

94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Endocrinol. , 27 June 2022
Sec. Diabetes: Molecular Mechanisms
Volume 13 - 2022 | https://doi.org/10.3389/fendo.2022.922825
This article is part of the Research Topic The Pancreatic Islet – a Multifaceted Hub of Inter-Cellular Communication View all 9 articles
 Marta Pablos1*
Marta Pablos1* Elena Casanueva-Álvarez2
Elena Casanueva-Álvarez2 Carlos M. González-Casimiro2
Carlos M. González-Casimiro2 Beatriz Merino2
Beatriz Merino2 Germán Perdomo2
Germán Perdomo2 Irene Cózar-Castellano1,2,3
Irene Cózar-Castellano1,2,3The primary cilium is a narrow organelle located at the surface of the cell in contact with the extracellular environment. Once underappreciated, now is thought to efficiently sense external environmental cues and mediate cell-to-cell communication, because many receptors, ion channels, and signaling molecules are highly or differentially expressed in primary cilium. Rare genetic disorders that affect cilia integrity and function, such as Bardet-Biedl syndrome and Alström syndrome, have awoken interest in studying the biology of cilium. In this review, we discuss recent evidence suggesting emerging roles of primary cilium and cilia-mediated signaling pathways in the regulation of pancreatic β- and α-cell functions, and its implications in regulating glucose homeostasis.
In 1676, Anton Van Leeuwenhoek was the first to discover the cilium and attribute it a motile function (1). However, the cilium was redefined as a critical organelle for development, homeostasis, regenerative processes, and regulation of signaling pathways in health and disease (2–4). Of note, studies in mammals have shown the relevance of cilia dysfunction in several pathologies. In humans, there are pathologies ranging from organ-specific disorders such as primary ciliary dyskinesia, hydrocephalus, polycystic liver and kidney disease, and retinal degeneration, to broad pleiotropic phenotypes such as Bardet-Biedl, Alström, and Meckel-Gruber syndromes (for a comprehensive review see ref. 5). Likewise, defects in cilia are associated with pathologies in rodents such as left-right asymmetry during mammals´ development (5), and kidney polycystic pathology (6). Consequently, the term “ciliopathy” refers to a spectrum of characteristic phenotypes with defects in ciliary structure and function (7).
The majority of differentiated cells present a single cilium at the apical surface, while some cells accumulate bundles of cilia consisting of 200-300 individual organelles (8). Structurally, the cilium consists of a microtubule backbone (axoneme) ensheathed by a ciliary membrane that is continuous with the plasma membrane, which typically projects from the apical surface of cells (9). There are four main ciliary types referring to the axonemal organization of microtubules pairs: primary nonmotile or sensory cilium (9 + 0), motile (9 + 2), nodal (9 + 0), and non-motile cilium (9 + 2) (Figure 1A and Table 1). Nonetheless, the classic distinction between sensory and motile cilia seems to be very simplistic because there are motile cilia with sensory roles (8, 16–19).
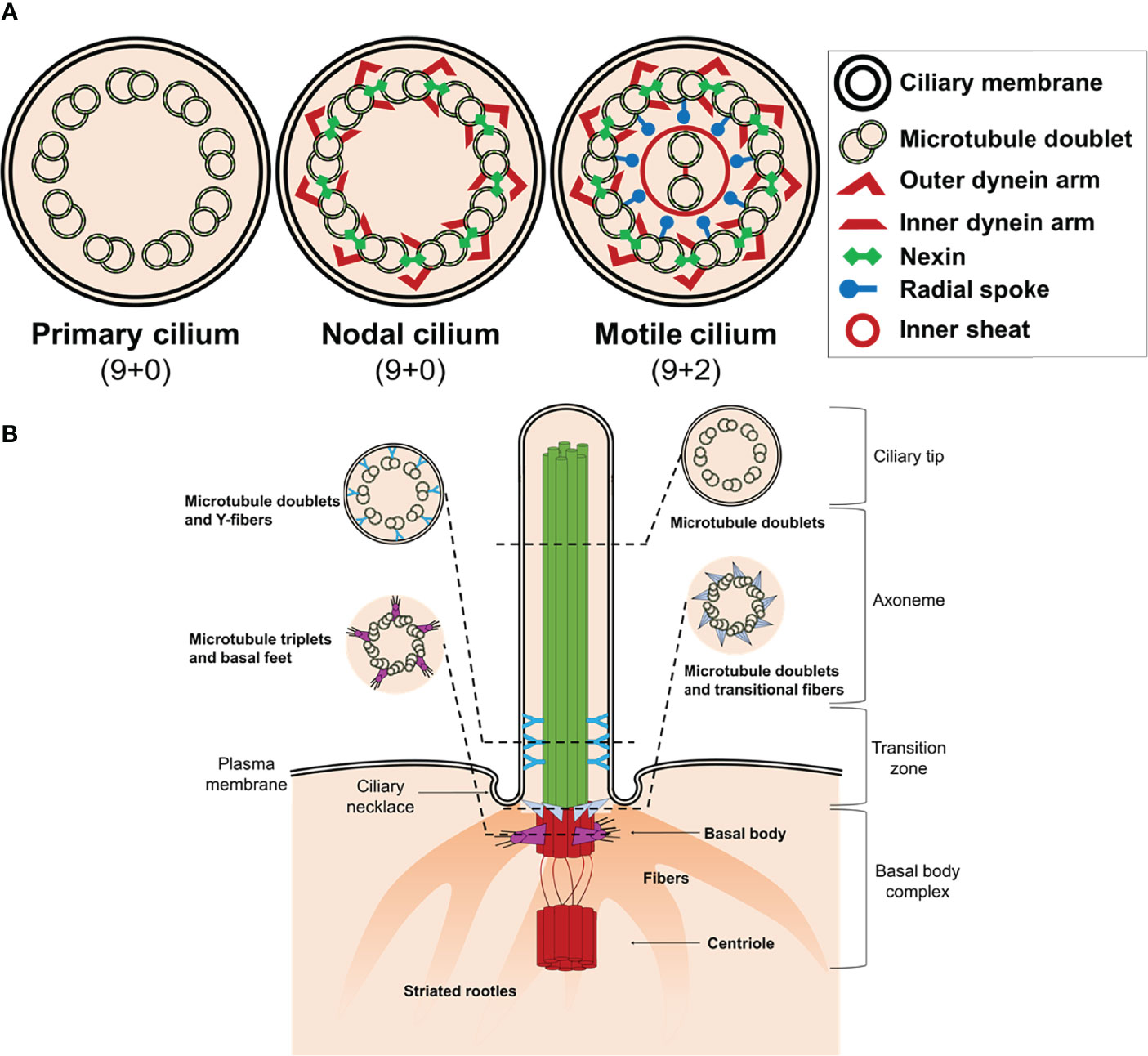
Figure 1 Structure of cilium. (A) Schematic representation of an axoneme cross section from a primary cilium, nodal cilium, and motile cilium. The axoneme cilium is composed of nine outer doublets of microtubules surrounding a central pair (9 + 2). The axoneme is ensheathed by a ciliary membrane. Inner and outer dynein arms, nexin, and radial spokes are responsible to link microtubules and form a cylindrical structure. (B) Schematic diagram of a typical non-motile primary cilium. The primary cilium is divided into the ciliary tip, the membrane bound axoneme extending from surface, the transition zone, and the basal body complex. The ciliary tip ends contain signaling molecules and can undergo morphological changes in response to signaling processes. The axoneme is the structural core of a cilium. The transition zone converts the triplet microtubular structure of the basal body into the axonemal doublet structure. The ciliary pocket (necklace) is an invagination of the plasma membrane at the root of cilium. The basal body complex comprises the basal body and its centriole. In most quiescent cells, the centrioles move to the apical plasma membrane and the basal body (mother centriole) functions as the microtubule-organizing centre to nucleate the axonemal microtubules. The centriole (daughter centriole) remains perpendicular to the basal body.
The basic structure of the primary cilium (9 + 0) is described elsewhere and it is out of the scope of this review (18, 20–22). Briefly, primary cilium consists of a ring of nine outer doublet microtubules (axoneme) which are devoid of dynein arms (Figure 1A). The framework of the doublet consists of a complete microtubule with 13 protofilaments (A-tubule) and an incomplete microtubule (B-tubule) with 10 protofilaments (21). The cilium also contains “matrix” proteins that are not tightly associated with either the membrane or the axoneme, including proteins required for assembly and maintenance of the organelle (23). The primary cilium structure can be considered an intricate network whose combination makes possible the integrity and function of the organelle. The literature describes almost 1,000 different proteins that exert some kind of function in the axoneme (24–26).
The cilium structure can be divided in ciliary and sub-ciliary compartments. The ciliary compartment consists in the axoneme and the ciliary membrane. The axoneme is a cylindrical structure of microtubules extended from the basal body, a specialized centriole structure, to the ciliary tip, which contains signaling molecules and can undergo morphological changes in response to signaling processes (Figure 1B). The ciliary membrane is continuous with the plasma membrane, but there are physiological differences between both. Thus, the ciliary membrane contains specific signaling molecules that are essential for the function of the cilium as antenna, including localization of ion channels and receptors at the base in an intra-membrane structure named ciliary necklace.
Microtubules in the axoneme are cylindrical polymers of α- and β-tubulin heterodimers. Post-translational modification of these tubulins (acetylation, methylation, tyrosination, phosphorylation, mono-glycylation, mono-glutamylation, poly-amination, poly-glycylation, and poly-glutamylation) may play a regulatory role on cilium mechanisms of action (27, 28). Most post-translational modifications occur on the external surface but, the Lys40 acetylation of α-tubulin (αK40) is located in the inner surface (or luminal side) of the microtubules (29, 30). Acetylation on αK40 most often stabilizes microtubules and it has been described to participate in the regulation of various signaling pathways by modulating the activity or the localization of plasma membrane proteins (28).
The structure and function of the sub-ciliary compartment is not fully understood. At the bottom of the cilium is found the basal body (commonly known as the mother centriole), a structure consisting of nine triplet microtubules arranged circumferentially (Figure 1B). The basal body maintains the transition zone between the axoneme and the cytoplasm of the cells and is attached to the membrane through the transitional fibers or alar sheets which go from the distal side of the triplets of the basal body to the cell membrane.
Another structure located to the sub-ciliary compartment is the striated rootlet, which is an array of periodically striated filamentous that radiate from the proximal end of the basal body to the cytoplasm of the cell (Figure 1B). The exact function of these rootlets is unknown, but it has been hypothesized to anchor the basal body/primary cilium complex with the cytoskeleton (31), participating as routes for the transport of proteins from the Golgi apparatus to the plasma membrane (32), and as structures that pull rapidly some primary cilia into the cell (33).
In addition, the protein turnover in primary cilium of pancreatic β-cells in mice is not uniform. Arrojo E Drigo and collaborators showed that the basal body contained high 15N levels, while the rest of the cilium was replaced by new components. These data suggest that long-lived structures are present in the basal body of β-cells leading to age mosaicism architecture within primary cilium (34).
The elongation of the cilium requires targeting of specific proteins from the cytoplasm to the basal body area where pre-assembly of axonemal structures occurs, as well as the selective transport of proteins at the ciliary base out to the ciliary tip by the intraflagellar transport (IFT) system (35). When IFT particles are transferred from base to tip is referred to anterograde transport; and vice versa (from tip to base) is named as retrograde transport (reviewed in ref. 30).
Two IFT complexes play complementary roles in the transport of ciliary proteins. IFT complex A is required for retrograde transport, but seems not to be necessary for ciliary assembly. On the other hand, IFT complex B participates in the anterograde transport and it is essential for the assembly and maintenance of cilia. To avoid collision, IFT-A use the A-tubule, and IFT-B move along the B-tubule (36–39).
Additionally, two heterodimerized Kinesin-2 motor proteins and an accessory subunit [kinesin-associated protein (KAP)] catalyze anterograde transport of IFT complexes along microtubules to the ciliary tip, whereas retrograde transport of cargo proteins from the ciliary tip to the cytoplasm is catalyzed by cytoplasmic dynein 2 motor complex (36, 37).
The existence of four different kind of cilia indicates that the diversity of this organelle is intimately linked to different cellular functions.
The motile function of 9 + 0 cilium at the embryonic node generate the nodal flow that is required for determine embryonic left-right asymmetry (5). On the other hand, the motile function of 9 + 2 cilium is required to move extracellular fluid. Thus, cilia of respiratory epithelial cells are responsible for mucociliary clearance (40). Likewise, the ependymal cilia facilitate ependymal flow (41), and the epithelial cilia in the female reproductive tract facilitate the movement of sperm to the site of fertilization (42).
There are non-motile functions such as those related with sensing environmental cues. In this case, cilia might act as antenna receiving signals due to the presence of receptors and ion channels in the ciliary membrane, which are transduced through intracellular signaling pathways. For example, monocilia from epithelial cells lining the mammalian kidney tubules have a mechanosensory role in sensing urine flow (43). Likewise, flow-sensing cilia of the periphery of the mammalian node seems to sense the leftward fluid flow generated by motile cilium within the node cavity (44). In addition, non-motile cilium are also important for the sensory apparatus of nose (45), eyes (46), and ears (47).
On the other hand, cilia and the cell cycle seems to be coordinately regulated in many cells. Thus, the presence of the cilium is associated with the establishment of polarity and differentiation of the cells (G0/G1 phase). Conversely, the ciliated cells undergo a resorption of its cilium just before the beginning of cell division, when the cell leaves G1 phase and entry in S phase (48).
Dissecting the interplay between primary cilium and the cell cycle is an emerging area of research. So far, it is not well understood how extracellular mitogens (including serum stimulation), and genetic or pharmacological inhibition of ciliary regulatory proteins, contribute to ciliary dynamics and control of cell cycle progression (49, 50).
Studies using different cell lines have shown that serum starvation synchronize cell cultures in the G0/G1 phase leading to ciliary assembly. In contrast, serum-supplemented medium triggers two waves (fist between 1-2 h and second between 18-24h) of ciliary disassembly (51). Serum-mediated activation of Aurora A kinase (AURKA) induces ciliary disassembling by activating histone deacetylase 6 (HDAC6), which in turn regulates deacetylation of α-tubulin and cortactin, and ubiquitin-binding activity-mediated regulation of autophagy (51, 52).
The phospholipid mitogen lysophosphatidic acid (LPA), which is present at high concentrations in serum, is the major serum factor driving ciliary disassembly (53, 54). LPA notably triggers ciliary disassembling and subsequent cell cycle re-entry in serum-starved cells, in fact LPA is as effective as serum at inducing ciliary disassembling (55, 56).
Cell cycle-associated ciliary disassembly seems to occur via resorption, through depolymerization of the axoneme and incorporating its constituents into the cell body. However, ciliary disassembly in response to stress or pharmacological induction is mediated by whole cilium shedding, a process in which the ciliary membrane and axoneme are excised near the base and released from the cell (57–59).
The primary cilium is formed from pre-existing centrioles, while multiciliated cells require new production of many centrioles (60). Once centrioles are formed in ciliary cells, they migrate to the cell surface, attach to the membrane and serve as basal bodies for ciliary elongation. During maturation, the centrioles acquire additional accessories such as transitional fibers and basal feet, which allow stabilization of the basal body/centriole (61, 62). The centriole was mostly known by its role in cell division, but its primary role maybe ciliogenesis, this idea is supported by the fact that cells can still divide without a centriole (48, 63).
Cilia are only assembled during the G0 (the period in which a cell remains in a quiescent and/or differentiated state). Conversely, entry into the cell cycle is preceded by ciliary resorption (48). Thus, the cilium has a dynamic structure, which is changing between growing and shrinking patterns and, is able to switch and quickly organize to other structures such as the mitotic spindle.
Ciliogenesis occurs via two different processes named extracellular and intracellular pathways (64–66). In the extracellular pathway, prior to axonemal growth, the mother centriole directly docks with the plasma membrane from where the ciliary shaft is formed and grows towards the extracellular environment (67, 68). In the intracellular pathway, the mother centriole docks and fuses with the plasma membrane of an intracellular primary ciliary vesicle, where the cilium is assembled within the ciliary vesicle. Then, the elongated intracellular ciliary vesicle docks and fuses with the plasma membrane and the ciliary shaft is released into the extracellular space (67, 68).
Membrane trafficking regulators, such as the small GTPases Rab and Arl family, regulate the intracellular pathway (69, 70). Other trafficking regulators, such as components of the exocyst and TRAPPI/II complexes and SNARE membrane fusion proteins also play a role in intracellular ciliogenesis (71–73).
Finally, as we previously discussed, IFT is required for assembly and maintenance of cilium. Therefore, IFT is another process that regulates ciliogenesis. However, scarce information about sub-cellular localization of the IFT components have limited our understanding of the role of IFT during ciliogenesis.
An important function of the primary cilium is the regulation of key signaling pathways such as Hedgehog, Wingless, and insulin-like growth factor 1 (IGF-1)/insulin.
The Hedgehog (Hh) signaling pathway allows communication between cells during development. Although the pathway was discovered in Drosophila melanogaster, core components are conserved across flies and mammalian, but mechanisms of signal transduction have diverged (74–76). The Hh signal transduction in vertebrates is completely dependent on the primary cilium, and alterations in cilium structure, changes in protein activity or function, and changes in the location of proteins can influence Hh signaling. Cells lacking cilium are unable to induce the pathway in response to exogenous Hh ligands (77, 78).
The functioning of the Hh pathway is based on the presence of the receptor Patched-1 (PTCH1) that is located in the surface of the ciliary membrane (79). In the absence of ligand, PTCH1 keeps the pathway off by inhibiting intracellular Smoothened (SMO; a Frizzled-Class-F G protein-coupled receptor). PTCH1 regulates the activity of SMO without direct interaction by changing the levels of cholesterol and/or cholesterol-derived molecules in the cell membrane (reviewed in refs. 74-76 (Figure 2A)). Without SMO activity, the suppressor of fused (SuFu) associates and represses the Glioma-associated oncogene (GLI; a zinc-finger transcription factor) at the ciliary tip, allowing that GLI protein undergoes proteasomal degradation leading to the formation of the transcriptional repressor (GLIR). The IFT machinery shuttles SuFu, GLI and GLIR proteins from the ciliary tip to the cell body and vice versa. GLIR enters the nucleus where represses the expression of genes under Hh control (Figure 2A) (80–82).
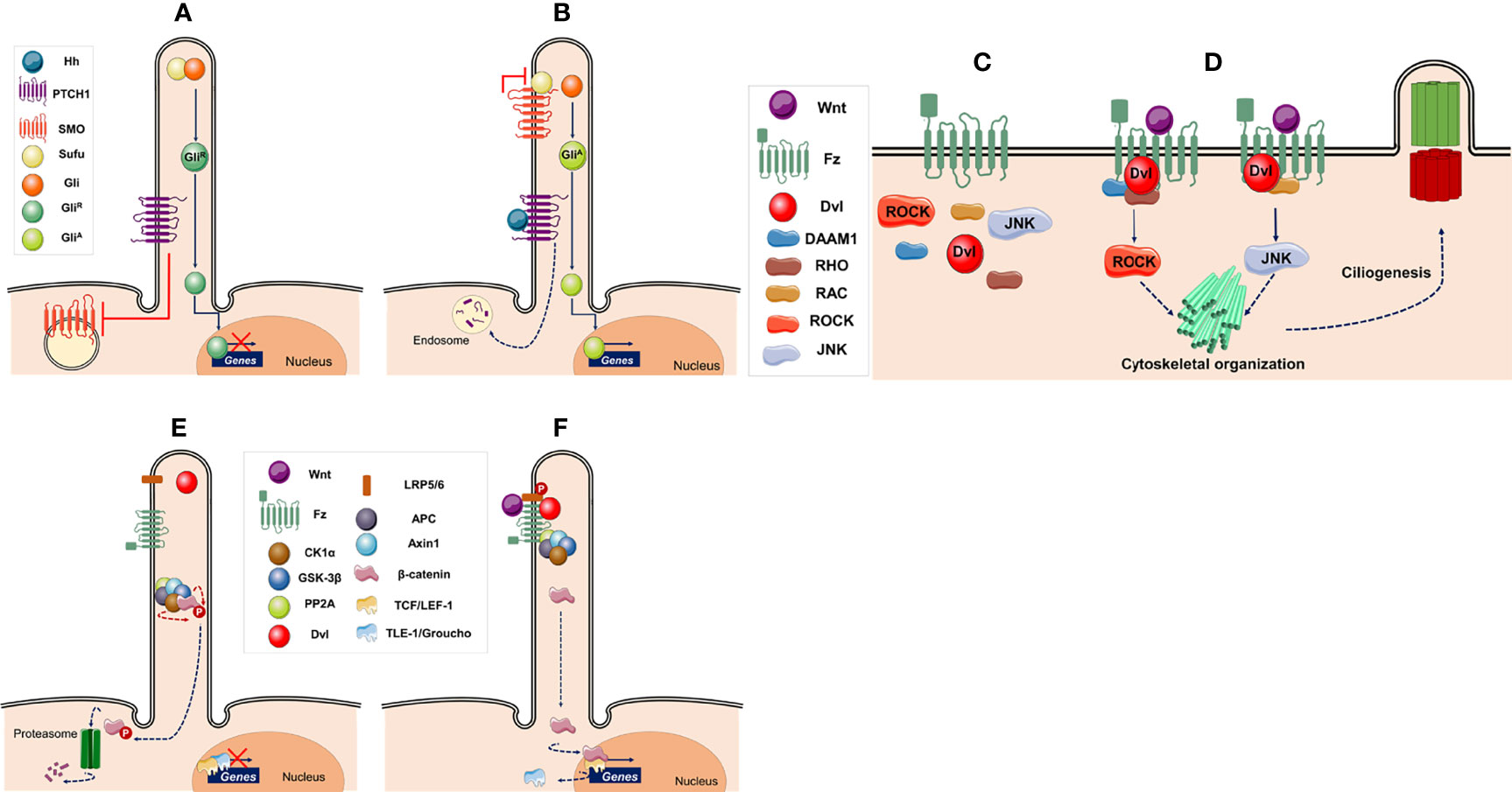
Figure 2 Ciliary signaling pathways. (A–B) Hedgehog signaling pathway (A) In the absence of hedgehog (Hh) ligand the intraflagellar transport machinery moves the transcription factor glioma-associated oncogene (GLI) and suppressor of fused (SuFu) to the ciliary tip. Patched-1 (PTCH1) located in the surface of the ciliary membrane inhibits Smoothened (SMO), which is located in the cytoplasm, keeping GLI in an inactive form (GLIR). The inactive transcription factor is transported back to the cell body and enters the nucleus where represses the expression of genes such as CCND1, N-MYC, GLI1, GLI2, and PATCH1. (B) Binding of ligand (Hh) to PTCH1 leads to the formation of oligomers, which are degraded in endosomes, reliving the repression of SMO, and causing its re-localization to the ciliary tip. SMO interacts with SuFu leading to the maturation of GLI into its active form (GLIA). GLIA is transported back to the cell body and enters the nucleus where activates transcription of target genes. (C, D) Canonical Wnt signaling pathway. (C) Without Wnt ligands, β-catenin is ubiquitinated and degraded by the proteasome. The initial events of this pathway are regulated by a destruction complex composed of casein kinase 1α (CK1α), glycogen synthase kinase 3β (GSK-3β), protein phosphatase 2A (PP2A), adenomatous polyposis coli (APC), and Axin 1. In the absence of β-catenin, the nuclear T cell-specific transcription factor/lymphoid enhancer factor-1 (TCF/LEF-1)-responsive elements are associated with transcriptional suppressors, such as Groucho (Gro) and Transducin-like enhancer of split-1 (TLE-1), keeping the pathway inactive. (D) When Wnt ligands bind the Frizzled (Fz) family receptors and its coreceptor low density lipoprotein receptor-related protein-5/6 (LRP5/6), Fz recruits Disheveled (Dvl) to inactivate the β-catenin destruction complex. Thus, β-catenin accumulates in the cytoplasm and translocates to the nucleus where replaces Gro/TLE, and acts as a transcriptional co-activator with TCF/LEF-1, inducing transcription of Wnt target genes. (E, F) Planar cell polarity signaling pathway. (E) In the absence of Wnt ligands the pathway is inactive. (F) When a signal is received by the Fz receptor a complex of proteins, including Dvl, is recruited at the plasma membrane. Dvl activates RHO-associated coiled-coil forming kinase (ROCK) and c-Jun N-terminal kinase (JUNK) in parallel, resulting in cytoskeletal organization and regulation of ciliogenesis. This figure was created using Servier Medical Art (available at https://smart.servier.com/).
By contrast, binding of ligand (Hh) to PTCH1 leads to the formation of oligomers, which are moved out of the cilium and degraded in endosomes (83). This alleviates SMO repression causing its re-localization to the cilium, where switches off GLI processing by interacting with SuFu, leading to the activation of GLI (GLIA) (80–82). GLIA is shuttle back to the nucleus allowing transcription of genes under Hh control. (Figure 2B) (79).
Early reports by Thomas and collaborators demonstrated the presence of Hh, PTCH and SMO in rat and mouse islets of Langerhans (84). In addition, PTCH co-expressed with insulin in β-cells lending support to the notion that Hh signaling is not restricted to early pancreas development but also to functions of differentiated β-cells. In fact, Hh gain-of-function (ectopic expression) increases insulin production at the transcriptional level in INS1 and MIN6 β-cell lines. Conversely, loss of Hh signaling (cyclopamine) decreased endogenous insulin mRNA expression leading to diminished insulin content and secretion in INS1 cells (84).
The relevance of Hh signaling in β-cell function has been shown by Lau and collaborators (85). Thus, pancreatic epithelium elimination of SMO function (Pdx1-Creearly;Smoflox/null mice) resulted in a transient delay in β-cell development leading to a temporary reduction in β-cell numbers that were recovered after birth. However, adult knockout mice exhibited a mild insulin-dependent diabetes associated with glucose intolerance, reduced insulin secretion, and increased insulin sensitivity (85).
On the other hand, pancreatic epithelium overexpression of an activated version of GLI2 (Pdx1-Cre;CLEG2 mice) failed to efficiently up-regulate the Hh pathway in the pancreas epithelium, suggesting the existence of a mechanism(s) that block inappropriate activation of the Hh pathway in epithelial cells (86). However, ablation of primary cilia in epithelial cells by specific deletion of Kif3a. resulted in strong activation of Hh signaling in mice harboring an activated version of GLI2 but not in SMO and PTCH1 mutant mice, indicating that primary cilia regulate Hh activity downstream of SMO in pancreas (86). Interestingly, the double transgenic mice (Pdx1-CreER;CLEG2;Kif3af/f) showed lower expression of mature β-cell transcription factors, such as Pdx1, MafA, Ngn3, NeuronD1, and Nkx6.1. In addition, Hh target genes that are normally excluded from β-cells increased its expression, such as the precursor markers Hes1 and Sox9. Augmented Hh signaling resulted in impaired β-cell function and insulin secretion leading to glucose intolerance in double transgenic mice. Over time, the majority of double transgenic β-cells regained their differentiation state by downregulating GLI2 levels. Sustained Hh activity in the remainder of double transgenic β-cells resulted in neoplastic transformation of insulin cells into insulin-negative pancreatic tumors (87). These studies underscore the relevance of Hh signaling for maintaining β-cell function and identity.
Hh signaling has also been implicated in protecting β-cells from cytokine-induced cytotoxicity. Thus, Umeda and collaborators showed that proinflammatory cytokines increased Hh expression in rat islets and INS1E cells. Interestingly, Hh overexpression reduced cytokine-mediated apoptosis by decreasing nuclear factor-κB (NF-κB) promoter, whereas cyclopamine-mediated loss of Hh signaling increased cytokine-mediated apoptosis (88).
Similarly, Yalcinkaya and collaborators demonstrated that Hh signaling is involved in the mechanism by which high-density lipoprotein (HDL) protects β-cells from thapsigargin-induced endoplasmic reticulum stress and apoptosis. They showed that HDL involves the generation and mobilization of specific oxysterols and subsequent activation of SMO to elicit GLIA nuclear translocation in INS1E cells (89).
The protective role of Hh signaling against cytokine- and –ER-induced apoptosis in β-cells await further confirmation in vivo, particularly in preclinical mouse models of type 2 diabetes (T2D), whereas both stimuli play important roles in the pathogenesis of the disease.
The Wingless-related integration site (Wnt) is an evolutionary conserved signaling pathway that regulates key features during development (90, 91).
Wnt signaling can be divided into two categories: canonical (or Wnt/β-catenin dependent) and non-canonical (or β-catenin-independent) (91, 92). The Wnt/β-catenin dependent pathway regulates stemness, cell differentiation and proliferation, whereas β-catenin-independent pathway regulates cytoskeleton, cell polarity, and cell movements (93, 94). Although for simplicity, Wnt signaling is often dichotomized in two arms, both pathways often overlap to organize complex cellular responses.
In the absence of Wnt ligands, β-catenin is ubiquitinated and degraded in the proteasome, keeping cytosolic β-catenin at low levels. Degradation of β-catenin is mediated by a “destruction complex” composed of casein kinase 1α (CK1α), glycogen synthase kinase 3β (GSK-3β), protein phosphatase 2A (PP2A), adenomatous polyposis coli (APC), and Axin 1. CK1α and GSK-3β sequentially phosphorylate the amino terminal region of β-catenin, resulting in β-catenin recognition by the F-box containing E3-ligase protein β-TrCP, an adaptor protein that forms a complex with the Skp1/Cullin machinery to attach ubiquitin to its binding partners, and subsequent ubiquitination and proteasomal degradation. In the absence of β-catenin, the nuclear T cell-specific transcription factor/lymphoid enhancer factor-1 (TCF/LEF-1)-responsive elements are associated with transcriptional suppressors, such as Groucho (Gro) and Transducin-like enhancer of split-1 (TLE-1), to keep the canonical Wnt pathway inactive (Figure 2C) (16, 95–98).
The outcome of canonical Wnt signaling is the expression of β-catenin target genes via β-catenin stabilization and subsequent nuclear translocation. Thus, extracellular Wnt binds to a complex of the Frizzled (Fz) receptor and the low-density lipoproteins receptor-related protein 5 (LRP5) or LRP6. Upon binding of Wnt, Fz receptor recruits the cytoplasmic phosphoprotein Disheveled (Dvl; A.K.A. Dsh) and GSK-3β is inactivated. In addition, Dvl recruitment by Fz leads to LRP5/6 phosphorylation, and Axin recruitment. These signaling events allow stabilization of β-catenin in the cytoplasm. Thus, β-catenin translocates to the nucleus, replaces Gro/TLE, and acts as a transcriptional co-activator with TCF/LEF-1, inducing transcription of Wnt target genes such as cMYC, AXIN2 or L1CAM (Figure 2D) (8, 99–101).
The non-canonical pathway refers to a group of Wnt-dependent signaling pathways that do not require LRP5/6 co-receptors and is β-catenin independent. The non-canonical pathway can be further classified into the planar cell polarity (PCP) and the Wnt/Ca2+ pathway (102). In this review, we only focus on the PCP pathway. The PCP pathway signals asymmetric cytoskeletal organization and coordinated polarization of cells within the plane of epithelial cells.
In the absence of Wnt ligands, components of the pathway are located in the cytoplasm (Figure 2E). When extracellular Wnt binds to Fz receptor, it recruits a complex of proteins at the plasma membrane that includes Dvl (103–105). Two independent and parallel pathways downstream Dvl led to the activation of the small GTPases RHO and RAC (Figure 2F) (106–110). The first pathway through the molecule Dishevelled associated activator of morphogenesis 1 (DAAM1) signals to RHO, which activates the RHO-associated coiled-coil forming kinase (ROCK) allowing regulation of cytoskeletal re-organization (109, 111, 112). In the second pathway, Dvl activates RAC, which in turn stimulates c-Jun N-terminal kinase (JNK) activity (Figure 2F), which also regulates cytoskeleton (110, 113).
Contrary to the connections between primary cilium and Hh signaling, the relationship between primary cilium and Wnt signaling is controversial (3, 114, 115) with the exception of the Wnt/PCP pathway, which affects cilia formation and functions via effects on cytoskeleton and basal body positioning (Figure 2F) (116, 117). There are reports showing that primary cilium disruption leads to upregulation of the pathway activity (118–121), and conversely, studies that refute any involvement of primary cilium in Wnt signaling (122, 123). Furthermore, two opposing models have been proposed regarding function of Wnt pathway in cilium formation: (i) a negative role of Wnt signaling in ciliogenesis (124, 125); and (ii) a direct role of the pathway in promotion of primary cilium (126–128). Even more puzzling, Bernatik and collaborators showed that neither activation nor deactivation of the canonical Wnt pathway affected the ciliogenesis (129). Thus, despite deep investigation, the function of the primary cilium in Wnt signaling remains unclear, particularly in pancreatic islets.
Corbit and collaborators have investigated the role of primary cilium in Wnt signaling generating three different transgenic mice in which Kif3a, ITF88/Polaris, and oral-facial-digital syndrome 1 (Ofd1) genes were knockdown resulting in impaired ciliogenesis (118). Genetic depletion of Kif3a, but not ITF88/Polaris or Ofd1, causes constitutive phosphorylation of Dvl and stabilization of β-catenin. Thus, primary cilium restricts the activity of the canonical Wnt pathway in mouse embryos, primary fibroblasts, and embryonic stem cells. Unciliated cells respond more robustly to Wnt stimulation than ciliated cells. Ciliary deficiency in pancreas also leads to activation of Wnt signaling (130). Thus, it appears that Wnt signaling is upregulated when cilia are absent, whereas cells with aberrant cilia structure downregulate the Wnt signaling pathway (131).
The impact of the canonical Wnt signaling pathway has been investigated using tissue-specific knockout mice in which β-catenin was ablated from β-cells. Thus, deletion of β-catenin early in development (Pdx1-Cre,Catnblox/lox mice) resulted in reduced islets numbers, in agreement with the notion that the Wnt pathway is active in endocrine cells during development. However, in adult mice, where the pathway is not active in islets, glucose tolerance was normal in mice lacking β-catenin, suggesting that the Wnt pathway is not necessary for function or to maintain islet architecture later in life (132, 133).
On the other hand, deletion of β-catenin in the maturing β-cells (RIP-Cre,Catnblox/lox mice) induced ~70% perinatal lethality and negatively impacts islets morphology and function as newborn mutant pancreas showed increased insulin content due to a defect in insulin release, and a reduction in total endocrine tissue. Nonetheless, the surviving mice showed mild glucose intolerance later in adulthood (134). These findings suggest that around the time of birth, where endogenous canonical Wnt signaling is activated in the endocrine pancreas, β-cells might be susceptible to loss of β-catenin signaling. However, this signaling might be dispensable in β-cells later in life.
The Wnt pathway is activated by numerous Wnt ligands generally divided into classical ligands (Wnt1, Wnt3a, and Wnt8) that activate the canonical pathway, and non-classical ligands (Wnt4, Wnt5, and Wnt11) that activate the non-canonical pathways. However, some Wnt ligands (e.g. Wnt3a) can activate both the classical and non-classical pathways underlying the cross talk between both pathways to organize complex cellular responses (135).
Krutzfeldt and collaborators analyzed activation of canonical Wnt signaling in adult pancreatic islets from wild-type (WT) and obese (ob/ob) mice in vivo. The canonical Wnt signaling did not occur in both WT and obese mice. Additionally, they identified the non-classical ligand Wnt4 as an abundant signaling molecule in adult mice islets that was upregulated in two different insulin-resistant mouse models [ob/ob and the lipodystrophic mice that lack adipose tissue (aP2-SREBP-1c)]. Furthermore, increased expression of Wnt4 inhibited canonical Wnt signaling in pancreatic islets and MIN6 cells (136, 137). These findings also highlight the complexity of the Wnt signaling in islets and β-cells since some non-classical Wnt ligands act as antagonists of the classical pathway.
Many hormones and growth factors, such as insulin and IGF-1/2, act through receptor tyrosine kinases (RTK), including the insulin receptor (IR) and insulin-like growth factor receptors 1 and 2 (IGF-1/2R). In vertebrates, the three receptors (IR and IGF-1/2R) can bind with different affinities to insulin and IGF-1/2 (138). Of note, the three receptors can form a dimeric structure, which can be either a homodimer or a heterodimer, e.g., IRαβ/IGF-1αβR (139). The RTKs play a critical role in regulating cell proliferation, differentiation, survival, metabolism, migration, and cell-cycle control (140, 141). In the last decades, IR and IGF-1R have been linked to primary cilium and coordination of signaling events (142, 143).
Zhu and collaborators demonstrated that disruption of primary cilium assembly, by knockdown of IFT88 or the anterograde IFT motor subunit KIF3a in 3T3-L1 pre-adipocytes, reduced the ability of insulin to phosphorylate IGF-1R and AKT at the base of the primary cilium, leading to a decreased cellular expression of adipocyte transcription factors C/EBPα and PPARγ. Of note, a fraction of the cellular pool of IGF-R1 was detected in primary cilia of differentiated preadipocytes, and the receptors localized in the plasma membrane were less sensitive to insulin stimulation than those present in primary cilium (142). Likewise, Dalbay and collaborators demonstrated that differentiation of human mesenchymal stem cells into adipocytes required primary cilium elongation associated with recruitment of IGF-1βR onto the cilium (144). These studies demonstrate a link between insulin signaling through the primary cilium and cell differentiation.
Additionally, ciliary IGF-1R activation in 3T3-L1 cells also induces ciliary resorption. Wang and collaborators demonstrated that insulin-mediated activation of IGF-1R recruited and activated IRS-1, followed its re-localization to the ciliary neck region, where the heat shock protein Hsp90α might function as a hub for activation of AKT (Figure 3A) (145). Similarly, Yeh and collaborators showed that ciliary IGF-1R activation mediated recruitment of phosphorylated dynein light chain Tctex-type 1 (TCTEX-1) to the transition zone, leading to a mitogenic signaling cascade that accelerates ciliary resorption and G1/S progression in RPE-1 cells and cultured embryonic fibroblasts (Figure 3B) (146).
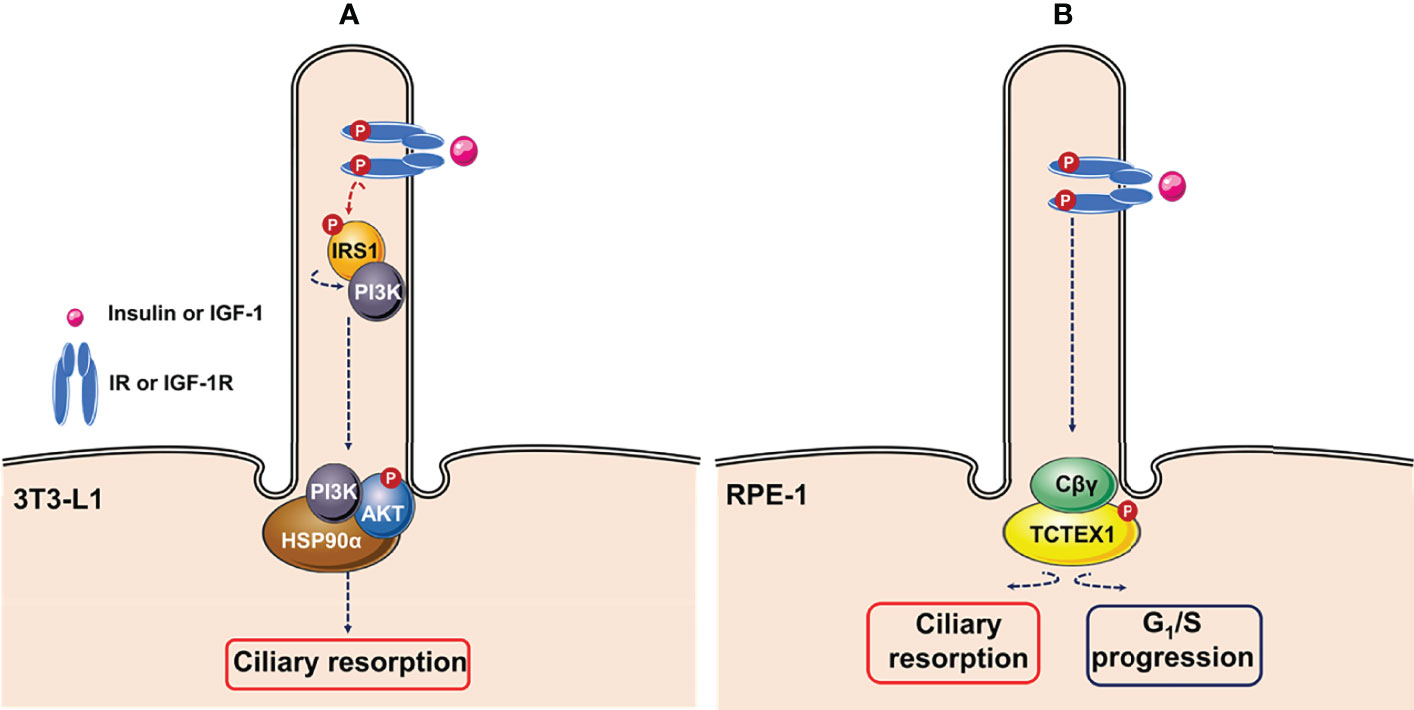
Figure 3 Ciliary insulin/IGF-1 signaling pathways. (A) Insulin stimulates resorption of cilia mouse in 3T3-L1 cells through activation of IGF-1R via recruitment of IRS1. Activated IRS1 is re-localized to the ciliary neck region, where heat shock protein Hsp90α might functions as a hub for activation of AKT. (B) Ciliary insulin growth factor-1 receptor (IGF-1R) activation induces ciliary resorption and G1/S progression via IGF-1-mediated recruitment of phosphorylated Tctex-type 1 (TCTEX-1) via non-canonical G-protein signaling (Cβγ) in RPE-1 cells. This figure was created using Servier Medical Art (available at https://smart.servier.com/).
The endocrine pancreas is an important secretory gland in the regulation of glucose homeostasis due its capacity to secrete two glucoregulatory and antagonistic hormones, insulin and glucagon, in response to higher or lower blood glucose levels, respectively. The discovery of the islets of Langerhans, more than 150 years ago, made it possible to identify several endocrine cell types, most notably insulin-producing β-cells, glucagon-producing α-cells, and somatostatin-producing δ-cells, among other cellular types (147). For many years, insufficient insulin secretion was considered a culprit in the pathophysiology of T2D, but with the bihormonal hypothesis proposed by Unger and Orci this paradigm changed, stating that T2D resulted of the combined effect of hypoinsulinemia and hyperglucagonemia (148).
Primary cilium is found in endocrine pancreas (α-, β-, and δ-cells) (149–152), and exocrine pancreas (ductal cells of the Chinese Hamster, and centroacinar cells of bat) (153, 154), with the exception of acinar cells (130, 155). Several animal models of T2D have shown evidence of ciliary defects, suggesting a link between primary cilium and the pathophysiology of the disease. Thus, in the diabetes model of Goto-Kakizaki (GK) rat was observed a 3-fold reduction in primary cilia in β-cells, which was associated with missexpression of several ciliary/basal body genes (143). Similarly, several ciliary/basal body genes have been shown to be misregulated in pancreatic islets of the obesity and diabetes model ob/ob (156). A panel with mutations in genes related to cilia and their phenotypes in different animal models is shown in Table 2.

Table 2 Mutations in genes related to cilia and their phenotypes in several cellular and animal models.
Genetic studies of mutated genes associated with the primary cilium have begun to unveil a role for cilia in endocrine cells. Thus, ciliary dysfunction can cause inherited ciliopathies such as the Bardet-Biedl syndrome (BBS) and the Alström syndrome (AMLS).
BBS is characterized by multi-organ dysfunctions (obesity, retinal degeneration, polydactyly, renal and gonadal malformations, and learning disabilities) (176). BBS is a genetic disease caused by the combination of at least 19 genes. Among them, the most commonly mutated are BBS1 and BBS10 (177, 178). The link between ciliary dysfunction and BBS was established by the discovery of BBS8, which codifies for a protein localized to centrosomes and basal bodies (179). BBS8 is one of the eight proteins [BBS1, BBS2, BBS4, BBS5, BBS7, BBS8, BBS9, and BBS18 (A.K.A BBIP10)] that form a complex named the BBSome, which is a component of the basal body involved in trafficking vesicles to the primary cilium (180, 181).
Obesity-associated BBS is paradoxically by related to lower susceptibility to develop T2D early in life (2-6% prevalence in childhood), and higher insulin sensitivity and glucose usage (160, 182). In the last decades, several studies using cell lines and animal models have begun to unveil how primary cilium defects in BBS syndrome can explain the associated phenotypes. Thus, the Bbs12-/- mouse model (BBS12; a chaperone protein required for ciliogenesis during adipogenic differentiation of human mesenchymal cells) showed increased obesity, normal glucose tolerance and increased insulin sensitivity in the fat, recapitulating clinical features of BBS (160). Intriguingly, plasma insulin levels (in fasting and during an oral glucose tolerance test) and pancreatic islets size in Bbs12-/- were similar to control mice despite enhanced in vivo insulin sensitivity (160).
On the other hand, the Bbs4 -/- mouse model exhibited impaired glucose homeostasis before the onset of obesity. As reported for Bbs12 null mouse, serum insulin levels and pancreas histomorphometry remained unchanged (143), although the mutant strain reported by Eichers and collaborators exhibited higher plasma insulin levels (158). Disruption of basal body integrity in the Bbs4 null mouse led to impaired first phase insulin secretion ex vivo and in vivo (143). Of note, insulin receptor is recruited to the cilium of insulin-stimulated β-cells. Ciliary integrity was required for activation of downstream targets of insulin signaling (PI3K/FoxO1), leading to a reduction of its targets Snap25 and Syntaxin1A (143).
Interestingly, downregulation of Bbs5, Bbs7, and Bbs9 in the Min6 mouse insulinoma cell line LED to ~2-fold increase in insulin secretion (159). By contrast, downregulation of Bbs4 resulted in a loss of first phase insulin release (143) in Min6 cells. Furthermore, unstimulated Min6 cells did not show the insulin receptor isoform A or B (IR-A or IR-B) in the cilium. Although IR-A, but not IR-B, was recruited to the cilium after insulin stimulation (143).
The ALMS is another ciliopathy resembling BBS that is characterized by obesity, insulin resistance, T2D, hypertriglyceridemia, hyperleptinemia, retinal dystrophy, hearing loss, short stature, pulmonary and renal dysfunctions, and cardiomyopathy (183–190). AMLS is characterized by a highly penetrant obesity, but unlike BBS displays higher childhood diabetes rates (75%). These discrepancies seem to be explained partly by severe insulin resistance observed in Alström patients, a condition that apparently is not present in BBS patients (184–186, 188, 189, 191, 192).
The progression from early impaired fasting glucose toward overt diabetes in Alström patients is mostly due to a progressive failure in β-cell insulin secretion without any further worsening of insulin resistance (183, 193, 194). AMLS is caused by loss-of-function mutations in the AMLS1 gene, which is highly expressed in adult and fetal pancreatic islets (164, 195). Genetic ablation of Amls1 in mice results in hyperplastic islets, partial degranulation of β-cells, and islets cysts (161, 162). AMLS1 is enriched at the basal body of primary cilium, and mutations result in stunted cilia, with cells showing a loss of calcium signaling (164, 196).
Interestingly, transient knockdown of ALMS1 (siRNA-Alms1) in the β-TC-6 mouse insulinoma cell line resulted in constitutive insulin secretion independently of glucose concentrations (163). This inappropriate insulin secretion was paralleled with altered expression of genes known to transmit signals downstream of glucose transport, suggesting a potential involvement of ALMS1 in glucose sensing (163).
Using zebrafish models of BBS and ALMS syndromes, Lohd and collaborators showed that loss of Bbs1 or Bbs4 resulted in a significant increase of β-cells mass. Conversely, loss of Alms1 gene led to a significant decrease in β-cells mass. Further investigations into the mechanisms underlying these phenotypes reveled increased susceptibility to cell death under prolonged exposure to high-glucose conditions in both disease models. Interestingly, although Bbs1-deficient β-cells were similarly susceptible to apoptosis, the overall maintenance of β-cells was likely due to compensatory increased proliferation. Of note, changes in β-cells mass were paralleled by a decrease in α- and δ-cell types during early developmental stages (157). These findings potentially implicate primary cilium as important regulator of β-cell plasticity, and if they would be translated into the clinical setting of BBS and ALMS, suggest that the differential susceptibility to suffer T2D in both syndromes may be related to the production and maintenance of β-cell mass (185, 186, 188, 189).
Consistent with hyperinsulinemia seen in ALMS patients (184, 188), loss of Alms1 in β-cells of zebrafish also exhibited hyperinsulinemia and an impairment in glucose-stimulated insulin secretion (GSIS) (163). This observation was also confirmed in mouse β-TC-6 cells (163). These data suggest a role for ALMS1 in both β-cell proliferation and function.
The Ift88/Polaris gene is an intraflagellar transport protein necessary for ciliary assembly whose expression is restricted to the pancreatic ducts and islets (2, 197, 198). In mice lacking expression of the Ift88/Polaris gene (the orpk mouse model) cilia were absent or shorter in the pancreas. Perturbation of excocrine and ductal cells cilia result in cystogenesis, formation of abnormal tubular structures, and appearance of endocrine cells in the duct. Unexpectedly, acinar cells undergo apoptosis, resulting in an overall loss of exocrine tissue and pancreas size. The endocrine islets cells, where IFT88/Polaris is highly expressed, looked normal except for increased β-cells clustering, due to the reduced mass of exocrine pancreas (10, 130).
The kinesin family member 3a (KIF3a) is required for the intraflagellar transport and cilia formation (199). It is highly expressed in non-diabetic human islets and in islets of obese non-diabetic mouse model when compared to their respective diabetic controls (199). Pancreas-specific ablation of Kif3a in mice (Pdx1-Creearly; Kif3af/f) resulted in conditional loss of cilia in ductal and endocrine cells, leading to acinar-to-ductal metaplasia, fibrosis, cyst formation, aberrant ductal cell morphology, and lipomatosis. The use of different pancreas-specific Cre strains to knockdown Kif3a suggests that phenotypes seen in Pdx1-Cre mice might be caused by the absence of cilia in ductal cells. The pancreatic lesions in this mouse model resemble those found in patients with chronic pancreatitis of cystic fibrosis (10). Finally, the shRNA-mediated knockdown of Kif3a decreased proliferation of Min6 cells as well as dispersed primary mouse and human islets, providing direct functional evidence for the involvement of cilia in β-cells proliferation (165).
The hepatocyte nuclear factor 6 (HNF6) is expressed in the pancreatic progenitor cells and regulates the expression of pancreatic and duodenal homeobox1 (Pdx1) and differentiation to ductal cells (166, 167, 200). In pancreatic ducts, HNF6 controls primary cilium formation by regulating hepatocyte nuclear factor 1 homeobox B/transcription factor 2 (HNF1B/TFC2) and genes associated with cilium such as fibrocystin and cystin (167, 168, 201). Genetic ablation of Hnf6 in mice resulted in an absence of primary cilium from pancreatic ducts and causing enlargement of the lumen and multiple cysts (167, 168). In addition, Hnf6 null mice exhibited delayed Pdx1 expression and a hypoplastic pancreas (166). This phenotype was not related to decreased proliferation or increased apoptosis, but from retarded pancreatic specification of endodermal cells (166).
Transcription factors belonging to the regulatory factor X (RFX) family are conserved along a wide range of species. In humans and mice have been identified five RFX factors (RFX1-5) (202). RFX3 is important for cilium formation by regulating expression of proteins necessary for cilium maintenance (155, 203). Rfx3 gene is expressed in developing and mature pancreatic endocrine cells during embryogenesis and in adult mice (155). Endocrine cells of Rfx3 null mice exhibited reduced number and severely stunned primary cilia. Consistently with the role of Rfx3 in ciliogenesis, null mice showed uniquely altered cellular composition of the islets of Langerhans with fewer β-cells, α-cells, and δ-cells, whereas pancreatic polypeptide-positive cells were markedly increased in number during perinatal stages (155). The adult mice showed small and disorganized islets, decreased insulin production, reduced glucose-stimulated insulin section, and impaired glucose tolerance (155).
The liver kinase B1 (LKB1/STK11) is a tumor suppressor that acts via the activation of AMP-activated protein kinase (AMPK) (169, 204). In humans, STK11 mutation is associated to Peutz-Jeghers syndrome, a condition characterized by high risk of pancreatic cancer and predisposition to gastrointestinal neoplasms (205). LKB1 is expressed in both - acinar and islets - cells during development and in early neonatal tissue, but in adults, it is expressed primarily in islets. Genetic depletion of LKB1 in mice (Pdx1-Cre; Lkb1L/L) resulted in pancreatitis, ductal cyst formation, abnormal cytoskeleton organization, defective acinar cell polarization, loss of tight junctions, and inactivated AMPK/MARK/SAD kinase family (170). Lkb1-null mice presented an exocrine phenotype that resembled the defect seen in the cilia of mice lacking Kif3a and IFT88/Polaris (10, 130). Additionally, Lkb1-/- mice showed an endocrine phenotype consisting in overall decrease in insulin-positive, glucagon–positive, and somatostatin-positive cells (170).
The primary cilia are generally found in the lateral surfaces of β-cells, which are normally arranged as rosettes around capillaries in islets. This organization of primary cilia relative to blood capillaries is important for β-cells insulin secretion as most exocytic processes happen in the vicinity of capillary beds (171). Lkb1-null β-cells presented altered localization of the cilia relative to islet capillaries, and they were not found in lateral surfaces of β-cells but localized to the cell surface opposite to the blood vessels (171, 172). Concordantly with this shift of cilia, Lkb1-deficient β-cells showed increased insulin secretion in vivo (169). Additionally, this change in cellular polarity resulted in hyperactivation of mTOR pathway, leading to a marked increase β-cell volume (65%) (169). Thus, LKB1 is not essential for ciliogenesis but instead influences cilia position regulating insulin secretion. On the other hand, LKB1 controls β-cell size independently of cilia polarization, because treatment of mice with rapamycin (an inhibitor or mTOR) restored normal β-cell size but did not reverse polarity defects of cilia (169). Finally, adult Lkb1-deficient mice exhibited faster glucose clearance in response to a bolus of glucose, which most likely is attributable to insulin hypersecretion (169, 170).
Insulin-degrading enzyme (IDE) is a ubiquitously expressed Zn2+-metalloendoprotease highly conserved and present in phylogenetically diverse organisms, ranging from viruses to humans. IDE is a multifunctional protein with proteolytic and non-proteolytic functions that was discovered more than 70 years ago by Mirsky and Broh–Kahn due its capacity to degrade insulin in vitro. In addition to this glucoregulatory hormone, IDE can also degrade glucagon and somatostatin (206, 207). Of note, genetic polymorphisms within or near the Ide locus have been linked to increased risk for T2D in different ethnicities, and mutations are associated with the development of T2D in the Goto-Kakizaki rat (206, 207). Also, genetic variations in and around the Hhex/Ide locus are associated with T2D incidence, decreased GSIS, and differential β-cell glucose sensitivity in response to an oral glucose challenge (207).
As an initial approach to help elucidating the function of IDE on insulin metabolism in vivo, several laboratories developed mice with pancellular deletion of IDE (IDE-KO) and found age-dependent hyperinsulinemia and glucose intolerance (208, 209). To further decipher the role of IDE in pancreatic β-cells, Steneberg and collaborators found that GSIS was impaired by deletion of Ide (173). Furthermore, they showed that IDE levels were diminished by 40% in whole islets from T2D donors compared to controls (173), a finding later corroborated by Fernández–Díaz and colleagues via immunostaining (210). Of note, patients under oral hypoglycemic treatment showed decreased IDE levels in pancreatic β-cells, whereas insulin-treated patients showed increased IDE levels in β-cells relative to patients treated with oral hypoglycemic agents (210).
Fernández–Díaz and colleagues demonstrated a key role of IDE in regulating insulin secretion in mouse β-cells. shRNA-mediated silencing of Ide in the INS1E insulinoma cell line (INS1E-shRNA-IDE cells) resulted in decreased insulin secretion in response to glucose. Likewise, transient inhibition of IDE, with the specific inhibitor NTE-2, in rat and human islets resulted in abolition of GSIS (174). Furthermore, islets isolated from B-IDE-KO mice (Ide-deficient β-cells) showed a hypersecretory basal state in glucose-unstimulated islets accompanied by an impairment in GSIS (174).
Interestingly, GSIS of B-IDE-KO isolated islets is similar to the observed in transient knockdown of Alms1 and Rfx3 in β-cells (155, 163, 164, 196, 203). Further findings support the notion that IDE may play a role on β-cell ciliogenesis. Thus, isolated islets from B-IDE-KO mice showed hallmarks of β-cell functional immaturity, such as constitutive insulin and pro-insulin secretion, decreased gene expression of Ins2, Ucn3, and Pcsk1, and decreased GLUT2 in plasma membrane (174). This phenotype also resembles the seen in the Pdx1-CreER;CLEG2;Kif3af/f double transgenic mouse, which was devoid of primary cilium in β-cells and showed lower expression of mature β-cell transcription factors (87). Further research is warranted to demonstrate weather IDE has a role on β-cell ciliogenesis. To this end, B-IDE-KO mice and INS1E-shRNA-IDE cells would be important research tools to advance our knowledge on primary cilium in β-cells.
Fernández–Díaz and colleagues showed that IDE is differentially expressed in pancreatic islet cells, being expressed to substantially higher levels in pancreatic α-cells relative to β-cells and all other islet cell types (210). This finding suggests that it may be relevant to investigate the role of this protease in glucagon-producing cells to understand the molecular mechanisms underlying glucagon secretion. Merino and colleagues developed a mouse model in which Ide was knockout in α-cells (A-IDE-KO) that resulted in metabolic phenotypes consisting of hyperglucagonemia and hyperinsulinemia, but normal glucose tolerance. Importantly, Ide-null α-cells triggers hyperplasia, hypertrophy and impairment in cilia formation (175). The diversity of phenotypes seen in the A-IDE-KO mouse model indicates that IDE participates directly or indirectly in a number of cellular and physiological processes in α-cells. Deciphering the cause-effect relationship between IDE and each one of these phenotypes is an exciting opportunity but also a major challenge.
Closely paralleling the phenotype of constitutive insulin secretion produced by deletion of Ide from β-cells (174), α-cell specific-Ide deletion resulted in hyperglucagonemia (175). Two observations could explain, at least in part, this phenotype: (a) high-glucose and insulin levels failed to inhibit glucagon secretion in A-IDE-KO islets, and (b) the α-cell hyperplasia and hypertrophy.
The A-IDE-KO mice also exhibited hyperinsulinemia (175). This metabolic phenotype could be attributed to an exacerbated paracrine effect wherein excess glucagon release by α-cells stimulates the glucagon receptor on β-cells, leading to the activation of cAMP/PKA/EPAC pathway and thereby stimulating insulin secretion (211–215).
Unexpectedly, the A-IDE-KO mice showed α-cell hyperplasia (175). Based on published studies, it is plausible to hypothesize that this phenotype might be mediated by an interaction between IDE and the retinoblastoma protein (pRb), a tumor suppressor that inhibits cell-cycle progression at the G1/S transition when interacting with E2F transcription factors (216). IDE co-purifies with pRb on proteasomal preparations of breast cancer and hepatoma cells (217). Similarly, IDE has been shown to co-immunoprecipitate with the tumor suppressor PTEN, accelerating its degradation by SIRT4 in response to nutritional starvation stresses (218). Further research is warranted to decipher internal machineries that lead to α-cell division in the absence of IDE.
Genetic depletion of Ide in α-cells (αTC1.9-shRNA-IDE) resulted in cytoskeleton disarrangement and a significant reduction in the number of cilia (Figure 4A) (175). Classically, IDE has been viewed as a protease of insulin and glucagon, but this finding points towards non-proteolytic functions that could be regulating cytoskeleton integrity in pancreatic endocrine cells.
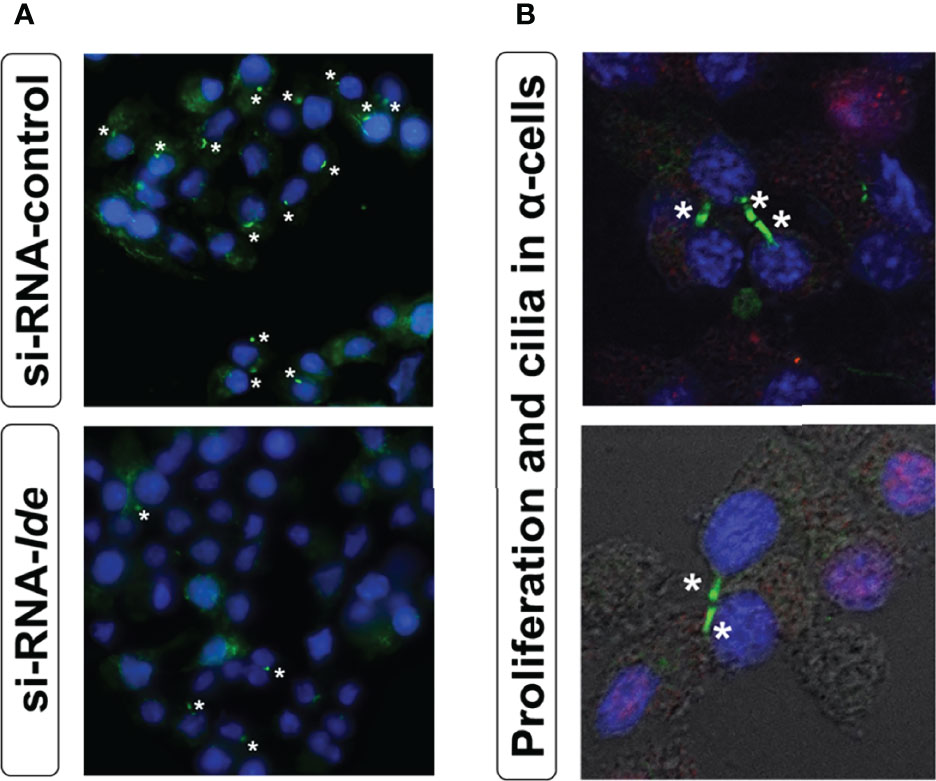
Figure 4 (A) Loss of IDE expression reduces ciliated α-cells number. Representative epifluorescence microscopy (40X zoom) images of cilia signal in siRNA-Ide- and control-treated α-cells. Acetylated α-tubulin (green) and DAPI (blue). Asterisks indicate the presence of cilia. (B) Primary cilium and proliferation in α-cells. Representative fluorescence microscopy images of cilia signal obtained with confocal microscopy (60X zoom) in non-permeabilized α-cells. As seen in the images, proliferation (BrdU staining) was associated with unciliated cells. Acetylated α-tubulin (green), DAPI (blue), and BrdU (red).
How non-proteolytic IDE function(s) may regulate ciliogenesis? IDE binds avidly to monomeric α-synuclein, leading to the formation of stable and irreversible complexes, thereby slowing the formation of higher-n aggregates of α-synuclein (173). In addition, IDE prevents α-synuclein aggregation in a non-proteolytical manner (219). Steneberg and collaborators first showed α-synuclein aggregation in IDE-KO β-cells. They assessed β-cell function in mice harboring pancellular deletion of Ide. They found impaired GSIS and reduced islet autophagic flux and microtubule content in IDE-KO islet cells. They also reported that IDE can form stable complexes with α-synuclein and that levels of α-synuclein were increased in islets from IDE-KO. Furthermore, IDE levels were inversely associated with increased levels of α-synuclein in islets of T2D patients (173).
Merino and collaborators showed that deletion of Ide in α-cells resulted in accumulation of oligomeric α-synuclein together with decreased levels of acetylated-α-tubulin (175). Acetylation of α-tubulin on Lys40 is a posttranslational modification associated with stable microtubules (220). The phenotype in α-cells is similar to that reported in β-cells from IDE-KO mice (173). Therefore, it is plausible to hypothesize that IDE loss of function causes α-synuclein oligomers formation that in turn reduces microtubule content and/or stabilization, which impairs assembly of axonema in primary cilia (221).
Hughes and collaborators demonstrated that primary cilia in β-cells mediate cross talk both within the islet and from islets to other metabolic tissues. β-cell specific depletion of cilia (INS1-Cre/IFT88-Flox mice) disrupts circulating hormone levels, impairs glucose homeostasis and fuel usage, and leads to the development of diabetes (222). In view of IDE-mediated regulation of primary cilium in α-cells, metabolic phenotypes of hyperglucagonemia and hyperinsulinemia seen in A-IDE-KO mice could be attributed to the absence of cilia leading to loss of cross regulation of β- and α-cells (175).
Similarly, the observed phenotype of α-cell hyperplasia and hypertrophy in A-IDE-KO mice could be related to loss of cilia. Merino and collaborators showed that in Ide depleted α-cells, lack of primary cilium was associated with increased proliferation (Figure 4B). Interestingly, proliferating α-cells exhibited a marked reduction in cilia abundance, an important hallmark of α-cell differentiation (175). The absence of cilia has been associated with increased proliferation in a variety of cell types, including pancreatic β-cells (223). The assembly and disassembly of primary cilium and lifecycle of centrosomes are tightly linked to cell division (165, 224–226). The absence of primary cilium could unleash the α-cells from a quiescent state, potentially triggering internal machineries that lead to α-cell division. Further research is warranted to unveil non-proteolytical roles of IDE in regulating cytoskeleton integrity, ciliogenesis and cilia-dependent cellular processes in pancreatic endocrine cells.
On the other hand, nonclassical chaperone activity mediated by synucleins is required for maintenance of continuous presynaptic SNARE-complex in neurons. α-synuclein directly binds to the SNARE-protein Vamp2 and promotes SNARE complex assembly (227). This evidence may explain why deletion of IDE in α-cells resulted in increased expression of genes coding for several members of the SNAREs protein complex, including snap25, syntaxin1A and vamp2 (175). Because of the SNARE complex plays a key role in facilitating the fusion of glucagon granules to the plasma membrane, regulating cellular exocytosis, it is reasonable to hypothesize that these genes would be upregulated to meet the demand of continuous glucagon secretion (228). It is worthy to mention that SNARE proteins also play a role in intracellular ciliogenesis (71–73). Therefore, IDE might indirectly regulate ciliogenesis through its interaction with α-synuclein, which promotes SNAREs complex assembly.
In summary, B-IDE-KO and A-IDE-KO mice have uncovered novel non-proteolytical functions of IDE on ciliogenesis possibly through regulation of α-synuclein aggregation and/or SNAREs complex assembly. IDE deficiency leading to cytoskeleton disarrangements and ciliogenesis impairment in pancreatic α-cells, and most likely in β-cells, results in dysregulation of hormone secretion and cellular immaturity, which maybe triggering insulin and glucagon imbalance seen in diabetes (Figure 5).
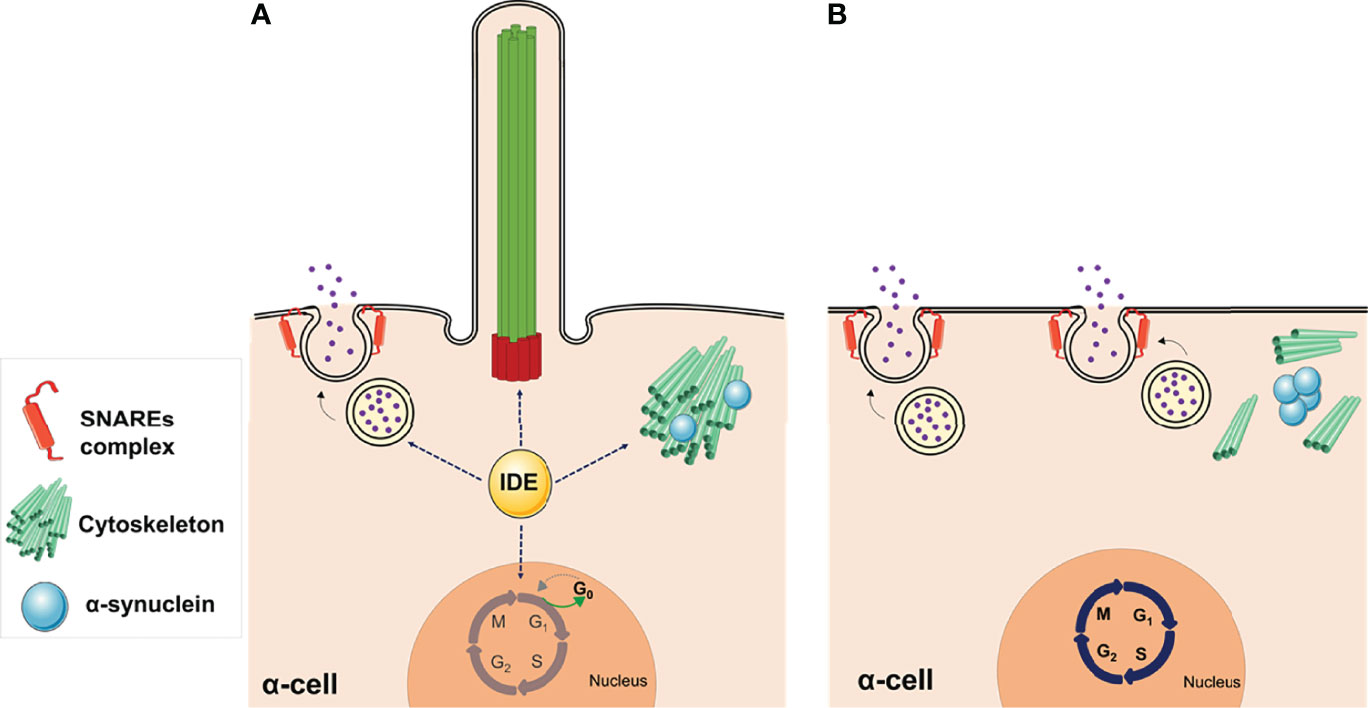
Figure 5 Non-proteolytic functions of IDE in α-cells. (A) The abundance of IDE in pancreatic α-cells is relevant for maintaining several cellular functions, such as glucagon secretion, cytoskeletal organization, and ciliogenesis, while cells are maintained in a quiescent state. (B) Deletion of IDE in mouse α-cells revealed multiple phenotypes, such as hyperglucagonemia, hyperplasia, and hypertrophy, suggesting different non-proteolytic functions of IDE in these cells. Lack of primary cilia along with increased proliferation, in Ide depleted α-cells, provide direct functional evidence for the involvement of cilia in α-cell proliferation. Likewise, loss of IDE causes α-synuclein aggregation, which might underlie the absence of cilia, cytoskeletal alterations, and augmented SNARE proteins of the secretory machinery. This figure was created using Servier Medical Art (available at https://smart.servier.com/).
A growing body of evidence over the past decades have uncovered the importance of cilia in the maintenance of tissue homeostasis through paracrine and autocrine cellular communication. The notion that pancreatic cilia act as an antenna in which hormone receptors (such as insulin, glucagon, and somatostatin receptor) and regulatory proteins may change over environmental cues, enabling the cell to carry out specific functions in the maintenance of glucose homeostasis, opens new avenues for uncovering the pathophysiological processes underlying diabetes.
Current evidence suggests that primary cilium coordinate a variety of signaling pathways to control pancreas development, islets plasticity, and cell-type specific functions. While some signaling pathways are considered to be the bona fide ciliary pathways (e.g., Hh), others might be associated with cilia and probably act in cell-type specific manner, such as the insulin, glucagon, or somatostatin signaling. Although the prevailing idea considers the cilium as a sensory antenna, cilia also can participate as structural mediators of α- and δ-cells cross talk (222). This is especially relevant in the context of communication between islets cells that coordinate hormonal responses and maintain glucose homeostasis. Further research is warranted to discover intracellular crosstalk between cilium-dependent and cilium-independent signaling pathways in pancreatic islets.
Primary cilia are highly dynamic organelles whose configuration is tightly coupled to cellular proliferation and differentiation states. A major debate in pancreatic β-cell biology is focused on mechanisms (proliferation, progenitor differentiation, or trans-differentiation) governing plasticity of pancreatic islets cells in response to physiological conditions (e.g., pregnancy) and diseases (e.g., diabetes and obesity). In both T1D and T2D, reduced and/or inadequately β-cell mass leads to insufficient insulin secretion and hyperglycemia. Restoring β-cell mass and/or function is a major challenge in diabetes. Beyond this challenge, the study of β-cell ciliary biology is a promising possibility for improving β-cell regeneration and/or function that requires additional research.
For more than 70 years, emphasis has been placed on the study of the proteolytic functions of IDE. However, the biology of IDE has proven to be considerably more complex than expected. Tissue-specific knockout mouse models of IDE in endocrine pancreas (A-IDE-KO and B-IDE-KO mice) have demonstrated non-proteolytic functions of this enzyme. Particularly, its capacity to regulate the formation of α-synuclein aggregates strongly implicated in cytoskeletal integrity and vesicular trafficking. Thus, IDE is emerging as an important regulator of ciliogenesis in pancreatic cells, albeit molecular and cellular mechanisms remain obscure. IDE-mediated regulation of cilium seems to be of relevance to the processes of hormone secretion, inter-cellular communication, and islets plasticity to adapt environmental cues.
In conclusion, the extraordinary complexity of the primary cilium promises exciting future discovering such us the potential of cilia-related signaling molecules as therapeutic targets for new treatments fighting diabetes.
Conceptualization, MP, GP, and IC-C; writing—original draft preparation, MP, GP, and IC-C; writing—review and editing, MP, EC-Á, CG-C, BM, GP, and IC-C; supervision, IC-C and GP; funding acquisition, GP and IC-C. All authors have read and agreed to the published version of the manuscript. We apologize to those authors whose work has not been cited because of space limitations.
The project leading to this Review has received funding from “La Caixa” Foundation, under agreement LCF/PR/PR18/51130007 to GP; Grants PID2019-110496RB-C21 and PID2019-110496RB-C22 funded by MCIN/AEI/10.13039/501100011033 to IC-C and GP, respectively; European Foundation for the Study of Diabetes Rising Star Fellowship to BM supported by EFSD-Novo Nordisk; This research was funded by the Programa Estratégico Instituto de Biología y Genética Molecular (IBGM), Junta de Castilla y León (CCVC8485). CG-C and EC-Á were supported by a fellowship from the Junta de Castilla y León and the European Social Fund (ORDER EDU/574/2018 and ORDER EDU/556/2019, respectively).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The authors would like to thank the microscopy and image analysis service of the IBGM for their support and assistance in this work.
1. Satir P. Landmarks in Cilia Research From Leeuwenhoek to Us. Cell Motil Cytoskeleton (1995) 32:90–4. doi: 10.1002/cm.970320203
2. Pazour GJ, Dickert BL, Vucica Y, Seeley ES, Rosenbaum JL, Witman GB, et al. Chlamydomonas Ift88 and Its Mouse Homologue, Polycystic Kidney Disease Gene Tg737, Are Required for Assembly of Cilia and Flagella. J Cell Biol (2000) 151(3):709–18. doi: 10.1083/jcb.151.3.709
3. Huangfu D, Liu A, Rakeman AS, Murcia NS, Niswander L, Anderson KV. Hedgehog Signalling in the Mouse Requires Intraflagellar Transport Proteins. Nature (2003) 426(6962):83–7. doi: 10.1038/nature02061
4. Singla V, Reiter JF. The Primary Cilium as the Cell's Antenna: Signaling at a Sensory Organelle. Science (New York NY) (2006) 313(5787):629–33. doi: 10.1126/science.1124534
5. Nonaka S, Tanaka Y, Okada Y, Takeda S, Harada A, Kanai Y, et al. Randomization of Left-Right Asymmetry Due to Loss of Nodal Cilia Generating Leftward Flow of Extraembryonic Fluid in Mice Lacking Kif3b Motor Protein. Cell (1998) 95(6):829–37. doi: 10.1016/s0092-8674(00)81705-5
6. Anvarian Z, Mykytyn K, Mukhopadhyay S, Pedersen LB, Christensen ST. Cellular Signalling by Primary Cilia in Development, Organ Function and Disease. Nat Rev Nephrol (2019) 15(4):199–219. doi: 10.1038/s41581-019-0116-9
7. Badano JL, Mitsuma N, Beales PL, Katsanis N. The Ciliopathies: An Emerging Class of Human Genetic Disorders. Annu Rev Genomics Hum Genet (2006) 7:125–48. doi: 10.1146/annurev.genom.7.080505.115610
8. Fliegauf M, Benzing T, Omran H. When Cilia Go Bad: Cilia Defects and Ciliopathies. Nat Rev Mol Cell Biol (2007) 8(11):880–93. doi: 10.1038/nrm2278
9. Witman GB. Introduction to Cilia and Flagella. In: Bloodgood RA, editor. Ciliary and Flagellar Membranes. Norwell, MA: Kluwer Acad (1990). p. 1–30.
10. Cano DA, Sekine S, Hebrok M. Primary Cilia Deletion in Pancreatic Epithelial Cells Results in Cyst Formation and Pancreatitis. Gastroenterology (2006) 131(6):1856–69. doi: 10.1053/j.gastro.2006.10.050
11. Teilmann SC, Christensen ST. Localization of the Angiopoietin Receptors Tie-1 and Tie-2 on the Primary Cilia in the Female Reproductive Organs. Cell Biol Int (2005) 29(5):340–6. doi: 10.1016/j.cellbi.2005.03.006
12. Dabdoub A, Kelley MW. Planar Cell Polarity and a Potential Role for a Wnt Morphogen Gradient in Stereociliary Bundle Orientation in the Mammalian Inner Ear. J Neurobiol (2005) 64(4):446–57. doi: 10.1002/neu.20171
13. Huang BQ, Masyuk TV, Muff MA, Tietz PS, Masyuk AI, Larusso NF. Isolation and Characterization of Cholangiocyte Primary Cilia. Am J Physiol Gastrointestinal Liver Physiol (2006) 291(3):G500–9. doi: 10.1152/ajpgi.00064.2006
14. Choksi SP, Lauter G, Swoboda P, Roy S. Switching on Cilia: Transcriptional Networks Regulating Ciliogenesis. Development (Cambridge England) (2014) 141(7):1427–41. doi: 10.1242/dev.074666
15. Falk N, Lösl M, Schröder N, Gießl A. Specialized Cilia in Mammalian Sensory Systems. Cells (2015) 4(3):500–19. doi: 10.3390/cells4030500
16. Berbari NF, O'Connor AK, Haycraft CJ, Yoder BK. The Primary Cilium as a Complex Signaling Center. Curr Biol (2009) 19(13):R526–35. doi: 10.1016/j.cub.2009.05.025
17. Ong AC, Wagner B. Detection of Proximal Tubular Motile Cilia in a Patient With Renal Sarcoidosis Associated With Hypercalcemia. Am J Kidney Dis (2005) 45(6):1096–9. doi: 10.1053/j.ajkd.2005.02.019
18. Silverman MA, Leroux MR. Intraflagellar Transport and the Generation of Dynamic, Structurally and Functionally Diverse Cilia. Trends Cell Biol (2009) 19(7):306–16. doi: 10.1016/j.tcb.2009.04.002
19. Praveen K, Davis EE, Katsanis N. Unique Among Ciliopathies: Primary Ciliary Dyskinesia, a Motile Cilia Disorder. F1000prime Rep (2015) 7:36. doi: 10.12703/p7-36
20. Besschetnova TY, Kolpakova-Hart E, Guan Y, Zhou J, Olsen BR, Shah JV. Identification of Signaling Pathways Regulating Primary Cilium Length and Flow-Mediated Adaptation. Curr Biol (2010) 20(2):182–7. doi: 10.1016/j.cub.2009.11.072
21. Downing KH, Sui H. Structural Insights Into Microtubule Doublet Interactions in Axonemes. Curr Opin Struct Biol (2007) 17(2):253–9. doi: 10.1016/j.sbi.2007.03.013
22. Ishikawa H, Marshall WF. Ciliogenesis: Building the Cell's Antenna. Nat Rev Mol Cell Biol (2011) 12(4):222–34. doi: 10.1038/nrm3085
23. Pazour GJ, Agrin N, Leszyk J, Witman GB. Proteomic Analysis of a Eukaryotic Cilium. J Cell Biol (2005) 170(1):103–13. doi: 10.1083/jcb.200504008
24. van Dam TJ, Wheway G, Slaats GG, Huynen MA, Giles RH. The Syscilia Gold Standard (Scgsv1) of Known Ciliary Components and Its Applications Within a Systems Biology Consortium. Cilia (2013) 2(1):7. doi: 10.1186/2046-2530-2-7
25. Boldt K, van Reeuwijk J, Lu Q, Koutroumpas K, Nguyen TM, Texier Y, et al. An Organelle-Specific Protein Landscape Identifies Novel Diseases and Molecular Mechanisms. Nat Commun (2016) 7:11491. doi: 10.1038/ncomms11491
26. Heydeck W, Fievet L, Davis EE, Katsanis N. The Complexity of the Cilium: Spatiotemporal Diversity of an Ancient Organelle. Curr Opin Cell Biol (2018) 55:139–49. doi: 10.1016/j.ceb.2018.08.001
27. Janke C, Bulinski JC. Post-Translational Regulation of the Microtubule Cytoskeleton: Mechanisms and Functions. Nat Rev Mol Cell Biol (2011) 12(12):773–86. doi: 10.1038/nrm3227
28. Roll-Mecak A. The Tubulin Code in Microtubule Dynamics and Information Encoding. Dev Cell (2020) 54(1):7–20. doi: 10.1016/j.devcel.2020.06.008
29. L'Hernault SW, Rosenbaum JL. Chlamydomonas Alpha-Tubulin Is Posttranslationally Modified by Acetylation on the Epsilon-Amino Group of a Lysine. Biochemistry (1985) 24(2):473–8. doi: 10.1021/bi00323a034
30. LeDizet M, Piperno G. Identification of an Acetylation Site of Chlamydomonas Alpha-Tubulin. Proc Natl Acad Sci USA (1987) 84(16):5720–4. doi: 10.1073/pnas.84.16.5720
31. Kobayashi N, Hirokawa N. Cytoskeletal Architecture and Immunocytochemical Localization of Fodrin in the Terminal Web of the Ciliated Epithelial Cell. Cell Motil Cytoskeleton (1988) 11(3):167–77. doi: 10.1002/cm.970110304
32. Fariss RN, Molday RS, Fisher SK, Matsumoto B. Evidence From Normal and Degenerating Photoreceptors That Two Outer Segment Integral Membrane Proteins Have Separate Transport Pathways. J Comp Neurol (1997) 387(1):148–56. doi: 10.1002/(sici)1096-9861(19971013)387:1<148::aid-cne12>3.0.co;2-q
33. Wheatley DN. Nanobiology of the Primary Cilium–Paradigm of a Multifunctional Nanomachine Complex. Methods Cell Biol (2008) 90:139–56. doi: 10.1016/s0091-679x(08)00807-8
34. Arrojo EDR, Lev-Ram V, Tyagi S, Ramachandra R, Deerinck T, Bushong E, et al. Age Mosaicism Across Multiple Scales in Adult Tissues. Cell Metab (2019) 30(2):343–51.e3. doi: 10.1016/j.cmet.2019.05.010
35. Fowkes ME, Mitchell DR. The Role of Preassembled Cytoplasmic Complexes in Assembly of Flagellar Dynein Subunits. Mol Biol Cell (1998) 9(9):2337–47. doi: 10.1091/mbc.9.9.2337
36. Rosenbaum JL, Witman GB. Intraflagellar Transport. Nat Rev Mol Cell Biol (2002) 3(11):813–25. doi: 10.1038/nrm952
37. Pedersen LB, Rosenbaum JL. Intraflagellar Transport (Ift) Role in Ciliary Assembly, Resorption and Signalling. Curr Topics Dev Biol (2008) 85:23–61. doi: 10.1016/s0070-2153(08)00802-8
38. Hao L, Scholey JM. Intraflagellar Transport at a Glance. J Cell Sci (2009) 122(Pt 7):889–92. doi: 10.1242/jcs.023861
39. Jordan MA, Pigino G. The Structural Basis of Intraflagellar Transport at a Glance. J Cell Sci (2021) 134(12):jcs247163. doi: 10.1242/jcs.247163
40. Hill DB, Button B, Rubinstein M, Boucher RC. Physiology and Pathophysiology of Human Airway Mucus. Physiol Rev (2022). doi: 10.1152/physrev.00004.2021
41. Ibañez-Tallon I, Pagenstecher A, Fliegauf M, Olbrich H, Kispert A, Ketelsen UP, et al. Dysfunction of Axonemal Dynein Heavy Chain Mdnah5 Inhibits Ependymal Flow and Reveals a Novel Mechanism for Hydrocephalus Formation. Hum Mol Genet (2004) 13(18):2133–41. doi: 10.1093/hmg/ddh219
42. Tung CK, Suarez SS. Co-Adaptation of Physical Attributes of the Mammalian Female Reproductive Tract and Sperm to Facilitate Fertilization. Cells (2021) 10(6):1297. doi: 10.3390/cells10061297
43. Praetorius HA, Leipziger J. Primary Cilium-Dependent Sensing of Urinary Flow and Paracrine Purinergic Signaling. Semin Cell Dev Biol (2013) 24(1):3–10. doi: 10.1016/j.semcdb.2012.10.003
44. Babu D, Roy S. Left-Right Asymmetry: Cilia Stir Up New Surprises in the Node. Open Biol (2013) 3(5):130052. doi: 10.1098/rsob.130052
45. Jenkins PM, McEwen DP, Martens JR. Olfactory Cilia: Linking Sensory Cilia Function and Human Disease. Chem Senses (2009) 34(5):451–64. doi: 10.1093/chemse/bjp020
46. Insinna C, Besharse JC. Intraflagellar Transport and the Sensory Outer Segment of Vertebrate Photoreceptors. Dev Dynamics an Off Publ Am Assoc Anatomists (2008) 237(8):1982–92. doi: 10.1002/dvdy.21554
47. Schwander M, Kachar B, Müller U. Review Series: The Cell Biology of Hearing. J Cell Biol (2010) 190(1):9–20. doi: 10.1083/jcb.201001138
48. Quarmby LM, Parker JD. Cilia and the Cell Cycle? J Cell Biol (2005) 169(5):707–10. doi: 10.1083/jcb.200503053
49. Pan J, Seeger-Nukpezah T, Golemis EA. The Role of the Cilium in Normal and Abnormal Cell Cycles: Emphasis on Renal Cystic Pathologies. Cell Mol Life Sci (2013) 70(11):1849–74. doi: 10.1007/s00018-012-1052-z
50. Izawa I, Goto H, Kasahara K, Inagaki M. Current Topics of Functional Links Between Primary Cilia and Cell Cycle. Cilia (2015) 4:12. doi: 10.1186/s13630-015-0021-1
51. Pugacheva EN, Jablonski SA, Hartman TR, Henske EP, Golemis EA. Hef1-Dependent Aurora a Activation Induces Disassembly of the Primary Cilium. Cell (2007) 129(7):1351–63. doi: 10.1016/j.cell.2007.04.035
52. Yu F, Ran J, Zhou J. Ciliopathies: Does Hdac6 Represent a New Therapeutic Target? Trends Pharmacol Sci (2016) 37(2):114–9. doi: 10.1016/j.tips.2015.11.002
53. Radeff-Huang J, Seasholtz TM, Matteo RG, Brown JH. G Protein Mediated Signaling Pathways in Lysophospholipid Induced Cell Proliferation and Survival. J Cell Biochem (2004) 92(5):949–66. doi: 10.1002/jcb.20094
54. Kasahara K, Aoki H, Kiyono T, Wang S, Kagiwada H, Yuge M, et al. Egf Receptor Kinase Suppresses Ciliogenesis Through Activation of Usp8 Deubiquitinase. Nat Commun (2018) 9(1):758. doi: 10.1038/s41467-018-03117-y
55. Walia V, Cuenca A, Vetter M, Insinna C, Perera S, Lu Q, et al. Akt Regulates a Rab11-Effector Switch Required for Ciliogenesis. Dev Cell (2019) 50(2):229–46.e7. doi: 10.1016/j.devcel.2019.05.022
56. Hu HB, Song ZQ, Song GP, Li S, Tu HQ, Wu M, et al. Lpa Signaling Acts as a Cell-Extrinsic Mechanism to Initiate Cilia Disassembly and Promote Neurogenesis. Nat Commun (2021) 12(1):662. doi: 10.1038/s41467-021-20986-y
58. Mirvis M, Stearns T, James Nelson W. Cilium Structure, Assembly, and Disassembly Regulated by the Cytoskeleton. Biochem J (2018) 475(14):2329–53. doi: 10.1042/bcj20170453
59. Mirvis M, Siemers KA, Nelson WJ, Stearns TP. Primary Cilium Loss in Mammalian Cells Occurs Predominantly by Whole-Cilium Shedding. PLoS Biol (2019) 17(7):e3000381. doi: 10.1371/journal.pbio.3000381
60. Dawe HR, Farr H, Gull K. Centriole/Basal Body Morphogenesis and Migration During Ciliogenesis in Animal Cells. J Cell Sci (2007) 120(Pt 1):7–15. doi: 10.1242/jcs.03305
61. Ryan JL, Henkart PA. Fc Receptor-Mediated Inhibition of Murine B-Lymphocyte Activation. J Exp Med (1976) 144(3):768–75. doi: 10.1084/jem.144.3.768
62. Kleene SJ, Van Houten JL. Electrical Signaling in Motile and Primary Cilia. Bioscience (2014) 64(12):1092–102. doi: 10.1093/biosci/biu181
63. Mahoney NM, Goshima G, Douglass AD, Vale RD. Making Microtubules and Mitotic Spindles in Cells Without Functional Centrosomes. Curr Biol (2006) 16(6):564–9. doi: 10.1016/j.cub.2006.01.053
64. Sorokin SP. Reconstructions of Centriole Formation and Ciliogenesis in Mammalian Lungs. J Cell Sci (1968) 3(2):207–30. doi: 10.1242/jcs.3.2.207
66. Sorokin S. Centrioles and the Formation of Rudimentary Cilia by Fibroblasts and Smooth Muscle Cells. J Cell Biol (1962) 15(2):363–77. doi: 10.1083/jcb.15.2.363
67. Molla-Herman A, Ghossoub R, Blisnick T, Meunier A, Serres C, Silbermann F, et al. The Ciliary Pocket: An Endocytic Membrane Domain at the Base of Primary and Motile Cilia. J Cell Sci (2010) 123(Pt 10):1785–95. doi: 10.1242/jcs.059519
68. Ghossoub R, Molla-Herman A, Bastin P, Benmerah A. The Ciliary Pocket: A Once-Forgotten Membrane Domain at the Base of Cilia. Biol Cell (2011) 103(3):131–44. doi: 10.1042/bc20100128
69. Knödler A, Feng S, Zhang J, Zhang X, Das A, Peränen J, et al. Coordination of Rab8 and Rab11 in Primary Ciliogenesis. Proc Natl Acad Sci USA (2010) 107(14):6346–51. doi: 10.1073/pnas.1002401107
70. Larkins CE, Aviles GD, East MP, Kahn RA, Caspary T. Arl13b Regulates Ciliogenesis and the Dynamic Localization of Shh Signaling Proteins. Mol Biol Cell (2011) 22(23):4694–703. doi: 10.1091/mbc.E10-12-0994
71. Westlake CJ, Baye LM, Nachury MV, Wright KJ, Ervin KE, Phu L, et al. Primary Cilia Membrane Assembly Is Initiated by Rab11 and Transport Protein Particle Ii (Trappii) Complex-Dependent Trafficking of Rabin8 to the Centrosome. Proc Natl Acad Sci USA (2011) 108(7):2759–64. doi: 10.1073/pnas.1018823108
72. Lobo GP, Fulmer D, Guo L, Zuo X, Dang Y, Kim SH, et al. The Exocyst Is Required for Photoreceptor Ciliogenesis and Retinal Development. J Biol Chem (2017) 292(36):14814–26. doi: 10.1074/jbc.M117.795674
73. Lu Q, Insinna C, Ott C, Stauffer J, Pintado PA, Rahajeng J, et al. Early Steps in Primary Cilium Assembly Require Ehd1/Ehd3-Dependent Ciliary Vesicle Formation. Nat Cell Biol (2015) 17(3):228–40. doi: 10.1038/ncb3109
74. Huangfu D, Anderson KV. Signaling From Smo to Ci/Gli: Conservation and Divergence of Hedgehog Pathways From Drosophila to Vertebrates. Development (Cambridge England) (2006) 133(1):3–14. doi: 10.1242/dev.02169
75. Varjosalo M, Li SP, Taipale J. Divergence of Hedgehog Signal Transduction Mechanism Between Drosophila and Mammals. Dev Cell (2006) 10(2):177–86. doi: 10.1016/j.devcel.2005.12.014
76. Nüsslein-Volhard C, Wieschaus E. Mutations Affecting Segment Number and Polarity in Drosophila. Nature (1980) 287(5785):795–801. doi: 10.1038/287795a0
77. Haycraft CJ, Banizs B, Aydin-Son Y, Zhang Q, Michaud EJ, Yoder BK. Gli2 and Gli3 Localize to Cilia and Require the Intraflagellar Transport Protein Polaris for Processing and Function. PLoS Genet (2005) 1(4):e53. doi: 10.1371/journal.pgen.0010053
78. Ocbina PJ, Anderson KV. Intraflagellar Transport, Cilia, and Mammalian Hedgehog Signaling: Analysis in Mouse Embryonic Fibroblasts. Dev Dynamics an Off Publ Am Assoc Anatomists (2008) 237(8):2030–8. doi: 10.1002/dvdy.21551
79. Bangs F, Anderson KV. Primary Cilia and Mammalian Hedgehog Signaling. Cold Spring Harbor Perspect Biol (2017) 9(5):a028175. doi: 10.1101/cshperspect.a028175
80. Qi X, Li X. Mechanistic Insights Into the Generation and Transduction of Hedgehog Signaling. Trends Biochem Sci (2020) 45(5):397–410. doi: 10.1016/j.tibs.2020.01.006
81. Daggubati V, Raleigh DR, Sever N. Sterol Regulation of Developmental and Oncogenic Hedgehog Signaling. Biochem Pharmacol (2022) 196:114647. doi: 10.1016/j.bcp.2021.114647
82. Kaushal JB, Batra SK, Rachagani S. Hedgehog Signaling and Its Molecular Perspective With Cholesterol: A Comprehensive Review. Cell Mol Life Sci (2022) 79(5):266. doi: 10.1007/s00018-022-04233-1
83. Tukachinsky H, Petrov K, Watanabe M, Salic A. Mechanism of Inhibition of the Tumor Suppressor Patched by Sonic Hedgehog. Proc Natl Acad Sci U S A (2016) 113(40):E5866–e75. doi: 10.1073/pnas.1606719113
84. Thomas MK, Rastalsky N, Lee JH, Habener JF. Hedgehog Signaling Regulation of Insulin Production by Pancreatic Beta-Cells. Diabetes (2000) 49(12):2039–47. doi: 10.2337/diabetes.49.12.2039
85. Lau J, Hebrok M. Hedgehog Signaling in Pancreas Epithelium Regulates Embryonic Organ Formation and Adult Beta-Cell Function. Diabetes (2010) 59(5):1211–21. doi: 10.2337/db09-0914
86. Cervantes S, Lau J, Cano DA, Borromeo-Austin C, Hebrok M. Primary Cilia Regulate Gli/Hedgehog Activation in Pancreas. Proc Natl Acad Sci USA (2010) 107(22):10109–14. doi: 10.1073/pnas.0909900107
87. Landsman L, Parent A, Hebrok M. Elevated Hedgehog/Gli Signaling Causes Beta-Cell Dedifferentiation in Mice. Proc Natl Acad Sci USA (2011) 108(41):17010–5. doi: 10.1073/pnas.1105404108
88. Umeda H, Ozaki N, Mizutani N, Fukuyama T, Nagasaki H, Arima H, et al. Protective Effect of Hedgehog Signaling on Cytokine-Induced Cytotoxicity in Pancreatic Beta-Cells. Exp Clin Endocrinol Diabetes (2010) 118(10):692–8. doi: 10.1055/s-0030-1254151
89. Yalcinkaya M, Kerksiek A, Gebert K, Annema W, Sibler R, Radosavljevic S, et al. Hdl Inhibits Endoplasmic Reticulum Stress-Induced Apoptosis of Pancreatic β-Cells in Vitro by Activation of Smoothened. J Lipid Res (2020) 61(4):492–504. doi: 10.1194/jlr.RA119000509
90. Yamaguchi TP. Heads or Tails: Wnts and Anterior-Posterior Patterning. Curr Biol CB (2001) 11(17):R713–24. doi: 10.1016/s0960-9822(01)00417-1
91. Komiya Y, Habas R. Wnt Signal Transduction Pathways. Organogenesis (2008) 4(2):68–75. doi: 10.4161/org.4.2.5851
92. Nusse R, Varmus H. Three Decades of Wnts: A Personal Perspective on How a Scientific Field Developed. EMBO J (2012) 31(12):2670–84. doi: 10.1038/emboj.2012.146
93. Steinhart Z, Angers S. Wnt Signaling in Development and Tissue Homeostasis. Development (Cambridge England) (2018) 145(11):dev146589. doi: 10.1242/dev.146589
94. Humphries AC, Mlodzik M. From Instruction to Output: Wnt/Pcp Signaling in Development and Cancer. Curr Opin Cell Biol (2018) 51:110–6. doi: 10.1016/j.ceb.2017.12.005
95. He X, Semenov M, Tamai K, Zeng X. Ldl Receptor-Related Proteins 5 and 6 in Wnt/Beta-Catenin Signaling: Arrows Point the Way. Development (Cambridge England) (2004) 131(8):1663–77. doi: 10.1242/dev.01117
96. Gordon MD, Nusse R. Wnt Signaling: Multiple Pathways, Multiple Receptors, and Multiple Transcription Factors. J Biol Chem (2006) 281(32):22429–33. doi: 10.1074/jbc.R600015200
97. Oh EC, Katsanis N. Context-Dependent Regulation of Wnt Signaling Through the Primary Cilium. J Am Soc Nephrol JASN (2013) 24(1):10–8. doi: 10.1681/asn.2012050526
98. Stamos JL, Weis WI. The β-Catenin Destruction Complex. Cold Spring Harbor Perspect Biol (2013) 5(1):a007898. doi: 10.1101/cshperspect.a007898
99. Itoh K, Brott BK, Bae GU, Ratcliffe MJ, Sokol SY. Nuclear Localization Is Required for Dishevelled Function in Wnt/Beta-Catenin Signaling. J Biol (2005) 4(1):3. doi: 10.1186/jbiol20
100. Gerdes JM, Davis EE, Katsanis N. The Vertebrate Primary Cilium in Development, Homeostasis, and Disease. Cell (2009) 137(1):32–45. doi: 10.1016/j.cell.2009.03.023
101. Song DK, Choi JH, Kim MS. Primary Cilia as a Signaling Platform for Control of Energy Metabolism. Diabetes Metab J (2018) 42(2):117–27. doi: 10.4093/dmj.2018.42.2.117
102. Habas R, Dawid IB. Dishevelled and Wnt Signaling: Is the Nucleus the Final Frontier? J Biol (2005) 4(1):2. doi: 10.1186/jbiol22
103. Wallingford JB, Harland RM. Neural Tube Closure Requires Dishevelled-Dependent Convergent Extension of the Midline. Dev (Cambridge England) (2002) 129(24):5815–25. doi: 10.1242/dev.00123
104. Veeman MT, Axelrod JD, Moon RT. A Second Canon. Functions and Mechanisms of Beta-Catenin-Independent Wnt Signaling. Dev Cell (2003) 5(3):367–77. doi: 10.1016/s1534-5807(03)00266-1
105. Klein TJ, Mlodzik M. Planar Cell Polarization: An Emerging Model Points in the Right Direction. Annu Rev Cell Dev Biol (2005) 21:155–76. doi: 10.1146/annurev.cellbio.21.012704.132806
106. Eaton S, Wepf R, Simons K. Roles for Rac1 and Cdc42 in Planar Polarization and Hair Outgrowth in the Wing of Drosophila. J Cell Biol (1996) 135(5):1277–89. doi: 10.1083/jcb.135.5.1277
107. Strutt DI, Weber U, Mlodzik M. The Role of Rhoa in Tissue Polarity and Frizzled Signalling. Nature (1997) 387(6630):292–5. doi: 10.1038/387292a0
108. Fanto M, Weber U, Strutt DI, Mlodzik M. Nuclear Signaling by Rac and Rho Gtpases Is Required in the Establishment of Epithelial Planar Polarity in the Drosophila Eye. Curr Biol CB (2000) 10(16):979–88. doi: 10.1016/s0960-9822(00)00645-x
109. Winter CG, Wang B, Ballew A, Royou A, Karess R, Axelrod JD, et al. Drosophila Rho-Associated Kinase (Drok) Links Frizzled-Mediated Planar Cell Polarity Signaling to the Actin Cytoskeleton. Cell (2001) 105(1):81–91. doi: 10.1016/s0092-8674(01)00298-7
110. Habas R, Dawid IB, He X. Coactivation of Rac and Rho by Wnt/Frizzled Signaling Is Required for Vertebrate Gastrulation. Genes Dev (2003) 17(2):295–309. doi: 10.1101/gad.1022203
111. Marlow F, Topczewski J, Sepich D, Solnica-Krezel L. Zebrafish Rho Kinase 2 Acts Downstream of Wnt11 to Mediate Cell Polarity and Effective Convergence and Extension Movements. Curr Biol (2002) 12(11):876–84. doi: 10.1016/s0960-9822(02)00864-3
112. Kim GH, Han JK. Jnk and Rokalpha Function in the Noncanonical Wnt/Rhoa Signaling Pathway to Regulate Xenopus Convergent Extension Movements. Dev dynamics (2005) 232(4):958–68. doi: 10.1002/dvdy.20262
113. Tahinci E, Symes K. Distinct Functions of Rho and Rac Are Required for Convergent Extension During Xenopus Gastrulation. Dev Biol (2003) 259(2):318–35. doi: 10.1016/s0012-1606(03)00206-9
114. Corbit KC, Aanstad P, Singla V, Norman AR, Stainier DY, Reiter JF. Vertebrate Smoothened Functions at the Primary Cilium. Nature (2005) 437(7061):1018–21. doi: 10.1038/nature04117
115. Rohatgi R, Milenkovic L, Scott MP. Patched1 Regulates Hedgehog Signaling at the Primary Cilium. Science (New York NY) (2007) 317(5836):372–6. doi: 10.1126/science.1139740
116. Wallingford JB, Mitchell B. Strange as It May Seem: The Many Links Between Wnt Signaling, Planar Cell Polarity, and Cilia. Genes Dev (2011) 25(3):201–13. doi: 10.1101/gad.2008011
117. Butler MT, Wallingford JB. Planar Cell Polarity in Development and Disease. Nat Rev Mol Cell Biol (2017) 18(6):375–88. doi: 10.1038/nrm.2017.11
118. Corbit KC, Shyer AE, Dowdle WE, Gaulden J, Singla V, Chen MH, et al. Kif3a Constrains Beta-Catenin-Dependent Wnt Signalling Through Dual Ciliary and Non-Ciliary Mechanisms. Nat Cell Biol (2008) 10(1):70–6. doi: 10.1038/ncb1670
119. Wiens CJ, Tong Y, Esmail MA, Oh E, Gerdes JM, Wang J, et al. Bardet-Biedl Syndrome-Associated Small Gtpase Arl6 (Bbs3) Functions at or Near the Ciliary Gate and Modulates Wnt Signaling. J Biol Chem (2010) 285(21):16218–30. doi: 10.1074/jbc.M109.070953
120. McDermott KM, Liu BY, Tlsty TD, Pazour GJ. Primary Cilia Regulate Branching Morphogenesis During Mammary Gland Development. Curr Biol (2010) 20(8):731–7. doi: 10.1016/j.cub.2010.02.048
121. Patnaik SR, Kretschmer V, Brücker L, Schneider S, Volz AK, Oancea-Castillo LDR, et al. Bardet-Biedl Syndrome Proteins Regulate Cilia Disassembly During Tissue Maturation. Cell Mol Life Sci (2019) 76(4):757–75. doi: 10.1007/s00018-018-2966-x
122. Huang P, Schier AF. Dampened Hedgehog Signaling But Normal Wnt Signaling in Zebrafish Without Cilia. Development (Cambridge England) (2009) 136(18):3089–98. doi: 10.1242/dev.041343
123. Ocbina PJ, Tuson M, Anderson KV. Primary Cilia Are Not Required for Normal Canonical Wnt Signaling in the Mouse Embryo. PLoS One (2009) 4(8):e6839. doi: 10.1371/journal.pone.0006839
124. Nakagawa N, Li J, Yabuno-Nakagawa K, Eom TY, Cowles M, Mapp T, et al. Apc Sets the Wnt Tone Necessary for Cerebral Cortical Progenitor Development. Genes Dev (2017) 31(16):1679–92. doi: 10.1101/gad.302679.117
125. Kawata K, Narita K, Washio A, Kitamura C, Nishihara T, Kubota S, et al. Odontoblast Differentiation Is Regulated by an Interplay Between Primary Cilia and the Canonical Wnt Pathway. Bone (2021) 150:116001. doi: 10.1016/j.bone.2021.116001
126. Steere N, Chae V, Burke M, Li FQ, Takemaru K, Kuriyama R. A Wnt/Beta-Catenin Pathway Antagonist Chibby Binds Cenexin at the Distal End of Mother Centrioles and Functions in Primary Cilia Formation. PLoS One (2012) 7(7):e41077. doi: 10.1371/journal.pone.0041077
127. Kyun ML, Kim SO, Lee HG, Hwang JA, Hwang J, Soung NK, et al. Wnt3a Stimulation Promotes Primary Ciliogenesis Through β-Catenin Phosphorylation-Induced Reorganization of Centriolar Satellites. Cell Rep (2020) 30(5):1447–62.e5. doi: 10.1016/j.celrep.2020.01.019
128. Xu J, Deng X, Wu X, Zhu H, Zhu Y, Liu J, et al. Primary Cilia Regulate Gastric Cancer-Induced Bone Loss Via Cilia/Wnt/β-Catenin Signaling Pathway. Aging (2021) 13(6):8989–9010. doi: 10.18632/aging.202734
129. Bernatik O, Paclikova P, Kotrbova A, Bryja V, Cajanek L. Primary Cilia Formation Does Not Rely on Wnt/β-Catenin Signaling. Front Cell Dev Biol (2021) 9:623753. doi: 10.3389/fcell.2021.623753
130. Cano DA, Murcia NS, Pazour GJ, Hebrok M. Orpk Mouse Model of Polycystic Kidney Disease Reveals Essential Role of Primary Cilia in Pancreatic Tissue Organization. Development (Cambridge England) (2004) 131(14):3457–67. doi: 10.1242/dev.01189
131. Abdelhamed ZA, Wheway G, Szymanska K, Natarajan S, Toomes C, Inglehearn C, et al. Variable Expressivity of Ciliopathy Neurological Phenotypes That Encompass Meckel-Gruber Syndrome and Joubert Syndrome Is Caused by Complex De-Regulated Ciliogenesis, Shh and Wnt Signalling Defects. Hum Mol Genet (2013) 22(7):1358–72. doi: 10.1093/hmg/dds546
132. Murtaugh LC, Law AC, Dor Y, Melton DA. Beta-Catenin Is Essential for Pancreatic Acinar But Not Islet Development. Development (Cambridge England) (2005) 132(21):4663–74. doi: 10.1242/dev.02063
133. Dessimoz J, Bonnard C, Huelsken J, Grapin-Botton A. Pancreas-Specific Deletion of Beta-Catenin Reveals Wnt-Dependent and Wnt-Independent Functions During Development. Curr Biol (2005) 15(18):1677–83. doi: 10.1016/j.cub.2005.08.037
134. Dabernat S, Secrest P, Peuchant E, Moreau-Gaudry F, Dubus P, Sarvetnick N. Lack of Beta-Catenin in Early Life Induces Abnormal Glucose Homeostasis in Mice. Diabetologia (2009) 52(8):1608–17. doi: 10.1007/s00125-009-1411-y
135. Bikkavilli RK, Feigin ME, Malbon CC. G Alpha O Mediates Wnt-Jnk Signaling Through Dishevelled 1 and 3, Rhoa Family Members, and Mekk 1 and 4 in Mammalian Cells. J Cell Sci (2008) 121(Pt 2):234–45. doi: 10.1242/jcs.021964
136. Bowen A, Kos K, Whatmore J, Richardson S, Welters HJ. Wnt4 Antagonises Wnt3a Mediated Increases in Growth and Glucose Stimulated Insulin Secretion in the Pancreatic Beta-Cell Line, Ins-1. Biochem Biophys Res Commun (2016) 479(4):793–9. doi: 10.1016/j.bbrc.2016.09.130
137. Krützfeldt J, Stoffel M. Regulation of Wingless-Type Mmtv Integration Site Family (Wnt) Signalling in Pancreatic Islets From Wild-Type and Obese Mice. Diabetologia (2010) 53(1):123–7. doi: 10.1007/s00125-009-1578-2
138. Hernández-Sánchez C, Mansilla A, de Pablo F, Zardoya R. Evolution of the Insulin Receptor Family and Receptor Isoform Expression in Vertebrates. Mol Biol Evol (2008) 25(6):1043–53. doi: 10.1093/molbev/msn036
139. Moxham CP, Duronio V, Jacobs S. Insulin-Like Growth Factor I Receptor Beta-Subunit Heterogeneity. Evidence for Hybrid Tetramers Composed of Insulin-Like Growth Factor I and Insulin Receptor Heterodimers. J Biol Chem (1989) 264(22):13238–44. doi: 10.1016/S0021-9258(18)51620-3
140. Christensen ST, Clement CA, Satir P, Pedersen LB. Primary Cilia and Coordination of Receptor Tyrosine Kinase (Rtk) Signalling. J Pathol (2012) 226(2):172–84. doi: 10.1002/path.3004
141. Christensen ST, Morthorst SK, Mogensen JB, Pedersen LB. Primary Cilia and Coordination of Receptor Tyrosine Kinase (Rtk) and Transforming Growth Factor β (Tgf-β) Signaling. Cold Spring Harbor Perspect Biol (2017) 9(6):a028167. doi: 10.1101/cshperspect.a028167
142. Zhu D, Shi S, Wang H, Liao K. Growth Arrest Induces Primary-Cilium Formation and Sensitizes Igf-1-Receptor Signaling During Differentiation Induction of 3t3-L1 Preadipocytes. J Cell Sci (2009) 122(Pt 15):2760–8. doi: 10.1242/jcs.046276
143. Gerdes JM, Christou-Savina S, Xiong Y, Moede T, Moruzzi N, Karlsson-Edlund P, et al. Ciliary Dysfunction Impairs Beta-Cell Insulin Secretion and Promotes Development of Type 2 Diabetes in Rodents. Nat Commun (2014) 5:5308. doi: 10.1038/ncomms6308
144. Dalbay MT, Thorpe SD, Connelly JT, Chapple JP, Knight MM. Adipogenic Differentiation of Hmscs Is Mediated by Recruitment of Igf-1r Onto the Primary Cilium Associated With Cilia Elongation. Stem Cells (Dayton Ohio) (2015) 33(6):1952–61. doi: 10.1002/stem.1975
145. Wang H, Zou X, Wei Z, Wu Y, Li R, Zeng R, et al. Hsp90α Forms a Stable Complex at the Cilium Neck for the Interaction of Signalling Molecules in Igf-1 Receptor Signalling. J Cell Sci (2015) 128(1):100–8. doi: 10.1242/jcs.155101
146. Yeh C, Li A, Chuang JZ, Saito M, Cáceres A, Sung CH. Igf-1 Activates a Cilium-Localized Noncanonical Gβγ Signaling Pathway That Regulates Cell-Cycle Progression. Dev Cell (2013) 26(4):358–68. doi: 10.1016/j.devcel.2013.07.014
147. Sakata N, Yoshimatsu G, Kodama S. Development and Characteristics of Pancreatic Epsilon Cells. Int J Mol Sci (2019) 20(8):1867. doi: 10.3390/ijms20081867
148. Unger RH, Orci L. The Essential Role of Glucagon in the Pathogenesis of Diabetes Mellitus. Lancet (London England) (1975) 1(7897):14–6. doi: 10.1016/s0140-6736(75)92375-2
149. Munger BL. A Light and Electron Microscopic Study of Cellular Differentiation in the Pancreatic Islets of the Mouse. Am J Anat (1958) 103(2):275–311. doi: 10.1002/aja.1001030207
150. Greider MH, Elliott DW. Electron Microscopy of Human Pancreatic Tumors of Islet Cell Origin. Am J Pathol (1964) 44(4):663–78.
151. Yamamoto M, Kataoka K. Electron Microscopic Observation of the Primary Cilium in the Pancreatic Islets. Archivum histologicum Japonicum = Nihon soshikigaku kiroku (1986) 49(4):449–57. doi: 10.1679/aohc.49.449
152. Aughsteen AA. The Ultrastructure of Primary Cilia in the Endocrine and Excretory Duct Cells of the Pancreas of Mice and Rats. Eur J Morphology (2001) 39(5):277–83. doi: 10.1076/ejom.39.5.277.7380
153. Boquist L. Cilia in Normal and Regenerating Islet Tissue. An Utlrastructural Study in the Chinese Hamster With Particular Reference to the B-Cells and the Ductular Epithelium. Z Zellforsch mikrosk Anat (1968) 89:519–32. doi: 10.1007/BF00336177
154. Lodh S, O'Hare EA, Zaghloul NA. Primary Cilia in Pancreatic Development and Disease. Birth Defects Res Part C Embryo Today Rev (2014) 102(2):139–58. doi: 10.1002/bdrc.21063
155. Ait-Lounis A, Baas D, Barras E, Benadiba C, Charollais A, Nlend Nlend R, et al. Novel Function of the Ciliogenic Transcription Factor Rfx3 in Development of the Endocrine Pancreas. Diabetes (2007) 56(4):950–9. doi: 10.2337/db06-1187
156. Keller MP, Choi Y, Wang P, Davis DB, Rabaglia ME, Oler AT, et al. A Gene Expression Network Model of Type 2 Diabetes Links Cell Cycle Regulation in Islets With Diabetes Susceptibility. Genome Res (2008) 18(5):706–16. doi: 10.1101/gr.074914.107
157. Lodh S, Hostelley TL, Leitch CC, O'Hare EA, Zaghloul NA. Differential Effects on β-Cell Mass by Disruption of Bardet-Biedl Syndrome or Alstrom Syndrome Genes. Hum Mol Genet (2016) 25(1):57–68. doi: 10.1093/hmg/ddv447
158. Eichers ER, Abd-El-Barr MM, Paylor R, Lewis RA, Bi W, Lin X, et al. Phenotypic Characterization of Bbs4 Null Mice Reveals Age-Dependent Penetrance and Variable Expressivity. Hum Genet (2006) 120(2):211–26. doi: 10.1007/s00439-006-0197-y
159. Lee BH, Liu J, Wong D, Srinivasan S, Ashrafi K. Hyperactive Neuroendocrine Secretion Causes Size, Feeding, and Metabolic Defects of C. Elegans Bardet-Biedl Syndrome Mutants. PLoS Biol (2011) 9(12):e1001219. doi: 10.1371/journal.pbio.1001219
160. Marion V, Mockel A, De Melo C, Obringer C, Claussmann A, Simon A, et al. Bbs-Induced Ciliary Defect Enhances Adipogenesis, Causing Paradoxical Higher-Insulin Sensitivity, Glucose Usage, and Decreased Inflammatory Response. Cell Metab (2012) 16(3):363–77. doi: 10.1016/j.cmet.2012.08.005
161. Collin GB, Cyr E, Bronson R, Marshall JD, Gifford EJ, Hicks W, et al. Alms1-Disrupted Mice Recapitulate Human Alström Syndrome. Hum Mol Genet (2005) 14(16):2323–33. doi: 10.1093/hmg/ddi235
162. Arsov T, Silva DG, O'Bryan MK, Sainsbury A, Lee NJ, Kennedy C, et al. Fat Aussie–a New Alström Syndrome Mouse Showing a Critical Role for Alms1 in Obesity, Diabetes, and Spermatogenesis. Mol Endocrinol (Baltimore Md) (2006) 20(7):1610–22. doi: 10.1210/me.2005-0494
163. Nesmith JE, Hostelley TL, Leitch CC, Matern MS, Sethna S, McFarland R, et al. Genomic Knockout of Alms1 in Zebrafish Recapitulates Alström Syndrome and Provides Insight Into Metabolic Phenotypes. Hum Mol Genet (2019) 28(13):2212–23. doi: 10.1093/hmg/ddz053
164. Li G, Vega R, Nelms K, Gekakis N, Goodnow C, McNamara P, et al. A Role for Alström Syndrome Protein, Alms1, in Kidney Ciliogenesis and Cellular Quiescence. PLoS Genet (2007) 3(1):e8. doi: 10.1371/journal.pgen.0030008
165. Kluth O, Stadion M, Gottmann P, Aga H, Jähnert M, Scherneck S, et al. Decreased Expression of Cilia Genes in Pancreatic Islets as a Risk Factor for Type 2 Diabetes in Mice and Humans. Cell Rep (2019) 26(11):3027–36.e3. doi: 10.1016/j.celrep.2019.02.056
166. Jacquemin P, Lemaigre FP, Rousseau GG. The Onecut Transcription Factor Hnf-6 (Oc-1) Is Required for Timely Specification of the Pancreas and Acts Upstream of Pdx-1 in the Specification Cascade. Dev Biol (2003) 258(1):105–16. doi: 10.1016/s0012-1606(03)00115-5
167. Pierreux CE, Poll AV, Kemp CR, Clotman F, Maestro MA, Cordi S, et al. The Transcription Factor Hepatocyte Nuclear Factor-6 Controls the Development of Pancreatic Ducts in the Mouse. Gastroenterology (2006) 130(2):532–41. doi: 10.1053/j.gastro.2005.12.005
168. Kaestner KH. Of Cilia and Cysts: Modeling Pancreatic Polycystic Disease. Gastroenterology (2006) 130(3):926–8. doi: 10.1053/j.gastro.2006.01.056
169. Granot Z, Swisa A, Magenheim J, Stolovich-Rain M, Fujimoto W, Manduchi E, et al. Lkb1 Regulates Pancreatic Beta Cell Size, Polarity, and Function. Cell Metab (2009) 10(4):296–308. doi: 10.1016/j.cmet.2009.08.010
170. Hezel AF, Gurumurthy S, Granot Z, Swisa A, Chu GC, Bailey G, et al. Pancreatic Lkb1 Deletion Leads to Acinar Polarity Defects and Cystic Neoplasms. Mol Cell Biol (2008) 28(7):2414–25. doi: 10.1128/mcb.01621-07
171. diIorio P, Rittenhouse AR, Bortell R, Jurczyk A. Role of Cilia in Normal Pancreas Function and in Diseased States. Birth Defects Res Part C Embryo Today (2014) 102(2):126–38. doi: 10.1002/bdrc.21064
172. Takahashi N, Kishimoto T, Nemoto T, Kadowaki T, Kasai H. Fusion Pore Dynamics and Insulin Granule Exocytosis in the Pancreatic Islet. Science (New York NY) (2002) 297(5585):1349–52. doi: 10.1126/science.1073806
173. Steneberg P, Bernardo L, Edfalk S, Lundberg L, Backlund F, Ostenson CG, et al. The Type 2 Diabetes-Associated Gene Ide Is Required for Insulin Secretion and Suppression of α-Synuclein Levels in β-Cells. Diabetes (2013) 62(6):2004–14. doi: 10.2337/db12-1045
174. Fernández-Díaz CM, Merino B, López-Acosta JF, Cidad P, de la Fuente MA, Lobatón CD, et al. Pancreatic β-Cell-Specific Deletion of Insulin-Degrading Enzyme Leads to Dysregulated Insulin Secretion and β-Cell Functional Immaturity. Am J Physiol Endocrinol Metab (2019) 317(5):E805–e19. doi: 10.1152/ajpendo.00040.2019
175. Merino B, Casanueva-Álvarez E, Quesada I, González-Casimiro CM, Fernández-Díaz CM, Postigo-Casado T, et al. Insulin-Degrading Enzyme Ablation in Mouse Pancreatic Alpha Cells Triggers Cell Proliferation, Hyperplasia and Glucagon Secretion Dysregulation. Diabetologia (2022). doi: 10.1007/s00125-022-05729-y
176. Biedl A. Ein Geschwisterpaar Mit Adiposo-Genitaler Dystrophie. Dtsch Med Wochenschr (1922) 48:1630.
177. Beales PL, Warner AM, Hitman GA, Thakker R, Flinter FA. Bardet-Biedl Syndrome: A Molecular and Phenotypic Study of 18 Families. J Med Genet (1997) 34(2):92–8. doi: 10.1136/jmg.34.2.92
178. Katsanis N, Ansley SJ, Badano JL, Eichers ER, Lewis RA, Hoskins BE, et al. Triallelic Inheritance in Bardet-Biedl Syndrome, a Mendelian Recessive Disorder. Science (New York NY) (2001) 293(5538):2256–9. doi: 10.1126/science.1063525
179. Ansley SJ, Badano JL, Blacque OE, Hill J, Hoskins BE, Leitch CC, et al. Basal Body Dysfunction Is a Likely Cause of Pleiotropic Bardet-Biedl Syndrome. Nature (2003) 425(6958):628–33. doi: 10.1038/nature02030
180. Katoh Y, Nozaki S, Hartanto D, Miyano R, Nakayama K. Architectures of Multisubunit Complexes Revealed by a Visible Immunoprecipitation Assay Using Fluorescent Fusion Proteins. J Cell Sci (2015) 128(12):2351–62. doi: 10.1242/jcs.168740
181. Jin H, White SR, Shida T, Schulz S, Aguiar M, Gygi SP, et al. The Conserved Bardet-Biedl Syndrome Proteins Assemble a Coat That Traffics Membrane Proteins to Cilia. Cell (2010) 141(7):1208–19. doi: 10.1016/j.cell.2010.05.015
182. Lodh S. Primary Cilium, an Unsung Hero in Maintaining Functional β-Cell Population. Yale J Biol Med (2019) 92(3):471–80.
183. Bettini V, Maffei P, Pagano C, Romano S, Milan G, Favaretto F, et al. The Progression From Obesity to Type 2 Diabetes in Alström Syndrome. Pediatr Diabetes (2012) 13(1):59–67. doi: 10.1111/j.1399-5448.2011.00789.x
184. Marshall JD, Maffei P, Collin GB, Naggert JK. Alström Syndrome: Genetics and Clinical Overview. Curr Genomics (2011) 12(3):225–35. doi: 10.2174/138920211795677912
185. Beales PL, Elcioglu N, Woolf AS, Parker D, Flinter FA. New Criteria for Improved Diagnosis of Bardet-Biedl Syndrome: Results of a Population Survey. J Med Genet (1999) 36(6):437–46.
186. Grace C, Beales P, Summerbell C, Jebb SA, Wright A, Parker D, et al. Energy Metabolism in Bardet-Biedl Syndrome. Int J Obes related Metab Disord (2003) 27(11):1319–24. doi: 10.1038/sj.ijo.0802420
187. Marshall JD, Beck S, Maffei P, Naggert JK. Alström Syndrome. Eur J Hum Genet (2007) 15(12):1193–202. doi: 10.1038/sj.ejhg.5201933
188. Girard D, Petrovsky N. Alström Syndrome: Insights Into the Pathogenesis of Metabolic Disorders. Nat Rev Endocrinol (2011) 7(2):77–88. doi: 10.1038/nrendo.2010.210
189. Feuillan PP, Ng D, Han JC, Sapp JC, Wetsch K, Spaulding E, et al. Patients With Bardet-Biedl Syndrome Have Hyperleptinemia Suggestive of Leptin Resistance. J Clin Endocrinol Metab (2011) 96(3):E528–35. doi: 10.1210/jc.2010-2290
190. Forsythe E, Beales PL. Bardet-Biedl Syndrome. Eur J Hum Genet (2013) 21(1):8–13. doi: 10.1038/ejhg.2012.115
191. Marshall JD, Bronson RT, Collin GB, Nordstrom AD, Maffei P, Paisey RB, et al. New Alström Syndrome Phenotypes Based on the Evaluation of 182 Cases. Arch Internal Med (2005) 165(6):675–83. doi: 10.1001/archinte.165.6.675
192. Minton JA, Owen KR, Ricketts CJ, Crabtree N, Shaikh G, Ehtisham S, et al. Syndromic Obesity and Diabetes: Changes in Body Composition With Age and Mutation Analysis of Alms1 in 12 United Kingdom Kindreds With Alstrom Syndrome. J Clin Endocrinol Metab (2006) 91(8):3110–6. doi: 10.1210/jc.2005-2633
193. Green JS, Parfrey PS, Harnett JD, Farid NR, Cramer BC, Johnson G, et al. The Cardinal Manifestations of Bardet-Biedl Syndrome, A Form of Laurence-Moon-Biedl Syndrome. New Engl J Med (1989) 321(15):1002–9. doi: 10.1056/nejm198910123211503
194. Moore SJ, Green JS, Fan Y, Bhogal AK, Dicks E, Fernandez BA, et al. Clinical and Genetic Epidemiology of Bardet-Biedl Syndrome in Newfoundland: A 22-Year Prospective, Population-Based, Cohort Study. Am J Med Genet Part A (2005) 132a(4):352–60. doi: 10.1002/ajmg.a.30406
195. Hearn T, Spalluto C, Phillips VJ, Renforth GL, Copin N, Hanley NA, et al. Subcellular Localization of Alms1 Supports Involvement of Centrosome and Basal Body Dysfunction in the Pathogenesis of Obesity, Insulin Resistance, and Type 2 Diabetes. Diabetes (2005) 54(5):1581–7. doi: 10.2337/diabetes.54.5.1581
196. Jagger D, Collin G, Kelly J, Towers E, Nevill G, Longo-Guess C, et al. Alström Syndrome Protein Alms1 Localizes to Basal Bodies of Cochlear Hair Cells and Regulates Cilium-Dependent Planar Cell Polarity. Hum Mol Genet (2011) 20(3):466–81. doi: 10.1093/hmg/ddq493
197. Taulman PD, Haycraft CJ, Balkovetz DF, Yoder BK. Polaris, a Protein Involved in Left-Right Axis Patterning, Localizes to Basal Bodies and Cilia. Mol Biol Cell (2001) 12(3):589–99. doi: 10.1091/mbc.12.3.589
198. Yoder BK, Tousson A, Millican L, Wu JH, Bugg CE Jr., Schafer JA, et al. Polaris, a Protein Disrupted in Orpk Mutant Mice, Is Required for Assembly of Renal Cilium. Am J Physiol Renal Physiol (2002) 282(3):F541–52. doi: 10.1152/ajprenal.00273.2001
199. Kotsis F, Janusch H, Li Y, Viau A, Epting D, Kramer-Zucker A, et al. Ift88, But Not Kif3a, Is Required for Establishment of the Periciliary Membrane Compartment. Biochem Biophys Res Commun (2021) 584:19–25. doi: 10.1016/j.bbrc.2021.10.075
200. Zhang H, Ables ET, Pope CF, Washington MK, Hipkens S, Means AL, et al. Multiple, Temporal-Specific Roles for Hnf6 in Pancreatic Endocrine and Ductal Differentiation. Mech Dev (2009) 126(11-12):958–73. doi: 10.1016/j.mod.2009.09.006
201. Gresh L, Fischer E, Reimann A, Tanguy M, Garbay S, Shao X, et al. A Transcriptional Network in Polycystic Kidney Disease. EMBO J (2004) 23(7):1657–68. doi: 10.1038/sj.emboj.7600160
202. Emery P, Durand B, Mach B, Reith W. Rfx Proteins, a Novel Family of DNA Binding Proteins Conserved in the Eukaryotic Kingdom. Nucleic Acids Res (1996) 24(5):803–7. doi: 10.1093/nar/24.5.803
203. Bonnafe E, Touka M, AitLounis A, Baas D, Barras E, Ucla C, et al. The Transcription Factor Rfx3 Directs Nodal Cilium Development and Left-Right Asymmetry Specification. Mol Cell Biol (2004) 24(10):4417–27. doi: 10.1128/mcb.24.10.4417-4427.2004
204. Alessi DR, Sakamoto K, Bayascas JR. Lkb1-Dependent Signaling Pathways. Annu Rev Biochem (2006) 75:137–63. doi: 10.1146/annurev.biochem.75.103004.142702
205. Beggs AD, Latchford AR, Vasen HF, Moslein G, Alonso A, Aretz S, et al. Peutz-Jeghers Syndrome: A Systematic Review and Recommendations for Management. Gut (2010) 59(7):975–86. doi: 10.1136/gut.2009.198499
206. Leissring MA, González-Casimiro CM, Merino B, Suire CN, Perdomo G. Targeting Insulin-Degrading Enzyme in Insulin Clearance. Int J Mol Sci (2021) 22(5). doi: 10.3390/ijms22052235
207. González-Casimiro CM, Merino B, Casanueva-Álvarez E, Postigo-Casado T, Cámara-Torres P, Fernández-Díaz CM, et al. Modulation of Insulin Sensitivity by Insulin-Degrading Enzyme. Biomedicines (2021) 9(1). doi: 10.3390/biomedicines9010086
208. Abdul-Hay SO, Kang D, McBride M, Li L, Zhao J, Leissring MA. Deletion of Insulin-Degrading Enzyme Elicits Antipodal, Age-Dependent Effects on Glucose and Insulin Tolerance. PLoS One (2011) 6(6):e20818. doi: 10.1371/journal.pone.0020818
209. Farris W, Mansourian S, Chang Y, Lindsley L, Eckman EA, Frosch MP, et al. Insulin-Degrading Enzyme Regulates the Levels of Insulin, Amyloid Beta-Protein, and the Beta-Amyloid Precursor Protein Intracellular Domain in Vivo. Proc Natl Acad Sci USA (2003) 100(7):4162–7. doi: 10.1073/pnas.0230450100
210. Fernández-Díaz CM, Escobar-Curbelo L, López-Acosta JF, Lobatón CD, Moreno A, Sanz-Ortega J, et al. Insulin Degrading Enzyme Is Up-Regulated in Pancreatic β Cells by Insulin Treatment. Histol histopathology (2018) 33(11):1167–80. doi: 10.14670/hh-11-997
211. Pipeleers D, in't Veld PI, Maes E, Van De Winkel M. Glucose-Induced Insulin Release Depends on Functional Cooperation Between Islet Cells. Proc Natl Acad Sci USA (1982) 79(23):7322–5. doi: 10.1073/pnas.79.23.7322
212. Huypens P, Ling Z, Pipeleers D, Schuit F. Glucagon Receptors on Human Islet Cells Contribute to Glucose Competence of Insulin Release. Diabetologia (2000) 43(8):1012–9. doi: 10.1007/s001250051484
213. Samols E, Marri G, Marks V. Promotion of Insulin Secretion by Glucagon. Lancet (London England) (1965) 2(7409):415–6. doi: 10.1016/s0140-6736(65)90761-0
214. Tengholm A, Gylfe E. Camp Signalling in Insulin and Glucagon Secretion. Diabetes Obes Metab (2017) 19 Suppl 1:42–53. doi: 10.1111/dom.12993
215. Rodriguez-Diaz R, Tamayo A, Hara M, Caicedo A. The Local Paracrine Actions of the Pancreatic α-Cell. Diabetes (2020) 69(4):550–8. doi: 10.2337/dbi19-0002
216. Rubin SM, Sage J, Skotheim JM. Integrating Old and New Paradigms of G1/S Control. Mol Cell (2020) 80(2):183–92. doi: 10.1016/j.molcel.2020.08.020
217. Radulescu RT, Duckworth WC, Levy JL, Fawcett J. Retinoblastoma Protein Co-Purifies With Proteasomal Insulin-Degrading Enzyme: Implications for Cell Proliferation Control. Biochem Biophys Res Commun (2010) 395(2):196–9. doi: 10.1016/j.bbrc.2010.03.157
218. Liu M, Wang Z, Ren M, Yang X, Liu B, Qi H, et al. Sirt4 Regulates Pten Stability Through Ide in Response to Cellular Stresses. FASEB J (2019) 33(4):5535–47. doi: 10.1096/fj.201801987R
219. Sharma SK, Chorell E, Steneberg P, Vernersson-Lindahl E, Edlund H, Wittung-Stafshede P. Insulin-Degrading Enzyme Prevents α-Synuclein Fibril Formation in a Nonproteolytical Manner. Sci Rep (2015) 5:12531. doi: 10.1038/srep12531
220. Eshun-Wilson L, Zhang R, Portran D, Nachury MV, Toso DB, Löhr T, et al. Effects of α-Tubulin Acetylation on Microtubule Structure and Stability. Proc Natl Acad Sci USA (2019) 116(21):10366–71. doi: 10.1073/pnas.1900441116
221. Li L, Yang XJ. Tubulin Acetylation: Responsible Enzymes, Biological Functions and Human Diseases. Cell Mol Life Sci (2015) 72(22):4237–55. doi: 10.1007/s00018-015-2000-5
222. Hughes JW, Cho JH, Conway HE, DiGruccio MR, Ng XW, Roseman HF, et al. Primary Cilia Control Glucose Homeostasis Via Islet Paracrine Interactions. Proc Natl Acad Sci U S A (2020) 117(16):8912–23. doi: 10.1073/pnas.2001936117
223. Phelps EA, Cianciaruso C, Santo-Domingo J, Pasquier M, Galliverti G, Piemonti L, et al. Advances in Pancreatic Islet Monolayer Culture on Glass Surfaces Enable Super-Resolution Microscopy and Insights Into Beta Cell Ciliogenesis and Proliferation. Sci Rep (2017) 7:45961. doi: 10.1038/srep45961
224. Tucker RW, Pardee AB, Fujiwara K. Centriole Ciliation Is Related to Quiescence and DNA Synthesis in 3t3 Cells. Cell (1979) 17(3):527–35. doi: 10.1016/0092-8674(79)90261-7
225. Plotnikova OV, Pugacheva EN, Golemis EA. Primary Cilia and the Cell Cycle. Methods Cell Biol (2009) 94:137–60. doi: 10.1016/s0091-679x(08)94007-3
226. Irigoín F, Badano JL. Keeping the Balance Between Proliferation and Differentiation: The Primary Cilium. Curr Genomics (2011) 12(4):285–97. doi: 10.2174/138920211795860134
227. Burré J, Sharma M, Tsetsenis T, Buchman V, Etherton MR, Südhof TC. Alpha-Synuclein Promotes Snare-Complex Assembly in Vivo and in Vitro. Science (New York NY) (2010) 329(5999):1663–7. doi: 10.1126/science.1195227
Keywords: primary cilium, insulin-degrading enzyme, pancreas, β-cell, α-cell, insulin signaling, insulin, proliferation
Citation: Pablos M, Casanueva-Álvarez E, González-Casimiro CM, Merino B, Perdomo G and Cózar-Castellano I (2022) Primary Cilia in Pancreatic β- and α-Cells: Time to Revisit the Role of Insulin-Degrading Enzyme. Front. Endocrinol. 13:922825. doi: 10.3389/fendo.2022.922825
Received: 18 April 2022; Accepted: 24 May 2022;
Published: 27 June 2022.
Edited by:
Mark O. Huising, University of California, Davis, United StatesReviewed by:
Xuelin Lou, Medical College of Wisconsin, United StatesCopyright © 2022 Pablos, Casanueva-Álvarez, González-Casimiro, Merino, Perdomo and Cózar-Castellano. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Marta Pablos, bWFydGFpc2FiZWwucGFibG9zQHV2YS5lcw==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.