 Stefano Marullo
Stefano Marullo Mark G. H. Scott
Mark G. H. Scott Hervé Enslen
Hervé Enslen Mathieu Coureuil
Mathieu Coureuil
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
PERSPECTIVE article
Front. Endocrinol. , 02 May 2022
Sec. Cellular Endocrinology
Volume 13 - 2022 | https://doi.org/10.3389/fendo.2022.883568
This article is part of the Research Topic Physiology and Pathophysiology of GPCR Signal Transduction View all 8 articles
More than 12 years have passed since the seminal observation that meningococcus, a pathogen causing epidemic meningitis in humans, occasionally associated with infectious vasculitis and septic shock, can promote the translocation of β-arrestins to the cell surface beneath bacterial colonies. The cellular receptor used by the pathogen to induce signalling in host cells and allowing it to open endothelial cell junctions and reach meninges was unknown. The involvement of β-arrestins, which are scaffolding proteins regulating G protein coupled receptor signalling and function, incited us to specifically investigate this class of receptors. In this perspective article we will summarize the events leading to the discovery that the β2-adrenergic receptor is the receptor that initiates the signalling cascades induced by meningococcus in host cells. This receptor, however, cannot mediate cell infection on its own. It needs to be pre-associated with an “early” adhesion receptor, CD147, within a hetero-oligomeric complex, stabilized by the cytoskeletal protein α-actinin 4. It then required several years to understand how the pathogen actually activates the signalling receptor. Once bound to the N-terminal glycans of the β2-adrenergic receptor, meningococcus provides a mechanical stimulation that induces the biased activation of β-arrestin-mediated signalling pathways. This activating mechanical stimulus can be reproduced in the absence of any pathogen by applying equivalent forces on receptor glycans. Mechanical activation of the β2-adrenergic receptor might have a physiological role in signalling events promoted in the context of cell-to-cell interaction.
Among the multiple pathogens causing infectious diseases in humans, some are more daunting than others. Neisseria meningitidis (Nm, also known as meningococcus) belongs to this category. Indeed, this Gram-negative extracellular bacterium can kill a perfectly healthy person in a few hours. Meningococcus behaves in most cases as a commensal non-pathogenic bacterium of the human nasopharynx (1). However, when it leaves its “ecological niche” to penetrate into the blood stream it can infect the meninges, the membranes which envelop the brain and spinal cord, causing cerebrospinal meningitis (2). Moreover, in cases of invasive disease often associated with high bacteraemia (3), meningococcus disseminates into tissues promoting inflammation and coagulation activation, leading to extensive necrotic purpura and sepsis (4).
A key event in meningococcus tissue dissemination is its capacity to interact with host endothelial cells, allowing the rapid colonization of microvessels. The interaction with endothelia largely relies on bacterial filamentous appendages known as type-4 pili (TFP) (5, 6). The chemical inhibition of TFP using drugs that promote their disassembly markedly reduces meningococcus pathogenicity (7). TFP are made of polymers of protein subunits called pilins, which assemble in long helical structures. PilE, the most abundant pilin within the polymers, can assemble with minor (less abundant) PilV, PilX and ComP pilins (8). The PilC protein, presumably located at the tip of TFP is involved in pili adhesiveness along with PilV (9, 10). Pili are submitted to cycles of extension and retraction that are controlled by 2 ATPases: the PilF ATPase regulates pilin assembly, whereas the PilT ATPase is responsible for TFP disassembly and retraction. The dynamics of TFP elongation and retraction, allows the bacterium to crawl at the surface of endothelial cells and to adapt to microvessel geometry (11). Recently, TFP retraction was shown to promote the release of TFP-dependent contacts between bacteria facilitating sustained bacteremia (12). Although not directly measured for Nm, it has been established in a study on the close pathogen N. gonorrhoeae that TFP retraction can generate proportionally high forces in the nanonewton range (13).
The interaction of TFP with both epithelial and endothelial cells promotes the activation of multiple signalling cascades, the visible hallmark of this phenomenon being the bulk accumulation under bacterial colonies of membrane-associated proteins, which form a honeycomb shaped “cortical plaque” (14). However, signalling pathways Induced by TFP in endothelial and epithelial cells are different, likely involving different receptors (15).
In endothelial cells, meningococcus induces the accumulation of adhesion molecules, junctional proteins, cytosolic signalling proteins and cytoskeletal proteins, such as ezrin and actin (16), with two principal pathophysiological consequences. The first is the creation of actin-containing microvilli-like structures perpendicular to the endothelial surface, which protrude between the bacteria of a growing colony and stabilize it under the blood flow (17). The second is the formation of ectopic intercellular junctional domains at the site of bacteria-host cell interaction and a subsequent depletion of junctional proteins at the cell-cell interface, with opening of the intercellular junctions of the brain-endothelial interface (18). Early studies identified several signalling mechanism upstream of these cellular changes, including: local activation of Cdc42 and Rho GTPases (16); Src activation (19) and subsequent Src-dependent phosphorylation of cortactin, an actin-binding protein that controls actin polymerization; activation of the phosphoinositide-3-kinase (PI3K)/Rac1 pathway (20). However, the host cell receptor(s) activated by the pathogen upstream of these signalling events remained elusive for many years.
The accumulation in the cortical plaque of different protein types was consistent with the involvement of one or more scaffolding protein(s) that would be recruited by a putative meningococcus-activated receptor. Two such scaffolding proteins, β-arrestin 1 and 2 (βarr1 and βarr2), which are principally involved in G protein coupled receptor (GPCR) regulation and signalling, appeared as plausible candidates. Previous studies had shown that βarrs were essential for the co-localization of stimulated GPCRs, the actin-binding protein filamin-A and active ERK in membrane ruffles (21). Moreover, βarrs were shown to participate in the activation of CDC42 (22), PI3K (23), and Src (24).
The hypothesis of a role for βarrs in Nm-induced signalling in host endothelial cells was validated. In monolayers of hCMEC/D3 cells, a human brain endothelial cell line, which stably maintains phenotypic features of BBB in culture (25, 26), capsulated (pathogenic) piliated meningococci induced the accumulation in cortical plaques of both exogenous βarr2-GFP and endogenous βarr2, together with the proteins that were commonly found under bacterial colonies (27). Inhibition of βarrs with specific siRNAs inhibited the formation of cortical plaques (visualized by ezrin staining), demonstrating that βarr recruitment to the putative unknown Nm receptor was actually an upstream signalling event. Interestingly, siRNA-mediated inhibition of GRK2, a kinase which specifically phosphorylates GPCRs thus providing docking sites for βarrs, prevented βarr translocation under bacterial colonies and cortical plaque formation. This finding represented a strong experimental argument for the involvement of a GPCR in meningococcus signalling. Multiple studies combining loss and gain of function assays demonstrated that the missing signalling receptor in hCMEC/D3 cells was the β2-adrenergic receptor (β2AR) and provided a comprehensive scenario of the signalling events elicited by Nm following its adhesion to human endothelial cells (27). After binding onto endothelial cells, Nm TFP interact with the host cell β2AR, leading to its βarr-biased activation, independently of the activation of cognate heterotrimeric Gαs protein and of its downstream adenylyl cyclase-cAMP pathway. The pilus components PilE and PilV were identified as the specific “bacterial ligands” involved in β2AR activation. Once translocated to β2ARs, βarrs drive the 2 major events involved in Nm tissue infection: docking and activation of Src, leading to the formation of actin-rich cellular protrusions, which stabilizes bacterial colonies under the blood flow; accumulation under bacterial colonies of proteins, such as VE-cadherin and p120-catenin that are normally located in intracellular junctions, which, once depleted, become permeable to bacteria. However, from the studies above it also emerged that the β2AR was not competent for the initial adhesion of the pathogen, corroborated by the fact that β2AR-depleted cells still support initial pilus-mediated Nm adhesion. These observations were confirming previous reports indicating that TFP-mediated adhesion and signalling are two independent events (14).
A differential, quantitative, large-scale analysis of gene expression was conducted in a cell line, which only become permissive for the attachment of piliated capsulated meningococci after phorbol ester 12-O-Tetradecanoylphorbol-13-acetate treatment. This strategy led to the identification of the immunoglobulin superfamily member CD147 (also known as basigin or emmprin) as a brain microvasculature early adhesion receptor for meningococcus (28). PilE and PilV pilins specifically mediate the interaction of the pathogen with the C-terminal part of the CD147 Ig domain, showing that the same bacterial ligands are involved in the interaction with the signalling and the adhesion receptor. These findings indicated that CD147 and β2AR might cooperate to promote firm Nm adhesion and subsequent activation of signalling events, and suggested a potential functional connection between these receptors that might pre-exist pathogen infection (Figure 1).
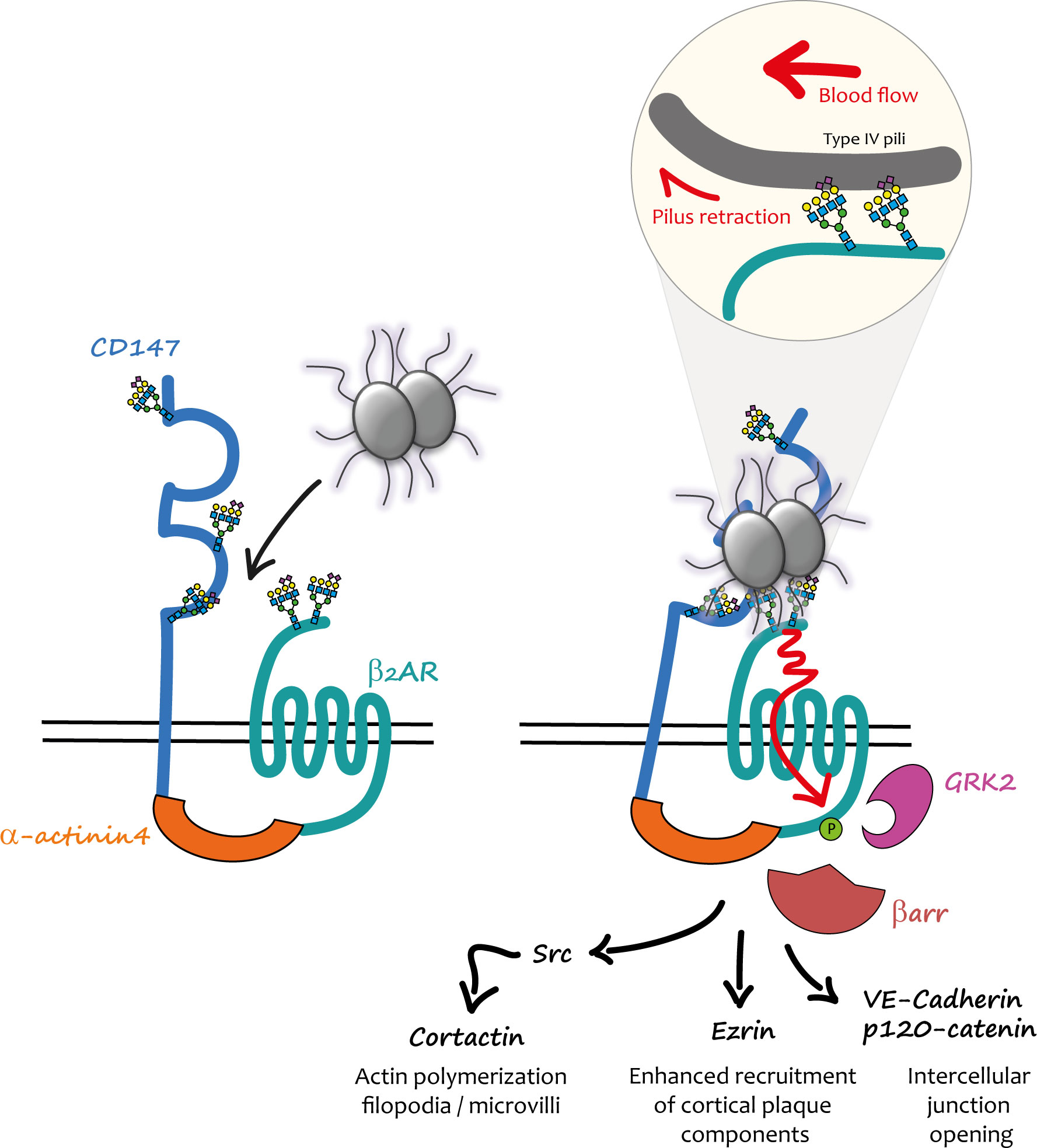
Figure 1 Mechanical activation of the CD147-β2AR heterodimer by meningococcus in endothelial cells. (Left) In endothelial cells CD147 and β2AR are pre-assembled into heterodimers stabilized by α-actinin4; the physiological role of these complexes is still unknown. When meningococci penetrate into the blood stream, they interact with these heterodimers via the PilE and PilV pilins of their TFP. (Right) The first interaction presumably involves CD147 and is accountable for its “early adhesion”. Terminal Neu5Ac (sialic acid) of the N-glycan chains present in the proximal IgG domain of CD147 constitutes the binding site of TFP pilins. Other TFP interact via PilE and PilV with the terminal Neu5Ac of close β2AR N-glycan chains. TFP retraction powered by PilT and forces generated by the blood flow concur to the mechanical activation of the β2AR (enlarged inset). The conformational change induced by this activation produces a cascade of signalling events promoted by βarr translocation to GRK2-phosphorylated β2AR. Once the “stable adhesion” of bacterial colonies is achieved meningococci cross the endothelium through intercellular spaces.
The hypothesis of a spatio-temporal coordination between adhesion to CD147 and β2AR stimulation by Nm TFP was specifically addressed in a subsequent study (29). CD147 and β2AR were found in pre-existing complexes at the cell surface of endothelial cells independently of bacterial infection. This interaction was stabilized by the cytoskeletal protein actinin-4, which in addition promoted the assembly of highly ordered clusters of receptor complexes in response to meningococcus adhesion. In vivo, under blood flow, this multi-molecular assembly process likely provides sufficient binding strength for the initial interaction of TFP with CD147 and then the subsequent rapid activation of the β2AR, which ultimately enhances and stabilizes bacterial adhesion.
Initial investigations showed that the βarr-biased activation of the β2AR by PilE and PilV components of TFP was allosteric, since a pre-incubated orthosteric antagonist of the receptor did not block it, and likely involved the extracellular N-terminal region of the receptor (27). Homogeneous time resolved FRET experiments with purified PilE and PilV confirmed their specific direct interaction with this region of the receptor (30). Intriguingly, the amino-acid composition of β2AR N-terminus is conserved in mammals and almost identical for some species to that of human β2AR, contrasting with the strict human specificity of meningococcus. Moreover, in a fully reconstituted cellular model of meningococcus signalling, the mouse β2AR was activated by the pathogen in cells of human origin, whereas in a symmetrical experiment the human β2AR was not stimulated in mouse cells. These findings suggested that host factors, independent of the amino-acid composition, were involved in meningococcus interaction/activation of β2AR. The N-terminus of the β2AR also contains two asparagine-branched glycan chains, which were then investigated as potential alternative pilin binding sites. In the β2AR sequence the asparagine residues from which the glycan chains arise are separated by 9 amino-acid positions. When the two consensus N-glycosylation sites of the β2AR were introduced in the sequence of the angiotensin AT1R receptor, which is also expressed in endothelial cells but cannot be stimulated by meningococcus, the resulting chimera became activatable by the pathogen. Interestingly, not only the number but also the distance of the glycan chains in the N-terminus sequence was critical for receptor activation (30). A panel of lectins (i.e. proteins that exhibit high avidity for glycoprotein- and/or glycolipid-associated carbohydrates) were then pre-incubated with endothelial cells to block the specific glycan(s) involved in the interaction between the β2AR and meningococcus. Nm signalling was exclusively inhibited by lectins with the capacity of binding to terminal sialic acid (or N-acetyl neuraminic acid). Supporting a role of sialic acid in the interaction of N-terminal glycans with meningococcus, biochemical studies on purified N-terminal β2AR domain confirmed that both glycan chains contain sialic acid, whereas the inhibition of sialyl transferases (the enzymes involved in the addition of sialic acid to the glycan chain) prevented Nm signalling. These findings were particularly interesting in the context of meningococcus species selectivity. Two principal forms of sialic acids are found in mammals, N-Acetylneuraminic acid (Neu5Ac) and N-Glycolylneuraminic acid (Neu5Gc), which differ by a single oxygen atom (31). In mammals, Neu5Gc is predominant, whereas humans mainly express Neu5Ac (32). Neu5Gc is actually synthesised from Neu5Ac by the cytidine monophosphate-N-acetylneuraminic acid hydrolase (CMAH), which is absent in humans, due to a mutation in the CMAH gene (33). The hypothesis that the difference in sialic acids might contribute to the species selectivity was confirmed experimentally. Indeed, the deletion of CMAH in mouse endothelial cells restored meningococcal signalling via the β2AR (30). The nature of the human N-glycan recognized by Nm was further investigated using recombinant CD147 receptors, confirming the role of a complex sialilated N-glycan devoid of fucose residues (34).
Although it had been established that host-pathogen interactions can involve host cell glycans (35) and that the signalling properties of some receptors can be modulated by glycosylation (36, 37), the activation of a GPCR via the interaction between a ligand and N-glycan chains had not been reported previously. On the other hand, in addition to their usual role of chemosensors, some GPCRs can be activated by mechanical cues that are transduced by the receptor into intracellular chemical signals (reviewed in (38)), raising the hypothesis that mechanical forces applied via meningococcal TFP to β2ARs might similarly produce their activation (Figure 1). Indeed, bacteria growing at the cell surface of endothelial cells are permanently submitted to forces exerted by blood flow. In addition, TFP retraction, powered by the PilT ATPase can generate traction forces independently of hemodynamic flow (13, 39). Wild type and mutant meningococci deficient in PilT activity [ΔpillT mutants (40)] were compared for their capacity to induce signalling in hCMEC/D3 cells. Under basal conditions ezrin recruitment (as a marker of the cortical plaque formation) was significantly impaired under ΔpillT mutants compared to wild type Nm. The application of centrifugal forces on bacteria enhanced ezrin accumulation under both wild type and ΔpillT colonies, although the effect was significantly larger for wild type bacteria. Maximal signalling was thus obtained with the additive effect of PilT-induced pilus retraction and exogenous forces applied to meningcocci (30).
To confirm that pulling forces transmitted via N-glycans are sufficient to activate β2AR signalling agarose beads coated with lectins that specifically recognize Neu5Ac were used in a reconstituted cell system. After incubation on cell monolayers to allow lectin binding to sialic acid-terminated N-glycan chains of the β2AR the beads were submitted to orbital rotation. Whereas under basal conditions, only background accumulation of ezrin (used as activation readout) was observed under the beads, rotation appliance significantly enhanced ezrin accumulation but only when exogenous β2AR and βarrs were co-expressed in the cells (30).
In addition to their well-established role of chemosensors, which transduce catecholamine stimulation, β2AR can also function as mechanosensors for the traction forces applied on their N-terminal glycan chains. The signalling output produced by mechanical stimulation appears biased toward βarr-dependent pathways, since the receptor cognate Gs protein is not activated. However, in the presence of particular ligands, GPGRs can switch their coupling to a non-cognate G protein, likely because these ligands stabilize a unique receptor conformation that is competent for this unusual coupling. For example, carvedilol, an inverse-agonist of the classically Gs-coupled β1-adrenergic receptor (β1AR), was reported to promote its coupling to the inhibitory G protein Gi, in addition to the recruitment of βarrs (41). Moreover, this study demonstrated the existence of interplay between Gi and βarrs in the activation of the ERK1/2 MAP kinase pathway. Cell pre-incubation with pertussis toxin (PTX), which prevents Gi from interacting with GPCRs, partially inhibited meningococcus-induced signalling (Figure 2), suggesting that the mechanical traction forces on glycan chains similarly stabilize a unique β2AR active conformation capable of synergistic Gi coupling and activation of βarr signalling. This or a close conformation is likely also obtained by applying traction forces via beads that are coated with lectins specific for the exposed sialic acid of receptor glycan chains. Finally, the effect of PTX is consistent with previous studies indicating that signalling events attributed to βarrs might actually require some G protein contribution (42).
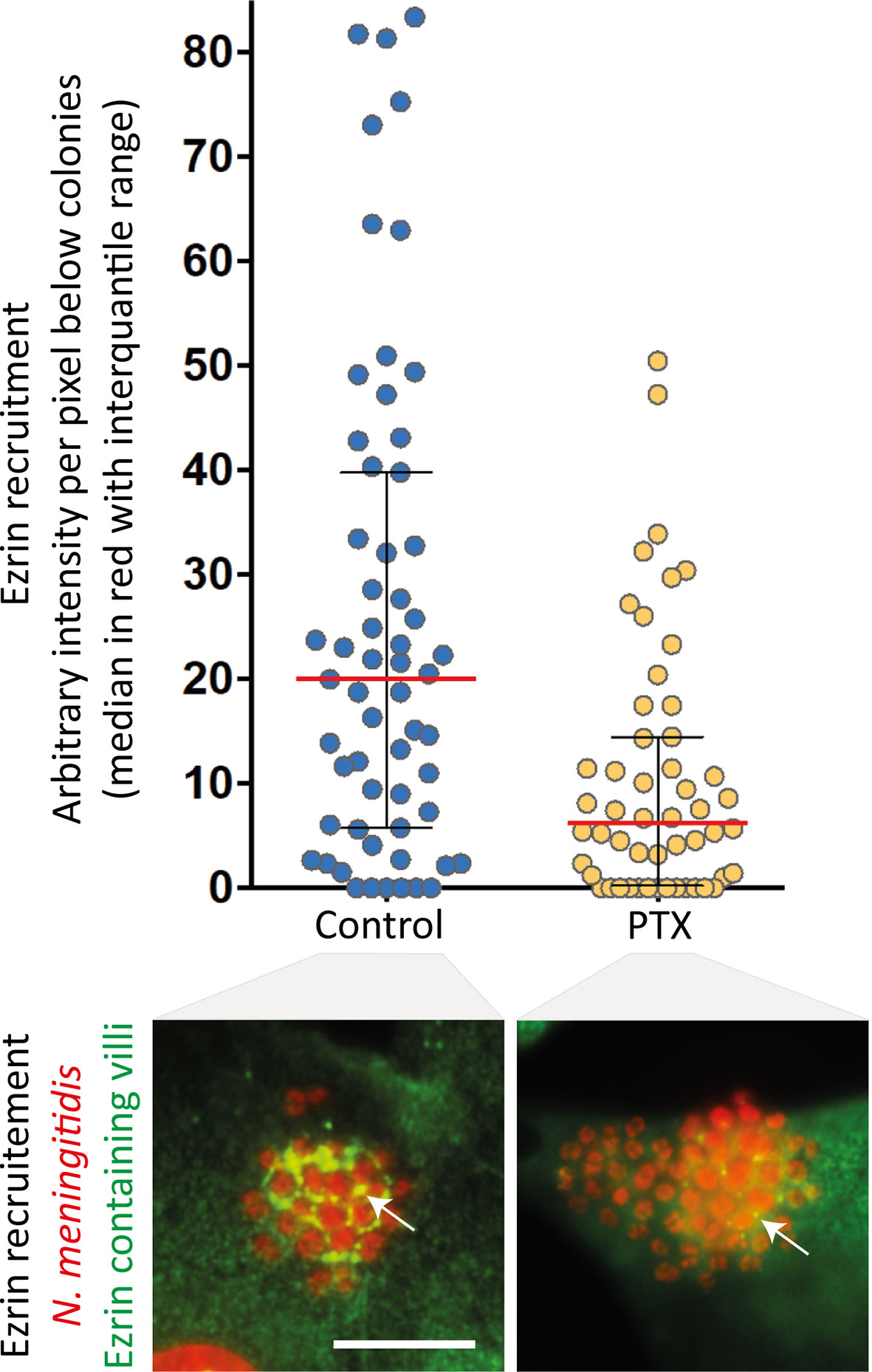
Figure 2 Pertussis toxin inhibits meningococcus-induced signalling. Endothelial hCMEC/D3 cells were infected with wild type N. meningitidis and submitted to centrifugal forces with or without pertussis toxin pre-incubation (PTX - 20ng/ml). Cells were then processed for the analysis of ezrin recruitment under bacterial colonies. Top panel: the intensity of ezrin recruitment was quantified for each individual colony and expressed as arbitrary units per pixel under colonies (as in 30). Each dot represents one colony. Median values (in red) with interquartile range are shown. A Mann-Whitney test was used to determine significant differences between median values. p=0.0002. Bottom panel: Representative immunofluorescence images. HCMEC/D3 cell nuclei and bacterial DNA (small, grouped dots) were labelled with DAPI (red); anti-ezrin antibodies stained ezrin (green). Arrows pointed at ezrin-enriched villi. Bar: 10µm.
Although there is no evidence at the moment that, in addition to the pathophysiological role in meningococcal infection, this mode of activation of the β2AR is involved in physiological processes, this hypothesis merits further investigations. Indeed, many cell types express integral lectins as structural components of their plasma membrane. Among them, I-type lectins are glycan-binding proteins in which the binding domain is homologous to immunoglobulin superfamily proteins. The Siglec family is a subgroup of I-type lectins, which all recognizes terminal sialic acids with specificity for adjacent carbohydrates within the glycan chain. Most Siglecs have one or more tyrosine-based signalling motifs in their cytoplasmic tails, or associate with membrane adaptor proteins containing cytosolic tyrosine motifs. Following their tyrosine phosphorylation by Src family kinases, they recruit and activate SH2-domain-containing effectors (43). Siglecs are expressed by blood cells, microglia, osteoclasts, myelin forming cells, and placental trophoblasts. Macrophages, adhere to and roll over endothelial cells before stopping under blood flow and penetrate into tissues. They express at the cell surface Siglec-1 (35), which displays the same glycan specificity as Mal-I, one of the lectins that block meningococcal signalling. In this context Siglec-1 might bind to the β2AR and induce signalling pathways contributing to macrophage diapedesis. Also, signalling promoted by cell-to-cell interactions are essential during development, during which sialic-acids are known to play a major role, as shown by early embryonic lethality in case of disruption of sialic acid synthesis (43).
More generally, GPCRs are almost ubiquitous and 80% of them display one or more N-glycan chains, which might similarly be involved in the interaction with surrounding cells through lectins. Although, it is difficult at the moment to evaluate how many GPCRs could actually be activated via a traction applied on their N-glycans, β2AR might not be a unique example, since the angiotensin AT1R engineered to express a second glycan chain in its N-terminus can be activated by meningococcus.
The raw data supporting the conclusions of this article will be made available by the authors, without undue reservation.
SM wrote the first version of the manuscript, which was reviewed and corrected by the co-authors. All authors contributed to the article and approved the submitted version.
This article was supported by ANR-14-IFEC-0006-01, ANR-15-CE15-0002 and ANR-19-CE14-0045 grants. SM and MC are also supported by the INSERM, the CNRS, and the Université de Paris.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Stephens DS. Biology and Pathogenesis of the Evolutionarily Successful, Obligate Human Bacterium Neisseria Meningitidis. Vaccine (2009) 27:B71–7. doi: 10.1016/j.vaccine.2009.04.070
2. Coureuil M, Join-Lambert O, Lecuyer H, Bourdoulous S, Marullo S, Nassif X. Mechanism of Meningeal Invasion by Neisseria Meningitidis. Virulence (2012) 3:164–72. doi: 10.4161/viru.18639
3. Darton T, Guiver M, Naylor S, Jack DL, Kaczmarski EB, Borrow R, et al. Severity of Meningococcal Disease Associated With Genomic Bacterial Load. Clin Infect Dis (2009) 48:587–94. doi: 10.1086/596707
4. Coureuil M, Bourdoulous S, Marullo S, Nassif X. Invasive Meningococcal Disease: A Disease of the Endothelial Cells. Trends Mol Med (2014) 20:571–8. doi: 10.1016/j.molmed.2014.08.002
5. Dos Santos Souza I, Maïssa N, Ziveri J, Morand PC, Coureuil M, Nassif X, et al. Meningococcal Disease: A Paradigm of Type-IV Pilus Dependent Pathogenesis. Cell Microbiol (2020) 22:e13185. doi: 10.1111/cmi.13185
6. Virji M, Kayhty H, Ferguson DJ, Alexandrescu C, Heckels JE, Moxon ER. The Role of Pili in the Interactions of Pathogenic Neisseria With Cultured Human Endothelial Cells. Mol Microbiol (1991) 5:1831–41. doi: 10.1111/j.1365-2958.1991.tb00807.x
7. Denis K, Le Bris M, Le Guennec L, Barnier JP, Faure C, Gouge A, et al. Targeting Type IV Pili as an Antivirulence Strategy Against Invasive Meningococcal Disease. Nat Microbiol (2019) 4:972–84. doi: 10.1038/s41564-019-0395-8
8. Craig L, Pique ME, Tainer JE. Type IV Pilus Structure and Bacterial Pathogenicity. Nat Rev Microbiol (2004) 2:363–78. doi: 10.1038/nrmicro885
9. Barnier JP, Meyer J, Kolappan S, Bouzinba-Ségard H, Gesbert G, Jamet A, et al. The Minor Pilin PilV Provides a Conserved Adhesion Site Throughout the Antigenically Variable Meningococcal Type IV Pilus. Proc Natl Acad Sci USA (2021) 118:e2109364118. doi: 10.1073/pnas.2109364118
10. Rudel T, Scheuerpflug I, Meyer TF. Neisseria PilC Protein Identified as Type-4 Pilus Tip-Located Adhesin. Nature (1995) 373:357–9. doi: 10.1038/373357a0
11. Bonazzi D, Lo Schiavo V, Machata S, Djafer-Cherif I, Nivoit P, Manriquez V, et al. Intermittent Pili-Mediated Forces Fluidize Neisseria Meningitidis Aggregates Promoting Vascular Colonization. Cell (2018) 174:143–55.e116. doi: 10.1016/j.cell.2018.04.010
12. Barnier JP, Euphrasie D, Join-Lambert O, Audry M, Schonherr-Hellec S, Schmitt T, et al. Type IV Pilus Retraction Enables Sustained Bacteremia and Plays a Key Role in the Outcome of Meningococcal Sepsis in a Humanized Mouse Model. PloS Pathog (2021) 17:e1009299. doi: 10.1371/journal.ppat.1009299
13. Biais N, Ladoux B, Higashi D, So M, Sheetz M. Cooperative Retraction of Bundled Type IV Pili Enables Nanonewton Force Generation. PloS Biol (2008) 6:e87. doi: 10.1371/journal.pbio.0060087
14. Merz AJ, Enns CA, So M. Type IV Pili of Pathogenic Neisseriae Elicit Cortical Plaque Formation in Epithelial Cells. Mol Microbiol (1999) 32:1316–32. doi: 10.1046/j.1365-2958.1999.01459.x
15. Lécuyer H, Nassif X, Coureuil M. Two Strikingly Different Signaling Pathways are Induced by Meningococcal Type IV Pili on Endothelial and Epithelial Cells. Infect Immun (2012) 80:175–86. doi: 10.1128/IAI.05837-11
16. Eugene E, Hoffmann I, Pujol C, Couraud PO, Bourdoulous S, Nassif X. Microvilli-Like Structures are Associated With the Internalization of Virulent Capsulated Neisseria Meningitidis Into Vascular Endothelial Cells. J Cell Sci (2002) 115:1231–41. doi: 10.1242/jcs.115.6.1231
17. Mikaty G, Soyer M, Mairey E, Henry N, Dyer D, Forest KT, et al. Extracellular Bacterial Pathogen Induces Host Cell Surface Reorganization to Resist Shear Stress. PloS Pathog (2009) 5:e1000314. doi: 10.1371/journal.ppat.1000314
18. Coureuil M, Mikaty G, Miller F, Lecuyer H, Bernard C, Bourdoulous S, et al. Meningococcal Type IV Pili Recruit the Polarity Complex to Cross the Brain Endothelium. Science (2009) 325:83–7. doi: 10.1126/science.1173196
19. Hoffmann I, Eugene E, Nassif X, Couraud PO, Bourdoulous S. Activation of ErbB2 Receptor Tyrosine Kinase Supports Invasion of Endothelial Cells by Neisseria Meningitidis. J Cell Biol (2001) 155:133–43. doi: 10.1083/jcb.200106148
20. Lambotin M, Hoffmann I, Laran-Chich MP, Nassif X, Couraud PO, Bourdoulous S. Invasion of Endothelial Cells by Neisseria Meningitidis Requires Cortactin Recruitment by a Phosphoinositide-3-Kinase/Rac1 Signalling Pathway Triggered by the Lipo-Oligosaccharide. J Cell Sci (2005) 118:3805–16. doi: 10.1242/jcs.02514
21. Scott MGH, Pierotti V, Storez H, Lindberg E, Thuret A, Muntaner O, et al. Cooperative Regulation of ERK Activation and Cell Shape Change by Filamin A and ß-Arrestins. Mol Cell Biol (2006) 26:3432–45. doi: 10.1128/MCB.26.9.3432-3445.2006
22. Mythreye K, Blobe GC. The Type III TGF-Beta Receptor Regulates Epithelial and Cancer Cell Migration Through Beta-Arrestin2-Mediated Activation of Cdc42. Proc Natl Acad Sci USA (2009) 106:8221–6. doi: 10.1073/pnas.0812879106
23. Povsic TJ, Kohout TA, Lefkowitz RJ. Beta-Arrestin1 Mediates Insulin-Like Growth Factor 1 (IGF-1) Activation of Phosphatidylinositol 3-Kinase (PI3K) and Anti-Apoptosis. J Biol Chem (2003) 278:51334–9. doi: 10.1074/jbc.M309968200
24. Luttrell LM, Ferguson SS, Daaka Y, Miller WE, Maudsley S, Della Rocca GJ, et al. Beta-Arrestin-Dependent Formation of Beta2 Adrenergic Receptor-Src Protein Kinase Complexes. Science (1999) 283:655–61. doi: 10.1126/science.283.5402.655
25. Poller B, Gutmann H, Krahenbuhl S, Weksler B, Romero I, Couraud PO, et al. The Human Brain Endothelial Cell Line hCMEC/D3 as a Human Blood-Brain Barrier Model for Drug Transport Studies. J Neurochem (2008) 107:1358–68. doi: 10.1111/j.1471-4159.2008.05730.x
26. Weksler BB, Subileau EA, Perriere N, Charneau P, Holloway K, Leveque M, et al. Blood-Brain Barrier-Specific Properties of a Human Adult Brain Endothelial Cell Line. FASEB J (2005) 19:1872–4. doi: 10.1096/fj.04-3458fje
27. Coureuil M, Lécuyer H, Scott MGH, Boularan C, Enslen H, Soyer M, et al. Meningococcus Hijack a ß2-Adrenoceptor-ß-Arrestin Pathway to Cross Brain Microvasculature Endothelium. Cell (2010) 143:1149–60. doi: 10.1016/j.cell.2010.11.035
28. Bernard SC, Simpson N, Join-Lambert O, Federici C, Laran-Chich MP, Maissa N, et al. Pathogenic Neisseria Meningitidis Utilizes CD147 for Vascular Colonization. Nat Med (2014) 20:725–31. doi: 10.1038/nm.3563
29. Maïssa N, Covarelli V, Janel S, Durel B, Simpson N, Bernard SC, et al. Strength of Neisseria Meningitidis Binding to Endothelial Cells Requires Highly-Ordered CD147/β2-Adrenoceptor Clusters Assembled by Alpha-Actinin-4. Nat Commun (2017) 8:15764. doi: 10.1038/ncomms15764
30. Virion Z, Doly S, Saha K, Lambert M, Bied C, Duke RM, et al. Sialic Acid-Mediated Mechanical Activation of β2 Adrenergic Receptors by Bacterial Pilins. Nat Commun (2019) 10:4752. doi: 10.1038/s41467-019-12685-6
31. Varki A. Loss of N-Glycolylneuraminic Acid in Humans: Mechanisms, Consequences, and Implications for Hominid Evolution. Am J Phys Anthropol (2001) 33:54–69. doi: 10.1002/ajpa.10018
32. Varki A. Glycan-Based Interactions Involving Vertebrate Sialic-Acid-Recognizing Proteins. Nature (2007) 446:1023–9. doi: 10.1038/nature05816
33. Kawano T, Koyama S, Takematsu H, Kozutsumi Y, Kawasaki H, Kawashima S, et al. Molecular Cloning of Cytidine Monophospho-N-Acetylneuraminic Acid Hydroxylase. Regulation of Species- and Tissue-Specific Expression of N-Glycolylneuraminic Acid. J Biol Chem (1995) 270:16458–63. doi: 10.1074/jbc.270.27.16458
34. Le Guennec L, Virion Z, Bouzinba-Ségard H, Robbe-Masselot C, Léonard R, Nassif X, et al. Receptor Recognition by Meningococcal Type IV Pili Relies on a Specific Complex N-Glycan. Proc Natl Acad Sci USA (2020) 117:2606–12. doi: 10.1073/pnas.1919567117
36. Luca VC, Jude KM, Pierce NW, Nachury MV, Fischer S, Garcia KC. Structural Basis for Notch1 Engagement Ofvdelta-Like 4. Science (2015) 347:847–53. doi: 10.1126/science.1261093
37. Yoon SJ, Nakayama K, Hikita T, Handa K, Hakomori SI. Epidermal Growth Factor Receptor Tyrosine Kinase is Modulated by GM3 Interaction With N-Linked GlcNAc Termini of the Receptor. Proc Natl Acad Sci USA (2006) 103:18987–91. doi: 10.1073/pnas.0609281103
38. Marullo S, Doly S, Saha K, Enslen H, Scott MGH, Coureuil M. Mechanical GPCR Activation by Traction Forces Exerted on Receptor N-Glycans. ACS Pharmacol Trans science (2020) 3:171–8. doi: 10.1021/acsptsci.9b00106
39. Merz AJ, So M, Sheetz MP. Pilus Retraction Powers Bacterial Twitching Motility. Nature (2000) 407:98–102. doi: 10.1038/35024105
40. Pujol C, Eugene E, Marceau M, Nassif X. The Meningococcal PilT Protein is Required for Induction of Intimate Attachment to Epithelial Cells Following Pilus-Mediated Adhesion. Proc Natl Acad Sci USA (1999) 96:4017–22. doi: 10.1073/pnas.96.7.4017
41. Wang J, Hanada K, Staus DP, Makara MA, Dahal GR, Chen Q, et al. Galphai is Required for Carvedilol-Induced Beta1 Adrenergic Receptor Beta-Arrestin Biased Signaling. Nat Commun (2017) 8:1706. doi: 10.1038/s41467-017-01855-z
42. Grundmann M, Merten N, Malfacini D, Inoue A, Preis P, Simon K, et al. Lack of Beta-Arrestin Signaling in the Absence of Active G Proteins. Nat Commun (2018) 9:341. doi: 10.1038/s41467-017-02661-3
43. Varki A, Schnaar RL, Schauer R. Sialic Acids and Other Nonulosonic Acids. In Essentials of Glycobiology. 3rd ed. C.R. Varki A, Esko JD, Stanley P, Hart GW, Aebi M, Darvill AG, Kinoshita T, Packer NH, Prestegard JH, Schnaar RL, Seeberger PH, editors. Cold Spring Harbor (NY: Cold Spring Harbor Laboratory Press (2017) p. 2015–7.
Keywords: meningococcus, β2-adrenergic receptor, β-arrestin, N-glycans, sialic acid, mechano-transduction, pilin, meningitis
Citation: Marullo S, Scott MGH, Enslen H and Coureuil M (2022) Mechanical Activation of the β2-Adrenergic Receptor by Meningococcus: A Historical and Future Perspective Analysis of How a Bacterial Probe Can Reveal Signalling Pathways in Endothelial Cells, and a Unique Mode of Receptor Activation Involving Its N-Terminal Glycan Chains. Front. Endocrinol. 13:883568. doi: 10.3389/fendo.2022.883568
Received: 25 February 2022; Accepted: 04 April 2022;
Published: 02 May 2022.
Edited by:
Marián Castro, University of Santiago de Compostela, SpainReviewed by:
David Poyner, Aston University, United KingdomCopyright © 2022 Marullo, Scott, Enslen and Coureuil. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Stefano Marullo, c3RlZmFuby5tYXJ1bGxvQGluc2VybS5mcg==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.