
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Endocrinol. , 19 April 2022
Sec. Cellular Endocrinology
Volume 13 - 2022 | https://doi.org/10.3389/fendo.2022.880002
This article is part of the Research Topic Peptide-binding GPCRs coming of age View all 7 articles
 David Speck1†
David Speck1† Gunnar Kleinau1†
Gunnar Kleinau1† Michal Szczepek1
Michal Szczepek1 Dennis Kwiatkowski1
Dennis Kwiatkowski1 Rusan Catar2
Rusan Catar2 Aurélie Philippe3,4
Aurélie Philippe3,4 Patrick Scheerer1,5*
Patrick Scheerer1,5*In conjunction with the endothelin (ET) type A (ETAR) and type B (ETBR) receptors, angiotensin (AT) type 1 (AT1R) and type 2 (AT2R) receptors, are peptide-binding class A G-protein-coupled receptors (GPCRs) acting in a physiologically overlapping context. Angiotensin receptors (ATRs) are involved in regulating cell proliferation, as well as cardiovascular, renal, neurological, and endothelial functions. They are important therapeutic targets for several diseases or pathological conditions, such as hypertrophy, vascular inflammation, atherosclerosis, angiogenesis, and cancer. Endothelin receptors (ETRs) are expressed primarily in blood vessels, but also in the central nervous system or epithelial cells. They regulate blood pressure and cardiovascular homeostasis. Pathogenic conditions associated with ETR dysfunctions include cancer and pulmonary hypertension. While both receptor groups are activated by their respective peptide agonists, pathogenic autoantibodies (auto-Abs) can also activate the AT1R and ETAR accompanied by respective clinical conditions. To date, the exact mechanisms and differences in binding and receptor-activation mediated by auto-Abs as opposed to endogenous ligands are not well understood. Further, several questions regarding signaling regulation in these receptors remain open. In the last decade, several receptor structures in the apo- and ligand-bound states were determined with protein X-ray crystallography using conventional synchrotrons or X-ray Free-Electron Lasers (XFEL). These inactive and active complexes provide detailed information on ligand binding, signal induction or inhibition, as well as signal transduction, which is fundamental for understanding properties of different activity states. They are also supportive in the development of pharmacological strategies against dysfunctions at the receptors or in the associated signaling axis. Here, we summarize current structural information for the AT1R, AT2R, and ETBR to provide an improved molecular understanding.
The high biological, medical, and pharmacological relevance of GPCRs (~830 in humans) is due to their key role in signal transduction across the cell membrane from the extracellular side toward the cell interior (1). They interact with a large number of stimulants (agonists), such as odors, peptides, metabolites, light, nucleotides, amines, or a variety of hormones and proteins (2). Generally, receptor interaction with agonists results in an increased capacity of intracellular coupling and subsequent activation of G-protein(s) or arrestin(s) (3). This causes induction of downstream pathways regulating e.g., ion channel activity or gene expression (4–7). GPCR signaling is linked with almost all physiological processes, such as growth, learning, memory, reproduction, or senses like taste and vision (7). More than 100 diseases or pathogenic conditions are linked to dysfunctional GPCRs (8), including viral infections, cancer, infertility, inflammation, and metabolic and neurological disorders (9–11), which, altogether, makes these receptors essential for pharmacological and structural studies [e.g (12)]. The angiotensin (ATRs) and endothelin receptors (ETRs) belong to class A GPCRs (13, 14). For the groups of ETRs and ATRs, respectively, much detailed physiological information, but also pathophysiological relations are known.
In brief, the AT1 receptor (AT1R) binds different angiotensin (Ang) subtypes Ang I, Ang II, Ang III, and Ang IV, which are the main effector peptide hormones of the renin-angiotensin system (15). AT1R can activate the G-protein subtypes Gi/o and Gq/11, and also β-arrestin, upon agonist action (16).
Pharmacologic interventions that either decrease Ang production or modulate Ang actions through AT1R blockade are the current mainstay of renoprotection, as documented by extensive experimental work and clinical trials of diabetic and non-diabetic renal diseases (17). AT1R dysfunction leads to several pathophysiological conditions, including hypertrophy, vascular inflammation, atherosclerosis, endothelial dysfunction, insulin resistance, angiogenesis, and cancer (18). Antibodies (Abs) are involved in the development of preeclampsia, acute graft rejection, and systemic sclerosis (19–22). Of note, the Ang II/AT1R signaling axis was identified recently to be involved in inflammatory processes, collateral tissue damage, and systemic failure related to COVID-19 infection (23). AT1R blockers or biased AT1R agonists are discussed to contribute potentially to treatment strategies against COVID-19 effects (24–26).
Endogenous ligands of the AT2 receptor (AT2R) are Ang II and Ang III with affinities in the nanomolar range (14). Of note, during the elucidation of AT2R related signaling pathways several hypotheses arised and were studied/confirmed, including G-protein independent signal transduction (27–30), G-protein subtype Gi/o activation (31), and also ligand-independent signaling crucial in apoptosis (32). AT2R is expressed in vessels (endothelial cells), heart, kidney (tubules, glomeruli, collecting ducts, arterioles, and interstitial cells), brain, and immune cells (33). In the kidney, physiological stimulation of the receptor causes diuresis and natriuresis by decreasing salt and water transport from the tubules to the capillaries, triggering sodium and water excretion (34). Chronic AT2R overexpression has deleterious effects on cardiomyocytes (35) and AT2R activation, as AT1R, is involved in neuropathic pain (36, 37).
The ETA receptor (ETAR) (38, 39) is localized mainly in vascular smooth muscle cells and, therefore, in all tissues supplied with blood, including the heart, lung, and brain, but are also present on other cell types, including myocytes within the heart (38, 40) or endothelial cells. ETAR has a stronger affinity for ET-1 and ET-2 than for ET-3, all three constituting the family of endothelin peptides (41). ETAR has been associated with the vasoconstrictive effects of ET-1 and is involved in different pathologies (6). Hence, it was shown that ETAR activation has detrimental effects on preeclampsia (42), heart failure (43), and pulmonary hypertension (44). In the kidney, ETAR induces natriuresis (45) and its inhibition can improve short-term lesions triggered by ischemia-reperfusion injury (46). Finally, point mutations in the gene coding for ETAR are responsible for mandibulofacial dysostosis with alopecia (47) and Oro-Oto-Cardiac syndrome (48), as the receptor is involved in craniofacial development. ETAR signaling activity is associated primarily with the G-protein subtypes Gq/11, but there are also indications for Gi/o signaling (16).
With the same affinity the ETB receptor (ETBR) interacts with all three endothelin (ET-1, ET-2, and ET-3) peptides. It resembles many actions of ATRs on renal cell types (49). This receptor couples to the G-protein subtypes Gs, Gi/o, and Gq/11 (16). ETBR is expressed in the lungs and brain (50), and conveys reversal effects as ETAR, mainly vasodilatation by stimulating nitric oxide (NO) production and clearing ET-1 (51). In the kidney, ETBR is involved in sodium excretion (52). The ETBR contains a metal-proteinase cleavage site at the long N-terminus around an A-G-x-P-P-R motif (Figure 1) (55). Interestingly, there are reports on endothelin receptors homo- or heterodimerization with other receptors (see chapter below for details). Depending on the particular receptor-receptor configuration, the resulting signaling effects can differ (56).
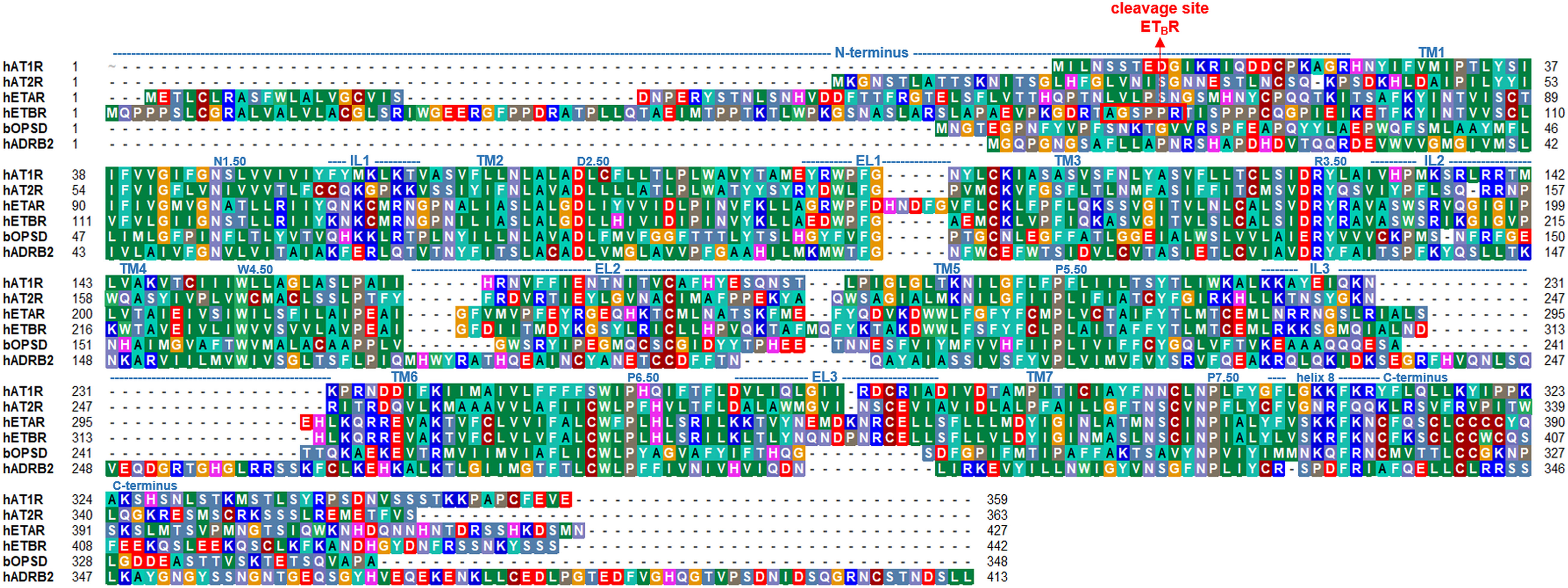
Figure 1 Sequence comparison between the ATRs, ETRs, and bovine rhodopsin (bOPSD) or human β-2 adrenergic receptor (hADRB2). The length of each transmembrane helix (TM1-7) or loops (IL, intracellular loop; EL, extracellular loop) are indicated above the sequence according to an AT1R structure [PDB ID: 4zud (53)] but can differ slightly in other structures. The overall sequence similarity between ETAR and ETBR is approximately 63%, whereas between AT1R and AT2R ~47%. Sequence similarities between ATRs and ETRs, respectively, are around 30%. The sequences of prototypical class A GPCRs bOPSD and hADRB2 are provided additionally for comparison. The alignment was visualized using the software BioEdit (54). Specific background colors reflect chemical properties of the amino acid side chains or the type of amino acid: black-proline; blue-positively charged; cyan/green-aromatic and hydrophobic; green- hydrophobic; red-negatively charged; gray-hydrophilic; dark red-cysteines; and magenta-histidine.
In summary, AT and ET receptors are of high physiological and medical importance, including e.g., renal effects, blood pressure (57), cell proliferation (6, 58, 59), or cancer development (60). Of note, an increasing amount of structural information has been published in recent years, complementing functional insights. Several structures in different activity states were determined by protein X-ray crystallography using conventional synchrotrons or XFELs (Table 1) for AT1R, AT2R, and ETBR. They reveal details of the signal transduction process at the molecular level. In this brief review, we summarise the current state of knowledge about these receptors and receptor complex structures. We aimed to provide a first systematic overview of structural insights into these receptors including ligand binding, dimerization, receptor activation, and inactivation. Thus, we will also identify open knowledge gaps that will aid in the identification of topics relevant for future studies.
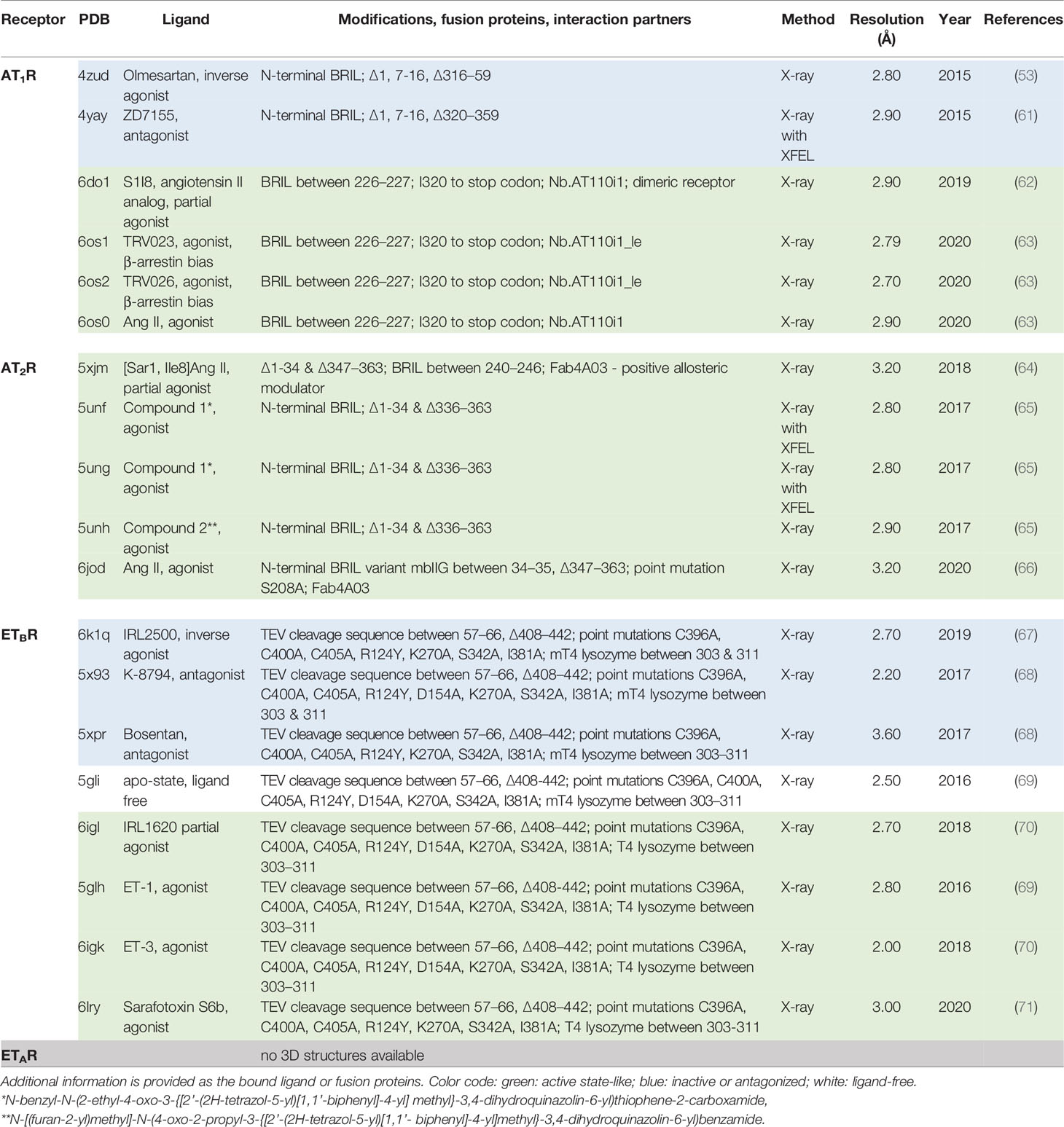
Table 1 Overview of ETR and ATR structures known so far (as of January 2022).
Two AT1R and three ETBR inactive state structures solved by X-ray crystallography have been published (as of January 2022; summarized in Table 1). They provide deeper insights into structural features associated with the inactive receptor states and how antagonists block the signaling process. Highly conserved amino acids (Figure 2A) significant for each GPCR class (74, 75) are generally important for expression and the folding of diverse receptor components, e.g., prolines defining weak points in helices because of steric conflicts with the preceding residue and the loss of a backbone H-bond, which can cause kinks (76, 77) as observed in the CWxP6.50 motif in transmembrane helix 6 (TM6) [superscripted numbers are provided additionally according to the unifying Ballesteros & Weinstein numbering for class A GPCRs (74)]. Conserved amino acids also play a fundamental role in maintaining an inactive state conformation(s), as, for example, in the AT1R the D742.50 in the transmembrane helix (TM) 2, or N2987.49 in TM7 (Figure 2A). They interact through hydrogen bonds with each other or with other hydrophilic amino acid side chains, or with water molecules constraining the inactive state between TM’s 1, 2, 3, and 7 (Figure 2B). In most of the inactive state structures of AT1R and ETBR, no water or sodium ions (region between D2.50-N7.49, as known from other GPCRs (78)) can be observed due to the low resolutions between 2.7 to 3.6 Å (Table 1). However, in the ETBR structure with a resolution of 2.2 Å [Protein Data Bank (79) (PDB) ID: 5x93 (68)], water molecules in tight interaction to hydrophilic amino acid side chains are visible (Figure 2B). This network of hydrogen bonds between hydrophilic residues in TM1, TM3, and TM7, as well as water molecules, is not observable in all active state structures of ATRs or ETBR receptors, nor in other active state GPCR structures (80), because they disappear in the course of receptor activation and related structural rearrangements. Of note, in an active state, such as the ETBR structure complexed with the partial agonist IRL1620, a few water molecules are still observed, and they are supposed to partly preserve the interaction network typical for inactive states (70). This might be related to the fact that in this structure, as for all ETBR structures with bound agonists so far, no intracellular transducer protein as a G-protein molecule stabilizes the active state conformation and, therefore, the TM6 orientation is different to known fully active state structures (restricted movement toward the membrane). In conclusion, such structures do not display a fully active receptor conformation.
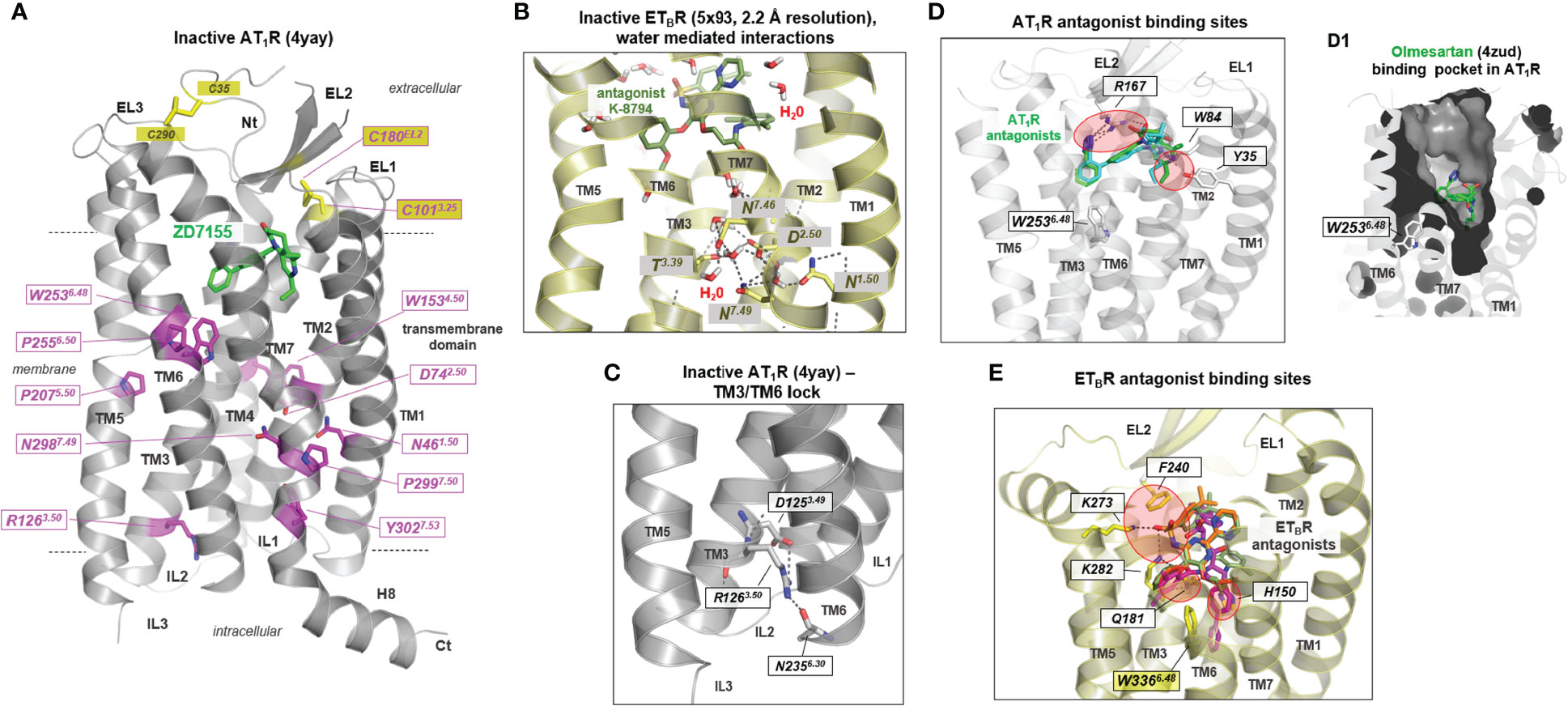
Figure 2 Structural features of inactive or antagonized AT1R and ETBR conformations. (A) Conserved residues in class A GPCRs (magenta sticks) important for receptor-fold, expression, and signaling are highlighted at the inactive state structure of AT1R (backbone cartoon) in complex with the antagonist ZD7155 (green sticks). Highly significant for non-active state conformations is the inward direction of the transmembrane helix (TM) 6 into the helical bundle, which closes the intracellular binding cavity for G-proteins or arrestin (see also Figure 4E). The antagonist ZD7155 (green) is bound in a pocket between the transmembrane helices and their transition to the extracellular loops. Notably, a disulfide bridge (yellow sticks) between the N-terminus and the EL3-TM7 transition forms and stabilizes the spatial region between the N-terminus and EL3, which is also present in the AT2R and the ETBR (not shown). (B) In the antagonized ETBR structure bound with the antagonist K-8794, water molecules solved at a high resolution of 2.2 Å. These water molecules are located centrally in the helical bundle, participating by H-bonds with hydrophilic residues in maintaining an inactive state conformation. (C) Of the currently known five inactive state structures for ETRs and ATRs, only one inactive state shows a H-bond between the intracellular parts of TM3 and TM6 involving the highly conserved R3.50. In several class A GPCRs, an “ionic lock” between this arginine and a negatively charged amino acid in TM6 has been postulated or shown to be essential for constraining the inactive state (72, 73). This cannot be perceived equally for most of the available inactive ETBR and AT1R structures. (D) AT1R antagonists olmesartan (inverse agonist, cyan sticks) and ZD7155 (green sticks, Table 1) are bound mainly between three residues in EL2, TM1, and TM2 in the upper part of the helical bundle (PDB IDs: 4zud and 4yay). Red circles indicate the main contact points. (D1) Visualized is the binding pocket of olmesartan by a clipped inner surface representation. (E) Superimposition of ETBR structures (PDB IDs: 6k1q, 5x93, 5xpr - only one backbone structure is visualized as cartoon because of high overlap between these structures) with antagonists K-8794 (green), bosentan (orange), and IRL2500 (inverse agonist, magenta) shows partially largely binding regions in the receptors, but also significant differences to antagonist binding sites of AT1R (red circles). While a residue of the N-terminal EL2 is involved in ligand binding in both receptors, several H-bonds to amino acids in TM3 and TM5 can be observed in the ETBR. The inverse agonist IRL2500 additionally contacts (blocks) the highly conserved tryptophan in TM6 (W336 in ETBR), which is part of the CWxP6.50 motif that participates in the activation mechanism of class A GPCRs. Red circles indicate the main contact points. All graphic representations in this article were created using the PyMol Molecular Graphics System Version 1.5 (Schrödinger, LLC, New York, NY). EL, extracellular loop; Nt, N terminus; IL, intracellular loop; H8, helix 8; TM1–7, transmembrane helices 1–7.
For diverse GPCRs a significant interaction (previously named “ionic lock”) between the highly conserved R3.50 in TM3 (Figure 2A) of the DR3.50Y motif and a negatively charged residue located at the intracellularly site of TM6 is known to be essential for maintaining the inactive state (72, 73). According to the available structures, such interaction has not yet been observed in AT1R or ETBR. Only in the case of an AT1R structure [PDB ID: 4yay (61)] a potential hydrogen bond interaction between R1263.50 and N2356.30 (backbone) is observable (Figure 2C), which may constrain the typical inactive state conformation of TM6 directed inward to the transmembrane core (Figure 2A) (1).
All previously known structures of inactivated or antagonized receptor states were obtained by binding antagonists (“antagonized”) or inverse agonists (“inactive”), in addition to specifically-directed mutations, which were usually necessary to stabilize an individual receptor state or improve receptor expression. (Table 1, Figures 2D, E). In the two inactive/antagonized AT1R structures, the ligands are bound mainly between residues located in the EL2, TM1, and TM2 (Figure 2D). This binding crevice (Figure 2D1) overlaps greatly with the binding sites of antagonists for the ETBR (Figure 2E). However, significant differences exist in binding details by an extended binding region of ETBR antagonists and the inverse agonist IRL2500 (Figure 2E). Here, specific residues in TM3 and TM5 are essentially involved in antagonist binding.
Of note, the inverse agonist IRL2500 in the inactive ETBR structure [PDB ID: 6k1q (67)] interacts, in addition to other residues, with an aromatic moiety directly at W3366.48 in TM6, which is known generally for class A GPCRs to be a crucial trigger for receptor activation. This W6.48 is located in the CWxP6.50 motif involved in activation-related TM6 outward movement as part of the “global toggle-switch” activation model (81, 82), also described as the “rotamer toggle switch” hypothesis (1, 83). The inverse agonistic activity of this ligand is assumed to be potentially associated with this interaction, which constraints tryptophan in a basally non-active state (67). However, independent of the antagonist or an inverse agonist status, these ligands (Figures 2D, E) occupy a receptor region that is also involved in agonist binding (next section, Figure 4) and therefore compete with agonist binding.
Notably, aside from diverse directed structural alterations for protein stabilization such as fusion with T4 lysozyme or deletions, the inactive, apo-, and agonist bound structural complexes of the ETBR are modified in their amino acid sequence (Table 1). Five combined particular substitutions were used to stabilize complexes with both antagonists, the apo state, and also with agonists, which is not unusual in GPCR preparation for crystallization studies (Supplementary Table S1). These mainly alanine substitutions are located in diverse receptor regions as TM’s 1, 2, 5, 6, and 7 (Figure 3A). Generally, individual or combined thermostabilizing mutations used in class A GPCRs (Supplementary Table S1, Figures 3B-D) can be localized at very diverse structural parts, either with side chains directed into the transmembrane core or with side chains directed toward the membrane. A statistical analysis of the distribution of thermostabilizing mutations used for class A GPCR crystallization (analysis of 17 different GPCRs; Supplementary Table S1 and Figure 3D) shows thermostabilization via mutations is principally feasible in each helix, including helix 8. The molecular effect of such mutations and their combinations is associated with, e.g., the stabilization of a certain conformational state (directed into the transmembrane core) as inactive or active, substitutions of residues facing lipids (directed toward the membrane or detergent), or mutations stabilizing local structural areas (e.g., helix-helix interface directed) (84, 85). In the case of the ETBR, a mixture of these “types” of substitutions can be postulated, whereby R124Y and I381A are directed to the membrane, D154A points into the helical core, K270A is in the interface between TM5 and EL2, and S342A is part of the TM6-TM7 interface (Figure 3A).
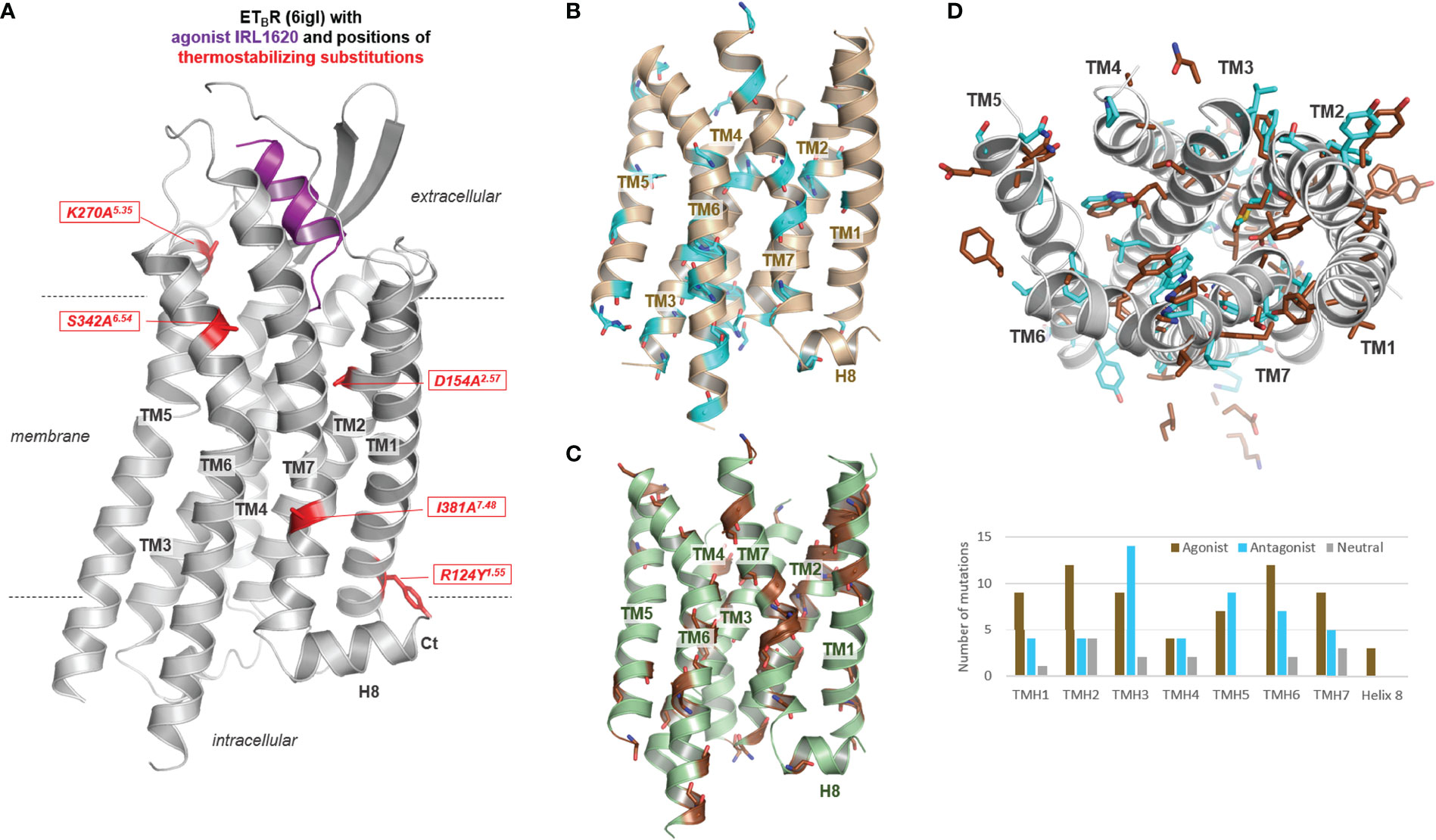
Figure 3 Thermostabilizing mutations of the ETBR and thermostabilizing mutant positions reported for class A GPCRs. (A) The five commonly and in combination used thermostabilizing ETBR substitutions are visualized at an active state structure bound with the agonist. These substitutions are also used for stabilizing inactive state and apo-state structures. (B) Positions (backbone as cyan-colored sticks) of thermostabilizing substitutions used for protein preparation of diverse class A GPCRs (Supplementary Table S1) are highlighted in a rhodopsin model (only transmembrane helices), as well as (C) of thermostabilizing substitutions in determined active state structures highlighted in an opsin model (brown backbone sticks). This mapping demonstrates that substitutions contributing to thermostability can be principally designed at each transmembrane helix [see also diagram in (D)]. (D) This depiction (top view from the extracellular side) of the rhodopsin/opsin wild-type side chains of positions in class A GPCRs used for thermostabilization of both states (~130 substitutions at 97 positions, Supplementary Table S1) demonstrates that they act in a contrasting manner, either by modulating the protein-membrane interaction or by changes of intramolecular interactions participating in the regulation of activity state-related conformations. The substitutions used for stabilizing class A GPCR structures either in the apo-, inactive-, or active-state (in the diagram termed as Agonist, Antagonist or Neutral according to the state of ligand occupancy) are located at each helix, however, more identified mutants to stabilize active than inactive state conformations are located in TM1, TM2, TM6, and TM7, whereby in TM3 and also in TM5/TM6 a high number of inactive state stabilizing mutations were identified.
GPCR activation commonly involves binding of an agonistic ligand or sensing of a physical trigger (e.g., light or mechanical forces), which induces alterations in the binding region and, subsequently, in specific helical adjustments relative to each other. This process finally enables intracellular binding of a transducer protein by enlargement of the crevice between the helices and ILs. The active state conformation is, therefore, stabilized by the ligand, the intracellular effector, and particular intramolecular side-chain interactions. In turn, this process, with the receptor as a central signaling hub of information, is primarily related to structural rearrangements, dependent on spatial-fit-in’s and biochemical recognition patterns [or “recognition barcodes” (86)] between the receptor-ligand complex and effector, such as the G-protein. How is this “activation process”, “signal transduction”, or “stabilization of the active state conformation” reflected by available ATR and ETBR structures?
More than ten ETBR and AT1R/AT2R structures (Table 1) with a bound agonist are known so far (Figure 4). These structures show specific features as intracellularly bound nanobodies (Figure 3A), extracellular bound antibody-fragments (Figure 4B), a non-canonical helix 8 orientation (Figure 4B1), or specificities in transmembrane helix conformations (Figure 4C). However, none of them is part of a complex with a G-protein or arrestin. However, when compared to inactive/antagonized conformations (Figures 4E, F), these active state-like conformations reveal how these GPCRs interact with agonists and how this binding process induces changes in receptor structure (Figure 5).
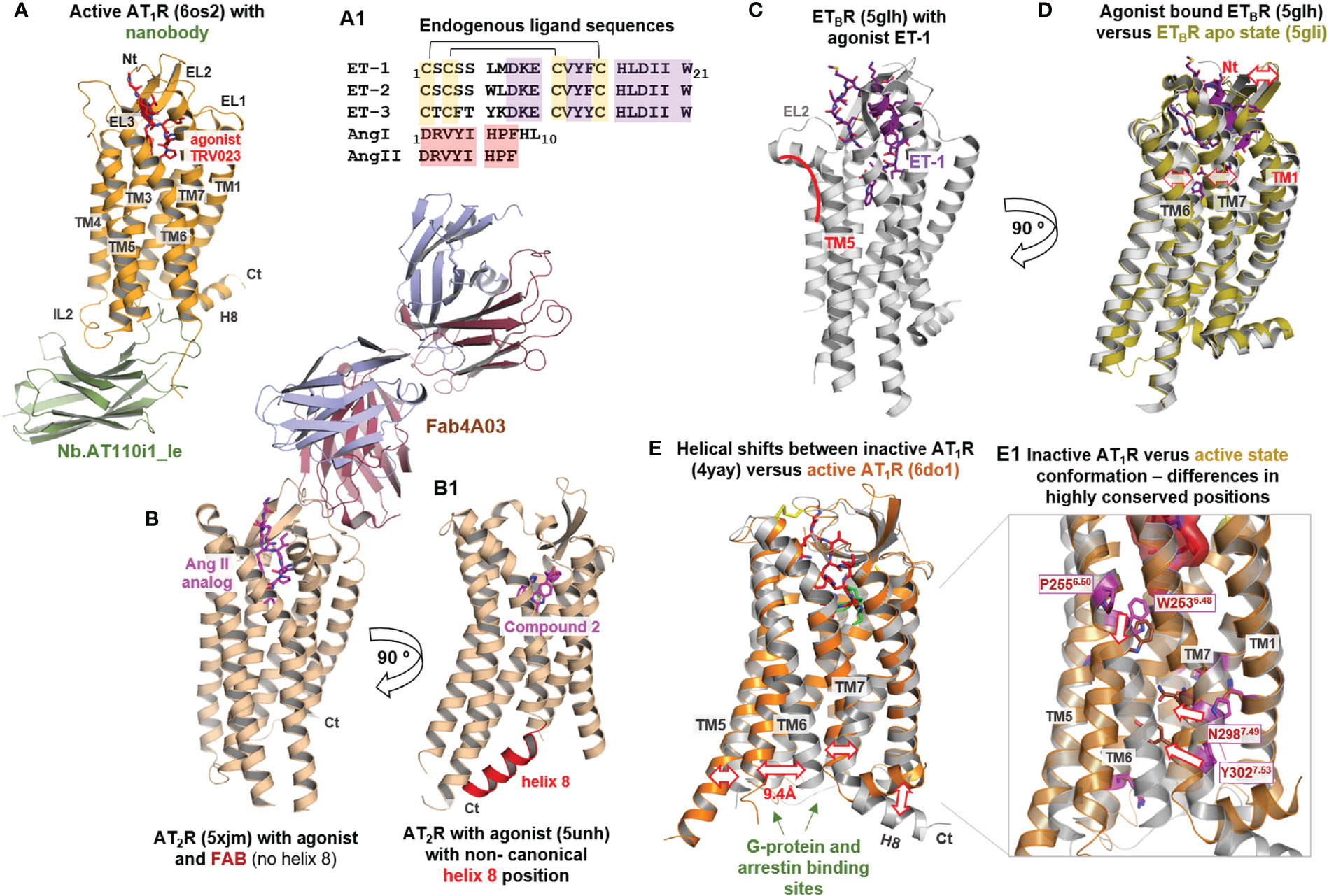
Figure 4 Agonist-bound and apo-state conformations of ATRs and ETBR. (A) Diverse AT1R structures in the agonist-bound state are already available (Table 1). The agonist [endogenous peptide agonist sequences are provided in (A1), including annotated disulfide bridges and conserved regions (colored background)] is bound extracellularly between the ELs and their transitions to the helices (Figure 5). Several AT1R structures are stabilized intracellularly by a bound nanobody (Table 1). The agonist-bound structures are not complexed yet with G-protein or arrestin. (B) The AT2R structures not only contain various agonists but have been further stabilized in some cases with Fabs (fragment antigen binding), which bind on the extracellular side. (B1) For AT2R, intracellular helix 8 has been observed to be directed inward to the transmembrane helix core and stabilizes the active state structure instead of a transducer protein like the G-protein. Generally, helix 8 is oriented parallel to the membrane and outside the helical bundle in GPCRs. (C) The active state ETBR structure bound with ET-1 represents endogenous ligand binding, whereby the ligand is buried deep within the ligand-binding pocket (see Figure 5). The helical transition from EL2 to TM5 is kinked (red line) in contrast to the ATR structures. (D) Comparison with the ligand-free apo-state conformation highlights structural differences in the extracellular region where the ligand is bound, mainly in TM6 and TM7, but also for EL2 (red arrows). A further difference is the helical transition between the N-terminus and helix 1 in the apo-state structure compared to an unfolded transition in the ligand-bound structure. (E) Agonist-bound AT1R and AT2R receptor conformations deviate from the inactive state structures in the intracellular orientation of TM6, but also relative spatial shifts are observed at the intracellular parts of TM5 and TM7 (red arrows). For the AT1R, strong deviations in the H8 orientation are observable in dependency of the activity state. (E1) The structural transitions between inactive and active state conformations are accompanied by re-organization of intramolecular interactions in the transmembrane helical core (62), as visualized here exemplarily at amino acid residues in TM6 and TM7. This re-organization and subsequent new interactions are involved in maintaining active state-like conformations.
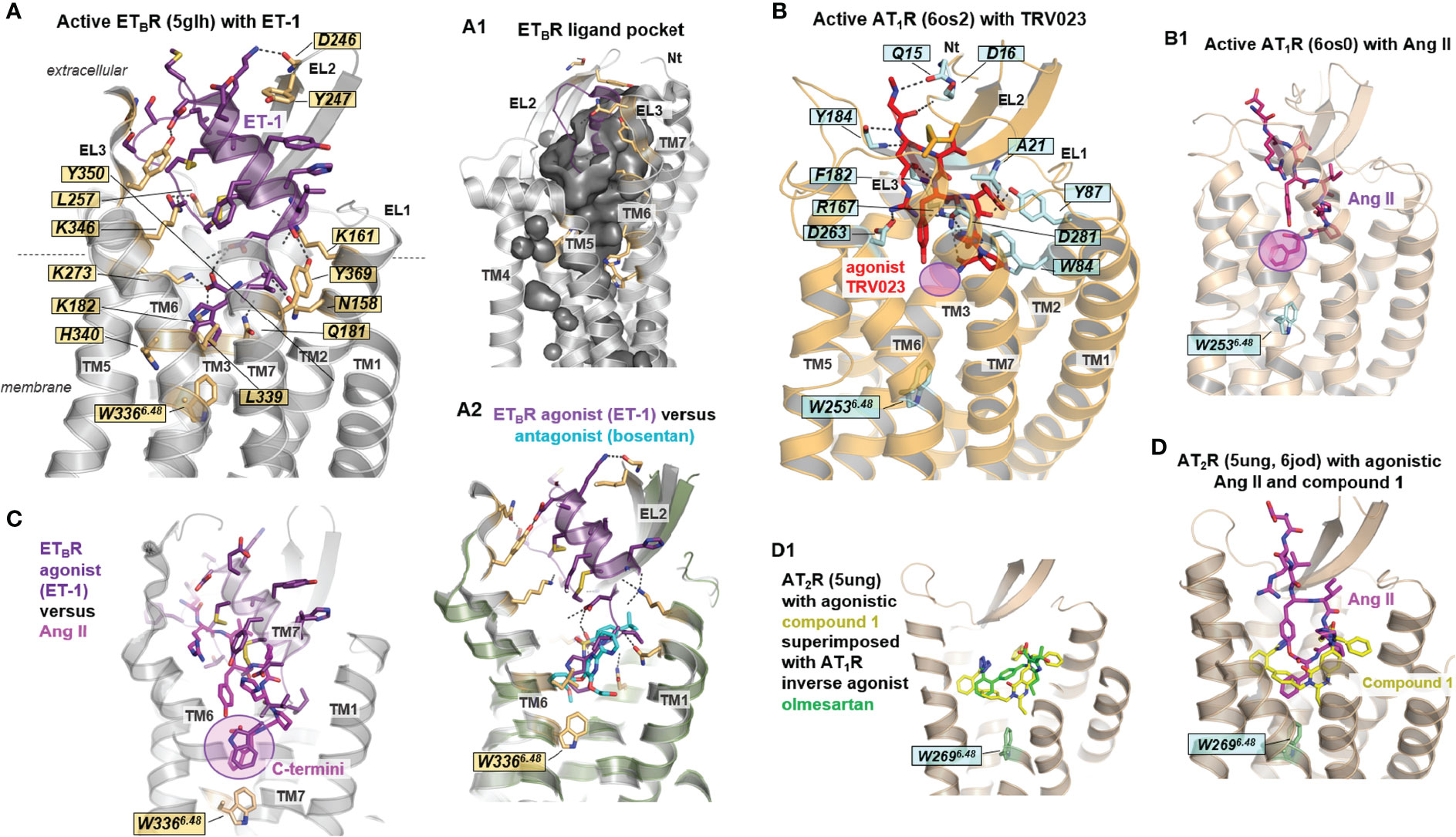
Figure 5 Details of agonist binding at AT1R, AT2R, and ETBR. (A) The ligands ET and Ang (or their derivatives) are bound mainly between the N-terminus, EL2, and several transmembrane helices, whereby the ligand-binding cavity is embedded deeply toward the transmembrane helical core close to tryptophan W3666.48 (ETBR). This essential tryptophan is in direct contact with the ligand-binding site of all receptors and their endogenous peptides [also (B, B1, C)]. Of note, the arrestin-biased angiotensin II analog TRV023 does not contact the W6.48 (B-B1) (magenta translucent circle). Many hydrophilic interactions between the receptors and the peptides can be observed, whereby four positively charged lysines and three tyrosines play a fundamental role in the corresponding ligand-receptor recognition in the ETBR/ET-1 complex. Generally, EL2, EL3, and the N-terminus cover the ligand-binding pocket [(A1), inner surface representation] extracellularly for both ETBR and ATRs. Several structures reveal direct interactions between the ligand and the N-terminus, for example, in the AT1R- β-arrestin biased agonist TRV026 (6os2) and ETBR- ET-3 (6igk) complexes. (A2) The bound peptide ET-1 in the ETBR (5glh) with superimposed non-peptidic antagonist bosentan (5xpr) shows partially overlapping binding pockets close to W6.48. (C) Superimposition of ET-1 (bound to ETBR, 5glh) and Ang II (bound to AT1R, 6os0) reveal structural differences between the ligands due to deviations in sequence composition and length (see also Figure 4A1); however, the C-terminally located aromatic residue in both ligands is close to the highly conserved W6.48, which is part of the activation-related toggle switch motif in helix 6. (D) Non-peptide AT2R agonists as compound 1 (Table 1) are bound deep within the ligand-binding region. This section is also occupied by the endogenous peptide agonist Ang II, indicating a region highly relevant for receptor activation. (D1) The non-peptide inverse agonist olmesartan for AT1R (4zud) is principally bound in the same region as the AT2R non-peptide agonist compound 1 (5ung) with identical interactions to EL2. The different effects of these ligands are attributed to their detailed interactions in corresponding receptors (not visualized in detail).
Generally, ATR and ETR agonists bind deep into an extracellular cleft formed between the EL1–3 and the adjacent TMs close to W6.48 (Figures 4, 5). The EL2, EL3, and the N-terminus cover the ligand-binding pocket extracellularly for both ETBR and ATRs (Figures 4A, 5A-A1). Receptor amino acids participating in ligand binding are located mainly at the C-terminal part of the receptor EL2, in TM2, TM6, and TM7 (Figures 5A, B). Further, direct interactions between the ligand and the N-terminus can be observed (AT1R- β-arrestin biased agonist TRV026 (PDB ID: 6os2) and ETBR/ET-3 (PDB ID: 6igk) complexes, Figure 5B).
Although no structure is available for the ETAR yet, it can be assumed that the binding mode of peptide-agonists at this receptor should be in principle similar to the binding mode observed at the agonist-bound ETBR structures. This hypothesis is based on comparison between receptor amino acids that are in direct contact to agonists (e.g. structure ETBR/ET-1, PDB ID: 5glh). Key contact (hydrogen bonds) amino acid residues from the receptor to the ligand are for instance K161 (TM2), K182 (TM3), E236 (TM5), R343 (TM6), K346 (TM6), Y350 (TM6), and they can be found also in the ETAR sequence at corresponding positions (K140, K166, E220, R326, K329, Y333). Based on this circumstance and the high overall sequence similarity of 62% between both receptor subtypes, it can be expected that the identified ETBR structures can serve as ideal templates to build ETAR homology-models. This is supported by experimental studies providing overlapping amino acids relevant for peptide-ligand binding (87). However, elucidation of potential differences in ligand binding properties (88), such as ligand affinity, definitely requires the determination of ETAR structures and structural complexes.
Together with W6.48, hydrophobic amino acids in TM3 (e.g., at positions 3.32 and 3.36) form a hydrophobic pocket that triggers receptor activation caused by endogenous ligand contact with an aromatic moiety (66). As mentioned above, this tryptophan is part of the CWxP6.50 motif that participates in the activation mechanism of class A GPCRs. Superimposition of ET-1 (bound to ETBR, PDB ID: 5glh) and Ang II (bound to AT1R, PDB ID: 6os0, Figure 5C) reveals structural differences between the ligands due to strong diversity in their sequence composition and length (Figure 4A1); however, the C-terminally located aromatic residues in both ligands are close to the highly conserved W6.48. Of note, the arrestin-biased Ang II analog ligand TRV023 with a shorter C-terminus does not interact with W6.48 (Figures 5B-B1), indicating selective receptor activation-dependent on specific ligand features.
What else can be observed via a comparison of structures with agonists vs. antagonists? Superimposing the structure of the agonistic peptide ET-1 in ETBR with that of the non-peptidic antagonist bosentan reveals a partially overlapping binding mode in the vicinity of W6.48, indicating that this region is important for receptor activation or inhibition of activation (Figure 5A2). In addition, several positively charged lysines are essential for ET-1 binding to the receptor in the ETBR/ET-1 complex (Figure 5A). These lysines are also key interaction partners for antagonist binding (Figure 2E), suggesting the importance of the inhibitory effect of antagonists on the binding of agonists. In the case of AT1R, the non-peptide inverse agonist olmesartan (PDB ID: 4zud) is bound in the same region as the AT2R non-peptide agonist compound 1 (PDB ID: 5ung, Figure 5D1), including identical interactions to the EL2. The different effects of these ligands can be attributed to their detailed interactions in corresponding receptors, namely an additional hydrogen-bond of the antagonist with a tyrosine in TM1 and a contact of the agonist with W6.48, which is blocked by a tyrosine in TM7 (Y2927.43) of the AT1R with an inverse agonist.
Interestingly, a comparison of the ETBR/ET-1 complex with the ligand-free apo-state conformation (Figure 4D) highlights structural differences specifically in the ligand-binding region at the extracellular ends of TM6, TM7, and in the EL2. Agonist binding causes structural modifications in the extracellular part, which, is, in strong contrast to observations from the comparison between agonist-bound and inactive/antagonized structures by antagonists (Figures 4E, E1). The agonist-bound structures of AT1R and AT2R deviate from the inactive state structures in the intracellular orientation of TM6 (shift of ~9Å), combined with relative spatial shifts at the intracellular parts of TM5 and TM7 (Figure 4E). These structural transitions between inactive and active state conformations are accompanied by re-organization of intramolecular interactions in the transmembrane helical core (62) (Figure 4E1).
As already noted, intracellular processes, such as G-protein binding or arrestin interactions concomitant to receptor-agonist complex formation, cannot yet be studied at available structures (Table 1). Usually, these molecules contribute toward stabilizing active state conformations. In the agonist-bound AT1R, a nanobody instead stabilizes the active state conformation [Figure 4A (63)] and, surprisingly, helix H8 is intracellularly directed inward to the transmembrane helix core of AT2R and stabilizes the active state receptor structure [Figure 4B1 (65)]. This non-canonical helix 8 orientation would impede binding of G-protein or arrestin and is assumed to be related to the finding of G-protein independent AT2R signaling (27–30). However, in a recent AT2R structure complexed with Ang II a regular helix 8 orientation as known to be canonical in GPCRs is observed (PDB ID: 6jod (66), shown in Figure 6), which evidences that this receptor can also adapt into a conformation able to bind G-protein or arrestin.

Figure 6 AT2R in complex with an antibody Fab-fragment and Ang II [PDB ID: 6jod (66)]. (A) The receptor is presented as a backbone cartoon, the ligand and the Fab are visualized with surfaces for clarity reasons. Amino acids involved in ligand and Fab binding are shown as sticks. Extracellular loops are colored red. (B) Ang II is bound deep within the helical bundle. Significant interactions can be observed with helices TM2, TM5, TM6, and EL2 (M197 backbone, R182). EL2 is involved simultaneously in Fab binding, namely with residues located in the central EL2 (E188, Y189, G191 backbone). In addition, Y106 (backbone) and D109 in the receptor EL1, contacting the Fab as well as amino acids Q37, P39 in the N-terminus. R107 in the EL1 is an important stabilizer of this constellation by several H-bonds to the transition between the N-terminus and TM1. The translucent-filled squares highlight distinguishable contact regions between the receptor with Fab and the receptor with the ligand.
In the agonist-bound ETBR structures (Table 1) without a nanobody, G-protein, or an inside orientated helix 8, the TM6 orientation is similar as in the inactive state conformations, whereby comparing the inactive state structure (PDB ID: 4zud) with the active state conformation (PDB ID: 6do1) of AT1R, a distance of intracellular TM6 of 9.4Å can be measured (Figure 4E). Moreover, in AT2R structures bound with a developed antibody Fab fragment without an intracellular stabilizer (PDB ID’s: 5xjm, 6jod), the extent of TM6 movement outside is smaller, only by approximately 7.8 Å compared to inactive AT1R structures, which indicates that these structures likely do not represent fully “active state conformations”.
The available AT2R-Fab complexes with Ang II or its derivative [Sar1, Ile8]-AngII (64, 66) show a specific binding epitope of the Fab fragment at the receptor, which is close to the ligand ‘core’ binding region, although not overlapping. The Fab fragment (Fab4A03) acts as a positive allosteric modulator without direct interaction with the ligands but increases the affinity of both agonists (64). Such a receptor/antibody interplay is known for many GPCRs (89). Recently, a human antibody (Ab) against human ETAR that exhibits antitumor potency has been published (90). Autoantibodies (auto-Abs) directed against AT1R acting as agonists or probably positive agonistic modulators inducing pathogenic conditions have been demonstrated several times (22, 91–93) as in women with preeclampsia (21), or in patients with acute vascular graft rejection (19, 94, 95). AT1R auto-Abs association with clinical features has also been studied extensively in the context of transplantation (96–100), or their effects on angiogenesis in preeclampsia (101–103). Binding of activating AT1R-Abs promotes specific downstream signaling via activation of AT1R (19, 20); however, while Ang II binding to the receptor has been already explored intensively (104–108), the binding mode(s) between auto-Abs and receptors have not yet been determined.
Based on current literature, only AT1R auto-Abs from patients with transplant rejection recognize epitopes that are located primarily in EL2 (19, 21). Accordingly, the known crystallized AT2R-Fab complexes (64, 66) (Table 1 and Figure 6) reveal that EL2 is involved in binding, namely with residues E188, Y189, and G191 located in the central EL2 (Figure 6). Furthermore, Y106 (backbone) and D109 in the receptor EL1 contribute to Fab binding as well as Q37 and P39 (backbone) in the N-terminus. This leads to the conclusion for ATRs that distinct receptor parts can interact simultaneously with Fabs and agonistic ligands (Figs. 4–6), whereby the concrete binding sites are distinct as at the N-terminus or EL2. This observation helps to explain how Fab fragments or antibodies mediate positive allosteric effects on signaling or directly trigger activation. The Abs may increase the predisposition of the receptor to bind Ang by a direct structural impact on the extended ligand-binding site (e.g., EL2), or/and increased signaling activity by bound Abs should lower the energetic barrier for the endogenous ligand to further stimulate the receptor. Of note, sequence comparison reveals that potential binding sites for antibodies in the EL1, EL2, and N-terminus are not conserved among ATRs and ETRs subtypes (Figure 1), with only a few amino acids at corresponding positions identical. This may support that so far known activating antibodies for both receptor subtypes could recognize specific structural conformations rather than binding-specific epitope residues at the receptor, which is in principle known from antibody studies at other proteins (109–111). However, different antibodies will bind naturally in a variety of ways and may differ in their receptor binding sites.
The term oligomerization indicates dimeric, trimeric, tetrameric, or higher-order complexes between GPCR protomers (monomers) and has been reported for numerous GPCRs not only in vitro (112) but also in native tissues (in vivo) (113–115). Homo- or hetero-oligomerization between single receptor protomers are mostly not a prerequisite for class A GPCR signaling capacity (116), but defines the spectrum of fine-tuning options in signaling, as they can act as a functional unit (117, 118). GPCR oligomerization has been reported for several GPCR classes, such as for class A, class B, taste receptors (119–121), or class D (122).
Dimerization describes interacting xGPCR/xGPCR (homodimer) or xGPCR-yGPCR (heterodimer) constellations. For defining relevant GPCR-GPCR dimers or oligomers, several aspects are of significance, such as direct intermolecular side-chain interactions or an impact on functionalities (e.g., expression, internalization, signaling, ligand binding) compared to monomeric receptors. In heterodimerization, GPCR expression in the same cell type and cell compartment, as well as simultaneous occurrence (time-dependent expression), are prerequisites (123, 124). A large amount of GPCR-GPCR protomer interfaces with intermolecular interactions between single amino acids or between several side chains have been reported under the involvement of TM4 (125–127), TM1, and TM5-6 (128, 129). Studying the available class A GPCR dimers in determined structures, specifically the TM1-TM1/helix8-helix8 and the TM4-TM4/TM5-TM5 interfaces, occur often (130). However, different oligomer GPCR interfaces for homo- and heterodimers can be assumed, whereby likely no universal interface exists. Supposedly, receptor interfaces are of dynamic character (131) and GPCRs are expressed as a mixture of monomers and homomers, whereby the two forms may interconvert dynamically (132). Several examples demonstrate that GPCR oligomerization can have a major impact on the signaling properties of interacting protomers, e.g., in ligand binding (133, 134), G-protein coupling specificity, and signal transduction mechanisms (114), or cell surface expression (135). In the event of a direct mutual effect of GPCRs organized in dimeric arrangements, a horizontal allosteric impact on each other, either positively or negatively, may occur (136).
For the ATRs and ETRs, a tremendous set of information is available, supporting a wide spectrum of oligomer formations. As exemplarily summarized from literature databases and a direct collection of GPCR oligomers (GPCR Interaction Network, http://www.gpcr-hetnet.com (137)), the following oligomers have been reported for ATRs or ETRs:
● AT1R with PAR1 (138), μOR (139), prostaglandin F2aR (140), ETBR (141), RXFP1 (in vivo (142, 143)), ADRB2 (144), AT2R (145), CB1R (146), secretin receptor (SCTR, class B) (147), bradykinin B2R (148);
● AT2R with AT2R (149), bradykinin B2R (150);
● ETBR with D3R (151), ETAR (56, 152–154); and
● ETAR with µOR (155).
Oligomerization of wild-type and a non-functional AT1R mutant inhibits Gαq-mediated signaling but not ERK activation, supporting a functional influence of a homo-oligomerization (156). Aldosterone-related effects activate AT1R and AT2R hetero-dimerizations (149), altering trafficking and arrestin recruitment profiles (145). Further functional effects reported to be associated with homo- or heterodimerization are, for example, transactivation and synergism [AT1R with PAR1 (138)], altered expression levels for AT1R - ETBR heteromers (141), or ATRs with RXFP1 show functional crosstalk in myofibroblasts (142, 143). AT2R heterodimerization with bradykinin B2R (150) has a strong impact on the signaling outcome and amplitude (NO production). ETBR-ETAR heterodimers are modified in internalization rates compared to the homo-dimerization of the wild-type receptors (152).
To date, only one report on the AT1R homodimer structure exists [PDB ID: 6do1 (62)]. The interface between the single protomers is constituted by hydrophobic and aromatic amino acid side chain contacts at EL1, TM1, TM2, TM3, and helix 8 (Figure 7A). Interestingly, this dimer is in an active state conformation, bound with an Ang II analog and with intracellularly stabilizing nanobodies at each protomer. The observable interface in the AT1R dimer is in agreement with interfaces in many other GPCR dimers (157), which might imply relevance also in vivo to cause a mutually allosteric (158) functional impact on ligand binding capacities or internalization rates. However, other interfaces were studied and recently proposed by atom molecular dynamics simulations (159), which is in line with the assumed multitude of feasible GPCR oligomer arrangements.

Figure 7 Dimer arrangement of the active state AT1R bound with an Ang II analog and nanobodies. (A) The complex between the Ang II analog, AT1R, and active state stabilizing nanobodies has been crystallized as a homodimer [PDB ID: 6do1 (62)]. The interface between the protomers is constituted by hydrophobic and aromatic amino acid side chain contacts at EL1 (M90, F96), TM1 (F55, intracellularly), TM2 (Y99), and helix 3 (L100). (B) In a putative scenario of a dimeric receptor arrangement with antibody binding at one protomer, the Fab fragment should also simultaneously contact the second receptor protomer. For this model, the AT2R structure (6jod), with and without a Fab, were arranged together as suggested by the AT1R homodimer.
As exemplified in Figure 6B in a dimeric receptor formation, a bound antibody at one protomer should simultaneously contact the second protomer (Figure 7B). This should be the case for homodimers of AT1R (156), AT2R (149), or heterodimers of ATRs (145) and ETRs (56, 141), which are known to be occupied endogenously by antibodies under pathogenic conditions (160, 161). As already mentioned above, an AT2R/Ang II analog complex was co-crystallized with a Fab. This Fab acts as a positive allosteric modulator (64), which might also be related to observed dimeric receptor constellations or might have consequences on the functional reactivity of receptor dimers.
Finally, if homo- or heterodimeric ATR and ETR arrangements are of functional and physiological relevance, pharmacological interventions may (must) target or consider these oligomers, especially with the aim of circumventing adverse effects mediated by allosteric heterodimer actions. Correspondingly, if the large number of putative heterodimers between ATRs/ETRs and other GPCRs are functionally relevant in vivo, any pharmacological intervention at their interaction partner should also have an impact on both receptor subtypes (ETR, ATR), which might be registered medically as unwanted adverse effects. Pharmacological strategies may profit from homo- or heterobivalent ligands specifically entering GPCR dimers (162, 163) in diverse ligand constellations, e.g., as bitopic and dualsteric ligands (164).
As summarized in this short review, an enormous amount of structural-functional information on ATRs and ETRs is available, with a clear boost on structure determination since 2015. These structures provide details and general insights into mechanisms of activation and features of nonactive or inactive states. An advantage of the high number of solved structures is the resulting capability for comparison, including diversities in ligand binding, and to study the spectrum of possibilities in structural arrangements, e.g., helix conformations or dimer formation. However, several gaps in knowledge are evident, with primary emphasis on not yet determined ETAR structures and missing structural information on G-protein or arrestin binding. Moreover, reflecting the high number of GPCR heteromer reports for ATRs and ETRs with functional impact, it also appears necessary to intensify further means of exploring ways to elucidate heteromer arrangements, both structurally and functionally for these receptors and binding partners. In addition, this is an area of utmost pharmacological importance (165, 166) and, therefore, must be of structural interest, especially given the increasing possibilities in the determination of complex structures (167). Finally, the relevance of autoantibody binding to both receptor groups require questions on antibody binding and its functional significance to be explored in-depth, intending to use improved understanding to tailor the design of optimal ligands useful for pharmacological intervention strategies or to recruit these receptors (as monomers or dimers) as hubs for precisely sought specific responses.
Manuscript writing: DS, GK, and PS. Figure and table preparation: DS and GK. Manuscript editing: MS, DK, RC, and AP. Data analyses: DS, GK, DK, and PS. Supervising: GK and PS Funding: PS. All authors contributed to the article and approved the submitted version.
This work was supported by the Deutsche Forschungsgemeinschaft (DFG) through CRC 1365, – Project-ID 394046635 – SFB 1365, subproject A03 (to PS); through CRC 1423, project number 421152132 – SFB 1423, subprojects A01 (to PS); and through the European Union’s Horizon 2020 MSCA Program under grant agreement 956314 [ALLODD] (to PS).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fendo.2022.880002/full#supplementary-material
1. Hofmann KP, Scheerer P, Hildebrand PW, Choe HW, Park JH, Heck M, et al. A G-Protein-Coupled Receptor at Work: The Rhodopsin Model. Trends Biochem Sci (2009) 34:540–52. doi: 10.1016/j.tibs.2009.07.005
2. Kristiansen K. Molecular Mechanisms of Ligand Binding, Signaling, and Regulation Within the Superfamily of G-Protein-Coupled Receptors: Molecular Modeling and Mutagenesis Approaches to Receptor Structure and Function. Pharmacol Ther (2004) 103:21–80. doi: 10.1016/j.pharmthera.2004.05.002
3. Weis WI, Kobilka BK. The Molecular Basis of G-Protein-Coupled Receptor Activation. Annu Rev Biochem (2018) 87:897–919. doi: 10.1146/annurev-biochem-060614-033910
4. Ho MK, Su Y, Yeung WW, Wong YH. Regulation of Transcription Factors by Heterotrimeric G-Proteins. Curr Mol Pharmacol (2009) 2:19–31. doi: 10.2174/1874467210902010019
5. Veldhuis NA, Poole DP, Grace M, McIntyre P, Bunnett NW. The G-Protein-Coupled Receptor-Transient Receptor Potential Channel Axis: Molecular Insights for Targeting Disorders of Sensation and Inflammation. Pharmacol Rev (2015) 67:36–73. doi: 10.1124/pr.114.009555
6. Horinouchi T, Terada K, Higashi T, Miwa S. Endothelin Receptor Signaling: New Insight Into its Regulatory Mechanisms. J Pharmacol Sci (2013) 123:85–101. doi: 10.1254/jphs.13R02CR
7. Limbird LE. The Receptor Concept: A Continuing Evolution. Mol Interv (2004) 4:326–36. doi: 10.1124/mi.4.6.6
8. Sriram K, Insel PA. G-Protein-Coupled Receptors as Targets for Approved Drugs: How Many Targets and How Many Drugs? Mol Pharmacol (2018) 93:251–8. doi: 10.1124/mol.117.111062
9. Dorsam RT, Gutkind JS. G-Protein-Coupled Receptors and Cancer. Nat Rev Cancer (2007) 7:79–94. doi: 10.1038/nrc2069
10. Schoneberg T, Schulz A, Biebermann H, Hermsdorf T, Rompler H, Sangkuhl K. Mutant G-Protein-Coupled Receptors as a Cause of Human Diseases. Pharmacol Ther (2004) 104:173–206. doi: 10.1016/j.pharmthera.2004.08.008
11. Heyder N, Kleinau G, Szczepek M, Kwiatkowski D, Speck D, Soletto L, et al. Signal Transduction and Pathogenic Modifications at the Melanocortin-4 Receptor: A Structural Perspective. Front Endocrinol (Lausanne) (2019) 10:515. doi: 10.3389/fendo.2019.00515
12. Heyder NA, Kleinau G, Speck D, Schmidt A, Paisdzior S, Szczepek M, et al. Structures of Active Melanocortin-4 Receptor-Gs-Protein Complexes With NDP-Alpha-MSH and Setmelanotide. Cell Res (2021) 31:1176–89. doi: 10.1038/s41422-021-00569-8
13. Hyndman KA, Miyamoto MM, Evans DH. Phylogeny, Taxonomy, and Evolution of the Endothelin Receptor Gene Family. Mol Phylogenet Evol (2009) 52:677–87. doi: 10.1016/j.ympev.2009.04.015
14. Singh KD, Karnik SS. Angiotensin Receptors: Structure, Function, Signaling and Clinical Applications. J Cell Signal 1 (2016) 1(2):111. doi: 10.4172/jcs.1000111
15. Ardaillou R, Chansel D. Synthesis and Effects of Active Fragments of Angiotensin II. Kidney Int (1997) 52:1458–68. doi: 10.1038/ki.1997.476
16. Armstrong JF, Faccenda E, Harding SD, Pawson AJ, Southan C, Sharman JL, et al. The IUPHAR/BPS Guide to PHARMACOLOGY in 2020: Extending Immunopharmacology Content and Introducing the IUPHAR/MMV Guide to MALARIA PHARMACOLOGY. Nucleic Acids Res (2020) 48:D1006–21. doi: 10.1093/nar/gkz951
17. Turner JM, Bauer C, Abramowitz MK, Melamed ML, Hostetter TH. Treatment of Chronic Kidney Disease. Kidney Int (2012) 81:351–62. doi: 10.1038/ki.2011.380
18. Karnik SS, Unal H, Kemp JR, Tirupula KC, Eguchi S, Vanderheyden PM, et al. International Union of Basic and Clinical Pharmacology. XCIX. Angiotensin Receptors: Interpreters of Pathophysiological Angiotensinergic Stimuli [Corrected]. Pharmacol Rev (2015) 67:754–819. doi: 10.1124/pr.114.010454
19. Dragun D, Muller DN, Brasen JH, Fritsche L, Nieminen-Kelha M, Dechend R, et al. Angiotensin II Type 1-Receptor Activating Antibodies in Renal-Allograft Rejection. N Engl J Med (2005) 352:558–69. doi: 10.1056/NEJMoa035717
20. Riemekasten G, Philippe A, Nather M, Slowinski T, Muller DN, Heidecke H, et al. Involvement of Functional Autoantibodies Against Vascular Receptors in Systemic Sclerosis. Ann Rheum Dis (2011) 70:530–6. doi: 10.1136/ard.2010.135772
21. Wallukat G, Homuth V, Fischer T, Lindschau C, Horstkamp B, Jupner A, et al. Patients With Preeclampsia Develop Agonistic Autoantibodies Against the Angiotensin AT1 Receptor. J Clin Invest (1999) 103:945–52. doi: 10.1172/JCI4106
22. Catar R, Herse-Naether M, Zhu N, Wagner P, Wischnewski O, Kusch A, et al. Autoantibodies Targeting AT1- and ETA-Receptors Link Endothelial Proliferation and Coagulation via Ets-1 Transcription Factor. Int J Mol Sci (2021) 23(1):244. doi: 10.3390/ijms23010244
23. Trougakos IP, Stamatelopoulos K, Terpos E, Tsitsilonis OE, Aivalioti E, Paraskevis D, et al. Insights to SARS-CoV-2 Life Cycle, Pathophysiology, and Rationalized Treatments That Target COVID-19 Clinical Complications. J BioMed Sci (2021) 28:9. doi: 10.1186/s12929-020-00703-5
24. Bellis A, Mauro C, Barbato E, Trimarco B, Morisco C. The Rationale for Angiotensin Receptor Neprilysin Inhibitors in a Multi-Targeted Therapeutic Approach to COVID-19. Int J Mol Sci (2020) 21(22):8621. doi: 10.3390/ijms21228612
25. Sharma T, Mehan S. Possible Therapeutic Interventions in COVID-19 Induced ARDS by Cotinine as an ACE-2 Promoter and AT-1R Blocker. Infect Disord Drug Targets (2020) 21(6):e170721189261. doi: 10.2174/1871526520666201218153554
26. Manglik A, Wingler LM, Rockman HA, Lefkowitz RJ. Beta-Arrestin-Biased Angiotensin II Receptor Agonists for COVID-19. Circulation (2020) 142:318–20. doi: 10.1161/CIRCULATIONAHA.120.048723
27. Bottari SP, Taylor V, King IN, Bogdal Y, Whitebread S, de Gasparo M. Angiotensin II AT2 Receptors Do Not Interact With Guanine Nucleotide Binding Proteins. Eur J Pharmacol (1991) 207:157–63. doi: 10.1016/0922-4106(91)90091-U
28. Feng YH, Sun Y, Douglas JG. Gbeta Gamma -Independent Constitutive Association of Galpha s With SHP-1 and Angiotensin II Receptor AT2 is Essential in AT2-Mediated ITIM-Independent Activation of SHP-1. Proc Natl Acad Sci U.S.A. (2002) 99:12049–54. doi: 10.1073/pnas.192404199
29. Mukoyama M, Horiuchi M, Nakajima M, Pratt RE, Dzau VJ. Characterization of a Rat Type 2 Angiotensin II Receptor Stably Expressed in 293 Cells. Mol Cell Endocrinol (1995) 112:61–8. doi: 10.1016/0303-7207(95)03586-V
30. Brechler V, Reichlin S, De Gasparo M, Bottari SP. Angiotensin II Stimulates Protein Tyrosine Phosphatase Activity Through a G-Protein Independent Mechanism. Recept Channels (1994) 2:89–98.
31. Zhang J, Pratt RE. The AT2 Receptor Selectively Associates With Gialpha2 and Gialpha3 in the Rat Fetus. J Biol Chem (1996) 271:15026–33. doi: 10.1074/jbc.271.25.15026
32. Miura S, Karnik SS. Ligand-Independent Signals From Angiotensin II Type 2 Receptor Induce Apoptosis. EMBO J (2000) 19:4026–35. doi: 10.1093/emboj/19.15.4026
33. Fatima N, Patel SN, Hussain T. Angiotensin II Type 2 Receptor: A Target for Protection Against Hypertension, Metabolic Dysfunction, and Organ Remodeling. Hypertension (2021) 77:1845–56. doi: 10.1161/HYPERTENSIONAHA.120.11941
34. Peluso AA, Santos RA, Unger T, Steckelings UM. The Angiotensin Type 2 Receptor and the Kidney. Curr Opin Nephrol Hypertens (2017) 26:36–42. doi: 10.1097/MNH.0000000000000289
35. Yan X, Price RL, Nakayama M, Ito K, Schuldt AJ, Manning WJ, et al. Ventricular-Specific Expression of Angiotensin II Type 2 Receptors Causes Dilated Cardiomyopathy and Heart Failure in Transgenic Mice. Am J Physiol Heart Circ Physiol (2003) 285:H2179–87. doi: 10.1152/ajpheart.00361.2003
36. Balogh M, Aguilar C, Nguyen NT, Shepherd AJ. Angiotensin Receptors and Neuropathic Pain. Pain Rep (2021) 6:e869. doi: 10.1097/PR9.0000000000000869
37. Matavelli LC, Siragy HM. AT2 Receptor Activities and Pathophysiological Implications. J Cardiovasc Pharmacol (2015) 65:226–32. doi: 10.1097/FJC.0000000000000208
38. Arai H, Hori S, Aramori I, Ohkubo H, Nakanishi S. Cloning and Expression of a cDNA Encoding an Endothelin Receptor. Nature (1990) 348:730–2. doi: 10.1038/348730a0
39. Hosoda K, Nakao K, Hiroshi A, Suga S, Ogawa Y, Mukoyama M, et al. Cloning and Expression of Human Endothelin-1 Receptor cDNA. FEBS Lett (1991) 287:23–6. doi: 10.1016/0014-5793(91)80007-P
40. Regard JB, Sato IT, Coughlin SR. Anatomical Profiling of G Protein-Coupled Receptor Expression. Cell (2008) 135:561–71. doi: 10.1016/j.cell.2008.08.040
41. Maguire JJ, Davenport AP. Endothelin@25 - New Agonists, Antagonists, Inhibitors and Emerging Research Frontiers: IUPHAR Review 12. Br J Pharmacol (2014) 171:5555–72. doi: 10.1111/bph.12874
42. Granger JP, Spradley FT, Bakrania BA. The Endothelin System: A Critical Player in the Pathophysiology of Preeclampsia. Curr Hypertens Rep (2018) 20:32. doi: 10.1007/s11906-018-0828-4
43. Zile MR, Bourge RC, Redfield MM, Zhou D, Baicu CF, Little WC. Randomized, Double-Blind, Placebo-Controlled Study of Sitaxsentan to Improve Impaired Exercise Tolerance in Patients With Heart Failure and a Preserved Ejection Fraction. JACC Heart Fail (2014) 2:123–30. doi: 10.1016/j.jchf.2013.12.002
44. Rubin LJ, Badesch DB, Barst RJ, Galie N, Black CM, Keogh A, et al. Bosentan Therapy for Pulmonary Arterial Hypertension. N Engl J Med (2002) 346:896–903. doi: 10.1056/NEJMoa012212
45. Nakano D, Pollock DM. Contribution of Endothelin A Receptors in Endothelin 1-Dependent Natriuresis in Female Rats. Hypertension (2009) 53:324–30. doi: 10.1161/HYPERTENSIONAHA.108.123687
46. Zager RA, Johnson AC, Andress D, Becker K. Progressive Endothelin-1 Gene Activation Initiates Chronic/End-Stage Renal Disease Following Experimental Ischemic/Reperfusion Injury. Kidney Int (2013) 84:703–12. doi: 10.1038/ki.2013.157
47. Gordon CT, Weaver KN, Zechi-Ceide RM, Madsen EC, Tavares AL, Oufadem M, et al. Mutations in the Endothelin Receptor Type A Cause Mandibulofacial Dysostosis With Alopecia. Am J Hum Genet (2015) 96:519–31. doi: 10.1016/j.ajhg.2015.01.015
48. Pritchard AB, Kanai SM, Krock B, Schindewolf E, Oliver-Krasinski J, Khalek N, et al. Loss-Of-Function of Endothelin Receptor Type A Results in Oro-Oto-Cardiac Syndrome. Am J Med Genet A (2020) 182:1104–16. doi: 10.1002/ajmg.a.61531
49. Czopek A, Moorhouse R, Webb DJ, Dhaun N. Therapeutic Potential of Endothelin Receptor Antagonism in Kidney Disease. Am J Physiol Regul Integr Comp Physiol (2016) 310:R388–97. doi: 10.1152/ajpregu.00478.2015
50. Davenport AP, Hyndman KA, Dhaun N, Southan C, Kohan DE, Pollock JS, et al. Endothelin. Pharmacol Rev (2016) 68:357–418. doi: 10.1124/pr.115.011833
51. Dhaun N, Webb DJ. Endothelins in Cardiovascular Biology and Therapeutics. Nat Rev Cardiol (2019) 16:491–502. doi: 10.1038/s41569-019-0176-3
52. Vignon-Zellweger N, Heiden S, Miyauchi T, Emoto N. Endothelin and Endothelin Receptors in the Renal and Cardiovascular Systems. Life Sci (2012) 91:490–500. doi: 10.1016/j.lfs.2012.03.026
53. Zhang H, Unal H, Desnoyer R, Han GW, Patel N, Katritch V, et al. Structural Basis for Ligand Recognition and Functional Selectivity at Angiotensin Receptor. J Biol Chem (2015) 290:29127–39. doi: 10.1074/jbc.M115.689000
54. Hall TA. BioEdit: A User-Friendly Biological Sequence Alignment Editor and Analysis Program for Windows 95/98/Nt. Nucleic Acids Symposium Ser Ser (1999) 41:95–8.
55. Saito Y, Mizuno T, Itakura M, Suzuki Y, Ito T, Hagiwara H, et al. Primary Structure of Bovine Endothelin ETB Receptor and Identification of Signal Peptidase and Metal Proteinase Cleavage Sites. J Biol Chem (1991) 266:23433–7. doi: 10.1016/S0021-9258(18)54515-4
56. Evans NJ, Walker JW. Endothelin Receptor Dimers Evaluated by FRET, Ligand Binding, and Calcium Mobilization. Biophys J (2008) 95:483–92. doi: 10.1529/biophysj.107.119206
57. Boesen EI. Endothelin Receptors, Renal Effects and Blood Pressure. Curr Opin Pharmacol (2015) 21:25–34. doi: 10.1016/j.coph.2014.12.007
58. Douglas SA, Ohlstein EH. Signal Transduction Mechanisms Mediating the Vascular Actions of Endothelin. J Vasc Res (1997) 34:152–64. doi: 10.1159/000159219
59. Takigawa M, Sakurai T, Kasuya Y, Abe Y, Masaki T, Goto K. Molecular Identification of Guanine-Nucleotide-Binding Regulatory Proteins Which Couple to Endothelin Receptors. Eur J Biochem (1995) 228:102–8. doi: 10.1111/j.1432-1033.1995.0102o.x
60. Wang R, Dashwood RH. Endothelins and Their Receptors in Cancer: Identification of Therapeutic Targets. Pharmacol Res (2011) 63:519–24. doi: 10.1016/j.phrs.2011.01.002
61. Zhang H, Unal H, Gati C, Han GW, Liu W, Zatsepin NA, et al. Structure of the Angiotensin Receptor Revealed by Serial Femtosecond Crystallography. Cell (2015) 161:833–44. doi: 10.1016/j.cell.2015.04.011
62. Wingler LM, McMahon C, Staus DP, Lefkowitz RJ, Kruse AC. Distinctive Activation Mechanism for Angiotensin Receptor Revealed by a Synthetic Nanobody. Cell (2019) 176:479–490 e12. doi: 10.1016/j.cell.2018.12.006
63. Wingler LM, Skiba MA, McMahon C, Staus DP, Kleinhenz ALW, Suomivuori CM, et al. Angiotensin and Biased Analogs Induce Structurally Distinct Active Conformations Within a GPCR. Science (2020) 367:888–92. doi: 10.1126/science.aay9813
64. Asada H, Horita S, Hirata K, Shiroishi M, Shiimura Y, Iwanari H, et al. Crystal Structure of the Human Angiotensin II Type 2 Receptor Bound to an Angiotensin II Analog. Nat Struct Mol Biol (2018) 25:570–6. doi: 10.1038/s41594-018-0079-8
65. Zhang H, Han GW, Batyuk A, Ishchenko A, White KL, Patel N, et al. Structural Basis for Selectivity and Diversity in Angiotensin II Receptors. Nature (2017) 544:327–32. doi: 10.1038/nature22035
66. Asada H, Inoue A, Ngako Kadji FM, Hirata K, Shiimura Y, Im D, et al. The Crystal Structure of Angiotensin II Type 2 Receptor With Endogenous Peptide Hormone. Structure (2020) 28:418–425 e4. doi: 10.1016/j.str.2019.12.003
67. Nagiri C, Shihoya W, Inoue A, Kadji FMN, Aoki J, Nureki O. Crystal Structure of Human Endothelin ETB Receptor in Complex With Peptide Inverse Agonist IRL2500. Commun Biol (2019) 2:236. doi: 10.1038/s42003-019-0482-7
68. Shihoya W, Nishizawa T, Yamashita K, Inoue A, Hirata K, Kadji FMN, et al. X-Ray Structures of Endothelin ETB Receptor Bound to Clinical Antagonist Bosentan and its Analog. Nat Struct Mol Biol (2017) 24:758–64. doi: 10.1038/nsmb.3450
69. Shihoya W, Nishizawa T, Okuta A, Tani K, Dohmae N, Fujiyoshi Y, et al. Activation Mechanism of Endothelin ETB Receptor by Endothelin-1. Nature (2016) 537:363–8. doi: 10.1038/nature19319
70. Shihoya W, Izume T, Inoue A, Yamashita K, Kadji FMN, Hirata K, et al. Crystal Structures of Human ETB Receptor Provide Mechanistic Insight Into Receptor Activation and Partial Activation. Nat Commun (2018) 9:4711. doi: 10.1038/s41467-018-07094-0
71. Izume T, Miyauchi H, Shihoya W, Nureki O. Crystal Structure of Human Endothelin ETB Receptor in Complex With Sarafotoxin S6b. Biochem Biophys Res Commun (2020) 528:383–8. doi: 10.1016/j.bbrc.2019.12.091
72. Park JH, Scheerer P, Hofmann KP, Choe HW, Ernst OP. Crystal Structure of the Ligand-Free G-Protein-Coupled Receptor Opsin. Nature (2008) 454:183–7. doi: 10.1038/nature07063
73. Schwartz TW, Frimurer TM, Holst B, Rosenkilde MM, Elling CE. Molecular Mechanism of 7TM Receptor Activation–A Global Toggle Switch Model. Annu Rev Pharmacol Toxicol (2006) 46:481–519. doi: 10.1146/annurev.pharmtox.46.120604.141218
74. Ballesteros JA, Weinstein H. Integrated Methods for the Construction of Three-Dimensional Models and Computational Probing of Structure-Function Relationships in G-Protein Coupled Receptors. Methods Neurosci (1995) 25:366–428. doi: 10.1016/S1043-9471(05)80049-7
75. Fredriksson R, Lagerstrom MC, Lundin LG, Schioth HB. The G-Protein-Coupled Receptors in the Human Genome Form Five Main Families. Phylogenetic Analysis, Paralogon Groups, and Fingerprints. Mol Pharmacol (2003) 63:1256–72. doi: 10.1124/mol.63.6.1256
76. Reiersen H, Rees AR. The Hunchback and its Neighbours: Proline as an Environmental Modulator. Trends Biochem Sci (2001) 26:679–84. doi: 10.1016/S0968-0004(01)01957-0
77. Yohannan S, Faham S, Yang D, Whitelegge JP, Bowie JU. The Evolution of Transmembrane Helix Kinks and the Structural Diversity of G-Protein-Coupled Receptors. Proc Natl Acad Sci USA (2004) 101:959–63. doi: 10.1073/pnas.0306077101
78. Katritch V, Fenalti G, Abola EE, Roth BL, Cherezov V, Stevens RC. Allosteric Sodium in Class A GPCR Signaling. Trends Biochem Sci (2014) 39:233–44. doi: 10.1016/j.tibs.2014.03.002
79. Berman HM, Battistuz T, Bhat TN, Bluhm WF, Bourne PE, Burkhardt K, et al. The Protein Data Bank. Acta Crystallogr D Biol Crystallogr (2002) 58:899–907. doi: 10.1107/S0907444902003451
80. Filipek S. Molecular Switches in GPCRs. Curr Opin Struct Biol (2019) 55:114–20. doi: 10.1016/j.sbi.2019.03.017
81. Visiers I, Ballesteros JA, Weinstein H. Three-Dimensional Representations of G-Protein-Coupled Receptor Structures and Mechanisms. Methods Enzymol (2002) 343:329–71. doi: 10.1016/S0076-6879(02)43145-X
82. Nygaard R, Frimurer TM, Holst B, Rosenkilde MM, Schwartz TW. Ligand Binding and Micro-Switches in 7TM Receptor Structures. Trends Pharmacol Sci (2009) 30:249–59. doi: 10.1016/j.tips.2009.02.006
83. Shi L, Liapakis G, Xu R, Guarnieri F, Ballesteros JA, Javitch JA. Beta2 Adrenergic Receptor Activation. Modulation of the Proline Kink in Transmembrane 6 by a Rotamer Toggle Switch. J Biol Chem (2002) 277:40989–96. doi: 10.1074/jbc.M206801200
84. Heydenreich FM, Vuckovic Z, Matkovic M, Veprintsev DB. Stabilization of G-Protein-Coupled Receptors by Point Mutations. Front Pharmacol (2015) 6:82. doi: 10.3389/fphar.2015.00082
85. Magnani F, Serrano-Vega MJ, Shibata Y, Abdul-Hussein S, Lebon G, Miller-Gallacher J, et al. A Mutagenesis and Screening Strategy to Generate Optimally Thermostabilized Membrane Proteins for Structural Studies. Nat Protoc (2016) 11:1554–71. doi: 10.1038/nprot.2016.088
86. Flock T, Hauser AS, Lund N, Gloriam DE, Balaji S, Babu MM. Selectivity Determinants of GPCR-G-Protein Binding. Nature (2017) 545:317–22. doi: 10.1038/nature22070
87. Han SG, Ko S, Lee WK, Jung ST, Yu YG. Determination of the Endothelin-1 Recognition Sites of Endothelin Receptor Type A by the Directed-Degeneration Method. Sci Rep (2017) 7:7577. doi: 10.1038/s41598-017-08096-6
88. Lattig J, Oksche A, Beyermann M, Rosenthal W, Krause G. Structural Determinants for Selective Recognition of Peptide Ligands for Endothelin Receptor Subtypes ETA and ETB. J Pept Sci (2009) 15:479–91. doi: 10.1002/psc.1146
89. Skiba MA, Kruse AC. Autoantibodies as Endogenous Modulators of GPCR Signaling. Trends Pharmacol Sci (2021) 42:135–50. doi: 10.1016/j.tips.2020.11.013
90. Ju MS, Ahn HM, Han SG, Ko S, Na JH, Jo M, et al. A Human Antibody Against Human Endothelin Receptor Type A That Exhibits Antitumor Potency. Exp Mol Med (2021) 53:1437–48. doi: 10.1038/s12276-021-00678-9
91. Lefaucheur C, Viglietti D, Bouatou Y, Philippe A, Pievani D, Aubert O, et al. Non-HLA Agonistic Anti-Angiotensin II Type 1 Receptor Antibodies Induce a Distinctive Phenotype of Antibody-Mediated Rejection in Kidney Transplant Recipients. Kidney Int (2019) 96:189–201. doi: 10.1016/j.kint.2019.01.030
92. Sas-Strozik A, Donizy P, Koscielska-Kasprzak K, Kaminska D, Gawlik K, Mazanowska O, et al. Angiotensin II Type 1 Receptor Expression in Renal Transplant Biopsies and Anti-AT1R Antibodies in Serum Indicates the Risk of Transplant Loss. Transplant Proc (2020) 52:2299–304. doi: 10.1016/j.transproceed.2020.01.126
93. Wozniak LJ, Hickey MJ, Chan AP, Venick RS, Farmer DG, Busuttil RW, et al. Angiotensin II Type-1 Receptor Antibodies Are Associated With Active Allograft Dysfunction Following Pediatric Liver Transplantation. Transplantation (2020) 104:2547–56. doi: 10.1097/TP.0000000000003206
94. Hinchcliff M, Varga J. Obliterative Vasculopathy in Systemic Sclerosis: Endothelial Precursor Cells as Novel Targets for Therapy. Expert Rev Clin Immunol (2007) 3:11–5. doi: 10.1586/1744666X.3.1.11
95. Kuwana M, Kaburaki J, Okazaki Y, Yasuoka H, Kawakami Y, Ikeda Y. Increase in Circulating Endothelial Precursors by Atorvastatin in Patients With Systemic Sclerosis. Arthritis Rheum (2006) 54:1946–51. doi: 10.1002/art.21899
96. Dragun D, Catar R, Philippe A. Non-HLA Antibodies Against Endothelial Targets Bridging Allo- and Autoimmunity. Kidney Int (2016) 90:280–8. doi: 10.1016/j.kint.2016.03.019
97. Sas-Strozik A, Krajewska M, Banasik M. The Significance of Angiotensin II Type 1 Receptor (AT1 Receptor) in Renal Transplant Injury. Adv Clin Exp Med (2020) 29:629–33. doi: 10.17219/acem/121510
98. Sorohan BM, Ismail G, Leca N, Tacu D, Obrisca B, Constantinescu I, et al. Angiotensin II Type 1 Receptor Antibodies in Kidney Transplantation: An Evidence-Based Comprehensive Review. Transplant Rev (Orlando) (2020) 34:100573. doi: 10.1016/j.trre.2020.100573
99. Zhang X, Reinsmoen NL. Angiotensin II Type I Receptor Antibodies in Thoracic Transplantation. Hum Immunol (2019) 80:579–82. doi: 10.1016/j.humimm.2019.04.007
100. Zhang X, Reinsmoen NL. Impact and Production of Non-HLA-Specific Antibodies in Solid Organ Transplantation. Int J Immunogenet (2020) 47:235–42. doi: 10.1111/iji.12494
101. Liu F, Wang YX, Wang XF, Zheng YQ, Jin Z, Zhi JM. Role of Agonistic Autoantibodies Against Type-1 Angiotensin II Receptor in the Pathogenesis of Retinopathy in Preeclampsia. Sci Rep (2016) 6:29036. doi: 10.1038/srep29036
102. Siddiqui AH, Irani RA, Zhang W, Wang W, Blackwell SC, Kellems RE, et al. Angiotensin Receptor Agonistic Autoantibody-Mediated Soluble Fms-Like Tyrosine Kinase-1 Induction Contributes to Impaired Adrenal Vasculature and Decreased Aldosterone Production in Preeclampsia. Hypertension (2013) 61:472–9. doi: 10.1161/HYPERTENSIONAHA.111.00157
103. Zhou CC, Ahmad S, Mi T, Abbasi S, Xia L, Day MC, et al. Autoantibody From Women With Preeclampsia Induces Soluble Fms-Like Tyrosine Kinase-1 Production via Angiotensin Type 1 Receptor and Calcineurin/Nuclear Factor of Activated T-Cells Signaling. Hypertension (2008) 51:1010–9. doi: 10.1161/HYPERTENSIONAHA.107.097790
104. Clement MJ, Fortune A, Phalipon A, Marcel-Peyre V, Simenel C, Imberty A, et al. Toward a Better Understanding of the Basis of the Molecular Mimicry of Polysaccharide Antigens by Peptides: The Example of Shigella Flexneri 5a. J Biol Chem (2006) 281:2317–32. doi: 10.1074/jbc.M510172200
105. Fillion D, Cabana J, Guillemette G, Leduc R, Lavigne P, Escher E. Structure of the Human Angiotensin II Type 1 (AT1) Receptor Bound to Angiotensin II From Multiple Chemoselective Photoprobe Contacts Reveals a Unique Peptide Binding Mode. J Biol Chem (2013) 288:8187–97. doi: 10.1074/jbc.M112.442053
106. Laporte SA, Boucard AA, Servant G, Guillemette G, Leduc R, Escher E. Determination of Peptide Contact Points in the Human Angiotensin II Type I Receptor (AT1) With Photosensitive Analogs of Angiotensin II. Mol Endocrinol (1999) 13:578–86. doi: 10.1210/mend.13.4.0270
107. Unal H, Jagannathan R, Bhat MB, Karnik SS. Ligand-Specific Conformation of Extracellular Loop-2 in the Angiotensin II Type 1 Receptor. J Biol Chem (2010) 285:16341–50. doi: 10.1074/jbc.M109.094870
108. Unal H, Jagannathan R, Bhatnagar A, Tirupula K, Desnoyer R, Karnik SS. Long Range Effect of Mutations on Specific Conformational Changes in the Extracellular Loop 2 of Angiotensin II Type 1 Receptor. J Biol Chem (2013) 288:540–51. doi: 10.1074/jbc.M112.392514
109. Dragic T, Trkola A, Lin SW, Nagashima KA, Kajumo F, Zhao L, et al. Amino-Terminal Substitutions in the CCR5 Coreceptor Impair Gp120 Binding and Human Immunodeficiency Virus Type 1 Entry. J Virol (1998) 72:279–85. doi: 10.1128/JVI.72.1.279-285.1998
110. Lee B, Sharron M, Blanpain C, Doranz BJ, Vakili J, Setoh P, et al. Epitope Mapping of CCR5 Reveals Multiple Conformational States and Distinct But Overlapping Structures Involved in Chemokine and Coreceptor Function. J Biol Chem (1999) 274:9617–26. doi: 10.1074/jbc.274.14.9617
111. Takagi J, Isobe T, Takada Y, Saito Y. Structural Interlock Between Ligand-Binding Site and Stalk-Like Region of Beta1 Integrin Revealed by a Monoclonal Antibody Recognizing Conformation-Dependent Epitope. J Biochem (1997) 121:914–21. doi: 10.1093/oxfordjournals.jbchem.a021673
112. Tadagaki K, Jockers R, Kamal M. History and Biological Significance of GPCR Heteromerization in the Neuroendocrine System. Neuroendocrinology (2012) 95:223–31. doi: 10.1159/000330000
113. Albizu L, Cottet M, Kralikova M, Stoev S, Seyer R, Brabet I, et al. Time-Resolved FRET Between GPCR Ligands Reveals Oligomers in Native Tissues. Nat Chem Biol (2010) 6:587–94. doi: 10.1038/nchembio.396
114. Bouvier M. Oligomerization of G-Protein-Coupled Transmitter Receptors. Nat Rev Neurosci (2001) 2:274–86. doi: 10.1038/35067575
115. Rozenfeld R, Devi LA. Exploring a Role for Heteromerization in GPCR Signalling Specificity. Biochem J (2011) 433:11–8. doi: 10.1042/BJ20100458
116. Whorton MR, Bokoch MP, Rasmussen SG, Huang B, Zare RN, Kobilka B, et al. A Monomeric G-Protein-Coupled Receptor Isolated in a High-Density Lipoprotein Particle Efficiently Activates its G-Protein. Proc Natl Acad Sci USA (2007) 104:7682–7. doi: 10.1073/pnas.0611448104
117. Ciruela F, Vilardaga JP, Fernandez-Duenas V. Lighting Up Multiprotein Complexes: Lessons From GPCR Oligomerization. Trends Biotechnol (2010) 28:407–15. doi: 10.1016/j.tibtech.2010.05.002
118. White JF, Grodnitzky J, Louis JM, Trinh LB, Shiloach J, Gutierrez J, et al. Dimerization of the Class A G-Protein-Coupled Neurotensin Receptor NTS1 Alters G-Protein Interaction. Proc Natl Acad Sci USA (2007) 104:12199–204. doi: 10.1073/pnas.0705312104
119. Harikumar KG, Morfis MM, Sexton PM, Miller LJ. Pattern of Intra-Family Hetero-Oligomerization Involving the G-Protein-Coupled Secretin Receptor. J Mol Neurosci (2008) 36:279–85. doi: 10.1007/s12031-008-9060-z
120. Li X, Staszewski L, Xu H, Durick K, Zoller M, Adler E. Human Receptors for Sweet and Umami Taste. Proc Natl Acad Sci USA (2002) 99:4692–6. doi: 10.1073/pnas.072090199
121. Ng HK, Chow BK. Oligomerization of Family B GPCRs: Exploration in Inter-Family Oligomer Formation. Front Endocrinol (Lausanne) (2015) 6:10. doi: 10.3389/fendo.2015.00010
122. Velazhahan V, Ma N, Pandy-Szekeres G, Kooistra AJ, Lee Y, Gloriam DE, et al. Structure of the Class D GPCR Ste2 Dimer Coupled to Two G-Proteins. Nature (2021) 589:148–53. doi: 10.1038/s41586-020-2994-1
123. Waldhoer M, Fong J, Jones RM, Lunzer MM, Sharma SK, Kostenis E, et al. A Heterodimer-Selective Agonist Shows In Vivo Relevance of G-Protein-Coupled Receptor Dimers. Proc Natl Acad Sci USA (2005) 102:9050–5. doi: 10.1073/pnas.0501112102
124. Pin JP, Neubig R, Bouvier M, Devi L, Filizola M, Javitch JA, et al. International Union of Basic and Clinical Pharmacology. LXVII. Recommendations for the Recognition and Nomenclature of G-Protein-Coupled Receptor Heteromultimers. Pharmacol Rev (2007) 59:5–13. doi: 10.1124/pr.59.1.5
125. Carrillo JJ, Lopez-Gimenez JF, Milligan G. Multiple Interactions Between Transmembrane Helices Generate the Oligomeric Alpha1b-Adrenoceptor. Mol Pharmacol (2004) 66:1123–37. doi: 10.1124/mol.104.001586
126. Guo W, Urizar E, Kralikova M, Mobarec JC, Shi L, Filizola M, et al. Dopamine D2 Receptors Form Higher Order Oligomers at Physiological Expression Levels. EMBO J (2008) 27:2293–304. doi: 10.1038/emboj.2008.153
127. Mancia F, Assur Z, Herman AG, Siegel R, Hendrickson WA. Ligand Sensitivity in Dimeric Associations of the Serotonin 5HT2c Receptor. EMBO Rep (2008) 9:363–9. doi: 10.1038/embor.2008.27
128. Hebert TE, Moffett S, Morello JP, Loisel TP, Bichet DG, Barret C, et al. A Peptide Derived From a Beta2-Adrenergic Receptor Transmembrane Domain Inhibits Both Receptor Dimerization and Activation. J Biol Chem (1996) 271:16384–92. doi: 10.1074/jbc.271.27.16384
129. Yanagawa M, Yamashita T, Shichida Y. Comparative Fluorescence Resonance Energy Transfer Analysis of Metabotropic Glutamate Receptors: Implications About the Dimeric Arrangement and Rearrangement Upon Ligand Bindings. J Biol Chem (2011) 286:22971–81. doi: 10.1074/jbc.M110.206870
130. Schiedel AC, Kose M, Barreto C, Bueschbell B, Morra G, Sensoy O, et al. Prediction and Targeting of Interaction Interfaces in G-Protein Coupled Receptor Oligomers. Curr Top Med Chem (2018) 18:714–46. doi: 10.2174/1568026618666180604082610
131. Hu J, Hu K, Liu T, Stern MK, Mistry R, Challiss RA, et al. Novel Structural and Functional Insights Into M3 Muscarinic Receptor Dimer/Oligomer Formation. J Biol Chem (2013) 288:34777–90. doi: 10.1074/jbc.M113.503714
133. Levoye A, Dam J, Ayoub MA, Guillaume JL, Couturier C, Delagrange P, et al. The Orphan GPR50 Receptor Specifically Inhibits MT1 Melatonin Receptor Function Through Heterodimerization. EMBO J (2006) 25:3012–23. doi: 10.1038/sj.emboj.7601193
134. Lohse MJ. Dimerization in GPCR Mobility and Signaling. Curr Opin Pharmacol (2010) 10:53–8. doi: 10.1016/j.coph.2009.10.007
135. Uberti MA, Hague C, Oller H, Minneman KP, Hall RA. Heterodimerization With Beta2-Adrenergic Receptors Promotes Surface Expression and Functional Activity of Alpha1d-Adrenergic Receptors. J Pharmacol Exp Ther (2005) 313:16–23. doi: 10.1124/jpet.104.079541
136. Ferre S, Ciruela F, Casado V, Pardo L. Oligomerization of G-Protein-Coupled Receptors: Still Doubted? Prog Mol Biol Transl Sci (2020) 169:297–321. doi: 10.1016/bs.pmbts.2019.11.006
137. Borroto-Escuela DO, Brito I, Romero-Fernandez W, Di Palma M, Oflijan J, Skieterska K, et al. The G-Protein-Coupled Receptor Heterodimer Network (GPCR-HetNet) and its Hub Components. Int J Mol Sci (2014) 15:8570–90. doi: 10.3390/ijms15058570
138. Al Zamel I, Palakkott A, Ayoub MA. Synergistic Activation of Thrombin and Angiotensin II Receptors Revealed by Bioluminescence Resonance Energy Transfer. FEBS Lett (2021) 595:2628-37. doi: 10.1002/1873-3468.14187
139. Sun GC, Wong TY, Chen HH, Ho CY, Yeh TC, Ho WY, et al. Angiotensin II Inhibits DDAH1-nNOS Signaling via AT1R and muOR Dimerization to Modulate Blood Pressure Control in the Central Nervous System. Clin Sci (Lond) (2019) 133:2401–13. doi: 10.1042/CS20191005
140. Fillion D, Devost D, Sleno R, Inoue A, Hebert TE. Asymmetric Recruitment of Beta-Arrestin1/2 by the Angiotensin II Type I and Prostaglandin F2alpha Receptor Dimer. Front Endocrinol (Lausanne) (2019) 10:162. doi: 10.3389/fendo.2019.00162
141. Zeng C, Hopfer U, Asico LD, Eisner GM, Felder RA, Jose PA. Altered AT1 Receptor Regulation of ETB Receptors in Renal Proximal Tubule Cells of Spontaneously Hypertensive Rats. Hypertension (2005) 46:926–31. doi: 10.1161/01.HYP.0000174595.41637.13
142. Chow BS, Kocan M, Bosnyak S, Sarwar M, Wigg B, Jones ES, et al. Relaxin Requires the Angiotensin II Type 2 Receptor to Abrogate Renal Interstitial Fibrosis. Kidney Int (2014) 86:75–85. doi: 10.1038/ki.2013.518
143. Chow BSM, Kocan M, Shen M, Wang Y, Han L, Chew JY, et al. AT1R-AT2R-RXFP1 Functional Crosstalk in Myofibroblasts: Impact on the Therapeutic Targeting of Renal and Cardiac Fibrosis. J Am Soc Nephrol (2019) 30:2191–207. doi: 10.1681/ASN.2019060597
144. Barki-Harrington L, Luttrell LM, Rockman HA. Dual Inhibition of Beta-Adrenergic and Angiotensin II Receptors by a Single Antagonist: A Functional Role for Receptor-Receptor Interaction In Vivo. Circulation (2003) 108:1611–8. doi: 10.1161/01.CIR.0000092166.30360.78
145. Porrello ER, Pfleger KD, Seeber RM, Qian H, Oro C, Abogadie F, et al. Heteromerization of Angiotensin Receptors Changes Trafficking and Arrestin Recruitment Profiles. Cell Signal (2011) 23:1767–76. doi: 10.1016/j.cellsig.2011.06.011
146. Rozenfeld R, Gupta A, Gagnidze K, Lim MP, Gomes I, Lee-Ramos D, et al. AT1R-Cb(1)R Heteromerization Reveals a New Mechanism for the Pathogenic Properties of Angiotensin II. EMBO J (2011) 30:2350–63. doi: 10.1038/emboj.2011.139
147. Lee LT, Ng SY, Chu JY, Sekar R, Harikumar KG, Miller LJ, et al. Transmembrane Peptides as Unique Tools to Demonstrate the In Vivo Action of a Cross-Class GPCR Heterocomplex. FASEB J (2014) 28:2632–44. doi: 10.1096/fj.13-246868
148. Quitterer U, AbdAlla S. Pathological AT1R-B2R Protein Aggregation and Preeclampsia. Cells 10 (2021) 10(10):2609. doi: 10.3390/cells10102609
149. Sinphitukkul K, Manotham K, Eiam-Ong S, Eiam-Ong S. Aldosterone Nongenomically Induces Angiotensin II Receptor Dimerization in Rat Kidney: Role of Mineralocorticoid Receptor and NADPH Oxidase. Arch Med Sci (2019) 15:1589–98. doi: 10.5114/aoms.2019.87135
150. Abadir PM, Periasamy A, Carey RM, Siragy HM. Angiotensin II Type 2 Receptor-Bradykinin B2 Receptor Functional Heterodimerization. Hypertension (2006) 48:316–22. doi: 10.1161/01.HYP.0000228997.88162.a8
151. Zeng C, Asico LD, Yu C, Villar VA, Shi W, Luo Y, et al. Renal D3 Dopamine Receptor Stimulation Induces Natriuresis by Endothelin B Receptor Interactions. Kidney Int (2008) 74:750–9. doi: 10.1038/ki.2008.247
152. Gregan B, Jurgensen J, Papsdorf G, Furkert J, Schaefer M, Beyermann M, et al. Ligand-Dependent Differences in the Internalization of Endothelin A and Endothelin B Receptor Heterodimers. J Biol Chem (2004) 279:27679–87. doi: 10.1074/jbc.M403601200
153. Yatawara A, Wilson JL, Taylor L, Polgar P, Mierke DF. C-Terminus of ETA/ETB Receptors Regulate Endothelin-1 Signal Transmission. J Pept Sci (2013) 19:257–62. doi: 10.1002/psc.2499
154. Wolf P, Mohr A, Gavins G, Behr V, Morl K, Seitz O, et al. Orthogonal Peptide-Templated Labeling Elucidates Lateral ETA R/ETB R Proximity and Reveals Altered Downstream Signaling. Chembiochem (2021) 23:e202100340. doi: 10.1002/cbic.202100340
155. Kuroda Y, Nonaka M, Kamikubo Y, Ogawa H, Murayama T, Kurebayashi N, et al. Inhibition of Endothelin A Receptor by a Novel, Selective Receptor Antagonist Enhances Morphine-Induced Analgesia: Possible Functional Interaction of Dimerized Endothelin A and Mu-Opioid Receptors. BioMed Pharmacother (2021) 141:111800. doi: 10.1016/j.biopha.2021.111800
156. Hansen JL, Theilade J, Haunso S, Sheikh SP. Oligomerization of Wild Type and Nonfunctional Mutant Angiotensin II Type I Receptors Inhibits Galphaq Protein Signaling But Not ERK Activation. J Biol Chem (2004) 279:24108–15. doi: 10.1074/jbc.M400092200
157. Stenkamp RE. Identifying G-Protein-Coupled Receptor Dimers From Crystal Packings. Acta Crystallogr D Struct Biol (2018) 74:655–70. doi: 10.1107/S2059798318008136
158. Pin JP, Kniazeff J, Prezeau L, Liu JF, Rondard P. GPCR Interaction as a Possible Way for Allosteric Control Between Receptors. Mol Cell Endocrinol (2019) 486:89–95. doi: 10.1016/j.mce.2019.02.019
159. Erol I, Cosut B, Durdagi S. Toward Understanding the Impact of Dimerization Interfaces in Angiotensin II Type 1 Receptor. J Chem Inf Model (2019) 59:4314–27. doi: 10.1021/acs.jcim.9b00294
160. Miedema J, Schreurs M, van der Sar-van der Brugge S, Paats M, Baart S, Bakker M, et al. Antibodies Against Angiotensin II Receptor Type 1 and Endothelin A Receptor Are Associated With an Unfavorable COVID19 Disease Course. Front Immunol (2021) 12:684142. doi: 10.3389/fimmu.2021.684142
161. Philogene MC, Johnson T, Vaught AJ, Zakaria S, Fedarko N. Antibodies Against Angiotensin II Type 1 and Endothelin A Receptors: Relevance and Pathogenicity. Hum Immunol (2019) 80:561–7. doi: 10.1016/j.humimm.2019.04.012
162. Berque-Bestel I, Lezoualc'h F, Jockers R. Bivalent Ligands as Specific Pharmacological Tools for G-Protein-Coupled Receptor Dimers. Curr Drug Discov Technol (2008) 5:312–8. doi: 10.2174/157016308786733591
163. Huang B, St Onge CM, Ma H, Zhang Y. Design of Bivalent Ligands Targeting Putative GPCR Dimers. Drug Discov Today (2021) 26:189–99. doi: 10.1016/j.drudis.2020.10.006
164. Mohr K, Schmitz J, Schrage R, Trankle C, Holzgrabe U. Molecular Alliance-From Orthosteric and Allosteric Ligands to Dualsteric/Bitopic Agonists at G-Protein Coupled Receptors. Angew Chem Int Ed Engl (2013) 52:508–16. doi: 10.1002/anie.201205315
165. George SR, O'Dowd BF, Lee SP. G-Protein-Coupled Receptor Oligomerization and its Potential for Drug Discovery. Nat Rev Drug Discovery (2002) 1:808–20. doi: 10.1038/nrd913
166. Rozenfeld R, Devi LA. Receptor Heteromerization and Drug Discovery. Trends Pharmacol Sci (2010) 31:124–30. doi: 10.1016/j.tips.2009.11.008
Keywords: angiotensin II type 1 receptor (AT1R), angiotensin II type 2 receptor (AT2R), endothelin type A receptor (ETAR), endothelin type B receptor (ETBR), G-protein coupled receptor (GPCR), autoantibodies, GPCR structures
Citation: Speck D, Kleinau G, Szczepek M, Kwiatkowski D, Catar R, Philippe A and Scheerer P (2022) Angiotensin and Endothelin Receptor Structures With Implications for Signaling Regulation and Pharmacological Targeting. Front. Endocrinol. 13:880002. doi: 10.3389/fendo.2022.880002
Received: 20 February 2022; Accepted: 18 March 2022;
Published: 19 April 2022.
Edited by:
Marcel Bermudez, Freie Universität Berlin, GermanyReviewed by:
David Poyner, Aston University, United KingdomCopyright © 2022 Speck, Kleinau, Szczepek, Kwiatkowski, Catar, Philippe and Scheerer. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Patrick Scheerer, cGF0cmljay5zY2hlZXJlckBjaGFyaXRlLmRl; orcid.org/0000-0001-5028-2075
†These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.