 Xinqian Geng1
Xinqian Geng1 Ying Yang
Ying Yang- 1Department of Endocrinology, The Affiliated Hospital of Yunnan University and the Second People’s Hospital of Yunnan Province, Kunming, China
- 2School of Pharmacy, Guangdong Pharmaceutical University, Guangzhou, China
Diabetes mellitus (DM) and its related complications are among the leading causes of disability and mortality worldwide. Substantial studies have explored epigenetic regulation that is involved in the modifications of DNA and proteins, but RNA modifications in diabetes are still poorly investigated. In recent years, posttranscriptional epigenetic modification of RNA (the so-called ‘epitranscriptome’) has emerged as an interesting field of research. Numerous modifications, mainly N6-methyladenosine (m6A), have been identified in nearly all types of RNAs and have been demonstrated to have an indispensable effect in a variety of human diseases, such as cancer, obesity, and diabetes. Therefore, it is particularly important to understand the molecular basis of RNA modifications, which might provide a new perspective for the pathogenesis of diabetes mellitus and the discovery of new therapeutic targets. In this review, we aim to summarize the recent progress in the epitranscriptomics involved in diabetes and diabetes-related complications. We hope to provide some insights for enriching the understanding of the epitranscriptomic regulatory mechanisms of this disease as well as the development of novel therapeutic targets for future clinical benefit.
Introduction
Diabetes mellitus (DM) is a complex metabolic disorder that is characterized by increased blood glucose concentrations; it has reached epidemic proportions worldwide (1). It is well established that high levels of glucose contribute to higher risks of microvascular and macrovascular diseases, including diabetic cardiomyopathy, retinopathy, nephropathy, neuropathy, and ulcers, which are major causes of disability and mortality in patients with diabetes (2–6). The global diabetes prevalence in adults aged 20-79 was estimated to be 10.5% (536.6 million people) in 2021, and it is estimated to rise to 12.2% (783.2 million) in 2045. Thus, it contributes to increasingly high health care costs (1). Diabetes-related health care expenditure was 966 billion USD in 2021 and is projected to reach 1,054 billion USD by 2045. This would represent an increase of 9.1% compared to that in 2021 (1). Substantial evidence has demonstrated that epigenetic regulatory mechanisms (e.g., histone modifications, DNA methylation and noncoding RNA) play a pivotal role in the occurrence and development of diabetes (7–9). However, the identification of posttranscriptional epigenetic modification of RNA (epitranscriptomic modification) in diabetes and its complications is still in its nascent stage.
Similar to DNA methylation modification, cellular RNA is also modified with diverse chemical modifications, such as N6-methyladenosine (m6A), 5-methylcytidine (m5C), inosine (I), and pseudouridine (Ψ) (10, 11). More than 160 types of chemical modifications have been discovered in RNAs to date (11). Such modifications can influence the metabolism and function of RNAs and have been regarded as key regulators of gene expression (12). The multitude of modifications in RNAs lead to an emerging scientific field of “RNA epigenetics” or “epitranscriptomics”, which could add a new layer of control of genetic information (11, 13–16). It is believed that epitranscriptomics, which are reversible modifications of RNAs, play a crucial role in various disease conditions, such as cancer, obesity, leukemia, neurological disorders and diabetes (17–20). For instance, methyltransferase-like 3 (Mettl3)-mediated m6A modification was reported to be connected to hepatogenous diabetes in mice fed a high-fat diet (HFD) (20).
In consideration of the influence on human health, abundant work has focused on understanding the epigenetic mechanisms involved in the pathophysiology of diabetes mellitus and how these processes are disrupted in the diabetic state. In this review, we provide a brief overview of epitranscriptomics and summarize the recent progresses in the research of epitranscriptomics involved in diabetes and diabetes-related complications, which may provide novel insights for understanding the pathogenesis of diabetes and indicate promising therapeutic targets for this disease.
Brief Overview of RNA Modifications
Many studies have reported that chemical modifications of RNA transcripts are also key regulators of gene expression that participate in RNA splicing, transcript stability, translation, nucleation, and RNA–protein interactions (12, 18, 21–26). Modifications on the 5’ cap and 3’ poly(A) tail of mRNA play crucial roles in mRNA metabolism; their roles have been extensively reviewed elsewhere (4). Thus, in the following sections, we briefly summarize several internal chemical modifications of coding and noncoding RNA (Figure 1). For a detailed discussion on this topic, readers can refer to several excellent reviews (10, 11, 13, 15, 27).
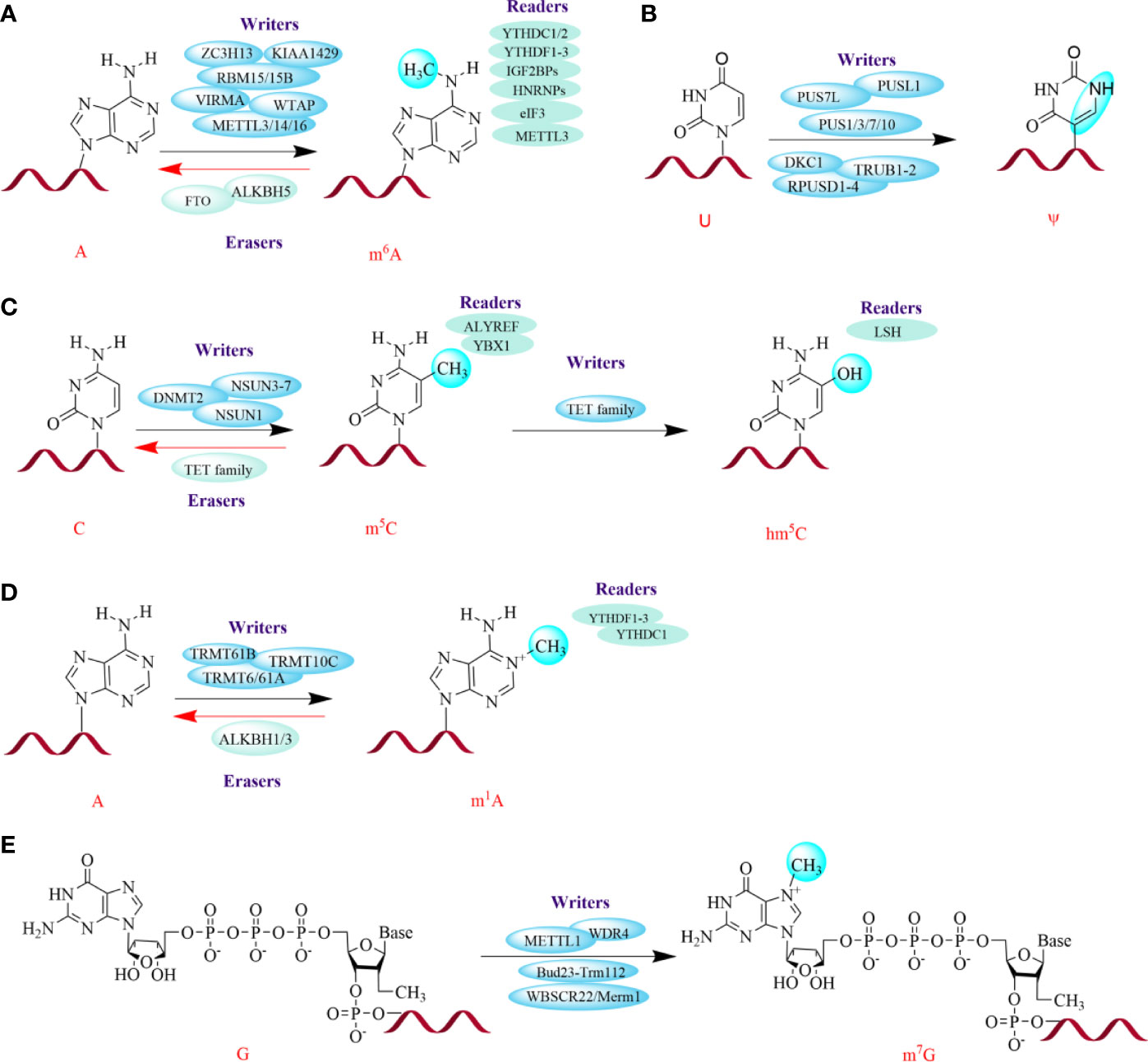
Figure 1 Several major chemical modifications in RNAs. The green spheres on the bases represent modification sites. The red helices represent RNA. The dynamic and reversible modifications are mediated by three groups of proteins: writers (install modifications on specific sites), erasers (remove modifications), and readers (recognize modifications). METTL3/14/16, Methyltransferase-like 3, 14, 16; WTAP, Wilms' tumor 1-associating protein; RBM15/15B, RNA-binding motif protein 15/15B; VIRMA, Vir-like m6A methyltransferase associated; ZC3H13, Zinc finger CCCH domain-containing protein 13; FTO, Fat mass and obesity-associated protein; ALKBH5, ALKB homolog 5; YTHDF1/2/3, YT521-B homology (YTH) protein families including YTH domain family 1, 2, 3; YTHDC1/2, YTH domain containing 1, 2; IGF2BPs, Insulin-like growth factor 2 mRNA-binding proteins; HNRNPs, Heterogeneous nuclear ribonucleoproteins; eIF3, Eukaryotic translation initiation factor; TRMT6/61A, tRNA methyltransferase 6, 61A; PUSs, Pseudouridine synthases; PUS7L, PUS7-like; PUSL1, PUS-like 1; DKC1, Dyskerin PUS1; RPUSD1/2/3/4, RNA PSU domain-covering 1, 2, 3, 4; TRUB1/2, TruB PSU class member 1, 2; NSUN3-7, NOL1/NOP2/SUN domain (NSUN) family of proteins 3, 4, 5, 6, 7; DNMT2, DNA methyltransferase (DNMT) homolog-2; TET family, Ten-eleven translocation family enzymes; ALYREF, Aly/REF export factor; YBX1, Y box binding protein 1; LSH, lymphoid specific helicase; WBSCR22, Williams-Beuren syndrome chromosomal region 22 protein, also called BUD23; Merm1, Metastasis-related methyltransferase 1; WDR4, WD 159 repeat domain 4.
N6-Methyladenosine (m6A)
m6A is the most abundant and prevalent internal mRNA modification in eukaryotic mRNA and is a critical posttranscriptional mRNA regulator (26, 28, 29). m6A is a stable and conserved chemical derivative of adenosine that accounts for more than 60% of all RNA modifications (26). It was initially identified in the 1970s in mRNA, and later reported to exist in non-coding RNAs, including lncRNA, rRNA, and circRNAs (20–23). The dynamic and reversible m6A patterns are mediated by three groups of proteins: writers, erasers, and readers (26, 30). m6A methyltransferase complexes (MTCs), termed “writers”, can selectively install m6A-modified sites upstream of information processing; these writers mainly include methyltransferase-like 3/14 (METTL3/14) and Wilms’ tumor 1-associating protein (WTAP) and KIAA1429 (31–33). Additionally, there are a handful of regulatory factors that contribute to the activity and specificity of the catalytic complex; these include METTL16, RNA-binding motif protein 15/15B (RBM15/15B), vir-like m6A methyltransferase associated (VIRMA), and zinc finger CCCH domain-containing protein 13 (ZC3H13) (34–37). The demethylases, also known as “erasers”, are mainly composed of fat mass and obesity-associated protein (FTO) and ALKB homolog 5 (ALKBH5) (38, 39). However, the role for FTO as an m6A eraser is controversial since several studies have showed that FTO preferentially demethylates N6,2’-O-dimethyladenosine (m6Am) rather than m6A (40–43). Further studies showed that FTO might target at nuclear RNA over mRNA; this was demonstrated by the phenomena that more RNA methylation accumulated in snRNAs and small nucleolar RNAs (snoRNAs) in Fto - knockout cells compared with mRNA (40, 41). Moreover, m6A can be recognized by m6A-binding proteins, also called “readers”; this group principally consists of YT521-B homology (YTH) protein families, including YTH domain family 1/2/3 (YTHDF1/2/3) and YTH domain containing 1/2 (YTHDC1/2), insulin-like growth Factor 2 mRNA-binding proteins (IGF2BPs), heterogeneous nuclear ribonucleoproteins (HNRNPs), eukaryotic translation initiation factor (eIF3), and METTL3 (44–50).
N1-Methyladenosine (m1A)
m1A methylation occurs at the N1 position of adenosine and accumulates in tRNA, rRNA and mRNA (13, 44, 51). In tRNA and rRNA, m1A maintains the tertiary structure by affecting translation (52, 53). In mRNA, m1A is primarily located in highly structured or GC-rich regions of 5’ untranslated regions (UTRs), although it is also found in the coding sequence (CDS) and 3’UTR (54). Similar to mRNA writers, tRNA methyltransferase 6/61A (TRMT6/61A), TRMT61B, and TRMT10C could contribute to m1A methylation in tRNA. Additionally, these tRNA methyltransferases can also regulate m1A methylation in mRNAs (55). Interestingly, YTHDF1-3 and YTHDC1 can also act as “readers” in m1A modifications by binding to m1A-bearing RNA (56). Subsequently, this modification can be demethylated by ALKBH1 and ALKBH3, which function as erasers (54, 57).
5-Methylcytosine (m5C)
Methylation at the 5 position of cytosine (m5C) was also first discovered to be gathered at the UTRs of mRNA, especially in GC-rich regions, which is similar to that of m1A (58–60). Subsequently, the NOL1/NOP2/SUN domain (NSUN) family of proteins (NSUN1, NSUN3/4/5/6/7) and DNA methyltransferase (DNMT) homolog-2 (DNMT2) were reported to serve as “writers” in tRNAs and rRNAs, but only NSUN2 can fix m5C on mRNA (27, 61–63). Later, a specific mRNA m5C-binding protein, Aly/REF export factor (ALYREF), was reported as a “reader” of m5C (59). Recently, Chen et al. found that YBX1 acts as another m5C reader that can maintain the stability of target mRNAs in bladder cancer (64). However, little is known about the “eraser” protein(s) for this modification.
5-Hydroxymethylcytosine (hm5C)
hm5C has been identified as a transformed form of m5C (13, 27). Similar to that in DNA, m5C in RNA can also be catalyzed into hm5C by ten-eleven translocation (TET) family enzymes (65, 66). Furthermore, methylated RNA immunoprecipitation sequencing (called MeRIP-seq) in Drosophila S2 cells illustrated that Tet-family enzymes tend to act on coding regions, thus promoting mRNA translation (67). In tRNAs, several studies have revealed that ALKBH1 is also involved in the biogenesis of hm5C and 5-formyl-2′-O-methylcytidine (f5Cm) (68, 69). Interestingly, a chromatin remodeling factor, lymphoid specific helicase (LSH), is considered a reader of hm5C in cancer. It has been reported that LSH can interact with TET2. This increases the expression of the latter and favors genome stability by silencing satellite expression by improving the levels of hm5C in metastasis (70).
N7-Methylguanosine (m7G)
m7G is the most ubiquitous RNA cap modification on the 5’ end of eukaryotic mRNAs and exists in mRNA, tRNA and rRNA (55, 71, 72). In humans, the 1639 site of 18S rRNA is m7G-modified by the Williams-Beuren syndrome chromosomal region 22 protein (WBSCR22, also called BUD23)/metastasis-related methyltransferase 1 (Merm1), whereas in yeast, the Bud23-Trm112 heterodimer is required for N7-methylation of G1575 in the small ribosomal subunit rRNA (73, 74). Similar to m7G modification in rRNA, m7G tRNA modification is catalyzed by METTL1 and WD repeat domain 4 (WDR4) in humans and the heterodimeric complex of Trm8p/Trm82p in yeast (75–77). Moreover, METTL1/WDR4 also act as “writers” for m7G modification on mRNA (72).
Pseudouridine (Ψ)
Ψ is the isomerization of uridine. It is the most abundant modification in cellular RNA and appears in various RNAs, including small nucleolar RNA, snRNA, and mRNA, especially in rRNA and tRNA (13, 78). This modification can be dynamically regulated by “writers”, pseudouridine synthases (PUSs), and 13 synthases have been discovered to date, including PUS7-like (PUS7L), PUS-like 1 (PUSL1), dyskerin PUS1 (DKC1), RNA PSU domain-covering 1/2/3/4 (RPUSD1/2/3/4), TruB PSU class member 1/2 (TRUB1/2), and PUS1/3/7/10 (79, 80). Notably, although Ψ can improve translation, it can negatively affect protein expression (27, 81).
RNA Editing
Adenosine-to-inosine RNA editing (A-to-I editing), also termed I, is a type of programmed alteration that has broad physiologic significance (27, 82). It has been demonstrated that A-to-I editing can be regulated by a family of adenosine deaminases acting on RNA (ADARs) (82–84). Although three ADAR genes have been found in mammals, only ADAR1 and ADAR2 possess deaminase activity in regions of RNA with double-stranded character (82). Since A-to-I editing can fasten pairs of nucleotides, this modification can affect the secondary structure of mRNA in metazoans and further influence RNA structure stability (85).
Cytidine-to-uridine RNA editing (C-to-U editing), namely, U, is the other kind of canonical but rare RNA editing in humans (86, 87). Previous studies have reported that U prefers to be located in 3’ UTRs and is catalyzed by apolipoprotein B-editing enzyme catalytic polypeptide-1 (APOBEC1) (88, 89). Subsequently, in a study of small intestine and liver mRNA in mice, the C-to-U editing of RNA was demonstrated to be associated with the protein level, although the exact relationship between this modification and the expression of transcripts is unknown (86).
Methods for Detecting RNA Modifications
Recent years, the development of new technologies dramatically promoted the understanding of functions of RNA modifications and the discovery of new modifications. Meanwhile, the increasing interest in epitranscriptomics has also prompted the progresses in developing new detecting tools. It is recognized that total level of m6A modification in RNA can be detected by liquid chromatography-tandem mass spectrometry (LC-MS/MS) and colorimetric method (90, 91). Many high-throughput sequencing methods have been developed to detected m6A modification based on different mechanisms, including antibody-dependent single-nucleotide resolution sequencing (PA-m6A-seq, m6A-CLIP and miCLIP), chemical labeling sequencing (m6A-label-seq and m6A-SEAL), MeRIP-m6A-seq (the most widely used method so far), endoribonuclease-based sequencing (m6A-REF-seq and MASTER-seq), and other methods (such as m6A-LAIC-seq, DART-seq and Nanopore-seq), endoribonuclease-based sequencing (m6A-REF-seq and MASTER-seq), and other methods (such as m6A-LAIC-seq, DART-seq and Nanopore-seq) (11, 92–96). Recently, an antibody-independent single-nucleotide-resolution method named m6A-SAC-seq has been developed (97). Similarly, m1A modification can be detected by LC-MS method as well as high-throughput sequencing strategies (including m1A-ID-seq, m1A-seq, m1A-IP-seq, m1A-seq-TGIRT, m1A-MAP, and m1A-quant-seq) (11, 98, 99). The level of m5C methylation can be detected by LC-MS, Bisulfite-seq and high-throughput sequencing methods (including m5C-RIP-seq, RNA-BisSeq, miCLIP-seq, TAWO-seq, Aza-IP-seq, and Nanopore-seq) (11, 99–102). LC-MS/MS method and MeRIP-seq are also available for the detection of hm5C and m7G modifications (11, 72, 103). Besides, other sequencing strategies, such as m7G-seq and miCLIP-seq, are used for quantitative detection of m7G modification (72, 104). As for Ψ site, it can not only be tested by Ψ-Seq, Pseudo-Seq, PSI-Seq, Nanopore-seq and CeU-Seq, but also be predicted by computational methods such as iRNA-PseU and XG-PseU (105, 106).
Although many detecting technologies have been developed, cautions should still be taken when detecting chemical modifications on RNAs by specific methods considering their limited sensitivity and accuracy. Moreover, to better understand the multiple detecting techniques of RNA modifications, readers can refer to several professional studies (11, 72, 98, 99, 106).
The Role of Epitranscriptomics in Diabetes
Normal blood glucose levels are achieved by a feedback loop between pancreatic β cells producing insulin and insulin-sensitive tissues (primarily liver, adipose, and muscle) (50). DM is characterized by chronic hyperglycemia resulting from impaired insulin action (i.e., insulin resistance (IR) and/or secretion) (50). It is well established that both genetics and the environment play major roles in the development of diabetes (107). Among environmental factors, reversible RNA modifications have recently raised increasing interest from researchers and have been demonstrated to affect glucose homeostasis (20, 108, 109). As one of the most widespread RNA modifications, m6A was first reported to stimulate glucose oxidation in rat adipocytes in 1982; this finding implies that m6A could affect glucose metabolism in fat cells (109). Later, it was discovered that both the m6A level and METTL3 expression were increased in the livers of patients with type 2 diabetes mellitus (T2DM), and their expression was positively associated with IR by promoting the expression of fatty acid synthase (Fasn) (110). Consistently, hepatocyte-specific knockout of Mettl3 mice feeding with HFD ameliorated insulin sensitivity and reduced fatty acid synthesis by down-regulating the expressions of fatty acid synthesis and oxidation genes (such as Fasn, Sirt1, Ehhadh, and Foxo1) in a m6A-mediated way (110). Moreover, Li et al. also revealed that the overexpression of Mettl3 aggravated IR in HFD-fed mice. RNA-seq and miCLIP-seq data showed that some candidate genes (such as Lpin1, Lpin2, G6pc, Pck1, and Ppara) were down-regulated in Mettl3Ctrl mice feeding with HFD (20). Significantly, lpin1 has been recognized as an important factor in lipid metabolism and IR (111, 112). It is demonstrated that silencing of lipin-1 in C2C12 myotubes resulted in elevated ceramide accumulation, but attenuated insulin-induced Akt phosphorylation and glucose uptake, thereby leading to IR (113). Correspondingly, Li et al. further demonstrated that knockdown of Mettl3 could stabilize Lpin1 mRNA by regulating m6A levels, thus improving IR. These findings reveal a key role of Mettl3-mediated m6A modification in hepatogenous diabetes (20, 113). IGF2BP2, an m6A reader, has been identified as a T2DM-associated gene. Single nucleotide polymorphisms (SNPs) in the human IGF2 mRNA binding protein 2 (IMP2/IGF2BP2) are associated with increased risk of T2DM in different populations, such as Chinese and Indians (114, 115). Further study demonstrated that variants in IMP2 locus exerted their effects in T2DM susceptibility predominantly by impairing insulin secretion rather than reducing insulin sensitivity (116–118). Moreover, a recent study showed that IMP2/IGF2BP2 working together with the IGF2-AKT-GSK3β-PDX1 pathway, could promote β-cell proliferation and enhance insulin secretion (119).
As the pivotal pathogenic mechanism of diabetes, β cell function has been associated with RNA modifications. It was shown that the deletion of Mettl3 in islet β cells may diminish the modification of m6A and suppress the expression of insulin secretion-related genes, thereby inducing β cell failure and hyperglycemia (120). Notably, a recent study of islet cells from patients with T2DM further suggested that m6A was significantly reduced in β-cells and broadly located in insulin secretion-related genes. Moreover, further analyses revealed that a lower level of m6A resulting from knockdown of Mettl3 or Mettl14 could impair glucose-stimulated insulin secretion (GSIS) by inhibiting the insulin/IGF1–AKT–PDX1 pathway (121). For METTL14, specific knockout of METTL14 in β-cells resulted in reduced insulin secretion and induced glucose intolerance by activating the IRE1α/sXBP-1 pathway (122). Collectively, it is obvious that METTL3/14 is vital for islet β-cell biology and glucose homeostasis. Thus, METTL3/14 might be potential therapeutic targets for the treatment of β-cell failure in diabetes.
Fan et al. found that a lower level of m6A was related to increased FTO expression instead of ALKBH5 expression in T2DM patients (108). However, accumulating evidence has demonstrated that both FTO and ALKBH5, two of the most widely known m6A demethylases, were important for regulating m6A levels despite their different mechanisms of demethylation (30, 39, 50, 123, 124). Bornaque et al. reported increased level of Alkbh5 mRNA and decreased m6A methylation under high glucose condition in pancreatic β cell line (Min6 cells) (50). It is recognized that obesity is the one of the risk factors for the development of IR resulting in T2DM (125). FTO has been reported as an obesity-risk gene. Silencing of Fto could lead to a decrease in adipose tissue and body mass index, which protected from obesity and metabolic syndrome in mice (125, 126). Mizuno et al. showed that glucose and insulin could keep glucose homeostasis by suppressing the expressions of hepatic Fto and gluconeogenic genes, such as phosphoenolpyruvate carboxynase (Pepck) and glucose-6-phosphatase (G6pase) in liver (127). Furthermore, FTO participates in glucose metabolism in both m6A-dependent and nonm6A-dependent manners (26). On the one hand, FTO modulates glucose metabolism by affecting forkhead box O1 (FOXO1), activating transcription factor 4 (ATF4), and glucose-6-phosphatase catalytic subunit (G6PC) expression in a m6A-dependent manner in response to external stimulation or disease conditions (128, 129). On the other hand, FTO affects glucose metabolism by regulating signal transducers of adipocyte and transcription activator 3 (STAT3), cAMP responsive element binding protein 1 (CREB1), CCAAT/enhancer‐binding protein‐beta (C/EBP‐β), and G6PC expression by a nonm6A-dependent pathway under normal conditions (130, 131). Additionally, in pancreatic β cells, overexpression of Fto accelerated ROS production and NF-κB activation and consequently inhibited insulin secretion (132) (Figure 2). Interestingly, variants in the FTO gene, e.g., rs9939609 and rs1421085, were also reported to be associated with IR and obesity (133, 134). However, some variants associated with BMI (such as rs9930506) in the FTO locus are correlated with increased IRX3 expression rather than FTO expression (135).
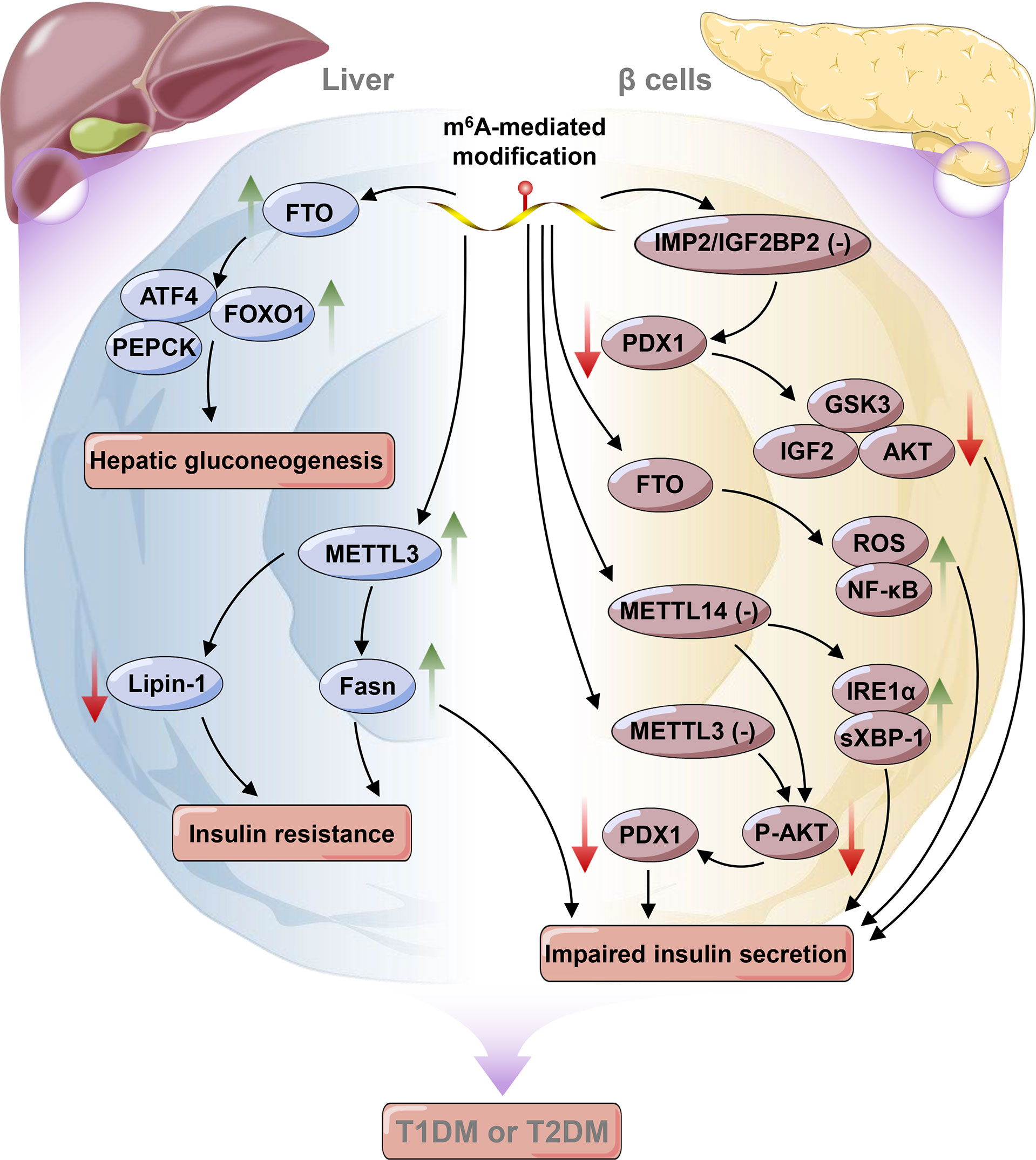
Figure 2 Epitranscriptomic regulatory mechanisms in liver and pancreatic β cells in diabetes mellitus. The green arrow indicates an increase in the change, whereas the red arrow indicates a decrease in the change. METTL3, Methyltransferase-like 3; FTO, Fat mass and obesity-associated protein; PEPCK, Phosphoenolpyruvate arboxynase; FOXO1, Forkhead box O1; ATF4, Activating transcription factor 4; Fasn, Fatty acid synthase; PEPCK, Phosphoenolpyruvate carboxynase; IGF2, Insulin-like growth factor 2; IMP2/IGF2BP2, IGF2 mRNA binding protein 2; PDX1, Pancreatic duodenal homeobox 1; GSK3, Glycogen synthase kinase-3; ROS, Reactive oxygen species; IRE1α, Inositol-requiring enzyme 1 alpha; XBP-1, X-box binding protein spliced.
Among all RNAs, tRNAs (both cytosolic and mitochondrial (mt)-tRNAs) experience the most extensive posttranscriptional modifications, with more than 80 chemical modifications on tRNAs manipulated by specific enzymes (136, 137). It has been demonstrated that disorders in tRNA biology are involved in the pathogenesis of many disease conditions, such as cancer and metabolic diseases (137–139). Recently, the crosstalk between abnormal tRNA modifications and pancreatic β-cell function and diabetes has attracted much attention (Figure 3). For example, it has been demonstrated that the knockdown of CDK5 regulatory subunit associated protein 1-like 1 (CDKAL1) can reduce CDKAL1-mediated ms2t6A37 tRNALys (UUU) modification and damage GSIS. Thus, it can lead to impaired β-cell function and increased risk for T2DM (140–142). Two kinds of mitochondrial DNA mutations have been discovered to cause maternally inherited diabetes and deafness (MIDD) and are associated with the tRNA epitranscriptome. These mutations are the heterogeneous m.3243A>G mutation and the homoplasmic m.14692A>G mutation. The m.3243A>G mutation can lower mt-tRNALeu (UUR) 5-taurinomethyluridine modification at position 34 and result in impaired energy synthesis and insulin secretion (138, 143). The m.14692A>G mutation causes the loss of Ψ modification in position 55 (Ψ55) of mt-tRNAGlu. This leads to the degradation of tRNAGlu, subsequently impairing mitochondrial function (144). tRNA methyltransferase 10 homolog A (TRMT10A), a tRNA modification enzyme, can methylate guanine at position 9 (m1G9) in several tRNAs, as previously identified (137, 145). Mutations in TRMT10A are recognized as contributors to young-onset diabetes and microcephaly (146–148). A study conducted in clonal human β-cells revealed that the silencing of TRMT10A could reduce m1G9 modification in TRMT10A substrates, including tRNAGln (UUG/CUG), thus resulting in pancreatic β-cell apoptosis (149, 150). Mutations in TRIT1 encoding tRNA isopentenyltransferase 1 cause a reduction in i6A37 modifications in mt-tRNASer (UCN) and cytoplasmic tRNASer (UGA) and further impair the mitochondrial respiratory chain (138, 151). Consequently, a previous study reported that the homozygous c.968G>A mutation in TRIT1 could result in encephalopathy, epilepsy, and diabetes (138, 151, 152). These results highlight the vital importance of the tRNA epitranscriptome in β-cell function. These findings may uncover a novel perspective of β-cell death and provide new therapeutic targets for diabetes.
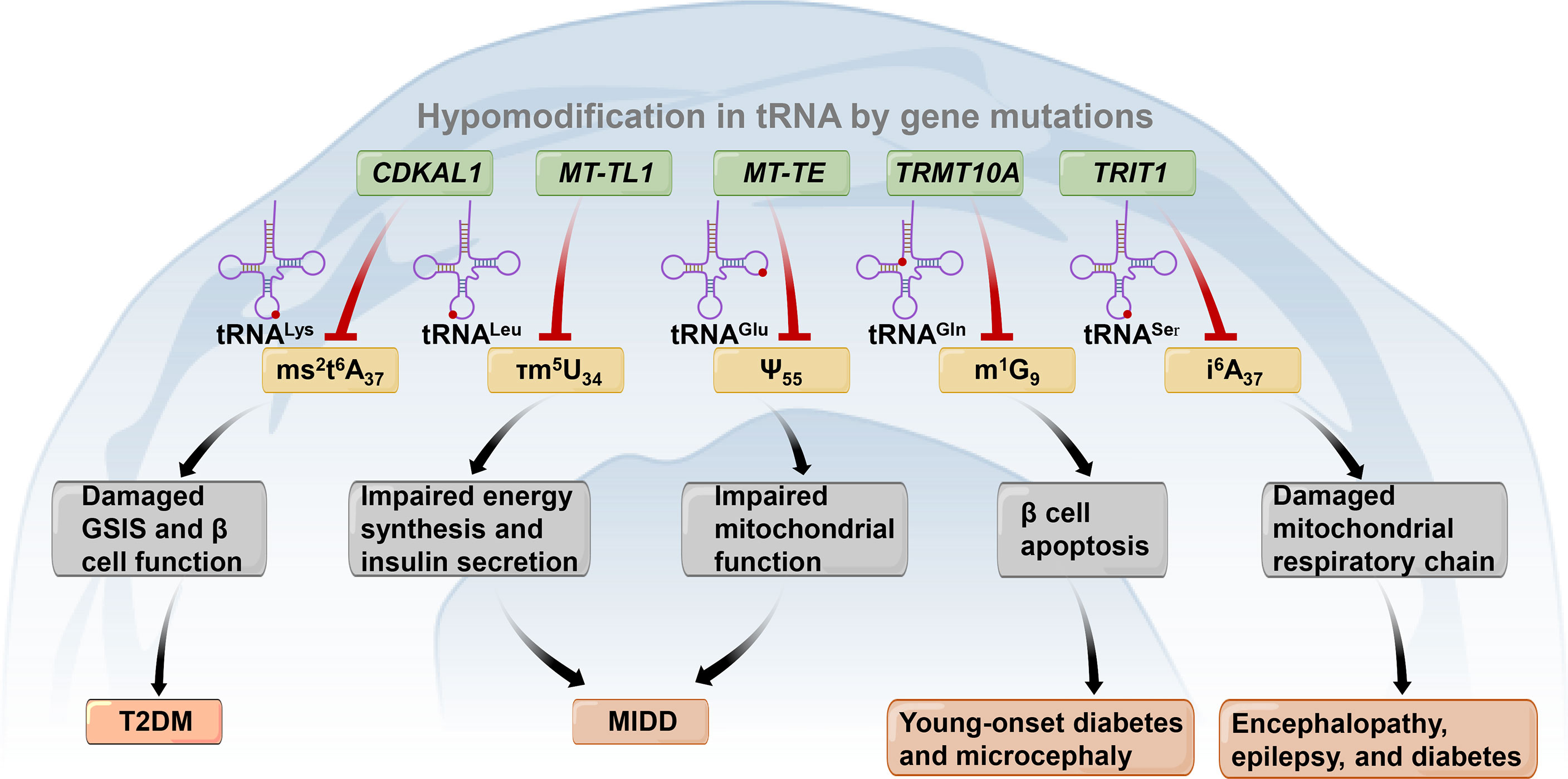
Figure 3 Gene mutations (shown in the blue box) lead to tRNA hypomodifications (shown in the yellow box) associated with diabetes and other metabolic diseases. CDKAL1, CDK5 regulatory subunit associated protein 1-like 1; TRMT10A, tRNA methyltransferase 10 homolog A; TRIT1, tRNA isopentenyltransferase 1; ms2t6A37, 2-methylthio-N6-threonylcarbamoyladenosine at adenosine at position 37 of tRNALys; τm5U34, 5-taurinomethyluridine at uridine at position 34 of tRNALeu; Ψ55, Pseudouridine modification at position 55 of tRNAGlu; m1G9, N1-methylguanine at guanosine at position 9 of tRNAGln. GSIS, Glucose-stimulated insulin secretion; T2DM, Type 2 diabetes mellitus; MIDD, Maternally inherited diabetes and deafness. .
Moreover, in consideration of the crucial relationships between mitochondrial dysfunction, IR and T2DM (153–155), it is also interesting to look at the intersection of the mitochondrion and epitranscriptome without genetic mutation in T2DM. Recently, METTL8, another RNA methyltransferase, was found to promote the 3-methyl-cytidine (m3C) methylation of mt-tRNASer/Thr (UCN) at position C32 (156). Schöller et al. further offered proof that a reduction in METTL8-dependent m3C32 methylation modifications might inhibit the activities of the components of complex I (mainly ND6 and ND1) and therefore influence the activity of the mitochondrial respiratory chain (157). Moreover, m1A modifications on mRNA lead to translational repression due to its destructive effect on Watson-Crick base pairing. This modification has been reported be catalyzed by TRMT61B in the cytosol and TRMT10C in the mitochondrion (158, 159). It is interesting that TRMT10C could facilitate m1A methylation in mitochondrial ND5 mRNA, albeit the latter exhibited tissue-specific expression (159). These results suggest that posttranscriptional modifications of RNA in mitochondria play a key role in T2DM and may in part explain mitochondrial dysfunction in this pathological condition. However, the identification of the mitochondrial epitranscriptome in diabetes is still in its infancy, and further studies are warranted.
The Role of Epitranscriptomics in Diabetic Cardiomyopathy
Diabetic cardiomyopathy (DCM) is a severe complication of diabetes, which is the major cause of mortality in patients with diabetes (160). Over 70% of individuals with diabetes will develop some form of heart disease, such as acute coronary syndrome, during their lifespan (161). It is estimated that 12% of patients with diabetes eventually develop severe heart failure (HF) and often die due to DCM (162, 163). Recent studies demonstrated that diabetes could increase the risk of HF 2-3 times and increase the death rate of HF by 2.5 times in individuals with diabetes compared with control individuals (164). DCM is characterized by myocardium fibrosis, cardiac remodeling and cardiac dysfunction (e.g., contractile dysfunction and reduced ejection fraction) (165). Numerous studies have indicated that the pathogenesis of DCM is complex and multifactorial. It includes the following processes: (1) the overproduction of advanced glycation end-products (AGEs), resulting in extracellular matrix (ECM) accumulation and cardiac fibrosis; (2) excess oxidative stress activating the NF-κB pathway, subsequently leading to myocardial fibrosis; (3) activation of various inflammatory cytokines, activating the proapoptotic signaling pathway and damaging cardiac function; (4) suppression of the insulin signaling pathway, promoting autophagy and apoptosis in myocardial cells; and (5) disturbances in calcium handling, contributing to contractile dysfunction (166, 167).
Previous studies have shown that epigenetic modifications, such as DNA methylation modification, histone modifications, and posttranscriptional RNA regulation (e.g., microRNAs and lncRNAs), play crucial roles in the complexity of the pathogenetic mechanisms underlying cardiovascular disease (CVD) (168, 169). In particular, it has been reported that m6A modification extensively participates in various CVDs, such as atherosclerosis (AS), cardiac hypertrophy and HF (12, 30). For instance, Mathiyalagan et al. found decreased FTO expression, cardiomyocyte contractile dysfunction and increased m6A RNA modification in failed hearts (170). Overexpression of Fto lowered m6A levels and improved cardiac contractile function in ischemia-induced HF. It also promoted angiogenesis and inhibited fibrosis in mice with myocardial infarction (MI) (170). In both in vivo and in vitro studies, increased METTL3 led to m6A RNA hypermethylation. It thus enhanced nucleotide-binding domain (NBD) and leucine-rich repeat (LRR) pyrin-domain containing protein 1 (NLRP1) expression and suppressed Kruppel-like Factor 4 (KLF4) expression in a YTHDF1- and YTHDF2-regulated way, respectively (171). These factors could affect atherogenic inflammatory cascades, such as NF-κB p65 Ser536 phosphorylation in the vascular endothelium, which ultimately impacts the initiation of atherosclerosis (171). Similarly, Xu et al. discovered that YTHDF2 could hinder cardiac hypertrophy by identifying the m6A sites on Myh7 (beta-myosin heavy chain, a marker of cardiac hypertrophy) mRNA and accelerating its degradation (172). Moreover, RNA editing, such as A-to-I editing, has been reported to modulate vascular contraction, arterial remodeling and diastolic blood pressure by editing the actin crosslinking protein filamin A (FLNA) mRNA (173). Nevertheless, exploration of the influence of RNA modifications on the development and progression of DCM is at an early age.
In a study conducted in mice with DCM, researchers identified 984 differentially m6A-modified transcripts (including Mef2a, Bcl2l2, Slc25a33, Cd36, and Klf15), which were closely related to myocardial fibrosis, cardiac hypertrophy, and energy metabolism in the myocardium (174). Interestingly, the AGEs-RAGE pathway has also been demonstrated to participate in DCM pathogenesis by Kyoto Encyclopedia of Genes and Genomes (KEGG) Pathway analysis and protein interaction network analysis of differentially methylated-RNAs in DCM samples. The level of total m6A was higher in DCM patients than in normal control individuals. Although several writers and erasers (such as METTL3, METTL14, and ALKBH5) of m6A modification were detected, the FTO levels were decreased in DCM. This may contribute to cardiac dysfunction by promoting cardiac fibrosis and cardiomyocyte hypertrophy (174). Thus, m6A-dependent epitranscriptomic modifications may be a promising therapeutic approach for DCM.
Pyroptosis is a type of genetically encoded proinflammatory cell death that is different from classic apoptosis. It has been recognized as an essential factor in the pathogenesis of cardiomyopathy and diabetes, especially in DCM (175, 176). For example, a high level of glucose could induce cardiac cell injury and pyroptosis by activating the NLRP3 inflammasome, which is a key regulator of pyroptosis (177). It is worth noting that not only hyperglycemia but also insulin resistance and hyperinsulinemia can lead to the overproduction of ROS and pyroptosis in DCM (6, 176, 178). Xie et al. discovered that chemerin, G-protein-coupled chemokine-like receptor 1 (CMKLR1) and NLRP3 were overexpressed in rats with DCM. They also found that the chemerin/CMKLR1 pathway could activate inflammation and induce pyroptosis in an NLRP3 inflammasome-mediated way, thereby resulting in DCM (179). Accumulating evidence indicates that changes in m6A levels can dynamically influence multiple biological processes, such as cell apoptosis, cell proliferation and the inflammatory response (180, 181). Indeed, a recent study found a reduction in METTL14 levels and an increase in the levels of pyroptosis-related markers, including caspase-1, the N-terminus of GSDMD (GSDMD-N), and NLRP3, in both rats with DCM and high glucose (HG)-treated cardiomyocytes (182). Further investigation revealed that METTL14 levels were negatively correlated with terminal differentiation-induced noncoding RNA (TINCR) levels and that the former could downregulate TINCR expression by enhancing m6A methylated sites in TINCR in a YTHDF2-dependent manner. Further study revealed that METTL14 could suppress the expression of lncRNA TINCR and NLRP3, thereby inhibiting pyroptosis and DCM (182). Collectively, these results provide a new perspective on the epigenetic mechanism of pyroptosis in DCM pathologies and suggest that the METTL14-TINCR-NLRP3 axis might be a potential therapeutic target for DCM (182).
Taken together, studies of RNA modifications in diabetic cardiomyopathy are currently lacking. Thus, as new technologies and methods are being developed, more studies on the alterations in levels of epitranscriptomic modifications (e.g., m6A and m5C, and their regulatory enzymes) in DCM are needed. These investigations could offer novel insights into the physiopathologic mechanisms of DCM and may provide therapeutic targets for this disease given its reversible property.
The Role of Epitranscriptomics in Diabetic Nephropathy
Diabetic nephropathy (DN), namely, diabetic kidney disease (DKD), is known to be a prevalent microvascular complication of diabetes mellitus (7, 90). It was estimated that more than 40% of individuals with T1DM or T2DM will suffer from DN in their lifespan, Moreover, half of these patients could progress to end-stage renal disease (ESRD), which would significantly increase the mortality rate of patients with diabetes (5, 183, 184). DN is characterized by glomerulosclerosis, epithelial-mesenchymal transition (EMT) of tubular cells, hypertrophy and proliferation of mesangial cells, apoptosis of podocytes, renal vascular disease and so on (185, 186). The pathogenesis of DKD is complex and involves genetic and environmental factors. In recent years, epigenetic regulation, especially RNA modifications, has played a key role in the initiation and progression of DN through diverse mechanisms (7).
For example, m6A and ψ were thought to be linked to DN several years ago (187, 188). In a more recent prospective cohort study, Niewczas et al. reported that the level of ψ in serum from patients with diabetes with renal dysfunction was related to damage to renal function and the risk of ESRD (189). Further cross-sectional studies were performed in four groups and revealed increases in m6A/C and pseU/U in the DN group. However, the level of I/C was dynamic among the groups. No significant difference in the value of 5-mdC/C and 5-mC/C was observed between the DN group and normal control group (90). Collectively, these candidate metabolites associated with epitranscriptomics may serve as potential biomarkers for the clinical diagnosis of DN and help explain the pathogenesis of DKD. In recent years, epigenetic research has further evaluated DN at the molecular level (190–193).
Klotho, an antiaging gene, has been identified to exert a protective role in DN by alleviating renal tubular and glomerular injury (194, 195). Despite epigenetic modifications, such as DNA methylation and noncoding RNA, METTL14-mediated hypermethylation modification exists in α-klotho mRNA and suppresses its expression (190). Li et al. found that the level of METTL14 was positively associated with the levels of ROS, IL-6 and TNF-α and apoptosis in human renal glomerular endothelial cells. Moreover, METTL14 could trigger glomerular endothelial cell damage and the inflammatory response in diabetic mice by inhibiting α-klotho expression in a m6A-mediated pathway (190).
Podocytes are a critical cell type in the glomerulus that play an essential role in the progression of DKD (196). Elevated expression of METTL14 was observed both in renal biopsy samples derived from patients with DN and in cultured human adriamycin (ADR)- or AGE-treated podocytes (191). Furthermore, specific Mettl14 deletion in podocytes in vitro could upregulate Sirt1 expression in a m6A-dependent manner. Thus, it could promote autophagy, ameliorate apoptosis, and ultimately improve podocyte injury and glomerular dysfunction (191). Similarly, as another writer enzyme of m6A, METTL3 was also shown to be a negative factor in the pathophysiology of podocytes (192). In mouse models of T1DM and T2DM, the level of m6A modification in the kidney was markedly increased. Moreover, in renal biopsies from patients with DKD, the expression of METTL3 in podocytes was upregulated. Further study identified that METTL3-modulated m6A modification of tissue inhibitor of metalloproteinases 2 (TIMP2) mRNA could promote TIMP2 expression through the IGF2BP2/Notch3/4 signaling pathway. This could aggravate the inflammatory response, podocyte damage and apoptosis, leading to diabetic nephropathy (192). Considering that posttranscriptional regulation has been considered a fundamental mechanism of podocyte injury in the development and progression of DN, these chemical modifications of RNAs and their related enzymes may be therapeutic targets for this disease. Indeed, a recent study demonstrated that the total flavones of Abelmoschus manihot (TFA), which is a kind of Chinese patented medicine extracted from Abelmoschus manihot, could improve injury and pyroptosis in HG-treated podocytes. Mechanistically, Liu et al. further demonstrated that TFA exerted this protective effect by activating the PTEN/PI3K/Akt pathway and NLRP3 inflammasome, which are modulated by m6A modifications in a METTL3-mediated manner (197).
As mentioned above, EMT of renal tubular cells is one of the classical pathological mechanisms of DKD. It was reported that the overexpression of METTL14 could suppress EMT in HG-treated human renal proximal tubular cells (HK2) by increasing PTEN expression but inhibiting histone deacetylase 5 (HDAC5) and TGF-β1 expression by regulating the PI3K/Akt pathway (193). These results indicate that inhibition of HDAC could alleviate some manifestations of DKD, such as EMT, fibrosis, and albuminuria (198, 199).
The Role of Epitranscriptomics in Diabetic Retinopathy
Diabetic retinopathy (DR) is a well-known microvascular complication of diabetes and a main reason for visual impairment and blindness in patients with diabetes (200, 201). The pathological mechanisms are complex and include retinal microvascular remodeling, vascular endothelial dysfunction, progressive fibrosis, pericyte dysfunction, neurodegeneration and so on (201, 202). Hyperglycemia-induced processes, such as inflammation, oxidative stress, activation of protein kinase C (PKC) and the renin-angiotensin system (RAS), are considered important factors involved in the pathophysiology of DR (4, 203). Of note, retinal angiogenesis, usually driven by hypoxia, is one of the most vital effects responsible for the pathogenesis and progression of DR (4, 203, 204). In response to hypoxic stimuli, increased levels of m6A modification and METTL3 were observed in the retinas of a mouse model as well as endothelial cells (204). Furthermore, METTL3-mediated m6A modification aggravates angiogenesis by interacting with YTHDF1 and promoting the expression of genes (LRP6 and DVL1) that take part in the Wnt signaling pathway. This pathway has been demonstrated to undergo significant changes in DR conditions and play a key role in the modulation of retinal homeostasis (204, 205). Interestingly, overexpression of METTL3 suppressed the EMT of human retinal pigment epithelial (RPE) cells and delayed the progression of proliferative vitreoretinopathy by modulating the Wnt/β-catenin signaling pathway (206).
Although m6A RNA modifications and their associated enzymes have been reported to participate in many possible mechanisms of DR, such as inflammation and oxidative stress, their roles in DR remain elusive. Compared to normal control individuals, the expression levels of METTL3 and miR-25-3p were lower in diabetic patients, which is consistent with the results observed in HG-induced RPE cells. An in vitro study showed that the overexpression of METTL3 could upregulate miR-25-3p, downregulate PTEN and elevate phosphorylated Akt levels in HG-treated RPE cells. These effects alleviated RPE cell pyroptosis and apoptosis under high glucose stress and attenuated the deterioration of DR (207). However, STAT3 signaling was shown to promote the expression of miR-25 in response to oxidative stress, subsequently resulting in RPE degeneration (208). More research on the role of miR-25 in different pathological mechanisms of DR is necessary. Additionally, microarray analysis of retinal tissues derived from streptozotocin-induced mice with DR revealed that the expression of lysine acetyltransferase 1 (KAT1) was markedly downregulated. In two types of retinal cells treated with high glucose medium, KAT1 promoted YTHDF2-mediated integrin β1 (ITGB1) mRNA degradation in a m6A-dependent manner by regulating the FAK/PI3K/AKT axis and thereby improving DR (209). Pericytes are mysterious cells that surround the endothelial cells (ECs) of capillaries. They are involved in microvascular remodeling, which results in diseases in several organs, such as the kidney and retina (201, 202, 210). Pericyte dysfunction has been recognized as the primary initiator of vascular impairment in the pathophysiology of DR (201). A recent study identified increased levels of m6A RNA modification and METTL3 mRNA expression in pericytes and retinas from mice with DR (202). Further study demonstrated that METTL3-modulated m6A modification aggravated pericyte dysfunction by suppressing the PKC-η/FAT4/PDGFRA signaling pathway in a YTHDF2-mediated manner (202). Dysregulation of microglia (tissue-specific macrophages in the retina) polarization was considered to be one of the primary pathogenesis of DR (211, 212). Indeed, a higher rate of M1 inflammatory type but lower rate of M2 anti-inflammatory type were observed in both diabetic rats and BV2 cells (a type of mouse microglia cell line) treated with glucose (213). High glucose repressed expression of Alkbh5 in microglia, which in turn contributed to lower A20 (also known as tumor necrosis factor-a induced protein 3 (TNFAIP3)) expression and increased M1 polarization; these changes were the results of higher m6A modification level and faster degradation rate of A20 mRNA, which ultimately contributed to the initiation and development of DR (213). Overall, epigenetic regulation of these targets may help control and delay the development and progression of DR, which provides new insights into the pathogenesis and therapy of this disease.
Moreover, upregulation of METTL3 was found in lens epithelial cells derived from patients with diabetes with cataracts. In HG-induced human lens epithelial cells (HLECs), the level of m6A RNA modification was increased, and the expression of METTL3 mRNA was upregulated. These effects aggravate HLEC apoptosis by governing the 3’ UTR of intercellular adhesion molecule-1 (ICAM-1) (214). Hence, despite the findings in DR, more research focusing on other diabetic-related ocular manifestations, such as glaucoma and ischemic optic neuropathy, is needed in the future.
The Role of Epitranscriptomics in Diabetic Wounds
Chronic nonhealing wounds are a major threat to patients with diabetes since they can lead to limb amputation (215). Diabetic foot ulcer (DFU), is a wound underneath the ankle that usually results from foot deformities, lower extremity vascular disease and diabetic peripheral neuropathy (216, 217). DFU is one of the most serious complications of diabetes mellitus and can result in high rates of limb loss and disability. Approximately 4–10% of patients with diabetes encounter foot ulcers in their lives, and one diabetic individual loses his or her lower limb due to amputation surgery every 30 seconds worldwide (3, 218). Since DFUs also pose huge threats to personal mental health and the domestic and social economy, it is of great importance to promote wound healing for the treatment of this diabetes-related complication. The etiology of DFUs is multifactorial, including an impaired inflammatory response, reepithelialization, granulation formation, angiogenesis and others, and these processes can be affected by genes, the environment, and nutrition (215, 219). Substantial evidence has demonstrated that epigenetic regulatory mechanisms (such as histone modifications and DNA methylation) play a pivotal role in epidermal homeostasis and participate in the processes of wound healing (215, 220).
It was reported that the expressions of sequestosome 1 (SQSTM1)/p62, a receptor that regulates autophagy, and the m6A reader protein YTHDC1, were greatly decreased in both HG-treated human keratinocyte cells and in the epidermis derived from diabetic mice and patients with diabetes. Moreover, high glucose led to reduced m6A level in keratinocytes (218). Furthermore, Liang et at. demonstrated that YTHDC1 could facilitate the instability of SQSTM1 mRNA and autophagy in HG-treated keratinocytes by interacting with ELAV-like RNA binding protein 1 (ELAVL1)/HuR in the progression of wound repair (218). A vast number of studies have demonstrated that keratinocyte dysfunction results in defective epithelialization and contributes to delayed wound healing in DFUs (220, 221). In recent years, stem cell therapy has emerged as a promising treatment for wound repair in diabetes (219). Adipose-derived mesenchymal stem cells (ADSCs) have been suggested to improve wound healing in DFUs and facilitate lymphangiogenesis in previous studies (222, 223). Recent research showed that ADSCs could accelerate vascular endothelial growth factor C (VEGF-C) expression through the METTL3/IGF2BP2-m6A pathway and promote lymphangiogenesis. Thus, they improved the delayed wound repair in mice with DFU (217). This provides a new potential mechanism for ADSCs involved in the regulation of wound healing and confirms their crucial role in promising therapies for DFU.
However, studies focusing on the relationships between RNA modifications (such as m6A) and wound repair in DFUs are very limited. It is recognized that epidermal progenitor cells, which exist in the basal layer of the epidermis, play a crucial role in wound healing and the self-renewal of mammalian skin (224). Transcript analysis of mouse skin epithelial progenitors indicated that transcripts that participate in hair follicle morphogenesis were almost hypermethylated by m6A (225). Losses of m6A and METTL3 expression contribute to severe defects in hair follicle morphogenesis by regulating several possible signaling pathways, such as NOTCH signaling, BMP signaling, SHH signaling and especially Wnt signaling (225, 226). Another study found that ablation of Mettl14 could suppress the interaction between Pvt1 and MYC in a m6A-dependent manner, thereby damaging epidermal stemness and wound repair in skin (224, 226). Collectively, the relationship between the epitranscriptome and epithelial homeostasis, especially wound repair in DFU, deserves more attention in the future. Additionally, it is particularly important to help understand the molecular basis of epigenetic regulation, which might provide a new perspective for the pathogenesis of diabetic wound healing and the discovery of new therapeutic targets for DFU.
The Role of Epitranscriptomics in Diabetic Neuropathy
Diabetic neuropathy is a prominent and progressive complication of DM that affects more than 50% of individuals with diabetes and lowers their quality of life (2, 227). The manifestations of neuropathies secondary to diabetes can present as diabetic autonomic neuropathy (DAN) (such as diabetic cystopathy), uremic neuropathy (UN) and diabetic peripheral neuropathy (DPN); of these, the most prevalent is distal symmetric polyneuropathy (DSPN) (2, 228, 229). Previous studies have shown that the occurrence and development of diabetic neuropathy are related to many factors, such as the course of diabetes, control of blood glucose, obesity, insulin resistance and chronic low-grade inflammation. These factors are implicated in various mechanisms, such as overproduction of ROS and AGEs, excessive polyol pathway flux, neurovascular impairments, and perturbations of immunomodulation (2, 230). In addition, gene polymorphisms and epigenomic regulation, such as DNA methylation and posttranscriptional histone modifications, have been shown to participate in diabetic neuropathy in recent studies (230).
Regarding RNA modifications, a growing amount of evidence has demonstrated that epitranscriptomics is involved in neurodevelopmental as well as neurodegenerative diseases in recent years (231–234). For example, in a mouse model with muscle atrophy, denervation resulted in decreased levels of m6A and increased levels of the m6A demethylase ALKBH5. Overexpression of Alkbh5 could reduce m6A levels and aggravate muscle atrophy after denervation by activating the HDAC4/FoxO3 signaling pathway (235). Furthermore, m6A modifications have been regarded as facilitators for epilepsy, Alzheimer’s disease (AD), and Parkinson’s disease (PD) (233, 234). Although the exact mechanisms remain unknown, other RNA modifications, such as m5C, pseudouridine, and RNA editing, have also been reported to be associated with Dubowitz-like syndrome, AD, and schizophrenia (231, 232, 234, 236).
Regrettably, links between RNA modifications and diabetic neuropathy have not been well confirmed. A recent study found that inosine could improve sciatic nerve histological structure and function in mice with DPN by regulating the GLO1/AGE/RAGE/NF-κB and TGF-β/PKC/TRPV1/SP axes (227). RNA editing exerts its effects on neurological diseases, as stated in previous studies. Thus, A-to-I editing, which is located in approximately 85% of all premRNAs, may likely play important roles in diabetic neuropathy (234, 237, 238). In short, investigations such as this have guided this hotpot of the current research to explore the potential relationships between RNA modifications and diabetic neuropathy in more extensive studies.
Specific Small-Molecule Inhibitors Targeting Epitranscriptome
Taking into consideration the dysregulation of the epitranscriptome in diseases such as obesity, diabetes, and cancer, the discovery and design of compounds that may reverse dysregulated epitranscriptome will fire a new and exciting scientific filed. Indeed, some specific activators and inhibitors selectively regulating m6A modification have been developed, especially in oncology (239–242). For instance, piperidine and piperazine derivatives can serve as activators of METTL3-METTL14-WTAP complex to promote m6A modification on mRNA (243). METTL3 inhibitors, such as UZH1a, have a good potential for the treatment of METTL3-related cancers (244, 245). R-2-hydroxyglutarate, a FTO inhibitor, could suppress the expression of FTO/PFKP/LDHB and impair aerobic glycolysis in leukemia (242). Rhein has been demonstrated to function as an inhibitor of FTO by competitively binding the FTO catalytic domain and increasing m6A modification in mRNA (246). More FTO inhibitors, such as CHTB, N-CDPCB, meclofenamic acid 2, CS1 and CS2 have shown anticancer effects in existing literature (239, 240). Furthermore, ALK-04, identified as a ALKBH5 inhibitor, together with GVAX/anti-programmed cell death protein 1 immunotherapy, have obvious antitumous effect against melanoma (247). Interestingly, C17 could inhibit PUS7 activity and tRNA pseudouridylation which may impede glioma stem cells growth and thereby suppress glioblastoma tumorigenesis (241, 248). Collectively, these studies highlight the therapeutic potential of compounds targeting epitranscriptomics in cancer therapy.
However, little is known about the effects of these activators and inhibitors in diabetes. And less small-molecule compounds targeting RNA modifying enzymes involved in diabetes and its complications is reported to date. Therefore, it is urgent to design, select and discover potent RNA methyltransferase, demethylase, or RNA-regulating protein inhibitors/activator based on present mechanism researches, which may contribute to the treatment of people with diabetes.
Conclusions and Future Perspectives
As an epidemic disease worldwide, diabetes mellitus and its complications are still undertreated without efficient therapies in most patients. With the rapid progress of new technologies, such as high-throughput sequencing, epitranscriptomics has recently emerged as a crucial regulatory mechanism involved in both normal physiology and many pathological conditions. Due to its dynamic and reversible nature in response to environmental stimuli, manipulation of RNA modifications might be a promising approach for the treatment and prevention of DM and its related complications.
However, several gaps in knowledge exist in the understanding of the connection between RNA modification and DM. First, the function of m6A and modulating enzymes in diabetes has recently become a topic of interest, and many groups have explored its involvement in various pathophysiologies of diabetes (Figure 4). Nonetheless, other RNA modifications, such as m1A, m5C and m7G, are less studied in relation to diabetes. Moreover, studies focusing on the interaction of epitranscriptomics and diabetes-induced complications, such as DCM and diabetic neuropathy, are still insufficient. Hence, future research is warranted to investigate which chemical modifications of RNAs are involved in the pathogenesis of diabetes as well as to determine the exact regulatory and functional mechanisms of these factors that contribute to the occurrence and development of this disease. Second, some RNA modifications are cell type-specific or organ-specific. That is, whether RNA modifications exhibit their protective or destructive effects in response to metabolic or environmental stress depends on the cellular context and specific transcripts involved. To this end, there is a challenge to understand the condition-specific influences of RNA modifications on a specific transcript as well as the precise mechanisms by which RNA modifying enzymes differentially regulate the given transcript into a specific functional protein. Moreover, it is urgent to identify more RNA modification writers, readers, and erasers and determine the specificity of these regulatory proteins. Such findings could be beneficial for understanding the mechanisms of these proteins involved in both physiological homeostasis and pathological conditions. Third, similar to the identification of small molecule inhibitors against DNA methylation and histone modifications, it is of utmost importance to discover small molecules that target RNA modifying enzymes. These may shed light on the exploration of epigenetic regulatory mechanisms underlying diabetes pathogenesis and the identification of “epidrugs” in the era of precision medicine. Collectively, it is essential to explore whether the overexpression or knockout of RNA modifying enzymes (writers, erasers, and readers) can be taken from bench to bedside for diabetes management in future studies.
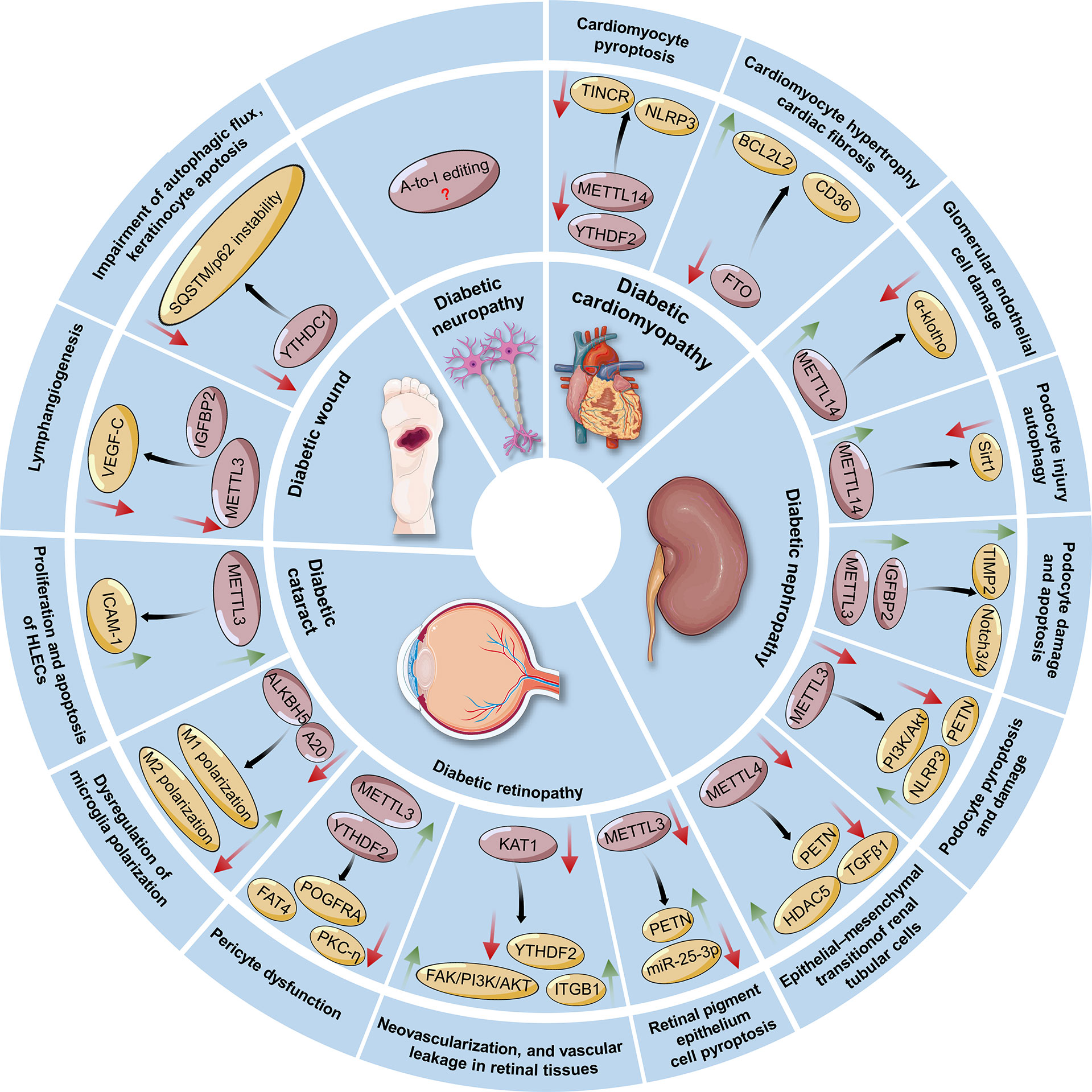
Figure 4 Regulatory roles and possible mechanisms of RNA modifications in the pathogenesis of diabetes-associated complications, including diabetic cardiomyopathy, nephropathy, retinopathy, cataracts, wounds, and neuropathy. The green arrow indicates an increase in the change, whereas the red arrow indicates a decrease in the change. METTL3/14/16, Methyltransferase-like 3, 14, 16; FTO, Fat mass and obesity-associated protein; ALKBH5, ALKB homolog 5; YTHDF2, YT521-B homology (YTH) protein families including YTH domain family 2; YTHDC1, YTH domain containing 1; TINCR: Terminal differentiation-induced noncoding RNA; NLRP3, Nucleotide-binding domain (NBD) and leucine-rich repeat (LRR) pyrin-domain containing protein 3; TIMP2, Tissue inhibitor of metalloproteinases 2; IGF2BPs, Insulin-like growth factor 2 mRNA-binding proteins; HDAC5, Histone deacetylase 5; ITGB1, Integrin β1; KAT1, Lysine acetyltransferase 1; ICAM-1, Intercellular adhesion molecule-1; SQSTM1, Sequestosome 1; VEGF-C, Vascular endothelial growth factor C. HLECs, Human lens epithelial cells; A20, Also known as tumor necrosis factor-a induced protein 3 (TNFAIP3); ALKBH5, ALKB homolog 5.
Data Availability Statement
The original contributions presented in the study are included in the article/supplementary material. Further inquiries can be directed to the corresponding author.
Author Contributions
XG contributed to the conception and first draft of the manuscript. ZL contributed to the figures and writing. YY contributed to the revision and editing of the manuscript. All authors reviewed the manuscript and approved the final draft.
Funding
The work was financially supported by funding from Basic Research Program of Yunnan Province (Kunming Medical University Joint Special Project) (No. 202101AY070001-276), the National Natural Science Foundation of China (No. 82160159), the Key Field R&D Plan Project of Guangdong province (No. 2019B020201002) and the Innovative Team of Precise Prevention and Treatment against Metabolic Diseases of Yunnan University.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The handling editor ZZ declared a shared parent affiliation with the authors XG and YY at the time of review.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Sun H, Saeedi P, Karuranga S, Pinkepank M, Ogurtsova K, Duncan BB, et al. IDF Diabetes Atlas: Global, Regional and Country-Level Diabetes Prevalence Estimates for 2021 and Projections for 2045. Diabetes Res Clin Pract (2022) 183:109119. doi: 10.1016/j.diabres.2021.109119
2. Feldman EL, Callaghan BC, Pop-Busui R, Zochodne DW, Wright DE, Bennett DL, et al. Diabetic Neuropathy. Nat Rev Dis Primers (2019) 5:42. doi: 10.1038/s41572-019-0097-9
3. Singh N, Armstrong DG, Lipsky BA. Preventing Foot Ulcers in Patients With Diabetes. JAMA (2005) 293:217–28. doi: 10.1001/jama.293.2.217
4. Cheung N, Mitchell P, Wong TY. Diabetic Retinopathy. Lancet (2010) 376:124–36. doi: 10.1016/S0140-6736(09)62124-3
5. Park CW. Diabetic Kidney Disease: From Epidemiology to Clinical Perspectives. Diabetes Metab J (2014) 38:252–60. doi: 10.4093/dmj.2014.38.4.252
6. Lee WS, Kim J. Diabetic Cardiomyopathy: Where We are and Where We are Going. Korean J Intern Med (2017) 32:404–21. doi: 10.3904/kjim.2016.208
7. Kato M, Natarajan R. Epigenetics and Epigenomics in Diabetic Kidney Disease and Metabolic Memory. Nat Rev Nephrol (2019) 15:327–45. doi: 10.1038/s41581-019-0135-6
8. Ling C, Ronn T. Epigenetics in Human Obesity and Type 2 Diabetes. Cell Metab (2019) 29:1028–44. doi: 10.1016/j.cmet.2019.03.009
9. Singh R, Chandel S, Dey D, Ghosh A, Roy S, Ravichandiran V, et al. Epigenetic Modification and Therapeutic Targets of Diabetes Mellitus. Biosci Rep (2020) 40:BSR20202160. doi: 10.1042/BSR20202160
10. Li X, Xiong X, Yi C. Epitranscriptome Sequencing Technologies: Decoding RNA Modifications. Nat Methods (2016) 14:23–31. doi: 10.1038/nmeth.4110
11. Zhao LY, Song J, Liu Y, Song CX, Yi C. Mapping the Epigenetic Modifications of DNA and RNA. Protein Cell (2020) 11:792–808. doi: 10.1007/s13238-020-00733-7
12. Longenecker JZ, Gilbert CJ, Golubeva VA, Martens CR, Accornero F. Epitranscriptomics in the Heart: A Focus on M(6)a. Curr Heart Fail Rep (2020) 17:205–12. doi: 10.1007/s11897-020-00473-z
13. Roundtree IA, Evans ME, Pan T, He C. Dynamic RNA Modifications in Gene Expression Regulation. Cell (2017) 169:1187–200. doi: 10.1016/j.cell.2017.05.045
14. Gilbert WV, Bell TA, Schaening C. Messenger RNA Modifications: Form, Distribution, and Function. Science (2016) 352:1408–12. doi: 10.1126/science.aad8711
15. Frye M, Jaffrey SR, Pan T, Rechavi G, Suzuki T. RNA Modifications: What Have We Learned and Where are We Headed? Nat Rev Genet (2016) 17:365–72. doi: 10.1038/nrg.2016.47
16. He C. Grand Challenge Commentary: RNA Epigenetics? Nat Chem Biol (2010) 6:863–5. doi: 10.1038/nchembio.482
17. Yanas A, Liu KF. RNA Modifications and the Link to Human Disease. Methods Enzymol (2019) 626:133–46. doi: 10.1016/bs.mie.2019.08.003
18. Zhao X, Yang Y, Sun BF, Shi Y, Yang X, Xiao W, et al. FTO-Dependent Demethylation of N6-Methyladenosine Regulates mRNA Splicing and is Required for Adipogenesis. Cell Res (2014) 24:1403–19. doi: 10.1038/cr.2014.151
19. Hess ME, Hess S, Meyer KD, Verhagen LA, Koch L, Bronneke HS, et al. The Fat Mass and Obesity Associated Gene (Fto) Regulates Activity of the Dopaminergic Midbrain Circuitry. Nat Neurosci (2013) 16:1042–8. doi: 10.1038/nn.3449
20. Li Y, Zhang Q, Cui G, Zhao F, Tian X, Sun BF, et al. M(6)A Regulates Liver Metabolic Disorders and Hepatogenous Diabetes. Genomics Proteomics Bioinf (2020) 18:371–83. doi: 10.1016/j.gpb.2020.06.003
21. Wang X, Zhao BS, Roundtree IA, Lu Z, Han D, Ma H, et al. N(6)-Methyladenosine Modulates Messenger RNA Translation Efficiency. Cell (2015) 161:1388–99. doi: 10.1016/j.cell.2015.05.014
22. Liu N, Dai Q, Zheng G, He C, Parisien M, Pan T. N(6)-Methyladenosine-Dependent RNA Structural Switches Regulate RNA-Protein Interactions. Nature (2015) 518:560–4. doi: 10.1038/nature14234
23. Wang T, Kong S, Tao M, Ju S. The Potential Role of RNA N6-Methyladenosine in Cancer Progression. Mol Cancer (2020) 19:88. doi: 10.1186/s12943-020-01204-7
24. Xiao W, Adhikari S, Dahal U, Chen YS, Hao YJ, Sun BF, et al. Nuclear M(6)A Reader YTHDC1 Regulates mRNA Splicing. Mol Cell (2016) 61:507–19. doi: 10.1016/j.molcel.2016.01.012
25. Wang X, Lu Z, Gomez A, Hon GC, Yue Y, Han D, et al. N6-Methyladenosine-Dependent Regulation of Messenger RNA Stability. Nature (2014) 505:117–20. doi: 10.1038/nature12730
26. Wu J, Frazier K, Zhang J, Gan Z, Wang T, Zhong X. Emerging Role of M(6) A RNA Methylation in Nutritional Physiology and Metabolism. Obes Rev (2020) 21:e12942. doi: 10.1111/obr.12942
27. Shi H, Chai P, Jia R, Fan X. Novel Insight Into the Regulatory Roles of Diverse RNA Modifications: Re-Defining the Bridge Between Transcription and Translation. Mol Cancer (2020) 19:78. doi: 10.1186/s12943-020-01194-6
28. Frye M, Harada BT, Behm M, He C. RNA Modifications Modulate Gene Expression During Development. Science (2018) 361:1346–49. doi: 10.1126/science.aau1646
29. Wang X, He C. Dynamic RNA Modifications in Posttranscriptional Regulation. Mol Cell (2014) 56:5–12. doi: 10.1016/j.molcel.2014.09.001
30. Chen YS, Ouyang XP, Yu XH, Novak P, Zhou L, He PP, et al. N6-Adenosine Methylation (M(6)A) RNA Modification: An Emerging Role in Cardiovascular Diseases. J Cardiovasc Transl Res (2021) 14:857–72. doi: 10.1007/s12265-021-10108-w
31. Ping XL, Sun BF, Wang L, Xiao W, Yang X, Wang WJ, et al. Mammalian WTAP is a Regulatory Subunit of the RNA N6-Methyladenosine Methyltransferase. Cell Res (2014) 24:177–89. doi: 10.1038/cr.2014.3
32. Wang X, Feng J, Xue Y, Guan Z, Zhang D, Liu Z, et al. Structural Basis of N(6)-Adenosine Methylation by the METTL3-METTL14 Complex. Nature (2016) 534:575–8. doi: 10.1038/nature18298
33. Schwartz S, Mumbach MR, Jovanovic M, Wang T, Maciag K, Bushkin GG, et al. Perturbation of M6a Writers Reveals Two Distinct Classes of mRNA Methylation at Internal and 5’ Sites. Cell Rep (2014) 8:284–96. doi: 10.1016/j.celrep.2014.05.048
34. Pendleton KE, Chen B, Liu K, Hunter OV, Xie Y, Tu BP, et al. The U6 snRNA M(6)A Methyltransferase METTL16 Regulates SAM Synthetase Intron Retention. Cell (2017) 169:824–35 e14. doi: 10.1016/j.cell.2017.05.003
35. Wen J, Lv R, Ma H, Shen H, He C, Wang J, et al. Zc3h13 Regulates Nuclear RNA M(6)A Methylation and Mouse Embryonic Stem Cell Self-Renewal. Mol Cell (2018) 69:1028–38 e6. doi: 10.1016/j.molcel.2018.02.015
36. Patil DP, Chen CK, Pickering BF, Chow A, Jackson C, Guttman M, et al. M(6)A RNA Methylation Promotes XIST-Mediated Transcriptional Repression. Nature (2016) 537:369–73. doi: 10.1038/nature19342
37. Yue Y, Liu J, Cui X, Cao J, Luo G, Zhang Z, et al. VIRMA Mediates Preferential M(6)A mRNA Methylation in 3’UTR and Near Stop Codon and Associates With Alternative Polyadenylation. Cell Discovery (2018) 4:10. doi: 10.1038/s41421-018-0019-0
38. Zheng G, Dahl JA, Niu Y, Fedorcsak P, Huang CM, Li CJ, et al. ALKBH5 is a Mammalian RNA Demethylase That Impacts RNA Metabolism and Mouse Fertility. Mol Cell (2013) 49:18–29. doi: 10.1016/j.molcel.2012.10.015
39. Jia G, Fu Y, Zhao X, Dai Q, Zheng G, Yang Y, et al. N6-Methyladenosine in Nuclear RNA is a Major Substrate of the Obesity-Associated FTO. Nat Chem Biol (2011) 7:885–7. doi: 10.1038/nchembio.687
40. Koh CWQ, Goh YT, Goh WSS. Atlas of Quantitative Single-Base-Resolution N(6)-Methyl-Adenine Methylomes. Nat Commun (2019) 10:5636. doi: 10.1038/s41467-019-13561-z
41. Mauer J, Sindelar M, Despic V, Guez T, Hawley BR, Vasseur JJ, et al. FTO Controls Reversible M(6)Am RNA Methylation During snRNA Biogenesis. Nat Chem Biol (2019) 15:340–47. doi: 10.1038/s41589-019-0231-8
42. Wei J, Liu F, Lu Z, Fei Q, Ai Y, He PC, et al. Differential M(6)A, M(6)Am, and M(1)A Demethylation Mediated by FTO in the Cell Nucleus and Cytoplasm. Mol Cell (2018) 71:973–85 e5. doi: 10.1016/j.molcel.2018.08.011
43. Mauer J, Luo X, Blanjoie A, Jiao X, Grozhik AV, Patil DP, et al. Reversible Methylation of M(6)Am in the 5’ Cap Controls mRNA Stability. Nature (2017) 541:371–75. doi: 10.1038/nature21022
44. Zhao Y, Shi Y, Shen H, Xie W. M(6)A-Binding Proteins: The Emerging Crucial Performers in Epigenetics. J Hematol Oncol (2020) 13:35. doi: 10.1186/s13045-020-00872-8
45. Huang H, Weng H, Sun W, Qin X, Shi H, Wu H, et al. Recognition of RNA N(6)-Methyladenosine by IGF2BP Proteins Enhances mRNA Stability and Translation. Nat Cell Biol (2018) 20:285–95. doi: 10.1038/s41556-018-0045-z
46. Liu N, Zhou KI, Parisien M, Dai Q, Diatchenko L, Pan T. N6-Methyladenosine Alters RNA Structure to Regulate Binding of a Low-Complexity Protein. Nucleic Acids Res (2017) 45:6051–63. doi: 10.1093/nar/gkx141
47. Choe J, Lin S, Zhang W, Liu Q, Wang L, Ramirez-Moya J, et al. mRNA Circularization by METTL3-Eif3h Enhances Translation and Promotes Oncogenesis. Nature (2018) 561:556–60. doi: 10.1038/s41586-018-0538-8
48. Zaccara S, Ries RJ, Jaffrey SR. Reading, Writing and Erasing mRNA Methylation. Nat Rev Mol Cell Biol (2019) 20:608–24. doi: 10.1038/s41580-019-0168-5
49. Feng ZY, Wang T, Su X, Guo S. Identification of the M(6)A RNA Methylation Regulators WTAP as a Novel Prognostic Biomarker and Genomic Alterations in Cutaneous Melanoma. Front Mol Biosci (2021) 8:665222. doi: 10.3389/fmolb.2021.665222
50. Bornaque F, Delannoy CP, Courty E, Rabhi N, Carney C, Rolland L, et al. Glucose Regulates M(6)A Methylation of RNA in Pancreatic Islets. Cells (2022) 11:291. doi: 10.3390/cells11020291
51. Boccaletto P, Machnicka MA, Purta E, Piatkowski P, Baginski B, Wirecki TK, et al. MODOMICS: a database of RNA modification pathways. 2017 update. Nucleic Acids Res (2018) 46:D303–D07. doi: 10.1093/nar/gkx1030
52. Sharma S, Watzinger P, Kotter P, Entian KD. Identification of a Novel Methyltransferase, Bmt2, Responsible for the N-1-Methyl-Adenosine Base Modification of 25S rRNA in Saccharomyces Cerevisiae. Nucleic Acids Res (2013) 41:5428–43. doi: 10.1093/nar/gkt195
53. El Yacoubi B, Bailly M, de Crecy-Lagard V. Biosynthesis and Function of Posttranscriptional Modifications of Transfer RNAs. Annu Rev Genet (2012) 46:69–95. doi: 10.1146/annurev-genet-110711-155641
54. Li X, Xiong X, Wang K, Wang L, Shu X, Ma S, et al. Transcriptome-Wide Mapping Reveals Reversible and Dynamic N(1)-Methyladenosine Methylome. Nat Chem Biol (2016) 12:311–6. doi: 10.1038/nchembio.2040
55. Zhou W, Wang C, Chang J, Huang Y, Xue Q, Miao C, et al. RNA Methylations in Cardiovascular Diseases, Molecular Structure, Biological Functions and Regulatory Roles in Cardiovascular Diseases. Front Pharmacol (2021) 12:722728. doi: 10.3389/fphar.2021.722728
56. Dai X, Wang T, Gonzalez G, Wang Y. Identification of YTH Domain-Containing Proteins as the Readers for N1-Methyladenosine in RNA. Anal Chem (2018) 90:6380–84. doi: 10.1021/acs.analchem.8b01703
57. Liu F, Clark W, Luo G, Wang X, Fu Y, Wei J, et al. ALKBH1-Mediated tRNA Demethylation Regulates Translation. Cell (2016) 167:816–28 e16. doi: 10.1016/j.cell.2016.09.038
58. Dubin DT, Taylor RH. The Methylation State of Poly A-Containing Messenger RNA From Cultured Hamster Cells. Nucleic Acids Res (1975) 2:1653–68. doi: 10.1093/nar/2.10.1653
59. Yang X, Yang Y, Sun BF, Chen YS, Xu JW, Lai WY, et al. 5-Methylcytosine Promotes mRNA Export - NSUN2 as the Methyltransferase and ALYREF as an M(5)C Reader. Cell Res (2017) 27:606–25. doi: 10.1038/cr.2017.55
60. Squires JE, Patel HR, Nousch M, Sibbritt T, Humphreys DT, Parker BJ, et al. Widespread Occurrence of 5-Methylcytosine in Human Coding and Non-Coding RNA. Nucleic Acids Res (2012) 40:5023–33. doi: 10.1093/nar/gks144
61. Schaefer M, Pollex T, Hanna K, Tuorto F, Meusburger M, Helm M, et al. RNA Methylation by Dnmt2 Protects Transfer RNAs Against Stress-Induced Cleavage. Genes Dev (2010) 24:1590–5. doi: 10.1101/gad.586710
62. Goll MG, Kirpekar F, Maggert KA, Yoder JA, Hsieh CL, Zhang X, et al. Methylation of Trnaasp by the DNA Methyltransferase Homolog Dnmt2. Science (2006) 311:395–8. doi: 10.1126/science.1120976
63. Reid R, Greene PJ, Santi DV. Exposition of a Family of RNA M(5)C Methyltransferases From Searching Genomic and Proteomic Sequences. Nucleic Acids Res (1999) 27:3138–45. doi: 10.1093/nar/27.15.3138
64. Chen X, Li A, Sun BF, Yang Y, Han YN, Yuan X, et al. 5-Methylcytosine Promotes Pathogenesis of Bladder Cancer Through Stabilizing mRNAs. Nat Cell Biol (2019) 21:978–90. doi: 10.1038/s41556-019-0361-y
65. Shen Q, Zhang Q, Shi Y, Shi Q, Jiang Y, Gu Y, et al. Tet2 Promotes Pathogen Infection-Induced Myelopoiesis Through mRNA Oxidation. Nature (2018) 554:123–27. doi: 10.1038/nature25434
66. Fu L, Guerrero CR, Zhong N, Amato NJ, Liu Y, Liu S, et al. Tet-Mediated Formation of 5-Hydroxymethylcytosine in RNA. J Am Chem Soc (2014) 136:11582–5. doi: 10.1021/ja505305z
67. Delatte B, Wang F, Ngoc LV, Collignon E, Bonvin E, Deplus R, et al. RNA Biochemistry. Transcriptome-Wide Distribution and Function of RNA Hydroxymethylcytosine. Science (2016) 351:282–5. doi: 10.1126/science.aac5253
68. Kawarada L, Suzuki T, Ohira T, Hirata S, Miyauchi K, Suzuki T. ALKBH1 is an RNA Dioxygenase Responsible for Cytoplasmic and Mitochondrial tRNA Modifications. Nucleic Acids Res (2017) 45:7401–15. doi: 10.1093/nar/gkx354
69. Haag S, Sloan KE, Ranjan N, Warda AS, Kretschmer J, Blessing C, et al. NSUN3 and ABH1 Modify the Wobble Position of Mt-Trnamet to Expand Codon Recognition in Mitochondrial Translation. EMBO J (2016) 35:2104–19. doi: 10.15252/embj.201694885
70. Jia J, Shi Y, Chen L, Lai W, Yan B, Jiang Y, et al. Decrease in Lymphoid Specific Helicase and 5-Hydroxymethylcytosine Is Associated With Metastasis and Genome Instability. Theranostics (2017) 7:3920–32. doi: 10.7150/thno.21389
71. Trotman JB, Schoenberg DR. A Recap of RNA Recapping. Wiley Interdiscip Rev RNA (2019) 10:e1504. doi: 10.1002/wrna.1504
72. Zhang LS, Liu C, Ma H, Dai Q, Sun HL, Luo G, et al. Transcriptome-Wide Mapping of Internal N(7)-Methylguanosine Methylome in Mammalian mRNA. Mol Cell (2019) 74:1304–16 e8. doi: 10.1016/j.molcel.2019.03.036
73. White J, Li Z, Sardana R, Bujnicki JM, Marcotte EM, Johnson AW. Bud23 Methylates G1575 of 18S rRNA and is Required for Efficient Nuclear Export of Pre-40S Subunits. Mol Cell Biol (2008) 28:3151–61. doi: 10.1128/MCB.01674-07
74. Haag S, Kretschmer J, Bohnsack MT. WBSCR22/Merm1 is Required for Late Nuclear Pre-Ribosomal RNA Processing and Mediates N7-Methylation of G1639 in Human 18S rRNA. RNA (2015) 21:180–7. doi: 10.1261/rna.047910.114
75. Deng Y, Zhou Z, Ji W, Lin S, Wang M. METTL1-Mediated M(7)G Methylation Maintains Pluripotency in Human Stem Cells and Limits Mesoderm Differentiation and Vascular Development. Stem Cell Res Ther (2020) 11:306. doi: 10.1186/s13287-020-01814-4
76. Alexandrov A, Martzen MR, Phizicky EM. Two Proteins That Form a Complex are Required for 7-Methylguanosine Modification of Yeast tRNA. RNA (2002) 8:1253–66. doi: 10.1017/s1355838202024019
77. Chen Z, Zhu W, Zhu S, Sun K, Liao J, Liu H, et al. METTL1 Promotes Hepatocarcinogenesis via M(7) G tRNA Modification-Dependent Translation Control. Clin Transl Med (2021) 11:e661. doi: 10.1002/ctm2.661
78. Ge J, Yu YT. RNA Pseudouridylation: New Insights Into an Old Modification. Trends Biochem Sci (2013) 38:210–8. doi: 10.1016/j.tibs.2013.01.002
79. Rintala-Dempsey AC, Kothe U. Eukaryotic Stand-Alone Pseudouridine Synthases - RNA Modifying Enzymes and Emerging Regulators of Gene Expression? RNA Biol (2017) 14:1185–96. doi: 10.1080/15476286.2016.1276150
80. Hamma T, Ferre-D’Amare AR. Pseudouridine Synthases. Chem Biol (2006) 13:1125–35. doi: 10.1016/j.chembiol.2006.09.009
81. Kariko K, Muramatsu H, Welsh FA, Ludwig J, Kato H, Akira S, et al. Incorporation of Pseudouridine Into mRNA Yields Superior Nonimmunogenic Vector With Increased Translational Capacity and Biological Stability. Mol Ther (2008) 16:1833–40. doi: 10.1038/mt.2008.200
82. Samuel CE. Adenosine Deaminases Acting on RNA (ADARs) are Both Antiviral and Proviral. Virology (2011) 411:180–93. doi: 10.1016/j.virol.2010.12.004
83. Licht K, Jantsch MF. Rapid and Dynamic Transcriptome Regulation by RNA Editing and RNA Modifications. J Cell Biol (2016) 213:15–22. doi: 10.1083/jcb.201511041
84. Nishikura K. A-To-I Editing of Coding and Non-Coding RNAs by ADARs. Nat Rev Mol Cell Biol (2016) 17:83–96. doi: 10.1038/nrm.2015.4
85. Nigita G, Veneziano D, Ferro A. A-To-I RNA Editing: Current Knowledge Sources and Computational Approaches With Special Emphasis on Non-Coding RNA Molecules. Front Bioeng Biotechnol (2015) 3:37. doi: 10.3389/fbioe.2015.00037
86. Blanc V, Park E, Schaefer S, Miller M, Lin Y, Kennedy S, et al. Genome-Wide Identification and Functional Analysis of Apobec-1-Mediated C-To-U RNA Editing in Mouse Small Intestine and Liver. Genome Biol (2014) 15:R79. doi: 10.1186/gb-2014-15-6-r79
87. Picardi E, Manzari C, Mastropasqua F, Aiello I, D’Erchia AM, Pesole G. Profiling RNA Editing in Human Tissues: Towards the Inosinome Atlas. Sci Rep (2015) 5:14941. doi: 10.1038/srep14941
88. Blanc V, Davidson NO. APOBEC-1-Mediated RNA Editing. Wiley Interdiscip Rev Syst Biol Med (2010) 2:594–602. doi: 10.1002/wsbm.82
89. Rosenberg BR, Hamilton CE, Mwangi MM, Dewell S, Papavasiliou FN. Transcriptome-Wide Sequencing Reveals Numerous APOBEC1 mRNA-Editing Targets in Transcript 3’ UTRs. Nat Struct Mol Biol (2011) 18:230–6. doi: 10.1038/nsmb.1975
90. Guo M, Liu D, Sha Q, Geng H, Liang J, Tang D. Succinic Acid Enhanced Quantitative Determination of Blood Modified Nucleosides in the Development of Diabetic Nephropathy Based on Hydrophilic Interaction Liquid Chromatography Mass Spectrometry. J Pharm BioMed Anal (2019) 164:309–16. doi: 10.1016/j.jpba.2018.10.042
91. Sun W, Song Y, Xia K, Yu L, Huang X, Zhao Z, et al. Transcriptome-Wide M(6)A Methylome During Osteogenic Differentiation of Human Adipose-Derived Stem Cells. Stem Cell Res Ther (2021) 12:489. doi: 10.1186/s13287-021-02508-1
92. Dominissini D, Moshitch-Moshkovitz S, Schwartz S, Salmon-Divon M, Ungar L, Osenberg S, et al. Topology of the Human and Mouse M6a RNA Methylomes Revealed by M6a-Seq. Nature (2012) 485:201–6. doi: 10.1038/nature11112
93. Meyer KD. DART-Seq: An Antibody-Free Method for Global M(6)A Detection. Nat Methods (2019) 16:1275–80. doi: 10.1038/s41592-019-0570-0
94. Garcia-Campos MA, Edelheit S, Toth U, Safra M, Shachar R, Viukov S, et al. Deciphering the “M(6)A Code” via Antibody-Independent Quantitative Profiling. Cell (2019) 178:731–47 e16. doi: 10.1016/j.cell.2019.06.013
95. Linder B, Grozhik AV, Olarerin-George AO, Meydan C, Mason CE, Jaffrey SR. Single-Nucleotide-Resolution Mapping of M6a and m6Am Throughout the Transcriptome. Nat Methods (2015) 12:767–72. doi: 10.1038/nmeth.3453
96. Chen K, Lu Z, Wang X, Fu Y, Luo GZ, Liu N, et al. High-Resolution N(6) -Methyladenosine (M(6) A) Map Using Photo-Crosslinking-Assisted M(6) A Sequencing. Angew Chem Int Ed Engl (2015) 54:1587–90. doi: 10.1002/anie.201410647
97. Hu L, Liu S, Peng Y, Ge R, Su R, Senevirathne C, et al. M(6)A RNA Modifications are Measured at Single-Base Resolution Across the Mammalian Transcriptome. Nat Biotechnol (2022). doi: 10.1038/s41587-022-01243-z
98. Song J, Yi C. Chemical Modifications to RNA: A New Layer of Gene Expression Regulation. ACS Chem Biol (2017) 12:316–25. doi: 10.1021/acschembio.6b00960
99. Chen YS, Yang WL, Zhao YL, Yang YG. Dynamic Transcriptomic M(5) C and its Regulatory Role in RNA Processing. Wiley Interdiscip Rev RNA (2021) 12:e1639. doi: 10.1002/wrna.1639
100. Yuan F, Bi Y, Siejka-Zielinska P, Zhou YL, Zhang XX, Song CX. Bisulfite-Free and Base-Resolution Analysis of 5-Methylcytidine and 5-Hydroxymethylcytidine in RNA With Peroxotungstate. Chem Commun (Camb) (2019) 55:2328–31. doi: 10.1039/c9cc00274j
101. Hussain S, Sajini AA, Blanco S, Dietmann S, Lombard P, Sugimoto Y, et al. NSun2-Mediated Cytosine-5 Methylation of Vault Noncoding RNA Determines its Processing Into Regulatory Small RNAs. Cell Rep (2013) 4:255–61. doi: 10.1016/j.celrep.2013.06.029
102. Schaefer M, Pollex T, Hanna K, Lyko F. RNA Cytosine Methylation Analysis by Bisulfite Sequencing. Nucleic Acids Res (2009) 37:e12. doi: 10.1093/nar/gkn954
103. Zhang Q, Liu F, Chen W, Miao H, Liang H, Liao Z, et al. The Role of RNA M(5)C Modification in Cancer Metastasis. Int J Biol Sci (2021) 17:3369–80. doi: 10.7150/ijbs.61439
104. Malbec L, Zhang T, Chen YS, Zhang Y, Sun BF, Shi BY, et al. Dynamic Methylome of Internal mRNA N(7)-Methylguanosine and its Regulatory Role in Translation. Cell Res (2019) 29:927–41. doi: 10.1038/s41422-019-0230-z
105. Begik O, Lucas MC, Pryszcz LP, Ramirez JM, Medina R, Milenkovic I, et al. Quantitative Profiling of Pseudouridylation Dynamics in Native RNAs With Nanopore Sequencing. Nat Biotechnol (2021) 39:1278–91. doi: 10.1038/s41587-021-00915-6
106. Liu K, Chen W, Lin H. XG-PseU: An Extreme Gradient Boosting Based Method for Identifying Pseudouridine Sites. Mol Genet Genomics (2020) 295:13–21. doi: 10.1007/s00438-019-01600-9
107. Pillon NJ, Loos RJF, Marshall SM, Zierath JR. Metabolic Consequences of Obesity and Type 2 Diabetes: Balancing Genes and Environment for Personalized Care. Cell (2021) 184:1530–44. doi: 10.1016/j.cell.2021.02.012
108. Shen F, Huang W, Huang JT, Xiong J, Yang Y, Wu K, et al. Decreased N(6)-Methyladenosine in Peripheral Blood RNA From Diabetic Patients is Associated With FTO Expression Rather Than ALKBH5. J Clin Endocrinol Metab (2015) 100:E148–54. doi: 10.1210/jc.2014-1893
109. Souness JE, Stouffer JE, Chagoya de Sanchez V. Effect of N6-Methyladenosine on Fat-Cell Glucose Metabolism. Evidence for two modes of action. Biochem Pharmacol (1982) 31:3961–71. doi: 10.1016/0006-2952(82)90642-6
110. Xie W, Ma LL, Xu YQ, Wang BH, Li SM. METTL3 Inhibits Hepatic Insulin Sensitivity via N6-Methyladenosine Modification of Fasn mRNA and Promoting Fatty Acid Metabolism. Biochem Biophys Res Commun (2019) 518:120–26. doi: 10.1016/j.bbrc.2019.08.018
111. Mlinar B, Pfeifer M, Vrtacnik-Bokal E, Jensterle M, Marc J. Decreased Lipin 1 Beta Expression in Visceral Adipose Tissue is Associated With Insulin Resistance in Polycystic Ovary Syndrome. Eur J Endocrinol (2008) 159:833–9. doi: 10.1530/EJE-08-0387
112. Donkor J, Sparks LM, Xie H, Smith SR, Reue K. Adipose Tissue Lipin-1 Expression is Correlated With Peroxisome Proliferator-Activated Receptor Alpha Gene Expression and Insulin Sensitivity in Healthy Young Men. J Clin Endocrinol Metab (2008) 93:233–9. doi: 10.1210/jc.2007-1535
113. Huang S, Huang S, Wang X, Zhang Q, Liu J, Leng Y. Downregulation of Lipin-1 Induces Insulin Resistance by Increasing Intracellular Ceramide Accumulation in C2C12 Myotubes. Int J Biol Sci (2017) 13:1–12. doi: 10.7150/ijbs.17149
114. Han L, Li Y, Tang L, Chen Z, Zhang T, Chen S, et al. IGF2BP2 Rs11705701 Polymorphisms are Associated With Prediabetes in a Chinese Population: A Population-Based Case-Control Study. Exp Ther Med (2016) 12:1849–56. doi: 10.3892/etm.2016.3554
115. Sanghera DK, Ortega L, Han S, Singh J, Ralhan SK, Wander GS, et al. Impact of Nine Common Type 2 Diabetes Risk Polymorphisms in Asian Indian Sikhs: PPARG2 (Pro12Ala), IGF2BP2, TCF7L2 and FTO Variants Confer a Significant Risk. BMC Med Genet (2008) 9:59. doi: 10.1186/1471-2350-9-59
116. Dai N. The Diverse Functions of IMP2/IGF2BP2 in Metabolism. Trends Endocrinol Metab (2020) 31:670–79. doi: 10.1016/j.tem.2020.05.007
117. Voight BF, Scott LJ, Steinthorsdottir V, Morris AP, Dina C, Welch RP, et al. Twelve Type 2 Diabetes Susceptibility Loci Identified Through Large-Scale Association Analysis. Nat Genet (2010) 42:579–89. doi: 10.1038/ng.609
118. Groenewoud MJ, Dekker JM, Fritsche A, Reiling E, Nijpels G, Heine RJ, et al. Variants of CDKAL1 and IGF2BP2 Affect First-Phase Insulin Secretion During Hyperglycaemic Clamps. Diabetologia (2008) 51:1659–63. doi: 10.1007/s00125-008-1083-z
119. Regue L, Zhao L, Ji F, Wang H, Avruch J, Dai N. RNA M6a Reader IMP2/IGF2BP2 Promotes Pancreatic Beta-Cell Proliferation and Insulin Secretion by Enhancing PDX1 Expression. Mol Metab (2021) 48:101209. doi: 10.1016/j.molmet.2021.101209
120. Li X, Jiang Y, Sun X, Wu Y, Chen Z. METTL3 is Required for Maintaining Beta-Cell Function. Metabolism (2021) 116:154702. doi: 10.1016/j.metabol.2021.154702
121. De Jesus DF, Zhang Z, Kahraman S, Brown NK, Chen M, Hu J, et al. M(6)A mRNA Methylation Regulates Human Beta-Cell Biology in Physiological States and in Type 2 Diabetes. Nat Metab (2019) 1:765–74. doi: 10.1038/s42255-019-0089-9
122. Men L, Sun J, Luo G, Ren D. Acute Deletion of METTL14 in Beta-Cells of Adult Mice Results in Glucose Intolerance. Endocrinology (2019) 160:2388–94. doi: 10.1210/en.2019-00350
123. Chen W, Zhang L, Zheng G, Fu Y, Ji Q, Liu F, et al. Crystal Structure of the RNA Demethylase ALKBH5 From Zebrafish. FEBS Lett (2014) 588:892–8. doi: 10.1016/j.febslet.2014.02.021
124. Yang Y, Shen F, Huang W, Qin S, Huang JT, Sergi C, et al. Glucose Is Involved in the Dynamic Regulation of M6a in Patients With Type 2 Diabetes. J Clin Endocrinol Metab (2019) 104:665–73. doi: 10.1210/jc.2018-00619
125. Ikels K, Kuschel S, Fischer J, Kaisers W, Eberhard D, Ruther U. FTO Is a Relevant Factor for the Development of the Metabolic Syndrome in Mice. PloS One (2014) 9:e105349. doi: 10.1371/journal.pone.0105349
126. Fischer J, Koch L, Emmerling C, Vierkotten J, Peters T, Bruning JC, et al. Inactivation of the Fto Gene Protects From Obesity. Nature (2009) 458:894–8. doi: 10.1038/nature07848
127. Mizuno TM, Lew PS, Luo Y, Leckstrom A. Negative Regulation of Hepatic Fat Mass and Obesity Associated (Fto) Gene Expression by Insulin. Life Sci (2017) 170:50–5. doi: 10.1016/j.lfs.2016.11.027
128. Zhou J, Wan J, Shu XE, Mao Y, Liu XM, Yuan X, et al. N(6)-Methyladenosine Guides mRNA Alternative Translation During Integrated Stress Response. Mol Cell (2018) 69:636–47 e7. doi: 10.1016/j.molcel.2018.01.019
129. Peng S, Xiao W, Ju D, Sun B, Hou N, Liu Q, et al. Identification of Entacapone as a Chemical Inhibitor of FTO Mediating Metabolic Regulation Through FOXO1. Sci Transl Med (2019) 11:eaau7116. doi: 10.1126/scitranslmed.aau7116
130. Guo F, Zhang Y, Zhang C, Wang S, Ni Y, Zhao R. Fatmass and Obesity Associated (FTO) Gene Regulates Gluconeogenesis in Chicken Embryo Fibroblast Cells. Comp Biochem Physiol A Mol Integr Physiol (2015) 179:149–56. doi: 10.1016/j.cbpa.2014.10.003
131. Bravard A, Vial G, Chauvin MA, Rouille Y, Bailleul B, Vidal H, et al. FTO Contributes to Hepatic Metabolism Regulation Through Regulation of Leptin Action and STAT3 Signalling in Liver. Cell Commun Signal (2014) 12:4. doi: 10.1186/1478-811X-12-4
132. Fan HQ, He W, Xu KF, Wang ZX, Xu XY, Chen H. FTO Inhibits Insulin Secretion and Promotes NF-kappaB Activation Through Positively Regulating ROS Production in Pancreatic Beta Cells. PloS One (2015) 10:e0127705. doi: 10.1371/journal.pone.0127705
133. Riffo B, Asenjo S, Saez K, Aguayo C, Munoz I, Bustos P, et al. FTO Gene Is Related to Obesity in Chilean Amerindian Children and Impairs HOMA-IR in Prepubertal Girls. Pediatr Diabetes (2012) 13:384–91. doi: 10.1111/j.1399-5448.2011.00834.x
134. Claussnitzer M, Dankel SN, Kim KH, Quon G, Meuleman W, Haugen C, et al. FTO Obesity Variant Circuitry and Adipocyte Browning in Humans. N Engl J Med (2015) 373:895–907. doi: 10.1056/NEJMoa1502214
135. Smemo S, Tena JJ, Kim KH, Gamazon ER, Sakabe NJ, Gomez-Marin C, et al. Obesity-Associated Variants Within FTO Form Long-Range Functional Connections With IRX3. Nature (2014) 507:371–5. doi: 10.1038/nature13138
136. Chan PP, Lowe TM. GtRNAdb 2.0: An Expanded Database of Transfer RNA Genes Identified in Complete and Draft Genomes. Nucleic Acids Res (2016) 44:D184–9. doi: 10.1093/nar/gkv1309
137. Arroyo MN, Green JA, Cnop M, Igoillo-Esteve M. tRNA Biology in the Pathogenesis of Diabetes: Role of Genetic and Environmental Factors. Int J Mol Sci (2021) 22:496. doi: 10.3390/ijms22020496
138. Cosentino C, Cnop M, Igoillo-Esteve M. The tRNA Epitranscriptome and Diabetes: Emergence of tRNA Hypomodifications as a Cause of Pancreatic Beta-Cell Failure. Endocrinology (2019) 160:1262–74. doi: 10.1210/en.2019-00098
139. Abbott JA, Francklyn CS, Robey-Bond SM. Transfer RNA and Human Disease. Front Genet (2014) 5:158. doi: 10.3389/fgene.2014.00158
140. Palmer CJ, Bruckner RJ, Paulo JA, Kazak L, Long JZ, Mina AI, et al. Cdkal1, a Type 2 Diabetes Susceptibility Gene, Regulates Mitochondrial Function in Adipose Tissue. Mol Metab (2017) 6:1212–25. doi: 10.1016/j.molmet.2017.07.013
141. Zeng H, Guo M, Zhou T, Tan L, Chong CN, Zhang T, et al. An Isogenic Human ESC Platform for Functional Evaluation of Genome-Wide-Association-Study-Identified Diabetes Genes and Drug Discovery. Cell Stem Cell (2016) 19:326–40. doi: 10.1016/j.stem.2016.07.002
142. Xie P, Wei FY, Hirata S, Kaitsuka T, Suzuki T, Suzuki T, et al. Quantitative PCR Measurement of tRNA 2-Methylthio Modification for Assessing Type 2 Diabetes Risk. Clin Chem (2013) 59:1604–12. doi: 10.1373/clinchem.2013.210401
143. Kirino Y, Suzuki T. Human Mitochondrial Diseases Associated With tRNA Wobble Modification Deficiency. RNA Biol (2005) 2:41–4. doi: 10.4161/rna.2.2.1610
144. Wang M, Liu H, Zheng J, Chen B, Zhou M, Fan W, et al. A Deafness- and Diabetes-Associated tRNA Mutation Causes Deficient Pseudouridinylation at Position 55 in Trnaglu and Mitochondrial Dysfunction. J Biol Chem (2016) 291:21029–41. doi: 10.1074/jbc.M116.739482
145. Chan CW, Chetnani B, Mondragon A. Structure and Function of the T-Loop Structural Motif in Noncoding RNAs. Wiley Interdiscip Rev RNA (2013) 4:507–22. doi: 10.1002/wrna.1175
146. Lin H, Zhou X, Chen X, Huang K, Wu W, Fu J, et al. tRNA Methyltransferase 10 Homologue A (TRMT10A) Mutation in a Chinese Patient With Diabetes, Insulin Resistance, Intellectual Deficiency and Microcephaly. BMJ Open Diabetes Res Care (2020) 8:e001601. doi: 10.1136/bmjdrc-2020-001601
147. Gillis D, Krishnamohan A, Yaacov B, Shaag A, Jackman JE, Elpeleg O. TRMT10A Dysfunction is Associated With Abnormalities in Glucose Homeostasis, Short Stature and Microcephaly. J Med Genet (2014) 51:581–6. doi: 10.1136/jmedgenet-2014-102282
148. Yew TW, McCreight L, Colclough K, Ellard S, Pearson ER. tRNA Methyltransferase Homologue Gene TRMT10A Mutation in Young Adult-Onset Diabetes With Intellectual Disability, Microcephaly and Epilepsy. Diabetes Med (2016) 33:e21–5. doi: 10.1111/dme.13024
149. Cosentino C, Toivonen S, Diaz Villamil E, Atta M, Ravanat JL, Demine S, et al. Pancreatic Beta-Cell tRNA Hypomethylation and Fragmentation Link TRMT10A Deficiency With Diabetes. Nucleic Acids Res (2018) 46:10302–18. doi: 10.1093/nar/gky839
150. Igoillo-Esteve M, Genin A, Lambert N, Desir J, Pirson I, Abdulkarim B, et al. tRNA Methyltransferase Homolog Gene TRMT10A Mutation in Young Onset Diabetes and Primary Microcephaly in Humans. PloS Genet (2013) 9:e1003888. doi: 10.1371/journal.pgen.1003888
151. Yarham JW, Lamichhane TN, Pyle A, Mattijssen S, Baruffini E, Bruni F, et al. Defective I6a37 Modification of Mitochondrial and Cytosolic tRNAs Results From Pathogenic Mutations in TRIT1 and its Substrate tRNA. PloS Genet (2014) 10:e1004424. doi: 10.1371/journal.pgen.1004424
152. Kernohan KD, Dyment DA, Pupavac M, Cramer Z, McBride A, Bernard G, et al. Matchmaking Facilitates the Diagnosis of an Autosomal-Recessive Mitochondrial Disease Caused by Biallelic Mutation of the tRNA Isopentenyltransferase (TRIT1) Gene. Hum Mutat (2017) 38:511–16. doi: 10.1002/humu.23196
153. Pinti MV, Fink GK, Hathaway QA, Durr AJ, Kunovac A, Hollander JM. Mitochondrial Dysfunction in Type 2 Diabetes Mellitus: An Organ-Based Analysis. Am J Physiol Endocrinol Metab (2019) 316:E268–E85. doi: 10.1152/ajpendo.00314.2018
154. Sangwung P, Petersen KF, Shulman GI, Knowles JW. Mitochondrial Dysfunction, Insulin Resistance, and Potential Genetic Implications. Endocrinology (2020) 161:bqaa017. doi: 10.1210/endocr/bqaa017
155. Rovira-Llopis S, Banuls C, Diaz-Morales N, Hernandez-Mijares A, Rocha M, Victor VM. Mitochondrial Dynamics in Type 2 Diabetes: Pathophysiological Implications. Redox Biol (2017) 11:637–45. doi: 10.1016/j.redox.2017.01.013
156. Kleiber N, Lemus-Diaz N, Stiller C, Heinrichs M, Mai MM, Hackert P, et al. The RNA Methyltransferase METTL8 Installs M(3)C32 in Mitochondrial tRNAs(Thr/Ser(UCN)) to Optimise tRNA Structure and Mitochondrial Translation. Nat Commun (2022) 13:209. doi: 10.1038/s41467-021-27905-1
157. Scholler E, Marks J, Marchand V, Bruckmann A, Powell CA, Reichold M, et al. Balancing of Mitochondrial Translation Through METTL8-Mediated M(3)C Modification of Mitochondrial tRNAs. Mol Cell (2021) 81:4810–25 e12. doi: 10.1016/j.molcel.2021.10.018
158. Li X, Xiong X, Zhang M, Wang K, Chen Y, Zhou J, et al. Base-Resolution Mapping Reveals Distinct M(1)A Methylome in Nuclear- and Mitochondrial-Encoded Transcripts. Mol Cell (2017) 68:993–1005 e9. doi: 10.1016/j.molcel.2017.10.019
159. Safra M, Sas-Chen A, Nir R, Winkler R, Nachshon A, Bar-Yaacov D, et al. The M1a Landscape on Cytosolic and Mitochondrial mRNA at Single-Base Resolution. Nature (2017) 551:251–55. doi: 10.1038/nature24456
160. Murtaza G, Virk HUH, Khalid M, Lavie CJ, Ventura H, Mukherjee D, et al. Diabetic Cardiomyopathy - A Comprehensive Updated Review. Prog Cardiovasc Dis (2019) 62:315–26. doi: 10.1016/j.pcad.2019.03.003
161. Anderson JL, Adams CD, Antman EM, Bridges CR, Califf RM, Casey DE Jr, et al. ACC/AHA 2007 Guidelines for the Management of Patients With Unstable Angina/Non-ST-Elevation Myocardial Infarction: A Report of the American College of Cardiology/American Heart Association Task Force on Practice Guidelines (Writing Committee to Revise the 2002 Guidelines for the Management of Patients With Unstable Angina/Non-ST-Elevation Myocardial Infarction) Developed in Collaboration With the American College of Emergency Physicians, the Society for Cardiovascular Angiography and Interventions, and the Society of Thoracic Surgeons Endorsed by the American Association of Cardiovascular and Pulmonary Rehabilitation and the Society for Academic Emergency Medicine. J Am Coll Cardiol (2007) 50:e1–e157. doi: 10.1016/j.jacc.2007.02.013
162. Deng J, Liao Y, Liu J, Liu W, Yan D. Research Progress on Epigenetics of Diabetic Cardiomyopathy in Type 2 Diabetes. Front Cell Dev Biol (2021) 9:777258. doi: 10.3389/fcell.2021.777258
163. Trachanas K, Sideris S, Aggeli C, Poulidakis E, Gatzoulis K, Tousoulis D, et al. Diabetic Cardiomyopathy: From Pathophysiology to Treatment. Hellenic J Cardiol (2014) 55:411–21.
164. Tan Y, Zhang Z, Zheng C, Wintergerst KA, Keller BB, Cai L. Mechanisms of Diabetic Cardiomyopathy and Potential Therapeutic Strategies: Preclinical and Clinical Evidence. Nat Rev Cardiol (2020) 17:585–607. doi: 10.1038/s41569-020-0339-2
165. Dillmann WH. Diabetic Cardiomyopathy. Circ Res (2019) 124:1160–62. doi: 10.1161/CIRCRESAHA.118.314665
166. Hu X, Bai T, Xu Z, Liu Q, Zheng Y, Cai L. Pathophysiological Fundamentals of Diabetic Cardiomyopathy. Compr Physiol (2017) 7:693–711. doi: 10.1002/cphy.c160021
167. Ghosh N, Katare R. Molecular Mechanism of Diabetic Cardiomyopathy and Modulation of microRNA Function by Synthetic Oligonucleotides. Cardiovasc Diabetol (2018) 17:43. doi: 10.1186/s12933-018-0684-1
168. Jin J, Wang X, Zhi X, Meng D. Epigenetic Regulation in Diabetic Vascular Complications. J Mol Endocrinol (2019) 63:R103–R15. doi: 10.1530/JME-19-0170
169. Ordovas JM, Smith CE. Epigenetics and Cardiovascular Disease. Nat Rev Cardiol (2010) 7:510–9. doi: 10.1038/nrcardio.2010.104
170. Mathiyalagan P, Adamiak M, Mayourian J, Sassi Y, Liang Y, Agarwal N, et al. FTO-Dependent N(6)-Methyladenosine Regulates Cardiac Function During Remodeling and Repair. Circulation (2019) 139:518–32. doi: 10.1161/CIRCULATIONAHA.118.033794
171. Chien CS, Li JY, Chien Y, Wang ML, Yarmishyn AA, Tsai PH, et al. METTL3-Dependent N(6)-Methyladenosine RNA Modification Mediates the Atherogenic Inflammatory Cascades in Vascular Endothelium. Proc Natl Acad Sci USA (2021) 118:e2025070118. doi: 10.1073/pnas.2025070118
172. Xu H, Wang Z, Chen M, Zhao W, Tao T, Ma L, et al. YTHDF2 Alleviates Cardiac Hypertrophy via Regulating Myh7 mRNA Decoy. Cell Biosci (2021) 11:132. doi: 10.1186/s13578-021-00649-7
173. Jain M, Mann TD, Stulic M, Rao SP, Kirsch A, Pullirsch D, et al. RNA Editing of Filamin A pre-mRNA Regulates Vascular Contraction and Diastolic Blood Pressure. EMBO J (2018) 37:e94813. doi: 10.15252/embj.201694813
174. Ju W, Liu K, Ouyang S, Liu Z, He F, Wu J. Changes in N6-Methyladenosine Modification Modulate Diabetic Cardiomyopathy by Reducing Myocardial Fibrosis and Myocyte Hypertrophy. Front Cell Dev Biol (2021) 9:702579. doi: 10.3389/fcell.2021.702579
175. Yang F, Li A, Qin Y, Che H, Wang Y, Lv J, et al. A Novel Circular RNA Mediates Pyroptosis of Diabetic Cardiomyopathy by Functioning as a Competing Endogenous RNA. Mol Ther Nucleic Acids (2019) 17:636–43. doi: 10.1016/j.omtn.2019.06.026
176. Lu Y, Lu Y, Meng J, Wang Z. Pyroptosis and Its Regulation in Diabetic Cardiomyopathy. Front Physiol (2021) 12:791848. doi: 10.3389/fphys.2021.791848
177. Cao R, Fang D, Wang J, Yu Y, Ye H, Kang P, et al. ALDH2 Overexpression Alleviates High Glucose-Induced Cardiotoxicity by Inhibiting NLRP3 Inflammasome Activation. J Diabetes Res (2019) 2019:4857921. doi: 10.1155/2019/4857921
178. Kar S, Shahshahan HR, Hackfort BT, Yadav SK, Yadav R, Kambis TN, et al. Exercise Training Promotes Cardiac Hydrogen Sulfide Biosynthesis and Mitigates Pyroptosis to Prevent High-Fat Diet-Induced Diabetic Cardiomyopathy. Antioxid (Basel) (2019) 8:638. doi: 10.3390/antiox8120638
179. Xie Y, Huang Y, Ling X, Qin H, Wang M, Luo B. Chemerin/CMKLR1 Axis Promotes Inflammation and Pyroptosis by Activating NLRP3 Inflammasome in Diabetic Cardiomyopathy Rat. Front Physiol (2020) 11:381. doi: 10.3389/fphys.2020.00381
180. Xu S, Li Y, Chen JP, Li DZ, Jiang Q, Wu T, et al. Oxygen Glucose Deprivation/Re-Oxygenation-Induced Neuronal Cell Death Is Associated With Lnc-D63785 M6a Methylation and miR-422a Accumulation. Cell Death Dis (2020) 11:816. doi: 10.1038/s41419-020-03021-8
181. Zhang C, Fu J, Zhou Y. A Review in Research Progress Concerning M6a Methylation and Immunoregulation. Front Immunol (2019) 10:922. doi: 10.3389/fimmu.2019.00922
182. Meng L, Lin H, Huang X, Weng J, Peng F, Wu S. METTL14 Suppresses Pyroptosis and Diabetic Cardiomyopathy by Downregulating TINCR lncRNA. Cell Death Dis (2022) 13:38. doi: 10.1038/s41419-021-04484-z
183. Jones CA, Krolewski AS, Rogus J, Xue JL, Collins A, Warram JH. Epidemic of End-Stage Renal Disease in People With Diabetes in the United States Population: Do We Know the Cause? Kidney Int (2005) 67:1684–91. doi: 10.1111/j.1523-1755.2005.00265.x
184. Alicic RZ, Rooney MT, Tuttle KR. Diabetic Kidney Disease: Challenges, Progress, and Possibilities. Clin J Am Soc Nephrol (2017) 12:2032–45. doi: 10.2215/CJN.11491116
185. Gnudi L, Coward RJM, Long DA. Diabetic Nephropathy: Perspective on Novel Molecular Mechanisms. Trends Endocrinol Metab (2016) 27:820–30. doi: 10.1016/j.tem.2016.07.002
186. Shi Y, Hu FB. The Global Implications of Diabetes and Cancer. Lancet (2014) 383:1947–8. doi: 10.1016/S0140-6736(14)60886-2
187. Song CX, Yi C, He C. Mapping Recently Identified Nucleotide Variants in the Genome and Transcriptome. Nat Biotechnol (2012) 30:1107–16. doi: 10.1038/nbt.2398
188. Schroder AS, Steinbacher J, Steigenberger B, Gnerlich FA, Schiesser S, Pfaffeneder T, et al. Synthesis of a DNA Promoter Segment Containing All Four Epigenetic Nucleosides: 5-Methyl-, 5-Hydroxymethyl-, 5-Formyl-, and 5-Carboxy-2’-Deoxycytidine. Angew Chem Int Ed Engl (2014) 53:315–8. doi: 10.1002/anie.201308469
189. Niewczas MA, Mathew AV, Croall S, Byun J, Major M, Sabisetti VS, et al. Circulating Modified Metabolites and a Risk of ESRD in Patients With Type 1 Diabetes and Chronic Kidney Disease. Diabetes Care (2017) 40:383–90. doi: 10.2337/dc16-0173
190. Li M, Deng L, Xu G. METTL14 Promotes Glomerular Endothelial Cell Injury and Diabetic Nephropathy via M6a Modification of Alpha-Klotho. Mol Med (2021) 27:106. doi: 10.1186/s10020-021-00365-5
191. Lu Z, Liu H, Song N, Liang Y, Zhu J, Chen J, et al. METTL14 Aggravates Podocyte Injury and Glomerulopathy Progression Through N(6)-Methyladenosine-Dependent Downregulating of Sirt1. Cell Death Dis (2021) 12:881. doi: 10.1038/s41419-021-04156-y
192. Jiang L, Liu X, Hu X, Gao L, Zeng H, Wang X, et al. METTL3-Mediated M(6)A Modification of TIMP2 mRNA Promotes Podocyte Injury in Diabetic Nephropathy. Mol Ther (2022) 30:1721–40. doi: 10.1016/j.ymthe.2022.01.002
193. Xu Z, Jia K, Wang H, Gao F, Zhao S, Li F, et al. METTL14-Regulated PI3K/Akt Signaling Pathway via PTEN Affects HDAC5-Mediated Epithelial-Mesenchymal Transition of Renal Tubular Cells in Diabetic Kidney Disease. Cell Death Dis (2021) 12:32. doi: 10.1038/s41419-020-03312-0
194. Xiong Y, Zhou L. The Signaling of Cellular Senescence in Diabetic Nephropathy. Oxid Med Cell Longev (2019) 2019:7495629. doi: 10.1155/2019/7495629
195. Kuro OM. The Klotho Proteins in Health and Disease. Nat Rev Nephrol (2019) 15:27–44. doi: 10.1038/s41581-018-0078-3
196. Shankland SJ. The Podocyte’s Response to Injury: Role in Proteinuria and Glomerulosclerosis. Kidney Int (2006) 69:2131–47. doi: 10.1038/sj.ki.5000410
197. Liu BH, Tu Y, Ni GX, Yan J, Yue L, Li ZL, et al. Total Flavones of Abelmoschus Manihot Ameliorates Podocyte Pyroptosis and Injury in High Glucose Conditions by Targeting METTL3-Dependent M(6)A Modification-Mediated NLRP3-Inflammasome Activation and PTEN/PI3K/Akt Signaling. Front Pharmacol (2021) 12:667644. doi: 10.3389/fphar.2021.667644
198. Hadden MJ, Advani A. Histone Deacetylase Inhibitors and Diabetic Kidney Disease. Int J Mol Sci (2018) 19:2630. doi: 10.3390/ijms19092630
199. Dong W, Jia Y, Liu X, Zhang H, Li T, Huang W, et al. Sodium Butyrate Activates NRF2 to Ameliorate Diabetic Nephropathy Possibly via Inhibition of HDAC. J Endocrinol (2017) 232:71–83. doi: 10.1530/JOE-16-0322
200. Wong TY, Cheung CM, Larsen M, Sharma S, Simo R. Diabetic Retinopathy. Nat Rev Dis Primers (2016) 2:16012. doi: 10.1038/nrdp.2016.12
201. Hammes HP. Diabetic Retinopathy: Hyperglycaemia, Oxidative Stress and Beyond. Diabetologia (2018) 61:29–38. doi: 10.1007/s00125-017-4435-8
202. Suo L, Liu C, Zhang QY, Yao MD, Ma Y, Yao J, et al. METTL3-Mediated N (6)-Methyladenosine Modification Governs Pericyte Dysfunction During Diabetes-Induced Retinal Vascular Complication. Theranostics (2022) 12:277–89. doi: 10.7150/thno.63441
203. Kumari N, Karmakar A, Ahamad Khan MM, Ganesan SK. The Potential Role of M6a RNA Methylation in Diabetic Retinopathy. Exp Eye Res (2021) 208:108616. doi: 10.1016/j.exer.2021.108616
204. Yao MD, Jiang Q, Ma Y, Liu C, Zhu CY, Sun YN, et al. Role of METTL3-Dependent N(6)-Methyladenosine mRNA Modification in the Promotion of Angiogenesis. Mol Ther (2020) 28:2191–202. doi: 10.1016/j.ymthe.2020.07.022
205. Yan M, Wang H, Gu Y, Li X, Tao L, Lu P. Melatonin Exerts Protective Effects on Diabetic Retinopathy via Inhibition of Wnt/beta-Catenin Pathway as Revealed by Quantitative Proteomics. Exp Eye Res (2021) 205:108521. doi: 10.1016/j.exer.2021.108521
206. Ma X, Long C, Wang F, Lou B, Yuan M, Duan F, et al. METTL3 Attenuates Proliferative Vitreoretinopathy and Epithelial-Mesenchymal Transition of Retinal Pigment Epithelial Cells via Wnt/Beta-Catenin Pathway. J Cell Mol Med (2021) 25:4220–34. doi: 10.1111/jcmm.16476
207. Zha X, Xi X, Fan X, Ma M, Zhang Y, Yang Y. Overexpression of METTL3 Attenuates High-Glucose Induced RPE Cell Pyroptosis by Regulating miR-25-3p/PTEN/Akt Signaling Cascade Through DGCR8. Aging (Albany NY) (2020) 12:8137–50. doi: 10.18632/aging.103130
208. Zhang J, Wang J, Zheng L, Wang M, Lu Y, Li Z, et al. miR-25 Mediates Retinal Degeneration Via Inhibiting ITGAV and PEDF in Rat. Curr Mol Med (2017) 17:359–74. doi: 10.2174/1566524018666171205122540
209. Qi Y, Yao R, Zhang W, Cui Q. KAT1 Triggers YTHDF2-Mediated ITGB1 mRNA Instability to Alleviate the Progression of Diabetic Retinopathy. Pharmacol Res (2021) 170:105713. doi: 10.1016/j.phrs.2021.105713
210. Park DY, Lee J, Kim J, Kim K, Hong S, Han S, et al. Plastic Roles of Pericytes in the Blood-Retinal Barrier. Nat Commun (2017) 8:15296. doi: 10.1038/ncomms15296
211. Kinuthia UM, Wolf A, Langmann T. Microglia and Inflammatory Responses in Diabetic Retinopathy. Front Immunol (2020) 11:564077. doi: 10.3389/fimmu.2020.564077
212. Fang M, Wan W, Li Q, Wan W, Long Y, Liu H, et al. Asiatic Acid Attenuates Diabetic Retinopathy Through TLR4/MyD88/NF-kappaB P65 Mediated Modulation of Microglia Polarization. Life Sci (2021) 277:119567. doi: 10.1016/j.lfs.2021.119567
213. Chen T, Zhu W, Wang C, Dong X, Yu F, Su Y, et al. ALKBH5-Mediated M(6)A Modification of A20 Regulates Microglia Polarization in Diabetic Retinopathy. Front Immunol (2022) 13:813979. doi: 10.3389/fimmu.2022.813979
214. Yang J, Liu J, Zhao S, Tian F. N(6)-Methyladenosine METTL3 Modulates the Proliferation and Apoptosis of Lens Epithelial Cells in Diabetic Cataract. Mol Ther Nucleic Acids (2020) 20:111–16. doi: 10.1016/j.omtn.2020.02.002
215. Dubey R, Prabhakar PK, Gupta J. Epigenetics: Key to Improve Delayed Wound Healing in Type 2 Diabetes. Mol Cell Biochem (2022) 477:371–83. doi: 10.1007/s11010-021-04285-0
216. Amin KN, Umapathy D, Anandharaj A, Ravichandran J, Sasikumar CS, Chandra SKR, et al. miR-23c Regulates Wound Healing by Targeting Stromal Cell-Derived Factor-1alpha (SDF-1alpha/CXCL12) Among Patients With Diabetic Foot Ulcer. Microvasc Res (2020) 127:103924. doi: 10.1016/j.mvr.2019.103924
217. Zhou J, Wei T, He Z. ADSCs Enhance VEGFR3-Mediated Lymphangiogenesis via METTL3-Mediated VEGF-C M(6)A Modification to Improve Wound Healing of Diabetic Foot Ulcers. Mol Med (2021) 27:146. doi: 10.1186/s10020-021-00406-z
218. Liang D, Lin WJ, Ren M, Qiu J, Yang C, Wang X, et al. M(6)A Reader YTHDC1 Modulates Autophagy by Targeting SQSTM1 in Diabetic Skin. Autophagy (2021) 17:1–20. doi: 10.1080/15548627.2021.1974175
219. Blumberg SN, Berger A, Hwang L, Pastar I, Warren SM, Chen W. The Role of Stem Cells in the Treatment of Diabetic Foot Ulcers. Diabetes Res Clin Pract (2012) 96:1–9. doi: 10.1016/j.diabres.2011.10.032
220. den Dekker A, Davis FM, Kunkel SL, Gallagher KA. Targeting Epigenetic Mechanisms in Diabetic Wound Healing. Transl Res (2019) 204:39–50. doi: 10.1016/j.trsl.2018.10.001
221. Lan CC, Liu IH, Fang AH, Wen CH, Wu CS. Hyperglycaemic Conditions Decrease Cultured Keratinocyte Mobility: Implications for Impaired Wound Healing in Patients With Diabetes. Br J Dermatol (2008) 159:1103–15. doi: 10.1111/j.1365-2133.2008.08789.x
222. Zhang W, Bai X, Zhao B, Li Y, Zhang Y, Li Z, et al. Cell-Free Therapy Based on Adipose Tissue Stem Cell-Derived Exosomes Promotes Wound Healing via the PI3K/Akt Signaling Pathway. Exp Cell Res (2018) 370:333–42. doi: 10.1016/j.yexcr.2018.06.035
223. Huang B, Huang LF, Zhao L, Zeng Z, Wang X, Cao D, et al. Microvesicles (MIVs) Secreted From Adipose-Derived Stem Cells (ADSCs) Contain Multiple microRNAs and Promote the Migration and Invasion of Endothelial Cells. Genes Dis (2020) 7:225–34. doi: 10.1016/j.gendis.2019.04.005
224. Lee J, Wu Y, Harada BT, Li Y, Zhao J, He C, et al. N(6) -Methyladenosine Modification of lncRNA Pvt1 Governs Epidermal Stemness. EMBO J (2021) 40:e106276. doi: 10.15252/embj.2020106276
225. Xi L, Carroll T, Matos I, Luo JD, Polak L, Pasolli HA, et al. M6a RNA Methylation Impacts Fate Choices During Skin Morphogenesis. Elife (2020) 9:e56980. doi: 10.7554/eLife.56980
226. Maldonado Lopez A, Capell BC. The METTL3-M(6)A Epitranscriptome: Dynamic Regulator of Epithelial Development, Differentiation, and Cancer. Genes (Basel) (2021) 12:1019. doi: 10.3390/genes12071019
227. Abdelkader NF, Ibrahim SM, Moustafa PE, Elbaset MA. Inosine Mitigated Diabetic Peripheral Neuropathy via Modulating GLO1/AGEs/RAGE/NF-Kappab/Nrf2 and TGF-Beta/PKC/TRPV1 Signaling Pathways. BioMed Pharmacother (2022) 145:112395. doi: 10.1016/j.biopha.2021.112395
228. Kallinikou D, Soldatou A, Tsentidis C, Louraki M, Kanaka-Gantenbein C, Kanavakis E, et al. Diabetic Neuropathy in Children and Adolescents With Type 1 Diabetes Mellitus: Diagnosis, Pathogenesis, and Associated Genetic Markers. Diabetes Metab Res Rev (2019) 35:e3178. doi: 10.1002/dmrr.3178
229. Witzel II, Jelinek HF, Khalaf K, Lee S, Khandoker AH, Alsafar H. Identifying Common Genetic Risk Factors of Diabetic Neuropathies. Front Endocrinol (Lausanne) (2015) 6:88. doi: 10.3389/fendo.2015.00088
230. Jankovic M, Novakovic I, Nikolic D, Mitrovic Maksic J, Brankovic S, Petronic I, et al. Genetic and Epigenomic Modifiers of Diabetic Neuropathy. Int J Mol Sci (2021) 22:4887. doi: 10.3390/ijms22094887
231. Satterlee JS, Basanta-Sanchez M, Blanco S, Li JB, Meyer K, Pollock J, et al. Novel RNA Modifications in the Nervous System: Form and Function. J Neurosci (2014) 34:15170–7. doi: 10.1523/JNEUROSCI.3236-14.2014
232. Kubota-Sakashita M, Iwamoto K, Bundo M, Kato T. A Role of ADAR2 and RNA Editing of Glutamate Receptors in Mood Disorders and Schizophrenia. Mol Brain (2014) 7:5. doi: 10.1186/1756-6606-7-5
233. Livneh I, Moshitch-Moshkovitz S, Amariglio N, Rechavi G, Dominissini D. The M(6)A Epitranscriptome: Transcriptome Plasticity in Brain Development and Function. Nat Rev Neurosci (2020) 21:36–51. doi: 10.1038/s41583-019-0244-z
234. Chatterjee B, Shen CJ, Majumder P. RNA Modifications and RNA Metabolism in Neurological Disease Pathogenesis. Int J Mol Sci (2021) 22:11870. doi: 10.3390/ijms222111870
235. Liu Y, Zhou T, Wang Q, Fu R, Zhang Z, Chen N, et al. M(6) A Demethylase ALKBH5 Drives Denervation-Induced Muscle Atrophy by Targeting HDAC4 to Activate FoxO3 Signalling. J Cachexia Sarcopenia Muscle (2022) 13:1210–23. doi: 10.1002/jcsm.12929
236. Boo SH, Kim YK. The Emerging Role of RNA Modifications in the Regulation of mRNA Stability. Exp Mol Med (2020) 52:400–08. doi: 10.1038/s12276-020-0407-z
237. Costa Cruz PH, Kawahara Y. RNA Editing in Neurological and Neurodegenerative Disorders. Methods Mol Biol (2021) 2181:309–30. doi: 10.1007/978-1-0716-0787-9_18
238. Tan MH, Li Q, Shanmugam R, Piskol R, Kohler J, Young AN, et al. Dynamic Landscape and Regulation of RNA Editing in Mammals. Nature (2017) 550:249–54. doi: 10.1038/nature24041
239. Sun T, Wu R, Ming L. The Role of M6a RNA Methylation in Cancer. BioMed Pharmacother (2019) 112:108613. doi: 10.1016/j.biopha.2019.108613
240. Lan Q, Liu PY, Bell JL, Wang JY, Huttelmaier S, Zhang XD, et al. The Emerging Roles of RNA M(6)A Methylation and Demethylation as Critical Regulators of Tumorigenesis, Drug Sensitivity, and Resistance. Cancer Res (2021) 81:3431–40. doi: 10.1158/0008-5472.CAN-20-4107
241. Zhang DY, Ming GL, Song H. PUS7: A Targetable Epitranscriptomic Regulator of Glioblastoma Growth. Trends Pharmacol Sci (2021) 42:976–78. doi: 10.1016/j.tips.2021.10.002
242. Qing Y, Dong L, Gao L, Li C, Li Y, Han L, et al. R-2-Hydroxyglutarate Attenuates Aerobic Glycolysis in Leukemia by Targeting the FTO/m(6)A/PFKP/LDHB Axis. Mol Cell (2021) 81:922–39 e9. doi: 10.1016/j.molcel.2020.12.026
243. Selberg S, Blokhina D, Aatonen M, Koivisto P, Siltanen A, Mervaala E, et al. Discovery of Small Molecules That Activate RNA Methylation Through Cooperative Binding to the METTL3-14-WTAP Complex Active Site. Cell Rep (2019) 26:3762–71 e5. doi: 10.1016/j.celrep.2019.02.100
244. Moroz-Omori EV, Huang D, Kumar Bedi R, Cheriyamkunnel SJ, Bochenkova E, Dolbois A, et al. METTL3 Inhibitors for Epitranscriptomic Modulation of Cellular Processes. ChemMedChem (2021) 16:3035–43. doi: 10.1002/cmdc.202100291
245. Bedi RK, Huang D, Eberle SA, Wiedmer L, Sledz P, Caflisch A. Small-Molecule Inhibitors of METTL3, the Major Human Epitranscriptomic Writer. ChemMedChem (2020) 15:744–48. doi: 10.1002/cmdc.202000011
246. Chen B, Ye F, Yu L, Jia G, Huang X, Zhang X, et al. Development of Cell-Active N6-Methyladenosine RNA Demethylase FTO Inhibitor. J Am Chem Soc (2012) 134:17963–71. doi: 10.1021/ja3064149
247. Li N, Kang Y, Wang L, Huff S, Tang R, Hui H, et al. ALKBH5 Regulates Anti-PD-1 Therapy Response by Modulating Lactate and Suppressive Immune Cell Accumulation in Tumor Microenvironment. Proc Natl Acad Sci USA (2020) 117:20159–70. doi: 10.1073/pnas.1918986117
Keywords: epitranscriptomics, diabetes, diabetic cardiomyopathy, diabetic nephropathy, diabetic retinopathy, diabetic wounds, diabetic neuropathy
Citation: Geng X, Li Z and Yang Y (2022) Emerging Role of Epitranscriptomics in Diabetes Mellitus and Its Complications. Front. Endocrinol. 13:907060. doi: 10.3389/fendo.2022.907060
Received: 29 March 2022; Accepted: 14 April 2022;
Published: 27 May 2022.
Edited by:
Zijie Zhang, Yunnan University, ChinaReviewed by:
Fan Shen, University of Alberta, CanadaRuifan Wu, South China Agricultural University, China
Copyright © 2022 Geng, Li and Yang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Ying Yang, eWFuZ3lpbmcyMDcyQDEyNi5jb20=