
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Endocrinol. , 16 August 2022
Sec. Adrenal Endocrinology
Volume 13 - 2022 | https://doi.org/10.3389/fendo.2022.921449
This article is part of the Research Topic Adrenal Related Hypertension: From Bench to Bedside View all 7 articles
 Celso E. Gomez-Sanchez1,2*
Celso E. Gomez-Sanchez1,2* Desmaré van Rooyen3,4
Desmaré van Rooyen3,4 William E. Rainey3,4
William E. Rainey3,4 Kazutaka Nanba3,5
Kazutaka Nanba3,5 Amy R. Blinder3
Amy R. Blinder3 Radhakrishna Baliga6
Radhakrishna Baliga6Aldosterone-producing adenoma is a rare cause of hypertension in children. Only a limited number of cases of aldosterone-producing adenomas with somatic KCNJ5 gene mutations have been described in children. Blacks are particularly more susceptible to developing long-standing cardiovascular effects of aldosterone-induced severe hypertension. Somatic CACNA1D gene mutations are particularly more prevalent in black males whereas KCNJ5 gene mutations are most frequently present in black females. We present here a novel somatic KCNJ5 p.I157S mutation in an aldosterone-producing adenoma from a 16-year-old black female whose severe drug-resistant hypertension significantly improved following unilateral adrenalectomy. Prompt diagnosis of aldosterone-producing adenoma and early identification of gene mutation would enable appropriate therapy and significantly reduce cardiovascular sequelae.
Primary aldosteronism (PA) is the most common secondary cause of hypertension (HTN) with a prevalence between 5-22% of adult hypertensive patients (1, 2). Two forms of PA are the most common presentations: namely aldosterone-producing adenomas (APA) accounting for 30-70% of patients and bilateral zona idiopathic hyperaldosteronism (IAH also accounting for 30-70% of patients with rare cases of unilateral hyperplasia, aldosterone-producing carcinomas and familial hyperaldosteronism accounting for 1-2% (1). Sporadic cases of PA are rare in the pediatric population (3–5). Only a limited number of cases have been reported in the pediatric literature, most of which have been of the familial type 3 (5–7) or presenting with a mosaicism (8) all bearing a mutation of the potassium voltage-gated channel subfamily J member 5 gene (KCNJ5). A somatic mutation of the KCNJ5 gene mutated at p.L168R was first described in a child with moderate to severe hypertension and hypokalemia and low renin with high aldosterone levels by Uchida et al. (3). In addition to mutations of the KCNJ5 channel, calcium (CACNA1D and CACNA1H) and chloride (CLCN2) channel gene mutations have been reported in adults and in familial cases and somatic mutations of the sodium ATPase pump (ATP1A1) and calcium pump (ATP2A3) mutations in adults have been described (9–12). Using a combination of CYP11B2 immunohistochemistry-guided DNA capture with sequencing has resulted in the identification of somatic mutations in approximately 94% of APAs (13). There is a racial and sex difference in the prevalence of the different mutations with KCNJ5 being more common in East Asians and women (10–13). We report here a novel somatic KCNJ5 mutation in a 16-year-old black female with APA whose HTN significantly improved following unilateral adrenalectomy.
16-year-old black women was noted to be hypertensive while being evaluated for depression. She was referred a year later for recurrent headaches, chronic drug resistant hypertension and persistent hypokalemia. Her therapeutic regimen consisted in lisinopril 40mg daily, amlodipine 10 mg daily, amiloride 15 mg daily, potassium chloride 40 mEq twice daily, labetalol 500 mg three times per day and minoxidil 5 mg. Family history was positive for a maternal uncle with hypertension. On examination, her weight was 98 kg [> 99%], height 178 cm [94%], BMI 36.00kg/m2, heart rate 88 beats per minute and blood pressure (BP) 155/94 mmHg [> 95%]. Pertinent laboratories: serum sodium 142, potassium 2.8, chloride 106, and CO2 content 26 mEq/L. Serum creatinine was 0.80 (estimated GFR 100 mL/mt/1.73m2), and BUN 10 mg/dL. Plasma aldosterone concentration (PAC) was 27.2 ng/dL and plasma renin activity (PRA) <0.6 ng/mL/h with PAC/PRA ratio of >45 [significant > 20]. Cortisol level was 8.7 [normal range (N) 1.7-14.1] μg/dl. Timed urine aldosterone for estimated urine creatinine of 1980 mg was 20 µg/d [N <12]. Liquid chromatography-tandem mass spectrometry (LC-MS/MS) quantification of 29 steroids in the pre-operatory serum was done (14). Serum levels of 18-oxocortisol and 18-hydroxycortisol were clearly elevated (Table 1) (15). Cardiac echocardiogram showed compaction cardiomyopathy. CT abdomen indicated a 1.3 x 2.7 cm right adrenal nodule measuring -16 Hounsfield units suggesting a benign adrenal adenoma. The contralateral adrenal had normal imagen characteristics. At the time of surgery her serum creatinine was 1.0 mg/dL (estimated GFR of 80 mL/mt/1.73m2). Written informed consent was obtained for surgery and pathological and genome studies. Robotic right adrenalectomy was performed, and pathology was consistent with an adrenocortical adenoma. Immunohistochemistry using a CYP11B2 monoclonal antibody (16) showed diffuse staining of the adenoma (APA) (Figures 1A, B) and the presence of at least one aldosterone-producing micronodule (Figure 1C). Genomic DNA samples from formalin-fixed paraffin embedded right adrenal tissue were sent for sequencing for germline or somatic mutations. CYP11B2 (aldosterone synthase) immunohistochemistry-guided targeted next-generation sequencing (NGS) of APA (11) revealed a somatic mutation in the KCNJ5 gene (nucleotide change c.T470G resulting in amino acid change p.I157S), with a variant allele frequency of 33%. The adjacent adrenal did not have the mutation. The results were confirmed by Sanger sequencing (Figure 1D) (11). Post-operatively PAC was < 3.0 ng/dL. Her headaches resolved; her blood pressure significantly improved to 124/69 mmHg [<90%] with normalization of her serum potassium. Six months after her right adrenalectomy BP was 108/63 mm Hg on a single antihypertensive agent and her serum potassium levels remain normal. Her repeat echocardiogram showed no significant change except for slight improvement in the left ventricular ejection fraction.
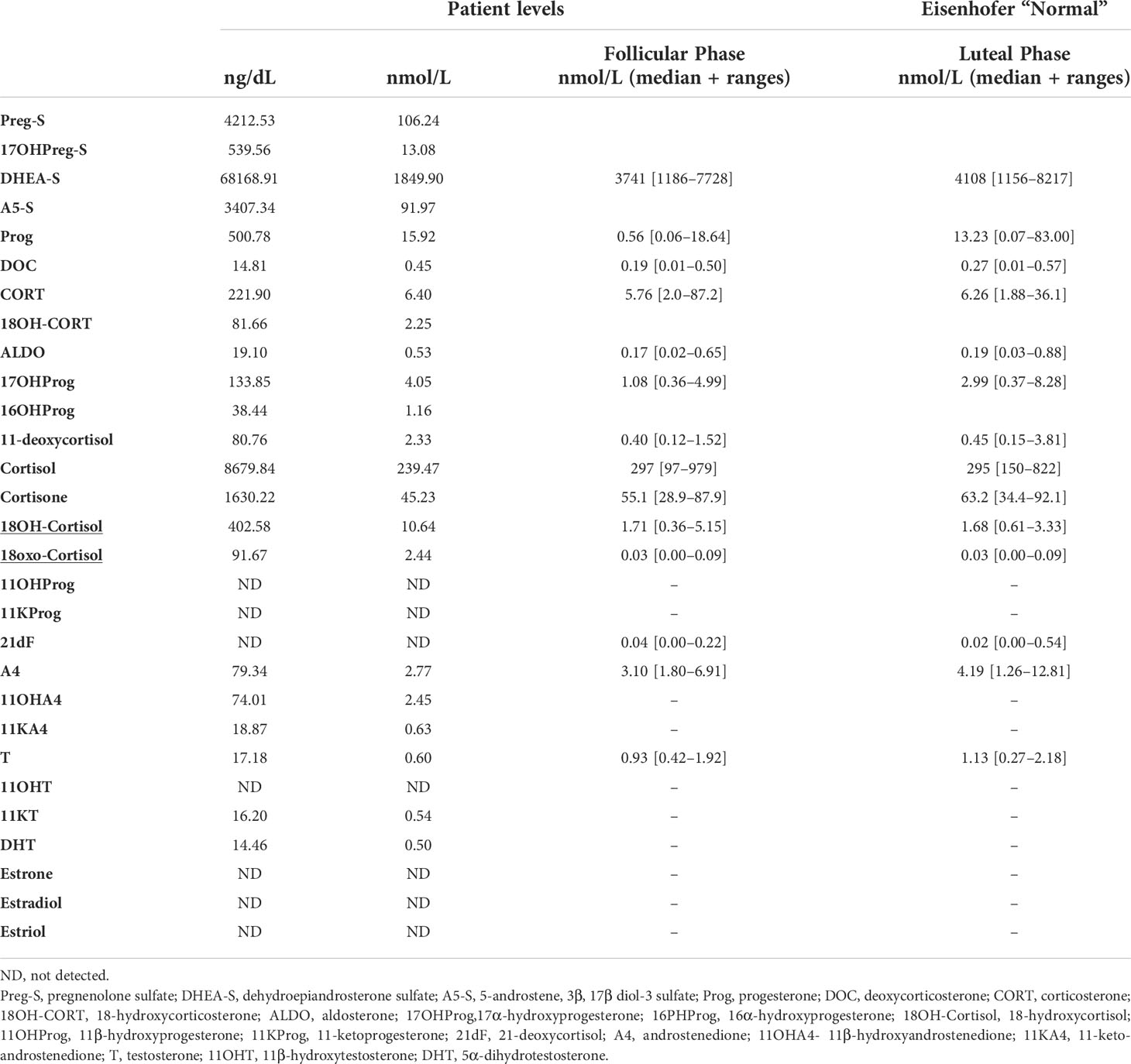
Table 1 LC-MS/MS quantification of 29 steroids in serum.
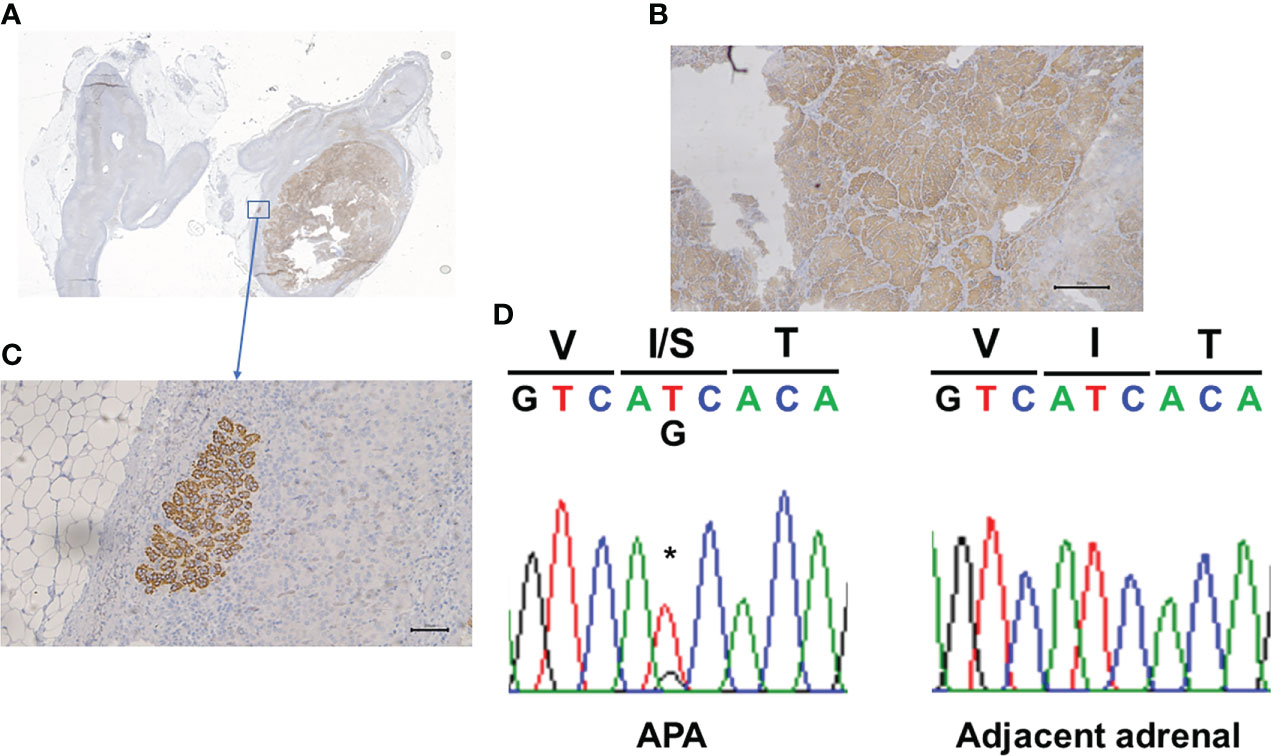
Figure 1 Panel (A, B) shows CYP11B2 immunohistochemistry demonstrating relative homogenous expression within the adrenal adenoma. Panel (C) shows the presence of an Aldosterone-Producing Cell Clusters. Panel (D) shows the Sanger demonstration of the mutation of nucleotide. *Site of the mutation.
HTN in the black population continues to be a significant cardiovascular (CV) risk factor with an overall increase in morbidity and mortality that has led to race specific clinical guidelines (17). African Americans appear to be more susceptible to the blood pressure effects of aldosterone (17). The prevalence of PA in African Americans in comparison to white individuals is not known. The pattern of somatic mutations in APA is different in the various ethnic groups and between sexes (10–13, 18). KCNJ5 mutations are more prevalent in East Asians, American and European white males and in females, including African Americans (10–13, 18), while CACNA1D mutations appear to be more prevalent in African American males (11).
Mutation in the KCNJ5 gene results in increased intracellular calcium concentration which activates the transcription of the aldosterone synthase gene CYP11B2 leading to autonomous aldosterone production (7, 19). Targeting the entire coding region for sequencing of genes mutated in APAs based on the tumor expression of CYP11B2, required for the final steps of aldosterone production, provides more accurate determination of the APA-related somatic mutations than the conventional mutation hot spot sequencing (10–12). Utilizing this technique, we sequenced DNA from the CYP11B2 positive tumor and CYP11B2 negative adjacent normal tissue and demonstrated the presence of somatic KCNJ5 mutation p.I157S in the tumor and not the adjacent normal area. This mutation has been previously characterized and reported only as a germline mutation in two patients from a single family. Both patients had severe primary hyperaldosteronism, early refractory HTN and bilateral massive adrenal hyperplasia (20) A single heterozygous thymine to guanine (T to G) substitution was noted at nucleotide position 470 (c.T470G) which resulted in isoleucine (I) to serine (S) substitution at the amino acid 157 (p.I157S). This mutation close to the channel pore has been shown to result in loss of potassium selectivity, cell membrane depolarization, increased Ca2 entry in adrenal glomerulosa cells and increased aldosterone production (20). A patient with uncontrolled bilateral aldosteronism treated with bilateral adrenalectomy was found to have 11 different adrenal nodules all of which had a disease-causing mutation in the KCNJ5 gene (p.G151R) and no family history of the mutation (8). Deep sequencing showed that 0.23% of germline DNA carried the same variant as the adrenal nodules (8) demonstrating the presence of a low grade mosaicism that manifested only in the adrenal nodules. We did not do deep sequencing but this is unlikely in this patient that had a single unilateral adenoma.
Kidney damage in APA can occur initially with the aldosterone-induced expansion of the intravascular fluid volume which can result in glomerular hyperfiltration. It can then result in intra glomerular hypertension leading to glomerular damage, albuminuria, and compromised kidney function. Our patient had prolonged persistent severe resistant HTN resulting in compromised kidney function that significantly improved following unilateral adrenalectomy. Mineralocorticoid receptor antagonist (MRA) has been shown to be reno-protective but was not initiated until prior to surgery due to the delay in diagnosis of her APA.
The outcome of patients following adrenalectomy for unilateral PA is found to be better in younger patients who were more likely to show complete clinical recovery when compared to older patients. Women as a whole have a greater chance of complete clinical success than men, however complete or partial success was limited in the presence of higher BMI and prior long standing untoward effects of vascular and renal changes resulting from long standing aldosterone induced HTN (21). In a study of Asian population with APA following unilateral adrenalectomy Nishikawa et al. (22) observed that those with a KCNJ5 mutation taking fewer anti hypertensives and with shorter duration of HTN had complete resolution of HTN with significant improvement in their LV hypertrophy when compared to the wild type. Our patient over a six-month period following unilateral adrenalectomy had significant improvement in her HTN requiring only Lisinopril. Her kidney function has also improved significantly being close to her baseline. However, her compaction cardiomyopathy and her LV ejection fraction continues to remain unchanged. Her BMI continues to remain high with her being black potentially contributing to the substantial delay in her successful clinical improvement.
In conclusion unilateral APA should be considered in any child presenting with drug resistant HTN with or without hypokalemia in order to ensure early diagnosis and adrenalectomy that would prevent significant CV sequelae. The identification of somatic and germline mutations will further provide insight into the mechanism of APA and assist in tailoring appropriate therapy particularly in blacks who appear to have higher CV disease morbidity and mortality resulting from autonomous aldosterone secretion.
The datasets for this article are not publicly available due to concerns regarding participant/patient anonymity. Requests to access the datasets should be directed to the corresponding author.
Ethical review and approval was not required for the study on human participants in accordance with the local legislation and institutional requirements. Written informed consent to participate in this study was provided by the participants’ legal guardian/next of kin.
CG-S conceived the study, performed experiments and wrote the paper. DR performed measurements, edited the paper, KN performed and interpreted the sequencing, edited the paper. AB performed and interpreted the sequenes and edited the paper, WR directed some of the experiments, edited the paper and RB studied the patient, wrote the initial paper and edited the final copy. All authors contributed to the article and approved the submitted version.
Research reported in this publication was supported by National Heart, Lung and Blood Institute grant R01 HL144847 (CEGS), the National Institute of General Medical Sciences grant U54 GM115428 (CEGS), Department of Veteran Affairs BX00468 (CEGS), the National Institute of Diabetes and Digestive and Kidney grant R01 DK43140 (WER), and a Japan Heart Foundation Grant (KN). The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
The authors would like to thank Dr. Aaron M. Udager and Chia-Jen Liu at the University of Michigan for next-generation sequencing.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Funder JW, Carey RM, Mantero F, Murad MH, Reincke M, Shibata H, et al. The management of primary aldosteronism: Case detection, diagnosis, and treatment: An endocrine society clinical practice guideline. J Clin Endocrinol Metab (2016) 101(5):1889–916. doi: 10.1210/jc.2015-4061
2. Brown JM, Siddiqui M, Calhoun DA, Carey RM, Hopkins PN, Williams GH, et al. The unrecognized prevalence of primary aldosteronism: A cross-sectional study. Ann Intern Med (2020) 173(1):10–20. doi: 10.7326/M20-0065
3. Uchida N, Amano N, Yamaoka Y, Uematsu A, Sekine Y, Suzuki M, et al. A novel case of somatic KCNJ5 mutation in pediatric-onset aldosterone-producing adenoma. J Endocr Soc (2017) 1(8):1056–61. doi: 10.1210/js.2017-00210
4. Tamura A, Nishimoto K, Seki T, Matsuzawa Y, Saito J, Omura M, et al. Somatic KCNJ5 mutation occurring early in adrenal development may cause a novel form of juvenile primary aldosteronism. Mol Cell Endocrinol (2017) 441:134–9. doi: 10.1016/j.mce.2016.07.031
5. Takizawa N, Tanaka S, Nishimoto K, Suguira Y, Suematsu M, Ohe C, et al. Familial hyperaldosteronism type 3 with a rapidly growing adrenal tumor: An In situ aldosterone imaging study. Curr Issues Mol Biol (2022) 44:128–38. doi: 10.3390/cimb44010010
6. Geller DS, Zhang J, Wisgerhof MV, Shackleton C, Kashgarian M, Lifton RP. A novel form of human mendelian hypertension featuring nonglucocorticoid-remediable aldosteronism. J Clin Endocrinol Metab (2008) 93(8):3117–23. doi: 10.1210/jc.2008-0594
7. Choi M, Scholl UI, Yue P, Bjorklund P, Zhao B, Nelson-Williams C, et al. K+ channel mutations in adrenal aldosterone-producing adenomas and hereditary hypertension. Sci (2011) 331(6018):768–72. doi: 10.1126/science.1198785
8. Maria AG, Suzuki M, Berthon A, Kamilaris C, Demidowich A, Lack J, et al. Mosaicism for KCNJ5 causing early-onset primary aldosteronism due to bilateral adrenocortical hyperplasia. Am J Hypertens (2019) 32(2):124–30. doi: 10.1093/ajh/hpz172
9. Mulatero P, Monticone S, Deinum J, Amar L, Prejbisz A, Zennaro MC, et al. Genetics, prevalence, screening and confirmation of primary aldosteronism: A position statement and consensus of the working group on endocrine hypertension of the European society of hypertension. J Hypertens (2020) 38(10):1919–28. doi: 10.1097/HJH.0000000000002510
10. Nanba K, Omata K, Else T, Beck PCC, Nanba AT, Turcu AF, et al. Targeted molecular characterization of aldosterone-producing adenomas in white americans. J Clin Endocrinol Metab (2018) 103(10):3869–76. doi: 10.1210/jc.2018-01004.
11. Nanba K, Omata K, Gomez-Sanchez CE, Stratakis CA, Demidowich AP, Suzuki M, et al. Genetic characteristics of aldosterone-producing adenomas in blacks. Hypertension (2019) 73(4):885–92. doi: 10.1161/HYPERTENSIONAHA.118.12070
12. Nanba K, Yamazaki Y, Bick N, Onodera K, Tezuka Y, Omata K, et al. Prevalence of somatic mutations in aldosterone-producing adenomas in Japanese patients. J Clin Endocrinol Metab (2020) 105(11):e4066–73. doi: 10.1210/clinem/dgaa595
13. De Sousa K, Boulkroun S, Baron S, Nanba K, Wack M, Rainey WE, et al. Genetic, cellular, and molecular heterogeneity in adrenals with aldosterone-producing adenoma. Hypertension (2020) 75(4):1034–44. doi: 10.1161/HYPERTENSIONAHA.119.14177
14. Turcu AF, Wannachalee T, Tsodikov A, Nanba AT, Ren J, Shields JJ, et al. Comprehensive analysis of steroid biomarkers for guiding primary aldosteronism subtyping. Hypertension (2020) 75(1):183–92. doi: 10.1161/HYPERTENSIONAHA.119.13866
15. Eisenhofer G, Duran C, Cannistraci CV, Peitzsch M, Williams TA, Riester A, et al. Use of steroid profiling combined with machine learning for identification and subtype classification in primary aldosteronism. JAMA Netw Open (2020) 3(9):e2016209. doi: 10.1001/jamanetworkopen.2020.16209
16. Gomez-Sanchez CE, Qi X, Velarde-Miranda C, Plonczynski MW, Parker CR, Rainey W, et al. Development of monoclonal antibodies against human CYP11B1 and CYP11B2. Mol Cell Endocrinol (2014) 383(1-2):111–7. doi: 10.1016/j.mce.2013.11.022
17. Tu W, Eckert GJ, Hannon TS, Liu H, Pratt LM, Wagner MA, et al. Racial differences in sensitivity of blood pressure to aldosterone. Hypertension (2014) 63(6):1212–8. doi: 10.1161/HYPERTENSIONAHA.113.02989
18. Lenzini L, Rossitto G, Maiolino G, Letizia C, Funder JW, Rossi GP. A meta-analysis of somatic KCNJ5 k(+) channel mutations in 1636 patients with an aldosterone-producing adenoma. J Clin Endocrinol Metab (2015) 100(8):E1089–95. doi: 10.1210/jc.2015-2149
19. Oki K, Plonczynski MW, Luis Lam M, Gomez-Sanchez EP, Gomez-Sanchez CE. Potassium channel mutant KCNJ5 T158A expression in HAC-15 cells increases aldosterone synthesis. Endocrinology (2012) 153(4):1774–82. doi: 10.1210/en.2011-1733
20. Charmandari E, Sertedaki A, Kino T, Merakou C, Hoffman DA, Hatch MM, et al. A novel point mutation in the KCNJ5 gene causing primary hyperaldosteronism and early-onset autosomal dominant hypertension. J Clin Endocrinol Metab (2012) 97(8):E1532–9. doi: 10.1210/jc.2012-1334
21. Williams TA, Lenders JWM, Mulatero P, Burrello J, Rottenkolber M, Adolf C, et al. Outcomes after adrenalectomy for unilateral primary aldosteronism: an international consensus on outcome measures and analysis of remission rates in an international cohort. Lancet Diabetes Endocrinol (2017) 5(9):689–99. doi: 10.1016/S2213-8587(17)30135-3
Keywords: primary aldosteronism, KCNJ5 mutation, aldosterone, pediatric hypertension, adrenal adenoma
Citation: Gomez-Sanchez CE, van Rooyen D, Rainey WE, Nanba K, Blinder AR and Baliga R (2022) Primary aldosteronism caused by a pI157S somatic KCNJ5 mutation in a black adolescent female with aldosterone-producing adenoma. Front. Endocrinol. 13:921449. doi: 10.3389/fendo.2022.921449
Received: 15 April 2022; Accepted: 26 July 2022;
Published: 16 August 2022.
Edited by:
Norlela Sukor, Universiti Kebangsaan Malaysia Medical Center (UKMMC), MalaysiaReviewed by:
Madson Almeida, University of São Paulo, BrazilCopyright © 2022 Gomez-Sanchez, van Rooyen, Rainey, Nanba, Blinder and Baliga. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Celso E. Gomez-Sanchez, Q2dvbWV6LXNhbmNoZXpAdW1jLmVkdQ==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.