adult Type 1 Diabetes Patients. J Diabetes Investig (2019) 10:62–6. doi: 10.1111/jdi.12862
32. Matsuo T, Miyagawa J, Kusunoki Y, Miuchi M, Ikawa T, Akagami T, et al. Postabsorptive Hyperglucagonemia in Patients With Type 2 Diabetes Mellitus Analyzed With a Novel Enzyme-Linked Immunosorbent Assay. J Diabetes Investig (2016) 7:324–31. doi: 10.1111/jdi.12400
33. Capozzi ME, Svendsen B, Encisco SE, Lewandowski SL, Martin MD, Lin H, et al. Beta Cell Tone Is Defined by Proglucagon Peptides Through cAMP Signaling. JCI Insight (2019) 4:e126742. doi: 10.1172/jci.insight.126742
34. Zhang Y, Han C, Zhu W, Yang G, Peng X, Mehta S, et al. Glucagon Potentiates Insulin Secretion Via Beta-Cell GCGR at Physiological Concentrations of Glucose. Cells (2021) 10:2495. doi: 10.3390/cells10092495
35. Thorens B, Weir GC, Leahy JL, Lodish HF, Bonner-Weir S. Reduced Expression of the Liver/Beta-Cell Glucose Transporter Isoform in Glucose-Insensitive Pancreatic Beta Cells of Diabetic Rats. Proc Natl Acad Sci USA (1990) 87:6492–6. doi: 10.1073/pnas.87.17.6492
36. Vuguin PM, Charron MJ. Novel Insight Into Glucagon Receptor Action: Lessons From Knockout and Transgenic Mouse Models. Diabetes Obes Metab (2011) 13 Suppl 1:144–50. doi: 10.1111/j.1463-1326.2011.01447.x
37. Cabrera O, Ficorilli J, Shaw J, Echeverri F, Schwede F, Chepurny OG, et al. Intra-Islet Glucagon Confers Beta-Cell Glucose Competence for First-Phase Insulin Secretion and Favors GLP-1R Stimulation by Exogenous Glucagon. J Biol Chem (2022) 298:101484. doi: 10.1016/j.jbc.2021.101484
38. Tian G, Sol ER, Xu Y, Shuai H, Tengholm A. Impaired cAMP Generation Contributes to Defective Glucose-Stimulated Insulin Secretion After Long-Term Exposure to Palmitate. Diabetes (2015) 64:904–15. doi: 10.2337/db14-1036
39. Dyachok O, Isakov Y, Sagetorp J, Tengholm A. Oscillations of Cyclic AMP in Hormone-Stimulated Insulin-Secreting Beta-Cells. Nature (2006) 439:349–52. doi: 10.1038/nature04410
40. Qiao A, Han S, Li X, Li Z, Zhao P, Dai A, et al. Structural Basis of Gs and Gi Recognition by the Human Glucagon Receptor. Science (2020) 367:1346–52. doi: 10.1126/science.aaz5346
41. Wewer Albrechtsen NJ. The Glucose-Mobilizing Effect of Glucagon at Fasting is Mediated by Cyclic AMP. Am J Physiol Endocrinol Metab (2021) 321:E571–4. doi: 10.1152/ajpendo.00172.2021
42. Lok S, Kuijper JL, Jelinek LJ, Kramer JM, Whitmore TE, Sprecher CA, et al. The Human Glucagon Receptor Encoding Gene: Structure, cDNA Sequence and Chromosomal Localization. Gene (1994) 140:203–9. doi: 10.1016/0378-1119(94)90545-2
43. Menzel S, Stoffel M, Espinosa R 3rd, Fernald AA, Le Beau MM, Bell GI. Localization of the Glucagon Receptor Gene to Human Chromosome Band 17q25. Genomics (1994) 20:327–8. doi: 10.1006/geno.1994.1179
44. Gragnoli C, Milord E, Habener JF. Linkage Study of the Glucagon Receptor Gene With Type 2 Diabetes Mellitus in Italians. Metabolism (2005) 54:786–7. doi: 10.1016/j.metabol.2005.01.022
45. Wewer Albrechtsen NJ, Pedersen J, Galsgaard KD, Winther-Sorensen M, Suppli MP, Janah L, et al. The Liver-Alpha-Cell Axis and Type 2 Diabetes. Endocr Rev (2019) 40:1353–66. doi: 10.1210/er.2018-00251
46. Bozadjieva Kramer N, Lubaczeuski C, Blandino-Rosano M, Barker G, Gittes GK, Caicedo A, et al. Glucagon Resistance and Decreased Susceptibility to Diabetes in a Model of Chronic Hyperglucagonemia. Diabetes (2021) 70:477–91. doi: 10.2337/db20-0440
47. Hager J, Hansen L, Vaisse C, Vionnet N, Philippi A, Poller W, et al. A Missense Mutation in the Glucagon Receptor Gene Is Associated With Non-Insulin-Dependent Diabetes Mellitus. Nat Genet (1995) 9:299–304. doi: 10.1038/ng0395-299
48. Gough SC, Saker PJ, Pritchard LE, Merriman TR, Merriman ME, Rowe BR, et al. Mutation of the Glucagon Receptor Gene and Diabetes Mellitus in the UK: Association or Founder Effect? Hum Mol Genet (1995) 4:1609–12. doi: 10.1093/hmg/4.9.1609
49. Fujisawa T, Ikegami H, Yamato E, Takekawa K, Nakagawa Y, Hamada Y, et al. A Mutation in the Glucagon Receptor Gene (Gly40Ser): Heterogeneity in the Association With Diabetes Mellitus. Diabetologia (1995) 38:983–5. doi: 10.1007/BF00400589
50. Odawara M, Tachi Y, Yamashita K. Absence of Association Between the Gly40–>Ser Mutation in the Human Glucagon Receptor and Japanese Patients With non-Insulin-Dependent Diabetes Mellitus or Impaired Glucose Tolerance. Hum Genet (1996) 98:636–9. doi: 10.1007/s004390050274
51. Ogata M, Iwasaki N, Ohgawara H, Karibe S, Omori Y. Absence of the Gly40-Ser Mutation in the Glucagon Receptor Gene in Japanese Subjects With NIDDM. Diabetes Res Clin Pract (1996) 33:71–4. doi: 10.1016/0168-8227(96)01284-3
52. Fujisawa T, Ikegami H, Babaya N, Ogihara TJH. Gly40Ser Mutation of Glucagon Receptor Gene and Essential Hypertension in Japanese. Hypertension (1996) 28:1100.
53. Huang X, Orho M, Lehto M, Groop L. Lack of Association Between the Gly40Ser Polymorphism in the Glucagon Receptor Gene and NIDDM in Finland. Diabetologia (1995) 38:1246–8. doi: 10.1007/BF00422376
54. Ristow M, Busch K, Schatz H, Pfeiffer A. Restricted Geographical Extension of the Association of a Glucagon Receptor Gene Mutation (Gly40Ser) With Non-Insulin-Dependent Diabetes Mellitus. Diabetes Res Clin Pract (1996) 32:183–5. doi: 10.1016/0168-8227(96)01261-2
55. Elbein SC, Hoffman MD. Role of Mitochondrial DNA tRNA Leucine and Glucagon Receptor Missense Mutations in Utah White Diabetic Patients. Diabetes Care (1996) 19:507–8. doi: 10.2337/diacare.19.5.507
56. Ambrosch A, Lobmann R, Dierkes J, König W, Luley C, Lehnert H. Analysis of the Gly40Ser Polymorphism in the Glucagon Receptor Gene in a German Non-Insulin-Dependent Diabetes Mellitus Population. Clin Chem Lab Med (1999) 37:719–21. doi: 10.1515/CCLM.1999.110
57. Babadjanova G, Reincke M, Mora P, Chuchalin A, Allolio B. Polymorphism of the Glucagon Receptor Gene and Non-Insulin-Dependent Diabetes Mellitus in the Russian Population. Exp Clin Endocrinol Diabetes (1997) 105:225–6. doi: 10.1055/s-0029-1211756
58. Leprêtre F, Vionnet N, Budhan S, Dina C, Powell KL, Génin E, et al. Genetic Studies of Polymorphisms in Ten Non-Insulin-Dependent Diabetes Mellitus Candidate Genes in Tamil Indians From Pondichery. Diabetes Metab (1998) 24:244–50.
59. Deng H, Tang WL, Pan Q. [Gly40Ser Mutation of Glucagon Receptor Gene and NIDDM in Han Nationality]. Hunan Yi Ke Da Xue Xue Bao (2001) 26:291–3.
60. Huang CN, Lee KC, Wu HP, Tai TY, Lin BJ, Chuang LM. Screening for the Gly40Ser Mutation in the Glucagon Receptor Gene Among Patients With Type 2 Diabetes or Essential Hypertension in Taiwan. Pancreas (1999) 18:151–5. doi: 10.1097/00006676-199903000-00006
61. Shiota D, Kasamatsu T, Dib SA, Chacra AR, Moisés RS. Role of the Gly40Ser Mutation in the Glucagon Receptor Gene in Brazilian Patients With Type 2 Diabetes Mellitus. Pancreas (2002) 24:386–90. doi: 10.1097/00006676-200205000-00010
62. Tonolo G, Melis MG, Ciccarese M, Secchi G, Atzeni MM, Maioli M, et al. Physiological and Genetic Characterization of the Gly40Ser Mutation in the Glucagon Receptor Gene in the Sardinian Population. The Sardinian Diabetes Genetic Study Group. Diabetologia (1997) 40:89–94. doi: 10.1007/s001250050647
63. Wang X, Wang F, Wu H, Chen X, Xie R, Chen T, et al. Detection and Analysis of Glucose Metabolism-Related Genes in Childhood Diabetes Using Targeted Next-Generation Sequencing: In Pediatric Population-a Hospital-Based Study. Exp Ther Med (2020) 19:3398–404. doi: 10.3892/etm.2020.8579
64. Hansen LH, Abrahamsen N, Hager J, Jelinek L, Kindsvogel W, Froguel P, et al. The Gly40Ser Mutation in the Human Glucagon Receptor Gene Associated With NIDDM Results in a Receptor With Reduced Sensitivity to Glucagon. Diabetes (1996) 45:725–30. doi: 10.2337/diab.45.6.725
65. Zhou C, Dhall D, Nissen NN, Chen CR, Yu R. Homozygous P86S Mutation of the Human Glucagon Receptor Is Associated With Hyperglucagonemia, Alpha Cell Hyperplasia, and Islet Cell Tumor. Pancreas (2009) 38:941–6. doi: 10.1097/MPA.0b013e3181b2bb03
66. Lindquist P, Madsen JS, Brauner-Osborne H, Rosenkilde MM, Hauser AS. Mutational Landscape of the Proglucagon-Derived Peptides. Front Endocrinol (2021) 12:698511. doi: 10.3389/fendo.2021.698511
67. van der Velden WJC, Lindquist P, Madsen JS, Stassen R, Wewer Albrechtsen NJ, Holst JJ, et al. Molecular and In Vivo Phenotyping of Missense Variants of the Human Glucagon Receptor. J Biol Chem (2022) 298:101413. doi: 10.1016/j.jbc.2021.101413
68. Yu R. The V369M Gcgr Knock-in Mice are a Precision Medicine Model of Mild Mahvash Disease. Biochem J (2020) 477:2873–4. doi: 10.1042/BCJ20200522
69. Lin G, Liu Q, Dai A, Cai X, Zhou Q, Wang X, et al. Characterization of a Naturally Occurring Mutation V368M in the Human Glucagon Receptor and Its Association With Metabolic Disorders. Biochem J (2020) 477:2581–94. doi: 10.1042/BCJ20200235
70. Nunez DJ, D’Alessio D. Glucagon Receptor as a Drug Target: A Witches’ Brew of Eye of Newt (Peptides) and Toe of Frog (Receptors). Diabetes Obes Metab (2018) 20:233–7. doi: 10.1111/dom.13102
71. Cheng C, Jabri S, Taoka BM, Sinz CJ. Small Molecule Glucagon Receptor Antagonists: An Updated Patent Review (2015-2019). Expert Opin Ther Pat (2020) 30:509–26. doi: 10.1080/13543776.2020.1769600
72. Patil M, Deshmukh NJ, Patel M, Sangle GV. Glucagon-Based Therapy: Past, Present and Future. Peptides (2020) 127:170296. doi: 10.1016/j.peptides.2020.170296
73. Wang MY, Yan H, Shi Z, Evans MR, Yu X, Lee Y, et al. Glucagon Receptor Antibody Completely Suppresses Type 1 Diabetes Phenotype Without Insulin by Disrupting a Novel Diabetogenic Pathway. Proc Natl Acad Sci USA (2015) 112:2503–8. doi: 10.1073/pnas.1424934112
74. Ali S, Drucker DJ. Benefits and Limitations of Reducing Glucagon Action for the Treatment of Type 2 Diabetes. Am J Physiol Endocrinol Metab (2009) 296:E415–421. doi: 10.1152/ajpendo.90887.2008
75. Bagger JI, Knop FK, Holst JJ, Vilsboll T. Glucagon Antagonism as a Potential Therapeutic Target in Type 2 Diabetes. Diabetes Obes Metab (2011) 13:965–71. doi: 10.1111/j.1463-1326.2011.01427.x
76. Scheen AJ, Paquot N, Lefebvre PJ. Investigational Glucagon Receptor Antagonists in Phase I and II Clinical Trials for Diabetes. Expert Opin Investig Drugs (2017) 26:1373–89. doi: 10.1080/13543784.2017.1395020
77. Guan HP, Yang X, Lu K, Wang SP, Castro-Perez JM, Previs S, et al. Glucagon Receptor Antagonism Induces Increased Cholesterol Absorption. J Lipid Res (2015) 56:2183–95. doi: 10.1194/jlr.M060897
78. Mu J, Jiang G, Brady E, Dallas-Yang Q, Liu F, Woods J, et al. Chronic Treatment With a Glucagon Receptor Antagonist Lowers Glucose and Moderately Raises Circulating Glucagon and Glucagon-Like Peptide 1 Without Severe Alpha Cell Hypertrophy in Diet-Induced Obese Mice. Diabetologia (2011) 54:2381–91. doi: 10.1007/s00125-011-2217-2
79. O’Harte FP, Franklin ZJ, Irwin N. Two Novel Glucagon Receptor Antagonists Prove Effective Therapeutic Agents in High-Fat-Fed and Obese Diabetic Mice. Diabetes Obes Metab (2014) 16:1214–22. doi: 10.1111/dom.12360
80. Kelly RP, Garhyan P, Raddad E, Fu H, Lim CN, Prince MJ, et al. Short-Term Administration of the Glucagon Receptor Antagonist LY2409021 Lowers Blood Glucose in Healthy People and in Those With Type 2 Diabetes. Diabetes Obes Metab (2015) 17:414–22. doi: 10.1111/dom.12446
81. Kazda CM, Ding Y, Kelly RP, Garhyan P, Shi C, Lim CN, et al. Evaluation of Efficacy and Safety of the Glucagon Receptor Antagonist LY2409021 in Patients With Type 2 Diabetes: 12- and 24-Week Phase 2 Studies. Diabetes Care (2016) 39:1241–9. doi: 10.2337/dc15-1643
82. Lang S, Yang J, Yang K, Gu L, Cui X, Wei T, et al. Glucagon Receptor Antagonist Upregulates Circulating GLP-1 Level by Promoting Intestinal L-Cell Proliferation and GLP-1 Production in Type 2 Diabetes. BMJ Open Diabetes Res Care (2020) 8:e001025. doi: 10.1136/bmjdrc-2019-001025
83. Engel SS, Teng R, Edwards RJ, Davies MJ, Kaufman KD, Goldstein BJ. Efficacy and Safety of the Glucagon Receptor Antagonist, MK-0893, in Combination With Metformin or Sitagliptin in Patients With Type 2 Diabetes Mellitus. Diabetologia (2011) 54:S86–7.
84. Engel SS, Xu L, Andryuk PJ, Davies MJ, Amatruda J, Kaufman K, et al. Efficacy and Tolerability of MK-0893, a Glucagon Receptor Antagonist (GRA), in Patients With Type 2 Diabetes (T2DM). Diabetes (2011) 60:A85–5. doi: 10.2337/db11-1-378
85. Engel SS, Reitman ML, Xu L, Andryuk PJ, Davies MJ, Kaufman KD, et al. Glycemic and Lipid Effects of the Short-Acting Glucagon Receptor Antagonist MK-3577 in Patients With Type 2 Diabetes. Diabetes (2012) 61:A266–6. doi: 10.2337/db12-836-1328
86. Xiong Y, Guo J, Candelore MR, Liang R, Miller C, Dallas-Yang Q, et al. Discovery of a Novel Glucagon Receptor Antagonist N-[(4-{(1S)-1-[3-(3, 5-Dichlorophenyl)-5-(6-Methoxynaphthalen-2-Yl)-1H-Pyrazol-1-Yl]Ethyl}Phenyl)Carbo Nyl]-Beta-Alanine (MK-0893) for the Treatment of Type II Diabetes. J Med Chem (2012) 55:6137–48. doi: 10.1021/jm300579z
87. Guzman CB, Zhang XM, Liu R, Regev A, Shankar S, Garhyan P, et al. Treatment With LY2409021, a Glucagon Receptor Antagonist, Increases Liver Fat in Patients With Type 2 Diabetes. Diabetes Obes Metab (2017) 19:1521–8. doi: 10.1111/dom.12958
88. Pettus J, Vajda EG, Pipkin J, Williamson G, Zangmeister MA, Li YX, et al. Glucagon Receptor Antagonist LGD-6972 Significantly Lowers HbA1c and Is Well Tolerated After 12-Week Treatment in Patients With Type 2 Diabetes Mellitus (T2DM) on Metformin. Diabetes (2018) 67:73-OR. doi: 10.2337/db18-73-OR
89. Vajda EG, Logan D, Lasseter K, Armas D, Plotkin DJ, Pipkin JD, et al. Pharmacokinetics and Pharmacodynamics of Single and Multiple Doses of the Glucagon Receptor Antagonist LGD-6972 in Healthy Subjects and Subjects With Type 2 Diabetes Mellitus. Diabetes Obes Metab (2017) 19:24–32. doi: 10.1111/dom.12752
90. Pettus J, Reeds D, Cavaiola TS, Boeder S, Levin M, Tobin G, et al. Effect of a Glucagon Receptor Antibody (REMD-477) in Type 1 Diabetes: A Randomized Controlled Trial. Diabetes Obes Metab (2018) 20:1302–5. doi: 10.1111/dom.13202
91. Yan H, Gu W, Yang J, Bi V, Shen Y, Lee E, et al. Fully Human Monoclonal Antibodies Antagonizing the Glucagon Receptor Improve Glucose Homeostasis in Mice and Monkeys. J Pharmacol Exp Ther (2009) 329:102–11. doi: 10.1124/jpet.108.147009
92. Okamoto H, Kim J, Aglione J, Lee J, Cavino K, Na E, et al. Glucagon Receptor Blockade With a Human Antibody Normalizes Blood Glucose in Diabetic Mice and Monkeys. Endocrinology (2015) 156:2781–94. doi: 10.1210/en.2015-1011
93. Gu L, Cui X, Lang S, Wang H, Hong T, Wei R. Glucagon Receptor Antagonism Increases Mouse Pancreatic Delta-Cell Mass Through Cell Proliferation and Duct-Derived Neogenesis. Biochem Biophys Res Commun (2019) 512:864–70. doi: 10.1016/j.bbrc.2019.03.148
94. Kostic A, King TA, Yang F, Chan KC, Yancopoulos GD, Gromada J, et al. A First-in-Human Pharmacodynamic and Pharmacokinetic Study of a Fully Human Anti-Glucagon Receptor Monoclonal Antibody in Normal Healthy Volunteers. Diabetes Obes Metab (2018) 20:283–91. doi: 10.1111/dom.13075
95. Liang Y, Osborne MC, Monia BP, Bhanot S, Gaarde WA, Reed C, et al. Reduction in Glucagon Receptor Expression by an Antisense Oligonucleotide Ameliorates Diabetic Syndrome in Db/Db Mice. Diabetes (2004) 53:410–7. doi: 10.2337/diabetes.53.2.410
96. Sloop KW, Cao JX, Siesky AM, Zhang HY, Bodenmiller DM, Cox AL, et al. Hepatic and Glucagon-Like Peptide-1-Mediated Reversal of Diabetes by Glucagon Receptor Antisense Oligonucleotide Inhibitors. J Clin Invest (2004) 113:1571–81. doi: 10.1172/JCI20911
97. Morgan ES, Tai LJ, Pham NC, Overman JK, Watts LM, Smith A, et al. Antisense Inhibition of Glucagon Receptor by IONIS-GCGRRx Improves Type 2 Diabetes Without Increase in Hepatic Glycogen Content in Patients With Type 2 Diabetes on Stable Metformin Therapy. Diabetes Care (2019) 42:585–93. doi: 10.2337/dc18-1343
98. van Dongen MG, Geerts BF, Morgan ES, Brandt TA, de Kam ML, Romijn JA, et al. First Proof of Pharmacology in Humans of a Novel Glucagon Receptor Antisense Drug. J Clin Pharmacol (2015) 55:298–306. doi: 10.1002/jcph.396
99. Kim W, Egan JM. The Role of Incretins in Glucose Homeostasis and Diabetes Treatment. Pharmacol Rev (2008) 60:470–512. doi: 10.1124/pr.108.000604
100. Ahren B. The Future of Incretin-Based Therapy: Novel Avenues–Novel Targets. Diabetes Obes Metab (2011) 13 Suppl 1:158–66. doi: 10.1111/j.1463-1326.2011.01457.x
101. Capozzi ME, Wait JB, Koech J, Gordon AN, Coch RW, Svendsen B, et al. Glucagon Lowers Glycemia When Beta-Cells Are Active. JCI Insight (2019) 5:e129954. doi: 10.1172/jci.insight.129954
102. Pierce KL, Premont RT, Lefkowitz RJ. Seven-Transmembrane Receptors. Nat Rev Mol Cell Biol (2002) 3:639–50. doi: 10.1038/nrm908
103. Perry SJ, Baillie GS, Kohout TA, McPhee I, Magiera MM, Ang KL, et al. Targeting of Cyclic AMP Degradation to Beta 2-Adrenergic Receptors by Beta-Arrestins. Science (2002) 298:834–6. doi: 10.1126/science.1074683
104. Jones B, McGlone ER, Fang Z, Pickford P, Correa IR Jr., Oishi A, et al. Genetic and Biased Agonist-Mediated Reductions in Beta-Arrestin Recruitment Prolong cAMP Signaling at Glucagon Family Receptors. J Biol Chem (2021) 296:100133. doi: 10.1074/jbc.RA120.016334
105. Finan B, Capozzi ME, Campbell JE. Repositioning Glucagon Action in the Physiology and Pharmacology of Diabetes. Diabetes (2020) 69:532–41. doi: 10.2337/dbi19-0004
106. Patel VJ, Joharapurkar AA, Kshirsagar SG, Patel KN, Shah GB, Jain MR. Therapeutic Potential of Coagonists of Glucagon and GLP-1. Cardiovasc Hematol Agents Med Chem (2014) 12:126–33. doi: 10.2174/1871525712666141019194025
107. Sanchez-Garrido MA, Brandt SJ, Clemmensen C, Muller TD, DiMarchi RD, Tschop MH. GLP-1/Glucagon Receptor Co-Agonism for Treatment of Obesity. Diabetologia (2017) 60:1851–61. doi: 10.1007/s00125-017-4354-8
108. Capozzi ME, DiMarchi RD, Tschop MH, Finan B, Campbell JE. Targeting the Incretin/Glucagon System With Triagonists to Treat Diabetes. Endocr Rev (2018) 39:719–38. doi: 10.1210/er.2018-00117
109. Rodriguez-Diaz R, Molano RD, Weitz JR, Abdulreda MH, Berman DM, Leibiger B, et al. Paracrine Interactions Within the Pancreatic Islet Determine the Glycemic Set Point. Cell Metab (2018) 27:549–58.e544. doi: 10.1016/j.cmet.2018.01.015
110. Tillner J, Posch MG, Wagner F, Teichert L, Hijazi Y, Einig C, et al. A Novel Dual Glucagon-Like Peptide and Glucagon Receptor Agonist SAR425899: Results of Randomized, Placebo-Controlled First-in-Human and First-in-Patient Trials. Diabetes Obes Metab (2019) 21:120–8. doi: 10.1111/dom.13494
Keywords: glucagon, diabetes, pathogenesis, glucagonocentric hypothesis, glucagon receptor, glucagon-like peptide 1
Citation: Jia Y, Liu Y, Feng L, Sun S and Sun G (2022) Role of Glucagon and Its Receptor in the Pathogenesis of Diabetes. Front. Endocrinol. 13:928016. doi: 10.3389/fendo.2022.928016
Received: 25 April 2022; Accepted: 13 May 2022;
Published: 16 June 2022.
Edited by:
Undurti Narasimha Das, UND Life Sciences LLC, United StatesReviewed by:
Bo Ahrén, Lund University, SwedenCopyright © 2022 Jia, Liu, Feng, Sun and Sun. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Siyu Sun, c3VuLXNpeXVAMTYzLmNvbQ==; Guangwei Sun, c3VucmlzZTEyNDEyNEAxNjMuY29t
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.