 Conghui Cao
Conghui Cao Xiaoli Wang
Xiaoli Wang Xiaojuan Zhao
Xiaojuan Zhao
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
CASE REPORT article
Front. Endocrinol., 25 July 2022
Sec. Clinical Diabetes
Volume 13 - 2022 | https://doi.org/10.3389/fendo.2022.914863
This article is part of the Research TopicClinical Aspects of Different Forms of Diabetes in Children and AdolescentsView all 9 articles
Background: Chromosome 8p11.2 includes several key genes in development such as the FGFR1, ANK1, KAT6A, and SLC20A2 genes. Deletion of this fragment causes a contiguous gene syndrome. Currently, few cases of interstitial deletion of whole 8p11.2 have been reported. We report a rare case of 8p11.2 deletion syndrome with the unique phenotypes, presenting with early-onset diabetes.
Case Description: A 20-year-old man with a 1-year history of diabetes mellitus was admitted to the Endocrinology Clinic. Physical examination revealed the dysmorphic facial features, and broad and foreshortened halluces. Laboratory examination indicated spherocytosis anemia, and hypogonadotropic hypogonadism. Bone mineral density analysis showed decreased bone density in the lumbar vertebrae. Brain CT showed calcification. Whole-exome sequencing revealed a 7.05-Mb deletion in 8p11 containing 43 OMIM genes, and a large in-frame deletion of exons 48–55 in the DMD gene. Metformin was given to the patient after which his blood glucose was well controlled. HCG was injected subcutaneously and was supplemented with calcium and vitamin D, which led to an improvement in the patient’s quality of life.
Conclusion: We report a rare case of 8p11.2 deletion syndrome with unique phenotypes, and early-onset diabetes. It is challenging for endocrinologists to simultaneously reconcile a combination of these diseases across multiple disciplines. We discussed the influencing factors of early-onset diabetes in this patient and speculated that it was caused by complex interactions of known and unknown genetic backgrounds and environmental factors.
Chromosome 8p11.2 includes several key genes in development such as the FGFR1, ANK1, KAT6A, and SLC20A2 genes. Deletion of this fragment causes a contiguous gene syndrome, including congenital hypogonadotropic hypogonadism (CHH) (1), dysmorphic facial features, hands, and feet (2, 3), spherocytosis (4), Arboleda-Tham syndrome (5), and brain calcification (6). Currently, few cases of interstitial deletion of whole 8p11.2 have been reported. Herein, we report a rare case of 8p11.2 deletion syndrome with the above unique phenotypes, presenting with early-onset diabetes.
A 20-year-old man with a 1-year history of diabetes mellitus was admitted to the Endocrinology Clinic of the First Affiliated Hospital of China Medical University. He was born after an uneventful 39-week gestation with a birth weight of 3.0 kg. His parents are non-consanguineous and healthy. His motor skills and language development were delayed. Physical examination revealed the following characteristics: height, 165.0 cm; weight, 50.0 kg; body mass index, 18.36 kg/m2; facial asymmetry; micrognathia; up-slanting palpebral fissure; malformed ear; dental dysplasia; high-arched palate (Figure 1A); and broad and foreshortened halluces (Figure 1B). Tanner’s staging showed prepubertal signs. A smell test excluded anosmia.
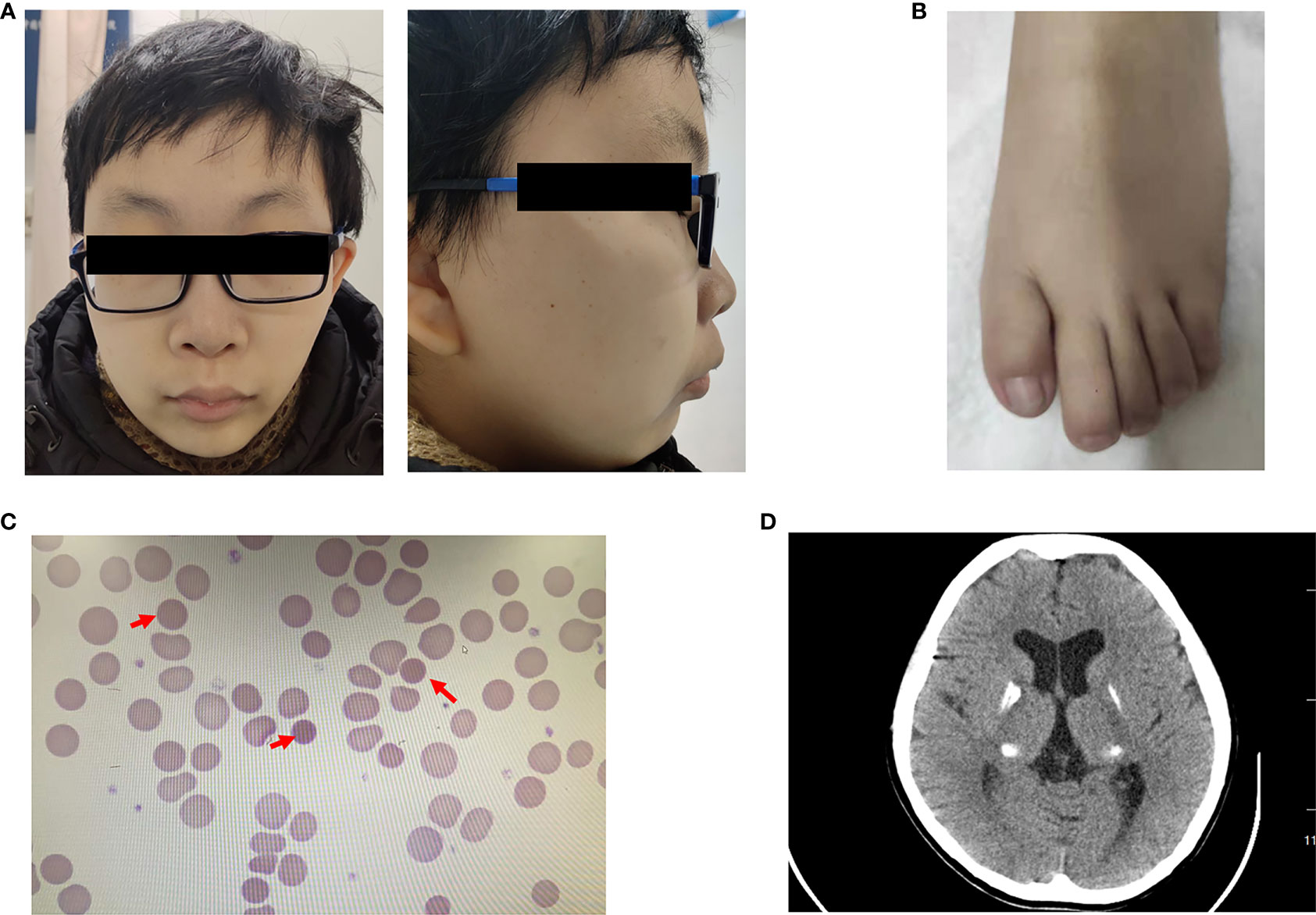
Figure 1 Clinical characters of the patient (A) Facial anomalies in the patient, including facial asymmetry, micrognathia, malformed ear, dental dysplasia, and a high-arched palate. (B) Feet anomalies showed broad and foreshortened halluces. (C) Peripheral blood smear revealed small spherocytes (red arrows). (D) Brain CT showed calcification in the bilateral lenticular nucleus, dorsal thalamus, and posterior horn of lateral ventricle.
Laboratory examination indicated anemia with a hemoglobin level of 101 g/L. Morphological examination of a peripheral blood smear revealed small spherocytes (Figure 1C). Total bilirubin was 71.2 μmol/L. The concentrations of luteinizing hormone and follicle-stimulating hormone were low at baseline and after gonadorelin stimulation, while the testosterone concentration was undetectable. His fasting glucose and insulin levels were 12.12 mmol/L and 9.93 mIU/L, respectively. T1D autoantibodies (GAD, ZnT8, IA-2, ICA, and IAA antibodies) were negative. His HbA1c was 5.8%. Detailed test results are listed in Tables 1, 2.
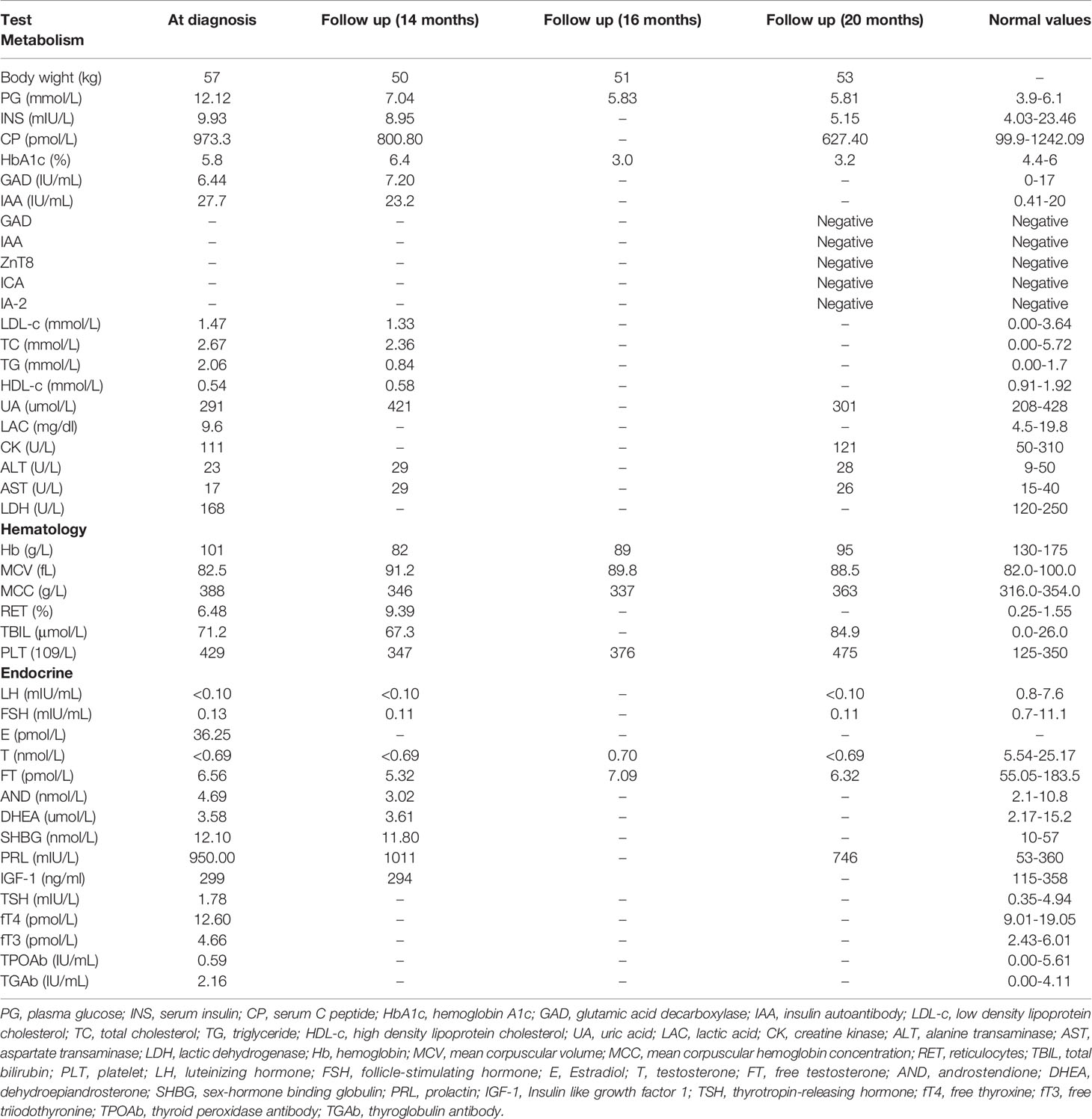
Table 1 Laboratory investigations.
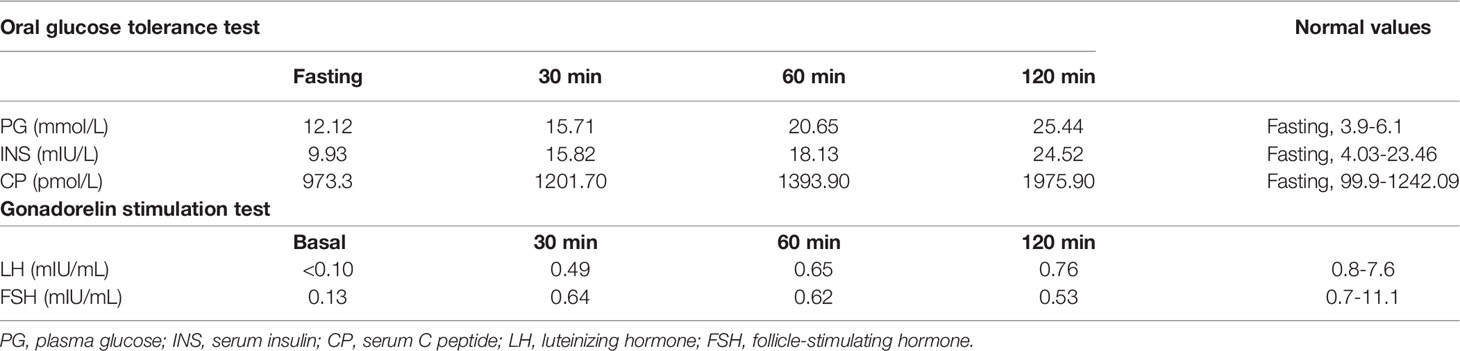
Table 2 Results of OGTT and gonadorelin stimulation test.
Bioelectrical impedance analysis showed visceral fat of 81 cm2, subcutaneous fat of 136.8 cm2, and a visceral-to-subcutaneous fat ratio of 59.2%. Testicular ultrasound revealed that the right testis was approximately 1.37 cm × 0.54 cm × 0.93 cm (0.36 ml) in size and that the left testis was approximately 1.29 cm × 0.52 cm × 0.93 cm (0.33 ml) in size. Bone mineral density analysis showed decreased bone density in the lumbar vertebrae (Z = -3.8). Cardiac color Doppler ultrasound indicated that cardiac function and structure were normal, whereas abdominal color Doppler ultrasound showed that the shape and size of the liver were normal, the liver surface was smooth, the edge of the liver was sharp, the echo of liver parenchyma was enhanced, and the contrast of the liver and kidney was increased. The length and diameter of spleen was approximately 10.61 cm, and the thickness of spleen was approximately 5.38 cm, indicating the presence of fatty liver and splenomegaly. Brain CT showed calcification in the bilateral lenticular nucleus, dorsal thalamus, and posterior horn of the lateral ventricle (Figure 1D). Ophthalmic examination revealed a cotton wool spot in the right eye retina, which was indicative of stage III diabetic retinopathy. Electromyography was normal, and electroaudiometry indicated normal hearing.
Whole-exome sequencing revealed a 7.05-Mb deletion in 8p11 (chr8: g.35541034–42587731) containing 43 Online Mendelian Inheritance in Man (OMIM) genes (Figures 2A, C and Table 3), and a large in-frame deletion of exons 48–55 in the DMD gene (chrX: g.31639827–31904558). These abnormalities were confirmed by copy number variation (CNV)-seq analysis (Figures 2A, B). Multiplex ligation probe amplification (MLPA)-DMD revealed that the proband’s mother is a heterozygous carrier of the mutant DMD gene.
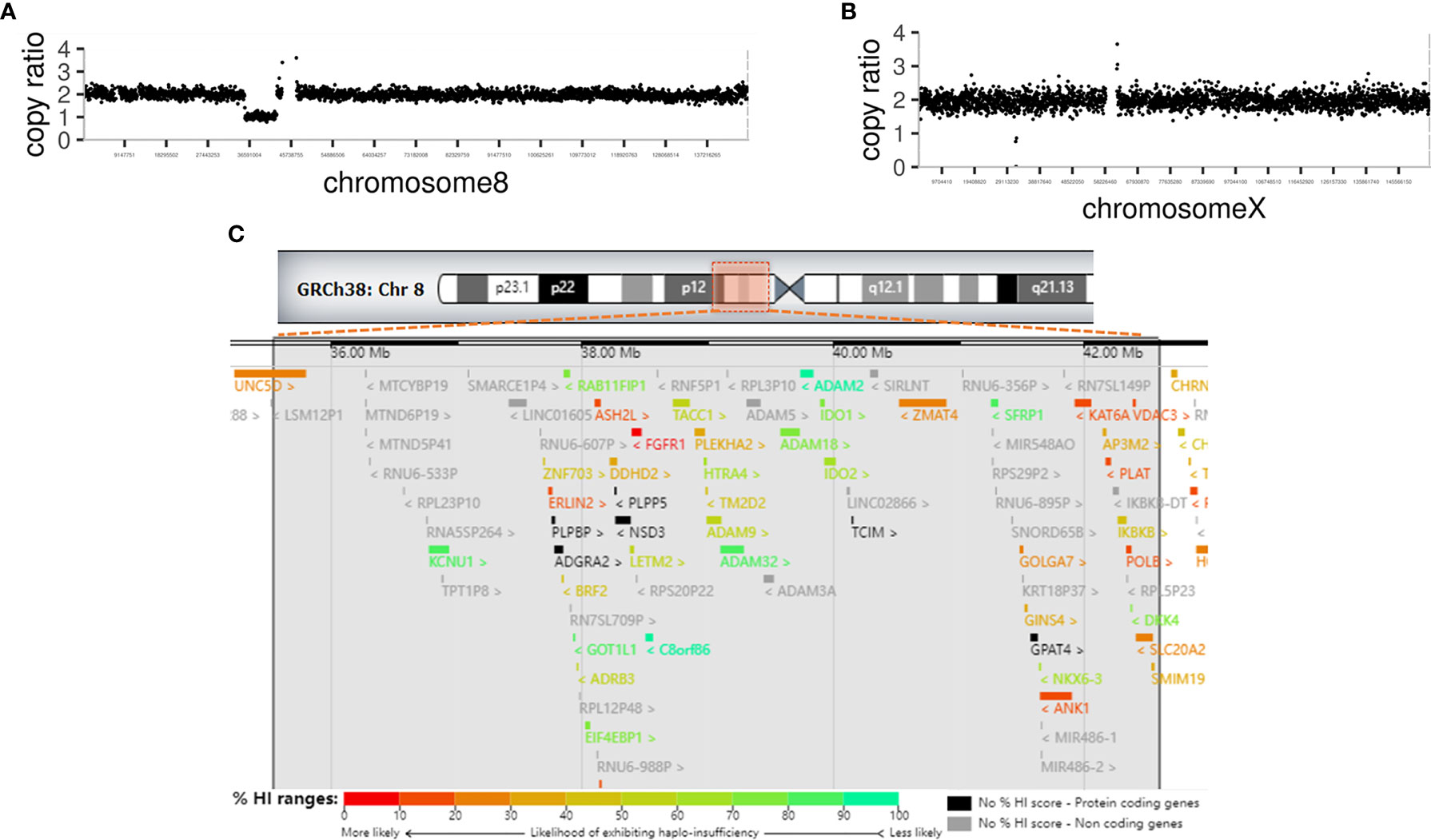
Figure 2 Genetic analysis of the patient (A) CNV-seq analysis revealed a large deletion of 7.05 Mb of 8p11 in chromosome 8. (B) CNV-seq analysis revealed an in-frame deletion of exons 48-55 in the DMD gene in chromosome X. (C) Chromosome ideogram. The impaired chromosomal region is highlighted, a 7.05-Mb region of hemizygous loss in Chr8p11.2 (Chr8: 35541034-42587731). Ninety-four genes are located in the hemizygous loss region of Chr8p11.2, including 43 OMIM genes. Genes are labeled by different colors from unlikely haploinsufficient to likely haploinsufficient, according to DECIPHER (http://decipher.sanger.ac.uk/). High ranks indicate that a gene is more likely to exhibit haploinsufficiency; low ranks indicate a gene is more likely not to exhibit haploinsufficiency.
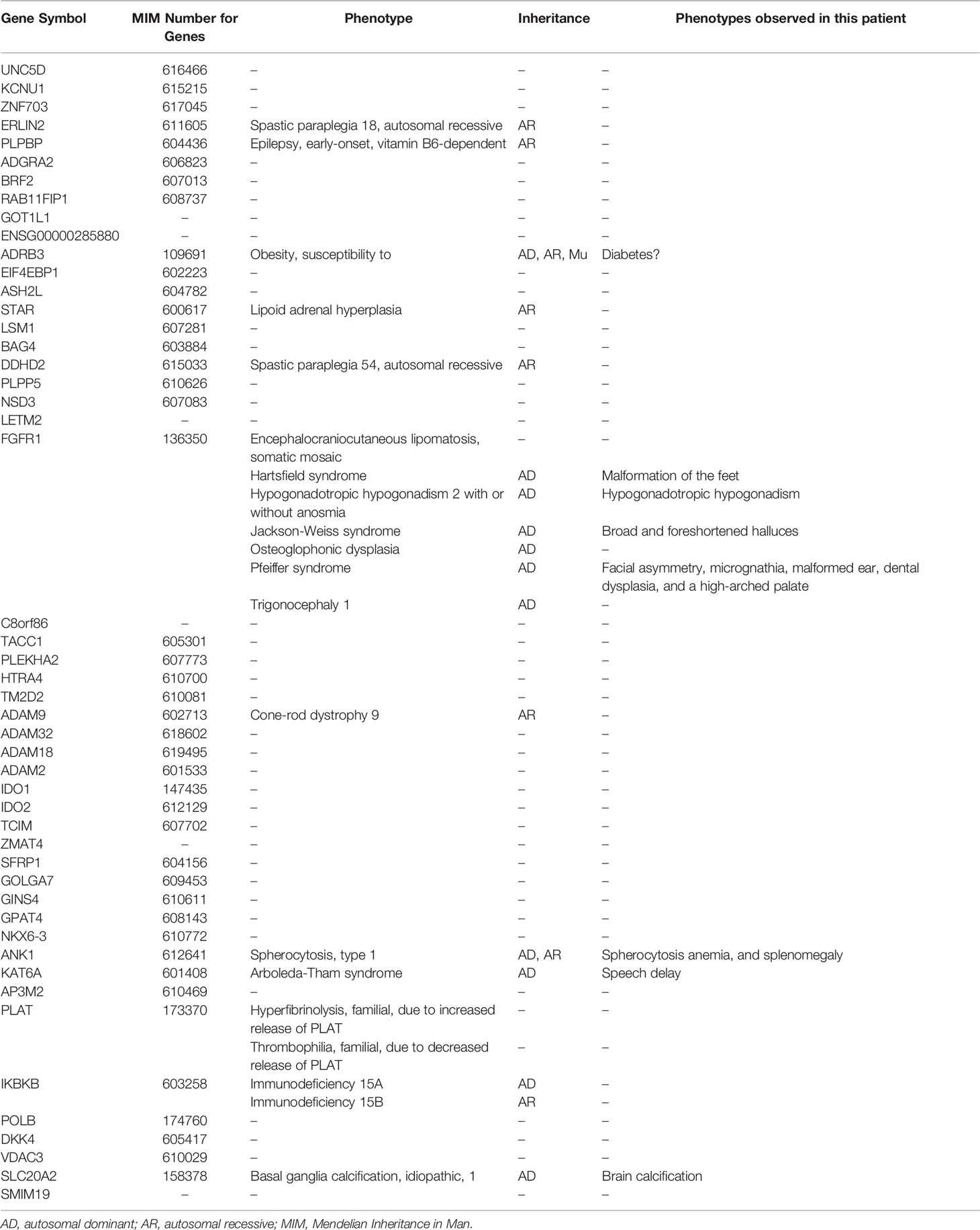
Table 3 List of genes in the deletion region.
Metformin at 1500 mg/d was given to the patient after which his blood glucose was well controlled. HCG 2000 IU was injected subcutaneously twice a week and was supplemented with calcium and vitamin D, which led to an improvement in the patient’s quality of life.
The deletion range of this patient covers almost the whole 8p11.2 area (chr8: 36,700,001– 43,200,000, 6.5 Mb), which resulted in a haploid deficiency of 94 genes, of which 49 were coding genes (43 OMIM genes); moreover, several genes had high ranks of haploinsufficiency (HI) (Figure 2C). Haploinsufficiency of the FGFR1, ANK1, KATA6, and the SLC20A2 genes causes a rare contiguous gene syndrome that includes CHH with or without anosmia (1), dysmorphic facial features, hands, and feet (Jackson-Weiss syndrome and Pfeiffer syndrome) (2, 3), spherocytosis (4), Arboleda-Tham syndrome (5), and brain calcification (6). These five groups of cardinal signs and symptoms could be named 8p11.2 deletion syndrome. Unlike previous cases reports, in which each patient had 1–3 cardinal symptoms depending on the interstitial deletion length (7–11), the present case developed early-onset diabetes. Interestingly, case no.421734 in decipher database also has diabetes mellitus, whose deletion region is very similar to our case (chr8:36909315-42578314) (12).
Other genes with high ranks of haploinsufficiency (0%–20%) were identified in the deleted region as follows: UNC5D (3.53%), STAR (11.9%), POLB (15.57%), PLAT (15.69%), ERLIN2 (17.19%), VDAC3 (19.05%), and ASH2L (19.21%). Among them, the UNC5D, POLB, VDAC3, and ASH2L genes have unknown functions. Biallelic pathogenic variants in the STAR gene is associated with lipoid congenital adrenal hyperplasia, which presented with symptoms such as salt wasting, hyponatremia, hypovolemia, hyperkalemia, acidosis, and death in infancy (13). PLAT is related to hyperfibrinolysis or thrombophilia with pending confirmation (14). ERLIN2 is related to autosomal recessive spastic paraplegia-18 (15). None of these genes related to the phenotypes were identified in this patient, which could be attributable to the fact that the haploinsufficiency of these genes is not enough to cause diseases.
In male individuals with CHH, low testosterone levels are strongly associated with an increased risk of developing type 2 diabetes mellitus and osteoporosis (16, 17). Specifically, FGF21/KLB/FGFR1 signaling related CHH is prone to metabolic defects (17). Furthermore, we cannot exclude other genes in this range that might contribute to the development of diabetes; for example, SNPs in the ADRB3 and ANK1 genes may be associated with obesity and T2DM (18, 19). However, at present, there is no direct evidence to prove that the deficiency of the ADRB3 and ANK1 genes is associated with human diabetes. Only indirect evidence has shown that SNP-induced ADRB3-mediated impairment in cAMP production was associated with T2DM susceptibility and development (20). In contrary, ANK1 gene SNPs causing increased promoter activity and sAnk1 expression in skeletal muscle might contribute to T2DM susceptibility (21). Some evidence also suggests that the EIF4EBP1 and SFRP1 genes are involved in mTORC1 signaling in cellular energy metabolism and Wnt signaling in adipogenesis, respectively (22, 23).
Becker muscular dystrophy (BMD, OMIM 300376) is a clinically heterogenous disorder caused by pathogenetic variants in the DMD gene. DMD is the largest known human gene comprising 79 exons and is inherited in an X-linked recessive manner (24). Patients with distal in-frame deletions showed a milder phenotype than those with deletions proximal to exon 45 (24–26). In particular, severe muscular involvement is reduced in patients sharing in-frame deletion of exons 45–55, and exon-skipping therapy has been developed to convert the severe phenotype to a more benign form (27, 28). The mild BMD phenotype of this patient could also be explained by the frame-shift theory (28). Usually, patients with DMD/BMD have fewer physiological activities, which is also a risk factor for obesity and diabetes (29). Due to the mildness of symptoms, this patient did not require glucocorticoid treatment, which was very fortunate for this patient because he avoided the endocrine problems caused by long-term use of glucocorticoids, such as aggravation of diabetes, osteoporosis, and decreased testosterone levels (30).
All of the above-involved genes are related to type 2 diabetes development but do not cause any known monogenic defects in diabetes. Thus, metformin will be the first choice for patients with 8p11.2 deletion syndrome with diabetes.
In addition, HbA1c levels in patients with hereditary spherocytosis are lower (31). When the patient’s blood sugar was high, the HbA1c was 5.8%, and after reasonable blood sugar control, the HbA1c was 3.0-3.2%. Glycosylated albumin and peripheral glycemic measurements are required to monitor this patient’s glycemic control.
Here, we report a rare case of 8p11.2 deletion syndrome with unique phenotypes involving developmental, hematological, endocrine, metabolic changes, and early-onset diabetes. It is challenging for endocrinologists to simultaneously reconcile a combination of these diseases across multiple disciplines. We discussed the influencing factors of early-onset diabetes in this patient and speculated that it was caused by complex interactions of known and unknown genetic and environmental factors.
The datasets for this article are not publicly available due to concerns regarding participant/patient anonymity. Requests to access the datasets should be directed to the corresponding author.
The studies involving human participants were reviewed and approved by First Affiliated Hospital of China Medical University. The patients/participants provided their written informed consent to participate in this study. Written informed consent was obtained from the individual(s) for the publication of any potentially identifiable images or data included in this article.
Study design, XW. Data collection, CC and XZ. Manuscript drafting, CC and XW. Data interpreting, XW. Revision of the manuscript, XZ and XW. Approval of final version of the manuscript, XW, CC, and XZ. All authors contributed to the article and approved the submitted version.
Sinocare Diabetes Foundation (2020SD05).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The authors thank the patient and his family members who agreed to participate in this study.
1. Dodé C, Levilliers J, Dupont JM, De Paepe A, Le Dû N, Soussi-Yanicostas N, et al. Loss-Of-Function Mutations in FGFR1 Cause Autosomal Dominant Kallmann Syndrome. Nat Genet (2003) 33(4):463–5. doi: 10.1038/ng1122
2. Muenke M, Schell U, Hehr A, Robin NH, Losken HW, Schinzel A, et al. A Common Mutation in the Fibroblast Growth Factor Receptor 1 Gene in Pfeiffer Syndrome. Nat Genet (1994) 8(3):269–74. doi: 10.1038/ng1194-269.
3. Roscioli T, Flanagan S, Kumar P, Masel J, Gattas M, Hyland VJ, et al. Clinical Findings in a Patient With FGFR1 P252R Mutation and Comparison With the Literature. Am J Med Genet (2000) 93(1):22–8. doi: 10.1002/1096-8628(20000703)93:1<22::AID-AJMG5>3.0.CO;2-U
4. Perrotta S, Gallagher PG, Mohandas N. Hereditary Spherocytosis. Lancet (Lond Engl) (2008) 372(9647):1411–26. doi: 10.1016/s0140-6736(08)61588-3
5. Tham E, Lindstrand A, Santani A, Malmgren H, Nesbitt A, Dubbs HA, et al. Dominant Mutations in KAT6A Cause Intellectual Disability With Recognizable Syndromic Features. Am J Hum Genet (2015) 96(3):507–13. doi: 10.1016/j.ajhg.2015.01.016
6. Guo XX, Su HZ, Zou XH, Lai LL, Lu YQ, Wang C, et al. Identification of SLC20A2 Deletions in Patients With Primary Familial Brain Calcification. Clin Genet (2019) 96(1):53–60. doi: 10.1111/cge.13540
7. Vermeulen S, Messiaen L, Scheir P, De Bie S, Speleman F, De Paepe A. Kallmann Syndrome in a Patient With Congenital Spherocytosis and an Interstitial 8p11.2 Deletion. Am J Med Genet (2002) 108(4):315–8. doi: 10.1002/ajmg.10295
8. Cau M, Congiu R, Origa R, Galanello R, Melis MA, Nucaro AL. New Case of Contiguous Gene Syndrome at Chromosome 8p11.2p12. Am J Med Genet A (2005) 136(2):221–2. doi: 10.1002/ajmg.a.30814
9. Miya K, Shimojima K, Sugawara M, Shimada S, Tsuri H, Harai-Tanaka T, et al. De Novo Interstitial Deletion of 8p11.2 Including ANK1 Identified in a Patient With Spherocytosis, Psychomotor Developmental Delay, and Distinctive Facial Features. Gene (2012) 506(1):146–9. doi: 10.1016/j.gene.2012.06.086
10. Wang D, Lai P. Global Retardation and Hereditary Spherocytosis Associated With a Novel Deletion of Chromosome 8p11.21 Encompassing KAT6A and ANK1. Eur J Med Genet (2020) 63(12):104082. doi: 10.1016/j.ejmg.2020.104082
11. Morris M, Kwon R, Chen L. Pediatric Idiopathic Basal Ganglia Calcification and Spherocytosis With Chromosome 8p11 Deletion. J Neuropathol Exp Neurol (2020) 79(2):238–41. doi: 10.1093/jnen/nlz133
12. Firth HV, Richards SM, Bevan AP, Clayton S, Corpas M, Rajan D, et al. DECIPHER: Database of Chromosomal Imbalance and Phenotype in Humans Using Ensembl Resources. Am J Hum Genet (2009) 84(4):524–33. doi: 10.1016/j.ajhg.2009.03.010
13. González AA, Reyes ML, Carvajal CA, Tobar JA, Mosso LM, Baquedano P, et al. Congenital Lipoid Adrenal Hyperplasia Caused by a Novel Splicing Mutation in the Gene for the Steroidogenic Acute Regulatory Protein. J Clin Endocrinol Metab (2004) 89(2):946–51. doi: 10.1210/jc.2003-030345
14. Ladenvall P, Nilsson S, Jood K, Rosengren A, Blomstrand C, Jern C. Genetic Variation at the Human Tissue-Type Plasminogen Activator (tPA) Locus: Haplotypes and Analysis of Association to Plasma Levels of tPA. Eur J Hum Genet (2003) 11(8):603–10. doi: 10.1038/sj.ejhg.5201011
15. Al-Saif A, Bohlega S, Al-Mohanna F. Loss of ERLIN2 Function Leads to Juvenile Primary Lateral Sclerosis. Ann Neurol (2012) 72(4):510–6. doi: 10.1002/ana.23641
16. Zarotsky V, Huang MY, Carman W, Morgentaler A, Singhal PK, Coffin D, et al. Systematic Literature Review of the Risk Factors, Comorbidities, and Consequences of Hypogonadism in Men. Andrology (2014) 2(6):819–34. doi: 10.1111/andr.274
17. Boehm U, Bouloux PM, Dattani MT, de Roux N, Dodé C, Dunkel L, et al. Expert Consensus Document: European Consensus Statement on Congenital Hypogonadotropic Hypogonadism–Pathogenesis, Diagnosis and Treatment. Nat Rev Endocrinol (2015) 11(9):547–64. doi: 10.1038/nrendo.2015.112
18. Coman OA, Păunescu H, Ghiţă I, Coman L, Bădărăru A, Fulga I. Beta 3 Adrenergic Receptors: Molecular, Histological, Functional and Pharmacological Approaches. Rom J Morphol Embryol (2009) 50(2):169–79.
19. Harder MN, Ribel-Madsen R, Justesen JM, Sparsø T, Andersson EA, Grarup N, et al. Type 2 Diabetes Risk Alleles Near BCAR1 and in ANK1 Associate With Decreased β-Cell Function Whereas Risk Alleles Near ANKRD55 and GRB14 Associate With Decreased Insulin Sensitivity in the Danish Inter99 Cohort. J Clin Endocrinol Metab (2013) 98(4):E801–6. doi: 10.1210/jc.2012-4169
20. Huang Q, Yang TL, Tang BS, Chen X, Huang X, Luo XH, et al. Two Novel Functional Single Nucleotide Polymorphisms of ADRB3 are Associated With Type 2 Diabetes in the Chinese Population. J Clin Endocrinol Metab (2013) 98(7):E1272–7. doi: 10.1210/jc.2013-1137
21. Yan R, Lai S, Yang Y, Shi H, Cai Z, Sorrentino V, et al. A Novel Type 2 Diabetes Risk Allele Increases the Promoter Activity of the Muscle-Specific Small Ankyrin 1 Gene. Sci Rep (2016) 6:25105. doi: 10.1038/srep25105
22. Morita M, Gravel SP, Chénard V, Sikström K, Zheng L, Alain T, et al. Mtorc1 Controls Mitochondrial Activity and Biogenesis Through 4E-BP-Dependent Translational Regulation. Cell Metab (2013) 18(5):698–711. doi: 10.1016/j.cmet.2013.10.001
23. Wang L, Wang Y, Meng Y, Zhang C, Di L. GSK3-Activated STAT5 Regulates Expression of SFRPs to Modulate Adipogenesis. FASEB J (2018) 32(9):4714–26. doi: 10.1096/fj.201701314R
24. Taglia A, Petillo R, D'Ambrosio P, Picillo E, Torella A, Orsini C, et al. Clinical Features of Patients With Dystrophinopathy Sharing the 45-55 Exon Deletion of DMD Gene. Acta Myol (2015) 34(1):9–13.
25. Magri F, Govoni A, D'Angelo MG, Del Bo R, Ghezzi S, Sandra G, et al. Genotype and Phenotype Characterization in a Large Dystrophinopathic Cohort With Extended Follow-Up. J Neurol (2011) 258(9):1610–23. doi: 10.1007/s00415-011-5979-z
26. Nakamura A, Yoshida K, Fukushima K, Ueda H, Urasawa N, Koyama J, et al. Follow-Up of Three Patients With a Large in-Frame Deletion of Exons 45-55 in the Duchenne Muscular Dystrophy (DMD) Gene. J Clin Neurosci (2008) 15(7):757–63. doi: 10.1016/j.jocn.2006.12.012
27. Nakamura A, Shiba N, Miyazaki D, Nishizawa H, Inaba Y, Fueki N, et al. Comparison of the Phenotypes of Patients Harboring in-Frame Deletions Starting at Exon 45 in the Duchenne Muscular Dystrophy Gene Indicates Potential for the Development of Exon Skipping Therapy. J Hum Genet (2017) 62(4):459–63. doi: 10.1038/jhg.2016.152
28. Nakamura A, Takeda S. Exon-Skipping Therapy for Duchenne Muscular Dystrophy. Neuropathology (2009) 29(4):494–501. doi: 10.1111/j.1440-1789.2009.01028.x
29. Aune D, Norat T, Leitzmann M, Tonstad S, Vatten LJ. Physical Activity and the Risk of Type 2 Diabetes: A Systematic Review and Dose-Response Meta-Analysis. Eur J Epidemiol (2015) 30(7):529–42. doi: 10.1007/s10654-015-0056-z
30. Birnkrant DJ, Bushby K, Bann CM, Apkon SD, Blackwell A, Brumbaugh D, et al. Diagnosis and Management of Duchenne Muscular Dystrophy, Part 1: Diagnosis, and Neuromuscular, Rehabilitation, Endocrine, and Gastrointestinal and Nutritional Management. Lancet Neurol (2018) 17(3):251–67. doi: 10.1016/s1474-4422(18)30024-3
Keywords: chromosome 8p11.2 deletion syndrome, Becker muscular dystrophy, case report, early onset diabetes mellitus, contiguous gene syndrome
Citation: Cao C, Wang X and Zhao X (2022) Early-Onset Diabetes Mellitus in Chromosome 8p11.2 Deletion Syndrome Combined With Becker Muscular Dystrophy - A Case Report. Front. Endocrinol. 13:914863. doi: 10.3389/fendo.2022.914863
Received: 07 April 2022; Accepted: 22 June 2022;
Published: 25 July 2022.
Edited by:
Riccardo Schiaffini, Bambino Gesù Children’s Hospital (IRCCS), ItalyReviewed by:
Abir Mukherjee, Royal Veterinary College (RVC), United KingdomCopyright © 2022 Cao, Wang and Zhao. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Xiaoli Wang, d2xpdHRsZXBlYXJAMTYzLmNvbQ==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.