

94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
SYSTEMATIC REVIEW article
Front. Endocrinol. , 08 September 2022
Sec. Clinical Diabetes
Volume 13 - 2022 | https://doi.org/10.3389/fendo.2022.920541
 Giuseppe Lisco1
Giuseppe Lisco1 Anna De Tullio1Olga Disoteo2Vincenzo De Geronimo3Giuseppina Piazzolla1Giovanni De Pergola4
Anna De Tullio1Olga Disoteo2Vincenzo De Geronimo3Giuseppina Piazzolla1Giovanni De Pergola4 Vito Angelo Giagulli1
Vito Angelo Giagulli1 Emilio Jirillo5Edoardo Guastamacchia1Carlo Sabbà1
Emilio Jirillo5Edoardo Guastamacchia1Carlo Sabbà1 Vincenzo Triggiani1*
Vincenzo Triggiani1*Tirzepatide, a dual agonist of Glucose-Dependent Insulinotropic Polypeptide (GIP) and Glucagon-Like Peptide 1 (GLP-1) receptors, improved glucose control and reduced body weight in different therapeutic approaches. Herein, we overviewed the role of GIP and GLP-1 in the pathophysiology of type 2 diabetes and systematically reviewed the efficacy and safety of injectable incretin-based therapy added to basal insulin in light of the results of the SURPASS-5 trial. We identified eleven randomized clinical trials. GLP-1 receptor agonists (GLP-1RAs) or Tirzepatide added to basal insulin than rigorously titrated basal insulin significantly ameliorates glucose control (Δ HbA1c = -1%, 95% CI -1.25; -0.74, I2 94%; Δ FPG = -14.6 mg/dL, 95% CI -21.6-; -7.6, I2 90%; chance to achieve HbA1c <7% = RR 2.62, 95% CI 2.10; 3.26, I2 89%), reduces body weight (Δ = -3.95 kg, 95% CI -5.1, -2.79, I2 96%) without increasing the risk of hypoglycemia (RR = 1.01, 95% CI 0.86; 1.18, I2 7.7%). Tirzepatide provides an impressive weight loss exceeding that observed with GLP-1RAs. Injectable incretin-based therapy plus basal insulin remains a potent and safe therapeutic approach in uncontrolled type 2 diabetes patients previously treated with basal insulin alone. Tirzepatide is expected to ameliorate the management of “diabesity” in this usually difficult-to-treat cluster of patients.
The endocrine role of the small intestine in the regulation of pancreatic secretion and glucose control dates back to the early 1900s when mechanistic studies demonstrated that chloride acid administration in the duodenum and jejunum stimulated pancreatic secretion, and intestine extracts administered to patients with type 2 diabetes mellitus (T2D) reduced glycosuria (1, 2). After numerous unsuccessful attempts to purify these substances, in 1930, the Belgian La Barre did it. Particularly, La Barre and coworkers isolated two fractions from animal duodenum that exhibited two different activities: the first stimulated the exocrine pancreas, while the second reduced glucose levels without affecting exocrine pancreas secretion. It was hypothesized that an intestinal peptide could regulate glucose levels by enhancing insulin secretion, hence introducing the concept of “intestine secretion insulin” or incretin as a possible therapeutic strategy in T2D (3).
McIntyre and Elrick formally demonstrated the previously hypothesized mechanism in the 1960s, both showing that significantly higher insulin response was observed after oral rather than intravenous glucose load at comparable blood glucose concentrations (4, 5). This phenomenon was defined as the incretin effect. Perley and Kipnis demonstrated that the incretin effect was responsible for at least 50%, and possibly two-thirds, of glucose-related insulin response after oral glycemic load in healthy individuals. At the same time, the contribution of other gastrointestinal peptides (e.g., cholecystokinin, secretin, and gastrin) in mediating the incretin effect was ruled-out and, in the early 1970s, Brown and Dupré isolated a new gastrointestinal peptide that affected gastric motility and secretion and induced a relevant insulin release in response to hyperglycemia but not euglycemia (6). Given these biological properties, the novel peptide was named Gastric Inhibitory Polypeptide, successively baptized as Glucose-dependent Insulinotropic Polypeptide or GIP (6).
In the 1960s, the widespread use of a new radioimmunoassay method led to the incidental discovery of glucagon-like immunoreactivity in the colonic mucosa, attributed to the so-named gut-glucagon successively identified as GIP. Although GIP was identified as a first contributor to the incretin effect, there was evidence that this effect persisted in experimental models of direct GIP antagonism. Hence, it was supposed that other gastrointestinal peptides could have contributed to the glucose-related insulin release. In the early 1980s, Bell and coworkers provided the first sequencing of the mammalian glucagon gene and demonstrated the existence of the so-named Glucagon-Like Peptide-1 (GLP-1) located in the encoding sequence of pro-glucagon (3). The incretin effect of GLP-1 was later demonstrated by Habener and Holst (3).
GIP is a gastrointestinal polypeptide composed of 42 amino acids secreted by K cells in the duodenum and jejunum mucosae. GLP-1 comprises 31 amino acids and is secreted at the level of L cells in the terminal ileum and colonic mucosae (3). GIP and GLP-1 receptors belong to the seven transmembrane G protein-coupled receptors and are abundantly expressed on the human ß-cell membrane (7, 8). GIP receptor (GIPR) has also been found on human α-, δ- and γ-cells, while GPL-1 receptor (GLP-1R) is expressed in a minority of human α- and δ-cells. GIPR has also been identified in human subcutaneous and visceral white adipose tissue and cultured osteoblasts. GLP-1R is moderately expressed in several brain areas in non-human primates, such as the nucleus accumbens, substantia nigra, and the amygdala, thus playing a role in regulating food intake. A weak expression of both receptors has been described in human cardiomyocytes (including sino-atrial node), intestine, pneumocytes, isolated endothelial cells, and seminiferous tubules in mice (7, 8).
Basal GIP and GLP-1 concentrations are very low at fast but increase remarkably after meal ingestion. GIP release is more evident after carbohydrates and lipids consumption (9), while GLP-1 release is enhanced by amino acids such as glutamine (7). GLP-1 release appears to be diphasic (10). The first phase of GLP-1 release occurs 10-15 minutes after meal intake. It is probably mediated by neuroendocrine mechanisms that include brainstem vagal stimulation (an anticipatory mechanism), mechanical stretching of the stomach and duodenum due to food transit, GIP release, and gastrin-related peptide from gastric and duodenal mucosae that, in turn, activate vagal efferences to distal intestine. The intestinal transit stimulates the second wave of GLP-1 release throughout L-cells in the distal intestine (10). GIP release occurs rapidly after meal intake and persists over time (up to four hours), with a serum and intestinal peak of concentration two hours later (11).
Insulin suppresses GIP secretion resulting from negative feedback, while several cytokines, including interleukins 1 and 6, enhance GIP and GLP-1 release (7, 8). Dipeptidyl-peptidase 4 (DPP-IV) activity is particularly intense, and the enzyme degrades both GIP and GLP-1 a few minutes after their release. Therefore, native GIP and GLP-1 have a short half-life (2-5 minutes), and almost all the action of these incretins takes place in the intestine and the portal vein system. However, a small amount (5-10%) of both incretins reaches the systemic circulation, where GIP and GLP-1 may carry out their pleiotropic effects (e.g., cardiovascular level). Therefore, a large amount of circulating GLP-1 is represented by its inactive peptide GLP-1 (8–35) with a null or mild antagonistic effect compared to the intact GLP-1.
A small amount of GLP-1 originates in the pancreas after the local breakdown of its precursor pro-glucagon. Both systemic and locally originated GLP-1 reach pancreatic islets and regulate insulin, glucagon, and somatostatin secretion (7). Both GIP and GLP-1 enhance insulin secretion in a glucose-dependent manner. Hyperglycemia is a permissive factor to GIP and GLP-1 action as the more the glucose concentration, the great the magnitude of ß-cells membrane depolarization and vice versa (7).
The underlying mechanisms by which GLP-1 suppresses glucagon secretion are still debated and include: 1) a direct GLP-1-mediated suppression of glucagon release, but this hypothesis appears controversial especially considering human α-cells that do not express GLP-1R; 2) an indirect effect due to GLP-1-mediated insulin release; 3) an indirect effect due to GLP-1-mediate somatostatin release (7, 8, 11, 12). GIP enhances glucagon secretion, and this action is directly mediated by GIPR agonism (via intracellular Protein Kinase A pathway) since GIPR is abundantly expressed on human α-cells (7, 8). This effect was described in experimental conditions in response to hypoglycemia and euglycemia but not hyperglycemia, suggesting that GIP probably does not contribute to hyperglucagonemia in T2D (7, 8).
The incretin effect is dampened in different metabolic conditions such as for overweight/obesity, insulin resistance, impaired glucose tolerance, and T2D, in which both incretin secretion and effect are disturbed (11). A GIP resistance has been described in T2D due to diminished GIPR expression or accelerated GIPR clearance. Higher or inappropriately normal levels of GIP are detected in T2D patients also during the fast, and the fine regulation of insulin and glucagon release in response to different glycemic levels is problematic. On the other side, GLP-1 response is preserved in T2D, but GLP-1 secretion in response to meal consumption weakens over time, making GLP-1-based therapy an effective strategy to manage hyperglycemia as replacement therapy (7, 13, 14).
GIP and GLP-1 exhibit extra glycemic effects. GLP-1 slows gastric and duodenal emptying, delays meal digestion, and reduces postprandial glycemic excursion, which, in turn, is an essential target of glycemic control in T2D (10). GLP-1 regulates gastrointestinal motility by several mechanisms that include direct activation of vagal efferences (peripheral pathway), cerebral regulation (central pathway), and modulation of local myenteric ganglion activity that controls circular muscle movement, such in a way to reduce evocated peristaltic bustle (10). In addition, GLP-1 reduces food intake by modulating the activity of cerebral areas involved in appetite control, such as the area postrema, amygdala, nucleus accumbens, and substantia nigra (7). The role of GIP in regulating food intake appears controversial. Mechanistic studies revealed that GIPR is expressed in cerebral areas that regulate food intake, such as the area postrema, arcuate, and paraventricular nuclei and GIPR expression overlaps GLP-1R expression (15). GIP signaling is expected to directly induce the activation of anorexigenic areas or facilitate GLP-1 action (15). Conversely, GIP seems not to affect gastrointestinal motility when administered in physiologic and supraphysiologic doses (7, 16), and no evidence, to date, has been provided for GIP to facilitate GLP-1-induced gastric empty delay (7). Incretins could reduce intestinal water resorption, and this mechanism is responsible for diarrhea, a well-recognized adverse effect of GLP-1 receptor agonists (GLP-1RAs).
GIP increases insulin sensibility of white adipose tissue, consequently improving lipid storage and ameliorating lipid profile (mainly triglycerides) and skeletal muscle (7). GIP-mediated actions appear to be additional than those exerted by GLP-1, which improves insulin sensibility in the liver (7).
GIP may stimulate osteoblast activity and bone remodeling, thus providing a possible reduction of fracture risk as similarly observed with GLP-1.
Other effects have been described for GLP-1 (and possibly GIP, with scantier evidence), including anti-inflammatory and vasoactive proprieties (17).
Gut-based therapy has become an attractive therapeutic approach for improving glucose control and the burden of related-comorbidity in T2D (18). In this field, GLP-1RAs are demonstrated to allow significant glycemic and extra-glycemic benefits when administered to this cluster of patients (19). Inversely, isolated GIP therapy is not currently deemed as medical therapy for T2D because of pathophysiological considerations. A marked GIP resistance has been described in T2D just in the early phase of the disease, and the contribution of pharmacological GIP administration to repristinate the incretin effect is scanty. In addition, despite controversial evidence, GIP may stimulate glucagon release even in hyperglycemic conditions, as observed in prediabetes and T2D, thus potentially contributing to a further increase of glycemia (20). However, GIP resistance is rapidly reversible when euglycemia is restored, and, in this setting, GLP-1 may significantly improve GIP signaling thanks to additive mechanisms. GLP-1 suppresses glucagon secretion and may completely hide GIP-related hyperglucagonemia in euglycemia but not in hypoglycemia hence maintaining counterregulatory response to hypoglycemia unaltered (20). Moreover, both GIP and GLP-1 repristinate the incretin effect, reduce appetite, and affect caloric intake by some synergic and affine mechanisms that could significantly contribute to achieving more therapeutic objectives (i.e., obesity) in people with T2D (20, 21).
Evidence suggests that dual agonists of GIPR and GLP-1RA can represent an attractive therapeutic opportunity shortly (20, 22–25). From this point of view, the biotechnological goal is to synthesize chimeric molecules that can activate both receptors alternatively. Different molecules have been engineered for this specific purpose in the last few years and are currently under investigation. Some have passed phases I and II clinical trials showing promising effects on glucose control and weight loss (20, 26–30).
Tirzepatide (LY3298176) successfully passed early phases of clinical investigation and showed relevant effects on glycemic control and body weight in phase III, randomized clinical trials (RCTs). Tirzepatide (TZP) is a 48 KDa synthetic peptide composed of 39 amino acids. The basic structure of TZP (Figure 1) comprises several highly affine fragments of other incretins, including GIP, GIP and GLP-1, Extendin-4, and glucagon. Moreover, two residues of alpha aminobutyric acid are placed in positions 2 and 13 with the strategic purposes to strengthen TZP resistance to DPP-IV cleavage and ameliorate molecular stability, respectively. A 20-carbon fatty acid linked to the lysine residue in position 20 of the primary structure allows TZP to be bound to circulating albumin. Acylation technology, previously adopted and developed for Liraglutide and Semaglutide, increases TZP half-life to around five days, making it administrable once weekly (28, 31–33).
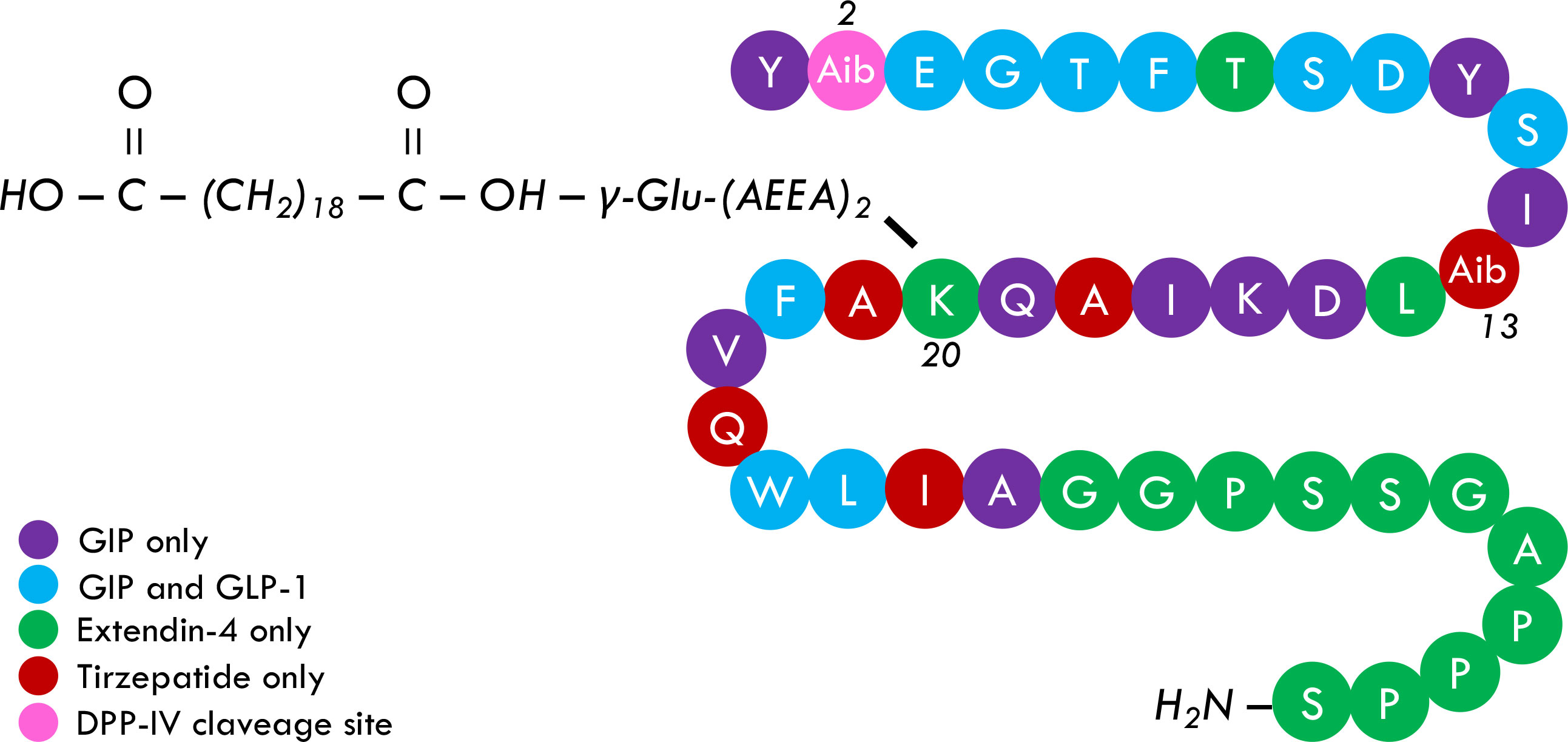
Figure 1 Tirzepatide primary structure. TZP is a 39-peptide containing different native and not-native incretins fragments, including GIP, GIP, GLP-1, and Extendin-4 (and glucagon, not shared). Alpha aminobutyric acid is placed in positions 2 and 13 and provides TZP an intrinsic resistance to DPP-IV attack and more structural stability, respectively. A 20-carbon fatty acid, namely eicosanedioic acid, is linked to Glu. A 2xAdo unit is attached to the lysine residue in position 20 and allows TZP to be bound to albumin, consequently increasing its half-life to five days. It is currently debated whether the acylation may also increase receptor bounding stability, hypothetically affecting the pharmacological potency of TZP.
TZP is an unbalanced dual agonist of GIPR and GLP-1R. Preclinical trials demonstrated that TZP (LY3298176) exhibited for GIPR the same affinity as native GIP and a five-fold weaker affinity for GLP-1R than the native GLP-1 (20). The potency of TZP for GIPR is similar to GIP and thirteen-fold lower for GLP-1R than GLP-1 (20). The potency of TZP for GLP-1R was also inferior compared to Semaglutide (20). In in vitro studies, TZP stimulated ß-cells response more efficiently than observed with GIP and GLP-1 alone. Also, in vivo, TZP enhanced insulin secretion by acting on both GIPR and GLP-1R (20).
Efficacy and safety of TZP were investigated in phase III, RCTs of the SURPASS program to verify its efficacy and safety in improving glycemic control in different therapeutic lines (Table 1). TZP monotherapy versus placebo was investigated in the SURPASS-1 (34). A head-to-head comparison between TZP and Semaglutide as an add-on to metformin in uncontrolled T2D was analyzed in the SURPASS-2 (35). The SURPASS-3 and SURPASS-4 examined the safety and efficacy of TZP as an intensification of previous oral treatment (metformin and sodium-glucose transporter 2 inhibitors in the SURPASS-3; metformin, sulfonylureas, or sodium-glucose transporter 2 inhibitors alone or in variable combinations in the SURPASS-4) versus insulin Degludec and Glargine respectively (36, 37). The latter trial was carried out in a population at high cardiovascular risk. TZP was administered subcutaneously in three doses (5, 10, and 15 mg once a week). Overall, TZP induced a relevant reduction in HbA1c with superiority versus direct comparators, and a relevant percentage of patients achieved optimal glucose control (i.e., HbA1c <7% or ≤6.5%) or reverted to euglycemia (HbA1c <5.7%). Also, weight loss was impressive at all tested doses.
The results of the 40-week double-blind randomized, placebo-controlled, phase III clinical trial SURPASS-5 (NCT04039503) have recently been published (38). This was a multicentric study involving 45 centers involved in diabetes clinical research from eight different countries (Czech Republic, Germany, Japan, Poland, Puerto Rico, Slovak Republic, Spain, and the USA) and assessing the efficacy and safety of TZP compared to placebo as add-on to insulin Glargine with or without metformin in uncontrolled T2D individuals.
The primary outcome was to test TZP 10 and 15 mg once weekly for superiority versus placebo in reducing HbA1c from baseline to 40 weeks. The secondary outcome included superiority of TZP 5 mg qw versus placebo in reducing HbA1c from baseline to 40 weeks. Additional outcomes were: superiority of TZP 5, 10, and 15 mg qw versus placebo in terms of weight loss; percentage of patients who would have achieved optimal glycemic control (HbA1c <7%); reduction of fasting blood glucose values, reduction of daily mean blood glucose assessed by 7-point glucose monitoring (fasting, pre-meal, post-meal and bedtime blood glucose); percentage of patients who would have achieved weight loss ≥5%; percentage of change in insulin Glargine dose; the number of severe hypoglycemia events (<54 mg/dL); percentage of patients who would have achieved blood glucose normalization (HbA1c <5.7%).
Patients were screened for eligibility and recruited based upon the following key elements: had received a diagnosis of T2D according to diagnostic standard by the World Health Organization; HbA1c 7-10.5%; body mass index >23 kg/m2 with irrelevant variation ( ± 5%) in last three months; stable (at least three months before) daily dose of insulin Glargine (>20 IU/day) plus daily metformin ≥ 1.500 g; age ≥18 years.
Exclusion criteria included: type 1 diabetes, either acute or chronic pancreatitis, proliferative diabetic retinopathy or macular diabetes, non-proliferative diabetic retinopathy requiring urgent treatment, either acute or chronic hepatopathy with clinic or laboratory evidence of liver injury (e.g., 3-fold elevation in plasmatic transaminases), severe hypoglycemia or unawareness hypoglycemia, gastroparesis, previous bariatric surgery, concomitant use of certain medications delaying gastric emptying, established coronary disease or severe chronic heart failure (NYHA class III or IV), glomerular filtration rate <30 mL/min/1.73 m2 in patients not taking metformin and glomerular filtration rate <45 mL/min/1.73 m2 in those on metformin, adrenal insufficiency or severe thyroid dysfunction, positive family history of medullary thyroid cancer or personal basal serum calcitonin ≥35 ng/L, active malignancy over the last five years, transplant recipients, alcoholism, and psychiatric disorders.
Four hundred seventy-five patients (44% women) were randomized, with a median age of 61 years and an average HbA1c of 8.3%. Among them, 95% concluded the trial. Patients were allocated in four groups and randomized (1:1:1:1) to receive TZP 5 mg (n: 166), TZP 10 mg (n: 119), TZP 15 mg (n:120), or placebo (n: 120).
TZP was started at 2.5 mg qw and up-titrated every four weeks until achieving the final dose. Insulin Glargine was administered once a day at bedtime and titrated every three days to maintain fasting glucose between 71 and 100 mg/dL. Insulin doses were increased by two, four, six, or eight units in case of fasting glycemia between 101 – 119, 120 – 139, 140 – 179, and more than 180 mg/dL, respectively. Insulin therapy was steadied within four weeks after the randomization (stabilization period) and then was managed with a treat-to-target purpose for the remaining 36 weeks.
After 40 weeks, HbA1c reduction was superior in the group treated with Tirzepatide 5 mg (-2.1%), 10 mg (-2.4%), and 15 mg (-2.3%) compared to placebo (-0.9%) with an estimated treatment difference of -1.24% in favor of TZP 5 mg, -1.53% in favor of TZP 10 mg, and -1.47% in favor of Tirzepatide 15 mg (p <.001 for superiority over placebo).
Around 85-90% of participants randomized to receive TZP achieved HbA1c <7%. This percentage was significantly higher compared to the placebo (34%). A normalization in glucose levels (HbA1c <5.7%) was obtained in 26-62% of patients on TZP (5-15 mg), with a relevant difference compared to placebo (2.5%).
A considerable body weight loss was reached after 40 weeks of treatment with TZP consisting of -5.4 kg (5 mg), -7.5 kg (10 mg), and -8.8 kg (15 mg). Patients randomized to placebo obtained a slight but relevant weight gain compared to baseline (+1.6 kg); therefore, the estimated treatment difference between TZP and placebo was -7 Kg (TZP 5 mg), -10 kg (TZP 10 mg), -8.8 Kg (TZP15 mg, all p <.001). Overall, a more significant proportion of participants (54-84%) randomized to TZP (5-15 mg) achieved a clinically relevant body weight loss (≥5%) compared to placebo (6%).
Fasting glucose levels decreased more evidently with TZP (-61 to -68 mg/dL) than placebo (-39 mg/dL). Similarly, average of daily glucose levels was lower in TZP (-67 to -74 mg/dL) than placebo (-39 mg/dL).
The daily dose of insulin Glargine increased by 75% from baseline to 40 weeks in the placebo group while increasing by 13% and 8% with TZP 5 and 10 mg, respectively. Patients randomized to TZP 15 mg exhibited a slight reduction in daily Glargine dose requirement by 11%.
Hypoglycemia was defined as glucose level <54 mg/dL. The hypoglycemic risk was low in all groups, and it did not change up to 44 weeks (including a 4-week study extension analysis aimed to assess safety end-points specifically). The incidence of hypoglycemia was 14-19% in TZP arms and 13% in the placebo group. Only three episodes of level 3 hypoglycemia were detected (two in the group TZP 10 mg and one in TPZ 15 mg).
Twenty-four patients withdrew from the study: seven of them assumed TZP 5 mg (three for adverse events and four because of personal decision); four were on TZP 10 mg (one due to protocol violation and three because of personal decision), ten were on Tirzepatide 15 mg (two for adverse events, one for missed follow-up, two because of protocol violation, and five because of personal decision). Safety monitoring was recorded for up to 17 months; the most common adverse events were nausea (13-18% with TZP vs. 3% with placebo), diarrhea (12-21% on TZP vs. 2% on placebo), loss of appetite (7-14% on TZP vs. 2% on placebo), vomiting (7-12% on TZP vs. 2% on placebo). Serious adverse events were uncommon, and no between-group differences were found.
Basal insulin intensification is necessary for managing specific situations such as T2D with long duration and poor glucometabolic control suggestive of reduced pancreatic reserve (39). A timely basal insulin prescription, when appropriate, is recommended to improve glucose control rapidly, reduce glucotoxicity, and ameliorate beta-cell reserve over time, facilitating further pharmacologic strategies to reach more appropriate therapeutic goals (40). Nonetheless, it is well-known that only a minority of insulin-treated patients have a further chance to achieve a tailored glycemic control (41) due to delayed or inadequate basal insulin titration over time or deferred therapeutic intensification (42).
GLP-1RAs added to basal insulin are demonstrated to improve glucometabolic control without increasing hypoglycemic risk and prevent weight gain compared to a further titration of basal insulin in T2D patients failing to reach adequate glycemic control (43–46). The mechanisms leading to favorable outcomes when adding a GLP-1RA to basal insulin include improving pancreatic reserve, ameliorating insulin sensibility, suppressing glucagon secretion, weight loss, cardiovascular and microvascular protection, and increasing treatment adherence, especially when considering once-weekly GLP-1RAs. In the SURPASS-5, TZP confirmed the importance of injectable incretin-based therapy in this cluster of patients and appeared to be a further therapeutic tool for T2D.
In light of this evidence, we carry out a rapid review and meta-analysis to review and summarize the effect of injectable incretin-based therapy compared to placebo when added to basal insulin therapy in uncontrolled T2D. PubMed/MEDLINE, Cochrane Library were searched by GL and ADT using the following strategy: “(exenatide) or (liraglutide) or (lixisenatide) or (dulaglutide) or (albiglutide) or (semaglutide) or (tirzepatide) or (glp-1ra) or (“glucagon-like peptide 1 receptor agonist”) and (glargine) or (glargine) or (degludec) or (basal insulin)”. Database search was performed until 15 February 2022, and the timeline of extracted data ranged from 01 January 2011 to 15 February 2022. After duplicate removals, articles were screened and extracted for the synthesis according to a hierarchical strategy that included title, abstract, and full-text appraisal. Studies were screened and included based upon the following criteria: population with established diagnosis of T2D; age ≥18 years; basal insulin treatment at stable dose ( ± 20%) for at least six weeks before screening visits (100% of participants) with or without oral antihyperglycemic agents such as metformin, sulfonylureas, pioglitazone, DPP-IV inhibitors, sodium glucose transporter type 2 inhibitors; inadequate glucose control (HbA1c ≥7%); clinically relevant follow-up period (≥24 weeks); intensification with a GLP-1RA (both short- and long-acting) or dual GIP/GLP-1RA specifically designed for the treatment of T2D (i.e. Lixisenatide 10-20 μg/day; Exenatide 5-10 μg/bid; Liraglutide up to 1.8 mg/day; Dulaglutide 1.5 mg/qw; Exenatide 2 mg/qw; Semaglutide 0.5 e 1 mg/qw; Tirzepatide 5, 10, 15 mg/qw) as an add-on to basal insulin and not vice versa; placebo as comparator group; primary and secondary outcomes including mean change from baseline to study end in HbA1c, fasting glycemia, body weight, insulin dose, number of individuals experiencing symptomatic hypoglycemia or glucose levels (<70 mg/dL); number of patients experiencing adverse events. Fixed-ratio formulations and separately administered basal insulin plus GPL-1RA were both included. The search was further restricted to RCTs written in English. Eleven RCTs have been included in the meta-analysis (38, 43–52) as show in Table 2. Studies focusing on specific ethnic groups were excluded (53, 54). Other trials exploring differences between GLP-1RAs added to basal insulins (free or fixed-ratio) and basal insulins plus placebo were excluded as participants were insulin naïve at baseline (55–57). One trial was excluded because Liraglutide 3 mg is formally approved for obesity (58). The identification of RCTs included in the systematic review is shown in Figure 2.
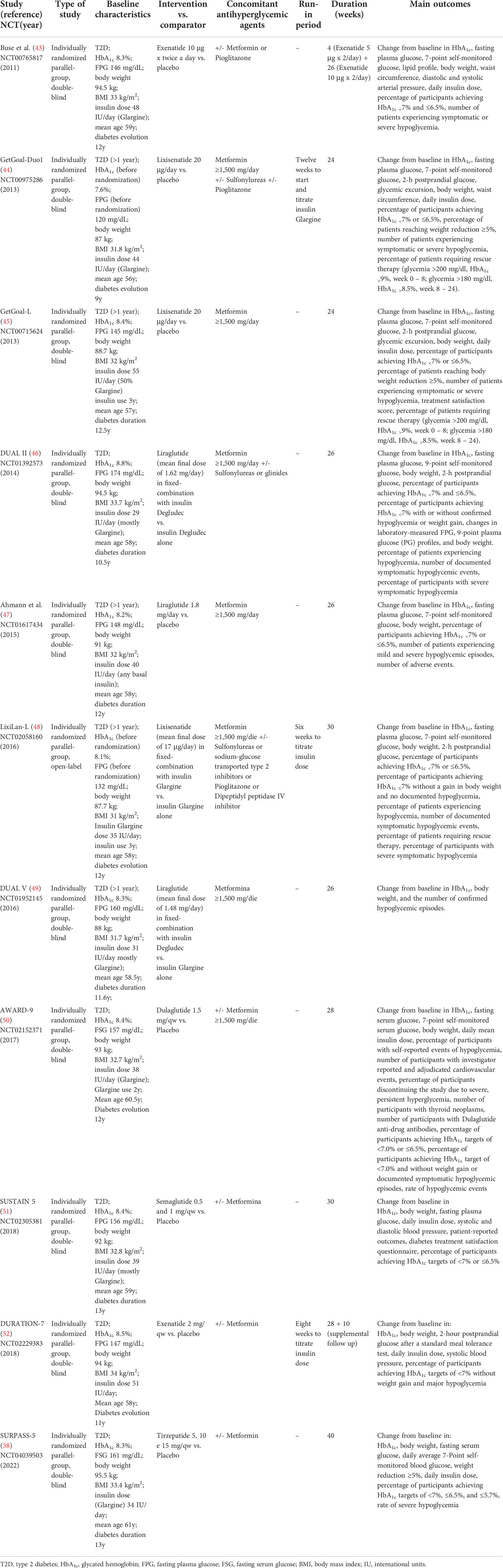
Table 2 Summary of RCTs included in the systematic review.
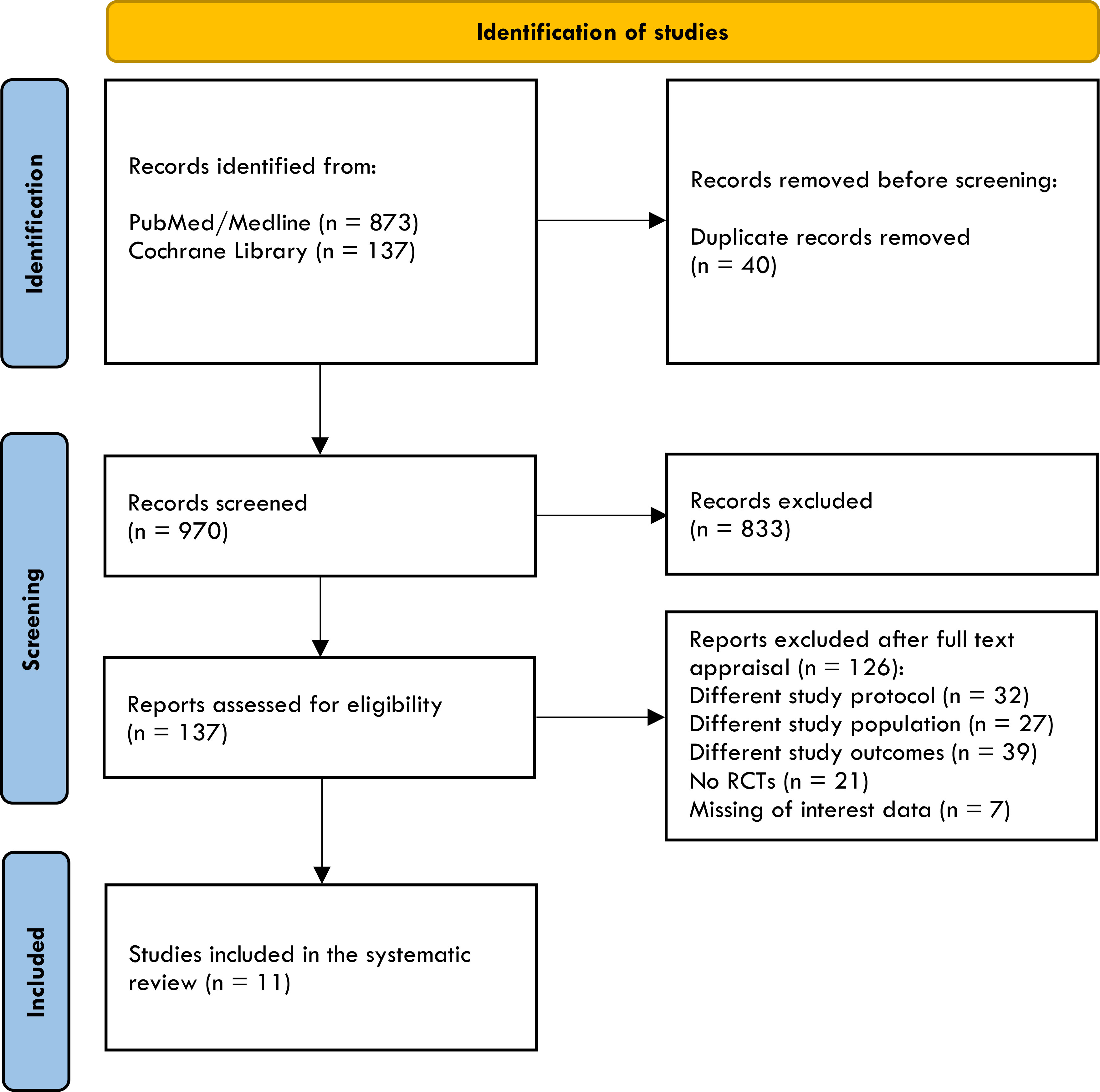
Figure 2 The PRISMA flow diagram of included studies.
The risk of bias was estimated by RoB2 for individual randomized, parallel-group trials (59). Six out of eleven trials have been considered at moderate risk of bias (some concerns) due to the randomization process for different baseline characteristics. In Buse et al., participants randomized to receive the placebo compared to those who received Exenatide had higher baseline HbA1c and fasting plasma glucose, lower body weight, and baseline insulin dose, and were predominantly men than women with a higher percentage of metformin users. In the GetGoal-Duo 1 trial, participants randomized to receive insulin Glargine had longer diabetes evolution than those assuming insulin Glargine/Lixisenatide, and this difference might have been responsible for different beta-cell reserve between the two study groups. In the GetGoal-L trial, participants randomized to placebo had higher insulin doses and higher body weight, while in Ahmann et al., patients on placebo were younger, more men than women, and had higher body weight. Moreover, a possible attrition bias might not be excluded because a significant percentage of patients withdrew in both groups. In the DUAL V, participants randomized to Liraglutide had higher body weight and HbA1c and were more hypertensive. All trials were double-blind except for the Lixilan-L, designed as open-label, but analyses were carried out blind. All trials were sponsored by the pharmaceutical industry (Eli Lilly and Company and Amylin Pharmaceuticals; Sanofi; Sanofi; NovoNordisk; NovoNordisk; Sanofi; NovoNordisk; Eli Lilly and Company; NovoNordisk; AstraZeneca; Eli Lilly and Company). The risk of bias in the included studies is shown in Figures 3A, B.
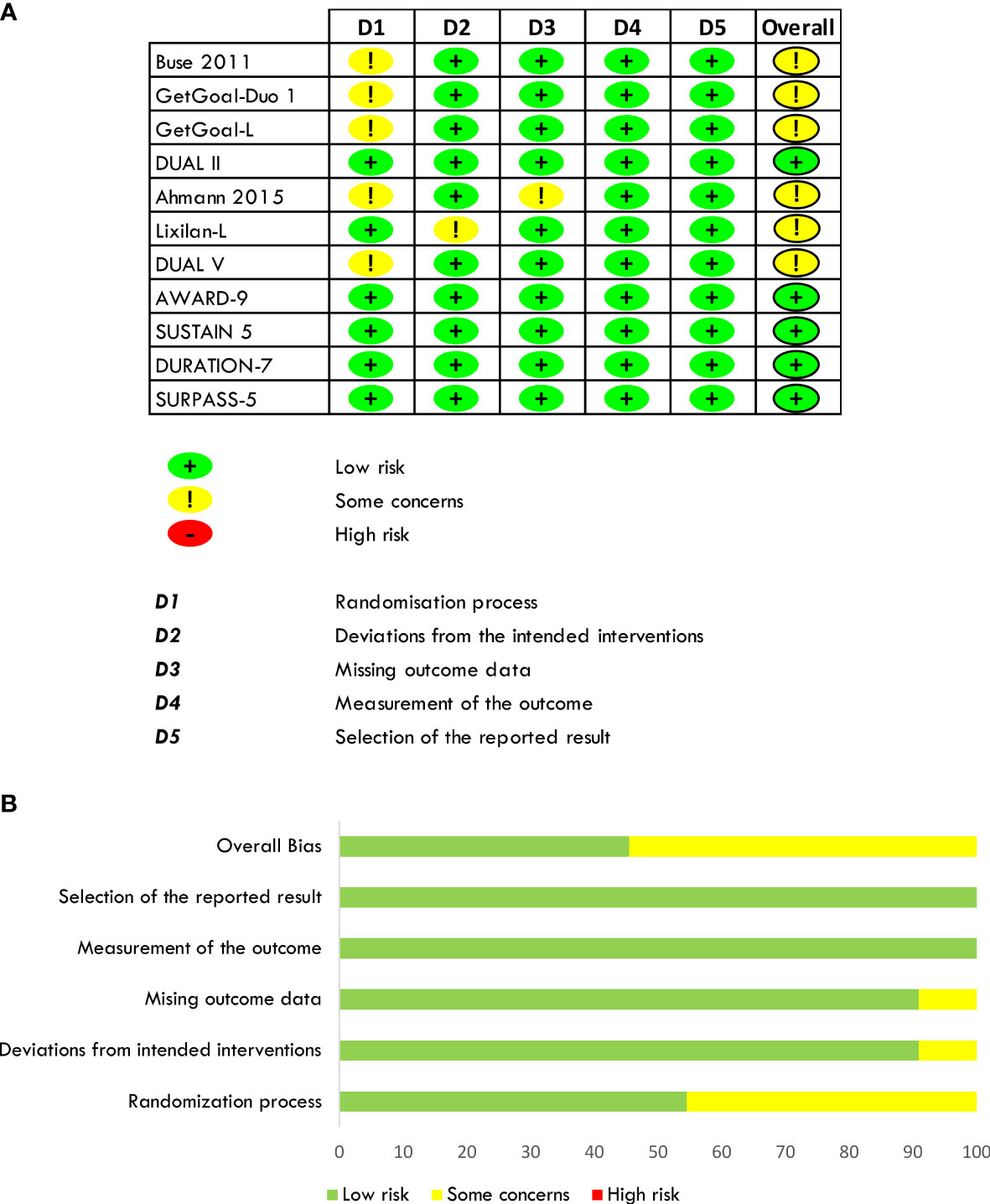
Figure 3 Quantification of the risk of bias of included studies (A, B).
All analyses were performed by RevMan 5.4.1 with a random-effect model, considering a p-value <.05 as statistically significant. Four outcomes (change in HbA1c, fasting plasma glucose, body weight, and basal insulin dose from baseline to study end) were analyzed as continuous variables, and results were summarized in weighted mean difference or standardized mean difference (change in basal insulin dose that was assessed by different scales). One outcome (percentage of participants achieving HbA1c<7%) was analyzed as a dichotomous variable, and results were synthesized in terms of risk ratio (RR). Heterogeneity was assessed by I2. The level of heterogeneity was considered substantial in the case of I2>50%. Standard error, 95% confidence interval (CI), and interquartile range were used alternatively to estimate the standard deviation when missed.
The metanalysis includes 5,323 patients (56-61 years) with T2D inadequately controlled (HbA1c 7-10.5%), baseline BMI 23-45 kg/m2, mean diabetes duration of 9-12 years, and randomized to receive a GLP-1RA or GIP/GLP-1RA versus placebo in add on to basal insulin for at least 24 weeks (24-40 weeks).
The weighted overage baseline HbA1c of included studies was 8.3% before the randomization. Combining a GLP-1RA or dual GIP/GLP-1RA to basal insulin provides a significant improvement in glucose control (Δ HbA1c = -1%, 95% CI -1.25; -0.74, I2 94%) compared to progressive basal insulin titration with a rigorous intention-to-treat approach. Also, the probability of reaching optimal glucose control (HbA1c <7%) is significantly higher with GLP-1RA or dual GIPR/GLP-1R added to basal insulin as compared to progressive basal insulin titration with a rigorous intention-to-treat approach (RR 2.62, 95% CI 2.10; 3.26, I2 89%). Baseline weighted overage fasting plasma glucose (FPG) was 149.6 mg/dL. Overall reduction in FPG is significantly higher when GLP-1RAs or dual GIPR/GLP-1R is added to basal insulin (Δ FPG = -14.6 mg/dL, 95% CI -21.6; -7.6, I2 90%). In this setting, daily administered GLP-1RAs added to basal insulin provides a less relevant improvement in glycemic control (Δ HbA1c = -0.68%, 95% CI -0.92; -0.43, I2 89%/RR HbA1c <7% = 2.03, 95% CI 1.59; 2.59, I2 86%/Δ FPG = -5.9 mg/dL, 95% CI -12.4; 0.8, I2 78%) than weekly administered analogues, including Tirzepatide (Δ HbA1c = -1.32%, 95% CI -1.63; -1.01, I2 92%/RR HbA1c <7% = 3.38, 95% CI 2.55; 4.46, I2 81%/Δ FPG = -23.4 mg/dL, 95% CI -29; -18, I2 66%). Results are shown in Figures 4A–C.
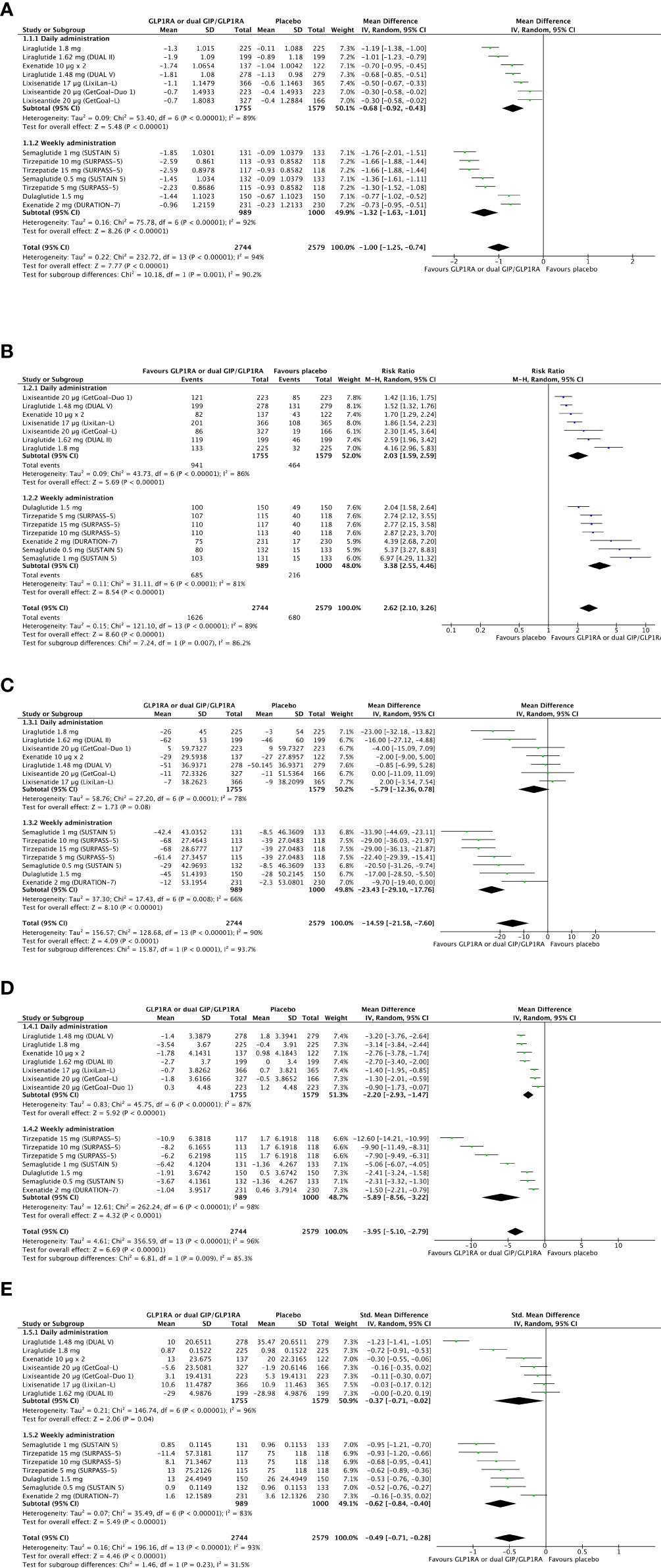
Figure 4 Forest plots of meta-analysis for change in HbA1c (A), chance of achieving optimal glucose control as HbA1c <7% (B), and change in fasting plasma glucose (C) from baseline to the last available follow-up (intention-to-treat analyses). Forest plot of meta-analysis for change in body weight (D) from baseline to the last available follow-up (intention-to-treat analysis). Forest plot of meta-analysis for change in the mean daily dose of basal insulin (E) from baseline to the last available follow-up (intention-to-treat analysis).
The weighted average body weight was 91.3 kg at baseline. GLP-1RA or dual GIP/GLP-1RA added to basal insulin provides a relevant weight reduction compared to basal insulin titration with a rigorous intention-to-treat approach (Δ weight = -3.95 kg, 95% CI -5.1, -2.79, I2 96%). The magnitude of this difference appears less evident with daily than weekly administered analogues (Δ weight = -2.2 kg, 95% CI -2.93, -1.47, I2 87% versus Δ weight = -5.9 kg, 95% CI -8.56; -3.22, I2 98%). Forest plot of meta-analysis for change in body weight from baseline to the last available follow-up is shown in Figure 4D.
The average weighted insulin dose was 41 IU/day. GLP-1RA or dual GIP/GLP-1RA added to basal insulin compared to basal insulin titrated with a rigorous intention-to-treat approach is moderately effective in preventing further increase in daily basal insulin dose from achieving treat-to-target glycemic goals (Δ daily insulin dose, SMD -0.49, 95% CI -0.71; -0.28, I2 93%) without specific difference between daily and weekly administered agonists and with a moderate intergroup heterogeneity (I2 31.5%). Forest plot of meta-analysis for change in basal insulin dose from baseline to the last available follow-up is shown in Figure 4E.
The overall hypoglycemic risk was unaffected by adding a GLP-1RA or TZP to basal insulin compared to basal insulin alone titrated with a rigorous treat-to-target approach (RR = 1.01, 95% CI 0.86; 1.18, I2 67%).
Adverse events were also registered and reported in RCTs, particularly those more frequently documented (usually with prevalence ≥5%). Reported descriptions are heterogeneous but are commonly imputable to gastrointestinal adverse events such as nausea (RR 4.45, 95% CI 3.19; 6.21, I2 49%), diarrhea (RR 1.96, 95% CI 1.58; 2.42, I2 0%), and vomiting (RR 4.44, 95% CI 3.15; 6.24, I2 3%) without specific differences between daily rather than weekly administered analogues. Data are shown in Table 3 with more details.
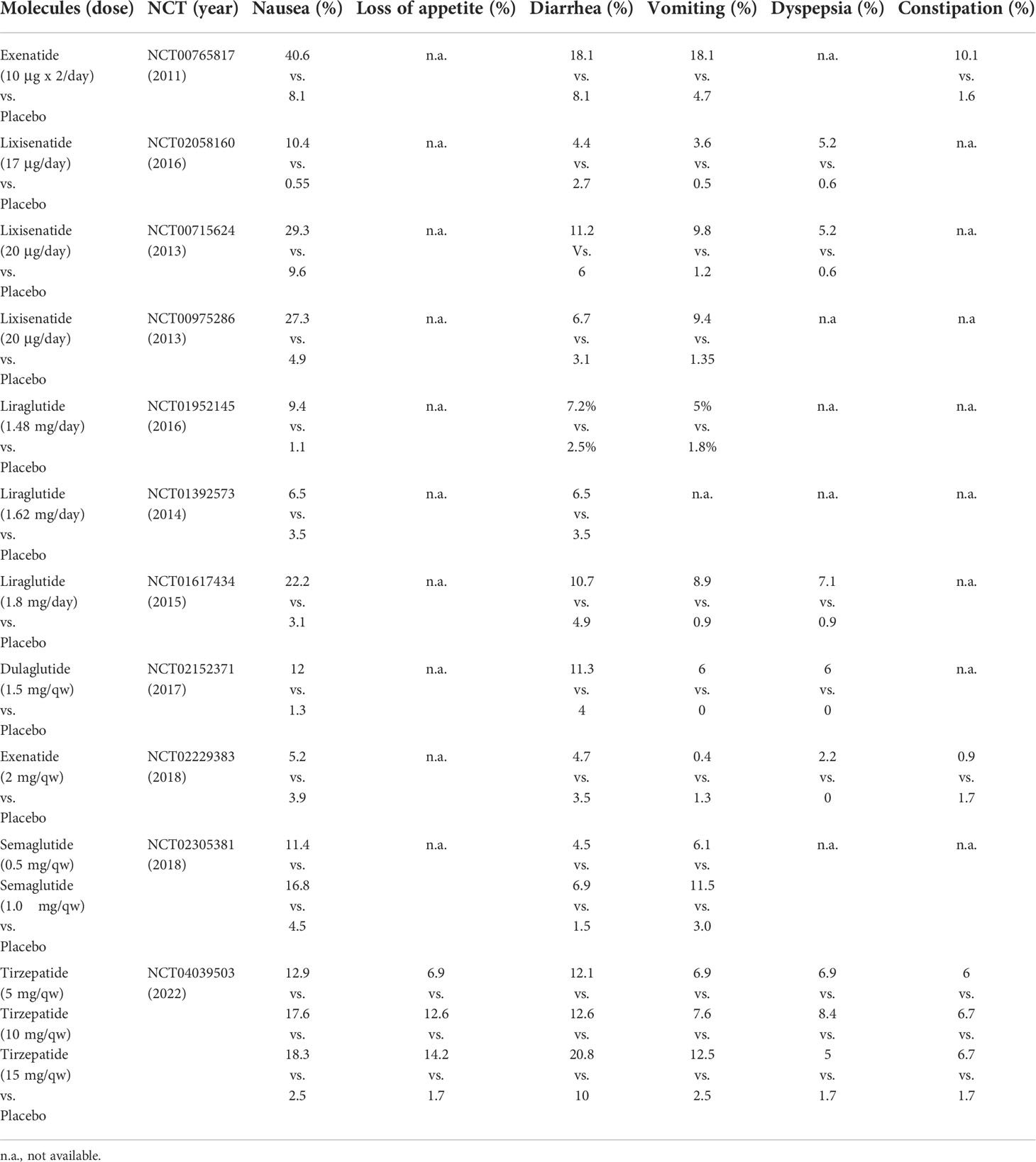
Table 3 Summary of the most common reported adverse events displayed by molecules and doses.
GLP-1RAs have changed the therapeutic paradigm of T2D as they significantly improve glucose control, also contributing to pleiotropic effects, especially at the cardiovascular level (60), in different clinical scenarios and therapeutic approaches. As some examples, GLP-1RAs are safer and slightly more effective in improving glucose control, significantly reducing body weight, and preventing hypoglycemia than basal insulin in poorly controlled T2D with oral antihyperglycemic agents (61). In addition, GLP-1RAs added to basal insulin, as both free and fixed-ratio, perform better than more composite insulin strategies, such as basal plus and basal-bolus regimens in uncontrolled T2D (62, 63).
Herein we examined the role of GLP-1RAs and the novel dual GIP/GLP-1RA TZP as an add-on to basal insulin therapy compared to basal insulin alone in improving glucose control. Three specific outcomes (change in HbA1c, FPG, and chance to achieve HbA1c <7% from baseline to study end) were included to estimate treatment differences in terms of glycemic control. Change in body weight was calculated, and safety data were also analyzed.
Intra and intergroup heterogeneities have characterized our results; therefore, some considerations and comments should be discussed to understand the findings better. First, six out of eleven RCTs had some concerns about the risk of randomization bias and some differences in terms of the baseline characteristics of the compared group. One RCT (Liraglutide 1.8 mg) was also at risk of attrition bias due to a significative and asymmetric withdrawal (placebo: 23%; liraglutide 16%). Second, study designs are formally similar, but there are some differences. In the GetGoal-Duo 1, insulin naïve patients started (with further titration) insulin Glargine during the twelve-week run-in period, and then they were randomized. Therefore, this study fell into inclusion criteria as patients received basal insulin during the last three months before randomization. However, as expected in insulin naïve, the response to insulin treatment was sudden and considerable; therefore, glucose control improved significantly during the run-in period (HbA1c from 8.6% to 7.6%). It is well-known that baseline HbA1c is positively related to the magnitude of HbA1c reduction upon an antihyperglycemic treatment is started (64); hence, the efficacy of Lixisenatide compared to the placebo might have been lessened. On the other hand, diabetes duration was slightly shorter compared to other RCTs, and this variable could have fostered better therapeutic response in both groups despite background different diabetes duration. Third, titration and final dose of GLP-1RAs and TZP were different in the included studies. Dulaglutide and Exenatide qw did not require any titration, and they were started at 1.5 and 2 mg from the first administration with global exposure to the specific GLP-1RA total dose of 28 weeks in both the AWARD-9 and DURATION-7. Lixisenatide was started at 10 μg once daily for two weeks and then continued to 20 μg/day for 22 weeks in GetGoal-Duo 1 and GetGoal-L. Exenatide bid was administered at the dose of 5 μg x 2/day for four weeks, and then it was up-titrated to 10 μg x 2/die for 26 weeks. Liraglutide was started at 0.6 mg once daily and up-treated by 0.6 mg every week up to 1.8 mg/day (24 weeks). In the DUAL II, DUAL V and Lixilan-L, Liraglutide and Lixisenatide were titrated depending on basal insulin requirement for obtaining fasting glucose control on the bases of a treat-to-target approach; therefore, the final dose of Liraglutide and Lixisenatide was approximately 1.6 mg/day (DUAL II), 1.5 mg/day (DUAL V) and 17 μg/day (Lixilan-L). In the DUAL V, 40% of patients assuming Liraglutide/Degludec received 50 dose steps after 26 weeks corresponding to 1.8 mg of Liraglutide, with a wide range of variability also depending on baseline BMI (65). In the Lixilan-L, around 27% of participants received the final dose of 60 IU of Lixisenatide/Glargine, equivalent to the entire daily dose of Lixisenatide. Semaglutide was started at 0.25 mg once a week, then increased to 0.5 mg and 1 mg every four weeks. Therefore, in the SUSTAIN-5, the final dose of Semaglutide was reached after four weeks in the arm randomized to 0.5 mg/qw and after eight weeks in those randomized to 1 mg/qw with global exposure to the intention-to-treat pre-established dose of 26 weeks (0.5 mg) and 22 weeks (1 mg). TZP was started at 2.5 mg once a week and progressively titrated by 2.5 mg every four weeks; thus, the final dose for each arm (5, 10, and 15 mg) was respectively achieved after four, twelve, and twenty weeks from randomization with a considerable difference in time of exposure to TZP 5 mg (36 weeks), TZP 10 mg (28 weeks), TZP 15 mg (20 weeks).
Once these considerations are discussed, GLP-1RAs or GIP/GLP-1RA added to basal insulin effectively improve glucose control, induce weight loss, and prevent unnecessary basal insulin over-titration with a reasonable chance to reduce the daily dose of basal insulin while improving glucose control. Most importantly, improvement in glucose control and weight reduction are obtained without increasing the overall risk of hypoglycemia and the number of severe hypoglycemic events. Safety data confirm that GLP-1RAs and TZP increase the risk of gastrointestinal adverse events (mostly mild-to-moderate), including nausea, diarrhea, and vomiting that are usually well-tolerated and resolved in a few weeks of treatment because of tachyphylaxis (especially with long-acting analogues and TZP). Weekly administered agonists performed better compared to daily ones. However, among daily administered analogues, Liraglutide appears to perform well, especially when administered separately in add-on to insulin Glargine (47). After excluding Liraglutide from the dataset, the levels of heterogeneity have been reduced moderately regarding the chance of reaching HbA1c <7% (I2 42%) and considerably for change in fasting plasma glucose (I2 0%), suggesting that Liraglutide compared to other daily administered GLP-1RAs is more effective in reducing those outcomes (data not shown). Short-acting GLP-1RAs exhibit a considerable fluctuation in circulating levels during the daytime that, in turn, produce an intermittent activation of GLP-1R, resulting in different pharmacodynamic effects compared to long-acting GLP-1RA (66). As a result, short-acting compared to long-acting GLP-1RAs are less effective in reducing fasting glucose and improving overall glucose control but are more effective in delaying gastric emptying and reducing glycemic excursion after meal ingestion (67). On the other hand, Liraglutide, Dulaglutide, and Exenatide LAR display similar efficacy in different therapeutic approaches, performing better than short-acting GLP-1RAs (68). The results of this metanalysis are in line with a previous one, thus confirming that therapeutic intensification with GLP-1RA is an effective and safe approach, especially using long-acting GLP-1RAs, mostly weekly than daily administered ones (69).
TZP looks like a further tool to improve glucose control as an add-on to basal insulin in this scenario. In addition to a potent antihyperglycemic effect, TZP leads to an impressive weight loss exceeding the results provided by other agents. When excluding TZP from the dataset, the overall efficacy of GLP-1RAs was reduced significantly and resulted in a slight body weight loss (-2.40 kg, 95% CI -3.04; -1.76) without subgroup differences (I2 0%) between daily and weekly administered analogues (data not shown).
Overweight and obesity are most commonly observed in T2D, and this unhealthy relationship is expected to rise over time with growing detrimental consequences (70). So far, body weight management should be addressed as one of the leading therapeutic goals in T2D, with stringent targets must be achieved (71). This issue has also been recently reviewed by De Block et. that emphasized the importance of reaching adequate glycemic control and body weight loss as a common goal to improve the cardiovascular safety of these individuals. For this purpose, they reviewed the role of high-dose GLP-1RAs, such as Liraglutide 3 mg, Semaglutide 2 and 2.4 mg, Dulaglutide 3 and 4.5 mg, TZP, and the dual agonist glucagon/GLP-1 Cotadutide (72).
Interestingly, the results of the SURPASS-5 tell us information about the impressive weight loss observed with TZP in insulin users. A decline in insulin dose requirement could explain weight reduction after intensification with the dual agonist in this setting. However, as observed in the SURPASS-5, daily insulin dose remained similar in participants randomized to TZP 5 and 10 mg and was mildly reduced in those assuming TZP 15 mg. Therefore, the contribution of daily insulin dose reduction to weight loss appeared almost negligible, and the mechanisms underlying this effect are not completely understood. However, they could be attributable to the GIP component of TZP that exerts some extra glycemic synergistic effects with GLP-1RA and some others over GLP-1RA. As an example, in experimental conditions with animal models (GLP-1R-null), TZP reduced appetite and food intake, liver glycogenolysis and gluconeogenesis, improved insulin sensibility (assessed by euglycemic clamp) and glucose disposal in white and brown adipose tissue, skeletal muscle, with body weight-dependent and independent effects (73). These actions resulted in a significant reduction in body weight, corresponding to liver and adipose tissue weight restrictions (73). These findings suggest that GIP but not GLP-1RA may improve glucose disposal by ameliorating peripheral insulin sensibility and glucose utilization, while GIP added on to GLP-1RA may reduce food intake with a combined effect resulting in improvement of glucose control and weight loss.
TZP appears a potent tool to improve glucose control without increasing hypoglycemic risk in poorly controlled T2D treated with basal insulin with or without other antihyperglycemic oral agents. Its impressive effect on body weight loss, despite background therapy, may be an important resource also for improving weight management in a usually difficult-to-treat cluster of patients.
The raw data supporting the conclusions of this article will be made available by the authors, on a reasonable request.
GL conceived the systematic review and meta-analysis. GL and ADT elaborated the search strategy. GL and ADT developed the selection criteria and assessed the risk of bias in included studies. GL provided statistical expertise. GL and ADT drafted the manuscript. GL, ADT, OD, VDG, GP, GDP, VAG, EJ, EG, CS, and VT read the text, provided feedback, and approved the manuscript.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
DPP-IV, Dipeptidyl-Peptidase 4; FPG, Fasting Plasma Glucose; GIP, Gastric Inhibitory Polypeptide/Glucose-dependent Insulinotropic Polypeptide; GIPR, GIP Receptor; GLP-1R, GLP-1 Receptor; GLP-1Ras, GLP-1 receptor agonists; GLP-1, Glucagon-Like Peptide-1; RCTs, Randomized Clinical Trials; RR, Risk Ratio; TZP, Tirzepatide; T2D, Type 2 Diabetes.
1. Bayliss WM, Starling EH. The mechanism of pancreatic secretion. J Physiol (1902) 28(5):325–53. doi: 10.1113/jphysiol.1902.sp000920
2. Moore B. On the treatment of diabetus mellitus by acid extract of duodenal mucous membrane. Biochem J (1906) 1(1):28–38. doi: 10.1042/bj0010028
3. Seino Y, Fukushima M, Yabe D. GIP and GLP-1, the two incretin hormones: Similarities and differences. J Diabetes Investig (2010) 1(1-2):8–23. doi: 10.1111/j.2040-1124.2010.00022.x
4. Elrick H, Stimmler L, Hlad CJ Jr, Arai Y. Plasma insulin response to oral and intravenous glucose administration. J Clin Endocrinol Metab (1964) 24:1076–82. doi: 10.1210/jcem-24-10-1076
5. Mcintyre N, Holdsworth CD, Turner DS. New interpretation of oral glucose tolerance. Lancet (1964) 2(7349):20–1. doi: 10.1016/S0140-6736(64)90011-X
6. Rehfeld JF. The origin and understanding of the incretin concept. Front Endocrinol (Lausanne) (2018) 9:387. doi: 10.3389/fendo.2018.00387
7. Nauck MA, Quast DR, Wefers J, Pfeiffer AFH. The evolving story of incretins (GIP and GLP-1) in metabolic and cardiovascular disease: A pathophysiological update. Diabetes Obes Metab (2021) 23 Suppl 3:5–29. doi: 10.1111/dom.14496
8. Andersen DB, Holst JJ. Peptides in the regulation of glucagon secretion. Peptides (2022) 148:170683. doi: 10.1016/j.peptides.2021.170683
9. Wolfe MM, Zhao KB, Glazier KD, Jarboe LA, Tseng CC. Regulation of glucose-dependent insulinotropic polypeptide release by protein in the rat. Am J Physiol Gastrointest Liver Physiol (2000) 279(3):G561–6. doi: 10.1152/ajpgi.2000.279.3.G561
10. Dailey MJ, Moran TH. Glucagon-like peptide 1 and appetite. Trends Endocrinol Metab (2013) 24(2):85–91. doi: 10.1016/j.tem.2012.11.008
11. Tseng CC, Jarboe LA, Wolfe MM. Regulation of glucose-dependent insulinotropic peptide gene expression by a glucose meal. Am J Physiol (1994) 266(5 Pt 1):G887–91. doi: 10.1152/ajpgi.1994.266.5.G887
12. El K, Campbell JE. The role of GIP in α-cells and glucagon secretion. Peptides (2020) 125:170213. doi: 10.1016/j.peptides.2019.170213
13. Zhang Y, Parajuli KR, Fava GE, Gupta R, Xu W, Nguyen LU, et al. GLP-1 receptor in pancreatic α-cells regulates glucagon secretion in a glucose-dependent bidirectional manner. Diabetes (20192020) 6869(2):267–8. doi: 10.2337/db20-er02c
14. Ahrén B. Incretin dysfunction in type 2 diabetes: clinical impact and future perspectives. Diabetes Metab (2013) 39(3):195–201. doi: 10.1016/j.diabet.2013.03.001
15. Adriaenssens AE, Biggs EK, Darwish T, Tadross J, Sukthankar T, Girish M, et al. Glucose-dependent insulinotropic polypeptide receptor-expressing cells in the hypothalamus regulate food intake. Cell Metab (2019) 30(5):987–96.e6. doi: 10.1016/j.cmet.2019.07.013
16. Meier JJ, Goetze O, Anstipp J, Hagemann D, Holst JJ, Schmidt WE, et al. Gastric inhibitory polypeptide does not inhibit gastric emptying in humans. Am J Physiol Endocrinol Metab (2004) 286(4):E621–5. doi: 10.1152/ajpendo.00499.2003
17. Nauck MA, Meier JJ. Incretin hormones: Their role in health and disease. Diabetes Obes Metab (2018) 20 Suppl 1:5–21. doi: 10.1111/dom.13129
18. Kamruzzaman M, Horowitz M, Jones KL, Marathe CS. Gut-based strategies to reduce postprandial glycaemia in type 2 diabetes. Front Endocrinol (Lausanne) (2021) 12:661877. doi: 10.3389/fendo.2021.661877
19. Nauck MA, Quast DR, Wefers J, Meier JJ. GLP-1 receptor agonists in the treatment of type 2 diabetes - state-of-the-art. Mol Metab (2021) 46:101102. doi: 10.1016/j.molmet.2020.101102
20. Coskun T, Sloop KW, Loghin C, Alsina-Fernandez J, Urva S, Bokvist KB, et al. LY3298176, a novel dual GIP and GLP-1 receptor agonist for the treatment of type 2 diabetes mellitus: From discovery to clinical proof of concept. Mol Metab (2018) 18:3–14. doi: 10.1016/j.molmet.2018.09.009
21. Skow MA, Bergmann NC, Knop FK. Diabetes and obesity treatment based on dual incretin receptor activation: ‘twincretins’. Diabetes Obes Metab (2016) 18(9):847–54. doi: 10.1111/dom.12685
22. Fisman EZ, Tenenbaum A. The dual glucose-dependent insulinotropic polypeptide (GIP) and glucagon-like peptide-1 (GLP-1) receptor agonist tirzepatide: a novel cardiometabolic therapeutic prospect. Cardiovasc Diabetol (2021) 20(1):225. doi: 10.1186/s12933-021-01412-5
23. Samms RJ, Coghlan MP, Sloop KW. How may GIP enhance the therapeutic efficacy of GLP-1? Trends Endocrinol Metab (2020) 31(6):410–21. doi: 10.1016/j.tem.2020.02.006
24. Min T, Bain SC. The role of tirzepatide Dual GIP And GLP-1 receptor agonist, in the management of type 2 diabetes: The SURPASS clinical trials. Diabetes Ther (2021) 12(1):143–57. doi: 10.1007/s13300-020-00981-0
25. Thomas MK, Nikooienejad A, Bray R, Cui X, Wilson J, Duffin K, et al. Dual GIP and GLP-1 receptor agonist tirzepatide improves beta-cell function and insulin sensitivity in type 2 diabetes. J Clin Endocrinol Metab (2021) 106(2):388–96. doi: 10.1210/clinem/dgaa863
26. Frias JP, 3rd BEJ, Vignati L, Tschöp MH, Schmitt C, Owen K, et al. The sustained effects of a dual GIP/GLP-1 receptor agonist, NNC0090-2746, in patients with type 2 diabetes. Cell Metab (2017) 26(2):343–52.e2. doi: 10.1016/j.cmet.2017.07.011
27. Schmitt C, Portron A, Jadidi S, Sarkar N, DiMarchi R. Pharmacodynamics, pharmacokinetics and safety of multiple ascending doses of the novel dual glucose-dependent insulinotropic polypeptide/glucagon-like peptide-1 agonist RG7697 in people with type 2 diabetes mellitus. Diabetes Obes Metab (2017) 19(10):1436–45. doi: 10.1111/dom.13024
28. Bastin M, Andreelli F. Dual GIP-GLP1-Receptor agonists in the treatment of type 2 diabetes: A short review on emerging data and therapeutic potential. Diabetes Metab Syndr Obes (2019) 12:1973–85. doi: 10.2147/DMSO.S191438
29. Mathiesen DS, Bagger JI, Bergmann NC, Lund A, Christensen MB, Vilsbøll T, et al. The effects of dual GLP-1/GIP receptor agonism on glucagon secretion-a review. Int J Mol Sci (2019) 20(17):4092. doi: 10.3390/ijms20174092
30. Holst JJ, Rosenkilde MM. GIP as a therapeutic target in diabetes and obesity: Insight from incretin Co-agonists. J Clin Endocrinol Metab (2020) 105(8):e2710–6. doi: 10.1210/clinem/dgaa327
31. Novikoff A, O’Brien SL, Bernecker M, Grandl G, Kleinert M, Knerr PJ, et al. Spatiotemporal GLP-1 and GIP receptor signaling and trafficking/recycling dynamics induced by selected receptor mono- and dual-agonists. Mol Metab (2021) 49:101181. doi: 10.1016/j.molmet.2021.101181
32. Østergaard S, Paulsson JF, Kofoed J, Zosel F, Olsen J, Jeppesen CB, et al. The effect of fatty diacid acylation of human PYY3-36 on Y2 receptor potency and half-life in minipigs. Sci Rep (2021) 11(1):21179. doi: 10.1038/s41598-021-00654-3
33. Zhao F, Zhou Q, Cong Z, Hang K, Zou X, Zhang C, et al. Structural insights into multiplexed pharmacological actions of tirzepatide and peptide 20 at the GIP, GLP-1 or glucagon receptors. Nat Commun (2022) 13(1):1057. doi: 10.1038/s41467-022-28683-0
34. Rosenstock J, Wysham C, Frías JP, Kaneko S, Lee CJ, Fernández Landó L, et al. Efficacy and safety of a novel dual GIP and GLP-1 receptor agonist tirzepatide in patients with type 2 diabetes (SURPASS-1): a double-blind, randomised, phase 3 trial. Lancet (2021) 398(10295):143–55. doi: 10.1016/S0140-6736(21)01324-6
35. Frías JP, Davies MJ, Rosenstock J, Pérez Manghi FC, Fernández Landó L, Bergman BK, et al. Tirzepatide versus semaglutide once weekly in patients with type 2 diabetes. N Engl J Med (2021) 385(6):503–15. doi: 10.1056/NEJMoa2107519
36. Ludvik B, Giorgino F, Jódar E, Frias JP, Fernández Landó L, Brown K, et al. Once-weekly tirzepatide versus once-daily insulin degludec as add-on to metformin with or without SGLT2 inhibitors in patients with type 2 diabetes (SURPASS-3): a randomised, open-label, parallel-group, phase 3 trial. Lancet (2021) 398(10300):583–98. doi: 10.1016/S0140-6736(21)01443-4
37. Del Prato S, Kahn SE, Pavo I, Weerakkody GJ, Yang Z, Doupis J, et al. Tirzepatide versus insulin glargine in type 2 diabetes and increased cardiovascular risk (SURPASS-4): a randomised, open-label, parallel-group, multicentre, phase 3 trial. Lancet (2021) 398(10313):1811–24. doi: 10.1016/S0140-6736(21)02188-7
38. Dahl D, Onishi Y, Norwood P, Huh R, Bray R, Patel H, et al. Effect of subcutaneous tirzepatide vs placebo added to titrated insulin glargine on glycemic control in patients with type 2 diabetes: The SURPASS-5 randomized clinical trial. JAMA (2022) 327(6):534–45. doi: 10.1001/jama.2022.0078
39. Chang-Chen KJ, Mullur R, Bernal-Mizrachi E. Beta-cell failure as a complication of diabetes. Rev Endocr Metab Disord (2008) 9(4):329–43. doi: 10.1007/s11154-008-9101-5
40. Hanefeld M, Fleischmann H, Siegmund T, Seufert J. Rationale for timely insulin therapy in type 2 diabetes within the framework of individualised treatment: 2020 update. Diabetes Ther (2020) 11(8):1645–66. doi: 10.1007/s13300-020-00855-5
41. Dalal MR, Grabner M, Bonine N, Stephenson JJ, DiGenio A, Bieszk N. Are patients on basal insulin attaining glycemic targets? characteristics and goal achievement of patients with type 2 diabetes mellitus treated with basal insulin and physician-perceived barriers to achieving glycemic targets. Diabetes Res Clin Pract (2016) 121:17–26. doi: 10.1016/j.diabres.2016.08.004
42. Mocarski M, Yeaw J, Divino V, DeKoven M, Guerrero G, Langer J, et al. Slow titration and delayed intensification of basal insulin among patients with type 2 diabetes. J Manag Care Spec Pharm (2018) 24(4):390–400. doi: 10.18553/jmcp.2017.17218
43. Buse JB, Bergenstal RM, Glass LC, Heilmann CR, Lewis MS, Kwan AY, et al. Use of twice-daily exenatide in basal insulin-treated patients with type 2 diabetes: a randomized, controlled trial. Ann Intern Med (2011) 154(2):103–12. doi: 10.7326/0003-4819-154-2-201101180-00300
44. Riddle MC, Forst T, Aronson R, Sauque-Reyna L, Souhami E, Silvestre L, et al. Adding once-daily lixisenatide for type 2 diabetes inadequately controlled with newly initiated and continuously titrated basal insulin glargine: a 24-week, randomized, placebo-controlled study (GetGoal-duo 1). Diabetes Care (2013) 36(9):2497–503. doi: 10.2337/dc12-2462
45. Riddle MC, Aronson R, Home P, Marre M, Niemoeller E, Miossec P, et al. Adding once-daily lixisenatide for type 2 diabetes inadequately controlled by established basal insulin: a 24-week, randomized, placebo-controlled comparison (GetGoal-l). Diabetes Care (2013) 36(9):2489–96. doi: 10.2337/dc12-2454
46. Buse JB, Vilsbøll T, Thurman J, Blevins TC, Langbakke IH, Bøttcher SG, et al. Contribution of liraglutide in the fixed-ratio combination of insulin degludec and liraglutide (IDegLira). Diabetes Care (2014) 37(11):2926–33. doi: 10.2337/dc14-0785
47. Ahmann A, Rodbard HW, Rosenstock J, Lahtela JT, de Loredo L, Tornøe K, et al. Efficacy and safety of liraglutide versus placebo added to basal insulin analogues (with or without metformin) in patients with type 2 diabetes: a randomized, placebo-controlled trial. Diabetes Obes Metab (2015) 17(11):1056–64. doi: 10.1111/dom.12539
48. Aroda VR, Rosenstock J, Wysham C, Unger J, Bellido D, González-Gálvez G, et al. Efficacy and safety of LixiLan, a titratable fixed-ratio combination of insulin glargine plus lixisenatide in type 2 diabetes inadequately controlled on basal insulin and metformin: The LixiLan-l randomized trial. Diabetes Care (2016) 39(11):1972–80. doi: 10.2337/dc16-1495
49. Lingvay I, Pérez Manghi F, García-Hernández P, Norwood P, Lehmann L, Tarp-Johansen MJ, et al. Effect of insulin glargine up-titration vs insulin Degludec/Liraglutide on glycated hemoglobin levels in patients with uncontrolled type 2 diabetes: The DUAL V randomized clinical trial. JAMA (2016) 315(9):898–907. doi: 10.1001/jama.2016.1252
50. Pozzilli P, Norwood P, Jódar E, Davies MJ, Ivanyi T, Jiang H, et al. Placebo-controlled, randomized trial of the addition of once-weekly glucagon-like peptide-1 receptor agonist dulaglutide to titrated daily insulin glargine in patients with type 2 diabetes (AWARD-9). Diabetes Obes Metab (2017) 19(7):1024–31. doi: 10.1111/dom.12937
51. Rodbard HW, Lingvay I, Reed J, de la Rosa R, Rose L, Sugimoto D, et al. Semaglutide added to basal insulin in type 2 diabetes (SUSTAIN 5): A randomized, controlled trial. J Clin Endocrinol Metab (2018) 103(6):2291–301. doi: 10.1210/jc.2018-00070
52. Guja C, Frías JP, Somogyi A, Jabbour S, Wang H, Hardy E, et al. Effect of exenatide QW or placebo, both added to titrated insulin glargine, in uncontrolled type 2 diabetes: The DURATION-7 randomized study. Diabetes Obes Metab (2018) 20(7):1602–14. doi: 10.1111/dom.13266
53. Seino Y, Min KW, Niemoeller E, Takami A, EFC10887 GETGOAL-L Asia Study Investigators. Randomized, double-blind, placebo-controlled trial of the once-daily GLP-1 receptor agonist lixisenatide in Asian patients with type 2 diabetes insufficiently controlled on basal insulin with or without a sulfonylurea (GetGoal-L-Asia). Diabetes Obes Metab (2012) 14(10):910–7. doi: 10.1111/j.1463-1326.2012.01618.x
54. Yang W, Min K, Zhou Z, Li L, Xu X, Zhu D, et al. Efficacy and safety of lixisenatide in a predominantly Asian population with type 2 diabetes insufficiently controlled with basal insulin: The GetGoal-L-C randomized trial. Diabetes Obes Metab (2018) 20(2):335–43. doi: 10.1111/dom.13072
55. Gough SC, Bode B, Woo V, Rodbard HW, Linjawi S, Poulsen P, et al. Efficacy and safety of a fixed-ratio combination of insulin degludec and liraglutide (IDegLira) compared with its components given alone: results of a phase 3, open-label, randomised, 26-week, treat-to-target trial in insulin-naive patients with type 2 diabetes. Lancet Diabetes Endocrinol (2014) 2(11):885–93. doi: 10.1016/S2213-8587(14)70174-3
56. Rosenstock J, Aronson R, Grunberger G, Hanefeld M, Piatti P, Serusclat P, et al. Benefits of LixiLan, a titratable fixed-ratio combination of insulin glargine plus lixisenatide, versus insulin glargine and lixisenatide monocomponents in type 2 diabetes inadequately controlled on oral agents: The LixiLan-O randomized trial. Diabetes Care (2016) 39(11):2026–35. doi: 10.2337/dc16-0917
57. Rosenstock J, Diamant M, Aroda VR, Silvestre L, Souhami E, Zhou T, et al. Efficacy and safety of LixiLan, a titratable fixed-ratio combination of lixisenatide and insulin glargine, versus insulin glargine in type 2 diabetes inadequately controlled on metformin monotherapy: The LixiLan proof-of-Concept randomized trial. Diabetes Care (2016) 39(9):1579–86. doi: 10.2337/dc16-0046
58. Garvey WT, Birkenfeld AL, Dicker D, Mingrone G, Pedersen SD, Satylganova A, et al. Efficacy and safety of liraglutide 3.0 mg in individuals with overweight or obesity and type 2 diabetes treated with basal insulin: The SCALE insulin randomized controlled trial. Diabetes Care (2020) 43(5):1085–93. doi: 10.2337/dc19-1745
59. Sterne JAC, Savović J, Page MJ, Elbers RG, Blencowe NS, Boutron I, et al. RoB 2: a revised tool for assessing risk of bias in randomised trials. BMJ (2019) 366:l4898. doi: 10.1136/bmj.l4898
60. Bethel MA, Patel RA, Merrill P, Lokhnygina Y, Buse JB, Mentz RJ, et al. Cardiovascular outcomes with glucagon-like peptide-1 receptor agonists in patients with type 2 diabetes: a meta-analysis. Lancet Diabetes Endocrinol (2018) 6(2):105–13. doi: 10.1016/S2213-8587(17)30412-6
61. Abd El Aziz MS, Kahle M, Meier JJ, Nauck MA. A meta-analysis comparing clinical effects of short- or long-acting GLP-1 receptor agonists versus insulin treatment from head-to-head studies in type 2 diabetic patients. Diabetes Obes Metab (2017) 19(2):216–27. doi: 10.1111/dom.12804
62. Castellana M, Cignarelli A, Brescia F, Laviola L, Giorgino F. GLP-1 receptor agonist added to insulin versus basal-plus or basal-bolus insulin therapy in type 2 diabetes: A systematic review and meta-analysis. Diabetes Metab Res Rev (2019) 35(1):e3082. doi: 10.1002/dmrr.3082
63. Maiorino MI, Chiodini P, Bellastella G, Scappaticcio L, Longo M, Esposito K, et al. Free and fixed-ratio combinations of basal insulin and GLP-1 receptor agonists versus basal insulin intensification in type 2 diabetes: A systematic review and meta-analysis of randomized controlled trials. Diabetes Obes Metab (2018) 20(9):2309–13. doi: 10.1111/dom.13343
64. DeFronzo RA, Stonehouse AH, Han J, Wintle ME. Relationship of baseline HbA1c and efficacy of current glucose-lowering therapies: a meta-analysis of randomized clinical trials. Diabetes Med (2010) 27(3):309–17. doi: 10.1111/j.1464-5491.2010.02941.x
65. Lingvay I, Harris S, Jaeckel E, Chandarana K, Ranthe MF, Jódar E. Insulin degludec/liraglutide (IDegLira) was effective across a range of dysglycaemia and body mass index categories in the DUAL V randomized trial. Diabetes Obes Metab (2018) 20(1):200–05. doi: 10.1111/dom.13043
66. Uccellatore A, Genovese S, Dicembrini I, Mannucci E, Ceriello A. Comparison review of short-acting and long-acting glucagon-like peptide-1 receptor agonists. Diabetes Ther (2015) 6(3):239–56. doi: 10.1007/s13300-015-0127-x
67. Miñambres I, Pérez A. Is there a justification for classifying GLP-1 receptor agonists as basal and prandial? Diabetol Metab Syndr (2017) 9:6. doi: 10.1186/s13098-017-0204-6
68. Htike ZZ, Zaccardi F, Papamargaritis D, Webb DR, Khunti K, Davies MJ. Efficacy and safety of glucagon-like peptide-1 receptor agonists in type 2 diabetes: A systematic review and mixed-treatment comparison analysis. Diabetes Obes Metab (2017) 19(4):524–36. doi: 10.1111/dom.12849
69. Huthmacher JA, Meier JJ, Nauck MA. Efficacy and safety of short- and long-acting glucagon-like peptide 1 receptor agonists on a background of basal insulin in type 2 diabetes: A meta-analysis. Diabetes Care (2020) 43(9):2303–12. doi: 10.2337/dc20-0498
70. Lin X, Li H. Obesity: Epidemiology, pathophysiology, and therapeutics. Front Endocrinol (Lausanne) (2021) 12:706978. doi: 10.3389/fendo.2021.706978
71. Lingvay I, Sumithran P, Cohen RV, le Roux CW. Obesity management as a primary treatment goal for type 2 diabetes: time to reframe the conversation. Lancet (2022) 399(10322):394–405. doi: 10.1016/S0140-6736(21)01919-X
72. De Block CEM, Dirinck E, Verhaegen A, Van Gaal LF. Efficacy and safety of high-dose glucagon-like peptide-1, glucagon-like peptide-1/glucose-dependent insulinotropic peptide, and glucagon-like peptide-1/glucagon receptor agonists in type 2 diabetes. Diabetes Obes Metab (2022) 24(5):788–805. doi: 10.1111/dom.14640
Keywords: GIP, GLP-1, tirzepatide, basal insulin, type 2 diabetes, obesity, body weight, hypoglycemia
Citation: Lisco G, De Tullio A, Disoteo O, De Geronimo V, Piazzolla G, De Pergola G, Giagulli VA, Jirillo E, Guastamacchia E, Sabbà C and Triggiani V (2022) Basal insulin intensification with GLP-1RA and dual GIP and GLP-1RA in patients with uncontrolled type 2 diabetes mellitus: A rapid review of randomized controlled trials and meta-analysis. Front. Endocrinol. 13:920541. doi: 10.3389/fendo.2022.920541
Received: 14 April 2022; Accepted: 19 August 2022;
Published: 08 September 2022.
Edited by:
Rishi Raj, Pikeville Medical Center, United StatesReviewed by:
Sudhanshu Kumar Bharti, Patna University, IndiaCopyright © 2022 Lisco, De Tullio, Disoteo, De Geronimo, Piazzolla, De Pergola, Giagulli, Jirillo, Guastamacchia, Sabbà and Triggiani. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Vincenzo Triggiani, dmluY2Vuem8udHJpZ2dpYW5pQHVuaWJhLml0
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.