 Linda A. Schuler*
Linda A. Schuler* Kathleen A. O’Leary
Kathleen A. O’Leary- Department of Comparative Biosciences, School of Veterinary Medicine, University of Wisconsin-Madison, Madison, WI, United States
Prolactin coordinates with the ovarian steroids to orchestrate mammary development and lactation, culminating in nourishment and an increasingly appreciated array of other benefits for neonates. Its central activities in mammary epithelial growth and differentiation suggest that it plays a role(s) in breast cancer, but it has been challenging to identify its contributions, essential for incorporation into prevention and treatment approaches. Large prospective epidemiologic studies have linked higher prolactin exposure to increased risk, particularly for ER+ breast cancer in postmenopausal women. However, it has been more difficult to determine its actions and clinical consequences in established tumors. Here we review experimental data implicating multiple mechanisms by which prolactin may increase the risk of breast cancer. We then consider the evidence for role(s) of prolactin and its downstream signaling cascades in disease progression and treatment responses, and discuss how new approaches are beginning to illuminate the biology behind the seemingly conflicting epidemiologic and experimental studies of prolactin actions across diverse breast cancers.
1 Introduction
Factors that regulate cell-specific proliferation and differentiation repeatedly have been shown to be significant actors in oncogenesis and potential therapeutic targets in established cancers. Prolactin (PRL) cooperates with the ovarian steroids, estrogen and progesterone, to orchestrate the cycles of mammary development and differentiation that lead to successful lactation, providing nourishment for the offspring. PRL-initiated signals that expand alveolar cells during pregnancy and coordinate their differentiation at the time of birth have been mechanistically defined [ (1–4) and references therein]. The essential actions of PRL in these physiological processes have suggested roles in breast cancer [ (5–14) and references therein], by analogy to the recognized roles of the two other major hormones that regulate mammary development and function, estrogen and progesterone. Yet understanding its activities and consequences across diverse clinical breast cancers in order to develop prevention or treatment strategies has been elusive.
While control of PRL expression by pituitary lactotrophs during pregnancy and lactation is well understood [reviewed in (15)], its expression outside of pregnancy has received less attention. Pituitary PRL secretion is influenced by many factors, and circulating levels in nonpregnant women vary considerably (16–18). In addition to physiological stimuli, estrogen-progestin menopausal hormone therapy (MHT) raises circulating PRL (18), and anti-psychotics that antagonize dopamine induce hyperprolactinemia (19, 20). Further, PRL also can be expressed by non-lactotrophs, including within the mammary gland (21–23), and by breast cancer cells themselves (24–27). COX-2 (PTGS2) can induce PRL expression in fibroblasts, including at potential metastatic sites, mediated by PGE2 induction of NR4A (28). Moreover, in contrast to growth hormone (GH) in nonprimates, hGH is also a potent PRL receptor agonist (29, 30). Like PRL, it can be produced locally by breast cancer cells (26), and hGH and PRL receptors can heterodimerize (31). Thus, PRL receptors (PRLR) in the breast may be exposed to agonists from local and circulating systemic sources, even in the absence of pregnancy.
Here we review the epidemiologic evidence linking PRL to oncogenesis in the breast, and recent experimental studies implicating multiple underlying mechanisms. We then address the more controversial role(s) for PRL in established breast cancers. PRLR is highly expressed in many breast cancers across all different subtypes, and epidemiologic analyses and experimental studies are revealing that PRL can elicit both pro-differentiation and pro-aggression outcomes. We discuss how new approaches are illuminating the factors that determine the responses to PRL, including intrinsic tumor cell properties and the microenvironment, and point to directions for future studies that will integrate our understanding of this hormone in breast cancer progression and therapeutic responses.
2 PRL Actions in Development of Breast Cancer
2.1 Epidemiological Studies
Multiple epidemiologic studies have examined the relationship between levels of circulating PRL and development of breast cancer [meta-analysis and review (32),]. Large prospective studies have linked higher levels of circulating PRL within the normal range to increased risk for breast cancers which express estrogen receptor alpha (ER+) in postmenopausal (16, 33), or premenopausal women (34). In the study nested within the Nurses’ Health Study, PRL levels predicted breast cancer risk independent of estrogen (35). Additional analyses of this cohort found that the association of circulating PRL in the highest quartile in postmenopausal women ten years prior to diagnosis was strongest for aggressive ER+ breast cancer (36). Furthermore, epidemiologic studies have linked PRL to mammographic density (34, 37, 38), a potent independent contributor to increased breast cancer risk (39, 40). Incorporation of PRL in risk prediction models improves their efficacy (34, 41). Conversely, the reduced PRL levels in parous compared to nulliparous women may play a role in the long term protection conferred by pregnancy (16, 18, 34, 42).
2.2 Experimental Studies
2.2.1 In Vivo Models
The ability of PRL to stimulate mammary tumorigenesis in rodent models has been recognized for some time. Many early studies manipulated pituitary PRL (5–7), especially using pituitary isografts transplanted to the kidney capsule to chronically elevate circulating PRL by removing the inhibitory effects of dopamine (43). This approach reveals effects of PRL in combination with progesterone; in rodents, PRL also supports the corpus luteum (44).
More recently, genetically modified mice have permitted interrogation of mechanisms by which PRL may increase risk of breast cancer, apart from ovarian steroids. Transgenic PRL under the control of several promoters leads to mammary cancers [reviewed in (45)], as does transgenic mammary STAT5A, the canonical mediator of PRL signals (46). PRL drives development of mammary cancers in mice with germline ablation of Stat1 secondary to somatic truncating mutations in Prlr, resulting in an alternatively spliced protein resembling the human “intermediate” isoform (47) (see Sections 3.1, 3.4.2). Our group generated the NRL-PRL mouse (48, 49), in which transgenic rat PRL is expressed by mammary epithelia, mimicking the local PRL synthesis in breasts of women (23). Unlike circulating PRL, this locally elevated PRL does not disturb estrous cycling, enabling study of the interactions of PRL with ovarian hormones, of particular importance when assessing models of pre- and post-menopausal breast cancer. Mammary glands of young adult NRL-PRL females exhibit elevated pERK1/2 and pAKT, in addition to pSTAT5 (50), reflecting the spectrum of PRL-initiated signaling cascades (22, 51). These mammary glands exhibit both ductal abnormalities (mammary intraepithelial neoplasias, resembling ductal carcinoma in situ, DCIS), and epithelial hyperplasias (48, 52). With age, nulliparous females spontaneously develop histologically diverse, metastatic ER+ carcinomas with long latencies, mirroring the epidemiologic link between PRL exposure and aggressive ER+ cancer (36). These tumors can develop without postpubertal ovarian steroids, similar to the observation that the increased risk conferred by PRL in women is independent from estrogen (16), although supplemental 17β-estradiol decreases tumor latency (50). Once established, tumors are no longer dependent on estrogen for growth, modeling clinical anti-estrogen resistant luminal B cancers (53–55). However, the ER remains functional; estrogen activity modulates tumor gene expression and behavior, including proliferation and cancer stem cell activity (54, 56).
In order to understand the molecular events underlying PRL-driven oncogenesis, we performed comprehensive genomic profiling over the course of disease (57). Similar to clinical ER+ breast cancers (58–60), end stage tumors exhibited few nonsynonymous somatic mutations. However, nearly 80% of tumors showed alterations in the Ras pathway, including canonical activating mutations and copy number amplifications of Kras. Interestingly, many aggressive clinical ER+ breast cancers exhibit elevated Ras pathway activity as a result of mutations in the Ras proteins, or reduced expression or somatic loss of Ras-GAP tumor suppressors (61–64). Many of the remaining 20% of experimental PRL-induced cancers exhibited elevated pAKT, but not pERK1/2, consistent with driver mutations in the phosphatidylinositol-3-kinase pathway, common in many clinical ER+ cancers (63, 65, 66). Transcriptomic analyses showed that tumors expressed variable transgenic PRL compared to preneoplastic tissue, suggesting divergent PRL influence once tumors are established. These analyses also revealed marked alterations in cell-intrinsic processes and the tumor microenvironment, including immune activity. Consistent with low numbers of intratumoral lymphocytes, including CD8+ effector T cells, but large numbers of infiltrating macrophages, tumors contained strikingly reduced transcripts for many chemokines and indicators of anti-tumor immunity. This immunosuppressed environment resembles that of clinical ER+ breast cancers [reviewed in (67, 68)].
2.2.2 Direct Actions on Mammary Epithelia
Extensive studies of the direct actions of PRL on breast cancer cells in vitro have demonstrated increased proliferation and cell turnover [reviewed in (22)], and these effects are also observed in normal mammary epithelia in the dynamic in vivo environment in multiple murine models (2, 45, 49). In addition, recent studies have revealed that PRL powerfully influences the mammary epithelial hierarchy, both independent of ovarian steroids and in concert with these hormones (69) (Figure 1).
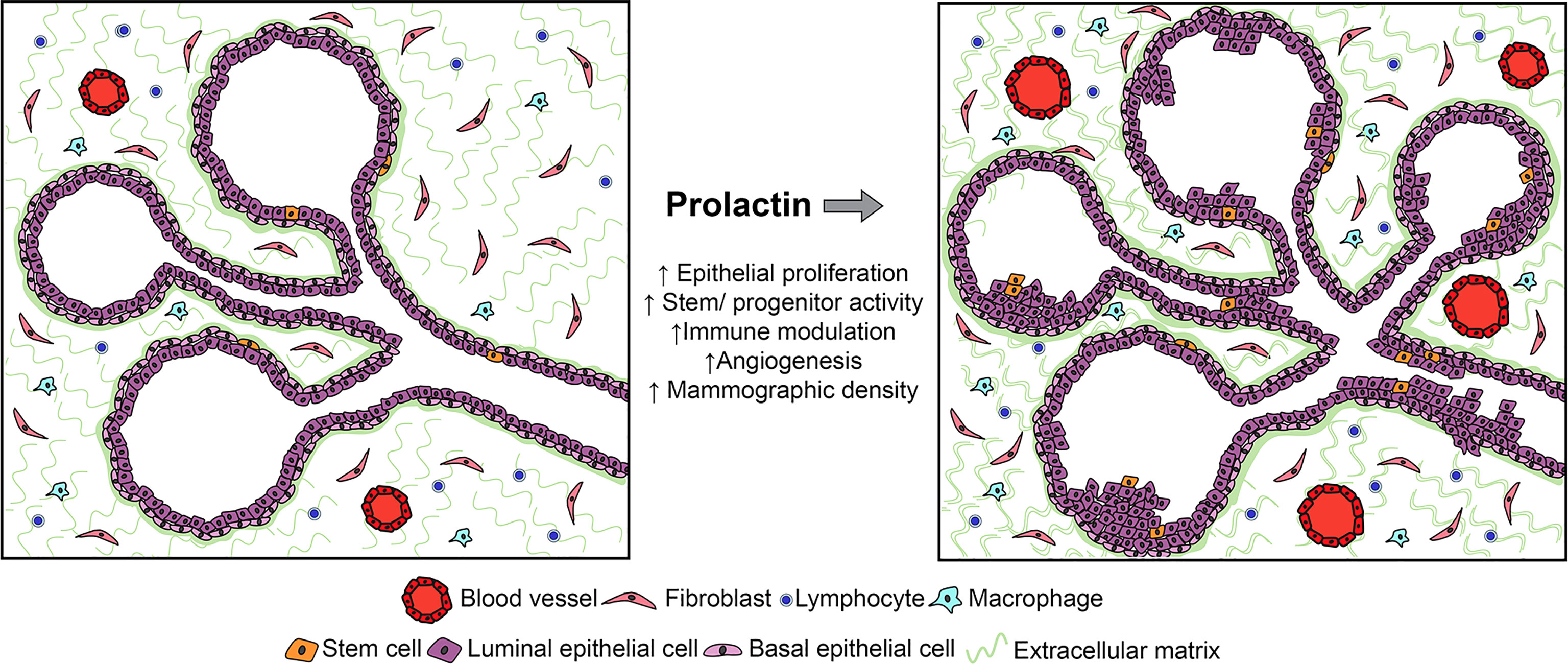
Figure 1 Studies of human samples and experimental models have shown that PRL can act on multiple target cells within the mammary environment, including not only epithelia, but also stromal cells, including immune and fibroblastic cell subpopulations. Although its effects on epithelia are best understood in the context of breast cancer, its actions on stromal targets which have been defined in other systems would also be predicted to increase the risk for breast cancer. See Section 2.2 for details.
In the NRL-PRL model, local PRL increased epithelial stem/progenitor activity and dampened the regulatory networks which drive differentiation (69). In ovariectomized young adult females, transgenic PRL increased luminal progenitors; in combination with estrogen and progesterone, PRL increased bilineage progenitors, and raised stem cell activity associated with augmented canonical Wnt signaling. However, PRL opposed steroid-driven luminal maturation, associated with reduced Gata3 and higher Sox9 transcripts (69). A growing literature supports stem/progenitor cell populations as cancer cells of origin (70), and mammary luminal progenitors have been implicated as precursors for multiple subtypes of breast cancer [reviewed in (71)]. The ability of PRL to expand these epithelial subpopulations would contribute to increased cancer risk.
2.2.3 Effects on Non-Epithelial Cells
Multiple non-epithelial cells in the mammary environment have been reported to express PRLR (72, 73). Although few actions of PRL at stromal targets have been addressed experimentally in the context of breast cancer, data from physiologic and other pathologic states suggest the need for additional study. The critical roles of the immune system in mammary development, lactation and involution are increasingly appreciated (74–76). PRL, like the ovarian steroids, can influence mammary immune cell content and activity, both indirectly by altering epithelial cytokine secretion (77), as well as directly acting on both innate and adaptive immune cell subpopulations (78–83). Studies of lymphocyte activation in vitro showed a bimodal concentration-dependent response to PRL (84, 85), suggesting the intriguing possibility that mammary PRL synthesis may influence local immune activity. Together, these reports suggest that PRL may modulate inflammation and/or immunotolerance during tumorigenesis, with potential to contribute to a permissive environment for development of breast cancer.
PRL-induced synthesis of components of the extracellular matrix (ECM) by both epithelial and non-epithelial target cells may complement mitogenic effects of PRL on mammary epithelium to augment mammographic density, suggesting another mechanism by which PRL may raise risk (86). In this regard, the ability of PRL to stimulate macrophages to augment fibrosis in pancreatic cancer is of interest (87). Furthermore, PRL can modulate angiogenesis. As an intact protein, it can promote vascularization (88, 89); in contrast, its proteolytic products (vasoinhibins) impede this process (90). These activities have suggested roles in normal mammary function as well as breast cancer (91).
2.2.4 Contributions to Growth of Early Lesions
These actions of PRL on breast epithelia and potentially on other stromal cells could support development of breast cancers. Moreover, these activities would fuel early lesions regardless of the initiating event (Figure 1). Many clinical DCIS lesions express PRLR (92), and the PRL antagonist, Δ1-9-G129R-hPRL, inhibited the mammosphere-forming activity of primary DCIS samples (93). The rich literature elevating systemic PRL using pituitary isografts in mouse models demonstrates that PRL in combination with progesterone can promote carcinogen- and p53 null-induced tumors [e.g., (6,94)]. Conversely, germline genetic ablation of Prl or Prlr slowed growth of lesions induced by viral oncogenes (95, 96). Antipsychotics that act by antagonizing dopamine, thereby raising circulating PRL, promoted tumorigenesis in experimental models initiated by RCAS-caErbB2, RCAS-HrasQ61L and MMTV-Wnt-1 (97). A recent study found that patients using these drugs had a significantly increased risk of breast cancer (20). Together, these observations in patients and murine models indicate a role(s) for PRL in progression of early lesions.
2.2.5 Cooperation With Other Factors
2.2.5.1 Estrogen, Progesterone
In patients, PRL would act in concert with other hormones and potentially carcinogenic factors, as well as dysregulation of multiple pathways as disease progressed. Prior to menopause, PRL would interact with ovarian steroids; after menopause, estrogen/progestin MHT would continue this crosstalk. In the European Prospective Investigation into Cancer and Nutrition cohort, postmenopausal women with higher circulating PRL who had used combined estrogen/progestin MHT had the most significant increase in incidence of ER+ breast cancer (33). Estrogen in the absence of progestins also would be an actor in postmenopausal women receiving either estrogen only MHT, or in untreated women by extraovarian estrogen synthesis (98, 99). PRL cooperates with estrogen, a well-recognized risk factor for breast cancer, by multiple mechanisms, including reciprocal upregulation of the other’s receptors (100, 101), and downstream crosstalk (102, 103). Supplemental estrogen accelerates PRL-driven mammary cancers in the NRL-PRL model (50). Furthermore, PRL induced pAKT and pERK1/2 can activate ERα in the absence of estrogen ligand in vivo as well as in vitro (104–106). PRL interaction with progesterone has been best studied in the context of pregnancy and lactation, where these hormones cooperatively drive expansion of alveolar cells during pregnancy, but oppose one another to initiate lactation (3). Outside of pregnancy, they regulate the other’s receptors, and as observed above, work together to increase mammary stem cells (69, 107, 108).
2.2.5.2 Other Oncogenic Factors
Locally elevated transgenic PRL also has revealed potent collaboration with other oncogenic pathways. Crosses between NRL-PRL mice and other murine models of mammary cancer, including elevated TGFα, loss of p53, and mutagen with increased canonical Wnt signals conferred by an inactivating mutation in the tumor suppressor APC, dramatically reduced tumor latency or increased tumor incidence (52, 104, 109–111). Transgenic PRL not only enhanced carcinogenesis, but also markedly influenced the resulting cancers in ways that would impact treatment responses. For example, transgenic local PRL increased the proportion of claudin low tumors in the absence of p53 (110), and in the presence of mutated APC, elevated PRL resulted in tumors with Notch-dependent cancer stem cell activity, compared to the β-catenin-dependence observed in tumors with mutant APC alone (111). Further, transgenic PRL and TGFα in combination sustained activation of the ERK1/2 and AKT signaling cascades (104, 109), reflecting the potent cooperation of PRL with growth factor-initiated signals (112, 113). This further activates ERα in the absence of estrogen ligand in vivo (104, 106, 109), one mechanism which underlies resistance of ER+ breast cancers to anti-estrogens (114, 115) (Section 3.3 below). In contrast to the positive interactions between PRL and growth factors in these transgenic murine and breast cancer models, PRL and EGF have been reported to oppose one another in “normal” mammary cell lines, such as HC11 and NMuMG; the phenotype of the target cell is likely to be critical in dictating the outcome of PRL signals and crosstalk with other signals (112, 116).
3 Role of PRL in Established Breast Cancers
In contrast to the strong epidemiologic data supporting a role for PRL in development of breast cancer, particularly of ER+ tumors, its role in established cancers continues to be actively debated. Much of the discussion revolves around the extent of PRLR expression by the tumor parenchyma, including which breast cancer subtypes and which PRLR isoforms, and importantly, whether PRL fuels tumor aggression or fosters a more differentiated phenotype.
3.1 PRL Receptors in Clinical Breast Cancers
PRLR isoforms with distinct intracellular domains and consequent differing signaling capacities are generated by alternative splicing. The full length “long” PRLR isoform is best studied, but as noted below, expression of the “intermediate” PRLR isoform in breast cancers is also recognized. Homo- and hetero-dimerization of these PRLR isoforms not only influences the repertoire of potential signaling pathways, but also stability of the receptors [reviewed in (22, 117)]. These isoforms have further confounded detection of PRLR across breast cancer subtypes, which is already complicated by the specificity and sensitivity of historically available antibodies (117, 118). However, multiple recent studies have reported PRLR protein expression in ER+, HER2+ and triple negative (TNBC) breast cancers, in sharp contrast to the epidemiologic link between PRL and development of only ER+ breast cancers. The relative proportion of tumors within each subtype that expressed PRLR varied with the cohort examined, antibody utilized [i.e., detecting the extracellular domain shared by most PRLR isoforms (119–121), intracellular domain of the full length “long” PRLR (92, 122), or unique intracellular domain of the “intermediate” PRLR isoform (117)], and other methodological differences. Given these variables, it is not surprising that the proportion of PRLR-expressing breast cancers varied from 25-83%.
Although PRLR expression was independent of ER (92, 119, 120, 122), PRLR levels were highest in ER+ tumors when analyzed (92, 123). Some of these studies found highest PRLR expression in more differentiated tumors in patients with longer metastasis free survival (92, 119). In contrast, Shemanko and her colleagues found that higher tumor PRLR protein levels correlated with a shorter time to bone metastasis, consistent with experimental PRL-induced osteoclast differentiation (120). Some of these reports included interrogation of relative transcript abundance and outcomes in various publicly available databases; with the caveat that transcript levels may not reflect protein expression, these results also differed (92, 123).
Although overall, TNBCs expressed less PRLR detected with an antibody to the intracellular domain of the “long” PRLR (92), a subset of these tumors expressed higher PRLR levels. This TNBC subset was found to be more differentiated, supporting the hypothesis that PRL drives a pro-differentiation program in these cancers (122). (See review by Ali and colleagues elsewhere in this series). Interestingly, however, Clevenger and colleagues reported that the “intermediate” isoform of the PRLR, which would not have been detected with the antibody used in the study above, was most highly expressed in TNBC (117). Moreover, they found that cancers with a high ratio of transcripts for the “intermediate” to the “long” PRLR isoform were associated with greater likelihood of distant metastases in the TCGA database. Experimentally, heterodimers of these PRLR isoforms were more stable, and less able to activate STAT5 (Section 3.4.2 below). Clinical TNBCs are very heterogeneous (124, 125); together, these reports suggest that different TNBC subsets may respond quite differently to PRL.
Although many of these studies correlated levels of PRLR protein or transcripts with prognosis, albeit with conflicting conclusions, the outcome of PRLR signaling has been directly addressed only in small Phase I/II studies of patients with advanced disease. A study of a PRLR neutralizing antibody as a monotherapy that included 34 breast cancer patients (all subtypes, but 75% ER+ cancers) found no significant effect on disease progression (126). In another small Phase II trial (20 breast cancer patients), the dopamine D2 receptor agonist, cabergoline, was used to inhibit secretion of pituitary PRL; two of these patients experienced extended disease control (127). The lack of definitive positive results dampened enthusiasm, although the small number of patients, the advanced stage of their disease, and extensive pretreatment regimens limit interpretation. However, interest in this area persists. Conjugates of other therapeutic agents to PRL antagonists or PRLR neutralizing antibodies are being developed, as discussed further in Section 3.3 below.
3.2 Experimental Studies Using Xenografts
Mouse PRL has little activity at the human PRLR (30), which has complicated experimental study of breast cancers in vivo. However, Rui and his colleagues have developed a mouse in which the mouse Prl gene has been replaced with the human PRL gene (NSG-Pro), resulting in physiologic regulation of hPRL expression (128). In these recipients, ER+ patient derived xenografts (PDXs) displayed a remarkable 15-20 fold higher transplantation rate than in wildtype NOD SCID gamma (NSG) mice. Moreover, the NSG-Pro mice facilitated metastatic dissemination and growth of distant lesions, genetic evolution and development of anti-estrogen resistance (128). These studies support an important role for PRL in the biology of ER+ tumors.
Well-characterized breast cancer cell lines modeling different breast cancer subtypes have been extensively studied in vitro to understand the outcomes of PRL actions, and to dissect its signals and mechanisms of interaction with other factors; these reports will not be further reviewed here. (See reviews by Ali and Clevenger and their colleagues elsewhere in this series). Some investigators have examined PRL responses in murine recipients of transplanted cell lines by providing another source of hPRL, with conflicting results. Primary tumors of transplanted MDA-MB-468 breast cancer cells that expressed hPRL grew more rapidly than tumors that did not in nu/nu (nude) mice (129). Reduction of the “long” PRLR isoform reduced pulmonary and hepatic metastatic burden in NOD-SCID recipients of HER2+ BT474 cells, which were supplemented with hPRL (130). In contrast, hPRL-treatment of NOD/SCID mice bearing xenografts of MDA-MB-453 breast cancer cells reduced tumor growth and dissemination (122), and growth of primary HER2+ SKBR3 tumors (131).
In a different approach, Ali and her colleagues used CRISPR/Cas9 to reduce PRLR expression in the ER+ MCF7 and HER2+ SKBR3 breast cancer cell lines (132). Loss of PRLR in MCF7 cells reduced ER expression, consistent with regulation of ESR1 by PRL, and promoted less differentiated, more aggressive tumors upon transplantation to NOD/SCID mice. In HER2+ SKBR3 cells, loss of PRLR increased HER2 expression, and ability to colonize lungs of NSG recipients. These findings, together with associated in vitro analyses, indicate beneficial actions of PRLR in these models (132). (See review by Ali and colleagues elsewhere in this series).
The basis for the disparate responses to PRL in these xenograft studies is unclear. There are many differences among these experiments. The transplanted cancer cells, whether PDXs or different breast cancer cell lines (133, 134), are quite distinct. Moreover, the design of these studies differs markedly, including placement of the transplanted cells, the extent that the murine hosts are immunocompromised, and method of manipulating PRL activity. Additional studies are necessary to understand how these findings reflect diverse clinical breast cancers.
Few studies have been performed in syngeneic experimental models. However, the findings are intriguing. In a murine model of HER2+ cancer (MMTV-neu), the PRL antagonist, G129R-hPRL, reduced metastases after removal of the primary tumor (135). In a subsequent study, the effects of G129R-hPRL on HER2 signaling in this model were found to be dependent on cancer associated fibroblasts (136), supporting the importance of study of complex systems with multiple cell types. Systemically reducing expression of the “long” PRLR isoform using a novel method reduced metastases in the aggressive 4T1 model, and PRL-supported immunosuppressive Tregs were identified as a major target (83, 130). These observations underscore the drawbacks of xenograft models. Most notably, currently available xenograft recipients are severely immunocompromised, lacking critical components of the host response. Ongoing efforts to develop mice with “humanized” immune systems will address this shortcoming. In addition, subtle differences in the structures of mouse/human proteins can obscure paracrine/systemic communication between tumor and stromal cells, e.g., PRL itself (30).
3.3 PRL Interactions With Other Treatment Approaches
Although PRL/PRLR inhibitors have not shown robust promise as monotherapies, there has been long term interest in their interaction with other treatment modalities, especially with anti-estrogens in ER+ breast cancers. As noted in Section 2.2.5, PRL cooperates with estrogen by multiple mechanisms, which have been dissected primarily in the well-differentiated ER+ breast cancer cell line, MCF7 (100–103). Not surprisingly, as for other aspects of cancer biology, this relationship evolves with disease progression. In an experimental rat model of hormonally-responsive ER+ mammary cancer, concomitant inhibition of PRL and aromatase cooperatively reduced tumor growth (137). Similarly, in therapy naïve ER+ PDXs transplanted to NSG-Pro recipients, PRL initially supported anti-estrogen responsiveness, but with time, the PRL environment facilitated development of resistance to tamoxifen (128). This was associated with increased growth factor signals, including ligand independent activation of ER, a potent outcome of PRL-growth factor crosstalk (See Section 2.2.5), and activation of the ERBB2 pathway (128). This relationship between PRL and resistance to anti-estrogens is reflected in some but not all small clinical studies [reviewed in (16)]. Interestingly, LAT1/SLC7A5, a transporter for branched chain amino acids which is regulated by PRL during lactation (138), is highly expressed by tamoxifen resistant cancers (139–141). The growing recognition of the importance of tumor metabolism, and role of PRL in regulation of metabolism during lactation, points to this area for further study.
The potent crosstalk of PRL with growth factor-initiated signals observed both in breast cancer cell lines (112, 142), and anti-estrogen resistant ER+ PDXs (128) has suggested that targeting PRL signaling in combination with these pathways may be an efficacious therapeutic strategy. PRL can initiate phosphorylation of HER2 in SKBR3 and BT474 breast cancer cells in vitro and in a murine MMTV-neu-derived tumor ex vivo (136, 143, 144). In light of the apparent conflict of these in vitro observations with the results of some but not all xenograft studies as noted in Section 3.2, this deserves additional investigation.
Several small clinical studies suggested that reduction of PRL might improve responses to chemotherapies [reviewed in (16, 145)]. The ability of PRL to promote survival of breast cancer cells in vitro has been appreciated for some time [reviewed in (22, 145)]. In breast cancer cell lines representing different cancer subtypes, PRL promoted to resistance to chemotherapies, including doxorubicin, paclitaxel, and cisplatin (93, 146, 147). Several related mechanisms have been identified, including PRL-induced expression of anti-apoptotic proteins (148), transcription of the multidrug resistance transporter ABCG2 (149), and activation of glutathione-S-transferase (147). Interaction with chemotherapies has not been directly revisited clinically, but these actions may contribute to the efficacy of compounds conjugated to anti-PRL agents, as noted below.
The relatively low toxicity of PRL antagonists and PRLR neutralizing antibodies and widespread PRLR expression across different breast cancer subtypes have prompted their development as delivery vehicles for other therapeutic agents, including cytotoxic compounds (121, 135, 150, 151), anti-HER2 (152), and immunomodulators to attract and/or activate CD8+ T cells (135, 153). In addition to targeting delivery of other treatments, testing of these molecules will also provide information on the efficacy of concomitant inhibition of PRL signals.
3.4 PRL Initiated Signals
3.4.1 Canonical JAK2-STAT5 Pathway
As described above, most studies have examined the outcome of the sum of all PRL-initiated signals in different breast cancer settings. However, it has long been appreciated that PRL can activate multiple signaling cascades (22, 51), with potentially different outcomes. The JAK2-STAT5A pathway mediates PRL-driven proliferation and differentiation that is essential for successful lactation (2, 154, 155), and binding of STAT5A to regulatory enhancer regions initiates chromatin remodeling, coordinating tissue specific gene expression (4, 156). (See review by Clevenger and colleagues elsewhere in this series). Transgenic overexpression of STAT5A leads to mammary carcinomas in the absence of other oncogenes (46). However, evidence for high STAT5A activity in clinical breast cancers has been repeatedly associated with more differentiated cancers and better prognoses [ (154, 157, 158) and references therein]. Consistently, the JAK2-STAT5A pathway also has been linked to PRL-induced pro-differentiation activities in various experimental models (53, 56, 159–161) (Figure 2). Of particular interest for premenopausal breast cancer, PRL-activated STAT5 suppressed a progestin-induced progenitor population in T47D breast cancer cells (162).
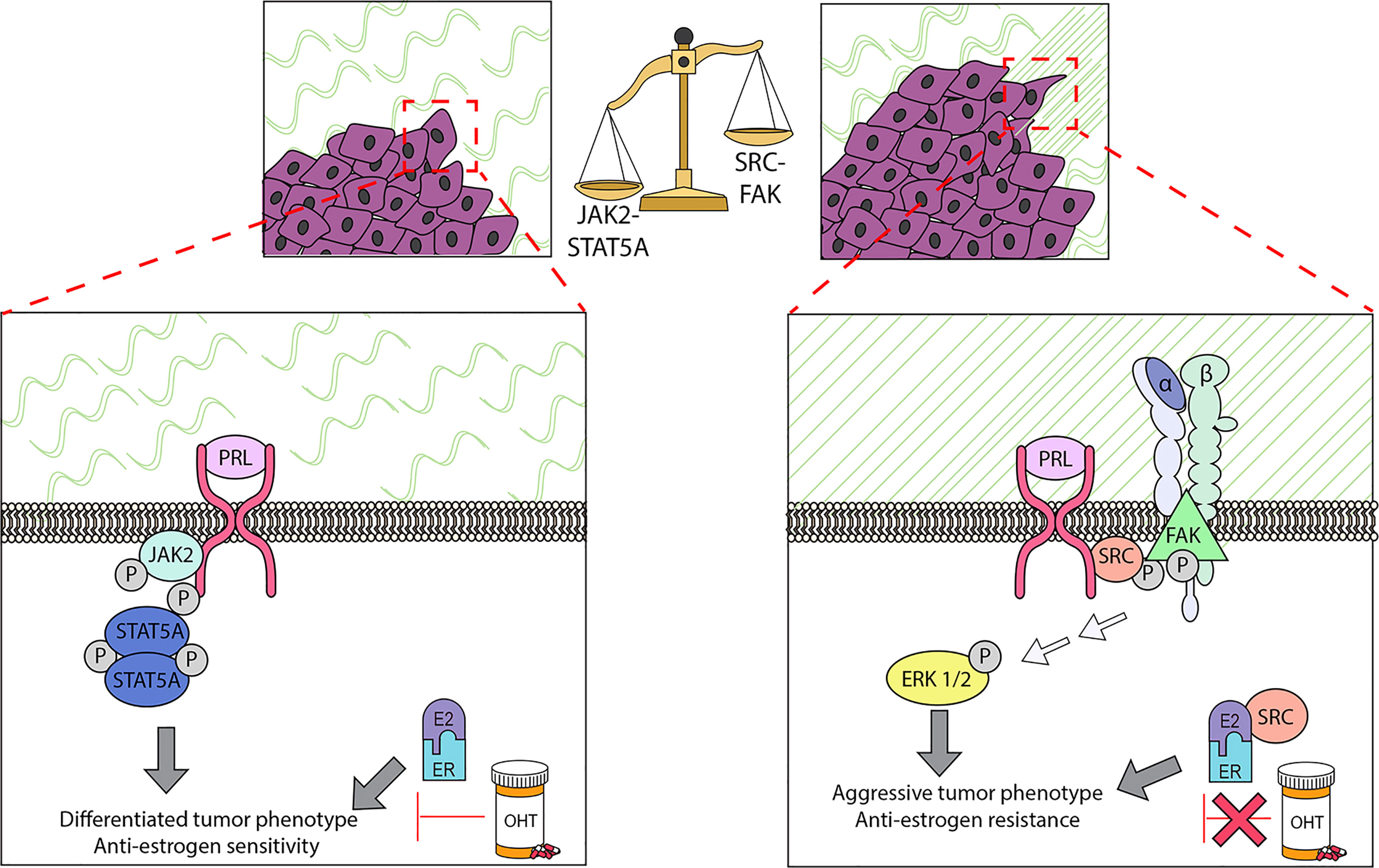
Figure 2 PRL can initiate multiple signaling cascades in established cancers, which can result in different biological outcomes. Determination of the repertoire of PRL signals can be modulated by multiple factors, including properties of the ECM. In ER+ cell lines, the stiffness and density of the extracellular matrix (ECM) strongly influences the balance of these signals: in stiff matrices, PRL signals are shifted away from the canonical JAK2/STAT5A pathway, and toward FAK/SFK/ERK1/2. This shift permits PRL to drive proliferation, invasion, and resistance to tamoxifen (OHT), and further remodel collagen fibers in the ECM. These experimental findings support the clinical observations that ER+ cancers with regions of aligned collagen perpendicular to the tumor boundary have a worse prognosis, and that activated STAT5A is strongly linked to more differentiated cancers and tamoxifen sensitivity. See Section 3.4 for details.
The highly homologous STAT5B remains a complication in these studies. In contrast to STAT5A, STAT5B is not associated with a more favorable prognosis in patients (158), and in vitro, STAT5B drives aggressive behavior in several models (56, 159, 161). Activities distinct from STAT5A are supported by different target genes (56, 158, 163), and divergent regulation by estrogen activity (56). Additional studies with more specific reagents are needed to resolve the roles of the STAT5 isoforms in PRL actions in breast cancer.
3.4.2 Other Signaling Cascades
PRL can also initiate activation of src family kinases, and multiple additional mediators, including AKT and MAP kinases (164–168). Interestingly, the alternatively spliced PRLR isoforms with distinct intracellular domains are less able to activate the JAK2/STAT5 pathway than the well-studied “long” PRLR isoform. Indeed, heterodimerization of the “long” PRLR isoform with the “intermediate” isoform, which was recently reported to be highly expressed in a subset of TNBC (see Section 3.1), inhibits phosphorylation of STAT5, without impacting other PRL-activated signaling pathways (117). AKT and MAP kinase cascades are linked to tumor progression for many cancers (169–173). Moreover, as noted above, they can activate ERα in the absence of ligand (105, 106), with implications for therapeutic responses to anti-estrogens in ER+ breast cancers. Importantly, they are also potent sites of cooperation with other oncogenic factors, including growth factors (112, 142). Together, these observations point to the potentially divergent outcomes of different arms of PRL signals, and raise the question of determinants of the repertoire of signaling options for PRL. Clearly intrinsic differences in tumor cells themselves, including relative levels of PRLR isoforms and other signaling components play a role; different breast cancer cell lines, even of the same breast cancer subtype, exhibit different spectra of PRL activated signals in vitro [e.g., (137, 174)]. In addition, as discussed below, environmental factors also can powerfully modulate the balance of PRL signals.
3.4.3 Features of the Extracellular Matrix Shift PRL-Initiated Signals in ER+ Tumor Cells and Alter Sensitivity To Anti-Estrogens
Accumulating data underscore the importance of the ECM in normal mammary function and tumor behavior (86, 175). A mechanically stiff matrix increases signaling through focal adhesions (176, 177). Aligned collagen fibers oriented perpendicularly to the tumor boundary have been linked to a poor prognosis, particularly in ER+ breast cancer (178). We have demonstrated that ECM structure can strongly influence the spectrum of PRL signals and PRL-estrogen crosstalk in ER+ breast cancer cells, and reciprocally, that these hormones can modify ECM structure (Figure 2). When well-characterized ER+ breast cancer cell lines were cultured in stiff ECM in vitro (MCF7 and T47D cells in 3-dimensional collagen cultures and tunable polyacrylamide substrates), PRL was less able to activate JAK2/STAT5, but more strongly activated FAK-SRC-ERK1/2, associated with increased localization of PRLR in focal adhesions (167, 179). These conditions augmented PRL-driven invasion and re-orientation of collagen fibers in vitro (167), and intravasation and metastasis of PRL-initiated ER+ tumors in a syngeneic model of increased COL1A1 density/stiffness in vivo (180). Moreover, a stiff/dense matrix enhanced PRL-estrogen crosstalk to increase invasion, reduce responsiveness to tamoxifen, and further modify ECM structure in vitro (181); these findings were supported using the syngeneic ER+ model above (55). Moreover, progesterone further augmented PRL induction of MMP3 RNA in stiff matrices (107). These studies indicate that desmoplasia, a feature of the microenvironment of many tumors, can alter the repertoire of PRL-initiated signals to favor pro-tumor pathways and anti-estrogen resistance, thus illuminating one mechanism underlying the apparent disparate reports of the outcomes of PRL signals in ER+ breast cancers. Extension of these studies of the effect of ECM characteristics on PRL signals to other breast cancer subtypes may further resolve some of the apparently contradictory reports.
4 Summary and Future Directions
Strong epidemiologic data linking higher levels of circulating PRL to increased risk for ER+ breast cancer are supported by multiple lines of experimental evidence. Independently from ovarian steroids, PRL can modulate the epithelial hierarchy and increase progenitor populations, drive development of ductal and alveolar abnormalities, and with time, promote aggressive metastatic ER+ carcinomas. PRL engages in complex crosstalk with estrogen and progesterone, cooperating with them by multiple mechanisms, but also opposing steroid-driven differentiation. Further understanding of these interactions apart from the hormonal milieu of pregnancy will provide additional insight into the impact of PRL on increased breast cancer risk in premenopausal women and postmenopausal women treated with estrogen-progesterone MHT (107, 162, 182). As disease progresses with dysregulation of multiple pathways, intrinsic tumor cell properties and the stromal environment are likely to alter the responses of target cells to PRL and its interactions with other potential oncogenic factors. Although not well understood, the literature suggests that PRL also may act directly on multiple mammary stromal cell types including immune cell subpopulations and/or modulate their activity via paracrine signals, which may further increase risk. The high PRLR expression in clinical DCIS and preneoplastic structures in preclinical models is reminiscent of ER expression in many of these lesions [reviewed in (183)], and suggests a role for PRL at this early stage of the disease process.
In contrast, the role(s) of PRL in the biology of established clinical breast cancers remains unclear. Although PRLR is highly expressed by many tumors across breast cancer subtypes, data from small clinical trials inhibiting PRL action are difficult to interpret, and studies of xenografts, particularly of breast cancer cell lines, are conflicting. Responses of phenotypically diverse heterogeneous cancers are complicated by different levels of PRLR isoforms with distinct signaling capabilities, selection and genomic evolution as tumors progress and respond to initial therapies, and environmental context, including site-specific responses of the metastatic niche [e.g., bone (120)], ECM properties and the steroid hormone and growth factor milieu (Figure 3). The emerging data support complex actions of PRL in breast cancer biology, resembling the major recognized hormonal actor in breast cancer, estrogen (184–186). This is illustrated in ER+ cancers, the breast cancer subtype in which PRL actions are currently best understood. Experimental evidence shows that PRL can activate STAT5A-driven differentiation, and maintain ERα expression, thereby facilitating anti-estrogen responsiveness (56, 128, 132, 159, 161). However, PRL can also drive proliferation and invasion, and support development of resistance to anti-estrogens (128, 181). Within the heterogeneous TNBCs, evidence indicates distinct subgroups which exhibit divergent responses to PRL (117, 122). Conflicting outcomes in a very limited number of different HER2+ breast cancer cell lines suggest similar possibilities. Together, these reports paint a more nuanced picture of PRL action in established cancers, and potential for very different outcomes depending on context. They underscore the need for additional study of PRL in diverse clinical breast cancers, changes with disease progression and therapeutic pressure, and influences of the metastatic sites.
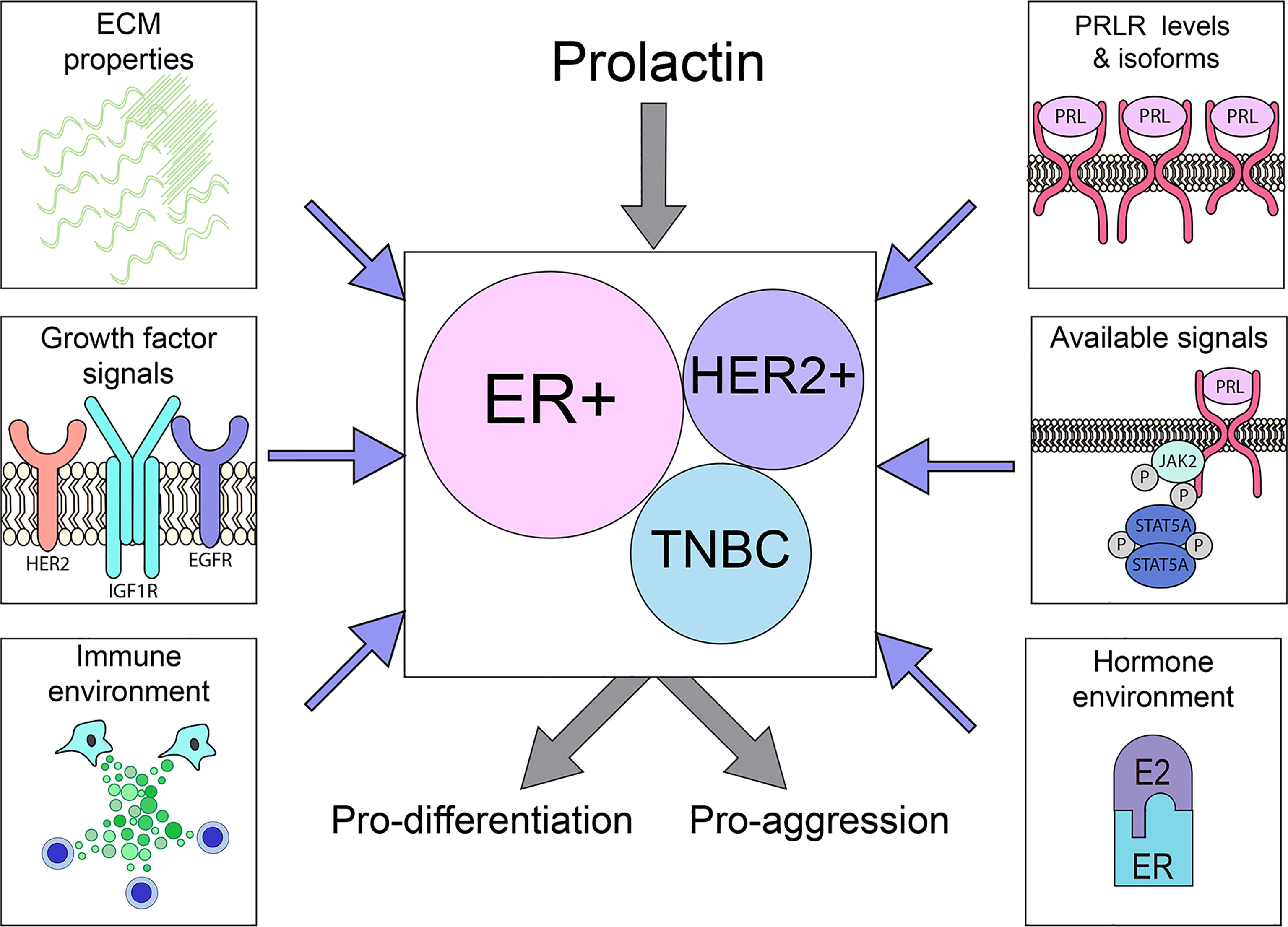
Figure 3 PRLR is expressed on a substantial subset of breast cancers across the major subtypes. The outcome of PRL signals may vary considerably, driving either differentiation or aggression, depending on tumor cell intrinsic properties (PRLR expression, available downstream signaling cascades) as well as extrinsic factors (ECM characteristics, hormone/growth factor milieu), as illustrated above. See Section 3 for details.
New technologies will assist in the resolution of these issues. The NSG-Pro mouse is a powerful tool to interrogate the actions of PRL in diverse clinical breast cancers in a dynamic in vivo environment (128). Already providing insights into ER+ cancers, this model will help resolve some of the conflicting studies observed with experimental xenografts of a relatively small number of breast cancer cell lines of other subtypes. It will enable dissection of the mechanisms underlying observed differences, and facilitate identification of biomarkers that predict beneficial or adverse responses to PRL and/or PRL inhibitors. Pending validation of humanized mice, syngeneic mouse models continue to be essential to reveal the impact of PRL as well as other agents on inflammation and suppression of anti-tumor immunity, a critical step toward employment of the promise of immunotherapies for hormonally responsive cancers. Further, as discussed in Section 2.2.3, many other stromal cell types which sculpt the tumor microenvironment are potential PRL targets, motivating additional study in the context of breast cancers. In addition, the paucity of inhibitors to interrogate PRL actions in clinical samples and experimental models is now addressed by small molecule inhibitors (129), the technology to reduce specific PRLR isoforms in vivo (130), and renewed interest in PRLR neutralizing antibodies (Sections 3.1, 3.3). Development of selective inhibitors of the JAK2 and src family kinase-mediated signals of PRL, taking advantage of our understanding of the closely related growth hormone receptor, will advance these studies (142, 187, 188). Together, these approaches will unravel the complex actions of PRL, permitting a new understanding of the role of this third hormone in breast cancer, with implications for prevention and treatment.
Author Contributions
Both LS and KO’L contributed to the original drafts and editing process. All authors have read and agreed to the published version of the manuscript.
Funding
This work was supported by NIH R01 CA179556, P30 CA014520 (University of Wisconsin Carbone Comprehensive Cancer Center), and More for Stage IV.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Acknowledgments
The authors are grateful to Dr. Molly Morgan for her thoughtful and generous assistance with the illustrations, and Dr. Fern Murdoch for helpful discussions.
Abbreviations
APC, adenomatous polyposis coli; COL1A1, collagen type I alpha 1; DCIS, ductal carcinoma in situ; ECM, extracellular matrix; ER, estrogen receptor alpha; FAK, focal adhesion kinase; GH, growth hormone; JAK2, janus kinase 2; MHT, menopausal hormone therapy; MMP, matrix metalloprotease; MMTV, mouse mammary tumor virus; NOD, non-obese diabetic; NR4A, nuclear receptor subfamily 4 group A member 1; NRL, neu-related lipocalin; NSG, NOD SCID gamma; OHT, hydroxytamoxifen; PDX, patient-derived xenograft; PGE2, prostaglandin E2; PRL, prolactin; PRLR, prolactin receptor; PTGS2, prostaglandin-endoperoxide synthase 2; RCAS, replication-competent ASLV long terminal repeat (LTR) with a splice acceptor; SCID, severe combined immunodeficiency; STAT, signal transducer and activator of transcription; TNBC, triple negative breast cancer.
References
1. Horseman ND. Prolactin and Mammary Gland Development. J Mammary Gland Biol Neoplasia (1999) 4:79–88. doi: 10.1023/A:1018708704335
2. Oakes SR, Rogers RL, Naylor MJ, Ormandy CJ. Prolactin Regulation of Mammary Gland Development. J Mammary Gland Biol Neoplasia (2008) 13:13–28. doi: 10.1007/s10911-008-9069-5
3. Obr AE, Grimm SL, Bishop KA, Pike JW, Lydon JP, Edwards DP. Progesterone Receptor and Stat5 Signaling Cross Talk Through RANKL in Mammary Epithelial Cells. Mol Endocrinol (2013) 27:1808–24. doi: 10.1210/me.2013-1077
4. Shin HY, Hennighausen L, Yoo KH. STAT5-Driven Enhancers Tightly Control Temporal Expression of Mammary-Specific Genes. J Mammary Gland Biol Neoplasia (2019) 24:61–71. doi: 10.1007/s10911-018-9418-y
5. Welsch CW, Gribler C. Prophylaxis of Spontaneously Developing Mammary Carcinoma in C3H- HeJ Female Mice by Suppression of Prolactin. Cancer Res (1973) 33:2939–46.
6. Nandi S, Guzman RC, Yang J. Hormones and Mammary Carcinogenesis in Mice, Rats, and Humans: A Unifying Hypothesis. Proc Natl Acad Sci USA (1995) 92:3650–7. doi: 10.1073/pnas.92.9.3650
7. Vonderhaar BK. Prolactin Involvement in Breast Cancer. Endocr Relat Cancer (1999) 6:389–404. doi: 10.1677/erc.0.0060389
8. Goffin V, Touraine P, Culler MD, Kelly PA. Drug Insight: Prolactin-Receptor Antagonists, a Novel Approach to Treatment of Unresolved Systemic and Local Hyperprolactinemia? Nat Clin Pract Endocrinol Metab (2006) 2:571–81. doi: 10.1038/ncpendmet0270
9. Muthuswamy SK. Autocrine Prolactin: An Emerging Market for Homegrown (Prolactin) Despite the Imports. Genes Dev (2012) 26:2253–8. doi: 10.1101/gad.204636.112
10. Harvey PW. Hypothesis: Prolactin is Tumorigenic to Human Breast: Dispelling the Myth That Prolactin-Induced Mammary Tumors are Rodent-Specific. J Appl Toxicol (2012) 32:1–9. doi: 10.1002/jat.1772
11. Damiano JS, Wasserman E. Molecular Pathways: Blockade of the PRLR Signaling Pathway as a Novel Antihormonal Approach for the Treatment of Breast and Prostate Cancer. Clin Cancer Res (2013) 19:1644–50. doi: 10.1158/1078-0432.CCR-12-0138
12. Gorvin CM. The Prolactin Receptor: Diverse and Emerging Roles in Pathophysiology. J Clin Transl Endocrinol (2015) 2:85–91. doi: 10.1016/j.jcte.2015.05.001
13. Chen WY. The Many Faces of Prolactin in Breast Cancer. Adv Exp Med Biol (2015) 846:61–81. doi: 10.1007/978-3-319-12114-7_3
14. Karayazi Atici O, Govindrajan N, Lopetegui-Gonzalez I, Shemanko CS. Prolactin: A Hormone With Diverse Functions From Mammary Gland Development to Cancer Metastasis. Sem Cell Dev Biol (2021) 114:159–70. doi: 10.1016/j.semcdb.2020.10.005
15. Phillipps HR, Yip SH, Grattan DR. Patterns of Prolactin Secretion. Mol Cell Endocrinol (2020) 502:110679. doi: 10.1016/j.mce.2019.110679
16. Tworoger SS, Hankinson SE. Prolactin and Breast Cancer Etiology: An Epidemiologic Perspective. J Mammary Gland Biol Neoplasia (2008) 13:41–53. doi: 10.1007/s10911-008-9063-y
17. Ben Jonathan N, LaPensee CR, LaPensee EW. What can We Learn From Rodents About Prolactin in Humans? Endocr Rev (2008) 29:1–41. doi: 10.1210/er.2007-0017
18. Tikk K, Sookthai D, Johnson T, Dossus L, Clavel-Chapelon F, Tjonneland A, et al. Prolactin Determinants in Healthy Women: A Large Cross-Sectional Study Within the EPIC Cohort. Cancer Epidemiol Biomarkers Prev (2014) 23:2532–42. doi: 10.1158/1055-9965.EPI-14-0613
19. Peuskens J, Pani L, Detraux J, De Hert M. The Effects of Novel and Newly Approved Antipsychotics on Serum Prolactin Levels: A Comprehensive Review. CNS Drugs (2014) 28:421–53. doi: 10.1007/s40263-014-0157-3
20. Rahman T, Sahrmann JM, Olsen MA, Nickel KB, Miller JP, Ma C, et al. Risk of Breast Cancer With Prolactin Elevating Antipsychotic Drugs: An Observational Study of US Women (Ages 18-64 Years). J Clin Psychopharmacol (2022) 42:7–16. doi: 10.1097/JCP.0000000000001513
21. Ben-Jonathan N, Mershon JL, Allen DL, Steinmetz RW. Extrapituitary Prolactin: Distribution, Regulation, Functions, and Clinical Aspects. Endocr Rev (1996) 17:639–69. doi: 10.1210/edrv-17-6-639
22. Clevenger CV, Furth PA, Hankinson SE, Schuler LA. Role of Prolactin in Mammary Carcinoma. Endocr Rev (2003) 24:1–27. doi: 10.1210/er.2001-0036
23. Marano RJ, Ben-Jonathan N. Minireview: Extrapituitary Prolactin: An Update on the Distribution, Regulation, and Functions. Mol Endocrinol (2014) 28:622–33. doi: 10.1210/me.2013-1349
24. Ginsburg E, Vonderhaar BK. Prolactin Synthesis and Secretion by Human Breast Cancer Cells. Cancer Res (1995) 55:2591–5.
25. McHale K, Tomaszewski JE, Puthiyaveettil R, Livolsi VA, Clevenger CV. Altered Expression of Prolactin Receptor-Associated Signaling Proteins in Human Breast Carcinoma. Mod Pathol (2008) 21:565–71. doi: 10.1038/modpathol.2008.7
26. Wu ZS, Yang K, Wan Y, Qian PX, Perry JK, Chiesa J, et al. Tumor Expression of Human Growth Hormone and Human Prolactin Predict a Worse Survival Outcome in Patients With Mammary or Endometrial Carcinoma. J Clin Endocrinol Metab (2011) 96:E1619–29. doi: 10.1210/jc.2011-1245
27. Hachim IY, Shams A, Lebrun JJ, Ali S. A Favorable Role of Prolactin in Human Breast Cancer Reveals Novel Pathway-Based Gene Signatures Indicative of Tumor Differentiation and Favorable Patient Outcome. Hum Pathol (2016) 53:142–52. doi: 10.1016/j.humpath.2016.02.010
28. Zheng Y, Comaills V, Burr R, Boulay G, Miyamoto DT, Wittner BS, et al. COX-2 Mediates Tumor-Stromal Prolactin Signaling to Initiate Tumorigenesis. Proc Natl Acad Sci USA (2019) 116:5223–32. doi: 10.1073/pnas.1819303116
29. Isaksson OGP, Eden S, Jansson JO. Mode of Action of Pituitary Growth Hormone on Target Cells. Ann Rev Physiol (1985) 47:483–99. doi: 10.1146/annurev.ph.47.030185.002411
30. Utama FE, Tran TH, Ryder A, LeBaron MJ, Parlow AF, Rui H. Insensitivity of Human Prolactin Receptors to non-Human Prolactins: Relevance for Experimental Modeling of Prolactin Receptor-Expressing Human Cells. Endocrinology (2009) 150:1782–90. doi: 10.1210/en.2008-1057
31. Xu J, Sun D, Jiang J, Deng L, Zhang Y, Yu H, et al. The Role of Prolactin Receptor in GH Signaling in Breast Cancer Cells. Mol Endocrinol (2013) 27:266–79. doi: 10.1210/me.2012-1297
32. Wang M, Wu X, Chai F, Zhang Y, Jiang J. Plasma Prolactin and Breast Cancer Risk: A Meta- Analysis. Sci Rep (2016) 6:25998. doi: 10.1038/srep25998
33. Tikk K, Sookthai D, Johnson T, Rinaldi S, Romieu I, Tjonneland A, et al. Circulating Prolactin and Breast Cancer Risk Among Pre- and Postmenopausal Women in the EPIC Cohort. Ann Oncol (2014) 25:1422–8. doi: 10.1093/annonc/mdu150
34. Gabrielson M, Ubhayasekera K, Ek B, Andersson Franko M, Eriksson M, Czene K, et al. Inclusion of Plasma Prolactin Levels in Current Risk Prediction Models of Premenopausal and Postmenopausal Breast Cancer. JNCI Cancer Spectr (2018) 2:pky055. doi: 10.1093/jncics/pky055
35. Tworoger SS, Eliassen AH, Sluss P, Hankinson SE. A Prospective Study of Plasma Prolactin Concentrations and Risk of Premenopausal and Postmenopausal Breast Cancer. J Clin Oncol (2007) 25:1482–8. doi: 10.1200/JCO.2006.07.6356
36. Tworoger SS, Eliassen AH, Zhang X, Qian J, Sluss PM, Rosner BA, et al. A 20-Year Prospective Study of Plasma Prolactin as a Risk Marker of Breast Cancer Development. Cancer Res (2013) 73:4810–9. doi: 10.1158/0008-5472.CAN-13-0665
37. Greendale GA, Huang MH, Ursin G, Ingles S, Stanczyk F, Crandall C, et al. Serum Prolactin Levels are Positively Associated With Mammographic Density in Postmenopausal Women. Breast Cancer Res Treat (2007) 105:337–46. doi: 10.1007/s10549-006-9454-y
38. Becker S, Kaaks R. Exogenous and Endogenous Hormones, Mammographic Density and Breast Cancer Risk: Can Mammographic Density be Considered an Intermediate Marker of Risk? Recent Results Cancer Res (2009) 181:135–57. doi: 10.1007/978-3-540-69297-3_14
39. Boyd NF, Martin LJ, Yaffe MJ, Minkin S. Mammographic Density and Breast Cancer Risk: Current Understanding and Future Prospects. Breast Cancer Res (2011) 13:223. doi: 10.1186/bcr2942
40. Huo CW, Chew GL, Britt KL, Ingman WV, Henderson MA, Hopper JL, Thompson EW, et al. Mammographic Density-A Review on the Current Understanding of its Association with Breast Cancer. Breast Cancer Res Treat (2014) 144:479-502. doi: 10.1007/s10549-014-2901-2
41. Tworoger SS, Zhang X, Eliassen AH, Qian J, Colditz GA, Willett WC, et al. Inclusion of Endogenous Hormone Levels in Risk Prediction Models of Postmenopausal Breast Cancer. J Clin Oncol (2014) 32:3111–7. doi: 10.1200/JCO.2014.56.1068
42. Schedin P. Pregnancy-Associated Breast Cancer and Metastasis. Nat Rev Cancer (2006) 6:281–91. doi: 10.1038/nrc1839
43. Christov K, Swanson SM, Guzman RC, Thordarson G, Jin E, Talamantes F, et al. Kinetics of Mammary Epithelial Cell Proliferation in Pituitary Isografted BALB/c Mice. Carcinogenesis (1993) 14:2019–25. doi: 10.1093/carcin/14.10.2019
44. Stocco C, Telleria C, Gibori G. The Molecular Control of Corpus Luteum Formation, Function, and Regression. Endocr Rev (2007) 28:117–49. doi: 10.1210/er.2006-0022
45. Arendt LM, Schuler LA. Transgenic Models to Study Actions of Prolactin in Mammary Neoplasia. J Mammary Gland Biol Neoplasia (2008) 13:29–40. doi: 10.1007/s10911-008-9073-9
46. Iavnilovitch E, Cardiff RD, Groner B, Barash I. Deregulation of STAT5 Expression and Activation Causes Mammary Tumors in Transgenic Mice. Int J Cancer (2004) 112:607–19. doi: 10.1002/ijc.20484
47. Griffith OL, Chan SR, Griffith M, Krysiak K, Skidmore ZL, Hundal J, et al. Truncating Prolactin Receptor Mutations Promote Tumor Growth in Murine Estrogen Receptor-Alpha Mammary Carcinomas. Cell Rep (2016) 17:249–60. doi: 10.1016/j.celrep.2016.08.076
48. Rose-Hellekant TA, Arendt LM, Schroeder MD, Gilchrist K, Sandgren EP, Schuler LA. Prolactin Induces ERα-Positive and ERα-Negative Mammary Cancer in Transgenic Mice. Oncogene (2003) 22:4664–74. doi: 10.1038/sj.onc.1206619
49. O'Leary KA, Shea MP, Schuler LA. Modeling Prolactin Actions in Breast Cancer In Vivo: Insights From the NRL-PRL Mouse. Adv Exp Med Biol (2015) 846:201–20. doi: 10.1007/978-3-319-12114-7_9
50. Arendt LM, Evans LC, Rugowski DE, Garcia-Barchino MJ, Rui H, Schuler LA. Ovarian Hormones are Not Required for PRL-Induced Mammary Tumorigenesis, But Estrogen Enhances Neoplastic Processes. J Endocrinol (2009) 203:99–110. doi: 10.1677/JOE-09-0221
51. Bole-Feysot C, Goffin V, Edery M, Binart N, Kelly PA. Prolactin (PRL) and its Receptor: Actions, Signal Transduction Pathways and Phenotypes Observed in PRL Receptor Knockout Mice. Endocr Rev (1998) 19:225–68. doi: 10.1210/edrv.19.3.0334
52. Arendt LM, Rose-Hellekant TA, Sandgren EP, Schuler LA. Prolactin Potentiates TGFα Induction of Mammary Neoplasia in Transgenic Mice. Am J Pathol (2006) 168:1365–74. doi: 10.2353/ajpath.2006.050861
53. Arendt LM, Rugowski DE, Grafwallner-Huseth TL, Garcia-Barchino MJ, Rui H, Schuler LA. Prolactin-Induced Mouse Mammary Carcinomas Model Estrogen Resistant Luminal Breast Cancer. Breast Cancer Res (2011) 13:R11–25. doi: 10.1186/bcr2819
54. Shea MP, O'Leary KA, Fakhraldeen SA, Goffin V, Friedl A, Wisinski KB, et al. Anti-Estrogen Therapy Increases Plasticity and Cancer Stemness of Prolactin-Induced ERα+ Mammary Carcinomas. Cancer Res (2018) 78:1672–84. doi: 10.1158/0008-5472.CAN-17-0985
55. Jallow F, O'Leary KA, Rugowski DE, Guerrero JF, Ponik SM, Schuler LA. Dynamic Interactions Between the Extracellular Matrix and Estrogen Activity in Progression of ER+ Breast Cancer. Oncogene (2019) 38:6913-25. doi: 10.1038/s41388-019-0941-0
56. Jallow F, Brockman JL, Helzer KT, Rugowski DE, Goffin V, Alarid ET, et al. 17beta-Estradiol and ICI182,780 Differentially Regulate STAT5 Isoforms in Female Mammary Epithelium, With Distinct Outcomes. J Endocr Soc (2018) 2:293–309. doi: 10.1210/js.2017-00399
57. Campbell KM, O'Leary KA, Rugowski DE, Mulligan WA, Barnell EK, Skidmore ZL, et al. A Spontaneous Aggressive Erα+ Mammary Tumor Model is Driven by Kras Activation. Cell Rep (2019) 28:1526–37. doi: 10.1016/j.celrep.2019.06.098
58. Perou CM, Borresen-Dale AL. Systems Biology and Genomics of Breast Cancer. Cold Spring Harb Perspect Biol (2011) 3:a003293. doi: 10.1101/cshperspect.a003293
59. Curtis C, Shah SP, Chin SF, Turashvili G, Rueda OM, Dunning MJ, et al. The Genomic and Transcriptomic Architecture of 2,000 Breast Tumours Reveals Novel Subgroups. Nature (2012) 486:346–52. doi: 10.1038/nature10983
60. Budczies J, Bockmayr M, Denkert C, Klauschen F, Lennerz JK, Gyorffy B, et al. Classical Pathology and Mutational Load of Breast Cancer - Integration of Two Worlds. J Pathol Clin Res (2015) 1:225–38. doi: 10.1002/cjp2.25
61. Olsen SN, Wronski A, Castano Z, Dake B, Malone C, De Raedt T, et al. Loss of RasGAP Tumor Suppressors Underlies the Aggressive Nature of Luminal B Breast Cancers. Cancer Discovery (2017) 7:202–17. doi: 10.1158/2159-8290.CD-16-0520
62. Wright KL, Adams JR, Liu JC, Loch AJ, Wong RG, Jo CE, et al. Ras Signaling is a Key Determinant for Metastatic Dissemination and Poor Survival of Luminal Breast Cancer Patients. Cancer Res (2015) 75:4960–72. doi: 10.1158/0008-5472.CAN-14-2992
63. Griffith OL, Spies NC, Anurag M, Griffith M, Luo J, Tu D, et al. The Prognostic Effects of Somatic Mutations in ER-Positive Breast Cancer. Nat Commun (2018) 9:3476. doi: 10.1038/s41467-018-05914-x
64. Bertucci F, Ng CKY, Patsouris A, Droin N, Piscuoglio S, Carbuccia N, et al. Genomic Characterization of Metastatic Breast Cancers. Nature (2019) 569:560–4. doi: 10.1038/s41586-019-1056-z
65. Stemke-Hale K, Gonzalez-Angulo AM, Lluch A, Neve RM, Kuo WL, Davies M, et al. An Integrative Genomic and Proteomic Analysis of PIK3CA, PTEN, and AKT Mutations in Breast Cancer. Cancer Res (2008) 68:6084–91. doi: 10.1158/0008-5472.CAN-07-6854
66. Vasan N, Toska E, Scaltriti M. Overview of the Relevance of PI3K Pathway in HR-Positive Breast Cancer. Ann Oncol (2019) 30:x3–x11. doi: 10.1093/annonc/mdz281
67. Schuler LA, Murdoch FE. Endogenous and Therapeutic Estrogens: Maestro Conductors of the Microenvironment of ER+ Breast Cancers. Cancers (2021) 13:3725. doi: 10.3390/cancers13153725
68. Goldberg J, Pastorello RG, Vallius T, Davis J, Cui YX, Agudo J, et al. The Immunology of Hormone Receptor Positive Breast Cancer. Front Immunol (2021) 12:674192. doi: 10.3389/fimmu.2021.674192
69. O'Leary KA, Shea MP, Salituro S, Blohm CE, Schuler LA. Prolactin Alters the Mammary Epithelial Hierarchy, Increasing Progenitors and Facilitating Ovarian Steroid Action. Stem Cell Rep (2017) 9:1167–79. doi: 10.1016/j.stemcr.2017.08.011
70. Sell S. On the Stem Cell Origin of Cancer. Am J Pathol (2010) 176:2584–494. doi: 10.2353/ajpath.2010.091064
71. Fu NY, Nolan E, Lindeman GJ, Visvader JE. Stem Cells and the Differentiation Hierarchy in Mammary Gland Development. Physiol Rev (2020) 100:489–523. doi: 10.1152/physrev.00040.2018
72. Camarillo IG, Thordarson G, Moffat JG, Van Horn KM, Binart N, Kelly PA, et al. Prolactin Receptor Expression in the Epithelia and Stroma of the Rat Mammary Gland. J Endocrinol (2001) 171:85–95. doi: 10.1677/joe.0.1710085
73. Hovey RC, Trott JF, Ginsburg E, Goldhar A, Sasaki MM, Fountain SJ, et al. Transcriptional and Spatiotemporal Regulation of Prolactin Receptor mRNA and Cooperativity With Progesterone Receptor Function During Ductal Branch Growth in the Mammary Gland. Dev Dyn (2001) 222:192–205. doi: 10.1002/dvdy.1179
74. Coussens LM, Pollard JW. Leukocytes in Mammary Development and Cancer. Cold Spring Harb Perspect Biol (2011) 3:a003285. doi: 10.1101/cshperspect.a003285
75. Need EF, Atashgaran V, Ingman WV, Dasari P. Hormonal Regulation of the Immune Microenvironment in the Mammary Gland. J Mammary Gland Biol Neoplasia (2014) 19:229–39. doi: 10.1007/s10911-014-9324-x
76. Plaks V, Boldajipour B, Linnemann JR, Nguyen NH, Kersten K, Wolf Y, et al. Adaptive Immune Regulation of Mammary Postnatal Organogenesis. Dev Cell (2015) 34:493–504. doi: 10.1016/j.devcel.2015.07.015
77. Dill R, Walker AM. Role of Prolactin in Promotion of Immune Cell Migration Into the Mammary Gland. J Mammary Gland Biol Neoplasia (2017) 22:13–26. doi: 10.1007/s10911-016-9369-0
78. Matera L. Action of Pituitary and Lymphocyte Prolactin. Neuroimmunomodulation (1997) 4:171–80. doi: 10.1159/000097335
79. Welniak LA, Richards SM, Murphy WJ. Effects of Prolactin on Hematopoiesis. Lupus (2001) 10:700–5. doi: 10.1191/096120301717164930
80. Savino W, Mendes-da-Cruz DA, Lepletier A, Dardenne M. Hormonal Control of T-Cell Development in Health and Disease. Nat Rev Endocrinol (2016) 12:77–89. doi: 10.1038/nrendo.2015.168
81. Recalde G, Moreno-Sosa T, Yudica F, Quintero CA, Sanchez MB, Jahn GA, et al. Contribution of Sex Steroids and Prolactin to the Modulation of T and B Cells During Autoimmunity. Autoimmun Rev (2018) 17:504–12. doi: 10.1016/j.autrev.2018.03.006
82. Tufa DM, Shank T, Yingst AM, Trahan GD, Shim S, Lake J, et al. Prolactin Acts on Myeloid Progenitors to Modulate SMAD7 Expression and Enhance Hematopoietic Stem Cell Differentiation Into the NK Cell Lineage. Sci Rep (2020) 10:6335. doi: 10.1038/s41598-020-63346-4
83. Chen KE, Ghosh M, Rivera L, Lin S, Kumar A, Swaminathan S, et al. Prolactin Enhances T Regulatory Cell Promotion of Breast Cancer Through the Long Form Prolactin Receptor. Transl Oncol (2021) 14:101195. doi: 10.1016/j.tranon.2021.101195
84. Spangelo BL, Hall NR, Ross PC, Goldstein AL. Stimulation of In Vivo Antibody Production and Concanavalin-A-Induced Mouse Spleen Cell Mitogenesis by Prolactin. Immunopharm (1987) 14:11–20. doi: 10.1016/0162-3109(87)90004-X
85. Matera L, Cesano A, Bellone G, Oberholtzer E. Modulatory Effect of Prolactin on the Resting and Mitogen-Induced Activity of T, B, and NK Lymphocytes. Brain Behav Immun (1992) 6:409–17. doi: 10.1016/0889-1591(92)90039-Q
86. Winkler J, Abisoye-Ogunniyan A, Metcalf KJ, Werb Z. Concepts of Extracellular Matrix Remodelling in Tumour Progression and Metastasis. Nat Commun (2020) 11:5120. doi: 10.1038/s41467-020-18794-x
87. Tandon M, Coudriet GM, Criscimanna A, Socorro M, Eliliwi M, Singhi AD, et al. Prolactin Promotes Fibrosis and Pancreatic Cancer Progression. Cancer Res (2019) 79:5316–27. doi: 10.1158/0008-5472.CAN-18-3064
88. Yang X, Meyer K, Friedl A. STAT5 and Prolactin Participate in a Positive Autocrine Feedback Loop That Promotes Angiogenesis. J Biol Chem (2013) 288:21184–96. doi: 10.1074/jbc.M113.481119
89. Vazquez-Membrillo M, Siqueiros-Marquez L, Nunez FF, Diaz-Lezama N, Adan-Castro E, Ramirez-Hernandez G, et al. Prolactin Stimulates the Vascularisation of the Retina in Newborn Mice Under Hyperoxia Conditions. J Neuroendocrinol (2020) 32:e12858. doi: 10.1111/jne.12858
90. Triebel J, Robles-Osorio ML, Garcia-Franco R, Martinez de la Escalera G, Clapp C, Bertsch T. From Bench to Bedside: Translating the Prolactin/Vasoinhibin Axis. Front Endocrinol (2017) 8:342. doi: 10.3389/fendo.2017.00342
91. Clapp C, Thebault S, Macotela Y, Moreno-Carranza B, Triebel J, Martinez de la Escalera G. Regulation of Blood Vessels by Prolactin and Vasoinhibins. Adv Exp Med Biol (2015) 846:83–95. doi: 10.1007/978-3-319-12114-7_4
92. Hachim IY, Hachim MY, Lopez VM, Lebrun JJ, Ali S. Prolactin Receptor Expression is an Independent Favorable Prognostic Marker in Human Breast Cancer. Appl Immunohistochem Mol Morphol (2016) 24:238–45. doi: 10.1097/PAI.0000000000000178
93. Howell SJ, Anderson E, Hunter T, Farnie G, Clarke RB. Prolactin Receptor Antagonism Reduces the Clonogenic Capacity of Breast Cancer Cells and Potentiates Doxorubicin and Paclitaxel Cytotoxicity. Breast Cancer Res (2008) 10:R68. doi: 10.1186/bcr2129
94. Jerry DJ, Kittrell FS, Kuperwasser C, Laucirica R, Dickinson ES, Bonilla PJ, et al. A Mammary-Specific Model Demonstrates the Role of the P53 Tumor Suppressor Gene in Tumor Development. Oncogene (2000) 19:1052–8. doi: 10.1038/sj.onc.1203270
95. Vomachka AJ, Pratt SL, Lockefeer JA, Horseman ND. Prolactin Gene-Disruption Arrests Mammary Gland Development and Retards T-Antigen-Induced Tumor Growth. Oncogene (2000) 19:1077–84. doi: 10.1038/sj.onc.1203348
96. Oakes SR, Robertson FG, Kench JG, Gardiner-Garden M, Wand MP, Green JE, et al. Loss of Mammary Epithelial Prolactin Receptor Delays Tumor Formation by Reducing Cell Proliferation in Low-Grade Preinvasive Lesions. Oncogene (2007) 26:543–53. doi: 10.1038/sj.onc.1209838
97. Johnston AN, Bu W, Hein S, Garcia S, Camacho L, Xue L, et al. Hyperprolactinemia-Inducing Antipsychotics Increase Breast Cancer Risk by Activating JAK-STAT5 in Precancerous Lesions. Breast Cancer Res (2018) 20:42. doi: 10.1186/s13058-018-0969-z
98. Barakat R, Oakley O, Kim H, Jin J, Ko CJ. Extra-Gonadal Sites of Estrogen Biosynthesis and Function. BMB Rep (2016) 49:488–96. doi: 10.5483/BMBRep.2016.49.9.141
99. Hetemaki N, Savolainen-Peltonen H, Tikkanen MJ, Wang F, Paatela H, Hamalainen E, et al. Estrogen Metabolism in Abdominal Subcutaneous and Visceral Adipose Tissue in Postmenopausal Women. J Clin Endocrinol Metab (2017) 102:4588–95. doi: 10.1210/jc.2017-01474
100. Gutzman JH, Miller KK, Schuler LA. Endogenous hPRL and Not Exogenous hPRL Induces ERa and PRLR Expression and Increases Estrogen Responsiveness in Breast Cancer Cells. J Steroid Biochem Mol Biol (2004) 88:69–77. doi: 10.1016/j.jsbmb.2003.10.008
101. Dong J, Tsai-Morris CH, Dufau ML. A Novel Estradiol/Estrogen Receptor Alpha-Dependent Transcriptional Mechanism Controls Expression of the Human Prolactin Receptor. J Biol Chem (2006) 281:18825–36. doi: 10.1074/jbc.M512826200
102. Gutzman JH, Nikolai SE, Rugowski DE, Watters JJ, Schuler LA. Prolactin and Estrogen Enhance the Activity of Activating Protein-1 in Breast Cancer Cells: Role of ERK1/2-Mediated Signals to C-Fos. Mol Endocrinol (2005) 19:1765–78. doi: 10.1210/me.2004-0339
103. Rasmussen LM, Frederiksen KS, Din N, Galsgaard E, Christensen L, Berchtold MW, et al. Prolactin and Oestrogen Synergistically Regulate Gene Expression and Proliferation of Breast Cancer Cells. Endocr Relat Cancer (2010) 17:809–22. doi: 10.1677/ERC-09-0326
104. Arendt LM, Schuler LA. Prolactin Drives ERα-Dependent Ductal Expansion and Synergizes With TGFα to Induce Mammary Tumors in Males. Am J Pathol (2008) 172:194–202. doi: 10.2353/ajpath.2008.070597
105. Gonzalez L, Zambrano A, Lazaro-Trueba I, Lopez E, Gonzalez JJ, Martin-Perez J, et al. Activation of the Unliganded Estrogen Receptor by Prolactin in Breast Cancer Cells. Oncogene (2009) 28:1298–308. doi: 10.1038/onc.2008.473
106. O'Leary KA, Jallow F, Rugowski DE, Sullivan R, Sinkevicius KW, Greene GL, et al. Prolactin Activates Erα in the Absence of Ligand in Female Mammary Development and Carcinogenesis In Vivo. Endocrinology (2013) 154:4483–92. doi: 10.1210/en.2013-1533
107. Leehy KA, Truong TH, Mauro LJ, Lange CA. Progesterone Receptors (PR) Mediate STAT Actions: PR and Prolactin Receptor Signaling Crosstalk in Breast Cancer Models. J Steroid Biochem Mol Biol (2018) 176:88–93. doi: 10.1016/j.jsbmb.2017.04.011
108. Trabert B, Sherman ME, Kannan N, Stanczyk FZ. Progesterone and Breast Cancer. Endocr Rev (2020) 41:320-44. doi: 10.1210/endrev/bnz001
109. Arendt LM, Grafwallner-Huseth TL, Schuler LA. Prolactin-Growth Factor Crosstalk Reduces Mammary Estrogen Responsiveness Despite Elevated ERα Expression. Am J Pathol (2009) 174:1065–74. doi: 10.2353/ajpath.2009.080719
110. O'Leary KA, Rugowski DE, Sullivan R, Schuler LA. Prolactin Cooperates With Loss of P53 to Promote Claudin Low Mammary Carcinomas. Oncogene (2014) 33:3075–82. doi: 10.1038/onc.2013.278
111. O'Leary KA, Rugowski DE, Shea MP, Sullivan R, Moser AR, Schuler LA. Prolactin Synergizes With Canonical Wnt Signals to Drive Development of ER+ Mammary Tumors via Activation of the Notch Pathway. Cancer Lett (2021) 503:231–9. doi: 10.1016/j.canlet.2021.01.012
112. Frank SJ. Mechanistic Aspects of Crosstalk Between GH and PRL and ErbB Receptor Family Signaling. J Mammary Gland Biol Neoplasia (2008) 13:119–29. doi: 10.1007/s10911-008-9065-9
113. Carver KC, Piazza TM, Schuler LA. Prolactin Enhances IGF-IR Phosphorylation by Decreasing its Association With the Tyrosine Phosphatase SHP-2 in MCF-7 Breast Cancer Cells. J Biol Chem (2010) 285:8003–12. doi: 10.1074/jbc.M109.066480
114. Osborne CK, Schiff R. Mechanisms of Endocrine Resistance in Breast Cancer. Ann Rev Med (2011) 62:233–47. doi: 10.1146/annurev-med-070909-182917
115. Roop RP, Ma CX. Endocrine Resistance in Breast Cancer: Molecular Pathways and Rational Development of Targeted Therapies. Future Oncol (2012) 8:273–92. doi: 10.2217/fon.12.8
116. Haines E, Minoo P, Feng Z, Resalatpanah N, Nie XM, Campiglio M, et al. Tyrosine Phosphorylation of Grb2: Role in Prolactin/Epidermal Growth Factor Cross Talk in Mammary Epithelial Cell Growth and Differentiation. Mol Cell Biol (2009) 29:2505–20. doi: 10.1128/MCB.00034-09
117. Grible JM, Zot P, Olex AL, Hedrick SE, Harrell JC, Woock AE, et al. The Human Intermediate Prolactin Receptor is a Mammary Proto-Oncogene. NPJ Breast Cancer (2021) 7:37. doi: 10.1038/s41523-021-00243-7
118. Galsgaard ED, Rasmussen BB, Folkesson CG, Rasmussen LM, Berchtold MW, Christensen L, et al. Re-Evaluation of the Prolactin Receptor Expression in Human Breast Cancer. J Endocrinol (2009) 201:115–28. doi: 10.1677/JOE-08-0479
119. Faupel-Badger JM, Duggan MA, Sherman ME, Garcia-Closas M, Yang XR, Lissowska J, et al. Prolactin Receptor Expression and Breast Cancer: Relationships With Tumor Characteristics Among Pre- and Post-Menopausal Women in a Population-Based Case-Control Study From Poland. Horm Cancer (2014) 5:42–50. doi: 10.1007/s12672-013-0165-7
120. Sutherland A, Forsyth A, Cong Y, Grant L, Juan TH, Lee JK, et al. The Role of Prolactin in Bone Metastasis and Breast Cancer Cell-Mediated Osteoclast Differentiation. J Natl Cancer Inst (2016) 108:djv338. doi: 10.1093/jnci/djv338
121. Kelly MP, Hickey C, Makonnen S, Coetzee S, Jalal S, Wang Y, et al. Preclinical Activity of the Novel Anti-Prolactin Receptor (PRLR) Antibody-Drug Conjugate REGN2878-DM1 in PRLR-Positive Breast Cancers. Mol Cancer Ther (2017) 16:1299–311. doi: 10.1158/1535-7163.MCT-16-0839
122. Lopez-Ozuna VM, Hachim IY, Hachim MY, Lebrun JJ, Ali S. Prolactin Pro-Differentiation Pathway in Triple Negative Breast Cancer: Impact on Prognosis and Potential Therapy. Sci Rep (2016) 6:30934. doi: 10.1038/srep30934
123. Fiorillo AA, Medler TR, Feeney YB, Wetz SM, Tommerdahl KL, Clevenger CV. The Prolactin Receptor Transactivation Domain is Associated With Steroid Hormone Receptor Expression and Malignant Progression of Breast Cancer. Am J Pathol (2013) 182:217–33. doi: 10.1016/j.ajpath.2012.09.021
124. Perou CM. Molecular Stratification of Triple-Negative Breast Cancers. Oncologist (2011) 16 Suppl 1:61–70. doi: 10.1634/theoncologist.2011-S1-61
125. Turner NC, Reis-Filho JS. Tackling the Diversity of Triple-Negative Breast Cancer. Clin Cancer Res (2013) 19:6380–8. doi: 10.1158/1078-0432.CCR-13-0915
126. Agarwal N, Machiels JP, Suarez C, Lewis N, Higgins M, Wisinski K, et al. Phase I Study of the Prolactin Receptor Antagonist LFA102 in Metastatic Breast and Castration-Resistant Prostate Cancer. Oncologist (2016) 21:535–6. doi: 10.1634/theoncologist.2015-0502
127. Costa R, Santa-Maria CA, Scholtens DM, Jain S, Flaum L, Gradishar WJ, et al. A Pilot Study of Cabergoline for the Treatment of Metastatic Breast Cancer. Breast Cancer Res Treat (2017) 165:585–92. doi: 10.1007/s10549-017-4370-x
128. Sun Y, Yang N, Utama FE, Udhane SS, Zhang J, Peck AR, et al. NSG-Pro Mouse Model for Uncovering Resistance Mechanisms and Unique Vulnerabilities in Human Luminal Breast Cancers. Sci Adv (2021) 7:eabc8145. doi: 10.1126/sciadv.abc8145
129. Borcherding DC, Hugo ER, Fox SR, Jacobson EM, Hunt BG, Merino EJ, et al. Suppression of Breast Cancer by Small Molecules That Block the Prolactin Receptor. Cancers (2021) 13:2662. doi: 10.3390/cancers13112662
130. Yonezawa T, Chen KH, Ghosh MK, Rivera L, Dill R, Ma L, et al. Anti-Metastatic Outcome of Isoform-Specific Prolactin Receptor Targeting in Breast Cancer. Cancer Lett (2015) 366:84–92. doi: 10.1016/j.canlet.2015.06.010
131. Hachim IY, Lopez-Ozuna VM, Hachim MY, Lebrun JJ, Ali S. Prolactin Hormone Exerts Anti-Tumorigenic Effects in HER-2 Overexpressing Breast Cancer Cells Through Regulation of Stemness. Stem Cell Res (2019) 40:101538. doi: 10.1016/j.scr.2019.101538
132. Shams A, Binothman N, Boudreault J, Wang N, Shams F, Hamam D, et al. Prolactin Receptor-Driven Combined Luminal and Epithelial Differentiation in Breast Cancer Restricts Plasticity, Stemness, Tumorigenesis and Metastasis. Oncogenesis (2021) 10:10. doi: 10.1038/s41389-020-00297-5
133. Neve RM, Chin K, Fridlyand J, Yeh J, Baehner FL, Fevr T, et al. A Collection of Breast Cancer Cell Lines for the Study of Functionally Distinct Cancer Subtypes. Cancer Cell (2006) 10:515–27. doi: 10.1016/j.ccr.2006.10.008
134. Kao J, Salari K, Bocanegra M, Choi YL, Girard L, Gandhi J, et al. Molecular Profiling of Breast Cancer Cell Lines Defines Relevant Tumor Models and Provides a Resource for Cancer Gene Discovery. PLos One (2009) 4:e6146. doi: 10.1371/journal.pone.0006146
135. Tomblyn S, Springs AE, Langenheim JF, Chen WY. Combination Therapy Using Three Novel Prolactin Receptor Antagonist-Based Fusion Proteins Effectively Inhibits Tumor Recurrence and Metastasis in HER2/neu Transgenic Mice. Int J Oncol (2009) 34:1139–46. doi: 10.3892/ijo_00000242
136. Xu C, Langenheim JF, Chen WY. Stromal-Epithelial Interactions Modulate Cross-Talk Between Prolactin Receptor and HER2/Neu in Breast Cancer. Breast Cancer Res Treat (2012) 134:157–69. doi: 10.1007/s10549-012-1954-3
137. Damiano JS, Rendahl KG, Karim C, Embry MG, Ghoddusi M, Holash J, et al. Neutralization of Prolactin Receptor Function by Monoclonal Antibody LFA102, a Novel Potential Therapeutic for the Treatment of Breast Cancer. Mol Cancer Ther (2013) 12:295–305. doi: 10.1158/1535-7163.MCT-12-0886
138. Osorio JS, Lohakare J, Bionaz M. Biosynthesis of Milk Fat, Protein, and Lactose: Roles of Transcriptional and Posttranscriptional Regulation. Physiol Genomics (2016) 48:231–56. doi: 10.1152/physiolgenomics.00016.2015
139. El Ansari R, Craze ML, Miligy I, Diez-Rodriguez M, Nolan CC, Ellis IO, et al. The Amino Acid Transporter SLC7A5 Confers a Poor Prognosis in the Highly Proliferative Breast Cancer Subtypes and is a Key Therapeutic Target in Luminal B Tumours. Breast Cancer Res (2018) 20:21. doi: 10.1186/s13058-018-0946-6
140. Saito Y, Li L, Coyaud E, Luna A, Sander C, Raught B, et al. LLGL2 Rescues Nutrient Stress by Promoting Leucine Uptake in ER(+) Breast Cancer. Nature (2019) 569:275–9. doi: 10.1038/s41586-019-1126-2
141. Shindo H, Harada-Shoji N, Ebata A, Sato M, Soga T, Miyashita M, et al. Targeting Amino Acid Metabolic Reprogramming via L-Type Amino Acid Transporter 1 (LAT1) for Endocrine-Resistant Breast Cancer. Cancers (2021) 13:4375. doi: 10.3390/cancers13174375
142. Carver KC, Arendt LM, Schuler LA. Complex Prolactin Crosstalk in Breast Cancer: New Therapeutic Implications. Mol Cell Endocrinol (2009) 307:1–7. doi: 10.1016/j.mce.2009.03.014
143. Yamauchi T, Yamauchi N, Ueki K, Sugiyama T, Waki H, Miki H, et al. Constitutive Tyrosine Phosphorylation of ErbB-2 via Jak2 by Autocrine Secretion of Prolactin in Human Breast Cancer. J Biol Chem (2000) 275:33937–44. doi: 10.1074/jbc.M000743200
144. Scotti ML, Langenheim JF, Tomblyn S, Springs AEB, Chen WY. Additive Effects of a Prolactin Receptor Antagonist, G129R, and Herceptin on Inhibition of HER2-Overexpressing Breast Cancer Cells. Breast Cancer Res Treat (2008) 111:241–50. doi: 10.1007/s10549-007-9789-z
145. LaPensee EW, Ben-Jonathan N. Novel Roles of Prolactin and Estrogens in Breast Cancer: Resistance to Chemotherapy. Endocr Relat Cancer (2010) 17:R91–107. doi: 10.1677/ERC-09-0253
146. Ramamoorthy P, Sticca R, Wagner TE, Chen WY. In Vitro Studies of a Prolactin Antagonist, hPRL-G129R in Human Breast Cancer Cells. IntJ Oncol (2001) 18:25–32. doi: 10.3892/ijo.18.1.25
147. LaPensee EW, Schwemberger SJ, LaPensee CR, Bahassi EM, Afton S, Ben Jonathan N. Prolactin Confers Resistance Against Cisplatin in Breast Cancer Cells by Activating Glutathione-S-Transferase. Carcinogenesis (2009) 30:1298–304. doi: 10.1093/carcin/bgp120
148. Peirce SK, Chen WY. Human Prolactin and its Antagonist, hPRL-G129R, Regulate Bax and Bcl-2 Gene Expression in Human Breast Cancer Cells and Transgenic Mice. Oncogene (2004) 23:1248–55. doi: 10.1038/sj.onc.1207245
149. Wu AM, Dalvi P, Lu X, Yang M, Riddick DS, Matthews J, et al. Induction of Multidrug Resistance Transporter ABCG2 by Prolactin in Human Breast Cancer Cells. Mol Pharmacol (2013) 83:377–88. doi: 10.1124/mol.112.082362
150. Lemech C, Woodward N, Chan N, Mortimer J, Naumovski L, Nuthalapati S, et al. A First-in-Human, Phase 1, Dose-Escalation Study of ABBV-176, an Antibody-Drug Conjugate Targeting the Prolactin Receptor, in Patients With Advanced Solid Tumors. Invest New Drugs (2020) 38:1815–25. doi: 10.1007/s10637-020-00960-z
151. Anderson MG, Zhang Q, Rodriguez LE, Hecquet CM, Donawho CK, Ansell PJ, et al. ABBV-176, a PRLR Antibody Drug Conjugate With a Potent DNA-Damaging PBD Cytotoxin and Enhanced Activity With PARP Inhibition. BMC Cancer (2021) 21:681. doi: 10.1186/s12885-021-08403-5
152. Andreev J, Thambi N, Perez Bay AE, Delfino F, Martin J, Kelly MP, et al. Bispecific Antibodies and Antibody-Drug Conjugates (ADCs) Bridging HER2 and Prolactin Receptor Improve Efficacy of HER2 ADCs. Mol Cancer Ther (2017) 16:681–93. doi: 10.1158/1535-7163.MCT-16-0658
153. Zhou Y, Zong H, Han L, Xie Y, Jiang H, Gilly J, et al. A Novel Bispecific Antibody Targeting CD3 and Prolactin Receptor (PRLR) Against PRLR-Expression Breast Cancer. J Exp Clin Cancer Res (2020) 39:87. doi: 10.1186/s13046-020-01564-4
154. Wagner KU, Rui H. Jak2/Stat5 Signaling in Mammogenesis, Breast Cancer Initiation and Progression. J Mammary Gland Biol Neoplasia (2008) 13:93–103. doi: 10.1007/s10911-008-9062-z
155. Yamaji D, Na R, Feuermann Y, Pechhold S, Chen W, Robinson GW, et al. Development of Mammary Luminal Progenitor Cells is Controlled by the Transcription Factor STAT5A. Genes Dev (2009) 23:2382–7. doi: 10.1101/gad.1840109
156. Craig JM, Turner TH, Harrell JC, Clevenger CV. Prolactin Drives a Dynamic STAT5A/HDAC6/HMGN2 Cis-Regulatory Landscape Exploitable in ER+ Breast Cancer. Endocrinology (2021) 162:bqab036. doi: 10.1210/endocr/bqab036
157. Peck AR, Witkiewicz AK, Liu C, Stringer GA, Klimowicz AC, Pequignot E, et al. Loss of Nuclear Localized and Tyrosine Phosphorylated Stat5 in Breast Cancer Predicts Poor Clinical Outcome and Increased Risk of Antiestrogen Therapy Failure. J Clin Oncol (2011) 29:2448–58. doi: 10.1200/JCO.2010.30.3552
158. Peck AR, Witkiewicz AK, Liu C, Klimowicz AC, Stringer GA, Pequignot E, et al. Low Levels of Stat5a Protein in Breast Cancer are Associated With Tumor Progression and Unfavorable Clinical Outcomes. Breast Cancer Res (2012) 14:R130. doi: 10.1186/bcr3328
159. Sultan AS, Xie J, LeBaron MJ, Ealley EL, Nevalainen MT, Rui H. Stat5 Promotes Homotypic Adhesion and Inhibits Invasive Characteristics of Human Breast Cancer Cells. Oncogene (2005) 24:746–60. doi: 10.1038/sj.onc.1208203
160. Nouhi Z, Chughtai N, Hartley S, Cocolakis E, Lebrun JJ, Ali S. Defining the Role of Prolactin as an Invasion Suppressor Hormone in Breast Cancer Cells. Cancer Res (2006) 66:1824–32. doi: 10.1158/0008-5472.CAN-05-2292
161. Tang JZ, Zuo ZH, Kong XJ, Steiner M, Yin Z, Perry JK, et al. Signal Transducer and Activator of Transcription (STAT)-5A and STAT5B Differentially Regulate Human Mammary Carcinoma Cell Behavior. Endocrinology (2010) 151:43–55. doi: 10.1210/en.2009-0651
162. Sato T, Tran TH, Peck AR, Girondo MA, Liu C, Goodman CR, et al. Prolactin Suppresses a Progestin-Induced CK5-Positive Cell Population in Luminal Breast Cancer Through Inhibition of Progestin-Driven BCL6 Expression. Oncogene (2014) 33:2215–24. doi: 10.1038/onc.2013.172
163. Tran TH, Utama FE, Lin J, Yang N, Sjolund AB, Ryder A, et al. Prolactin Inhibits BCL6 Expression in Breast Cancer Through a Stat5a-Dependent Mechanism. Cancer Res (2010) 70:1711–21. doi: 10.1158/0008-5472.CAN-09-2314
164. Canbay E, Norman M, Kilic E, Goffin V, Zachary I. Prolactin Stimulates the JAK2 and Focal Adhesion Kinase Pathways in Human Breast Carcinoma T47-D Cells. Biochem J (1997) 324:231–6. doi: 10.1042/bj3240231
165. Acosta JJ, Munoz RM, Gonzalez L, Subtil-Rodriguez A, Dominguez-Caceres MA, Garcia-Martinez JM, et al. Src Mediates Prolactin-Dependent Proliferation of T47D and MCF7 Cells via the Activation of Focal Adhesion Kinase/Erk1/2 and Phosphatidylinositol 3-Kinase Pathways. Mol Endocrinol (2003) 17:2268–82. doi: 10.1210/me.2002-0422
166. Piazza TM, Lu JC, Carver KC, Schuler LA. Src Family Kinases Accelerate Prolactin Receptor Internalization, Modulating Trafficking and Signaling in Breast Cancer Cells. Mol Endocrinol (2009) 23:202–12. doi: 10.1210/me.2008-0341
167. Barcus CE, Keely PJ, Eliceiri KW, Schuler LA. Stiff Collagen Matrices Increase Tumorigenic Prolactin Signaling in Breast Cancer Cells. J Biol Chem (2013) 288:12722–32. doi: 10.1074/jbc.M112.447631
168. Haxholm GW, Nikolajsen LF, Olsen JG, Fredsted J, Larsen FH, Goffin V, et al. Intrinsically Disordered Cytoplasmic Domains of Two Cytokine Receptors Mediate Conserved Interactions With Membranes. Biochem J (2015) 468:495–506. doi: 10.1042/BJ20141243
169. Dhillon AS, Hagan S, Rath O, Kolch W. MAP Kinase Signalling Pathways in Cancer. Oncogene (2007) 26:3279–90. doi: 10.1038/sj.onc.1210421
170. Saini KS, Loi S, de Azambuja E, Metzger-Filho O, Saini ML, Ignatiadis M, et al. Targeting the PI3K/AKT/mTOR and Raf/MEK/ERK Pathways in the Treatment of Breast Cancer. Cancer Treat Rev (2013) 39:935–46. doi: 10.1016/j.ctrv.2013.03.009
171. Simanshu DK, Nissley DV, McCormick F. RAS Proteins and Their Regulators in Human Disease. Cell (2017) 170:17–33. doi: 10.1016/j.cell.2017.06.009
172. Song M, Bode AM, Dong Z, Lee MH. AKT as a Therapeutic Target for Cancer. Cancer Res (2019) 79:1019–31. doi: 10.1158/0008-5472.CAN-18-2738
173. Braicu C, Buse M, Busuioc C, Drula R, Gulei D, Raduly L, et al. A Comprehensive Review on MAPK: A Promising Therapeutic Target in Cancer. Cancers (2019) 11:1618. doi: 10.3390/cancers11101618
174. Gutzman JH, Rugowski DE, Nikolai SE, Schuler LA. Stat5 Activation Inhibits Prolactin-Induced AP-1 Activity: Distinct Prolactin Initiated Signals in Tumorigenesis Dependent on Cell Context. Oncogene (2007) 26:6341–8. doi: 10.1038/sj.onc.1210454
175. Schedin P, Keely PJ. Mammary Gland ECM Remodeling, Stiffness, and Mechanosignaling in Normal Development and Tumor Progression. Cold Spring Harb Perspect Biol (2011) 3:a003228. doi: 10.1101/cshperspect.a003228
176. Keely PJ. Mechanisms by Which the Extracellular Matrix and Integrin Signaling Act to Regulate the Switch Between Tumor Suppression and Tumor Promotion. J Mammary Gland Biol Neoplasia (2011) 16:205–19. doi: 10.1007/s10911-011-9226-0
177. Piersma B, Hayward MK, Weaver VM. Fibrosis and Cancer: A Strained Relationship. Biochim Biophys Acta Rev Cancer (2020) 1873:188356. doi: 10.1016/j.bbcan.2020.188356
178. Conklin MW, Eickhoff JC, Riching KM, Pehlke CA, Eliceiri KW, Provenzano PP, et al. Aligned Collagen is a Prognostic Signature for Survival in Human Breast Carcinoma. Am J Pathol (2011) 178:1221–32. doi: 10.1016/j.ajpath.2010.11.076
179. Barcus CE, Keely PJ, Eliceiri KW, Schuler LA. Prolactin Signaling Through Focal Adhesion Complexes is Amplified by Stiff Extracellular Matrices in Breast Cancer Cells. Oncotarget (2016) 7:48093–106. doi: 10.18632/oncotarget.10137
180. Barcus CE, O'Leary KA, Brockman JL, Rugowski DE, Liu Y, Garcia N, et al. Elevated Collagen-I Augments Tumor Progressive Signals, Intravasation and Metastasis of Prolactin-Induced Estrogen Receptor Alpha Positive Mammary Tumor Cells. Breast Cancer Res (2017) 19:9. doi: 10.1186/s13058-017-0801-1
181. Barcus CE, Holt EC, Keely PJ, Eliceiri KW, Schuler LA. Dense Collagen-I Matrices Enhance Pro-Tumorigenic Estrogen-Prolactin Crosstalk in MCF-7 and T47D Breast Cancer Cells. PLos One (2015) 10:e0116891. doi: 10.1371/journal.pone.0116891
182. Horwitz KB, Sartorius CA. Progestins in Hormone Replacement Therapies Reactivate Cancer Stem Cells in Women With Preexisting Breast Cancers: A Hypothesis. J Clin Endocrinol Metab (2008) 93:3295–8. doi: 10.1210/jc.2008-0938
183. Villanueva H, Grimm S, Dhamne S, Rajapakshe K, Visbal A, Davis CM, et al. The Emerging Roles of Steroid Hormone Receptors in Ductal Carcinoma in Situ (DCIS) of the Breast. J Mammary Gland Biol Neoplasia (2018) 23:237–48. doi: 10.1007/s10911-018-9416-0
184. Guttilla IK, Adams BD, White BA. ERalpha, microRNAs, and the Epithelial-Mesenchymal Transition in Breast Cancer. Trends Endocrinol Metab (2012) 23:73–82. doi: 10.1016/j.tem.2011.12.001
185. Maynadier M, Chambon M, Basile I, Gleizes M, Nirde P, Gary-Bobo M, et al. Estrogens Promote Cell-Cell Adhesion of Normal and Malignant Mammary Cells Through Increased Desmosome Formation. Mol Cell Endocrinol (2012) 364:126–33. doi: 10.1016/j.mce.2012.08.016
186. Simoes BM, Alferez DG, Howell SJ, Clarke RB. The Role of Steroid Hormones in Breast Cancer Stem Cells. Endocr Relat Cancer (2015) 22:T177–86. doi: 10.1530/ERC-15-0350
187. Rowlinson SW, Yoshizato H, Barclay JL, Brooks AJ, Behncken SN, Kerr LM, et al. An Agonist-Induced Conformational Change in the Growth Hormone Receptor Determines the Choice of Signalling Pathway. Nat Cell Biol (2008) 10:740–7. doi: 10.1038/ncb1737
Keywords: prolactin (PRL), breast cancer, mammary cancer, luminal breast cancer, HER2+ breast cancer, STAT 5 transcription factor, triple negative breast cancer
Citation: Schuler LA and O’Leary KA (2022) Prolactin: The Third Hormone in Breast Cancer. Front. Endocrinol. 13:910978. doi: 10.3389/fendo.2022.910978
Received: 04 April 2022; Accepted: 16 May 2022;
Published: 16 June 2022.
Edited by:
Vincent Goffin, INSERM U1151 Institut Necker Enfants Malades, FranceReviewed by:
Carrie S Shemanko, University of Calgary, CanadaCarmen Clapp, National Autonomous University of Mexico, Mexico
Copyright © 2022 Schuler and O’Leary. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Linda A. Schuler, bGluZGEuc2NodWxlckB3aXNjLmVkdQ==