 Maria Eugenia Sabatino
Maria Eugenia Sabatino Ezequiel Grondona
Ezequiel Grondona Ana Lucía De Paul
Ana Lucía De Paul- 1Universidad Nacional de Córdoba, Facultad de Ciencias Químicas, Córdoba, Argentina
- 2Consejo Nacional de Investigaciones Científicas y Técnicas, Instituto de Ciencia y Tecnología de Alimentos Córdoba (ICYTAC), Córdoba, Argentina
- 3Universidad Nacional de Córdoba, Facultad de Ciencias Médicas, Centro de Microscopía Electrónica, Córdoba, Argentina
- 4Consejo Nacional de Investigaciones Científicas y Técnicas, Instituto de Investigaciones en Ciencias de la Salud (INICSA), Córdoba, Argentina
The pituitary is a master gland responsible for the modulation of critical endocrine functions. Pituitary neuroendocrine tumours (PitNETs) display a considerable prevalence of 1/1106, frequently observed as benign solid tumours. PitNETs still represent a cause of important morbidity, due to hormonal systemic deregulation, with surgical, radiological or chronic treatment required for illness management. The apparent scarceness, uncommon behaviour and molecular features of PitNETs have resulted in a relatively slow progress in depicting their pathogenesis. An appropriate interpretation of different phenotypes or cellular outcomes during tumour growth is desirable, since histopathological characterization still remains the main option for prognosis elucidation. Improved knowledge obtained in recent decades about pituitary tumorigenesis has revealed that this process involves several cellular routes in addition to proliferation and death, with its modulation depending on many signalling pathways rather than being the result of abnormalities of a unique proliferation pathway, as sometimes presented. PitNETs can display intrinsic heterogeneity and cell subpopulations with diverse biological, genetic and epigenetic particularities, including tumorigenic potential. Hence, to obtain a better understanding of PitNET growth new approaches are required and the systematization of the available data, with the role of cell death programs, autophagy, stem cells, cellular senescence, mitochondrial function, metabolic reprogramming still being emerging fields in pituitary research. We envisage that through the combination of molecular, genetic and epigenetic data, together with the improved morphological, biochemical, physiological and metabolically knowledge on pituitary neoplastic potential accumulated in recent decades, tumour classification schemes will become more accurate regarding tumour origin, behaviour and plausible clinical results.
Introduction
Pituitary Tumorigenesis Unintelligibility
The pituitary is a master gland responsible for the modulation of critical endocrine functions. This entails a subtle responsiveness to dynamic cell signalling, resulting in a fine physiological adjustment for homeostasis preservation. Pituitary neuroendocrine tumours (PitNETs) display a considerable prevalence of 1/1106 in the general population (1), which are frequently observed as benign solid tumours.
Traditionally, it has been proposed that anterior PitNET growth starts from a monoclonal origin and propagates slowly, becoming more aggressive while transiting from microtumour to early macrotumour and, subsequently, expressing an invasive profile that eventually transforms into a carcinoma via metastasis (2). Angiogenesis and invasive performance are presumed to be determinant for pituitary carcinoma development, and are assessed by VEGF, EGF, COX-2, HIF-1a expression and RSUME up-regulation (2–4).Histologic typification is the most common method used by far for determining particular growth patterns and prognosis. The actual WHO classification provides detailed histological subtyping of a PitNET, based on the tumour cell lineage, cell type, and related characteristics (5). Yet, no reliable aggressive predictor has been defined for most PitNET, although some histologic subtypes can present aggressive behaviour (6, 7). The high-risk PitNETs recognised are: sparsely granulated somatotroph tumours, lactotroph tumours in men, Crooke’s cell tumours, silent corticotroph tumours, and Pit-1 positive plurihormonal tumours (8). The driveline responsible for the rarely invasive and metastatic profile is unclear, even when oncogenic pathways are triggered or tumour suppressor pathways are deactivated.
The apparent scarceness, uncommon behaviour and molecular features of PitNETs has resulted in a relatively slow improvement in depicting their pathogenesis. A better understanding of the several phenotypes or cellular responses during tumorigenesis is needed, as histological typification has a limited forecasting potency, although it is still the main option for prognosis determination.
New Approaches for Pituitary Tumorigenic Models
Despite their generally benign growth, PitNETs still represent a cause of important morbidity due to hormonal systemic deregulation, which requires surgical, radiological or chronic treatment for illness management (1, 9). Most PitNETs appear as macrotumours (1, 10), which are submitted to surgical removal as the first-line treatment, except for dopamine agonist treated lactotroph tumours (7, 9). However, entire surgical resection can be arduous due to the pituitary location. In addition, considering that PitNETs show relatively lower remission rates (10, 11), novel models with greater forecasting efficacy continue to be necessary.
Currently, PitNET diagnosis and classification are in routine practice still based on hormone immunohistochemical examination. As every cell type within the pituitary gland is able to yield tumours, a varied group of neoplasms can occur, usually related to overly secreted hormones. This subclassification could become more complex since it is now recognised that there are also tumours with varied secretory properties, either due to plurihormonal or multicellular pituitary neoplasia (8, 12, 13). Some of these discrepancies have been resolved by molecular studies, through typification of tumour origin by the transcription factors involved in the differentiation of anterior pituitary cells, with PIT1, TPIT, SF1, GATA3, and ERα providing histological subtyping of PitNETs (9). However, the numerous stages of pituitary tumorigenesis are still poorly understood.
We envisage that through a combination of molecular, genetic and epigenetic data, together with morphological, biochemical, physiological and metabolically knowledge on pituitary neoplastic potential accumulated in recent decades, tumour classification schemes will become more enlightened regarding tumour origin, behaviour and plausible clinical results (14, 15). Thus, with the aim of improving pituitary tumorigenesis aetiology comprehension, new integrative perspectives should help to unravel the underlying mechanisms of PitNET tumour growth.
Models of Unconstrained Cell Proliferation in Pituitary Neuroendocrine Tumours
Many efforts have been made to identify the mechanisms and agents involved at either the beginning or evolution of PitNETs. Typical oncogene mutations such as Ras or p53 genes, have not been effectively associated with unconstrained pituitary cell proliferation (16–19). As an alternative, other reports have pointed out that PTTG, abundantly expressed in most human PitNETs, with its role in initial pituitary tumorigenesis having been experimentally established and related to invasiveness, recurrence, metastasis (20–26). Subsequent reports have recognized PTTG as being the human homolog of securin, which acts in sister chromatid separation during mitosis (27), thereby accounting for PitNET aneuploidy (28). Nevertheless, no substantial association has been determined between PTTG expression and tumour size, grade, or even prognosis or treatment responses (25, 29). Related to this, unconstrained PitNET growth has also been associated with disrupted cell cycle regulation through the alteration of cyclins D1, D3, and E, or cyclin-dependent kinase inhibitor family-like CDKN1A (p21Cip1), CDKN1B (p27Kip1), CDKN2A (p16INK4a p14Arf), CDKN2B (p15INK4b), CDKN2C (p18INK4c) and pRb expression (7, 30–33).
Cell Death Contribution to Pituitary Neuroendocrine Tumour Development
Despite there being few reports referring to apoptosis in PitNETs, its contribution in tumour growth and as a prognosis biomarker have been explored (34, 35). Although apoptotic cells are practically absent or difficult to identify in PitNETs (36, 37), a greater apoptotic activity has been reported in aggressive and drug-resistant tumours (34, 38–41), particularly in corticotroph tumours (39, 40). However, since no association with growth rate or recurrence has been noted, there is no support for using the apoptosis index as a prognostic indicator (29, 37). On the other hand, from a molecular point of view, deregulation of apoptosis-related proteins might be a relevant marker of tumorigenesis, with the BCL2/BAX ratio having been proposed (34, 42, 43).
A programmed non-apoptotic cell death was described in PitNET, paraptosis or parapoptosis induced by EGF in a pituitary cell line (44), or by bromocriptine in experimental tumours (45). Furthermore, “dark cells”, referring to a cell death type thus named due to its electron-dense morphological features, was described in dopamine-treated lactotroph turmors and oncocytomas, alongside apoptotic cells (40) and in functional pituitary glands (46). Furthermore, bromocriptine treatment also provokes another cell death type recognised as programmed necrosis or necroptosis, in human lactotroph turmours and in a pituitary cell line (47, 48). To date, however, little information has been combined regarding different types of cell death in the regulation of PitNET formation and progression. Moreover, certain contradictions about the precise role of cell death pathways in pituitary tumorigenesis require further elucidation (7, 29, 37). Thus, apoptosis has not been shown to be a decisive factor in PitNET growth.
Beyond “to Live or to Die” Cellular Decisions During Tumorigenesis
With the aim of understanding tumorigenesis, many models have principally projected two major cellular fates leading to an extended dichotomised analysis: to proliferate or to die, usually presenting both as excluding outcomes (49–59). Current knowledge has established the intricacy of the signalling networks that guide and preserve tumours, implying coordination of the intra- and extracellular cues that trigger various pathways, either simultaneously or in a spatio-temporal dynamic. Cell survival and proliferation are interrelated with cell death, acting as combined interdependent processes at several points by molecular links responsible for the coordination of cell growth (53, 60, 61). Remarkably, tumour cells harbour the possibility of eliciting intrinsic suppressor programmes, thus permitting tumour progression once this interlinked molecular network between proliferation and growth suppression gets uncoupled (62). Consequently, several mechanisms might be triggered to thwart uncontrolled cell division, such as autophagy, cellular senescence, programmed cell death and necrosis; all of which actually appear as crucial responses to tumoral alterations (61, 63, 64).
Pituitary neoplasm behaviour presents a significantly inconsistent and unpredictable growth performance (7–9, 14, 15, 29), so that events such as mitosis and apoptosis have ended up being unhelpful measures (36, 52), thereby requiring models based on proliferation and cell death to be reconsidered.
Achieving successful tumour development not only entails sustained cell division, but also their survival and thriving, circumstances that may require cellular physiology and metabolic reprogramming to cope with a changing environment and cellular damage (62). All these adjustments might transform a unified group of cells into a small tumour ecosystem, in which different cell phenotypes compete and may eventually collaborate for available space and resources (65). Accordingly, tumour progression represents the coevolution of a heterogeneous group of cells, which instead of acting as an individual uncontrolled cell, needs to coordinate and obtain the cooperation of the neighbourhood (66).
Evidence accumulated about pituitary tumorigenesis reveals it involves more than just the rate of cell multiplication and loss, with it also depending on more than a univocal abnormality factor in a central proliferation pathway (7). PTs can display intrinsic heterogeneity and cell subpopulations with diverse biological, genetic and epigenetic particularities, including tumorigenic potential (15, 55, 67). Hence, PitNET growth understanding requires new approaches and systematisation of the available data.
The role of cell death programs, autophagy, stem cells, cellular senescence, microenvironment, inflammation, mitochondrial function and metabolic reprogramming are still emerging fields in pituitary research. Future morphological and molecular studies also need to establish spatio-temporal dynamics, cellular heterogeneity, cell physiology adaptation and the ability to cope with cellular damage. To date, few studies have been based on integrative analysis, where proteomic data is used for defining which cell-signalling and metabolic pathways could be the most relevant during PitNET pathogenesis (14, 68–72).
Genetic and Epigenetic Regulation in Pituitary Neuroendocrine Tumours
Whole genomic sequencing has exposed numerous mutations in PitNETs. However, they display comparatively less genetic anomalies than other tumour types or cancers (73). In general terms, genetic anomalies associated with PitNET tumorigenesis progression may not be conclusive (73). Consequently, as only a small number of pituitary neuroendocrine tumours may be correlated with recurrent somatic mutations and unusual hereditary variations (74), some evidence has suggested that epigenetic modifications may participate in pituitary tumorigenesis (73, 75, 76).
Several reports have identified epigenetic modifications in PitNETs, and DNA methylation has been designated a major strategy for epigenetic modification, (77–79) in addition to the aberrant expression of DNMT enzymes (80, 81). However, even if increased amounts of methylation could be connected with more aggressive PitNETs (73), a correlation between gene expression and promoter methylation may not always be detected (74).
Recent studies have explored multiple dysregulated histone acetylation in PitNETs (82–85), which may lead to acetylation of the PTTG promoter (86), suggesting the presence of different arrangements of histone modifications (73). In addition, fluctuations in miRNA expression in several tumours reveal that this kind modification could be involved in essential decisions throughout tumour progression (87, 88). Indeed, altered miRNA expression has been shown to be related with increased or decreased tumour diameter, invasiveness, tumour subtype and therapeutic outcomes (89–97).
Coping With Cellular Stress: Cellular Physiology and Metabolic Status in Pituitary Neuroendocrine Tumour Cells
DNA Damage
Genomic instability is an extended feature of almost all tumour cells (98), Concerning pituitary neoplasia, it has been stated that genomic instability and oxidative DNA damage often occurs and could be associated with an early biomarker of invasive and aggressive behaviour (99–101). Moreover, loss of heterozygosity and an altered number of somatic copies of genes were reported in secreting and aggressive sporadic pituitary neuroendocrine tumours, and may correlate with clinical phenotypes (99, 102–104).
DNA repair systems is a main constituent of DNA damage response (DDR) in normal conditions, with ineffectiveness in this process being linked with the susceptibility to tumour growth through the occurrence of genomic instability (105, 106). Conversely, DDR also serves as a physiological barrier against tumour initiation or progression (107, 108). However, information about the DDR contribution to pituitary tumorigenesis is scarce. Gene mutations involved in DNA mismatch repair have been recognised in Lynch syndrome patients presenting aggressive corticotropin-secreting tumours (109, 110). Also, missense mutations have also been reported in mismatch repair genes in non-secreting tumours (111). DNA damage signs have appeared in association with cellular senescence in somatotroph tumours (112) and also in experimental lactotroph turmors (113). Moreover, PTTG was related to aneuploidy and DNA damage senescent GH-secreting cells, which are potentially responsible for growth constraint (114). Recently, it was shown that cAMP and Fanconi anemia DNA damage repair pathways were affected by alterations in the somatic copy number in somatotroph tumours, which could act as pathogenetic drivers of tumorigenesis (104).
Cellular Bioenergetics
The mitochondria’s mandatory role as an energy provider establishes it as a crucial link of cellular metabolism and oxidative stress management, thus supporting processes such as proliferation, apoptosis, autophagy, senescence and immunity response (115). In addition, mitochondrial proteins can regulate numerous signalling pathway networks and cellular behaviours, with this organelle being involved in an extensive range of diseases, including tumorigenesis (116). Molecular network studies have revealed that mitochondrial dysfunction, oxidative stress and mitochondria-mediated ROS-mitogen-activated protein kinase (MAPK) signalling abnormality are significantly associated with the pathogenesis of PitNETs (69, 70, 72, 117–120).
The modification in energy metabolism needed for tumour formation or progression is related to mitochondrial adaptation and seems to play an imperative role in PitNETs by influencing cell proliferation, growth, and angiogenesis. Increases in the mitochondria number (121–124), fusion process and biogenesis have been found during experimental pituitary tumorigenesis (124), with the volume of mitochondria varying between diverse tumour subtypes (118). In addition, an augmented production of lactate dehydrogenase toward aerobic glycolysis (124, 125) and a modification in the fatty acid metabolism have also been observed (97).
Amplified ROS and RNS actions and oxidative stress have been regarded as critical contributors in the pathogenesis of PitNETs (124–130). Furthermore, mitochondrial dysfunction has been described showing morphological and functional changes, such as bigger mitochondria with irregular swelling and fragmented cristaes (124). An activation of the nuclear factor erythroid 2 like 2 (Nrf2) pathways, a main regulator of oxidative stress response along with oxidative damage signal reduction, have been reported during PitNET development, which might provide cellular survival advantages (120, 124). Indeed, elevated mitophagy and mitochondrial dysfunction may favour resistance to chemotherapy in the pituitary GH3 cell line (131). Conversely, activation of mitochondria-mediated apoptosis has been proposed, which might favour novel therapy drugs (132–134). Although this evidence validates the significant roles of mitochondrial functions and adaptability in pituitary tumorigenesis, their molecular mechanisms still need to be clarified.
Surviving or Thriving: Senescence and Autophagy in Pituitary Neuroendocrine Tumours
Cellular Senescence
Cellular senescence (CS) is considered a stress response determined by stable cell cycle arrest in which cells remain viable and metabolically active (135, 136), with many studies having reported the presence of CS in PitNETs. As it is understood to be a spontaneous initial barrier in tumorigenesis (137), CS might constitute a conceivable explanation for the slow and benign growth of PTs (2, 138).
Significant differences in CS marker expression have been detected in human PitNETs (139–141). Moreover, a certain specificity of this cellular phenomenon to the tumour subtype has been suggested, as frequently aggressive ACTH tumours present lower senescence signs (141). Several experimental models have reported the contribution of CS during pituitary tumorigenesis (112–114, 142–144), supporting the idea it may be an impediment against oncogenic stimulation and prevent cellular transformation (145).
The driver forces underlying pituitary senescence are not entirely deciphered, as various cellular pathways and cytokines seem to contribute in triggering and modifying CS acquisition, such as PTTG, which displays oncogene activities and is overexpressed in many PitNETs (146). Yet, its deletion or overexpression promotes pituitary p53/p21-dependent senescence in GH-secreting cells (22, 112, 114, 147). Furthermore, the involvement of tissue-specific pathways has been proposed of intra-nuclear p21Cip1 diverse expression, p16Ink4a and p15Ink4b (112, 114, 139–141).
CS also develops a complex senescence-associated secretory phenotype (SASP) that emulates an inflammatory response. In particular, IL-6 contributes to maintaining pituitary senescence during tumorigenesis by its autocrine action, providing an IL-6-mediated benign tumour senescence model (67, 113, 138, 144, 148, 149). However, detrimental functions of senescent cells have also been uncovered in cancer development, mainly through pro-tumorigenic factor secretion inducing paracrine tumorigenesis (150). Indeed, pituitary IL-6 may provoke contrary effects (inhibitory or stimulatory) in different tumours such as ACTH-, PRL-, GH-secreting and non-functioning tumours (148). Furthermore, paracrine IL-6 triggered by surrounding folliculo-stellate cells is capable of stimulating tumour development (151).
Autophagy
Autophagy is an intracellular catabolic pathway based on self-degradation and recycling of the cellular components that collaborate to physiological homeostasis. However, as autophagy is also implicated under pathological conditions, it is considered a ‘double-edged sword’ for being a tumour suppressor as well as a pro-survival factor (152–154). It is triggered in response to a variety of stimuli and also connected with CS, due to both factors protecting cells from external and internal stressors (63). Autophagy could also be permissive for tumour survival in the face of stress (155, 156).
Concerning autophagy research in PitNETs, crinophagy, a specific form of autophagy of secretory granules, has been reported to have a role in intracellular hormone level modulation (157–159). Autophagy participates in lactotroph turmors cell survival and proliferation by the action of a long non-coding RNA CLRN1-AS1 affecting the Wnt/β-catenin signalling pathway (160).
Several reports have related autophagy to PitNET clinical behaviour and drug therapy sensitivity (161, 162). Dopamine agonists, cabergoline and bromocriptine, the first choice treatments for lactotrophs tumours, trigger autophagy in tumoral cells (163–165). Also, somatostatin analogue (octreotide, lanreotide or pasireotide) treatment induces concomitantly apoptosis and autophagy in GH tumours of acromegaly patients (166). The contribution of autophagy to radiotherapy and its manifestation in pituitary carcinomas has yet not been examined. Therefore, it is possible to suggest that the role of autophagy in PitNET cell biology might be context-dependent (161, 162, 167), although its mechanism elucidation requires further investigation.
A Novel Cellular Population Susceptible to Contributing to Pituitary Neuroendocrine Tumours: Pituitary Stem Cells
Resident pituitary stem cells (PSCs) exist at the marginal zone (MZ) of the intermediate lobe, dorsal anterior lobe (AL) and throughout the AL parenchyma (168–170). This group of cells are involved, at least in part, in tissue remodelling and hormone-producing cell generation during embryonic and postnatal life (171–173). This long-term pool of undifferentiated progenitors shares stemness-related factors which confer on them self-renewal and pluripotency properties responsive to homeostatic balance and injury (174–179). In addition, the in vivo expression of multiple markers indicates the existence of PSC population subsets or heterogeneity, which act as a cellular niche driving physiological pituitary plasticity (179–181).
Stem cells and cancer stem cells have been described in PitNETs (CSCs) (182–184) displaying SOX2 and NANOG expression, two pluripotency-associated transcription factors (185), possibly representing a tumour-initiating cell population (173, 186). In addition, the presence of plurihormonal and null cell-type tumours and the low mitotic rate present in the hyperplastic pituitary suggest that PSC are a potential cellular source of PitNETs (187). Pituitary neuroendocrine tumour stem cells (PASCs) expressing GFRa2, Sox9, Nestin, CD133 and CD44, identified in normal and experimental PitNETs, along with variations in PSC/CSC marker expression, were notably detected at tumour initiation (184). Further characterizations of human adult PSCs are now necessary to obtain better understanding of the physiological and pathological roles of these cell subsets. Innovative in vitro investigations, such as PSC-derived organoid models (188, 189), should provide a deeper insight into the role of PSC/CSC in pathophysiological contexts, thereby contributing to PitNET growth control.
Perspectives and Final Considerations
Tissues execute a continuous counterbalance between proliferation, differentiation and death in order to preserve a normal and healthy structure and function. These tasks involve an unceasing choreography, as evidenced by cellular architecture, and are coordinated by paracrine interactions. The loss of homeostatic dynamics can arise by mutations, cellular damage or stress, leading to aberrant proliferation, an essential step for tumour formation.
Many decades of tumorigenesis research have been devoted to identifying the genetic and molecular players, central keys or pathways that are mainly responsible for a particular cell fate decision, frequently within the proliferate/die binary axiom. However, the cellular decision process in tumour biology may result from redundant, interconnected and double-edged sword molecular signalling pathways. Biological networks are characterised by multiple feed-forward, feedback, and cross-talk characteristics that compensate for perturbations affecting individual components and provide them with great robustness. These are intricate dialogs entailing soluble molecules that comprise growth factors, cytokines, hormones and proteases, and also insoluble factors such as extracellular matrix components or cell-cell interactions. Figuring out how single components of such a complex and multifaceted network collaborate to the output of each programme network is a key requirement, because analysing components separately cannot provide a whole picture of the network dynamics.
As intratumoral heterogeneity might exist in many forms, the alteration of multiple, sometimes superposed molecular pathways can be condensed and understood as an array of phenotypes or behaviours, which can then be incorporated into a cellular interaction model. This involves not just the determination of individual genetic or epigenetic subtypes, but also the integration of consequent phenotypic features with the microenvironment to reflect this complex interplay, which may help to dissect definable tumour outcomes. We should emphasise the incorporation of cellular physiology and metabolic status and reprogramming examination as a tumour fitness depiction, thereby providing a measure of the ability of tumour cell phenotypes to survive and grow.
We have only lately begun to understand the variety and complexity of machinery by which tumorigenic lesions develop. The deficiency of many long-used models to faithfully represent the complexity of systemic tumour behaviour has generated a greater necessity for combining several viewpoints, to produce a wider comprehension of the critical objective of interventional therapies. By considering an ecological perspective for tumour cells, it is possible to define grouped or collective phenotypes beyond searching for individual mutations. In this cellular ecosystem, the relations among confined contributors will progressively transform, creating a vast net of cellular cross-talking and structural components that can promote growth (Figure 1). We suggest that the tissue architecture and microenvironment could play vital roles in neoplasms. New models that envisage a major complexity will be able to generate a tumour fitness interpretation, and allow the gradation of pituitary trophic plasticity to be discerned in order to bring boosted responses to regular stimuli throughout life and to the suboptimal reactions or homeostasis restoration that possibly influence trophic anomalies.
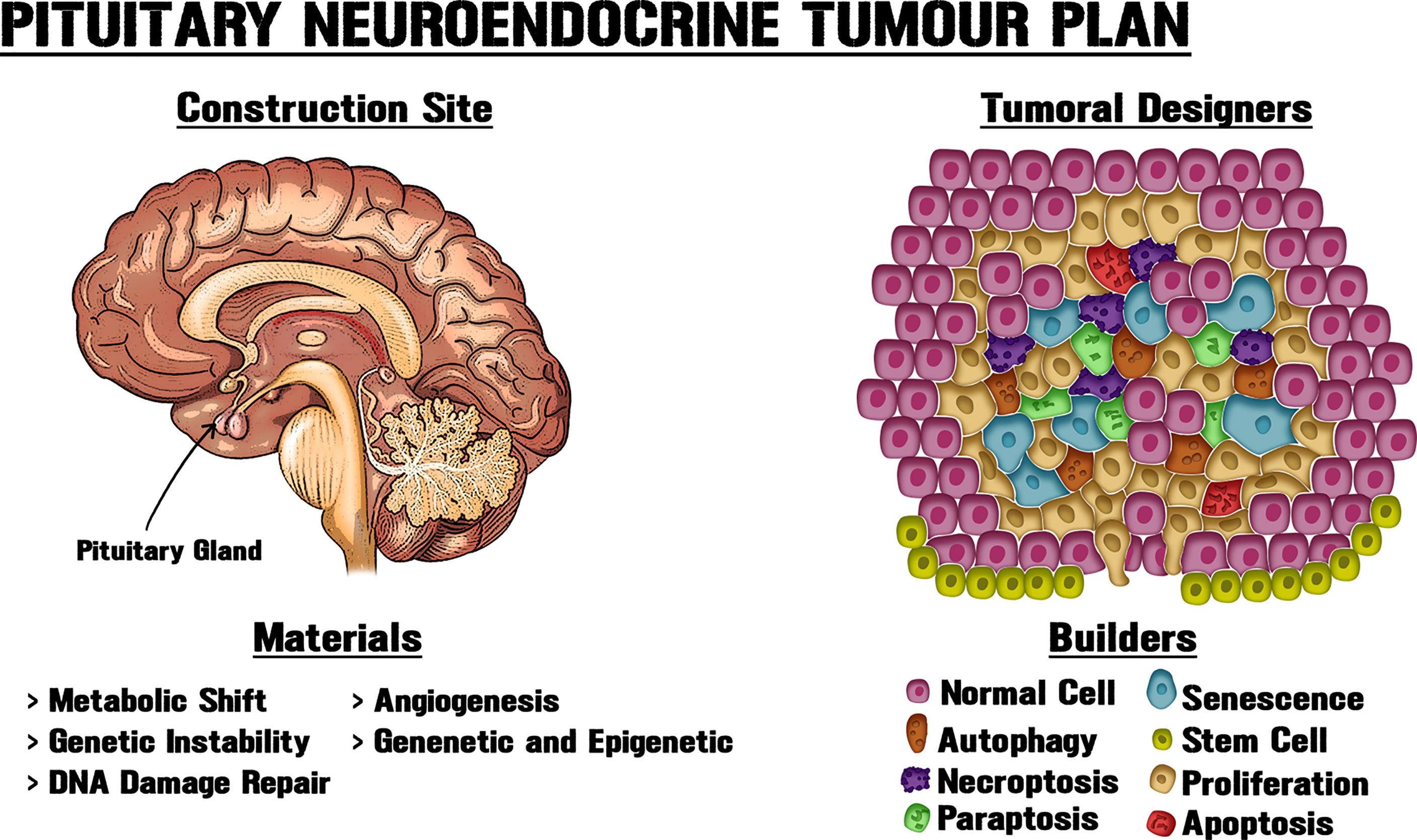
Figure 1 New approaches for pituitary tumorigenic models. PitNETs can display intrinsic heterogeneity and cell subpopulations with diverse biological, genetic and epigenetic particularities, including tumorigenic potential. Tissue architecture and microenvironment could play vital roles in neoplasms. Like an architect who determines the use of materials at a building site, different cellular processes modify the structure and interactions within the gland, thereby shaping tumour growth. Obtaining a better understanding of PitNET growth requires new approaches and systematization of the available data through the combination of molecular, genetic and epigenetic data, together with the utilization of morphological, biochemical, physiological and metabolic knowledge about pituitary neoplastic potential.
Author Contributions
MS, EG and ADP contributed to conception and design of the review. MS, EG and ADP organized the articles included. MS wrote the first draft of the manuscript. MS, EG and ADP wrote sections of the manuscript. EG designed the included figure. All authors contributed to manuscript revision, read, and approved the submitted version.
Funding
This work was funded by Consejo Nacional de Investigaciones Científicas y Técnicas (CONICET PIP 2020-2023 No. 11220200102210), Secretaría de Ciencia y Tecnología, Universidad Nacional de Córdoba (SECyT-UNC 2018-2022 Nº33620180100675CB), and Agencia Nacional de Promoción Científica y Tecnológica - Ministerio de Ciencia y Tecnología (FONCYT-PICT 0950-2020) to ALDP.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as being a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Daly AF, Beckers A. The Epidemiology of Pituitary Adenomas. Endocrinol Metab Clin North Am (2020) 49:347–55. doi: 10.1016/j.ecl.2020.04.002
2. Stalla GK, Renner U, Elguero MB, Arzt E. Molecular and Cellular Pathogenesis of Pituitary Tumors. Curr Opin Endocrine Metab Res (2018) 1:8. doi: 10.1016/j.coemr.2018.01.004
3. Shan B, Gerez J, Haedo M, Fuertes M, Theodoropoulou M, Buchfelder M, et al. RSUME is Implicated in HIF-1-Induced VEGF-A Production in Pituitary Tumour Cells. Endocr Relat Cancer (2012) 19:13–27. doi: 10.1530/ERC-11-0211
4. He W, Huang L, Shen X, Yang Y, Wang D, Yang Y, et al. Relationship Between RSUME and HIF-1alpha/VEGF-A With Invasion of Pituitary Adenoma. Gene (2017) 603:54–60. doi: 10.1016/j.gene.2016.12.012
5. Asa SL, Mete O, Perry A, Osamura RY. Overview of the 2022 WHO Classification of Pituitary Tumors. Endocr Pathol (2022) 33:6–26. doi: 10.1007/s12022-022-09703-7
6. Wierinckx A, Auger C, Devauchelle P, Reynaud A, Chevallier P, Jan M, et al. A Diagnostic Marker Set for Invasion, Proliferation, and Aggressiveness of Prolactin Pituitary Tumors. Endocr Relat Cancer (2007) 14:887–900. doi: 10.1677/ERC-07-0062
7. Jaffrain-Rea ML, Rotondi S, Alesse E. "New Insights in the Pathogenesis of Pituitary Tumours,". In: Fedele M, editor. Hot Topics in Endocrine and Endocrine-Related Diseases (2013) London: IntechOpen. doi: 10.5772/56028
8. Kontogeorgos G. Update on Pituitary Adenomas in the 2017 World Health Organization Classification: Innovations and Perspectives. Hormones (Athens) (2021) 20:287–91. doi: 10.1007/s42000-020-00269-9
9. Asa SL, Mete O, Cusimano MD, Mccutcheon IE, Perry A, Yamada S, et al. Pituitary Neuroendocrine Tumors: A Model for Neuroendocrine Tumor Classification. Mod Pathol (2021) 34:1634–50. doi: 10.1038/s41379-021-00820-y
10. Kim JH, Dho YS, Kim YH, Lee JH, Lee JH, Hong AR, et al. Developing an Optimal Follow-Up Strategy Based on the Natural History of Nonfunctioning Pituitary Adenomas. J Neurosurg (2018) 131:500–6. doi: 10.3171/2018.4.JNS172148
11. Kreutzer J, Vance ML, Lopes MB, Laws ER Jr. Surgical Management of GH-Secreting Pituitary Adenomas: An Outcome Study Using Modern Remission Criteria. J Clin Endocrinol Metab (2001) 86:4072–7. doi: 10.1210/jcem.86.9.7819
12. Clayton RN, Pfeifer M, Atkinson AB, Belchetz P, Wass JA, Kyrodimou E, et al. Different Patterns of Allelic Loss (Loss of Heterozygosity) in Recurrent Human Pituitary Tumors Provide Evidence for Multiclonal Origins. Clin Cancer Res (2000) 6:3973–82.
13. Lloyd Rv, Osamura Ry, Kloppel G, Rosai J. WHO Classification of Tumours of Endocrine Organs. Lion: IARC Press (2017).
14. Wierinckx A, Raverot G, Nazaret N, Jouanneau E, Auger C, Lachuer J, et al. Proliferation Markers of Human Pituitary Tumors: Contribution of a Genome-Wide Transcriptome Approach. Mol Cell Endocrinol (2010) 326:30–9. doi: 10.1016/j.mce.2010.02.043
15. Manojlovic-Gacic E, Bollerslev J, Casar-Borota O. Invited Review: Pathology of Pituitary Neuroendocrine Tumours: Present Status, Modern Diagnostic Approach, Controversies and Future Perspectives From a Neuropathological and Clinical Standpoint. Neuropathol Appl Neurobiol (2020) 46:89–110. doi: 10.1111/nan.12568
16. Cai WY, Alexander JM, Hedley-Whyte ET, Scheithauer BW, Jameson JL, Zervas NT, et al. Ras Mutations in Human Prolactinomas and Pituitary Carcinomas. J Clin Endocrinol Metab (1994) 78:89–93. doi: 10.1210/jc.78.1.89
17. Pei L, Melmed S, Scheithauer B, Kovacs K, Prager D. H-Ras Mutations in Human Pituitary Carcinoma Metastases. J Clin Endocrinol Metab (1994) 78:842–6. doi: 10.1210/jc.78.4.842
18. Gejman R, Swearingen B, Hedley-Whyte ET. Role of Ki-67 Proliferation Index and P53 Expression in Predicting Progression of Pituitary Adenomas. Hum Pathol (2008) 39:758–66. doi: 10.1016/j.humpath.2007.10.004
19. Aran V, Heringer M, Da Mata PJ, Kasuki L, Miranda RL, Andreiuolo F, et al. Identification of Mutant K-RAS in Pituitary Macroadenoma. Pituitary (2021) 24:746–53. doi: 10.1007/s11102-021-01151-6
20. Heaney AP, Horwitz GA, Wang Z, Singson R, Melmed S. Early Involvement of Estrogen-Induced Pituitary Tumor Transforming Gene and Fibroblast Growth Factor Expression in Prolactinoma Pathogenesis. Nat Med (1999) 5:1317–21. doi: 10.1038/15275
21. Zhang X, Horwitz GA, Heaney AP, Nakashima M, Prezant TR, Bronstein MD, et al. Pituitary Tumor Transforming Gene (PTTG) Expression in Pituitary Adenomas. J Clin Endocrinol Metab (1999) 84:761–7. doi: 10.1210/jcem.84.2.5432
22. Chesnokova V, Kovacs K, Castro AV, Zonis S, Melmed S. Pituitary Hypoplasia in Pttg-/- Mice is Protective for Rb+/- Pituitary Tumorigenesis. Mol Endocrinol (2005) 19:2371–9. doi: 10.1210/me.2005-0137
23. Filippella M, Galland F, Kujas M, Young J, Faggiano A, Lombardi G, et al. Pituitary Tumour Transforming Gene (PTTG) Expression Correlates With the Proliferative Activity and Recurrence Status of Pituitary Adenomas: A Clinical and Immunohistochemical Study. Clin Endocrinol (Oxf) (2006) 65:536–43. doi: 10.1111/j.1365-2265.2006.02630.x
24. Vlotides G, Eigler T, Melmed S. Pituitary Tumor-Transforming Gene: Physiology and Implications for Tumorigenesis. Endocr Rev (2007) 28:165–86. doi: 10.1210/er.2006-0042
25. Sanchez-Ortiga R, Sanchez Tejada L, Peiro Cabrera G, Moreno-Perez O, Arias Mendoza N, Aranda Lopez FI, et al. [Rol of Pituitary Tumour-Transforming Gene (PTTG) in the Pituitary Adenomas]. Endocrinol Nutr (2010) 57:28–34. doi: 10.1016/S1575-0922(10)70006-1
26. Fuertes M, Sapochnik M, Tedesco L, Senin S, Attorresi A, Ajler P, et al. Protein Stabilization by RSUME Accounts for PTTG Pituitary Tumor Abundance and Oncogenicity. Endocr Relat Cancer (2018) 25:665–76. doi: 10.1530/ERC-18-0028
27. Prezant TR, Kadioglu P, Melmed S. An Intronless Homolog of Human Proto-Oncogene hPTTG is Expressed in Pituitary Tumors: Evidence for hPTTG Family. J Clin Endocrinol Metab (1999) 84:1149–52. doi: 10.1210/jcem.84.3.5658
28. Kim DS, Fong J, Read ML, Mccabe CJ. The Emerging Role of Pituitary Tumour Transforming Gene (PTTG) in Endocrine Tumourigenesis. Mol Cell Endocrinol (2007) 278:1–6. doi: 10.1016/j.mce.2007.08.006
29. Sav A, Rotondo F, Syro LV, Scheithauer BW, Kovacs K. Biomarkers of Pituitary Neoplasms. Anticancer Res (2012) 32:4639–54.
30. Simpson DJ, Hibberts NA, Mcnicol AM, Clayton RN, Farrell WE. Loss of pRb Expression in Pituitary Adenomas is Associated With Methylation of the RB1 CpG Island. Cancer Res (2000) 60:1211–6.
31. Korbonits M, Chahal HS, Kaltsas G, Jordan S, Urmanova Y, Khalimova Z, et al. Expression of Phosphorylated P27(Kip1) Protein and Jun Activation Domain-Binding Protein 1 in Human Pituitary Tumors. J Clin Endocrinol Metab (2002) 87:2635–43. doi: 10.1210/jcem.87.6.8517
32. Quereda V, Malumbres M. Cell Cycle Control of Pituitary Development and Disease. J Mol Endocrinol (2009) 42:75–86. doi: 10.1677/JME-08-0146
33. Musat M, Morris DG, Korbonits M, Grossman AB. Cyclins and Their Related Proteins in Pituitary Tumourigenesis. Mol Cell Endocrinol (2010) 326:25–9. doi: 10.1016/j.mce.2010.03.017
34. Kulig E, Jin L, Qian X, Horvath E, Kovacs K, Stefaneanu L, et al. Apoptosis in Nontumorous and Neoplastic Human Pituitaries: Expression of the Bcl-2 Family of Proteins. Am J Pathol (1999) 154:767–74. doi: 10.1016/S0002-9440(10)65323-0
35. Ibrahim AE, Pickering RM, Gawne-Cain ML, King S, Lees PD, Ellison DW. Indices of Apoptosis and Proliferation as Potential Prognostic Markers in non-Functioning Pituitary Adenomas. Clin Neuropathol (2004) 23:8–15.
36. Kontogeorgos G. Predictive Markers of Pituitary Adenoma Behavior. Neuroendocrinology (2006) 83:179–88. doi: 10.1159/000095526
37. Guzzo MF, Carvalho LR, Bronstein MD. Apoptosis: Its Role in Pituitary Development and Neoplastic Pituitary Tissue. Pituitary (2014) 17:157–62. doi: 10.1007/s11102-013-0481-5
38. Kontogeorgos G, Sambaziotis D, Piaditis G, Karameris A. Apoptosis in Human Pituitary Adenomas: A Morphologic and in Situ End-Labeling Study. Mod Pathol (1997) 10:921–6.
39. Vidal S, Horvath E, Kovacs K, Scheithauer BW, Lloyd RV, Kontogeorgos G. Ultrastructural Features of Apoptosis in Human Pituitary Adenomas. Ultrastruct Pathol (2001) 25:85–92. doi: 10.1080/01913120121070
40. Kapranos N, Kontogeorgos G, Horvath E, Kovacs K. Morphology, Molecular Regulation and Significance of Apoptosis in Pituitary Adenomas. Front Horm Res (2004) 32:217–34. doi: 10.1159/000079047
41. Seilicovich A. Cell Life and Death in the Anterior Pituitary Gland: Role of Oestrogens. J Neuroendocrinol (2010) 22:758–64. doi: 10.1111/j.1365-2826.2010.02010.x
42. Ozer E, Canda MS, Ulukus C, Guray M, Erbayraktar S. Expression of Bcl-2, Bax and P53 Proteins in Pituitary Adenomas: An Immunohistochemical Study. Tumori (2003) 89:54–9. doi: 10.1177/030089160308900112
43. Sambaziotis D, Kapranos N, Kontogeorgos G. Correlation of Bcl-2 and Bax With Apoptosis in Human Pituitary Adenomas. Pituitary (2003) 6:127–33. doi: 10.1023/B:PITU.0000011173.04191.37
44. Fombonne J, Padron L, Enjalbert A, Krantic S, Torriglia A. A Novel Paraptosis Pathway Involving LEI/L-DNaseII for EGF-Induced Cell Death in Somato-Lactotrope Pituitary Cells. Apoptosis (2006) 11:367–75. doi: 10.1007/s10495-006-4568-6
45. Palmeri CM, Petiti JP, Sosa Ldel V, Gutierrez S, De Paul AL, Mukdsi JH, et al. Bromocriptine Induces Parapoptosis as the Main Type of Cell Death Responsible for Experimental Pituitary Tumor Shrinkage. Toxicol Appl Pharmacol (2009) 240:55–65. doi: 10.1016/j.taap.2009.07.002
46. Aoki MP, Aoki A, Maldonado CA. Sexual Dimorphism of Apoptosis in Lactotrophs Induced by Bromocryptine. Histochem Cell Biol (2001) 116:215–22. doi: 10.1007/s004180100307
47. Zhang SL, Tang HB, Hu JT, Zang ZL, Ding X, Li S, et al. PGAM5-CypD Pathway is Involved in Bromocriptine-Induced RIP3/MLKL-Dependent Necroptosis of Prolactinoma Cells. BioMed Pharmacother (2019) 111:638–48. doi: 10.1016/j.biopha.2018.12.128
48. Khamseh ME, Sheikhi A, Shahsavari Z, Ghorbani M, Akbari H, Imani M, et al. Evaluation of the Expression of Necroptosis Pathway Mediators and its Association With Tumor Characteristics in Functional and non-Functional Pituitary Adenomas. BMC Endocr Disord (2022) 22:1. doi: 10.1186/s12902-021-00919-y
49. Evan GI, Vousden KH. Proliferation, Cell Cycle and Apoptosis in Cancer. Nature (2001) 411:342–8. doi: 10.1038/35077213
50. Ameisen JC. On the Origin, Evolution, and Nature of Programmed Cell Death: A Timeline of Four Billion Years. Cell Death Differ (2002) 9:367–93. doi: 10.1038/sj.cdd.4400950
51. Green DR, Evan GI. A Matter of Life and Death. Cancer Cell (2002) 1:19–30. doi: 10.1016/S1535-6108(02)00024-7
52. Levy A, Lightman S. Molecular Defects in the Pathogenesis of Pituitary Tumours. Front Neuroendocrinol (2003) 24:94–127. doi: 10.1016/S0091-3022(03)00012-8
53. Lowe SW, Cepero E, Evan G. Intrinsic Tumour Suppression. Nature (2004) 432:307–15. doi: 10.1038/nature03098
54. Vogelstein B, Kinzler KW. Cancer Genes and the Pathways They Control. Nat Med (2004) 10:789–99. doi: 10.1038/nm1087
55. Di Ieva A, Rotondo F, Syro LV, Cusimano MD, Kovacs K. Aggressive Pituitary Adenomas–Diagnosis and Emerging Treatments. Nat Rev Endocrinol (2014) 10:423–35. doi: 10.1038/nrendo.2014.64
56. Wiman KG, Zhivotovsky B. Understanding Cell Cycle and Cell Death Regulation Provides Novel Weapons Against Human Diseases. J Intern Med (2017) 281:483–95. doi: 10.1111/joim.12609
57. Jarrett AM, Lima E, Hormuth DA 2nd, Mckenna MT, Feng X, Ekrut DA, et al. Mathematical Models of Tumour Cell Proliferation: A Review of the Literature. Expert Rev Anticancer Ther (2018) 18:1271–86. doi: 10.1080/14737140.2018.1527689
58. Voss AK, Strasser A. The Essentials of Developmental Apoptosis. F1000Res (2020) 9:148. doi: 10.12688/f1000research.21571.1
59. Loftus LV, Amend SR, Pienta KJ. Interplay Between Cell Death and Cell Proliferation Reveals New Strategies for Cancer Therapy. Int J Mol Sci (2022) 23:4723. doi: 10.3390/ijms23094723
60. Maddika S, Ande SR, Panigrahi S, Paranjothy T, Weglarczyk K, Zuse A, et al. Cell Survival, Cell Death and Cell Cycle Pathways are Interconnected: Implications for Cancer Therapy. Drug Resist Update (2007) 10:13–29. doi: 10.1016/j.drup.2007.01.003
61. Chaabane W, User SD, El-Gazzah M, Jaksik R, Sajjadi E, Rzeszowska-Wolny J, et al. Autophagy, Apoptosis, Mitoptosis and Necrosis: Interdependence Between Those Pathways and Effects on Cancer. Arch Immunol Ther Exp (Warsz) (2013) 61:43–58. doi: 10.1007/s00005-012-0205-y
62. Aktipis CA, Boddy AM, Jansen G, Hibner U, Hochberg ME, Maley CC, et al. Cancer Across the Tree of Life: Cooperation and Cheating in Multicellularity. Philos Trans R Soc Lond B Biol Sci (2015) 370:1673. doi: 10.1098/rstb.2014.0219
63. Gewirtz DA. Autophagy and Senescence: A Partnership in Search of Definition. Autophagy (2013) 9:808–12. doi: 10.4161/auto.23922
64. Childs BG, Baker DJ, Kirkland JL, Campisi J, Van Deursen JM. Senescence and Apoptosis: Dueling or Complementary Cell Fates? EMBO Rep (2014) 15:1139–53. doi: 10.15252/embr.201439245
65. Kenny PA, Nelson CM, Bissell MJ. The Ecology of Tumors: By Perturbing the Microenvironment, Wounds and Infection may be Key to Tumor Development. Scientist (2006) 20:30.
66. Strand DW, Franco OE, Basanta D, Anderson AR, Hayward SW. Perspectives on Tissue Interactions in Development and Disease. Curr Mol Med (2010) 10:95–112. doi: 10.2174/156652410791065363
67. Sapochnik M, Nieto LE, Fuertes M, Arzt E. Molecular Mechanisms Underlying Pituitary Pathogenesis. Biochem Genet (2016) 54:107–19. doi: 10.1007/s10528-015-9709-6
68. Zhan X, Desiderio DM. A Reference Map of a Human Pituitary Adenoma Proteome. Proteomics (2003) 3:699–713. doi: 10.1002/pmic.200300408
69. Zhan X, Desiderio DM. Signaling Pathway Networks Mined From Human Pituitary Adenoma Proteomics Data. BMC Med Genomics (2010) 3:13. doi: 10.1186/1755-8794-3-13
70. Zhan X, Wang X, Long Y, Desiderio DM. Heterogeneity Analysis of the Proteomes in Clinically Nonfunctional Pituitary Adenomas. BMC Med Genomics (2014) 7:69. doi: 10.1186/s12920-014-0069-6
71. Zhan X, Wang X, Cheng T. Human Pituitary Adenoma Proteomics: New Progresses and Perspectives. Front Endocrinol (Lausanne) (2016) 7:54. doi: 10.3389/fendo.2016.00054
72. Long Y, Lu M, Cheng T, Zhan X, Zhan X. Multiomics-Based Signaling Pathway Network Alterations in Human Non-Functional Pituitary Adenomas. Front Endocrinol (Lausanne) (2019) 10:835. doi: 10.3389/fendo.2019.00835
73. Hauser BM, Lau A, Gupta S, Bi WL, Dunn IF. The Epigenomics of Pituitary Adenoma. Front Endocrinol (Lausanne) (2019) 10:290. doi: 10.3389/fendo.2019.00290
74. Shariq AO, Lines KE. Epigenetic Dysregulation in Pituitary Tumors. Int J Endocrine Oncol (2019) 6:3. doi: 10.2217/ije-2019-0006
75. Chang M, Yang C, Bao X, Wang R. Genetic and Epigenetic Causes of Pituitary Adenomas. Front Endocrinol (Lausanne) (2020) 11:596554. doi: 10.3389/fendo.2020.596554
76. Srirangam Nadhamuni V, Korbonits M. Novel Insights Into Pituitary Tumorigenesis: Genetic and Epigenetic Mechanisms. Endocrine Rev (2020) 41:17. doi: 10.1210/endrev/bnaa006
77. Yacqub-Usman K, Duong CV, Clayton RN, Farrell WE. Epigenomic Silencing of the BMP-4 Gene in Pituitary Adenomas: A Potential Target for Epidrug-Induced Re-Expression. Endocrinology (2012) 153:3603–12. doi: 10.1210/en.2012-1231
78. Yacqub-Usman K, Richardson A, Duong CV, Clayton RN, Farrell WE. The Pituitary Tumour Epigenome: Aberrations and Prospects for Targeted Therapy. Nat Rev Endocrinol (2012) 8:486–94. doi: 10.1038/nrendo.2012.54
79. Ezzat S, Cheng S, Asa SL. Epigenetics of Pituitary Tumors: Pathogenetic and Therapeutic Implications. Mol Cell Endocrinol (2018) 469:70–6. doi: 10.1016/j.mce.2017.07.011
80. Zhu X, Mao X, Hurren R, Schimmer AD, Ezzat S, Asa SL. Deoxyribonucleic Acid Methyltransferase 3B Promotes Epigenetic Silencing Through Histone 3 Chromatin Modifications in Pituitary Cells. J Clin Endocrinol Metab (2008) 93:3610–7. doi: 10.1210/jc.2008-0578
81. Ma HS, Wang EL, Xu WF, Yamada S, Yoshimoto K, Qian ZR, et al. Overexpression of DNA (Cytosine-5)-Methyltransferase 1 (DNMT1) And DNA (Cytosine-5)-Methyltransferase 3a (DNMT3A) Is Associated With Aggressive Behavior and Hypermethylation of Tumor Suppressor Genes in Human Pituitary Adenomas. Med Sci Monit (2018) 24:4841–50. doi: 10.12659/MSM.910608
82. Tateno T, Zhu X, Asa SL, Ezzat S. Chromatin Remodeling and Histone Modifications in Pituitary Tumors. Mol Cell Endocrinol (2010) 326:66–70. doi: 10.1016/j.mce.2009.12.028
83. Ebrahimi A, Schittenhelm J, Honegger J, Schluesener HJ. Histone Acetylation Patterns of Typical and Atypical Pituitary Adenomas Indicate Epigenetic Shift of These Tumours. J Neuroendocrinol (2011) 23:525–30. doi: 10.1111/j.1365-2826.2011.02129.x
84. Wang W, Fu L, Li S, Xu Z, Li X. Histone Deacetylase 11 Suppresses P53 Expression in Pituitary Tumor Cells. Cell Biol Int (2017) 41:1290–5. doi: 10.1002/cbin.10834
85. Grande IPP, Amorim P, Freire A, Jallad RS, Musolino NR, Cescato VA, et al. Differential Gene Expression of Sirtuins Between Somatotropinomas and Nonfunctioning Pituitary Adenomas. Pituitary (2018) 21:355–61. doi: 10.1007/s11102-018-0881-7
86. Li T, Huang H, Huang B, Huang B, Lu J. Histone Acetyltransferase P300 Regulates the Expression of Human Pituitary Tumor Transforming Gene (hPTTG). J Genet Genomics (2009) 36:335–42. doi: 10.1016/S1673-8527(08)60122-8
87. Deng S, Calin GA, Croce CM, Coukos G, Zhang L. Mechanisms of microRNA Deregulation in Human Cancer. Cell Cycle (2008) 7:2643–6. doi: 10.4161/cc.7.17.6597
88. Croce CM. Causes and Consequences of microRNA Dysregulation in Cancer. Nat Rev Genet (2009) 10:704–14. doi: 10.1038/nrg2634
89. Bottoni A, Piccin D, Tagliati F, Luchin A, Zatelli MC, Degli Uberti EC. miR-15a and miR-16-1 Down-Regulation in Pituitary Adenomas. J Cell Physiol (2005) 204:280–5. doi: 10.1002/jcp.20282
90. Bottoni A, Zatelli MC, Ferracin M, Tagliati F, Piccin D, Vignali C, et al. Identification of Differentially Expressed microRNAs by Microarray: A Possible Role for microRNA Genes in Pituitary Adenomas. J Cell Physiol (2007) 210:370–7. doi: 10.1002/jcp.20832
91. Yuen T, Ruf F, Chu T, Sealfon SC. Microtranscriptome Regulation by Gonadotropin-Releasing Hormone. Mol Cell Endocrinol (2009) 302:12–7. doi: 10.1016/j.mce.2008.12.013
92. Mao ZG, He DS, Zhou J, Yao B, Xiao WW, Chen CH, et al. Differential Expression of microRNAs in GH-Secreting Pituitary Adenomas. Diagn Pathol (2010) 5:79. doi: 10.1186/1746-1596-5-79
93. Zhang Z, Florez S, Gutierrez-Hartmann A, Martin JF, Amendt BA. MicroRNAs Regulate Pituitary Development, and microRNA 26b Specifically Targets Lymphoid Enhancer Factor 1 (Lef-1), Which Modulates Pituitary Transcription Factor 1 (Pit-1) Expression. J Biol Chem (2010) 285:34718–28. doi: 10.1074/jbc.M110.126441
94. Sivapragasam M, Rotondo F, Lloyd RV, Scheithauer BW, Cusimano M, Syro LV, et al. MicroRNAs in the Human Pituitary. Endocr Pathol (2011) 22:134–43. doi: 10.1007/s12022-011-9167-6
95. D'angelo D, Palmieri D, Mussnich P, Roche M, Wierinckx A, Raverot G, et al. Altered microRNA Expression Profile in Human Pituitary GH Adenomas: Down-Regulation of miRNA Targeting HMGA1, HMGA2, and E2F1. J Clin Endocrinol Metab (2012) 97:E1128–1138. doi: 10.1210/jc.2011-3482
96. Palmieri D, D'angelo D, Valentino T, De Martino I, Ferraro A, Wierinckx A, et al. Downregulation of HMGA-Targeting microRNAs has a Critical Role in Human Pituitary Tumorigenesis. Oncogene (2012) 31:3857–65. doi: 10.1038/onc.2011.557
97. Wu S, Gu Y, Huang Y, Wong TC, Ding H, Liu T, et al. Novel Biomarkers for Non-Functioning Invasive Pituitary Adenomas Were Identified by Using Analysis of microRNAs Expression Profile. Biochem Genet (2017) 55:253–67. doi: 10.1007/s10528-017-9794-9
98. Yao Y, Dai W. Genomic Instability and Cancer. J Carcinog Mutagen (2014) 5:1000165. doi: 10.4172/2157-2518.1000165
99. Szymas J, Schluens K, Liebert W, Petersen I. Genomic Instability in Pituitary Adenomas. Pituitary (2002) 5:211–9. doi: 10.1023/a:1025313214951
100. Song ZJ, Reitman ZJ, Ma ZY, Chen JH, Zhang QL, Shou XF, et al. The Genome-Wide Mutational Landscape of Pituitary Adenomas. Cell Res (2016) 26:1255–9. doi: 10.1038/cr.2016.114
101. Bitgen N, Bayram F, Hamurcu Z, Ozturk F, Simsek Y, Baskol G, et al. Chromosomal and Oxidative DNA Damage in non-Functioning Pituitary Adenomas. Endokrynol Pol (2021) 72:97–103. doi: 10.5603/EP.a2020.0084
102. Boggild MD, Jenkinson S, Pistorello M, Boscaro M, Scanarini M, Mcternan P, et al. Molecular Genetic Studies of Sporadic Pituitary Tumors. J Clin Endocrinol Metab (1994) 78:387–92. doi: 10.1210/jc.78.2.387
103. Simpson DJ, Bicknell EJ, Buch HN, Cutty SJ, Clayton RN, Farrell WE. Genome-Wide Amplification and Allelotyping of Sporadic Pituitary Adenomas Identify Novel Regions of Genetic Loss. Genes Chromosomes Cancer (2003) 37:225–36. doi: 10.1002/gcc.10216
104. Ben-Shlomo A, Deng N, Ding E, Yamamoto M, Mamelak A, Chesnokova V, et al. DNA Damage and Growth Hormone Hypersecretion in Pituitary Somatotroph Adenomas. J Clin Invest (2020) 130:5738–55. doi: 10.1172/JCI138540
105. Shiloh Y. ATM and Related Protein Kinases: Safeguarding Genome Integrity. Nat Rev Cancer (2003) 3:155–68. doi: 10.1038/nrc1011
106. Hoeijmakers JH. Genome Maintenance Mechanisms are Critical for Preventing Cancer as Well as Other Aging-Associated Diseases. Mech Ageing Dev (2007) 128:460–2. doi: 10.1016/j.mad.2007.05.002
107. Bartkova J, Horejsi Z, Koed K, Kramer A, Tort F, Zieger K, et al. DNA Damage Response as a Candidate Anti-Cancer Barrier in Early Human Tumorigenesis. Nature (2005) 434:864–70. doi: 10.1038/nature03482
108. Bartek J, Bartkova J, Lukas J. DNA Damage Signalling Guards Against Activated Oncogenes and Tumour Progression. Oncogene (2007) 26:7773–9. doi: 10.1038/sj.onc.1210881
109. Bengtsson D, Joost P, Aravidis C, Askmalm Stenmark M, Backman AS, Melin B, et al. Corticotroph Pituitary Carcinoma in a Patient With Lynch Syndrome (LS) and Pituitary Tumors in a Nationwide LS Cohort. J Clin Endocrinol Metab (2017) 102:3928–32. doi: 10.1210/jc.2017-01401
110. Uraki S, Ariyasu H, Doi A, Furuta H, Nishi M, Sugano K, et al. Atypical Pituitary Adenoma With MEN1 Somatic Mutation Associated With Abnormalities of DNA Mismatch Repair Genes; MLH1 Germline Mutation and MSH6 Somatic Mutation. Endocr J (2017) 64:895–906. doi: 10.1507/endocrj.EJ17-0036
111. Salomon MP, Wang X, Marzese DM, Hsu SC, Nelson N, Zhang X, et al. The Epigenomic Landscape of Pituitary Adenomas Reveals Specific Alterations and Differentiates Among Acromegaly, Cushing's Disease and Endocrine-Inactive Subtypes. Clin Cancer Res (2018) 24:4126–36. doi: 10.1158/1078-0432.CCR-17-2206
112. Chesnokova V, Zonis S, Kovacs K, Ben-Shlomo A, Wawrowsky K, Bannykh S, et al. P21(Cip1) Restrains Pituitary Tumor Growth. Proc Natl Acad Sci U S A (2008) 105:17498–503. doi: 10.1073/pnas.0804810105
113. Sabatino ME, Petiti JP, Sosa Ldel V, Perez PA, Gutierrez S, Leimgruber C, et al. Evidence of Cellular Senescence During the Development of Estrogen-Induced Pituitary Tumors. Endocr Relat Cancer (2015) 22:299–317. doi: 10.1530/ERC-14-0333
114. Chesnokova V, Zonis S, Rubinek T, Yu R, Ben-Shlomo A, Kovacs K, et al. Senescence Mediates Pituitary Hypoplasia and Restrains Pituitary Tumor Growth. Cancer Res (2007) 67:10564–72. doi: 10.1158/0008-5472.CAN-07-0974
116. Magalhaes J, Venditti P, Adhihetty PJ, Ramsey JJ, Ascensao A. Mitochondria in Health and Disease. Oxid Med Cell Longev (2014) 2014:814042. doi: 10.1155/2014/814042
117. Tanase CP, Neagu M, Albulescu R. Key Signaling Molecules in Pituitary Tumors. Expert Rev Mol Diagn (2009) 9:859–77. doi: 10.1586/erm.09.60
118. Li N, Zhan X. Mitochondrial Dysfunction Pathway Networks and Mitochondrial Dynamics in the Pathogenesis of Pituitary Adenomas. Front Endocrinol (Lausanne) (2019) 10:690. doi: 10.3389/fendo.2019.00690
119. Lu M, Wang Y, Zhan X. The MAPK Pathway-Based Drug Therapeutic Targets in Pituitary Adenomas. Front Endocrinol (Lausanne) (2019) 10:330. doi: 10.3389/fendo.2019.00330
120. Zhan X, Li J, Zhou T. Targeting Nrf2-Mediated Oxidative Stress Response Signaling Pathways as New Therapeutic Strategy for Pituitary Adenomas. Front Pharmacol (2021) 12:565748. doi: 10.3389/fphar.2021.565748
121. Saeger W, Rubenach-Gerz K, Caselitz J, Ludecke DK. Electron Microscopical Morphometry of GH Producing Pituitary Adenomas in Comparison With Normal GH Cells. Virchows Arch A Pathol Anat Histopathol (1987) 411:467–72. doi: 10.1007/BF00735228
122. Yamada S, Asa SL, Kovacs K. Oncocytomas and Null Cell Adenomas of the Human Pituitary: Morphometric and In Vitro Functional Comparison. Virchows Arch A Pathol Anat Histopathol (1988) 413:333–9. doi: 10.1007/BF00783026
123. Fujisawa H, Tohma Y, Muramatsu N, Kida S, Kaizaki Y, Tamamura H. Spindle Cell Oncocytoma of the Adenohypophysis With Marked Hypervascularity. Case Report. Neurol Med Chir (Tokyo) (2012) 52:594–8. doi: 10.2176/nmc.52.594
124. Sabatino ME, Grondona E, Sosa LDV, Mongi Bragato B, Carreno L, Juarez V, et al. Oxidative Stress and Mitochondrial Adaptive Shift During Pituitary Tumoral Growth. Free Radic Biol Med (2018) 120:41–55. doi: 10.1016/j.freeradbiomed.2018.03.019
125. An J, Zhang Y, He J, Zang Z, Zhou Z, Pei X, et al. Lactate Dehydrogenase A Promotes the Invasion and Proliferation of Pituitary Adenoma. Sci Rep (2017) 774734. doi: 10.1038/s41598-017-04366-5
126. Pawlikowski M, Winczyk K, Jaranowska M. Immunohistochemical Demonstration of Nitric Oxide Synthase (NOS) in the Normal Rat Pituitary Gland, Estrogen-Induced Rat Pituitary Tumor and Human Pituitary Adenomas. Folia Histochem Cytobiol (2003) 41:87–90.
127. Onishi K, Kamida T, Momii Y, Abe T, Fujiki M. The Clinical and Pathological Significance of Nitric Oxide Synthase in Human Pituitary Adenomas: A Comparison With MIB-1. Endocrine (2014) 46:154–9. doi: 10.1007/s12020-013-0046-4
128. Wang D, Wong HK, Feng YB, Zhang ZJ. 18beta-Glycyrrhetinic Acid Induces Apoptosis in Pituitary Adenoma Cells via ROS/MAPKs-Mediated Pathway. J Neurooncol (2014) 116:221–30. doi: 10.1007/s11060-013-1292-2
129. Huang D, Cui L, Guo P, Xue X, Wu Q, Hussain HI, et al. Nitric Oxide Mediates Apoptosis and Mitochondrial Dysfunction and Plays a Role in Growth Hormone Deficiency by Nivalenol in GH3 Cells. Sci Rep (2017) 7:17079. doi: 10.1038/s41598-017-16908-y
130. Babula D, Horecka A, Luchowska-Kocot D, Kocot J, Kurzepa J. Decreased Nitric Oxide Serum Level After Pituitary Adenoma Resection. J Neurosurg Sci (2020) 64:452–5. doi: 10.23736/S0390-5616.17.04083-8
131. Deyu H, Luqing C, Xianglian L, Pu G, Qirong L, Xu W, et al. Protective Mechanisms Involving Enhanced Mitochondrial Functions and Mitophagy Against T-2 Toxin-Induced Toxicities in GH3 Cells. Toxicol Lett (2018) 295:41–53. doi: 10.1016/j.toxlet.2018.05.041
132. Tanase C, Albulescu R, Codrici E, Calenic B, Popescu ID, Mihai S, et al. Decreased Expression of APAF-1 and Increased Expression of Cathepsin B in Invasive Pituitary Adenoma. Onco Targets Ther (2015) 8:81–90. doi: 10.2147/OTT.S70886
133. Fatima Z, Guo P, Huang D, Lu Q, Wu Q, Dai M, et al. The Critical Role of P16/Rb Pathway in the Inhibition of GH3 Cell Cycle Induced by T-2 Toxin. Toxicology (2018) 400-401:28–39. doi: 10.1016/j.tox.2018.03.006
134. Wei Y, Zhou X, Ren L, Wang C, Li Y. The Prolactin-Release Inhibitor Paeoniflorin Suppresses Proliferation and Induces Apoptosis in Prolactinoma Cells via the Mitochondria-Dependent Pathway. J Cell Biochem (2018) 119:5704–14. doi: 10.1002/jcb.26752
135. Campisi J. Suppressing Cancer: The Importance of Being Senescent. Science (2005) 309:886–7. doi: 10.1126/science.1116801
136. Campisi J, D'adda Di Fagagna F. Cellular Senescence: When Bad Things Happen to Good Cells. Nat Rev Mol Cell Biol (2007) 8:729–40. doi: 10.1038/nrm2233
137. Campisi J. Cellular Senescence as a Tumor-Suppressor Mechanism. Trends Cell Biol (2001) 11:S27–31. doi: 10.1016/S0962-8924(01)82148-6
138. Arzt E, Chesnokova V, Stalla GK, Melmed S. Pituitary Adenoma Growth: A Model for Cellular Senescence and Cytokine Action. Cell Cycle (2009) 8:677–8. doi: 10.4161/cc.8.5.8065
139. Chesnokova V, Zonis S, Zhou C, Ben-Shlomo A, Wawrowsky K, Toledano Y, et al. Lineage-Specific Restraint of Pituitary Gonadotroph Cell Adenoma Growth. PLoS One (2011) 6:e17924. doi: 10.1371/journal.pone.0017924
140. Alexandraki KI, Munayem Khan M, Chahal HS, Dalantaeva NS, Trivellin G, Berney DM, et al. Oncogene-Induced Senescence in Pituitary Adenomas and Carcinomas. Hormones (Athens) (2012) 11:297–307. doi: 10.14310/horm.2002.1358
141. Manojlovic Gacic E, Skender-Gazibara M, Soldatovic I, Dundjerovic D, Boricic N, Raicevic S, et al. Immunohistochemical Expression of P16 and P21 in Pituitary Tissue Adjacent to Pituitary Adenoma Versus Pituitary Tissue Obtained at Autopsy: Is There a Difference? Endocr Pathol (2015) 26:104–10. doi: 10.1007/s12022-015-9358-7
142. Lazzerini Denchi E, Attwooll C, Pasini D, Helin K. Deregulated E2F Activity Induces Hyperplasia and Senescence-Like Features in the Mouse Pituitary Gland. Mol Cell Biol (2005) 25:2660–72. doi: 10.1128/MCB.25.7.2660-2672.2005
143. Zhang T, Zhao B, Li J, Zhang C, Li H, Wu J, et al. Pituitary Gene Expression Differs in D-Galactose-Induced Cell Senescence and Steroid-Induced Prolactinomas. Mol Med Rep (2015) 11:3027–32. doi: 10.3892/mmr.2014.3062
144. Sapochnik M, Haedo MR, Fuertes M, Ajler P, Carrizo G, Cervio A, et al. Autocrine IL-6 Mediates Pituitary Tumor Senescence. Oncotarget (2017) 8:4690–702. doi: 10.18632/oncotarget.13577
145. Collado M, Serrano M. Senescence in Tumours: Evidence From Mice and Humans. Nat Rev Cancer (2010) 10:51–7. doi: 10.1038/nrc2772
146. Chesnokova V, Melmed S. Pituitary Senescence: The Evolving Role of Pttg. Mol Cell Endocrinol (2010) 326(1–2):55–9. doi: 10.1016/j.mce.2010.02.012
147. Chesnokova V, Melmed S. Pituitary Tumour-Transforming Gene (PTTG) and Pituitary Senescence. Horm Res (2009) 71 Suppl 2:82–7. doi: 10.1159/000192443
148. Haedo MR, Gerez J, Fuertes M, Giacomini D, Paez-Pereda M, Labeur M, et al. Regulation of Pituitary Function by Cytokines. Horm Res (2009) 72:266–74. doi: 10.1159/000245928
149. Sapochnik M, Fuertes M, Arzt E. Programmed Cell Senescence: Role of IL-6 in the Pituitary. J Mol Endocrinol (2017) 58:R241–53. doi: 10.1530/JME-17-0026
150. Gonzalez-Meljem JM, Martinez-Barbera JP. Senescence Drives non-Cell Autonomous Tumorigenesis in the Pituitary Gland. Mol Cell Oncol (2018) 5:e1435180. doi: 10.1080/23723556.2018.1435180
151. Renner U, Gloddek J, Pereda MP, Arzt E, Stalla GK. Regulation and Role of Intrapituitary IL-6 Production by Folliculostellate Cells. Domest Anim Endocrinol (1998) 15:353–62. doi: 10.1016/S0739-7240(98)00027-7
152. Shintani T, Klionsky DJ. Autophagy in Health and Disease: A Double-Edged Sword. Science (2004) 306:990–5. doi: 10.1126/science.1099993
153. Mathew R, Karantza-Wadsworth V, White E. Role of Autophagy in Cancer. Nat Rev Cancer (2007) 7:961–7. doi: 10.1038/nrc2254
154. White E, Dipaola RS. The Double-Edged Sword of Autophagy Modulation in Cancer. Clin Cancer Res (2009) 15:5308–16. doi: 10.1158/1078-0432.CCR-07-5023
155. Mizushima N. Autophagy: Process and Function. Genes Dev (2007) 21:2861–73. doi: 10.1101/gad.1599207
156. Mizushima N, Levine B, Cuervo AM, Klionsky DJ. Autophagy Fights Disease Through Cellular Self-Digestion. Nature (2008) 451:1069–75. doi: 10.1038/nature06639
157. Kovacs K, Horvath E, Bayley TA, Hassaram ST, Ezrin C. Silent Corticotroph Cell Adenoma With Lysosomal Accumulation and Crinophagy. A Distinct Clinicopathologic Entity. Am J Med (1978) 64:492–9. doi: 10.1016/0002-9343(78)90236-X
158. Poole MC, Mahesh VB, Costoff A. Morphometric Analysis of the Autophagic and Crinophagic Lysosomal Systems in Mammotropes Throughout the Estrous Cycle of the Rat. Cell Tissue Res (1981) 220:131–7. doi: 10.1007/BF00209972
159. Kuriakose NR, Reifel CW, Bendayan M, Elce JS, Shin SH. Prolactin Crinophagy is Induced in the Estrogen-Stimulated Male Rat Pituitary. Histochemistry (1989) 92:499–503. doi: 10.1007/BF00524761
160. Wang C, Tan C, Wen Y, Zhang D, Li G, Chang L, et al. FOXP1-Induced lncRNA CLRN1-AS1 Acts as a Tumor Suppressor in Pituitary Prolactinoma by Repressing the Autophagy via Inactivating Wnt/beta-Catenin Signaling Pathway. Cell Death Dis (2019) 10:499. doi: 10.1038/s41419-019-1694-y
161. Wu Z, Gu W. Autophagy and Pituitary Adenoma. Adv Exp Med Biol (2020) 1207:183–94. doi: 10.1007/978-981-15-4272-5_12
162. Tulipano G, Giustina A. Autophagy in Normal Pituitary and Pituitary Tumor Cells and its Potential Role in the Actions of Somatostatin Receptor Ligands in Acromegaly. Rev Endocr Metab Disord (2021) 22:147–60. doi: 10.1007/s11154-021-09649-x
163. Lin SJ, Leng ZG, Guo YH, Cai L, Cai Y, Li N, et al. Suppression of mTOR Pathway and Induction of Autophagy-Dependent Cell Death by Cabergoline. Oncotarget (2015) 6:39329–41. doi: 10.18632/oncotarget.5744
164. Geng X, Ma L, Li Z, Li Z, Li J, Li M, et al. Bromocriptine Induces Autophagy-Dependent Cell Death in Pituitary Adenomas. World Neurosurg (2017) 100:407–16. doi: 10.1016/j.wneu.2017.01.052
165. Tang C, Sun R, Wen G, Zhong C, Yang J, Zhu J, et al. Bromocriptine and Cabergoline Induce Cell Death in Prolactinoma Cells via the ERK/EGR1 and AKT/mTOR Pathway Respectively. Cell Death Dis (2019) 10:335. doi: 10.1038/s41419-019-1526-0
166. Dagistanli FK, Ozkaya HM, Kucukyoruk B, Biceroglu H, Metin D, Gazioglu N, et al. Preoperative Somatostatin Analogue Treatment Might Trigger Apoptosis and Autophagy in Tumor Tissues of Patients With Acromegaly: A Pilot Study. Exp Clin Endocrinol Diabetes (2018) 126:168–75. doi: 10.1055/s-0042-107243
167. Weckman A, Rotondo F, Di Ieva A, Syro LV, Butz H, Cusimano MD, et al. Autophagy in Endocrine Tumors. Endocr Relat Cancer (2015) 22:R205–218. doi: 10.1530/ERC-15-0042
168. Chen J, Crabbe A, Van Duppen V, Vankelecom H. The Notch Signaling System is Present in the Postnatal Pituitary: Marked Expression and Regulatory Activity in the Newly Discovered Side Population. Mol Endocrinol (2006) 20:3293–307. doi: 10.1210/me.2006-0293
169. Fauquier T, Rizzoti K, Dattani M, Lovell-Badge R, Robinson IC. SOX2-Expressing Progenitor Cells Generate All of the Major Cell Types in the Adult Mouse Pituitary Gland. Proc Natl Acad Sci U S A (2008) 105:2907–12. doi: 10.1073/pnas.0707886105
170. Haston S, Manshaei S, Martinez-Barbera JP. Stem/progenitor Cells in Pituitary Organ Homeostasis and Tumourigenesis. J Endocrinol (2018) 236:R1–R13. doi: 10.1530/JOE-17-0258
171. Garcia-Lavandeira M, Diaz-Rodriguez E, Bahar D, Garcia-Rendueles AR, Rodrigues JS, Dieguez C, et al. Pituitary Cell Turnover: From Adult Stem Cell Recruitment Through Differentiation to Death. Neuroendocrinology (2015) 101:175–92. doi: 10.1159/000375502
172. Yoshida S, Kato T, Kato Y. Regulatory System for Stem/Progenitor Cell Niches in the Adult Rodent Pituitary. Int J Mol Sci (2016) 17:75. doi: 10.3390/ijms17010075
173. Vankelecom H, Roose H. The Stem Cell Connection of Pituitary Tumors. Front Endocrinol (Lausanne) (2017) 8:339. doi: 10.3389/fendo.2017.00339
174. Lepore DA, Roeszler K, Wagner J, Ross SA, Bauer K, Thomas PQ. Identification and Enrichment of Colony-Forming Cells From the Adult Murine Pituitary. Exp Cell Res (2005) 308:166–76. doi: 10.1016/j.yexcr.2005.04.023
175. Vankelecom H. Pituitary Stem/Progenitor Cells: Embryonic Players in the Adult Gland? Eur J Neurosci (2010) 32:2063–81. doi: 10.1111/j.1460-9568.2010.07523.x
176. Rizzoti K, Akiyama H, Lovell-Badge R. Mobilized Adult Pituitary Stem Cells Contribute to Endocrine Regeneration in Response to Physiological Demand. Cell Stem Cell (2013) 13:419–32. doi: 10.1016/j.stem.2013.07.006
177. Tan CL, Alavi SA, Baldeweg SE, Belli A, Carson A, Feeney C, et al. The Screening and Management of Pituitary Dysfunction Following Traumatic Brain Injury in Adults: British Neurotrauma Group Guidance. J Neurol Neurosurg Psychiatry (2017) 88:971–81. doi: 10.1136/jnnp-2016-315500
178. Peculis R, Mandrika I, Petrovska R, Dortane R, Megnis K, Nazarovs J, et al. Pituispheres Contain Genetic Variants Characteristic to Pituitary Adenoma Tumor Tissue. Front Endocrinol (Lausanne) (2020) 11:313. doi: 10.3389/fendo.2020.00313
179. Wurth R, Thellung S, Corsaro A, Barbieri F, Florio T. Experimental Evidence and Clinical Implications of Pituitary Adenoma Stem Cells. Front Endocrinol (Lausanne) (2020) 11:54. doi: 10.3389/fendo.2020.00054
180. Perez Millan MI, Brinkmeier ML, Mortensen AH, Camper SA. PROP1 Triggers Epithelial-Mesenchymal Transition-Like Process in Pituitary Stem Cells. Elife (2016) 5:e14470. doi: 10.7554/eLife.14470
181. Vaca AM, Guido CB, Sosa Ldel V, Nicola JP, Mukdsi J, Petiti JP, et al. The Expansion of Adult Stem/Progenitor Cells and Their Marker Expression Fluctuations are Linked With Pituitary Plastic Adaptation During Gestation and Lactancy. Am J Physiol Endocrinol Metab (2016) 311:E367–79. doi: 10.1152/ajpendo.00077.2016
182. Carreno G, Gonzalez-Meljem JM, Haston S, Martinez-Barbera JP. Stem Cells and Their Role in Pituitary Tumorigenesis. Mol Cell Endocrinol (2017) 445:27–34. doi: 10.1016/j.mce.2016.10.005
183. Caffarini M, Orciani M, Trementino L, Di Primio R, Arnaldi G. Pituitary Adenomas, Stem Cells, and Cancer Stem Cells: What's New? J Endocrinol Invest (2018) 41:745–53. doi: 10.1007/s40618-017-0803-y
184. Guido CB, Sosa LDV, Perez PA, Zlocoswki N, Velazquez FN, Gutierrez S, et al. Changes of Stem Cell Niche During Experimental Pituitary Tumor Development. J Neuroendocrinol (2021) 33:e13051. doi: 10.1111/jne.13051
185. Fu Q, Vankelecom H. Regenerative Capacity of the Adult Pituitary: Multiple Mechanisms of Lactotrope Restoration After Transgenic Ablation. Stem Cells Dev (2012) 21:3245–57. doi: 10.1089/scd.2012.0290
186. Lloyd RV, Hardin H, Montemayor-Garcia C, Rotondo F, Syro LV, Horvath E, et al. Stem Cells and Cancer Stem-Like Cells in Endocrine Tissues. Endocr Pathol (2013) 24:1–10. doi: 10.1007/s12022-013-9235-1
187. Melmed S. Mechanisms for Pituitary Tumorigenesis: The Plastic Pituitary. J Clin Invest (2003) 112:1603–18. doi: 10.1172/JCI20401
188. Cox B, Laporte E, Vennekens A, Kobayashi H, Nys C, Van Zundert I, et al. Organoids From Pituitary as a Novel Research Model Toward Pituitary Stem Cell Exploration. J Endocrinol (2019) 240:287–308. doi: 10.1530/JOE-18-0462
Keywords: pituitary gland, tumour growth, tumour growth suppression, cellular fates, cellular physiology
Citation: Sabatino ME, Grondona E and De Paul AL (2022) Architects of Pituitary Tumour Growth. Front. Endocrinol. 13:924942. doi: 10.3389/fendo.2022.924942
Received: 20 April 2022; Accepted: 27 May 2022;
Published: 28 June 2022.
Edited by:
Haim Werner, Tel Aviv University, IsraelReviewed by:
Rati Chkheidze, University of Alabama at Birmingham, United StatesCopyright © 2022 Sabatino, Grondona and De Paul. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Ana Lucía De Paul, YWRlcGF1bEBjbWVmY20udW5jb3IuZWR1