 Danielle L. Overton
Danielle L. Overton Teresa L. Mastracci
Teresa L. Mastracci
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Endocrinol. , 13 June 2022
Sec. Diabetes: Molecular Mechanisms
Volume 13 - 2022 | https://doi.org/10.3389/fendo.2022.904004
This article is part of the Research Topic Look who's Talking: Dialogues with Beta Cells View all 5 articles
Diabetes mellitus, a disease that affects nearly 536.6 million people worldwide, is characterized by the death or dysfunction of insulin-producing beta cells of the pancreas. The beta cells are found within the islets of Langerhans, which are composed of multiple hormone-producing endocrine cells including the alpha (glucagon), delta (somatostatin), PP (pancreatic polypeptide), and epsilon (ghrelin) cells. There is direct evidence that physical and paracrine interactions between the cells in the islet facilitate and support beta cell function. However, communication between endocrine and exocrine cells in the pancreas may also directly impact beta cell growth and function. Herein we review literature that contributes to the view that “crosstalk” between neighboring cells within the pancreas influences beta cell growth and function and the maintenance of beta cell health.
Diabetes mellitus is a chronic condition marked by elevated blood glucose levels, which can result from either the death or dysfunction of the insulin-producing beta cells in the pancreas. As of 2021, the global prevalence of diabetes cases was estimated to be 536.6 million with expectations to reach 783.2 million by 2045 (1). Research investigating how beta cells grow and function in the normal setting has provided a greater understanding of the pathways leading to beta cell dysfunction as well as possible avenues to reverse beta cell-related disease.
The insulin-producing beta cells are found within structures known as the islets of Langerhans, which are composed of multiple endocrine hormone-producing cell types including alpha (glucagon), delta (somatostatin), PP (pancreatic polypeptide), and epsilon (ghrelin) cells. As is made obvious by the complications resulting from diabetes, beta cells perform a function critical for the maintenance of whole-body metabolism. These cells are responsible for the secretion of insulin in response to elevated blood glucose levels, a function necessary to maintain whole-body metabolism. Interestingly, there is significant evidence that physical and paracrine interactions within the pancreas contribute to the maintenance of beta cell function. Moreover, there is mounting evidence that the islets/beta cells are influenced by the surrounding pancreatic exocrine acinar cells [reviewed in (2, 3)], which function to produce and secrete zymogens into the ductal system to facilitate digestion [reviewed in (4)]. Therefore, evaluating how cells within the pancreas communicate and interact can assist our understanding of organ growth, development and function.
The molecular and cellular communications that occur between cells and facilitate biological responses can loosely be categorized as “crosstalk”. In the pancreas, the physical proximity of endocrine cells to exocrine cells or to other endocrine cells in the islet, supports the idea that crosstalk may be possible between different pancreatic cell types. Animal models with defects in pancreas development as well as studies of pancreatic pathologies have also assisted our understanding of both pancreatic cell growth, function and dysfunction. Ultimately, investigating the cellular connections within the pancreas in both the healthy and diseased states provides a greater appreciation for the complexity of disease progression and the potential cellular opportunities that could be exploited to slow or reverse dysfunction. In this review, we explore pancreatic cellular crosstalk by examining the developmental cues that can instruct and manipulate certain pancreatic lineage decisions, the role of intra-islet communication and extracellular connections in beta cell function, and how exocrine pancreas function and dysfunction can influence the endocrine pancreas.
Endocrine and exocrine cells develop from a common progenitor cell population (Figure 1). In mammals, pancreas development begins when a region of the foregut endoderm receives signals from the aorta, notochord, and surrounding mesenchyme, leading to the emergence of the dorsal pancreatic progenitor cell domain (the “dorsal bud”) (5). Almost simultaneously, another portion of the endoderm receives inductive signals from the cardiac mesoderm, lateral plate mesoderm, and septum transversum mesenchyme to stimulate development of the ventral pancreatic progenitor cell domain (the “ventral bud”) (5). Once the pancreatic domains are specified in the embryo, development proceeds in two distinct stages: the primary transition and the secondary transition (5).

Figure 1 Pancreas development. Embryonic day (E) 9.5 marks the initiation of pancreas development in the mouse and the stage known as the primary transition. This first stage of development is characterized by the emergence of the dorsal and ventral pancreatic buds (purple) from the posterior foregut endoderm. These pancreatic buds are composed of progenitor cells that express the transcription factors Pdx1 and Ptf1a. The subsequent morphogenesis of the pancreatic ducts leads to an organization of the multipotent progenitor cells within the trunk (green) and tip (yellow) domains of the ductal epithelium. During the secondary transition (E12.5 – E15.5), the trunk cells become highly proliferative and push the tip cells to the exterior. Progenitor cells within the trunk express Neurog3 and subsequently differentiate into all islet cell types. Progenitor cells at the ductal tips, expressing Cpa1 and Ptf1a, and differentiate into acinar cells. By birth, the acinar cell mass has expanded and the islet cells begin to coalesce into the structures known as the islets of Langerhans.
The primary transition begins around embryonic day (E) 9.5 and concludes around E12.5. During this time, the progenitor cells within the dorsal and ventral buds expand by proliferation and organize to form the pancreatic epithelium (5) and the two distinct pancreatic domains fuse to become one organ (5). During the secondary transition, which spans E12.5 to E15.5, progenitor cells within the pancreatic epithelium differentiate, with the exocrine lineage deriving from progenitor cells in the “tip” domain of the epithelium and the endocrine lineage deriving from progenitor cells within the “trunk” domain (5). Changes in the expression of transcription factor networks regulate this pancreatic cellular differentiation, driving the formation of the distinct endocrine and exocrine cell lineages. To that end, transcription factor mutant mouse models have identified many genes responsible for these cell fate decisions. By resolving the factors or mechanisms necessary to instruct cell fate, we have gained significant insight into the cellular interactions that contribute to pancreas and beta cell development and function.
The transcription factors pancreas transcription factor 1 alpha (Ptf1a), pancreatic duodenal homeobox 1 (Pdx1), and NK6 homeobox 1 (Nkx6.1) are critical for the exocrine versus endocrine cell fate decision. Ptf1a is required for the specification and expansion of the pancreatic progenitor cells, as well as their differentiation into acinar cells (6–9). Pdx1 is also necessary for pancreatic organogenesis and then subsequently for proper beta cell development and function. In fact, Pdx1 is considered a master transcriptional regulator in the pancreas due to its roles not only in development but also insulin secretion, cell proliferation, and mitochondrial metabolism (10–14). The co-expression of Pdx1 and Ptf1a specifies the pancreatic progenitor cell pool and defines the initial cellular connection between the endocrine and exocrine pancreas (9). Similar to Pdx1, Nkx6.1 is required for proper development and function of the beta cells and is involved in the transcription factor network that instructs the endocrine versus exocrine fate decision (15–19). Moreover, a relationship between Nkx6 and Ptf1a was discovered wherein these transcription factors function to repress the alternative lineage program, driving the progenitors toward an endocrine or exocrine cell fate, respectively. A study by Schaffer et al. (20) demonstrated that changing the timing and localization of expression of Ptf1a and Nkx6.1 altered the differentiation of exocrine and endocrine cells. This study emphasizes the complex gene expression patterns necessary for proper pancreas development. Furthermore, it suggests that transcription factor expression could be exploited in certain circumstances to tip the scale toward increased endocrine cell production as a means to regenerate cells lost as a result of disease.
The creation of mutant animal models has also demonstrated how the exocrine pancreas can be altered. One example of this was shown in the study by Bonal et al. (21), which investigated the role of the basic helix-loop-helix protein c-Myc during pancreas development. C-Myc is known to direct progenitor cell expansion and differentiation, and loss of pancreatic c-Myc lead to decreased embryonic pancreas mass and postnatal transdifferentiation of acinar cell into adipocytes (21). The interruption of proper exocrine pancreas development was also observed with genetic deletion of the enzyme deoxyhypusine synthase (DHPS) in the embryonic pancreas (22). Loss of pancreatic Dhps resulted in a significant reduction in acinar cell mass due to an alteration in the translation of genes critical for exocrine pancreas development and function. The structural changes observed in these two studies alone demonstrate the importance of correct gene expression and signaling pathway integrity for the maintenance of proper pancreatic form and function.
Defining the timing and factors that direct critical lineages decisions in the pancreas can present theoretical opportunities for therapeutic manipulation in the setting of disease. To that end, inducing the expression of certain transcription factors can drive the trans-differentiation of exocrine (acinar) cells or other endocrine cell types into insulin-expressing beta-like cells. Specifically, overexpression of MafA, Pdx1, and Neurog3 redirected acinar cells toward a beta cell-like fate (23). Furthermore, loss of the gene Arx in alpha cells was shown to convert these glucagon-expressing cells into functional beta cells (24, 25). Similarly, pancreatic delta cells were manipulated to show the beta cell characteristic of insulin secretion, and thus aid in diabetes recovery, due to the action of FOXO1 (26). Whereas the goal to target a specific endogenous cell population to direct therapeutic trans-differentiation in the setting of diabetes has not yet been achieved, these studies have greatly assisted our understanding of the genes that regulate pancreatic cell growth. Further refining the cellular interactions that contribute to pancreas and beta cell development and function can also come from studying the islets themselves. By examining intra-islet cell communication and the external influences on the beta cells we obtain a greater understanding of the functional and dysfunctional beta cell.
Functionally, the endocrine and the exocrine compartments of the pancreas work in concert to achieve metabolic homeostasis. Succinctly, the endocrine cells, housed in the islets of Langerhans, function to secrete hormones that regulate glucose homeostasis and the exocrine cells function to secrete zymogens that become the digestive enzymes in the intestine required for digestion (27). As mentioned above, the physical location of the islets within the exocrine pancreas facilitates possible crosstalk between these cell types. Similarly, the organization of the islets of Langerhans, which bring all of the hormone-producing endocrine cells (alpha, beta, delta, PP and epsilon cells) into contact with each other, facilitates communication and thus there exists the possibility for positive and negative impact on cellular function. In particular, understanding intra-islet paracrine signaling in the developing and postnatal pancreas as well as the interactions of islets with the surrounding extracellular matrix (ECM) provides an opportunity to determine how these signals impact pancreatic beta cell function and/or contribute to beta cell dysfunction.
The communication that exists between the alpha and beta cells is commonly described as both counterregulatory and antagonistic, establishing a relationship wherein alpha cells are essential to beta cell function (Figure 2). This stems from the fact that beta cells are active during times of hyperglycemia whereas alpha cells are active during times of hypoglycemia [reviewed in (28)]. In essence, the hormones secreted from the alpha and beta cells oppose one another to maintain glucose homeostasis. However, this view of alpha and beta cell communication does not encompass the entirety of their relationship. Beyond their counterregulatory association, alpha and beta cells communicate with one another via paracrine signaling in order to maintain functionality. Svendsen et al. how insulin secretion is dependent upon intra-islet glucagon signaling. Specifically, beta cells that lack both the glucagon and GLP-1 receptors secrete significantly reduced levels of insulin, suggesting that paracrine signaling from alpha cells is necessary for proper beta cell function (29). Conversely, Kawamori et al. showed that deletion of the insulin receptor on alpha cells resulted in glucose intolerance, hyperglycemia, and hyperglucagonemia in male mice during the fed state, suggesting that paracrine signaling from beta cells is necessary for alpha cell function (30). Despite these studies, the hormonal interconnection between glucagon (alpha cells) and insulin (beta cells) remains an open question. Understanding the intricacies for how other secreted factors from these cells affect islet cell function may provide an additional endogenous avenue to positively impact beta cell functional integrity.
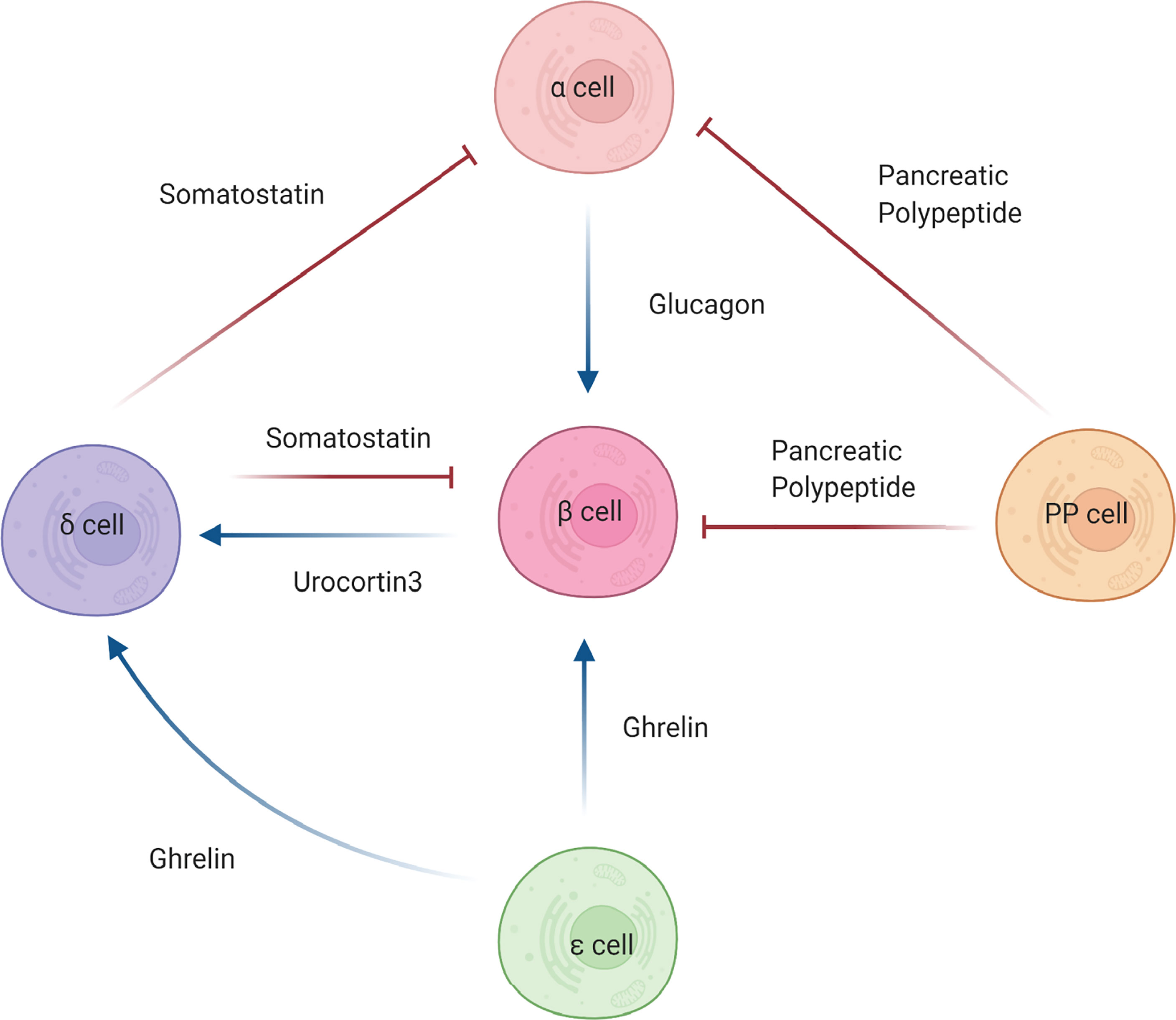
Figure 2 Intra-islet cell communications. Beta (β) cell function is influenced by the actions of other pancreatic islet cells. Alpha cells (α) secrete glucagon, which antagonistically effects insulin secretion from the beta cells. The action of glucagon can can be blunted by somatostatin from delta cells (δ) or pancreatic polypeptide by PP cells, leading to increased insulin secretion. Somatostatin and pancreatic polypeptide can also directly affect beta cells by negatively regulating insulin secretion. Ghrelin, secreted by epsilon cells (ε), positively regulates somatostatin release, which in turn negatively regulates insulin secretion. Furthermore, ghrelin can directly bind beta cells and, in certain forms, promote secretion of insulin. Urocortin3, secreted from the beta cells can mediates insulin secretion via somatostain. Blue arrows denote stimulatory hormonal effects. Red blunted arrows denote inhibitory hormonal effects.
Alpha and beta cells also interact closely with another endocrine cell type: delta cells. Delta cells primarily secrete somatostatin, which has been identified as a very strong inhibitor of both alpha cell-mediated glucagon release and beta cell-mediated insulin release (31) (Figure 2). Impaired secretion of somatostatin by delta cells has been shown to produce an exaggerated insulin release suggesting that beta cells work concurrently with delta cells via paracrine crosstalk to achieve glycemic control (32). The loss of the somatostatin receptor 2 (SSTR2) has been demonstrated to cause significantly higher levels of glucagon secretion, which further supports the necessity for delta cells in the regulation of alpha and beta cell function (33). Interestingly, delta cells were also discovered to express the ghrelin-receptor, which permits communication with ghrelin-expressing epsilon cells thus promoting an inhibitory feedback signal on insulin release (34, 35). Delta cells directly communicate with alpha, beta, and PP cells to maintain glucose homeostasis, showcasing their underrecognized importance and contribution to islet and beta cell function (36). Further investigation of the effects of somatostatin and other delta cell-derived factors is needed to define more completely the intra-islet signaling components emanating from the delta cells that influence beta cell function.
An even more underappreciated cell type in the islet is the pancreatic polypeptide-producing PP cells. Pancreatic polypeptide acts primarily as a digestive enzyme, inhibiting acid secretion and generally regulating digestive pancreatic secretions (37). However, the additional role PP cells may play in glucose metabolism is now becoming recognized (Figure 2). PP cells have been shown to be not as distantly related to the rest of the islet cell types as once thought (38). Rather, these cells have the potential to secrete hormones other than pancreatic polypeptide including insulin, glucagon and somatostatin, and exhibit the same identity markers as other islet cell types such as beta cells, alpha cells, and delta cells (38). Most excitingly, these cells display the ability to engage in insulin secretion in instances where the beta cells of the islet were damaged, despite originally lacking beta cell identity (38). This discovery connects back to a topic discussed above – the specific circumstances necessary to induce trans-differentiation and whether this can be harnessed to reverse the cellular dysfunction that results from disease. A potential caveat, however, lies in PP cell abundance, as they account for less than 1% of the total islet mass in the pancreatic tail but compose 70% of the total islet mass in the uncinate region and pancreatic head (39). As observed by Furuyama et al. (25), directing PP cells to function as insulin-secreting beta cells would also provide a potential therapeutic option in the setting of diabetes.
Epsilon cells are the fifth islet cell type found within the islets of Langerhans, although these ghrelin-expressing endocrine cells are found almost exclusively in the embryonic pancreas (40). Previous to their discovery in the pancreas, ghrelin-expressing cells were only identified in the stomach, where they function to secrete ghrelin, “the hunger hormone”, to increase appetite (41). Whereas there are limited studies of the pancreas, ghrelin has been shown to increase blood glucose levels while simultaneously decreasing plasma insulin levels (42) (Figure 2). Furthermore, it has been found that the loss of the ghrelin gene (Ghrl) in pancreatic epsilon cells has resulted in a significant increase in glucose-stimulated insulin secretion (43). Whereas ghrelin negatively impacts beta cell insulin secretion, in a context dependent manner, it has also been shown to positively impact islet cell mass by promoting cellular proliferation and growth (44). Granata et al. (45) demonstrated that in streptozotocin-treated rats, ghrelin gene products increased beta cell survival and prevented diabetes onset. Beta cells and other islet cells possess a ghrelin receptor known as GSH-R1a, through which ghrelin exerts its effects (46). Commonly denoted as the active form, acylated ghrelin specifically utilizes this binding site to activate the GTP-binding protein Gai2, which inhibits cAMP signaling and calcium influx. The result is less insulin release and an increase in blood glucose levels (42, 47). Its counterpart, un-acylated ghrelin, has not been shown to impact insulin sensitivity and is incapable of utilizing the GSH-R1a receptor, therefore demonstrating that it is not active in glucose metabolism (48). However, when acylated ghrelin and un-acylated ghrelin are combined, the result is increased insulin sensitivity, bringing into question the mechanisms by which this occurs (49). Obestatin, the third ghrelin gene product, is the most structurally distinct but arguably the most relevant to beta cell function. It has been shown to inhibit the apoptosis of beta cells, promote insulin release, and aid in sustaining beta cell longevity (50). Most notably, obestatin may serve as a regenerative stimulus for insulin-secreting cells derived from mesenchymal stem cells (51, 52). These studies showcase a positive effect of ghrelin and its derivatives on beta cell function and mass and support the idea that intra-islet derived factors exist that provide a direct benefit to beta cell health.
Although ghrelin alone is able to exert direct effects on beta cells, it should be noted that delta cells are also important in mediating the effect of ghrelin on insulin secretion. Transcriptomic profiling of pancreatic islet cells revealed that the ghrelin receptor gene was highly expressed and enriched in delta cells (35). Furthermore, activation of the ghrelin receptor on delta cells resulted in increased somatostatin secretion concomitant with decreased insulin secretion (35). This study confirms that there exists a complex endocrine regulatory network that is essential for proper insulin secretion and whole-body metabolism.
In addition to intra-islet communications, islet cells are physically connected with and therefore influenced by their surrounding environment. In particular, the extracellular connections that directly impact islet cell function include (but are not limited to) cell adhesion molecules (CAMs), calcium-dependent adhesion molecules (cadherins), integrins, and connexins (53). Whereas CAMs and cadherins act to ensure cell adhesion, connexins function to allow the transport of cytosolic molecules between connected cells [reviewed in (54)]. Furthermore, integrins act as a scaffold that connects beta cells, and other islet cells, to the extracellular basal lamina in order to receive internal and external signals. Specifically in beta cells, integrins play a direct role in glucose-stimulated insulin secretion by facilitating sensitivity to the presence of glucose (55), and the presence of integrins along with the ECM has been correlated with an increased rate of insulin secretion (56–58). Beta cells also exhibit direct cell-to-cell communication with their fellow islet cells via CAMs, which permit molecular communication between adjacent cells. For the mouse islet, cadherins, a subtype of CAMs mediated by calcium signaling, serve to arrange the cells in a distinct layout, with beta cells in the core and other islet cell types composing the mantle (59). In humans, cadherins play a similar structural role while also serving to protect beta cells from apoptosis by maintaining viability (60). It Is also known that beta cells interact with alpha cells by way of E-cadherin, a protein known to be integral to alpha and beta cell communication. To that end, in a beta cell-specific E-cadherin (CDH1) mutant animal model, the beta cells were unable to properly cluster and therefore did not receive regulatory signaling from alpha cells leading to disrupted glucose homeostasis (61). In general, this collective group of extracellular connections, which facilitate islet cell communication, may represent a possible avenue to manipulate hormone (e.g. insulin) production and possibly resolve beta cell dysfunction.
In general, the influence on the beta cell of other islet cell types or extracellular connections can be directly linked to physical proximity. With that in mind, the organization of the pancreas as a whole is such that the islets are embedded within the exocrine. Therefore, does form beget function? Can these two functionally distinct compartments directly influence their respective growth and function?
“Exocrine-endocrine crosstalk” can be defined as the cellular communications i.e. paracrine signals and/or external connections from the exocrine pancreas that influence growth and function of the endocrine pancreas (and vice versa) (Figure 3). Whereas the endocrine and exocrine compartments are considered to be functionally distinct, there are direct and indirect connections between the compartments that promote cellular growth, differentiation, viability, and function. Investigating these connections may unearth exogenous factors that could stimulate beta cell growth, differentiation, or insulin secretion. For example, the inhibition of pancreatic elastase, which is produced by the exocrine acinar cells, has been shown to cause increased beta cell proliferation (62). Thus, evidence for direct communication between the two functional compartments of the pancreas is slowly coming to light. This direct communication is likely assisted by the physical proximity and interconnections that exist between the exocrine and endocrine compartments, which are established during embryonic pancreas development. As described above, the conclusion of pancreas development results in the formation of a structured organ wherein the endocrine cells begin to coalesce into islets, which are distributed throughout the exocrine pancreas. This architecture is likely supportive in a healthy state; however, in the setting of pancreatic disease and inflammation, the close proximity of islets to acinar cells often results in collateral damage and islet dysfunction.
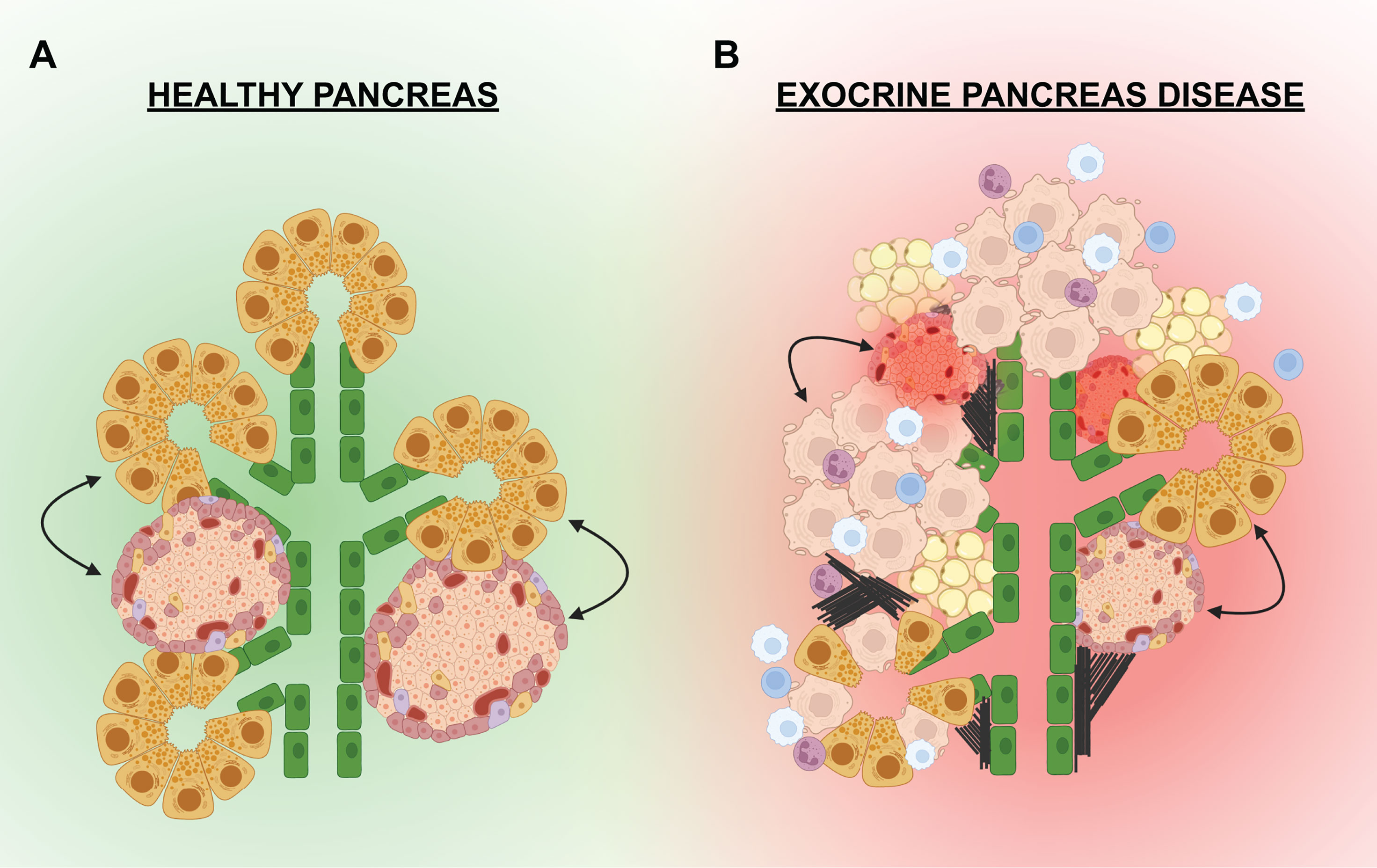
Figure 3 Exocrine-endocrine crosstalk in health and disease. (A) In a healthy pancreas, the islets are in physical proximity to the pancreatic epithelium (green) and the exocrine acinar cells (yellow). Paracrine factors or external cellular connections from the exocrine may influence the growth and function of the endocrine (and visa versa). (B) In the setting of exocrine disease, the pancreas can be infiltrated by immune cells (white, blue, purple) that promote inflammation and apoptosis of exocrine cells (beige). Fibrosis (black lines) and fatty deposits (tan) have also been noted in the pancreas of some exocrine diseases. Signals from the dying acinar cells, infiltrating immune cells, adipocytes, or external connections can negatively effect the islets leading to dysfunction and/or death.
Given the physical location of the islets within a sea of acinar cells in the pancreas, it is not surprising that pathological connections have been identified between the endocrine and exocrine pancreas. Both type 1 diabetes (T1D) and type 2 diabetes (T2D) have been described as having a high prevalence of exocrine insufficiency (63), with “exocrine insufficiency” being used as an umbrella term to describe an array of changes including general loss of pancreas size, acinar cell atrophy, and/or a reduction in exocrine enzymes (64–70). In addition to these changes, fibrosis, arteriosclerosis, and fatty infiltration have been observed in the pancreata of individuals with T1D (64). Furthermore, studies revealed that the clinical parameters of exocrine pancreas function including secretion of lipase, amylase, bicarbonate, trypsinogen, and fecal elastase, were decreased in individuals with T1D, which indicates the onset of exocrine insufficiency (64, 67, 68). Interestingly, the trend of reduced pancreas volume in T1D patients extends to first degree relatives, who also exhibit less total pancreas volume compared with healthy controls (66). Interestingly, individuals that express at least one T1D-associated autoantibody showed a similar trend of reduced pancreas volume compared with autoantibody negative controls, likely contributing to the increased risk of T1D (65). In studies examining pancreas size prior to the onset of T1D, it was revealed that the pancreas exhibits a decline in volume that begins with the appearance of autoantibodies and continues at least one year into clinical diagnosis (71). Of note, this study also determined that the reduced pancreatic mass was accompanied by a loss of structural integrity (71), which poses a possible explanation for decreased volume given that structural integrity is necessary for cell viability and proper function (72). What drives the loss of exocrine mass is not fully understood; however, there is data to support a mechanism that includes decreased exocrine cell number. Specifically, individuals with T1D displayed a striking 57% fewer total acinar cells compared with healthy controls (70). Interestingly, in this study the exocrine cell reduction could not be attributed solely to an increase in apoptosis, suggesting that there may be other factors that contribute to changes in pancreas size. Although the mechanisms driving exocrine loss and dysfunction remain unresolved, exocrine pancreatic insufficiency has become a significant characteristic associated with diabetes.
Direct evidence that endocrine function is impacted by exocrine dysfunction can be found in diseases wherein diabetes is observed following exocrine pancreas damage. Most significantly, Type 3c diabetes (T3cD) is a disease that begins in the exocrine pancreas due to pathologies such as chronic pancreatitis and pancreatic cancer; the disrupted exocrine pancreas then induces endocrine/beta cell dysfunction (73). This disease pathology underscores the impact of exocrine enzymes and other exocrine-associated proteins on islet and beta cell function. Studies suggest the loss of exocrine pancreas, as in chronic pancreatitis, contributes to T3cD indirectly by impacting insulin secretion via the incretin effect (73). Without nutrient absorption (mediated by pancreatic enzymes), the postprandial incretin response and insulin secretion are reduced, possibly impacting long-term beta cell health. Moreover, a study by Hostelley et al. (74) observed that exocrine pancreas-derived proteases regulate beta cell proliferation, specifically showcasing that an increase in certain pancreatic protease mRNAs correlated with an increase in beta cell mass. This would suggest that the changes in pancreatic enzyme levels observed in T3cD may be a confounding factor in beta cell health and progression to diabetes.
T3cD onset has also observed following pancreas cancer. Pancreatic ductal adenocarcinoma (PDAC) is a form of pancreatic cancer that originates from the epithelial cells of the pancreatic duct and results in the blockage of bicarbonate and enzyme secretion into the digestive tract (75, 76). PDAC has been reported to indirectly inhibit the secretion of insulin from pancreatic beta cells, which subsequently results in diabetes onset (77). The inhibition of beta cell secretion has been linked to tumor-derived exosomes that secrete micro-RNAs, which upregulate the PI3K/AKT/FOXO1 pathway, eventually leading to insulin resistance and beta cell failure (78). Interestingly, diabetes has been identified as a risk factor for pancreatic cancer, as later life diagnosis (after age 40) may predispose to PDAC (79, 80). Altogether, these studies point to a pathological connection between the endocrine and exocrine that influences pancreatic function. Exploring this connection may provide a greater understanding of the exocrine-derived factors and pathways that support proper endocrine health.
There exists clinical evidence from exocrine pathologies that exocrine-endocrine crosstalk can be a driver of disease. These include hereditary pancreatitis induced by mutations in PRSS1, metabolic syndrome caused by mutations in CELA2A, Cystic Fibrosis (CF), and Wolcott-Rallison Syndrome (81–84) (Table 1). In additional to genetic predisposition, these diseases may provide further evidence that diabetes onset following exocrine pancreatic disease stems from the physical proximity of the islets within a sea of dysfunctional acinar cells. Liu et al. (94) demonstrated that the prevalence of T3cD in patients with pancreatitis was increased in those patients with mutations in PRSS1, which cause improper production and secretion of the protease trypsinogen. This connection between diabetes onset and dysregulated protease production provides additional evidence that acinar cell secretions may play a role in the maintenance of beta cell health. Similarly, pancreatic elastase (encoded by CELA2A) is known to enhance insulin signaling by both promoting insulin secretion and mediating degradation (82). In the presence of CELA2A gene mutations and therefore nonfunctional CELA2A protein, dysregulated insulin secretion, hyperglycemia, and the eventual onset of T2D results (82, 95). Without this essential exocrine-derived protein, proper insulin signaling is unachievable. In essence, these studies identify factors that connect the exocrine pancreas function directly to islet health.
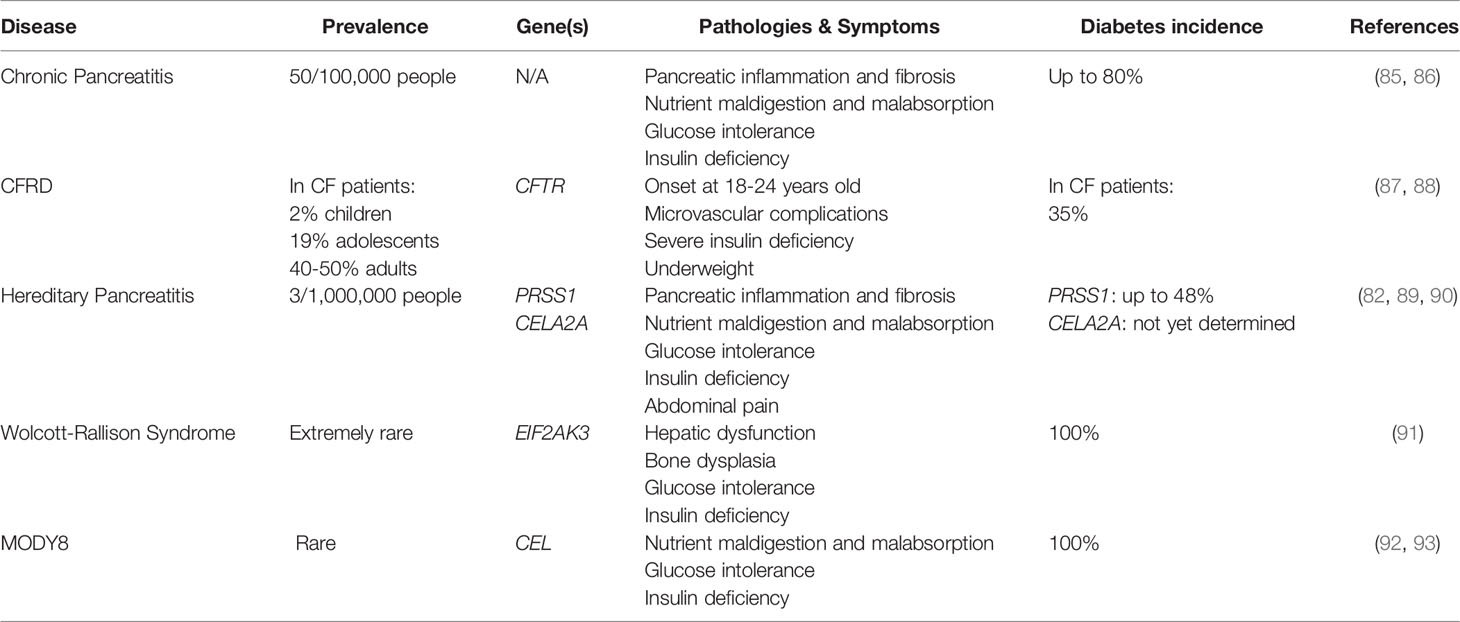
Table 1 Exocrine pathologies that progress to endocrine dysfunction.
Cystic fibrosis related diabetes (CFRD) is a form of diabetes caused by the monogenic disease CF (96). CF is a disease that arises from mutations in the cystic fibrosis transmembrane conductance regulator (CFTR) gene, resulting in disruptions of bicarbonate and salt flux and an accumulation of mucus in exocrine glands (96). For individuals with CF, the incidence of CFRD is estimated to be as high as 35%, with anecdotal clinical data suggesting that the prevalence is steadily increasing (81). CFRD is commonly recognized as a pancreas injury-driven form of diabetes, as studies in both humans and animal models demonstrate that it occurs as a result of pancreatic inflammation and autolysis (97, 98). However, there is also evidence that CFRD occurs as a result of altered CFTR directly impacting general beta cell function (99–101), cAMP regulated exocytosis of insulin (100), and glucagon secretion (102). Given that not all individuals with CF develop CFRD raised the question as to the role of CFTR mutations in diabetes development. Interestingly, Hart et al. (103) demonstrated that the loss of CTFR in the pancreatic beta cells of mice did not alter oral glucose tolerance or islet insulin secretion. Therefore, this study refutes the claims that CTFR in islet cells is critical to beta and alpha cell function and supports the data that beta cell dysfunction occurs as an indirect result of pancreatic inflammation. When considered alongside the clinical observations from patients with CF, CFRD likely occurs as a consequence of the islet/beta cell location within a dysfunctional exocrine pancreas. Therefore, this disease underscores the influence of the surrounding environment on beta cell integrity and function.
Like CF, Wolcott-Rallison Syndrome is a rare genetic disorder in which one of the prevalent comorbidities is diabetes. Diabetes resulting from Wolcott-Rallison Syndrome manifests in early infancy and individuals with the disease often do not survive to adulthood (83). Interestingly, the origin of this disease has been linked to mutations in EIF2AK3, the gene that encodes PKR-like ER kinase (PERK), which functions in the Unfolded Protein Response (UPR) to ameliorate endoplasmic reticulum (ER) stress (104, 105). EIF2AK3 expression levels are significantly higher in the pancreas (106, 107). Consistent with the importance of EIF2AK3 in the pancreas, Harding et al. demonstrated that mice lacking PERK developed exocrine pancreatic dysfunction and diabetes due to increased ER stress in pancreatic secretory cells (108). Interestingly, this effect was most prominent in the beta cells and acinar cells. Studying monogenic diseases such as Wolcott-Rallison Syndrome, where disease consequences impact both the exocrine and endocrine, highlights the crosstalk between these cell types as well as the potential mechanistic connections between acinar cells and beta cells.
Maturity Onset Diabetes of the Young type 8 (MODY8) is a form of diabetes that results from mutations in Carboxyl Ester Lipase (CEL) (92). Secreted by acinar cells, CEL functions to breakdown fats and allow their proper absorption into the intestines [reviewed in (109)]. Mutations in the CEL gene result in exocrine pancreatic insufficiency and eventual progression to diabetes. The mechanism underlying the progression to diabetes is not entirely clear; however, exocrine-endocrine crosstalk has been implicated. Kahraman et al. (110) demonstrated that CEL mutations produce dysfunctional CEL that is endocytosed by beta cells. Upon internalization, mutant CEL forms aggregates that disrupt beta cell secretion, ultimately hindering the beta cell’s capacity to maintain whole-body glucose metabolism. In essence, this supports the idea that exocrine pancreatic enzymes directly influence beta cell function and adds to the list of clinical diseases that provide evidence for proper endocrine/beta cell function being dependent upon proper acinar cell function.
In addition to physical proximity and the indirect effects from organ dysfunction, there is evidence that crosstalk between cells of the exocrine and endocrine pancreas can be facilitated by endogenous factors. In fact, the direct effects of exocrine secretions on beta cell health, function, and longevity have become a focus of significant investigation. Serine protease inhibitors (Serpins) and pancreatic enzymes such as carboxyl ester lipase (CEL), have been identified to influence beta growth and function, demonstrating another avenue through which the exocrine pancreas can exert effects on the endocrine. In particular, SerpinB1 is a pancreatic elastase inhibitor shown to promote beta cell proliferation by modulating growth and survival signaling (62). Derived from the liver, this inhibitor promotes the proliferation of beta cells in zebrafish, mice, and humans, indicating a general rather than organism-specific role (62). SerpinB1 inhibits pancreatic elastase, which suggests that in the normal setting certain acinar cell-derived digestive enzymes negatively impact beta cell growth, indicating direct exocrine-endocrine communication. Similar to SerpinB1, SerpinB13 influences beta cell growth by inhibiting a pancreatic protease. Specifically, SerpinB13 inhibits cathepsin L, known for its prominent role in pancreatic cancer metastasis; it is stored in and secreted from pancreatic lysosomes and ultimately functions by aiding in the modification of the extracellular matrix (111). Kryvalap et al. (112) demonstrated that the presence of SerpinB13 antibodies promoted beta cell development and imparted resistance to T1D. This occurred as a result of increased Notch1 cleavage leading to an upregulation in Notch signaling and growth (112). Excitingly, a decrease in SerpinB13 has also been found to be clinically relevant. Children with T1D who are given SerpinB13 antibodies showed a slower decline in residual beta cell function, which demonstrated the therapeutic potential for targeted protease inhibition (113). Taken together, these studies support the continued investigation of endogenous exocrine secretion in the promotion of beta cell growth and function. Moreover, the communication between acinar cells and beta cells via paracrine signals, physical connections, or environmental consequences of dysfunction demonstrates the importance of “exocrine-endocrine crosstalk” on beta cell health.
Cell-cell communication can influence organ development, function, and disease. Physical and molecular interactions between the cells in the pancreatic islet are known to facilitate and support beta cell function (Figure 2). However, the communication between exocrine and endocrine cells and the influence of this signaling on growth and function is not well understood. There is a growing body of literature from studies of both development and disease that suggest islets/beta cells are influenced by the surrounding pancreatic exocrine acinar cells. The study of “exocrine-endocrine crosstalk” (Figure 3) has therefore become a burgeoning area of investigation in the diabetes research field.
The need to decipher the mechanisms that drive intra-pancreatic cell communication and the influence of this crosstalk on cell and organ function has been driven by clinical observations. In particular, the occurrence of endocrine dysfunction, specifically diabetes, after exocrine pancreas damage or loss is significantly observed in many diseases (Table 1). Conversely, donor pancreas tissue from individuals with type 1 diabetes has been analyzed and shows a reduction in exocrine pancreas weight, acinar cell number, and exocrine-derived enzyme levels (63–70). Despite these convincing clinical observations supporting the idea of crosstalk, it remains unknown if the observed impact on organ function and size results from direct, targeted, cell-cell communication or simply occurs as collateral damage from disease. As noted above, the study of the origin of beta cell dysfunction in CFRD epitomizes this debate. There is research suggesting that CFTR loss-of-function drives impaired insulin secretion (99, 100); however, there is also evidence that the resultant diabetes occurs solely as a consequence of pancreatic inflammation (103, 114). Whether there are signals directly from the dysfunctional/dying exocrine cells that influence beta cell function remains a critical gap in knowledge.
Understanding the environment necessary for beta cell survival and proper insulin secretion is necessary for the generation of effective and long-lasting diabetes therapies. Current research demonstrates that islet transplantation is a successful therapy (115, 116). However, given the mounting evidence that the exocrine pancreas provides critical support and possibly direct maintenance of islet health, the question could be posed as to whether there would be even greater success if donor islets were transplanted into a more “pancreas-like” environment instead of the liver. The liver is clearly devoid of acinar cell and therefore the islets do not receive the expected endogenous exocrine signals. Perhaps donor islet health could be maintained for a greater period of time if transplanted into a more native environment or if whole pancreas transplant were performed. Clearly, further investigation is needed to fully understand the interactions between beta cells and exocrine tissue, and how these interactions might positively benefit islet transplant protocols.
To that end, the study of “exocrine-endocrine crosstalk” has produced novel findings related to the influence on pancreatic cell growth. In particular, studies of exocrine-derived secreted factors revealed that certain digestive enzymes can influence beta cell growth. For example, the inhibition of pancreatic elastase was found to directly stimulate beta cell growth (112). This finding alone has sparked excitement for the development of therapies; however, it is not without potential caveats. The mechanisms that drive beta cell expansion after treatment with digestive enzymes or enzyme inhibitors remains a gap in knowledge. It will be critical to understand the mechanism of action driving pancreatic enzymes to stimulate beta cell growth in the disease setting. As developmental mechanisms can often be coopted by diseased cells to drive pathology, we could look to the study of normal pancreatic development (Figure 1) to determine if exocrine enzymes serve a critical function during organ formation. It may be that exocrine signals (possibly in the form of secreted enzymes) function to restrict islet cell growth during development and, thus, inhibiting these enzymes in either a healthy or diseased state would stimulate beta cell growth. Altogether, defining the factors that direct beta cells to thrive, the influence of the exocrine on their differentiation, and the intra-pancreatic signals that promote their proper function may reveal new avenues for the development of therapeutics that can reverse diabetes.
All authors listed have made a substantial, direct, and intellectual contribution to the work and approved it for publication.
Funding provided by a Juvenile Diabetes Research Foundation (JDRF) Career Development Award (5-CDA-2016-194-A-N), an NIH R01 (1R01DK121987-01A1) and NIH R01 supplement (R01DK121987-S1) to TLM.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The authors would like to thank Craig Connors and Tanner Jackson for thoughtful discussions. Figures were made using BioRender.com.
1. Sun H, Saeedi P, Karuranga S, Pinkepank M, Ogurtsova K, Duncan BB, et al. IDF Diabetes Atlas: Global, Regional and Country-Level Diabetes Prevalence Estimates for 2021 and Projections for 2045. Diabetes Res Clin Pract (2022) 183:109119. doi: 10.1016/j.diabres.2021.109119
2. Wynne K, Devereaux B, Dornhorst A. Diabetes of the Exocrine Pancreas. J Gastroenterol Hepatol (2019) 34(2):346–54. doi: 10.1111/jgh.14451
3. Price S, Cole D, Alcolado JC. Diabetes Due to Exocrine Pancreatic Disease—a Review of Patients Attending a Hospital-Based Diabetes Clinic. QJM Int J Med (2010) 103(10):759–63. doi: 10.1093/qjmed/hcq127
4. Pandol SJ. The Exocrine Pancreas. In: Colloquium Series on Integrated Systems Physiology: From Molecule to Function to Disease. San Rafael (CA: Morgan & Claypool Life Sciences (2010). Available at: http://www.ncbi.nlm.nih.gov/books/NBK54128/.
5. Pan FC, Wright C. Pancreas Organogenesis: From Bud to Plexus to Gland. Dev Dyn Off Publ Am Assoc Anat (2011) 240(3):530–65. doi: 10.1002/dvdy.22584
6. Jin K, Xiang M. Transcription Factor Ptf1a in Development, Diseases and Reprogramming. Cell Mol Life Sci (2019) 76(5):921–40. doi: 10.1007/s00018-018-2972-z
7. Sellick GS, Barker KT, Stolte-Dijkstra I, Fleischmann C, Coleman RJ, Garrett C, et al. Mutations in PTF1A Cause Pancreatic and Cerebellar Agenesis. Nat Genet (2004) 36(12):1301–5. doi: 10.1038/ng1475
8. Fukuda A, Kawaguchi Y, Furuyama K, Kodama S, Horiguchi M, Kuhara T, et al. Reduction of Ptf1a Gene Dosage Causes Pancreatic Hypoplasia and Diabetes in Mice. Diabetes (2008) 57(9):2421–31. doi: 10.2337/db07-1558
9. Burlison JS, Long Q, Fujitani Y, Wright CVE, Magnuson MA. Pdx-1 and Ptf1a Concurrently Determine Fate Specification of Pancreatic Multipotent Progenitor Cells. Dev Biol (2008) 316(1):74–86. doi: 10.1016/j.ydbio.2008.01.011
10. Spaeth JM, Gupte M, Perelis M, Yang Y-P, Cyphert H, Guo S, et al. Defining a Novel Role for the Pdx1 Transcription Factor in Islet β-Cell Maturation and Proliferation During Weaning. Diabetes (2017) 66(11):2830–9. doi: 10.2337/db16-1516
11. Offield MF, Jetton TL, Labosky PA, Ray M, Stein RW, Magnuson MA, et al. PDX-1 is Required for Pancreatic Outgrowth and Differentiation of the Rostral Duodenum. Dev Camb Engl (1996) 122(3):983–95. doi: 10.1242/dev.122.3.983
12. Ahlgren U, Jonsson J, Edlund H. The Morphogenesis of the Pancreatic Mesenchyme Is Uncoupled From That of the Pancreatic Epithelium in IPF1/PDX1-Deficient Mice. Dev Camb Engl (1996) 122(5):1409–16. doi: 10.1242/dev.122.5.1409
13. Ahlgren U, Jonsson J, Jonsson L, Simu K, Edlund H. Beta-Cell-Specific Inactivation of the Mouse Ipf1/Pdx1 Gene Results in Loss of the Beta-Cell Phenotype and Maturity Onset Diabetes. Genes Dev (1998) 12(12):1763–8. doi: 10.1101/gad.12.12.1763
14. Gannon M, Ables ET, Crawford L, Lowe D, Offield MF, Magnuson MA, et al. Pdx-1 Function Is Specifically Required in Embryonic Beta Cells to Generate Appropriate Numbers of Endocrine Cell Types and Maintain Glucose Homeostasis. Dev Biol (2008) 314(2):406–17. doi: 10.1016/j.ydbio.2007.10.038
15. Taylor BL, Benthuysen J, Sander M. Postnatal β-Cell Proliferation and Mass Expansion Is Dependent on the Transcription Factor Nkx6.1. Diabetes (2015) 64(3):897–903. doi: 10.2337/db14-0684
16. Taylor BL, Liu F-F, Sander M. Nkx6.1 is Essential for Maintaining the Functional State of Pancreatic Beta Cells. Cell Rep (2013) 4(6):1262–75. doi: 10.1016/j.celrep.2013.08.010
17. Nelson SB, Schaffer AE, Sander M. The Transcription Factors Nkx6.1 and Nkx6.2 Possess Equivalent Activities in Promoting Beta-Cell Fate Specification in Pdx1+ Pancreatic Progenitor Cells. Dev Camb Engl (2007) 134(13):2491–500. doi: 10.1242/dev.002691
18. Pedersen JK, Nelson SB, Jorgensen MC, Henseleit KD, Fujitani Y, Wright CVE, et al. Endodermal Expression of Nkx6 Genes Depends Differentially on Pdx1. Dev Biol (2005) 288(2):487–501. doi: 10.1016/j.ydbio.2005.10.001
19. Sander M, Sussel L, Conners J, Scheel D, Kalamaras J, Dela Cruz F, et al. Homeobox Gene Nkx6.1 Lies Downstream of Nkx2.2 in the Major Pathway of Beta-Cell Formation in the Pancreas. Dev Camb Engl (2000) 127(24):5533–40. doi: 10.1242/dev.127.24.5533
20. Schaffer AE, Freude KK, Nelson SB, Sander M. Nkx6 Transcription Factors and Ptf1a Function as Antagonistic Lineage Determinants in Multipotent Pancreatic Progenitors. Dev Cell (2010) 18(6):1022–9. doi: 10.1016/j.devcel.2010.05.015
21. Bonal C, Thorel F, Ait-Lounis A, Reith W, Trumpp A, Herrera PL. Pancreatic Inactivation of C-Myc Decreases Acinar Mass and Transdifferentiates Acinar Cells Into Adipocytes in Mice. Gastroenterology (2009) 136(1):309–319.e9. doi: 10.1053/j.gastro.2008.10.015
22. Padgett LR, Robertson MA, Anderson-Baucum EK, Connors CT, Wu W, Mirmira RG, et al. Deoxyhypusine Synthase, an Essential Enzyme for Hypusine Biosynthesis, Is Required for Proper Exocrine Pancreas Development. FASEB J (2021) 35(5):e21473. doi: 10.1096/fj.201903177R
23. Clayton HW, Osipovich AB, Stancill JS, Schneider JD, Vianna PG, Shanks CM, et al. Pancreatic Inflammation Redirects Acinar to β Cell Reprogramming. Cell Rep (2016) 17(8):2028–41. doi: 10.1016/j.celrep.2016.10.068
24. Courtney M, Gjernes E, Druelle N, Ravaud C, Vieira A, Ben-Othman N, et al. The Inactivation of Arx in Pancreatic α-Cells Triggers Their Neogenesis and Conversion Into Functional β-Like Cells. PloS Genet (2013) 9(10):e1003934. doi: 10.1371/journal.pgen.1003934
25. Furuyama K, Chera S, van Gurp L, Oropeza D, Ghila L, Damond N, et al. Diabetes Relief in Mice by Glucose-Sensing Insulin-Secreting Human α-Cells. Nature (2019) 567(7746):43–8. doi: 10.1038/s41586-019-0942-8
26. Chera S, Baronnier D, Ghila L, Cigliola V, Jensen JN, Gu G, et al. Diabetes Recovery By Age-Dependent Conversion of Pancreatic δ-Cells Into Insulin Producers. Nature (2014) 514(7523):503–7. doi: 10.1038/nature13633
27. A-Kader HH, Ghishan FK. The Pancreas. Textb Clin Pediatr (2012) 1:1925–36. doi: 10.1007/978-3-642-02202-9_198
28. Moede T, Leibiger IB, Berggren P-O. Alpha Cell Regulation of Beta Cell Function. Diabetologia (2020) 63(10):2064–75. doi: 10.1007/s00125-020-05196-3
29. Insulin Secretion Depends on Intra-Islet Glucagon Signaling | Elsevier Enhanced Reader. Available at: https://reader.elsevier.com/reader/sd/pii/S2211124718315948?token=A7449C0A2B3F5785E7D9B8403A6EDFB0C558E89561A0D9AABEB6A008156FA2C4D9206816E74C532D81238E8C5DD432F6&originRegion=us-east-1&originCreation=20210719183607.
30. Kawamori D, Kurpad AJ, Hu J, Liew CW, Shih JL, Ford EL, et al. Insulin Signaling in α Cells Modulates Glucagon Secretion In Vivo. Cell Metab (2009) 9(4):350–61. doi: 10.1016/j.cmet.2009.02.007
31. Rorsman P, Huising MO. The Somatostatin-Secreting Pancreatic δ-Cell in Health and Disease. Nat Rev Endocrinol (2018) 14(7):404–14. doi: 10.1038/s41574-018-0020-6
32. van der Meulen T, Donaldson CJ, Cáceres E, Hunter AE, Cowing-Zitron C, Pound LD, et al. Urocortin3 Mediates Somatostatin-Dependent Negative Feedback Control of Insulin Secretion. Nat Med (2015) 21(7):769–76. doi: 10.1038/nm.3872
33. Strowski MZ, Parmar RM, Blake AD, Schaeffer JM. Somatostatin Inhibits Insulin and Glucagon Secretion via Two Receptors Subtypes: An In Vitro Study of Pancreatic Islets From Somatostatin Receptor 2 Knockout Mice. Endocrinology (2000) 141(1):111–7. doi: 10.1210/endo.141.1.7263
34. DiGruccio MR, Mawla AM, Donaldson CJ, Noguchi GM, Vaughan J, Cowing-Zitron C, et al. Comprehensive Alpha, Beta and Delta Cell Transcriptomes Reveal That Ghrelin Selectively Activates Delta Cells and Promotes Somatostatin Release From Pancreatic Islets. Mol Metab (2016) 5(7):449–58. doi: 10.1016/j.molmet.2016.04.007
35. Adriaenssens AE, Svendsen B, Lam BYH, Yeo GSH, Holst JJ, Reimann F, et al. Transcriptomic Profiling of Pancreatic Alpha, Beta and Delta Cell Populations Identifies Delta Cells as a Principal Target for Ghrelin in Mouse Islets. Diabetologia (2016) 59(10):2156–65. doi: 10.1007/s00125-016-4033-1
36. Kim W, Fiori JL, Shin Y-K, Okun E, Kim JS, Rapp PR, et al. Pancreatic Polypeptide Inhibits Somatostatin Secretion. FEBS Lett (2014) 588(17):3233–9. doi: 10.1016/j.febslet.2014.07.005
37. Lonovics J, Devitt P, Watson LC, Rayford PL, Thompson JC. Pancreatic Polypeptide. Rev Arch Surg Chic Ill 1960 (1981) 116(10):1256–64. doi: 10.1001/archsurg.1981.01380220010002
38. Perez-Frances M, van Gurp L, Abate MV, Cigliola V, Furuyama K, Bru-Tari E, et al. Pancreatic Ppy-Expressing γ-Cells Display Mixed Phenotypic Traits and the Adaptive Plasticity to Engage Insulin Production. Nat Commun (2021) 12(1):4458. doi: 10.1038/s41467-021-24788-0
39. Dolenšek J, Rupnik MS, Stožer A. Structural Similarities and Differences Between the Human and the Mouse Pancreas. Islets (2015) 7(1):e1024405. doi: 10.1080/19382014.2015.1024405
40. Wierup N, Svensson H, Mulder H, Sundler F. The Ghrelin Cell: A Novel Developmentally Regulated Islet Cell in the Human Pancreas. Regul Pept (2002) 107(1–3):63–9. doi: 10.1016/S0167-0115(02)00067-8
41. Pradhan G, Samson SL, Sun Y. Ghrelin: Much More Than a Hunger Hormone. Curr Opin Clin Nutr Metab Care (2013) 16(6):619–24. doi: 10.1097/MCO.0b013e328365b9be
42. Dezaki K, Hosoda H, Kakei M, Hashiguchi S, Watanabe M, Kangawa K, et al. Endogenous Ghrelin in Pancreatic Islets Restricts Insulin Release by Attenuating Ca2+ Signaling in Beta-Cells: Implication in the Glycemic Control in Rodents. Diabetes (2004) 53(12):3142–51. doi: 10.2337/diabetes.53.12.3142
43. Sun Y, Asnicar M, Saha PK, Chan L, Smith RG. Ablation of Ghrelin Improves the Diabetic But Not Obese Phenotype of Ob/Ob Mice. Cell Metab (2006) 3(5):379–86. doi: 10.1016/j.cmet.2006.04.004
44. Granata R, Volante M, Settanni F, Gauna C, Ghé C, Annunziata M, et al. Unacylated Ghrelin and Obestatin Increase Islet Cell Mass and Prevent Diabetes in Streptozotocin-Treated Newborn Rats. J Mol Endocrinol (2010) 45(1):9–17. doi: 10.1677/JME-09-0141
45. Granata R, Settanni F, Julien M, Nano R, Togliatto G, Trombetta A, et al. Des-Acyl Ghrelin Fragments and Analogues Promote Survival of Pancreatic β-Cells and Human Pancreatic Islets and Prevent Diabetes in Streptozotocin-Treated Rats. J Med Chem (2012) 55(6):2585–96. doi: 10.1021/jm201223m
46. Kageyama H, Funahashi H, Hirayama M, Takenoya F, Kita T, Kato S, et al. Morphological Analysis of Ghrelin and Its Receptor Distribution in the Rat Pancreas. Regul Pept (2005) 126(1):67–71. doi: 10.1016/j.regpep.2004.08.031
47. Dezaki K, Kakei M, Yada T. Ghrelin Uses Gαi2 and Activates Voltage-Dependent K+ Channels to Attenuate Glucose-Induced Ca2+ Signaling and Insulin Release in Islet β-Cells. Diabetes (2007) 56(9):2319–27. doi: 10.2337/db07-0345
48. Tong J, Davis HW, Summer S, Benoit SC, Haque A, Bidlingmaier M, et al. Acute Administration of Unacylated Ghrelin Has No Effect on Basal or Stimulated Insulin Secretion in Healthy Humans. Diabetes (2014) 63(7):2309–19. doi: 10.2337/db13-1598
49. Gauna C, Delhanty PJD, van Aken MO, Janssen JAMJL, Themmen APN, Hofland LJ, et al. Unacylated Ghrelin is Active on the INS-1E Rat Insulinoma Cell Line Independently of the Growth Hormone Secretagogue Receptor Type 1a and the Corticotropin Releasing Factor 2 Receptor. Mol Cell Endocrinol (2006) 251(1):103–11. doi: 10.1016/j.mce.2006.03.040
50. Li W, Chang M, Qiu M, Chen Y, Zhang X, Li Q, et al. Exogenous Obestatin Decreases Beta-Cell Apoptosis and Alfa-Cell Proliferation in High Fat Diet and Streptozotocin Induced Type 2 Diabetic Rats. Eur J Pharmacol (2019) 851:36–42. doi: 10.1016/j.ejphar.2019.02.028
51. El-Asfar RK, Kamal MM, Abd EL-Razek RS, EL-Demerdash E, El-Mesallamy HO. Obestatin can Potentially Differentiate Wharton’s Jelly Mesenchymal Stem Cells Into Insulin-Producing Cells. Cell Tissue Res (2018) 372(1):91–8. doi: 10.1007/s00441-017-2725-6
52. Yada T, Damdindorj B, Rita RS, Kurashina T, Ando A, Taguchi M, et al. Ghrelin Signalling in β-Cells Regulates Insulin Secretion and Blood Glucose. Diabetes Obes Metab (2014) 16(S1):111–7. doi: 10.1111/dom.12344
53. Meda P. Protein-Mediated Interactions of Pancreatic Islet Cells. Scientifica (2013) 2013:621249. doi: 10.1155/2013/621249
54. Su V, Lau AF. Connexins: Mechanisms Regulating Protein Levels and Intercellular Communication. FEBS Lett (2014) 588(8):1212–20. doi: 10.1016/j.febslet.2014.01.013
55. Peart J, Li J, Lee H, Riopel M, Feng Z-C, Wang R. Critical Role of β1 Integrin in Postnatal Beta-Cell Function and Expansion. Oncotarget (2017) 8(38):62939–52. doi: 10.18632/oncotarget.17969
56. Kaido T, Yebra M, Cirulli V, Rhodes C, Diaferia G, Montgomery AM. Impact of Defined Matrix Interactions on Insulin Production by Cultured Human β-Cells: Effect on Insulin Content, Secretion, and Gene Transcription. Diabetes (2006) 55(10):2723–9. doi: 10.2337/db06-0120
57. Beattie GM, Lappi DA, Baird A, Hayek A. Functional Impact of Attachment and Purification in the Short Term Culture of Human Pancreatic Islets. J Clin Endocrinol Metab (1991) 73(1):93–8. doi: 10.1210/jcem-73-1-93
58. Wang RN, Rosenberg L. Maintenance of Beta-Cell Function and Survival Following Islet Isolation Requires Re-Establishment of the Islet-Matrix Relationship. J Endocrinol (1999) 163(2):181–90. doi: 10.1677/joe.0.1630181
59. Halban PA, Powers SL, George KL, Bonner-Weir S. Spontaneous Reassociation of Dispersed Adult Rat Pancreatic Islet Cells Into Aggregates With Three-Dimensional Architecture Typical of Native Islets. Diabetes (1987) 36(7):783–90. doi: 10.2337/diab.36.7.783
60. Parnaud G, Gonelle-Gispert C, Morel P, Giovannoni L, Muller YD, Meier R, et al. Cadherin Engagement Protects Human β-Cells From Apoptosis. Endocrinology (2011) 152(12):4601–9. doi: 10.1210/en.2011-1286
61. Dahl U, Sjødin A, Semb H. Cadherins Regulate Aggregation of Pancreatic Beta-Cells In Vivo. Dev Camb Engl (1996) 122(9):2895–902. doi: 10.1242/dev.122.9.2895
62. El Ouaamari A, Dirice E, Gedeon N, Hu J, Zhou J-Y, Shirakawa J, et al. SerpinB1 Promotes Pancreatic β Cell Proliferation. Cell Metab (2016) 23(1):194–205. doi: 10.1016/j.cmet.2015.12.001
63. Hardt PD, Hauenschild A, Nalop J, Marzeion AM, Jaeger C, Teichmann J, et al. High Prevalence of Exocrine Pancreatic Insufficiency in Diabetes Mellitus. A Multicenter Study Screening Fecal Elastase 1 Concentrations in 1,021 Diabetic Patients. Pancreatol Off J Int Assoc Pancreatol IAP Al (2003) 3(5):395–402. doi: 10.1159/000073655
64. Foster TP, Bruggeman B, Campbell-Thompson M, Atkinson MA, Haller MJ, Schatz DA. Exocrine Pancreas Dysfunction in Type 1 Diabetes. Endocr Pract (2020) 26(12):1505–13. doi: 10.4158/EP-2020-0295
65. Campbell-Thompson M, Wasserfall C, Montgomery EL, Atkinson MA, Kaddis JS. Pancreas Organ Weight in Individuals With Disease-Associated Autoantibodies at Risk for Type 1 Diabetes. JAMA (2012) 308(22):2337–9. doi: 10.1001/jama.2012.15008
66. Campbell-Thompson ML, Filipp SL, Grajo JR, Nambam B, Beegle R, Middlebrooks EH, et al. Relative Pancreas Volume Is Reduced in First-Degree Relatives of Patients With Type 1 Diabetes. Diabetes Care (2019) 42(2):281–7. doi: 10.2337/dc18-1512
67. Lankisch PG, Manthey G, Otto J, Koop H, Talaulicar M, Willms B, et al. Exocrine Pancreatic Function in Insulin-Dependent Diabetes Mellitus. Digestion (1982) 25(3):211–6. doi: 10.1159/000198833
68. Li X, Campbell-Thompson M, Wasserfall CH, McGrail K, Posgai A, Schultz AR, et al. Serum Trypsinogen Levels in Type 1 Diabetes. Diabetes Care (2017) 40(4):577–82. doi: 10.2337/dc16-1774
69. Löhr M, Klöppel G. Residual Insulin Positivity and Pancreatic Atrophy in Relation to Duration of Chronic Type 1 (Insulin-Dependent) Diabetes Mellitus and Microangiopathy. Diabetologia (1987) 30(10):757–62. doi: 10.1007/BF00275740
70. Wright JJ, Saunders DC, Dai C, Poffenberger G, Cairns B, Serreze DV, et al. Decreased Pancreatic Acinar Cell Number in Type 1 Diabetes. Diabetologia (2020) 63(7):1418–23. doi: 10.1007/s00125-020-05155-y
71. Virostko J, Williams J, Hilmes M, Bowman C, Wright JJ, Du L, et al. Pancreas Volume Declines During the First Year After Diagnosis of Type 1 Diabetes and Exhibits Altered Diffusion at Disease Onset. Diabetes Care (2018) 42(2):248–57. doi: 10.2337/dc18-1507
72. Dias C, Nylandsted J. Plasma Membrane Integrity in Health and Disease: Significance and Therapeutic Potential. Cell Discovery (2021) 7(1):1–18. doi: 10.1038/s41421-020-00233-2
73. Hart PA, Bellin MD, Andersen DK, Bradley D, Cruz-Monserrate Z, Forsmark CE, et al. Type 3c (Pancreatogenic) Diabetes Mellitus Secondary to Chronic Pancreatitis and Pancreatic Cancer. Lancet Gastroenterol Hepatol (2016) 1(3):226–37. doi: 10.1016/S2468-1253(16)30106-6
74. Hostelley TL, Nesmith JE, Larkin E, Jones A, Boyes D, Leitch CC, et al. Exocrine Pancreas Proteases Regulate β-Cell Proliferation in Zebrafish Ciliopathy Models and in Murine Systems. Biol Open (2021) 10(6):bio046839. doi: 10.1242/bio.046839
75. Eibl AS, G. Pancreatic Ductal Adenocarcinoma. Pancreapedia Exocrine Pancreas Knowl Base (2015). Available at: https://www.pancreapedia.org/reviews/pancreatic-ductal-adenocarcinoma.
76. Rawla P, Sunkara T, Gaduputi V. Epidemiology of Pancreatic Cancer: Global Trends, Etiology and Risk Factors. World J Oncol (2019) 10(1):10–27. doi: 10.14740/wjon1166
77. Deng J, Guo Y, Du J, Gu J, Kong L, Tao B, et al. The Intricate Crosstalk Between Insulin and Pancreatic Ductal Adenocarcinoma: A Review From Clinical to Molecular. Front Cell Dev Biol (2022) 10:844028. doi: 10.3389/fcell.2022.844028
78. Wang L, Zhang B, Zheng W, Kang M, Chen Q, Qin W, et al. Exosomes Derived From Pancreatic Cancer Cells Induce Insulin Resistance in C2C12 Myotube Cells Through the PI3K/Akt/FoxO1 Pathway. Sci Rep (2017) 7(1):5384. doi: 10.1038/s41598-017-05541-4
79. Illés D, Ivány E, Holzinger G, Kosár K, Adam MG, Kamlage B, et al. New Onset of DiabetEs in Association With Pancreatic Ductal Adenocarcinoma (NODES Trial): Protocol of a Prospective, Multicentre Observational Trial. BMJ Open (2020) 10(11):e037267. doi: 10.1016/S1470-2045(08)70337-1
80. Ben Q, Xu M, Ning X, Liu J, Hong S, Huang W, et al. Diabetes Mellitus and Risk of Pancreatic Cancer: A Meta-Analysis of Cohort Studies. Eur J Cancer (2011) 47(13):1928–37. doi: 10.1016/j.ejca.2011.03.003
81. Granados A, Chan CL, Ode KL, Moheet A, Moran A, Holl R. Cystic Fibrosis Related Diabetes: Pathophysiology, Screening and Diagnosis. J Cyst Fibros (2019) 18:S3–9. doi: 10.1016/j.jcf.2019.08.016
82. Esteghamat F, Broughton JS, Smith E, Cardone R, Tyagi T, Guerra M, et al. CELA2A Mutations Predispose to Early-Onset Atherosclerosis and Metabolic Syndrome and Affect Plasma Insulin and Platelet Activation. Nat Genet (2019) 51(8):1233–43. doi: 10.1038/s41588-019-0470-3
83. Iyer S, Korada M, Rainbow L, Kirk J, Brown RM, Shaw N, et al. Wolcott-Rallison Syndrome: A Clinical and Genetic Study of Three Children, Novel Mutation in EIF2AK3 and a Review of the Literature. Acta Paediatr Oslo Nor 1992 (2004) 93(9):1195–201. doi: 10.1111/j.1651-2227.2004.tb02748.x
84. Zou W-B, Tang X-Y, Zhou D-Z, Qian Y-Y, Hu L-H, Yu F-F, et al. SPINK1, PRSS1, CTRC, and CFTR Genotypes Influence Disease Onset and Clinical Outcomes in Chronic Pancreatitis. Clin Transl Gastroenterol (2018) 9(11):204. doi: 10.1038/s41424-018-0069-5
85. Ewald N, Hardt PD. Diagnosis and Treatment of Diabetes Mellitus in Chronic Pancreatitis. World J Gastroenterol WJG. (2013) 19(42):7276–81. doi: 10.3748/wjg.v19.i42.7276
86. Yadav D, Lowenfels AB. The Epidemiology of Pancreatitis and Pancreatic Cancer. Gastroenterology (2013) 144:1252–61. doi: 10.1053/j.gastro.2013.01.068
87. Iafusco F, Maione G, Rosanio FM, Mozzillo E, Franzese A, Tinto N. Cystic Fibrosis-Related Diabetes (CFRD): Overview of Associated Genetic Factors. Diagn Basel Switz (2021) 11(3):572. doi: 10.3390/diagnostics11030572
88. Moran A, Dunitz J, Nathan B, Saeed A, Holme B, Thomas W. Cystic Fibrosis–Related Diabetes: Current Trends in Prevalence, Incidence, and Mortality. Diabetes Care (2009) 32(9):1626–31. doi: 10.2337/dc09-0586
89. Shelton C, Solomon S, LaRusch J, Whitcomb DC, Adam MP, Ardinger HH, et al. PRSS1-Related Hereditary Pancreatitis. In: Adam MP, Ardinger HH, Pagon RA, Wallace SE, Bean LJ, Gripp KW, et al, editors. GeneReviews®. Seattle (WA: University of Washington, Seattle (1993). Available at: http://www.ncbi.nlm.nih.gov/books/NBK84399/.
90. Rebours V, Lévy P, Ruszniewski P. An Overview of Hereditary Pancreatitis. Dig Liver Dis Off J Ital Soc Gastroenterol Ital Assoc Study Liver (2012) 44(1):8–15. doi: 10.1016/j.dld.2011.08.003
91. Julier C, Nicolino M. Wolcott-Rallison Syndrome. Orphanet J Rare Dis (2010) 5:29. doi: 10.1186/1750-1172-5-29
92. El Jellas K, Dušátková P, Haldorsen IS, Molnes J, Tjora E, Johansson BB, et al. Two New Mutations in the CEL Gene Causing Diabetes and Hereditary Pancreatitis: How to Correctly Identify MODY8 Cases. J Clin Endocrinol Metab (2021) 107:dgab864. doi: 10.1210/clinem/dgab864
93. Firdous P, Nissar K, Ali S, Ganai BA, Shabir U, Hassan T, et al. Genetic Testing of Maturity-Onset Diabetes of the Young Current Status and Future Perspectives. Front Endocrinol (2018) 9:253. doi: 10.3389/fendo.2018.00253
94. Liu Q, Zhuang Z, Zeng K, Cheng Z, Gao F, Wang Z. Prevalence of Pancreatic Diabetes in Patients Carrying Mutations or Polymorphisms of the PRSS1 Gene in the Han Population. Diabetes Technol Ther (2009) 11(12):799–804. doi: 10.1089/dia.2009.0051
95. Gloyn AL. Exocrine or Endocrine? A Circulating Pancreatic Elastase That Regulates Glucose Homeostasis. Nat Metab (2019) 1(9):853–5. doi: 10.1038/s42255-019-0107-y
96. Shteinberg M, Haq IJ, Polineni D, Davies JC. Cystic Fibrosis. Lancet (2021) 397(10290):2195–211. doi: 10.1016/S0140-6736(20)32542-3
97. Yi Y, Sun X, Gibson-Corley K, Xie W, Liang B, He N, et al. A Transient Metabolic Recovery From Early Life Glucose Intolerance in Cystic Fibrosis Ferrets Occurs During Pancreatic Remodeling. Endocrinology (2016) 157(5):1852–65. doi: 10.1210/en.2015-1935
98. Soejima K, Landing BH. Pancreatic Islets in Older Patients With Cystic Fibrosis With and Without Diabetes Mellitus: Morphometric and Immunocytologic Studies. Pediatr Pathol (1986) 6(1):25–46. doi: 10.3109/15513818609025923
99. Guo JH, Chen H, Ruan YC, Zhang XL, Zhang XH, Fok KL, et al. Glucose-Induced Electrical Activities and Insulin Secretion in Pancreatic Islet β-Cells are Modulated by CFTR. Nat Commun (2014) 5(1):4420. doi: 10.1038/ncomms5420
100. Edlund A, Esguerra JLS, Wendt A, Flodström-Tullberg M, Eliasson L. CFTR and Anoctamin 1 (ANO1) Contribute to cAMP Amplified Exocytosis and Insulin Secretion in Human and Murine Pancreatic Beta-Cells. BMC Med (2014) 12:87. doi: 10.1186/1741-7015-12-87
101. Ntimbane T, Mailhot G, Spahis S, Rabasa-Lhoret R, Kleme M-L, Melloul D, et al. CFTR Silencing in Pancreatic β-Cells Reveals a Functional Impact on Glucose-Stimulated Insulin Secretion and Oxidative Stress Response. Am J Physiol Endocrinol Metab (2016) 310(3):E200–12. doi: 10.1152/ajpendo.00333.2015
102. Edlund A, Pedersen MG, Lindqvist A, Wierup N, Flodström-Tullberg M, Eliasson L. CFTR is Involved in the Regulation of Glucagon Secretion in Human and Rodent Alpha Cells. Sci Rep (2017) 7(1):90. doi: 10.1038/s41598-017-00098-8
103. Hart NJ, Aramandla R, Poffenberger G, Fayolle C, Thames AH, Bautista A, et al. Cystic Fibrosis–Related Diabetes is Caused by Islet Loss and Inflammation. JCI Insight (2018) 3(8):e98240. doi: 10.1172/jci.insight.98240
104. Harding HP, Zhang Y, Ron D. Protein Translation and Folding are Coupled by an Endoplasmic-Reticulum-Resident Kinase. Nature (1999) 397(6716):271–4. doi: 10.1038/16729
105. Harding HP, Zhang Y, Bertolotti A, Zeng H, Ron D. Perk is Essential for Translational Regulation and Cell Survival During the Unfolded Protein Response. Mol Cell (2000) 5(5):897–904. doi: 10.1016/S1097-2765(00)80330-5
106. Brickwood S. Wolcott-Rallison Syndrome: Pathogenic Insights Into Neonatal Diabetes From New Mutation and Expression Studies of EIF2AK3. J Med Genet (2003) 40(9):685–9. doi: 10.1136/jmg.40.9.685
107. Tissue Expression of EIF2AK3 - Summary - The Human Protein Atlas. Available at: https://www.proteinatlas.org/ENSG00000172071-EIF2AK3/tissue.
108. Harding HP, Zeng H, Zhang Y, Jungries R, Chung P, Plesken H, et al. Diabetes Mellitus and Exocrine Pancreatic Dysfunction in Perk Ϫ/Ϫ Mice Reveals a Role for Translational Control in Secretory Cell Survival. Mol Cell (2001) 11:1153–63. doi: 10.1016/s1097-2765(01)00264-7
109. Johansson BB, Fjeld K, El Jellas K, Gravdal A, Dalva M, Tjora E, et al. The Role of the Carboxyl Ester Lipase (CEL) Gene in Pancreatic Disease. Pancreatol Off J Int Assoc Pancreatol IAP Al (2018) 18(1):12–9. doi: 10.1016/j.pan.2017.12.001
110. Kahraman S, Dirice E, Basile G, Diegisser D, Alam J, Johansson BB, et al. Abnormal Exocrine–Endocrine Cell Cross-Talk Promotes β-Cell Dysfunction and Loss in MODY8. Nat Metab (2022) 4(1):76–89. doi: 10.1038/s42255-021-00516-2
111. Singh N, Das P, Gupta S, Sachdev V, Srivasatava S, Datta Gupta S, et al. Plasma Cathepsin L: A Prognostic Marker for Pancreatic Cancer. World J Gastroenterol WJG. (2014) 20(46):17532–40. doi: 10.3748/wjg.v20.i46.17532
112. Kryvalap Y, Jiang ML, Kryvalap N, Hendrickson C, Czyzyk J. SerpinB13 Antibodies Promote β Cell Development and Resistance to Type 1 Diabetes. Sci Transl Med (2021) 13(588):eabf1587. doi: 10.1126/scitranslmed.abf1587
113. Kryvalap Y, Lo C-W, Manuylova E, Baldzizhar R, Jospe N, Czyzyk J. Antibody Response to Serpin B13 Induces Adaptive Changes in Mouse Pancreatic Islets and Slows Down the Decline in the Residual Beta Cell Function in Children With Recent Onset of Type 1 Diabetes Mellitus. J Biol Chem (2016) 291(1):266–78. doi: 10.1074/jbc.M115.687848
114. Sheikh S, Gudipaty L, De Leon DD, Hadjiliadis D, Kubrak C, Rosenfeld NK, et al. Reduced β-Cell Secretory Capacity in Pancreatic-Insufficient, But Not Pancreatic-Sufficient, Cystic Fibrosis Despite Normal Glucose Tolerance. Diabetes (2017) 66(1):134–44. doi: 10.2337/db16-0394
115. Rickels MR, Robertson RP. Pancreatic Islet Transplantation in Humans: Recent Progress and Future Directions. Endocr Rev (2019) 40(2):631–68. doi: 10.1210/er.2018-00154
Keywords: beta cell, exocrine pancreas, crosstalk, diabetes, exocrine pancreas dysfunction, pancreas development
Citation: Overton DL and Mastracci TL (2022) Exocrine-Endocrine Crosstalk: The Influence of Pancreatic Cellular Communications on Organ Growth, Function and Disease. Front. Endocrinol. 13:904004. doi: 10.3389/fendo.2022.904004
Received: 25 March 2022; Accepted: 26 April 2022;
Published: 13 June 2022.
Edited by:
Noelia Tellez, Universitat de Vic - Universitat Central de Catalunya, SpainReviewed by:
Meritxell Rovira, Institut d’Investigacio Biomedica de Bellvitge (IDIBELL), SpainCopyright © 2022 Overton and Mastracci. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Teresa L. Mastracci, dG1hc3RyYWNAaXUuZWR1
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.