
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Endocrinol. , 29 June 2022
Sec. Clinical Diabetes
Volume 13 - 2022 | https://doi.org/10.3389/fendo.2022.892677
 Robert Wagner1,2,3,4,5
Robert Wagner1,2,3,4,5 Sabine S. Eckstein1,2
Sabine S. Eckstein1,2 Louise Fritsche1,2
Louise Fritsche1,2 Katsiaryna Prystupa1,2,3
Katsiaryna Prystupa1,2,3 Sebastian Hörber1,2,6Hans-Ulrich Häring1,2,3
Sebastian Hörber1,2,6Hans-Ulrich Häring1,2,3 Andreas L. Birkenfeld1,2,3
Andreas L. Birkenfeld1,2,3 Andreas Peter1,2,6
Andreas Peter1,2,6 Andreas Fritsche1,2,3
Andreas Fritsche1,2,3 Martin Heni1,2,3,6,7*
Martin Heni1,2,3,6,7*Introduction: While oral glucose ingestion typically leads to a decrease in circulating glucagon levels, a substantial number of persons display stable or rising glucagon concentrations when assessed by radioimmunoassay (RIA). However, these assays show cross-reactivity to other proglucagon cleavage products. Recently, more specific assays became available, therefore we systematically assessed glucagon and other proglucagon cleavage products and their relation to metabolic health.
Research Design and Methods: We used samples from 52 oral glucose tolerance tests (OGTT) that were randomly selected from persons with different categories of glucose tolerance in an extensively phenotyped study cohort.
Results: Glucagon concentrations quantified with RIA were non-suppressed at 2 hours of the OGTT in 36% of the samples. Non-suppressors showed lower fasting glucagon levels compared to suppressors (p=0.011). Similar to RIA measurements, ELISA-derived fasting glucagon was lower in non-suppressors (p<0.001). Glucagon 1-61 as well as glicentin and GLP-1 kinetics were significantly different between suppressors and non-suppressors (p=0.004, p=0.002, p=0.008 respectively) with higher concentrations of all three hormones in non-suppressors. Levels of insulin, C-peptide, and free fatty acids were comparable between groups. Non-suppressors were leaner and had lower plasma glucose concentrations (p=0.03 and p=0.047, respectively). Despite comparable liver fat content and insulin sensitivity (p≥0.3), they had lower 2-hour post-challenge glucose (p=0.01).
Conclusions: Glucagon 1-61, glicentin and GLP-1 partially account for RIA-derived glucagon measurements due to cross-reactivity of the assay. However, this contribution is small, since the investigated proglucagon cleavage products contribute less than 10% to the variation in RIA measured glucagon. Altered glucagon levels and higher post-challenge incretins are associated with a healthier metabolic phenotype.
Glucagon, glicentin, and glucagon-like peptide (GLP)-1 as proglucagon cleavage products all originate from the same preproglucagon gene (see Figure 1). Differential expression of the preproglucagon gene in varying tissues is accompanied by differential processing of the proglucagon transcript by prohormone convertases.
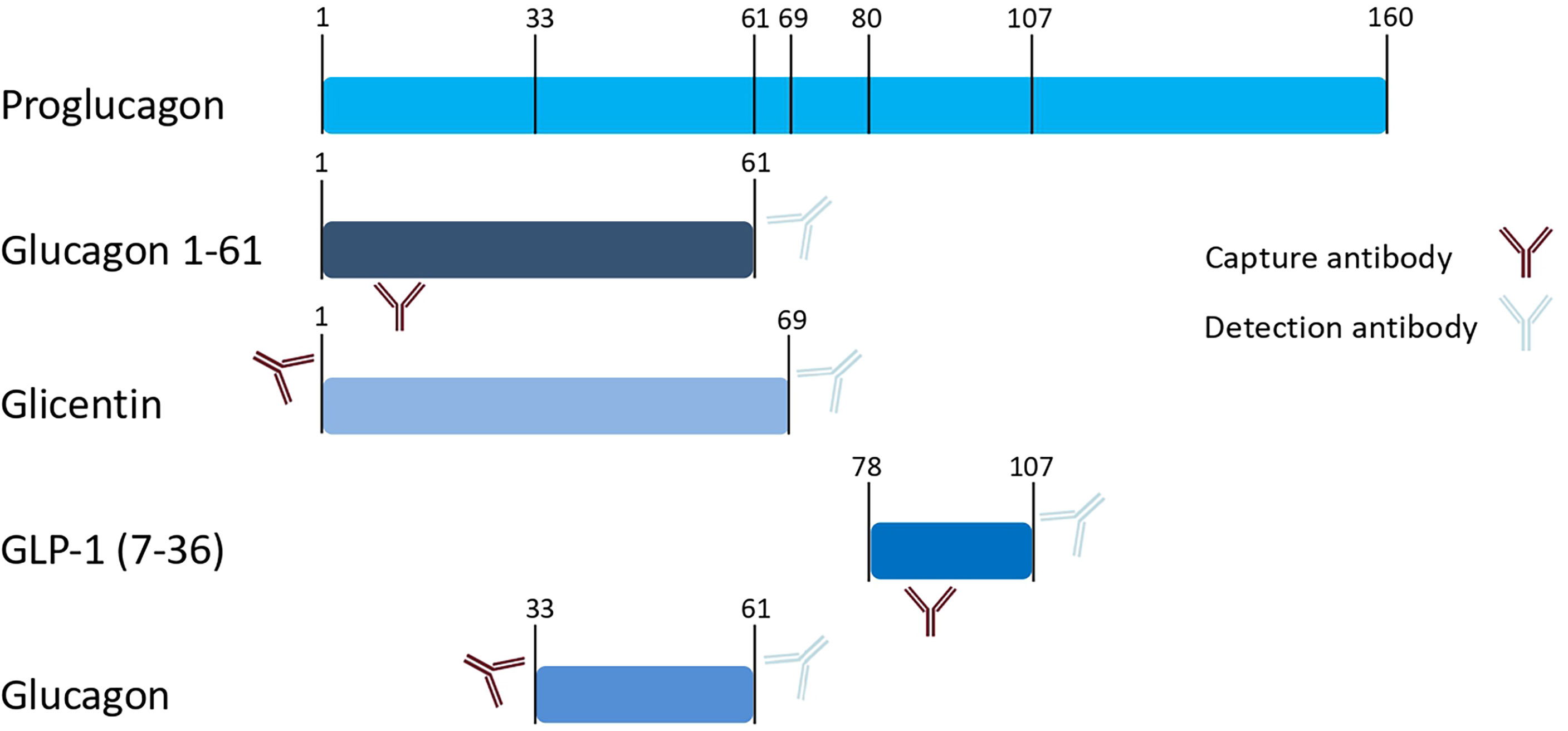
Figure 1 Schematic presentation of proglucagon and proglucagon cleavage products. Numbers indicate amino acid positions of cleavage sites. Antibodies schematically indicate epitopes that are used by the commercial immunoassays applied in our measurements (as provided by the manufacturer).
For a long time, counteracting insulin was thought to be the main function of glucagon. Since patients with type 2 diabetes show elevated fasting glucagon levels (1), the hormone has been believed to be a relevant contributor to hyperglycemia. Research over the last years, however, revealed a much more complex role of glucagon with potentially beneficial effects for whole-body metabolism (2, 3).
GLP-1 is an insulinotropic peptide that potentiates insulin secretion and mainly originates from the L cells in the distal small bowel and colon (4, 5). Glicentin is also produced in the intestinal L cells and its exact functions are still enigmatic. Studies in animal models and in vitro human tissues suggest a regulatory function on intestine, gastric acid production and insulin production (6–12).
Studies have shown that bariatric surgery for the treatment of obesity results in dramatic metabolic changes which can at least partially be attributed to incretins such as GLP-1 and glicentin (13–15). Uncovering the complex regulation that underlies the beneficial effects of bariatric surgery requires a better understanding of different incretins and their interplay on metabolism.
In a multi-cohort study with more than 4000 participants who underwent oral glucose tolerance tests, we found non-suppressed glucagon levels at 2-hours after glucose load in 21-34% of the study populations (16). Surprisingly, this non-suppression of glucagon was associated with a metabolically healthier phenotype. One limitation of the findings was the radioimmunoassay used to quantify glucagon in the study as this assay is known for substantial cross-reactivity with other proglucagon cleavage products (17), at least in an updated version marketed since around 1999 (18). This is caused by the overlapping amino acid sequences of glucagon with other incretin peptides, resulting in assay cross-reactivity when the capture antibody binds to an epitope within these shared sequences. The proglucagon sequence from amino acid position 33-61 corresponds to glucagon. The alternative gene product glicentin spans amino acids 1-69 of the proglucagon gene, whereas GLP-1 comprises amino acids 78-107 (Figure 1). Wewer-Albrechtsen et al. investigated glucagon 1-61, which is another circulating proglucagon fragment (spanning the proglucagon sequence 1-61). Glucagon 1-61 has been shown to stimulate insulin secretion and act on human hepatocytes in a series of experiments (19). Glucagon 1 – 61 comprises the amino acid sequence of glucagon and glicentin but lacks the eight C-terminal amino acids of glicentin (19) (Figure 1).
Due to a cross-reactivity of the radioimmunoassay, it is plausible that the presumed positive effects of glucagon on metabolism we observed in our previous study result from effects of other proglucagon fragments. Therefore, we aimed to investigate the post-load dynamics of glucagon and it’s with other proglucagon fragments quantified with a highly specific glucagon immunoassay.
Glucagon concentrations were first measured by radioimmunoassay (Figure 2A). Analyzing these measurements, 36% of the participants showed stable or rising RIA-derived glucagon concentrations during the OGTT. We defined them as non-suppressors. Of note, the non-suppressors had significantly lower fasting levels of glucagon compared to the suppressors (p=0.011). In parallel, samples were measured with the highly specific glucagon ELISA, showing lower values compared to RIA. Bland-Altman plots revealed reasonable agreement between the two methods with on average 15 ± 6 pmol/l higher concentrations from RIA measurements (Figure 3).
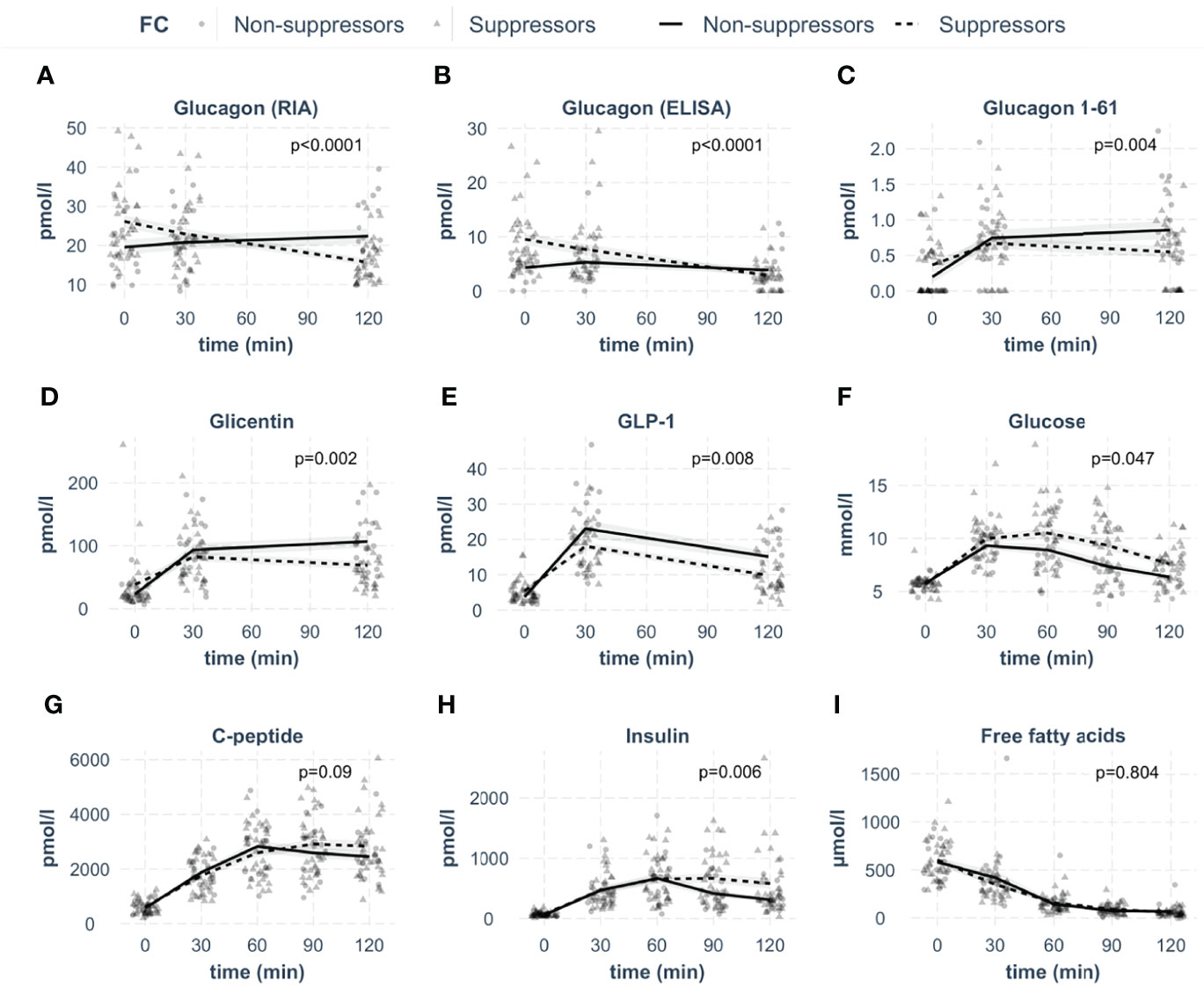
Figure 2 Concentrations of investigated analytes in the groups of glucagon suppressors and non-suppressors during the OGTT. The respective analyte is indicated in the box. (A: glucagon measured by radioimmunoassay, B: glucagon measured by ELISA, C: glucagon 1-61, D: glicentin, E: GLP-1, F: glucose, G: C-peptide, H: insulin, I: free fatty acids). Lines represent means with standard errors. Circles indicate data points for suppressors, triangles for non-suppressors, p-values were calculated with linear mixed models. N=52.
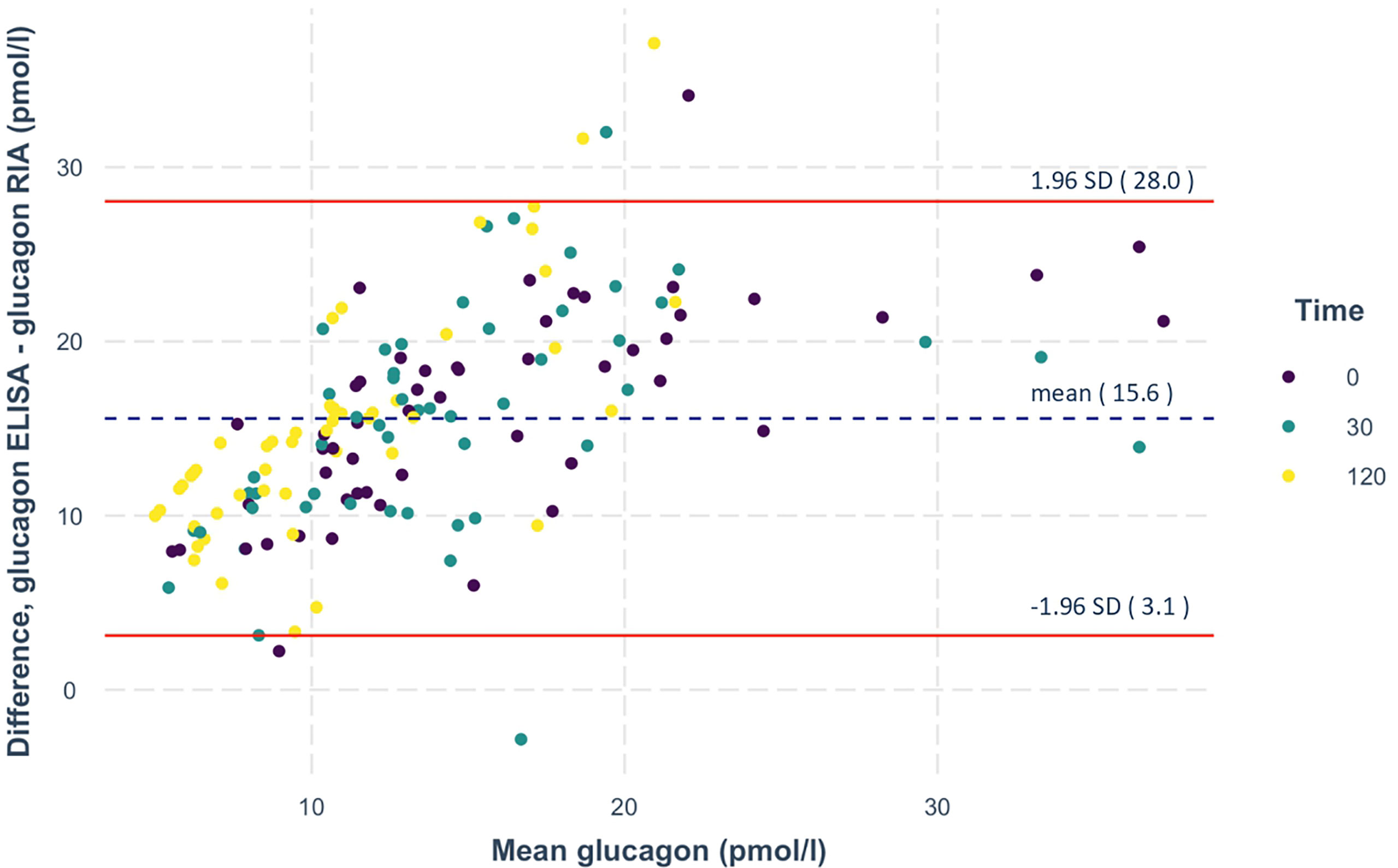
Figure 3 Bland-Altman plot of the RIA- and ELISA-measured glucagon. Differences in glucagon measurements between the two assays are plotted against mean glucagon values. The dashed lines represent the mean, the solid lines depict the lines of agreement calculated as mean ± 1.96 times the SD of this difference. N=156 measurements from 52 oGTTs.
The ELISA also detected persons with stable or rising glucagon. Similar to RIA measurements, fasting glucagon was lower in the non-suppressors (p<0.001). The RIA- and ELISA-derived glucagon suppressors and non-suppressors showed similar kinetics, regardless of which assay the hormone was measured with (Figures 2A, B). As the applied glucagon RIA is known to have cross-reactivity with other proglucagon cleavage products (18, 20, 21), we quantified these hormones by highly specific ELISAs. We measured glucagon 1-61, glicentin and GLP-1 from the same samples (Figures 2C–E).
To investigate the relative contribution of these proglucagon cleavage products to RIA-derived glucagon measurements, we modeled glucagon RIA levels at all available OGTT time points using glucagon ELISA, glicentin, glucagon 1-61 and GLP-1. The share of total variance explained by this model was 82.2%. The combination of the above-mentioned variables explained 43.6% as fixed effects. By removing each factor separately, we determined their relative contributions. In Figure 4, we show the relative contribution of glicentin, GLP-1 and ELISA-based glucagon to the total variance of the RIA-based glucagon measurement (total variance was set to 100%). ELISA measured glucagon explained 93% of the variance, whereas glicentin explained 5% of it. The contribution of GLP-1 (2%) was not statistically significant. Inclusion of Glucagon1-61 in the model deteriorated its performance and has been therefore removed from the final model.
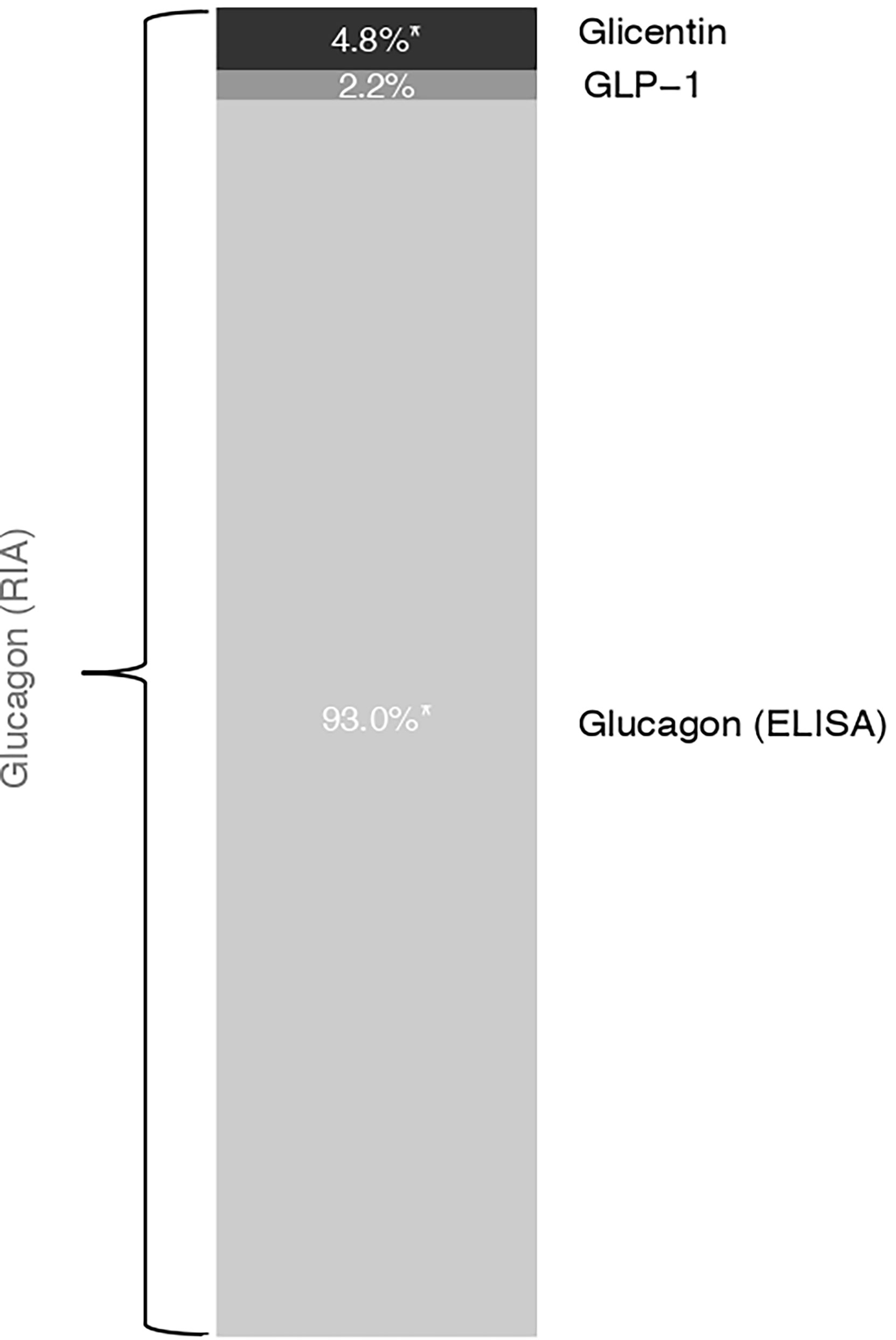
Figure 4 Estimated model-based relative contribution of proglucagon cleavage products to the variance of RIA-derived glucagon measurements. We used linear mixed models with the participant as a random intercept and the OGTT-time point as fixed effects. Marginal R squared was calculated to describe the proportion of variance in the outcome variable explained by the fixed effect. Removing each factor separately, we determined the percentage of its respective contribution to RIA-derived glucagon measurements that are presented here as a bar chart.
While the fasting concentration of glucagon 1-61 and glicentin were comparable between groups (p≥0.1), GLP-1 was significantly lower in non-suppressors (p=0.02).
Glucagon 1-61 as well as glicentin kinetics were significantly different between suppressors and non-suppressors with higher concentrations of both hormones in non-suppressors (Figures 2C, D). For GLP-1, the concentrations were not significantly different (Figure 2E).
As incretins stimulate pancreatic insulin release, we next analyzed insulin and C-peptide levels as well as plasma glucose. Insulin and C-peptide levels were comparable between the two groups (Figures 2G, H). However, insulin concentrations were lower in the non-suppressors in the last 30 minutes of the OGTT, resulting in a statistical difference between groups. In line with comparable insulin secretion, suppression of free fatty acids was not different between groups (Figure 2I). However, plasma glucose concentrations were significantly lower in the non-suppressors (Figure 2F), even after adjustment for BMI (p=0.04).
The non-suppressors were leaner (p=0.03) and had a lower waist circumference (p=0.04). Despite comparable liver fat content and insulin sensitivity (p≥0.3), they had lower post-challenge glucose concentrations (p=0.01), which remained significant after adjustment for BMI (p=0.002).
In a meta-analysis with over 4000 participants, we previously identified non-suppression of glucagon in response to oral glucose intake, to be linked to a favorable whole-body metabolism (16). One limitation of that study was quantification of glucagon by RIA that likely cross-reacts with additional proglucagon cleavage products. We therefore performed comparative analyses of glucagon kinetics assessed by RIA and a highly sensitive ELISA. In addition, we investigated the kinetics of the major proglucagon cleavage products glucagon 1-61, glicentin and GLP-1. Regardless of the measurement approach, we identified persons with glucagon suppression during the OGTT and such with stable or even rising concentrations. The persons that did not suppress RIA-measured glucagon in response to oral glucose intake showed higher incretin responses during the glucose challenge. Of note, these differences in glucagon and incretin kinetics were accompanied by a metabolically healthier phenotype.
Our data suggest that glucagon 1-61, glicentin and GLP-1 could partially account for RIA-derived glucagon measurements. However, this contribution appears to be small, because (i) there is a reasonable agreement between RIA and ELISA measurements and (ii) the investigated proglucagon cleavage products explain less than 10% of total glucagon measured with RIA.
There is substantial controversy on the role of glucagon in the pathogenesis of diabetes (22, 23). While glucagon promotes hepatic glucose production, this effect is self-limited, and compensated by glucagon’s stimulation of insulin secretion, inhibition of hepatic triglyceride synthesis, increase in basal energy expenditure and central inhibitory effects on appetite (3, 23, 24). Glucagon also stimulates GLP-1 receptors, though, to a lesser extent than its primary ligand (25), that has been proven beneficial in the treatment of both diabetes and obesity. Thus, although there is a clear association of fasting hyperglucagonemia and type 2 diabetes (26, 27), this might be secondary to compensatory processes without playing a causal role in diabetes development. When it comes to postprandial changes in glucagon concentrations, results are even more puzzling (16, 28–30) and the detailed contribution in the pathogenesis of type 2 diabetes is still unclear (22). While glucagon non-suppression in the postprandial state was long thought to be a hallmark of impaired glucose tolerance, based on smaller studies (31, 32), this did not hold true in our large meta-analysis (16), where post-load non-suppression of glucagon was detected in those with a healthier phenotype.
In our current study, we replicated this finding with the highly specific glucagon immunoassay and extended these findings by quantifying additional proglucagon cleavage products. Higher post-load levels of glucagon 1-61, glicentin and GLP-1 in the glucagon non-suppressor group were associated with lower blood glucose levels. Given the insulinotropic effect of incretins, the most obvious explanation could be a difference in insulin secretion between the two groups. However, our data argue against this, as we detected no significant difference in C-peptide levels, which are indicative for pancreatic insulin secretion. This is further underlined by comparable insulin-induced suppression of lipolysis, which was assessed by free fatty acid concentrations.
Fasting glucose, that strongly relies on endogenous glucose production (33) is not different between the suppressor and non-suppressor glucagon groups. This suggests that endogenous glucose production does not have a relevant contribution to differences in glycaemia between the two groups. Comparable whole-body insulin sensitivity argues against major differences in hepatic insulin sensitivity that would result in different suppression of endogenous glucose production during the OGTT.
One possible explanation for the altered glucose levels is therefore a difference in non-insulin dependent glucose disposal. Non-insulin dependent glucose disposal is also referred to as glucose sensitivity or glucose effectiveness. This term describes the ability of glucose to regulate glucose disposal and gluconeogenesis by itself under basal insulin conditions (34). Tissues such as fat and muscle usually take up glucose in an insulin-dependent fashion. However, insulin-independent glucose uptake is also present (35, 36). Glucagon has direct glucoregulatory effects through the brain (24). Thus, one possibility is that glucose effectiveness is regulated via the brain, though the molecular mechanisms are still unclear (37). Our results suggest that glucagon or incretins could be involved and may promote non-insulin dependent glucose disposal. This mechanism could contribute to the glucose-lowering effect of these hormones and to recent pharmacotherapies that target these pathways.
Apart from its classical hyperglycemic potency, glucagon has pleiotropic effects on appetite, energy metabolism and hepatic triglyceride synthesis. To exploit the beneficial effects while compensating for hyperglycemic effects, GLP-1 and glucagon coagonists are already being clinically tested for the treatment of diabetes and non-alcoholic steatohepatitis. Since levels increase postprandially, one mechanistic explanation for the association of increased post-challenge glucagon with metabolic health could be a physiologic co-agonism of GLP-1 and glucagon.
Our study is limited by the sample size and the fact that we did not apply mass spectrometry as gold standard for the identification of peptide hormones. We also did not measure all proglucagon cleavage products that could cross-react with the glucagon radioimmunoassay, including oxyntomodulin (18). In addition, we did not test additional stimuli that potentially trigger gastrointestinal secretion of proglucagon cleavage products, e.g. mixed meal test.
In summary, we verified that in some persons oral glucose intake does not result in a suppression of glucagon. Of note, these persons additionally displayed higher post-load concentrations of further preproglucagon cleavage-products, including glicentin. As these persons are leaner and have lower blood glucose, our results indicate that proglucagon cleavage-products could potentially contribute to the development of a healthier metabolic phenotype.
Samples from 52 randomly selected participants of the prediabetes lifestyle intervention study (38) (PLIS, registered with clinical trial.gov NCT01947595) were investigated in this project. None of the study participants took any kind of medication that interferes with glucose or energy metabolism. The study was approved by the local ethics board (Ethics Committee of the Medical Faculty of the Eberhard Karls University and the University Hospital Tübingen) and conducted in accordance with the declaration of Helsinki. All participants provided written informed consent.
Oral glucose tolerance tests (OGTT) were performed after an overnight fasting period of 12 hours. Venous blood was drawn at baseline and 30, 60, 90 and 120 minutes after drinking a 75 g glucose solution (Accu-Check Dextrose O.G.T., Roche Diagnostics, Mannheim, Germany). Plasma glucose and free fatty acids were measured from sodium fluoride plasma with an ADVIA chemistry XPT autoanalyzer (Siemens Healthineers). Serum insulin and C-peptide were analyzed using ADVIA Centaur XPT immunoassay system (Siemens Healthineers). Study participants were categorized into glycemic groups: normal fasting glucose, impaired fasting glucose, impaired glucose tolerance, impaired fasting glucose + impaired glucose tolerance, and type 2 diabetes according to ADA criteria (39). For details see Table 1.
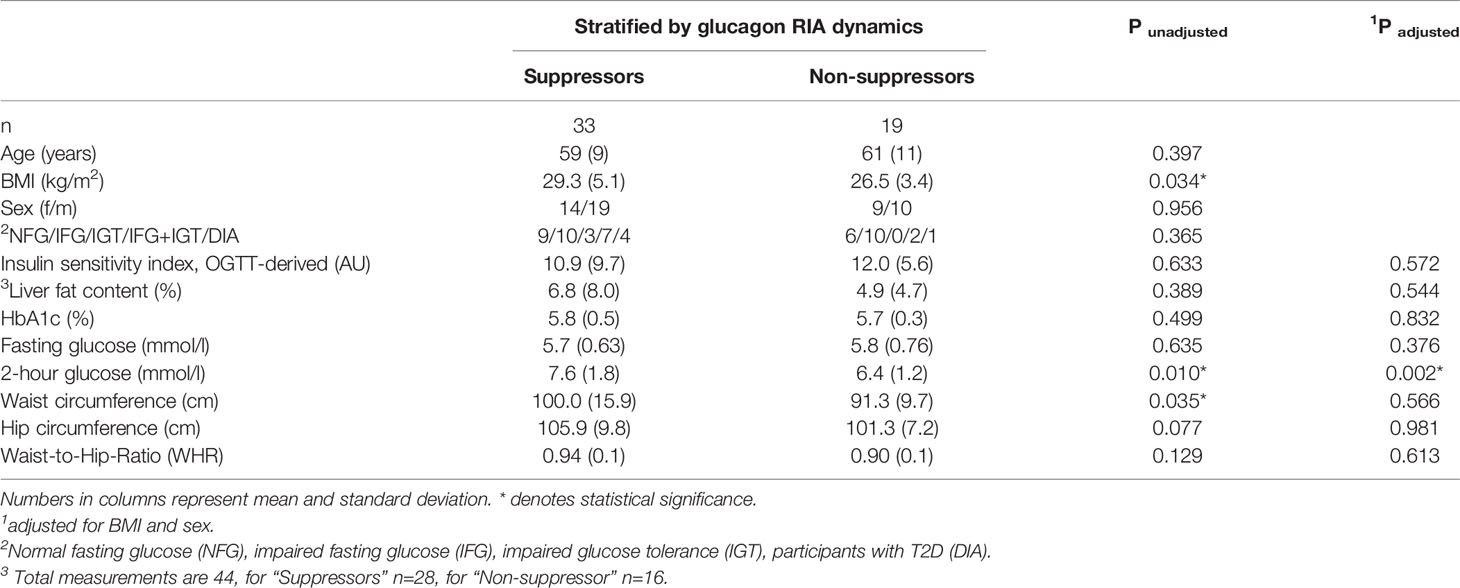
Table 1 Subject characteristics.
Glucagon was measured by a commercially available radioimmunoassay (Linco Research/Millipore, St. Charles, MO). Glucagon, glicentin and GLP-1 were also measured with commercially available ELISA assays (Mercodia, Uppsala, Sweden) according to the manufacturer’s instructions. We did not use the novel extended washing protocol that had been recommended for glucagon measurements in patients after bariatric surgery. The antibody binding sites (19, 40) are indicated in Figure 1. Glucagon 1-61 was measured with a prototype ELISA from Mercodia (Uppsala, Sweden) according to the manufacturer’s instructions. The prototype ELISA shows 0.5% cross-reactivity with glicentin and < 0.2% with glucagon respectively. To ensure optimal sample quality, EDTA plasma was stabilized with 300 ng/ml protease inhibitor aprotinin (Sigma, Merck, Darmstadt, Germany) und subsequently processed at 4°C and kept frozen at -80°C until batch measurement.
HbA1c was measured by HPLC. Height, weight, waist and hip circumference were measured according to standard operating procedures. Liver fat content was measured by localized (1)H-MR spectroscopy using a 1.5 T MR scanner (Magnetom Sonata, Siemens Healthcare, Erlangen, Germany) and the distribution of lean and adipose tissues was measured by whole body MR imaging as previously described (41).
Insulin sensitivity was assessed with the index proposed by Matsuda and De Fronzo (42).
The relative change of glucagon RIA from 0 to 120 min was used to define the groups of suppressors and non-suppressors. The suppressors were determined as participants with the fold change glucagon RIA (glucagon RIA 120 min/glucagon RIA 0 min) less than 1, while non-suppressors were those with equal or higher than 1.
To analyze measurements from the same participants at different time-points of the OGTT, linear mixed models with the participant as random intercept and the OGTT-time point as fixed effects were used. By constructing a model with RIA glucagon as outcome and glicentin, ELISA-glucagon, GLP-1 as explanatory variables, also accommodating the time-point of measurement (i.e. 0, 30 or 120 minutes) in a linear mixed model, we estimated the contribution of these variables to the measured RIA glucagon in vivo, during OGTT. To this end, the marginal coefficient of determination (pseudo R-squared) was calculated for mixed models to describe the proportion of variance in the outcome variable explained by the fixed effect using the MuMIn package in R. Removing each factor separately, we estimated the percentage of its respective contribution to RIA-derived glucagon measurements.
The Wilcoxon rank-sum/Kruskal-Wallis test was used to perform nonparametric tests on continuous variables, for comparing two independent samples. Categorical variables were compared by chi-squared test. P-values < 0.05 were considered statistically significant. Statistical analyses were performed with R (version 4.0. 3) (43).
The data is not publicly available due to containing information that could compromise research participant privacy/consent.
The studies involving human participants were reviewed and approved by Ethics Committee of the Medical Faculty of the Eberhard Karls University and the University Hospital Tübingen. The patients/participants provided their written informed consent to participate in this study.
RW, SE, LF and MH designed the analysis strategy, supervised the project and contributed to discussion. SE, LF, SH and AP performed measurements and contributed to discussion. LF and KP did statistical analysis. H-UH, AB and AF contributed to discussion. RW, SE and MH drafted the manuscript. All authors approved the final version of the manuscript prior to submission.
We acknowledge support by the Deutsche Forschungsgemeinschaft and the Open Access Publishing Fund of the University of Tübingen. This work was supported in part by grant 01GI0925 from the German Federal Ministry of Education and Research (BMBF) to the German Center for Diabetes Research (DZD).
Outside of the current work, RW does report lecture fees from Novo Nordisk, Sanofi-Aventis and travel grants from Eli Lilly. He served on the advisory board for Akcea Therapeutics, Daiichi Sankyo, Sanofi-Aventis and NovoNordisk. Outside of the current work, AF reports lecture fees and advisory board membership from Sanofi, Novo Nordisk, Eli Lilly, and AstraZeneca. In addition to his current work, AB reports lecture fees from AstraZeneca, Boehringer Ingelheim, and NovoNordisk. He served on advisory boards of AstraZeneca, Boehringer Ingelheim and NovoNordisk. Outside of the current work, MH, reports research grants from Boehringer Ingelheim and Sanofi (both to the University Hospital of Tübingen), advisory board for Boehringer Ingelheim, and lecture fees from Amryt, Novo Nordisk and Boehringer Ingelheim.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
We gratefully acknowledge the excellent technical assistance of Dorothee Neuscheler and Henrike Peuker (both University Hospital Tübingen). We thank Dr. Monica Vilhelmsson (Mercodia, Uppsala, Sweden) for scientific discussions about specificity and sensitivity and about the antibody binding areas of the used ELISAs. We furthermore thank Mercodia (Uppsala, Sweden) for making their prototype glucagon 1-61 ELISA available for purchase and for technical input.
1. D’Alessio D. The Role of Dysregulated Glucagon Secretion in Type 2 Diabetes. Diabetes Obes Metab (2011) 13(Suppl 1):126–32. doi: 10.1111/j.1463-1326.2011.01449.x
2. Habegger KM, Heppner KM, Geary N, Bartness TJ, DiMarchi R, Tschöp MH. The Metabolic Actions of Glucagon Revisited. Nat Rev Endocrinol (2010) 6:689–97. doi: 10.1038/nrendo.2010.187
3. Zeigerer A, Sekar R, Kleinert M, Nason S, Habegger KM, Müller TD. Glucagon’s Metabolic Action in Health and Disease. Compr Physiol (2021) 11:1759–83. doi: 10.1002/cphy.c200013
5. Müller TD, Finan B, Bloom SR, D’Alessio D, Drucker DJ, Flatt PR, et al. Glucagon-Like Peptide 1 (GLP-1). Mol Metab (2019) 30:72–130. doi: 10.1016/j.molmet.2019.09.010
6. Sasaki M, Fitzgerald AJ, Mandir N, Sasaki K, Wright NA, Goodlad RA. Glicentin, an Active Enteroglucagon, has a Significant Trophic Role on the Small Intestine But Not on the Colon in the Rat. Aliment Pharmacol Ther (2001) 15:1681–6. doi: 10.1046/j.1365-2036.2001.01082.x
7. Ohneda A, Ohneda K, Nagsaki T, Sasaki K. Insulinotropic Action of Human Glicentin in Dogs. Metabolism (1995) 44:47–51. doi: 10.1016/0026-0495(95)90288-0
8. Yamada T, Solomon TE, Petersen H, Levin SR, Lewin K, Walsh JH, et al. Effects of Gastrointestinal Polypeptides on Hormone Content of Endocrine Pancreas in the Rat. Am J Physiol Gastrointest Liver Physiol (1980) 238:G526–30. doi: 10.1152/ajpgi.1980.238.6.G526
9. Kirkegaard P, Moody A, Holst J, Loud F, Skov Olsen P, Christiansen J. Glicentin Inhibits Gastric Acid Secretion in the Rat | Nature. Nature (1982) 297:156–7. doi: 10.1038/297156a0
10. Myojo S, Tsujikawa T, Sasaki M, Fujiyama Y, Bamba T. Trophic Effects of Glicentin on Rat Small-Intestinal Mucosa In Vivo and In Vitro. J Gastroenterol (1997) 32:300–5. doi: 10.1007/BF02934484
11. Pellissier S, Sasaki K, Le-Nguyen D, Bataille D, Jarrousse C. Oxyntomodulin and Glicentin are Potent Inhibitors of the Fed Motility Pattern in Small Intestine. Neurogastroenterol Motil (2004) 16:455–63. doi: 10.1111/j.1365-2982.2004.00528.x
12. Tomita R, Igarashi S, Tanjoh K, Fujisaki S. Role of Recombinant Human Glicentin in the Normal Human Jejunum: An In Vitro Study. Hepatogastroenterology (2005) 52:1459–62.
13. Perakakis N, Kokkinos A, Peradze N, Tentolouris N, Ghaly W, Pilitsi E, et al. Circulating Levels of Gastrointestinal Hormones in Response to the Most Common Types of Bariatric Surgery and Predictive Value for Weight Loss Over One Year: Evidence From Two Independent Trials. Metabolism (2019) 101:153997. doi: 10.1016/j.metabol.2019.153997
14. Raffort J, Pana7iuml;a-Ferrari P, Lareyre F, Bayer P, Staccini P, Fénichel P, et al. Fasting Circulating Glicentin Increases After Bariatric Surgery. Obes Surg (2017) 27:1581–8. doi: 10.1007/s11695-016-2493-5
15. Jensen CZ, Bojsen-Møller KN, Svane MS, Holst LM, Hermansen K, Hartmann B, et al. Responses of Gut and Pancreatic Hormones, Bile Acids, and Fibroblast Growth Factor-21 Differ to Glucose, Protein, and Fat Ingestion After Gastric Bypass Surgery. Am J Physiol Gastrointest Liver Physiol (2020) 318:G661–72. doi: 10.1152/ajpgi.00265.2019
16. Wagner R, Hakaste LH, Ahlqvist E, Heni M, Machann J, Schick F, et al. Nonsuppressed Glucagon After Glucose Challenge as a Potential Predictor for Glucose Tolerance. Diabetes (2017) 66:1373–9. doi: 10.2337/db16-0354
17. Miyachi A, Kobayashi M, Mieno E, Goto M, Furusawa K, Inagaki T, et al. Accurate Analytical Method for Human Plasma Glucagon Levels Using Liquid Chromatography-High Resolution Mass Spectrometry: Comparison With Commercially Available Immunoassays. Anal Bioanal Chem (2017) 409:5911–8. doi: 10.1007/s00216-017-0534-0
18. Wu T, Rayner CK, Jones KL, Horowitz M, Feinle-Bisset C, Standfield SD, et al. Measurement of Plasma Glucagon in Humans: A Shift in the Performance of a Current Commercially Available Radioimmunoassay Kit. Diabetes Obes Metab (2022) 24:1182–4. doi: 10.1111/dom.14673
19. Wewer Albrechtsen NJ, Kuhre RE, Hornburg D, Jensen CZ, Hornum M, Dirksen C. Circulating Glucagon 1-61 Regulates Blood Glucose by Increasing Insulin Secretion and Hepatic Glucose Production. Cell Rep (2017) 21:1452–60. doi: 10.1016/j.celrep.2017.10.034
20. Matsuo T, Miyagawa J, Kusunoki Y, Miuchi M, Ikawa T, Akagami T, et al. Postabsorptive Hyperglucagonemia in Patients With Type 2 Diabetes Mellitus Analyzed With a Novel Enzyme-Linked Immunosorbent Assay. J Diabetes Invest (2016) 7:324–31. doi: 10.1111/jdi.12400
21. Wewer Albrechtsen NJ, Hartmann B, Veedfald S, Windeløv JA, Plamboeck A, Bojsen-Møller KN, et al. Hyperglucagonaemia Analysed by Glucagon Sandwich ELISA: Nonspecific Interference or Truly Elevated Levels? Diabetologia (2014) 57:1919–26. doi: 10.1007/s00125-014-3283-z
22. Geary N. Postprandial Suppression of Glucagon Secretion: A Puzzlement. Diabetes (2017) 66:1123–5. doi: 10.2337/dbi16-0075
23. Finan B, Capozzi ME, Campbell JE. Repositioning Glucagon Action in the Physiology and Pharmacology of Diabetes. Diabetes (2020) 69:532–41. doi: 10.2337/dbi19-0004
24. Abraham MA, Lam TKT. Glucagon Action in the Brain. Diabetologia (2016) 59:1–5. doi: 10.1007/s00125-016-3950-3
25. Sandoval D. Updating the Role of α-Cell Preproglucagon Products on GLP-1 Receptor–Mediated Insulin Secretion. Diabetes (2020) 69:2238–45. doi: 10.2337/dbi19-0027
26. Baron AD, Schaeffer L, Shragg P, Kolterman OG. Role of Hyperglucagonemia in Maintenance of Increased Rates of Hepatic Glucose Output in Type II Diabetics. Diabetes (1987) 36:274–83. doi: 10.2337/diabetes.36.3.274
27. Raju B, Cryer PE. Maintenance of the Postabsorptive Plasma Glucose Concentration: Insulin or Insulin Plus Glucagon? Am J Physiol Endocrinol Metab (2005) 289:E181–6. doi: 10.1152/ajpendo.00460.2004
28. Gar C, Rottenkolber M, Sacco V, Moschko S, Banning F, Hesse N, et al. Patterns of Plasma Glucagon Dynamics Do Not Match Metabolic Phenotypes in Young Women. J Clin Endocrinol Metab (2018) 103:972–82. doi: 10.1210/jc.2017-02014
29. Tripathy D, Wessman Y, Gullström M, Tuomi T, Groop L. Importance of Obtaining Independent Measures of Insulin Secretion and Insulin Sensitivity During the Same Test: Results With the Botnia Clamp. Diabetes Care (2003) 26:1395–401. doi: 10.2337/diacare.26.5.1395
30. Færch K, Vistisen D, Pacini G, Torekov SS, Johansen NB, Witte DR, et al. Insulin Resistance Is Accompanied by Increased Fasting Glucagon and Delayed Glucagon Suppression in Individuals With Normal and Impaired Glucose Regulation. Diabetes (2016) 65:3473–81. doi: 10.2337/db16-0240
31. Borghi VC, Wajchenberg BL, Cesar FP. Plasma Glucagon Suppressibility After Oral Glucose in Obese Subjects With Normal and Impaired Glucose Tolerance. Metabolism (1984) 33:1068–74. doi: 10.1016/0026-0495(84)90089-1
32. Knop FK, Vilsbøll T, Højberg PV, Larsen S, Madsbad S, Vølund A, et al. Reduced Incretin Effect in Type 2 Diabetes: Cause or Consequence of the Diabetic State? Diabetes (2007) 56:1951–9. doi: 10.2337/db07-0100
33. Sharabi K, Tavares CDJ, Rines AK, Puigserver P. Molecular Pathophysiology of Hepatic Glucose Production. Mol Aspects Med (2015) 46:21–33. doi: 10.1016/j.mam.2015.09.003
34. Best JD, Kahn SE, Ader M, Watanabe RM, Ni TC, Bergman RN. Role of Glucose Effectiveness in the Determination of Glucose Tolerance. Diabetes Care (1996) 19:1018–30. doi: 10.2337/diacare.19.9.1018
35. Galante P, Mosthaf L, Kellerer M, Berti L, Tippmer S, Bossenmaier B, et al. Acute Hyperglycemia Provides an Insulin-Independent Inducer for GLUT4 Translocation in C2C12 Myotubes and Rat Skeletal Muscle. Diabetes (1995) 44:646–51. doi: 10.2337/diab.44.6.646
36. Hayashi T, Wojtaszewski JFP, Goodyear LJ. Exercise Regulation of Glucose Transport In Skeletal Muscle. Am J Physiol Endocrinol Metab (1997) 273:E1039–51. doi: 10.1152/ajpendo.1997.273.6.E1039
37. Schwartz MW, Seeley RJ, Tschöp MH, Woods SC, Morton GJ, Myers MG, et al. Cooperation Between Brain and Islet in Glucose Homeostasis and Diabetes. Nature (2013) 503:59–66. doi: 10.1038/nature12709
38. Fritsche A, Wagner R, Heni M, Kantartzis K, Machann J, Schick F, et al. Risk-Stratified Lifestyle Intervention to Prevent Type 2 Diabetes. medRxiv (2021) 70(12):2785–95. doi: 10.1101/2021.01.26.21249582. 01.26.21249582.
39. American Diabetes Association. 2. Classification and Diagnosis of Diabetes: Standards of Medical Care in Diabetes-2018. Diabetes Care (2018) 41:S13–27. doi: 10.2337/dc18-S002
40. Mercodia Glicentin Elisa Immunoassay Kit. Available at: https://www.mercodia.com/product/glicentin-elisa/.
41. Stefan N, Schick F, Häring H-U. Causes, Characteristics, and Consequences of Metabolically Unhealthy Normal Weight in Humans. Cell Metab (2017) 26:292–300. doi: 10.1016/j.cmet.2017.07.008
42. Matsuda M, DeFronzo RA. Insulin Sensitivity Indices Obtained From Oral Glucose Tolerance Testing: Comparison With the Euglycemic Insulin Clamp. Diabetes Care (1999) 22:1462–70. doi: 10.2337/diacare.22.9.1462
43. R Core Team. R: A Language and Environment for Statistical Computing. Vienna, Austria: R Foundation for Statistical Computing (2020). Available at: https://www.r-project.org.
Keywords: glucagon, Glucagen-like peptides, insulin, metabolism, glicentin
Citation: Wagner R, Eckstein SS, Fritsche L, Prystupa K, Hörber S, Häring H-U, Birkenfeld AL, Peter A, Fritsche A and Heni M (2022) Postprandial Dynamics of Proglucagon Cleavage Products and Their Relation to Metabolic Health. Front. Endocrinol. 13:892677. doi: 10.3389/fendo.2022.892677
Received: 09 March 2022; Accepted: 24 May 2022;
Published: 29 June 2022.
Edited by:
Åke Sjöholm, Gävle Hospital, SwedenReviewed by:
Michael Nauck, Katholisches Klinikum Bochum, GermanyCopyright © 2022 Wagner, Eckstein, Fritsche, Prystupa, Hörber, Häring, Birkenfeld, Peter, Fritsche and Heni. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Martin Heni, bWFydGluLmhlbmlAbWVkLnVuaS10dWViaW5nZW4uZGU=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.