 Xiaosen Ma1
Xiaosen Ma1 Chao Ling2
Chao Ling2 Meng Zhao3
Meng Zhao3 Fen Wang1,4
Fen Wang1,4 Yunying Cui1
Yunying Cui1 Jin Wen5
Jin Wen5 Zhigang Ji5Caili Zhang6
Zhigang Ji5Caili Zhang6 Shi Chen1
Shi Chen1 Anli Tong1*Yuxiu Li1
Anli Tong1*Yuxiu Li1- 1Key Laboratory of Endocrinology, Department of Endocrinology, National Health Commission of the People’s Republic of China, Peking Union Medical College Hospital, Peking Union Medical College, Chinese Academy of Medical Sciences, Beijing, China
- 2The Laboratory of Clinical Genetics, Peking Union Medical College Hospital, Peking Union Medical College, Chinese Academy of Medical Sciences, Beijing, China
- 3Bioinformatics Institute, Novogene Co., Ltd., Beijing, China
- 4Department of Endocrinology, Tongji Hospital, Tongji Medical College, Huazhong University of Science and Technology, Wuhan, China
- 5Department of Urology, Peking Union Medical College Hospital, Peking Union Medical College, Chinese Academy of Medical Sciences, Beijing, China
- 6Department of Technical Support, Novogene Co., Ltd., Beijing, China
Purpose: Pheochromocytoma/paraganglioma (PCC/PGL; collectively known as PPGL) can be driven by germline and somatic mutations in susceptibility genes. We aimed to investigate the mutation profile and clinical features of pathogenic genes in highly genetically heterogeneous PPGL and to preliminary explore molecular therapeutic targets in PPGL.
Methods: We established a panel of 260 genes, including susceptibility genes of PPGL and other important tumorigenic genes to sequence 107 PPGL tissues.
Results: Overall, 608 genomic mutations were identified in 107 PPGL tissues. Almost 57% of PPGL tissue samples exhibited pathogenic mutations, and the most frequently mutated gene was SDHB (15/107, 14%). SDHB and HRAS were the most commonly mutated genes in germline-mutated PPGL (25/107, 23%) and nongermline-mutated PPGL (36/107, 34%), respectively. In addition, novel pathogenic mutations were detected in sporadic PPGL. PPGL with mutations in the hypoxia pathway had an earlier onset and higher norepinephrine level than those in the kinase pathway. Receptor tyrosine kinase (RTK; 22%, 24/107), mitogen-activated protein kinase (MAPK; 14%, 15/107), and tyrosine kinase (TK; 2%, 2/107) pathways were the most frequently mutated pathways in PPGL.
Conclusion: Our results provided the genetic mutation profile in PPGL tissues. Genetic mutations in PPGL were mainly concentrated in the RTK, TK, and MAPK pathways, suggesting potential molecular therapeutic targets for PPGL.
Introduction
Pheochromocytoma and paraganglioma (PCC/PGL; collectively known as PPGL) is a rare neuroendocrine tumor originating from chromaffin cells of the adrenal medulla or extra-adrenal paragangliomas. It represents a subset of tumors with high genetic heterogeneity. Approximately 40% cases of PPGL are caused by germline mutations in more than 20 known susceptibility genes (1). The inherent genetic mutations in PPGL fall into two major clusters in terms of their gene expression profile in tumors. Cluster I includes VHL, SDHx, IDH, EPAS1, MDH2, PHD2, and FH genes. Mutations in these genes cause activation of pseudo-hypoxic pathway and accumulation of hypoxia-inducible factor-1α, characteristically leading to a high expression of vasoendothelial growth factor and its receptors. Cluster II involves RET, NF1, MAX, and TMEM127 genes, which are associated with abnormal activation of the kinase signaling pathway, such as the phosphatidylinositol 3-kinase (PI3K)-Akt-mechanistic target of rapamycin (mTOR) pathways (2). Although the hereditary profile of PPGL has been well characterized, the clinical and genetic features of PPGL in Chinese population are not clearly described.
Apart from the studies on germline-susceptibility-associated mutations in PPGL, some studies have examined somatic mutations in PPGL cases. Somatic mutations, including NF1, RET, VHL, EPAS1, ATRX, HRAS, CSDE1, IDH1, BRAF, FGFR1, TP53, SETD2, and ARNT, were reported to be related to the development of PPGL (3, 4). Castro-Vega et al. conducted whole-exome sequencing (WES) of 30 tumor–normal DNA pairs and identified 672 somatic variants in coding regions or at exon/intron boundaries, with few nongermline mutations in genes that were not reported previously in PPGL (4). It has been widely accepted that PPGL is mainly driven by distinct germline or somatic mutations in known susceptibility genes.
Moreover, there is still a substantial fraction of PPGL whose etiology is largely unknown. WES allows direct measurement of the status of tumor mutations. However, it remains unsuitable for screening of large quantities of samples because it is expensive, labor-intensive, and time-consuming. Most of the recurrent somatically mutated genes in PPGL were reported previously, and few novel gene mutations were identified in some studies (4–7). Therefore, the limited number of genes involved in the tumorigenesis of PPGL render it possible to establish a gene panel that is equally accurate as WES (8, 9). In this study, we established a panel of 260 genes; this included recurrently reported genes of PPGL, genes that were probably relevant for PPGL but were not reported (such as genes involved in the tricarboxylic acid cycle, pseudohypoxia, and kinase signaling pathways), and reported genes in neuroendocrine and other tumors. We sequenced the genes in 107 PPGL tissues to assess the mutational and clinical profile of PPGL and explored the possible molecular therapeutic targets of PPGL.
Materials and Methods
Patients and Sample Collection
A total of 107 PPGL tissue samples were collected from 107 patients who had undergone surgical resection at the Peking Union Medical College Hospital, Beijing, China. None of the patients had received chemotherapy or radiotherapy before the surgery. All the pathological diagnoses were reviewed by two pathologists. PPGL tissues were collected after obtaining informed consent from patients. Approval was obtained from the medical ethics committee of the said hospital.
Genomic DNA Preparation and Target Sequencing
DNA was extracted from freshly frozen tumor tissues (wet weight 20–25 mg) using the QIAamp®Genomic DNA mini kit (QIAGEN, CA, USA) as per the manufacturer’s instructions. The probes of target regions for 260 genes (Supplementary Table S1) were designed against Agilent available online (http://www.agilent.com). The sequence library was generated using the Agilent SureSelectXT custom kit (Agilent Technologies, Palo Alto, CA) as per the manufacturer’s instructions. Briefly, fragmentation was performed using a hydrodynamic shearing system (Covaris, Massachusetts, USA) to generate 180–280 bp fragments. The DNA fragments were end-repaired, A-tailed, and adapter-ligated for Illumina sequencing. Further, size selection, PCR amplification, and library hybridization were performed. Each captured library with an index was loaded onto the Illumina HiSeq X platform (Illumina Inc., San Diego, CA), and 150-bp paired-end reads were generated.
Quality Control, Detection and Filtering of Genomic Alterations
The fastq files were subjected to quality control to exclude undesirable sequences, including those with adapter contamination or low-quality or unrecognizable nucleotides. The fastq files were aligned against the Human Reference Genome (UCSC hg19) using the Burrows-Wheeler Aligner. Further, SAM (Sequence Alignment/Map) tools (10) were used to sort the BAM files and to perform duplicate marking, local realignment, and base quality recalibration to generate the final BAM file (11) for computing the sequence coverage and depth. ANNOVAR was used to annotate the Variant Call Format file obtained in the previous step (12).
The genomic mutations were screened as follows:
1) Mutations with coverage less than 10× were excluded.
2) Variants with mutant allele frequency of ≥ 0.01 in the 1,000 Genomes databases (1000g2015aug_Chinese, 1000g2015aug_eas, 1000g2015aug_all, esp6500siv2_all, ExAC_ALL, ExAC_EAS, NovoDb_WES and NovoDb_WGS) were excluded (13, 14).
3) The variants in the exonic or splicing region (10 bp upstream and downstream of splicing sites) were retained.
4) The synonymous mutations were excluded.
5) Nonsynonymous single nucleotide variants (SNVs) were retained if at least two of the functional predictions by PolyPhen-2, SIFT, MutationTaster, and CADD revealed that the SNV was pathogenic or likely pathogenic, was included in the COSMIC database, or was predicted by ACMG to be “pathogenic or likely pathogenic” (15–18).
6) “Benign” or “likely benign” variants in ClinVar were excluded.
In addition, actionable mutations, defined as variants potentially linked to drugs in registered clinical trials, were detected by searching My Cancer Genome database (https://www.mycancergenome.org).
Mutation Validation in Blood Samples
Since susceptibility germline mutations were prevalent in patients with PPGL, we identified germline mutations in the cohort. If the mutated genes were among the 25 germline susceptibility genes (Supplementary Table S2), Sanger sequencing was performed to confirm whether the mutations in tumor tissues were also present in the corresponding blood samples. Accordingly, samples with known pathogenic germline mutations were detected from 107 PPGL tissues (Supplementary Figure 1).
Statistical Analysis
Continuous variables with normal distribution were expressed as means ± standard deviation, and those with non-normal distribution were expressed in median (25%, 75%). Differences between two groups with parametric and nonparametric variables were assessed using Student’s t and Mann–Whitney U tests, respectively. Categorical data were reported as numbers or corresponding percentages, and two groups were compared using χ2 test. P < 0.05 was considered statistically significant. SPSS version 23.0 for Windows was used for all statistical analyses.
Results
Clinical and Sequencing Data Obtained From PPGL Tissues
A total of 107 PPGL tissues were analyzed, involving tumors from 50 male and 57 female patients, with a mean age of 40 ± 14 years (range: 6–64 years). Overall, 8 multiple PPGL, 54 PGL, and 45 PCC samples were included in our cohort. In total, 8 of 107 cases had PPGL with distant metastasis (Table 1 and Supplementary Table S3).
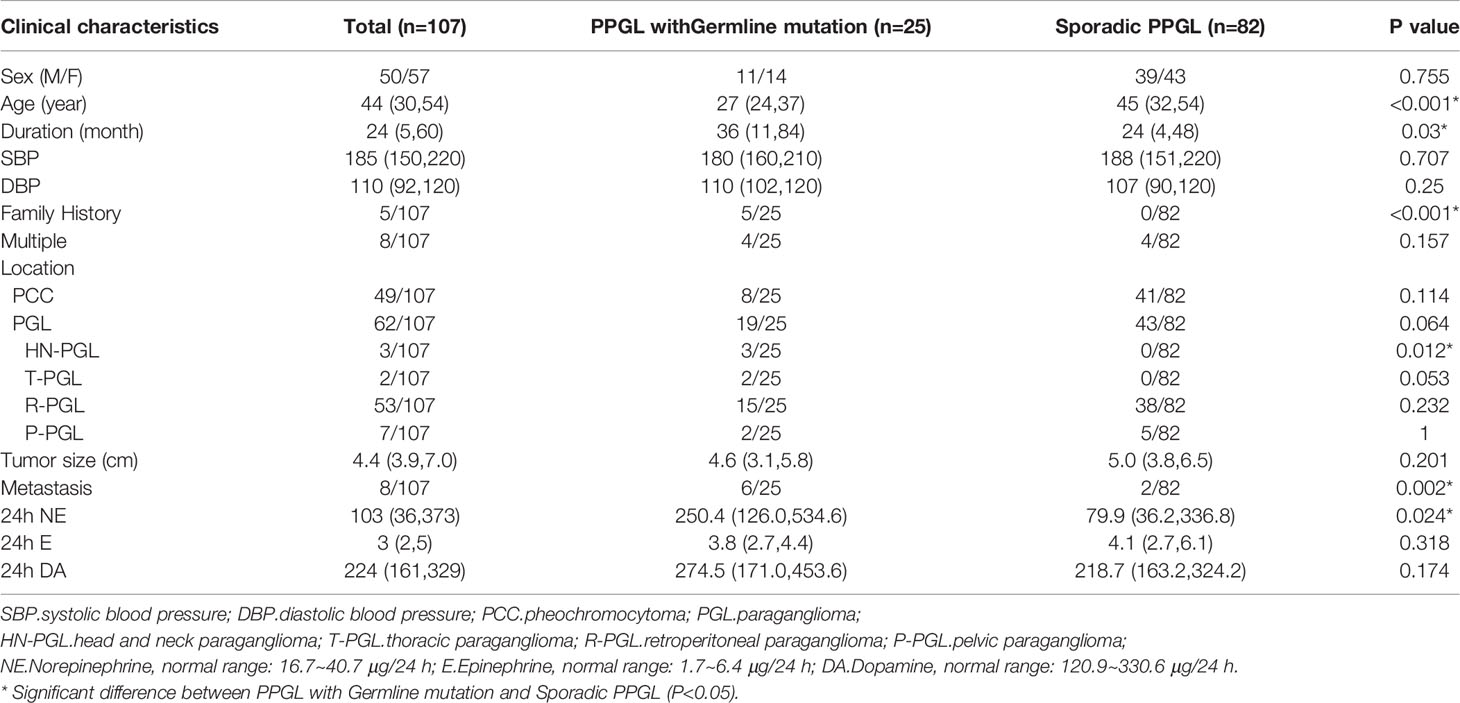
Table 1 Clinical features of PPGL.
We achieved an average sequencing depth of 346× (range 265–515×) in all samples. After multiple-step filtering, 608 genomic mutations were identified in the PPGL samples, including 528 missense, 17 stop-gain, 23 splicing, and 39 indel mutations (Supplementary Table S4). Moreover, one synonymous SNV (VHL: c.A414G, p.P138P) that was reported to be pathogenic was included (19). The overall frequency was 3.8 (range 0–7) mutations per Mb in tumor tissues upon the aforementioned filtration.
Germline Mutations and Nongermline Mutations in PPGL
Predisposing germline mutations were identified in 25 (23%) of 107 patients. The most frequent mutations in 25 patients occurred in SDHB (15/25, 60%) and followed by RET (2/25, 8%), SDHA (2/25, 8%). Germline mutations in VHL, SDHD, NF1, FH, MDH2, and TMEM127 were rare, standing at 4% (1/25) each (Table 2 and Figure 1).
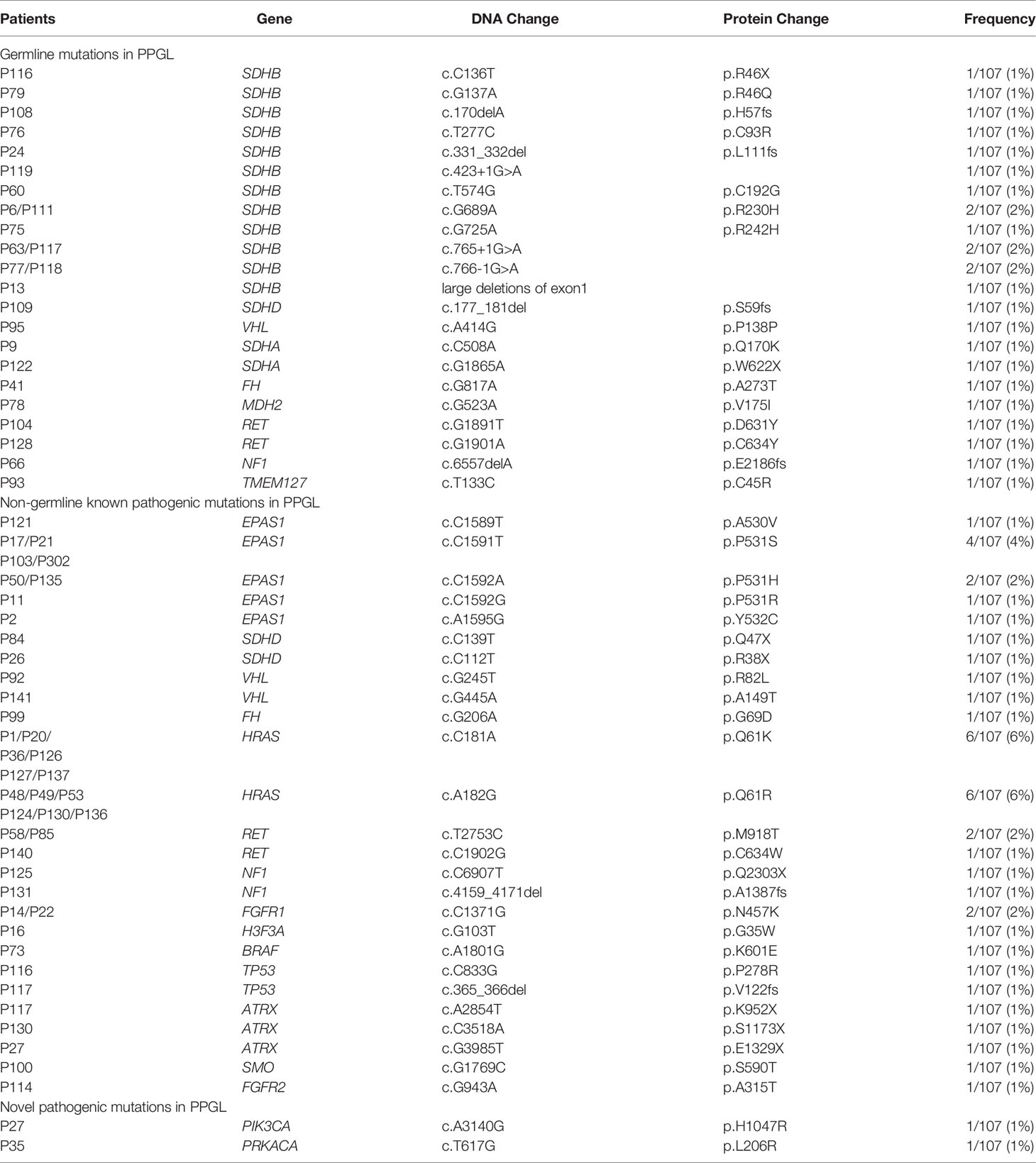
Table 2 Pathogenic mutations in PPGL.
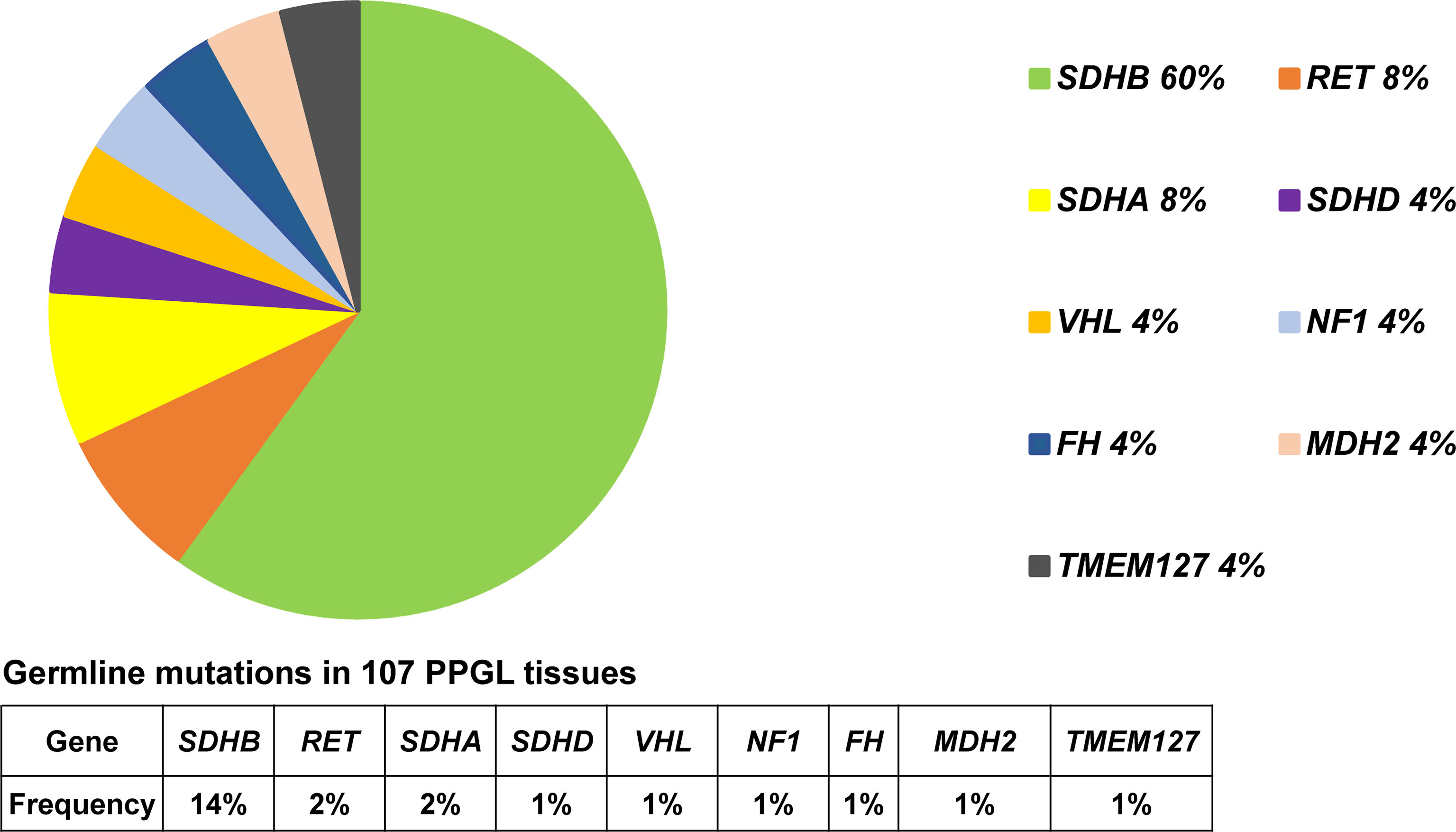
Figure 1 Pie diagram showing the distribution of germline mutations in PPGL. Circular statistical graphic was used to illustrate the proportion of germline mutations.
Clinical features of PPGL with or without germline mutation were shown in Table 1. PPGL with germline mutations often exhibited a family history of PPGL and a younger age of onset and were more likely to develop metastasis.
In 36 PPGL tissues without a recognizable germline mutation, 42 nongermline pathogenic variants were detected. The most frequently mutated gene was HRAS at a rate of 12/36 (33%), followed by EPAS1 (9/36, 25%), RET (3/36, 8%), ATRX (3/36, 8%), NF1 (2/36, 6%), SDHD (2/36, 6%), VHL (2/36, 6%), FGFR1 (2/36, 6%), TP53 (2/36, 6%), H3F3A (1/36, 3%), FH (1/36, 3%), FGFR2 (1/36, 3%), BRAF (1/36, 3%), and SMO (1/36, 3%; Table 2 and Figure 2).
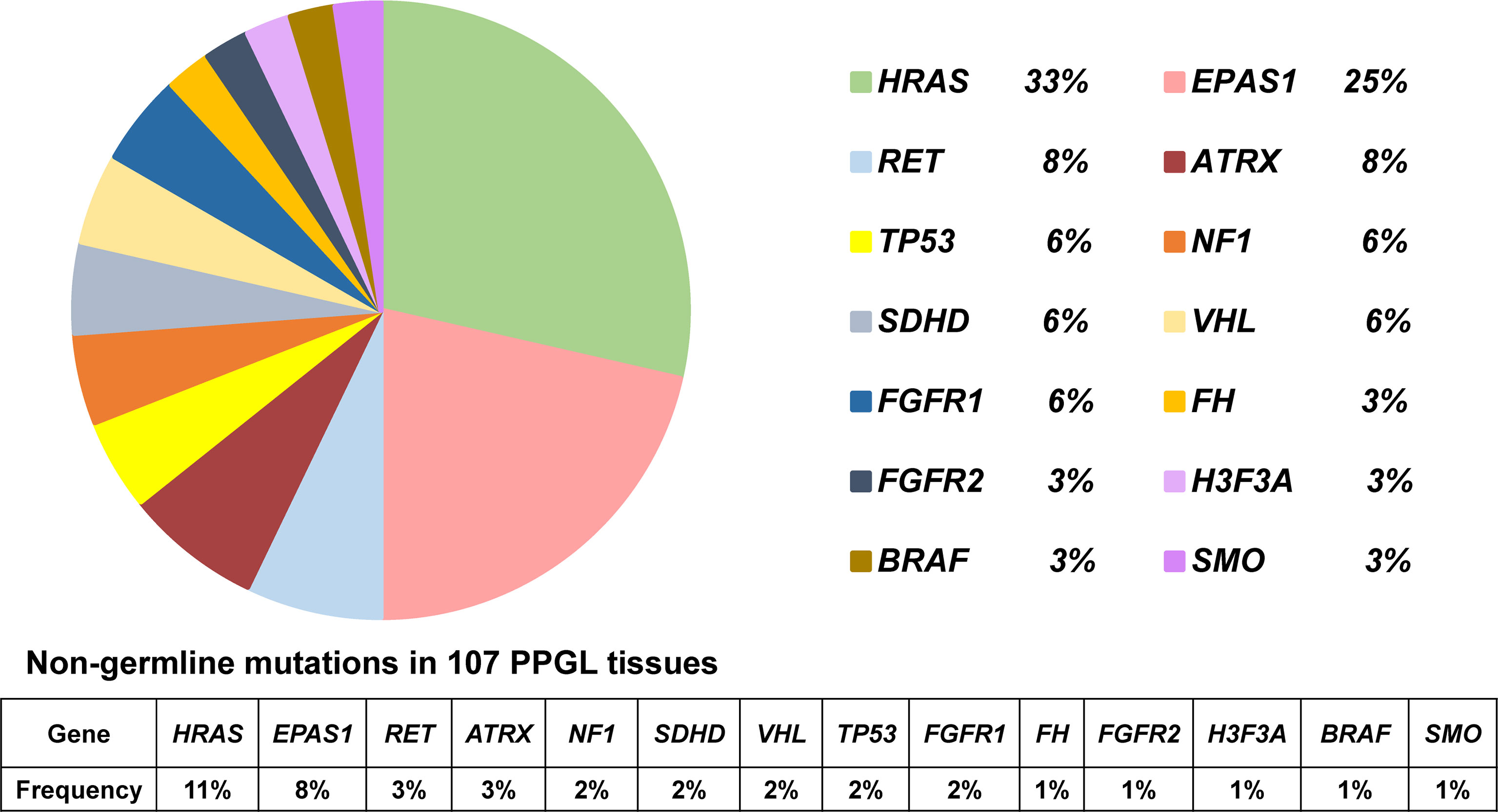
Figure 2 Pie diagram showing the distribution of nongermline mutations in PPGL. Circular statistical graphic was used to illustrate the proportion of nongermline mutations.
It should be noted that both a pathogenic SDHB germline mutation and TP53 somatic mutation were detected in PPGL lung metastases. SDHB germline pathogenic, TP53 somatic, and ATRX somatic variants were simultaneously detected in a primary PPGL tissue, which further developed distant metastasis. In addition, both PIK3CA and ATRX somatic variants were detected in a bladder PGL tissue. HRAS, ATRX, and BRAF somatic variants were concurrently detected in a retroperitoneal PGL tissue.
Novel Pathogenic Genes in PPGL
Novel pathogenic mutations such as PIK3CA (1/107, 1%) and PRKACA (1/107, 1%) were detected in two different patients with bladder PGL, respectively (Tables 1, 2). Both these mutations in PIK3CA and PRKACA were verified as somatic heterozygous variants with a mutation rate of 50.5% (95/188) and 36.9% (24/65), respectively. Interestingly, both PIK3CA and PRKACA were involved in the tyrosine kinase (TK) pathway.
Mutation Profile of PPGL With SDHB Germline Mutation
SDHB mutations were detected in 15 of 107 PPGL (Table 2 and Figures 1 and 2) tissues. Overall, 6 patients with SDHB mutations exhibited metastasis. Among the metastatic cases, 5 and 1 sample were from primary lesions and pulmonary metastatic focus, respectively. SDHB germline mutations were mainly concentrated in exon 2 and exon 7, including 7 missense, 2 frameshift, 1 stop-gain, and 5 splicing mutations.
Interestingly, mutations of TP53 (2/107, 1.9%) exclusively appeared in SDHB-mutated PPGL. All cases (2/107, 1.9%) with combined SDHB and TP53 mutations exhibited metastatic PPGL. In addition, ATRX, a risk factor associated with metastatic PPGL, was detected at a rate of 2.8% (3/107). ATRX somatic and SDHB germline mutations were both detected in a case of metastatic PGL. Simultaneous mutations in ATRX and PIK3CA were detected in a case of bladder PGL. Additionally, pathogenic mutations (ATRX, HRAS, and BRAF) were detected in the same PGL case with high levels of 24-h urinary norepinephrine and epinephrine.
Clinical Features of PPGL in Different Pathways
Pathogenic mutations of PPGL were clustered in the hypoxia (n = 35) and kinase signaling (n = 26) pathways. PPGL with mutations in the hypoxia pathway had an earlier onset, higher norepinephrine levels, and was more likely to develop PGL than that with mutations in the kinase signaling pathway. PPGL with mutations in the kinase signaling pathway exhibited a higher epinephrine level (Table 3).
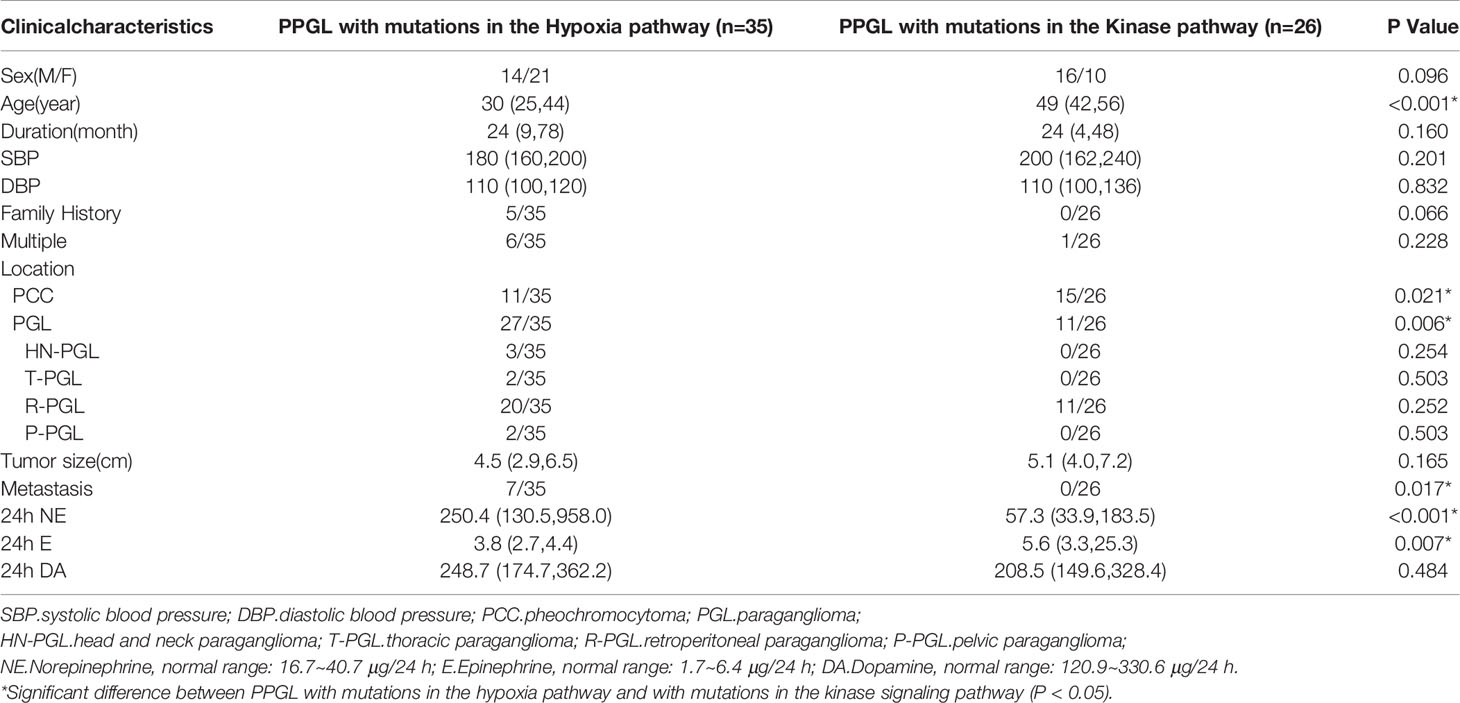
Table 3 Comprison of clinical characteristics of PPGL in different pathways.
To explore whether the differences were due to the high proportion of germline mutations in the hypoxia pathway, we compared the two pathways in nongermline-mutated PPGL. Surprisingly, the disparities in onset age and norepinephrine level persisted (Table 4). Therefore, we concluded that PPGL associated with the hypoxia pathway had an earlier onset and higher norepinephrine level than that with the kinase signaling pathway irrespective of germline or nongermline level. However, in the hypoxia pathway, no significant differences were detected between PPGL with germline and somatic mutation (Table 5).
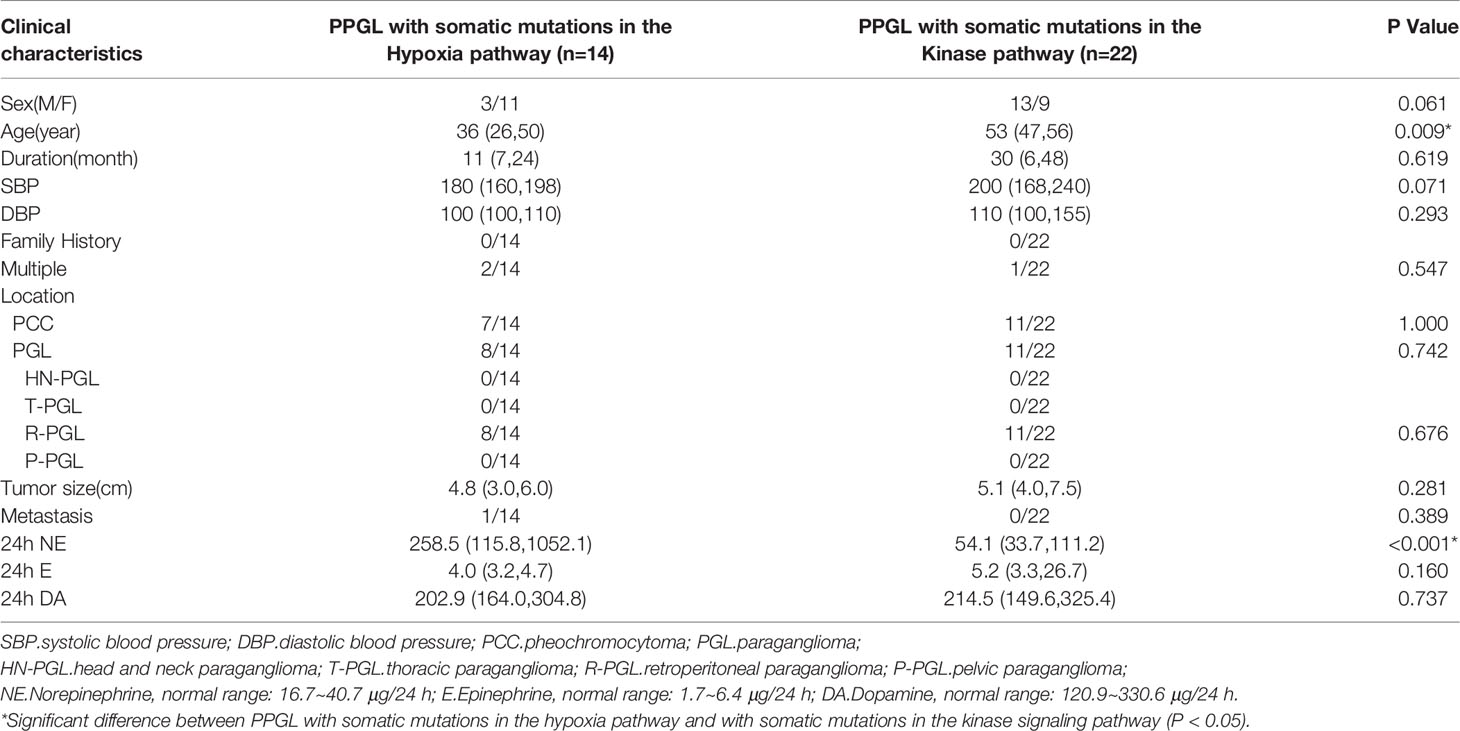
Table 4 Comparison of clinical characteristics of PPGL with somatic mutations in different pathways.
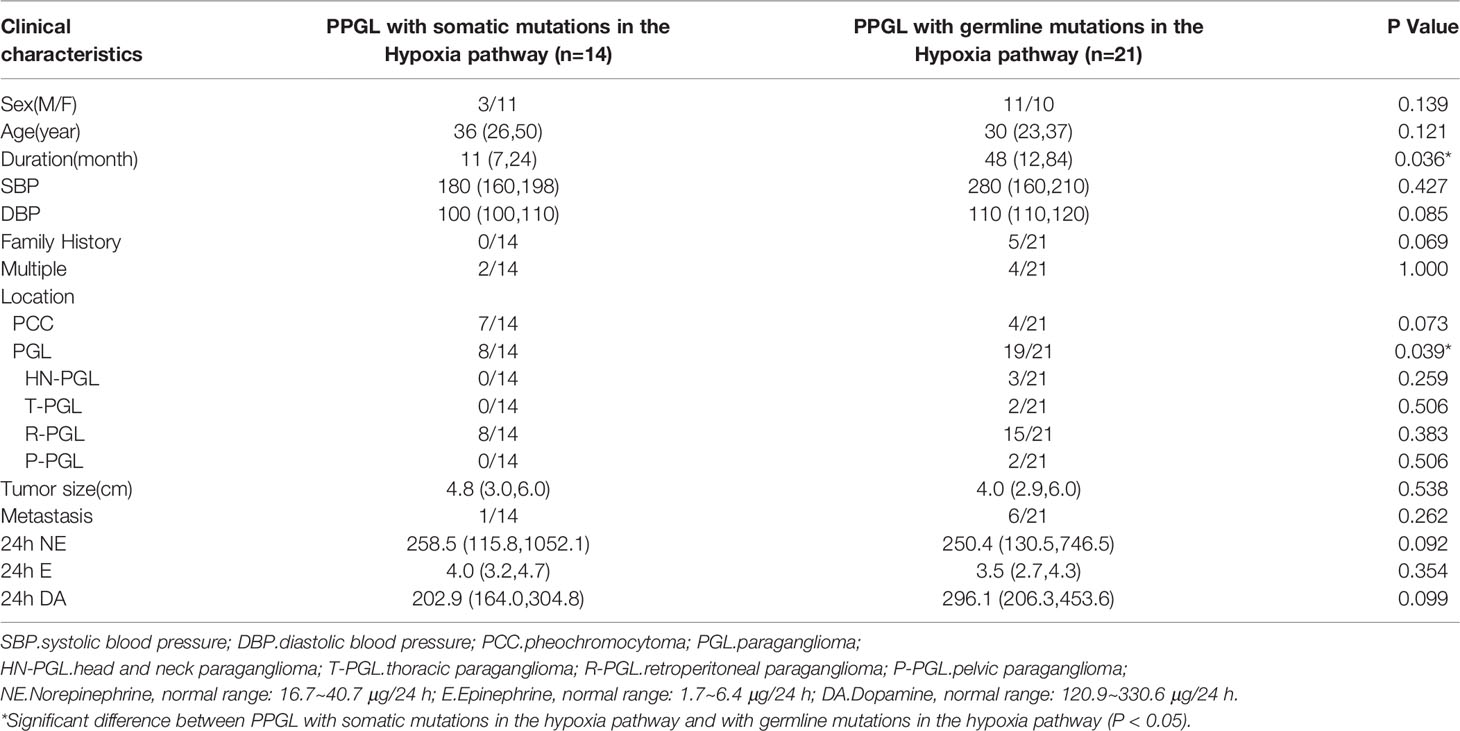
Table 5 Comparison of clinical characteristics of PPGL in the hypoxia pathway.
Actionable Gene Mutations in PPGL
Overall, 90 cases of PPGL (84%) exhibited at least one potentially actionable mutation in genes, including MUC16 (33/107, 31%), HRAS (13/107, 12%), LRP1B (9/107, 8%), TCS1 (8/107, 7%), RET (7/107, 7%), TCS2 (7/107, 7%), KMT2D (7/107, 7%), ACO2 (6/107, 6%), and NTRK1 (6/107, 6%). Only 17 (16%) PPGL cases exhibited no actionable mutations (Supplementary Figure 2). Actionable mutations in 13 genes were proved to be linked to targeted therapy in clinical trials in other tumor types (not PPGL). Interestingly, receptor tyrosine kinase (RTK), TK, and mitogen-activated protein kinase (MAPK) pathways were the most frequently involved pathways with a rate of 39% (42/107; Table 6).

Table 6 Actionable genomic variants in 107 PPGL.
Discussion
We established an expanded panel of 260 genes from known pathogenic pathways associated with PPGL, neuroendocrine tumors, and other cancers to explore the mutation spectrum in PPGL tissues. Our study helped to shed light on the mutation characteristics of PPGL in Chinese population. Furthermore, novel pathogenic mutations in PPGL tissues were observed. Clinical characteristics of PPGL with or without germline mutation and PPGL with mutated genes from various pathways were analyzed. Besides, molecular findings that could possibly serve for targeted therapy were identified.
Mutation profiling was different in several cohorts (Table 7). It was worth noting that SDHx was more frequently mutated, whereas FGFR1 was less frequently mutated in our study than the preceding cohort from China (18.7% vs. 4.4%, P < 0.05; 1.9% vs. 9.2%, P < 0.05, respectively) (20). SDHx mutations were common in PGL and metastatic PPGL, whereas FGFR1 mutations were expected in sporadic PGL. The proportion of PGL and metastatic PPGL in cohorts could explain the disparities in mutation frequency. Furthermore, by integrating our results with those of Jiang et al. (Table 7), we discovered that the mutation frequency of EPAS1 was much higher and that of NF1 was significantly lower in Chinese cohorts than in European cohorts. The different mutation rates of EPAS1 and NF1 might arise from racial differences.
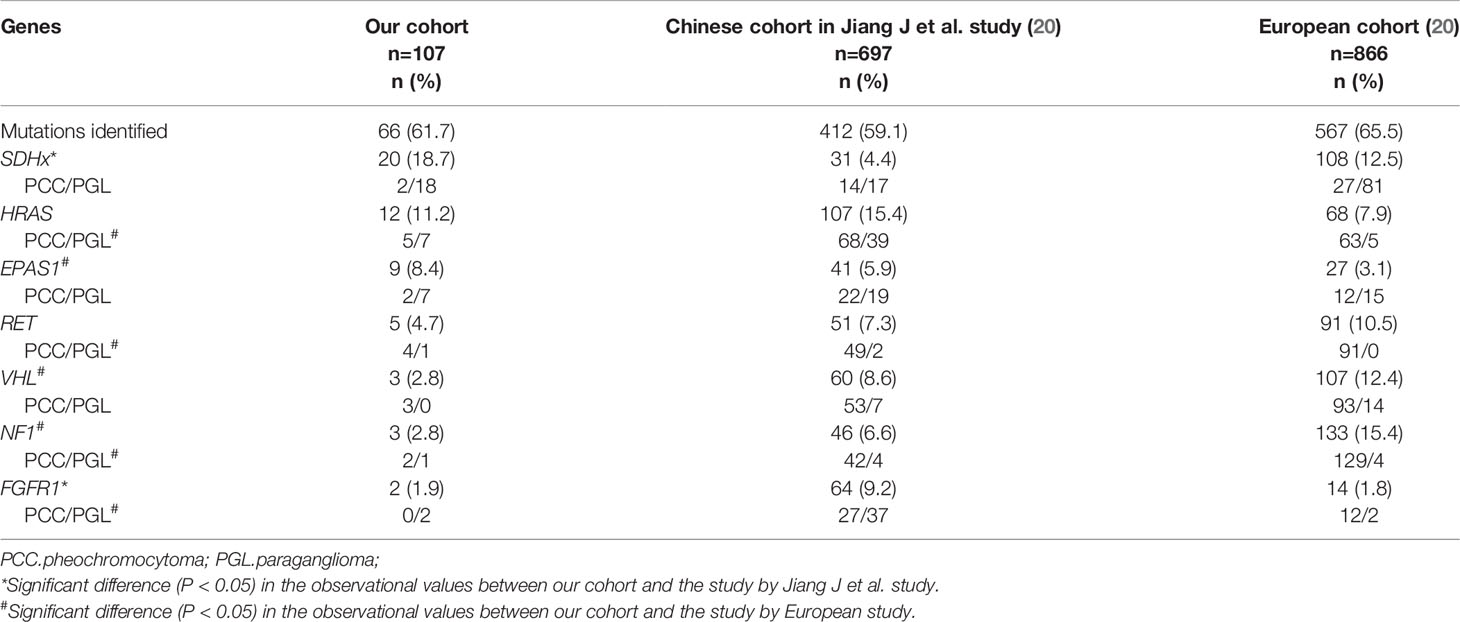
Table 7 Comprison of gene mutation frequency between different cohorts.
In addition, some hotspot mutations that were common in other solid tumors were detected in PPGL in our study. These genes were mutually exclusive with germline mutations in disease-causing genes. The hotspot mutation H1047R in oncogenic PIK3CA was often detected in breast cancer (21), which enhances the enzymatic activity of PI3K and activates the AKT/mTOR signaling pathway. Hotspot mutation L206R in PRKACA, which encodes the catalytic subunit of cyclic-adenosine-monophosphate-dependent protein kinase, was previously reported in adrenocortical adenomas associated with corticotropin-independent Cushing’s syndrome (22). This suggested that the pathogenic mechanism in some common solid tumors should not be ignored in PPGL to provide more possibilities for selecting the treatments for PPGL.
By comparing PPGL cases with mutations in different pathways, we discovered that PPGL with mutations in the hypoxia pathway had an earlier onset age and a higher norepinephrine level at the germline and somatic levels. Indeed, although the kinase signaling pathway were proved to be interconnected with the hypoxia pathway in many cancers (23), PPGL with mutations in the kinase signaling pathway still exhibited different clinical characteristics, such as the tumor location, metastasis, and catecholamine secretion, compared with PPGL with mutations in the hypoxia pathway. Recent studies have reported that mutated genes in the hypoxia pathway reduced the expression of PNMT (24), leading to reduced production of epinephrine, which could account for the lower epinephrine level in PPGL with mutations in the hypoxia pathway.
TP53 pathogenic mutations only occurred in SDHB-mutated metastatic PPGL. Overall, 2 patients with a combination of SDHB and TP53 mutations exhibited local recurrence and distant metastasis after surgical resection. Similarly, several studies detected simultaneous occurrence of SDHB and TP53 mutations in metastatic PPGL (4, 25). In addition, ATRX loss of function mutations, which were associated with poor prognosis in PPGL (26, 27), were detected in a case of metastatic, bladder, and abdominal PGLs each. In the follow-up, the outcome (especially recurrence or metastasis) of these patients with PPGL should be closely observed.
In our study, RTK, TK, and MAPK were the most commonly activated pathways in PPGL. These pathways are involved in cell proliferation and transformation and are closely associated with tumorigenesis. Our results indicated that the inhibitors of TK and MAPK could be the most effective targeted therapy for PPGL. At present, targeted therapy for PPGL has been a research hotspot; however, only a few clinical trials or studies have been completed (28–30). Among them, sunitinib was the most intensively studied and reported to have good efficacy. In 2012, Ayala-Ramirez et al. retrospectively studied 17 patients with metastatic PPGL treated with sunitinib. Imaging evaluation revealed that 21.4% patients attained partial response and 35.7% had stable disease (31). Several clinical trials are now underway to further confirm the efficacy of sunitinib therapy in patients with progressive PPGL (NCT01371201, NCT00843037). Moreover, phase II clinical trials are underway for axitinib and lenvatinib therapy for PPGL (NCT01967576, NCT03008369 and NCT03839498) (32).
Conclusion
We detected genetic mutations in 107 cases of PPGL and obtained the specific mutational profile of PPGL at our center. Our results indicated that mutations in pathogenic genes in the common tumors could also be detected in rare PPGL tumors. In addition, PPGL with mutated genes in the hypoxia pathway had earlier onset age and higher level of norepinephrine than PPGL with mutated genes in the kinase signaling pathway. This feature was of great significance at the germline and somatic levels. Finally, new molecular findings that could provide important information for targeted therapy were identified. Mutations in PPGL were mainly concentrated in the RTK, TK, and MAPK pathways, suggesting the potential molecular therapeutic targets in PPGL.
Data Availability Statement
The data presented in the study are deposited in the Genome Sequence Archive in National Genomics Data Center, Beijing Institute of Genomics (China National Center for Bioinformation), Chinese Academy of Sciences, accession number HRA000415.
Ethics Statement
The studies involving human participants were reviewed and approved by Ethics Committee of Peking Union Medical College Hospital. Written informed consent to participate in this study was provided by the participants’ legal guardian/next of kin.
Author Contributions
AT was contributed to the study conception and design. Material preparation, data collection were performed by AT, XM, FW, YC, JW, ZJ, SC and YL. Data analysis was performed by XM, CL, MZ, CZ, and AT. The first draft of the manuscript was written by XM and revised by AT. All authors commented on previous versions of the manuscript. All authors read and approved the final manuscript. All authors contributed to the article and approved the submitted version.
Funding
This research was funded by CAMS Innovation Fund for Medical Sciences (CIFMS), grant number 2021-I2M-C&T-B-002, 2017-I2M-1-001.
Conflict of Interest
Authors MZ and CZ were employed by the company Novogene Bioinformatics Technology Co., Ltd.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Acknowledgments
The authors are indebted to Dr. Zhengpei Zeng in Department of Endocrinology and Dr. Hanzhong Li in Department of Urology in Peking Union Medical College Hospital for providing expert advice.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fendo.2022.921645/full#supplementary-material
Supplementary Figure 1 | Study flow of 107 PPGL cases.
Supplementary Figure 2 | Actionable mutations in 107 PPGL cases.
References
1. Dahia PL. Pheochromocytoma and Paraganglioma Pathogenesis: Learning From Genetic Heterogeneity. Nat Rev Cancer (2014) 14:108–19. doi: 10.1038/nrc3648
2. Pillai S, Gopalan V, Smith RA, Lam AK. Updates on the Genetics and the Clinical Impacts on Phaeochromocytoma and Paraganglioma in the New Era. Crit Rev Oncol Hematol (2016) 100:190–208. doi: 10.1016/j.critrevonc.2016.01.022
3. Fishbein L, Leshchiner I, Walter V, Danilova L, Robertson AG, Johnson AR, et al. Comprehensive Molecular Characterization of Pheochromocytoma and Paraganglioma. Cancer Cell (2017) 31:181–93. doi: 10.1016/j.ccell.2017.01.001
4. Castro-Vega LJ, Letouze E, Burnichon N, Buffet A, Disderot PH, Khalifa E, et al. Multi-Omics Analysis Defines Core Genomic Alterations in Pheochromocytomas and Paragangliomas. Nat Commun (2015) 6:6044. doi: 10.1038/ncomms7044
5. Remacha L, Currás-Freixes M, Torres-Ruiz R, Schiavi F, Torres-Pérez R, Calsina B, et al. Gain-Of-Function Mutations in DNMT3A in Patients With Paraganglioma. Genet Med (2018) 20:1644–51. doi: 10.1038/s41436-018-0003-y
6. Urbini M, Nannini M, Astolfi A, Indio V, Vicennati V, De Luca M, et al. Whole Exome Sequencing Uncovers Germline Variants of Cancer-Related Genes in Sporadic Pheochromocytoma. Int J Genomics (2018) 2018:6582014. doi: 10.1155/2018/6582014
7. Remacha L, Pirman D, Mahoney CE, Coloma J, Calsina B, Curras-Freixes M, et al. Recurrent Germline DLST Mutations in Individuals With Multiple Pheochromocytomas and Paragangliomas. Am J Hum Genet (2019) 104:1008–10. doi: 10.1016/j.ajhg.2019.04.010
8. Rattenberry E, Vialard L, Yeung A, Bair H, McKay K, Jafri M, et al. A Comprehensive Next Generation Sequencing-Based Genetic Testing Strategy to Improve Diagnosis of Inherited Pheochromocytoma and Paraganglioma. J Clin Endocrinol Metab (2013) 98:E1248–56. doi: 10.1210/jc.2013-1319
9. Toledo RA, Burnichon N, Cascon A, Benn DE, Bayley JP, Welander J, et al. Consensus Statement on Next-Generation-Sequencing-Based Diagnostic Testing of Hereditary Phaeochromocytomas and Paragangliomas. Nat Rev Endocrinol (2017) 13:233–47. doi: 10.1038/nrendo.2016.185
10. Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, et al. The Sequence Alignment/Map Format and SAMtools. Bioinformatics (2009) 25:2078–9. doi: 10.1093/bioinformatics/btp352
11. Li H, Durbin R. Fast and Accurate Short Read Alignment With Burrows-Wheeler Transform. Bioinformatics (2009) 25:1754–60. doi: 10.1093/bioinformatics/btp324
12. Wang K, Li M, Hakonarson H. ANNOVAR: Functional Annotation of Genetic Variants From High-Throughput Sequencing Data. Nucleic Acids Res (2010) 38:e164. doi: 10.1093/nar/gkq603
13. Sherry ST, Ward MH, Kholodov M, Baker J, Phan L, Smigielski EM, et al. dbSNP: The NCBI Database of Genetic Variation. Nucleic Acids Res (2001) 29:308–11. doi: 10.1093/nar/29.1.308
14. Abecasis GR, Auton A, Brooks LD, DePristo MA, Durbin RM, Handsaker RE, et al. An Integrated Map of Genetic Variation From 1,092 Human Genomes. Nature (2012) 491:56–65. doi: 10.1038/nature11632
15. Adzhubei I, Jordan DM, Sunyaev SR. Predicting Functional Effect of Human Missense Mutations Using PolyPhen-2. Curr Protoc Hum Genet Chapter 7 (2013) 76:7.20.1-7.20.41. doi: 10.1002/0471142905.hg0720s76
16. Ng PC, Henikoff S. SIFT: Predicting Amino Acid Changes That Affect Protein Function. Nucleic Acids Res (2003) 31:3812–4. doi: 10.1093/nar/gkg509
17. Schwarz JM, Rodelsperger C, Schuelke M, Seelow D. MutationTaster Evaluates Disease-Causing Potential of Sequence Alterations. Nat Methods (2010) 7:575–6. doi: 10.1038/nmeth0810-575
18. Kircher M, Witten DM, Jain P, O'Roak BJ, Cooper GM, Shendure J. A General Framework for Estimating the Relative Pathogenicity of Human Genetic Variants. Nat Genet (2014) 46:310–5. doi: 10.1038/ng.2892
19. Flores SK, Cheng Z, Jasper AM, Natori K, Okamoto T, Tanabe A, et al. A Synonymous VHL Variant in Exon 2 Confers Susceptibility to Familial Pheochromocytoma and Von Hippel-Lindau Disease. J Clin Endocrinol Metab (2019) 104:3826–34. doi: 10.1210/jc.2019-00235
20. Jiang J, Zhang J, Pang Y, Bechmann N, Li M, Monteagudo M, et al. Sino-European Differences in the Genetic Landscape and Clinical Presentation of Pheochromocytoma and Paraganglioma. J Clin Endocrinol Metab (2020) 105:3295–307. doi: 10.1210/clinem/dgaa502
21. Liu P, Cheng H, Santiago S, Raeder M, Zhang F, Isabella A, et al. Oncogenic PIK3CA-Driven Mammary Tumors Frequently Recur via PI3K Pathway-Dependent and PI3K Pathway-Independent Mechanisms. Nat Med (2011) 17:1116–20. doi: 10.1038/nm.2402
22. Sato Y, Maekawa S, Ishii R, Sanada M, Morikawa T, Shiraishi Y, et al. Recurrent Somatic Mutations Underlie Corticotropin-Independent Cushing's Syndrome. Science (2014) 344:917–20. doi: 10.1126/science.1252328
23. Jochmanová I, Yang C, Zhuang Z, Pacak K. Hypoxia-Inducible Factor Signaling in Pheochromocytoma: Turning the Rudder in the Right Direction. J Natl Cancer Inst (2013) 105:1270–83. doi: 10.1093/jnci/djt201
24. Wang H, Cui J, Yang C, Rosenblum JS, Zhang Q, Song Q, et al. A Transgenic Mouse Model of Pacak⁻Zhuang Syndrome With An Epas1 Gain-Of-Function Mutation. Cancers (Basel) (2019) 11:667. doi: 10.3390/cancers11050667
25. Gniado E, Carracher CP, Sharma S. Simultaneous Occurrence of Germline Mutations of SDHB and TP53 in a Patient With Metastatic Pheochromocytoma. J Clin Endocrinol Metab (2019) 105:991–95. doi: 10.1210/clinem/dgz269
26. Job S, Draskovic I, Burnichon N, Buffet A, Cros J, Lépine C, et al. Telomerase Activation and ATRX Mutations Are Independent Risk Factors for Metastatic Pheochromocytoma and Paraganglioma. Clin Cancer Res (2019) 25:760–70. doi: 10.1158/1078-0432.Ccr-18-0139
27. Monteagudo M, Martínez P, Leandro-García LJ, Martínez-Montes Á M, Calsina B, Pulgarín-Alfaro M, et al. Analysis of Telomere Maintenance Related Genes Reveals NOP10 as a New Metastatic-Risk Marker in Pheochromocytoma/Paraganglioma. Cancers (Basel) (2021) 13:4758. doi: 10.3390/cancers13194758
28. Economides MP, Shah AY, Jimenez C, Habra MA, Desai M, Campbell MT. A Durable Response With the Combination of Nivolumab and Cabozantinib in a Patient With Metastatic Paraganglioma: A Case Report and Review of the Current Literature. Front Endocrinol (Lausanne) (2020) 11:594264. doi: 10.3389/fendo.2020.594264
29. Heregger R, Huemer F, Hutarew G, Hecht S, Cheveresan L, Kotzot D, et al. Sustained Response to Brigatinib in a Patient With Refractory Metastatic Pheochromocytoma Harboring R1192P Anaplastic Lymphoma Kinase Mutation: A Case Report From the Austrian Group Medical Tumor Therapy Next-Generation Sequencing Registry and Discussion of the Literature. ESMO Open (2021) 6:100233. doi: 10.1016/j.esmoop.2021.100233
30. Moog S, Salgues B, Braik-Djellas Y, Viel T, Balvay D, Autret G, et al. Preclinical Evaluation of Targeted Therapies in Sdhb-Mutated Tumors. Endocr Relat Cancer (2022) 29:375–88. doi: 10.1530/erc-22-0030
31. Ayala-Ramirez M, Chougnet CN, Habra MA, Palmer JL, Leboulleux S, Cabanillas ME, et al. Treatment With Sunitinib for Patients With Progressive Metastatic Pheochromocytomas and Sympathetic Paragangliomas. J Clin Endocrinol Metab (2012) 97:4040–50. doi: 10.1210/jc.2012-2356
Keywords: pheochromocytoma, paraganglioma, gene, mutational profile, potential molecular therapeutic targets
Citation: Ma X, Ling C, Zhao M, Wang F, Cui Y, Wen J, Ji Z, Zhang C, Chen S, Tong A and Li Y (2022) Mutational Profile and Potential Molecular Therapeutic Targets of Pheochromocytoma. Front. Endocrinol. 13:921645. doi: 10.3389/fendo.2022.921645
Received: 16 April 2022; Accepted: 07 June 2022;
Published: 28 July 2022.
Edited by:
Henrik Falhammar, Karolinska Institutet (KI), SwedenReviewed by:
Sergei Tevosian, University of Florida, United StatesCristina Lamas, Complejo Hospitalario Universitario de Albacete, Spain
Copyright © 2022 Ma, Ling, Zhao, Wang, Cui, Wen, Ji, Zhang, Chen, Tong and Li. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Anli Tong, dG9uZ2FubGlAaG90bWFpbC5jb20=