
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Endocrinol. , 18 July 2022
Sec. Systems Endocrinology
Volume 13 - 2022 | https://doi.org/10.3389/fendo.2022.874608
This article is part of the Research Topic Genetic Variants and Metabolic Diseases View all 10 articles
 Shuping Wang1,2,3†
Shuping Wang1,2,3† Yiping Cheng1,2†
Yiping Cheng1,2† Yingzhou Shi1,2
Yingzhou Shi1,2 Wanyi Zhao1,2
Wanyi Zhao1,2 Ling Gao4,5,6,7Li Fang1,2Xiaolong Jin1,2Xiaoyan Han1,2Qiuying Sun1,2
Ling Gao4,5,6,7Li Fang1,2Xiaolong Jin1,2Xiaoyan Han1,2Qiuying Sun1,2 Guimei Li8*
Guimei Li8* Jiajun Zhao1,2*
Jiajun Zhao1,2* Chao Xu1,2*
Chao Xu1,2*Background: Type I hyperlipoproteinemia, characterized by severe hypertriglyceridemia, is caused mainly by loss-of-function mutation of the lipoprotein lipase (LPL) gene. To date, more than 200 mutations in the LPL gene have been reported, while only a limited number of mutations have been evaluated for pathogenesis.
Objective: This study aims to explore the molecular mechanisms underlying lipoprotein lipase deficiency in two pedigrees with type 1 hyperlipoproteinemia.
Methods: We conducted a systematic clinical and genetic analysis of two pedigrees with type 1 hyperlipoproteinemia. Postheparin plasma of all the members was used for the LPL activity analysis. In vitro studies were performed in HEK-293T cells that were transiently transfected with wild-type or variant LPL plasmids. Furthermore, the production and activity of LPL were analyzed in cell lysates or culture medium.
Results: Proband 1 developed acute pancreatitis in youth, and her serum triglycerides (TGs) continued to be at an ultrahigh level, despite the application of various lipid-lowering drugs. Proband 2 was diagnosed with type 1 hyperlipoproteinemia at 9 months of age, and his serum TG levels were mildly elevated with treatment. Two novel compound heterozygous variants of LPL (c.3G>C, p. M1? and c.835_836delCT, p. L279Vfs*3, c.188C>T, p. Ser63Phe and c.662T>C, p. Ile221Thr) were identified in the two probands. The postheparin LPL activity of probands 1 and 2 showed decreases of 72.22 ± 9.46% (p<0.01) and 54.60 ± 9.03% (p<0.01), respectively, compared with the control. In vitro studies showed a substantial reduction in the expression or enzyme activity of LPL in the LPL variants.
Conclusions: Two novel compound heterozygous variants of LPL induced defects in the expression and function of LPL and caused type I hyperlipoproteinemia. The functional characterization of these variants was in keeping with the postulated LPL mutant activity.
Type I hyperlipoproteinemia, also known as familial lipoprotein lipase (LPL) deficiency, is characterized by very severe hypertriglyceridemia with episodes of abdominal pain, recurrent acute pancreatitis, eruptive cutaneous xanthomata, and hepatosplenomegaly (1). The prevalence of type I hyperlipoproteinemia in the general population is estimated to be 1–2 per million (2, 3). Inherited in an autosomal recessive manner, type I hyperlipoproteinemia is caused mainly by the loss-of-function mutation of the LPL gene (4).
The LPL gene encodes a secreted glycoprotein, LPL, containing 448 amino acids (5). Synthesized by cells such as fat cells, macrophages, and muscle cells, LPL is an important rate-limiting enzyme for triglyceride degradation, which can hydrolyze triglycerides into fatty acids and glycerol to regulate lipid metabolism (6, 7). Defective LPL can cause the accumulation of triglyceride-rich chylomicrons and very low-density lipoproteins and further lead to severe hypertriglyceridemia. To date, more than 200 mutations in the LPL gene have been reported to result in type I hyperlipoproteinemia in the Human Gene Mutation Database (HGMD), while only a limited number of mutations have been evaluated for pathogenesis (8, 9).
In this study, two patients with severe hypertriglyceridemia were confirmed to have type I hyperlipoproteinemia by whole-exome sequencing (WES), and two novel compound heterozygous variants of the LPL gene were identified. We showed through bioinformatics analysis and in vitro experiments that these variants can affect the enzymatic activity, production, and/or secretion of LPL and cause type I hyperlipoproteinemia in both cases. Our research provides evidence for elucidating the molecular mechanisms of mutant LPL and helps improve the genetic diagnosis rate and precise treatment of this disease.
This study was approved by the Ethics Committee of Shandong Provincial Hospital affiliated with Shandong University. The study protocol was in line with the Declaration of Helsinki (as revised in Brazil 2013). The consent obtained from all the participants was both informed and written.
Two patients diagnosed with severe hypertriglyceridemia from our hospital were involved in the study. All patient data were collected at the first visit. All family members received specific physical and laboratory examinations in Shandong Provincial Hospital. Peripheral blood specimens were collected from each member for genetic analysis.
We followed these two patients from their first presence and closely tracked their clinical and biochemical information.
Using a QIAamp DNA Mini Kit (Qiagen, Hilden, Germany), genomic DNA was isolated from peripheral blood leukocytes. Next, DNA from peripheral blood was used for WES. We performed genomic DNA fragmentation, paired-end adaptor ligation, amplification, and purification and then captured human exons using a SeqCap EZ Med Exome Enrichment Kit (Roche NimbleGen, Madison, WI, USA). By postcapture amplification and purification, a DNA library was generated and then sequenced with the Illumina HiSeq sequencing platform. To obtain the coverage and mean read depth of target regions, sequence data alignment to the human genome reference (hg19) and variant calling were performed using NextGene V2.3.4 software. The average coverage of the exome was >100×, which allowed a deep examination of the target region to accurately match >99% of the target exons. Mutations with low coverage in the target area were screened and filtered to ensure the accuracy of data analysis.
In addition, the frequency of normal populations [data from Genome Aggregation Database (GnomAD), Exome Aggregation Consortium (ExAC), Trans-Omics for Precision Medicine (TOPMED), Human Gene Mutation Database (HGMD), Clinvar and Online Mendelian Inheritance in Man (OMIM) databases] was obtained by NextGene V2.3.4 and our in-house scripts. A variant was identified as a mutation when it was not found in 500 Chinese controls, in dbSNP (http://www.ncbi.nlm.nih.gov/snp/) and the exome variant server (http://evs.gs.washington.edu/EVS/), or when the allele frequency was <0.001 in the database. According to Standards and Guidelines for the Interpretation of Sequence Variants published by the American College of Medical Genetics (ACMG) in 2015, pathogenic variants were determined with the Human Genome Variation Society (HGVS) nomenclature.
Candidate variants were detected by WES. Pathogenic or suspected pathogenic variants were verified by Sanger sequencing. Universally tagged sequencing primers were designed by using Primer3 version 1.1.4 (http://www.sourceforge.net) and GeneDistiller 2014 (http://www.genedistiller.org/). Polymerase chain reaction (PCR) was performed in a 50-μl system including 5 μl 10 × PCR buffer, 4 μl dNTPs, 4 μl genomic DNA, 1 μl forward and reverse primers, and 0.3 μl Taq Hot Start (Takara Bio, Otsu, Japan). The PCR conditions were as previously described (10). Amplicons were sequenced using an ABI 3730 system (Applied Biosystems, Foster City, CA, USA) and were analyzed by the autoassembler software Chromas 2.6 and visual inspection.
Preheparin blood samples were taken after fasting overnight. Then, heparin was injected intravenously (60 IU/kg), and the postheparin blood was collected from the contralateral arm 10 min later. The subjects stopped taking insulin and other medications in the morning to eliminate the interference caused by medications. The blood sample was centrifuged at 3,000 rpm for 10 min at 4°C to obtain plasma. A Lipoprotein Lipase Activity Assay Kit (Fluorometric, Biovision, Milpitas, CA, USA) was used to measure LPL activity. Mouse (C57BL/6) postheparin plasma was used as a positive control.
By GeneArt Gene Synthesis (Thermo Fisher Scientific, Rockford, IL, USA), wild-type LPL cDNA (pcDNA3.1- LPL-WT) was synthesized and cloned into the pcDNA3.1 vector with a V5 epitope tag. LPL variants (pcDNA3.1-LPL-MU: p. M1?, p. L279Vfs*3, p. Ser63Phe, or p. Ile221Thr) were obtained by site-directed mutagenesis. To obtain the mutant LPL cDNA, we used the following primers: forward (wild type) GACCCAATAAGCTTCGTCAGAATTTTGTAATACGACTCACTATAGG, reverse (wild type) ATTGGGTCAAGCTTATGTTTTGTAAAAGTTACTTCCTCCACT; forward (p. M1)? CAGAGGGACGCGCCCCGAGATCGAGAGCAAAGCCCTGCTCGTGC, primer reverse (p. M1)? AGCACGAGCAGGGCTTTGCTCTCGATCTCGGGGCGCGTCCCTCTG; primer forward (p. L279Vfs*3) GACCCAATAAGCTTCGTCAGAATTTTGTAATACGACTCACTATAGG, primer reverse (p. L279Vfs*3) ATTGGGTCAAGCTTGGAACTGCACCTGTAGGCCTTACTTGGA; primer forward (p. Ser63Phe) CACCTCATTCCCGGAGTAGCAGAGTTCGTGGCTACCTGTCATTTCA, primer reverse (p. Ser63Phe) TGAAATGACAGGTAGCCACGAACTCTGCTACTCCGGGAATGAGGTG; primer forward (p. Ile221Thr) GAGGGTCCCCTGGTCGAAGCACTGGAATCCAGAAACCAGTTGG, primer reverse (p. Ile221Thr) CCCAACTGGTTTCTGGATTCCAGTGCTTCGACCAGGGGACCCTCT. All the primers used were purchased from GenScript (Cayman Islands, UK). Polymerase chain reaction (PCR) was carried out in a 50-μl reaction system including 5 μl 10 × PCR buffer, 4 μl genomic DNA, 4 μl dNTPs, 1 μl forward and reverse primers, and 0.3 μl Taq Hot Start (Takara Bio, Otsu, Japan). The PCR conditions consisted of an initial denaturation step (95°C for 5 min), followed by 40 cycles of denaturation (95°C, 30 s), annealing (55°C, 30 s), and elongation (68°C, 30 s). The presence and fidelity of pcDNA3.1-LPL-MU were confirmed by the ABI 3730 sequencing system (Applied Biosystems, Foster City, CA, USA).
In high-glucose Dulbecco’s modified Eagle’s medium (DMEM) containing 10% fetal bovine serum (FBS), 5% penicillin–streptomycin, and 2 mM L-glutamine, human embryonic kidney 293T/17 (HEK293T/17) cells obtained from the National Collection of Authenticated Cell Cultures were cultured. Next, HEK293T/17 cells were transiently transfected with plasmids containing pcDNA3.1- LPL-WT and pcDNA3.1- LPL-MU by using Lipofectamine 3000 transfection reagent (Thermo Fisher Scientific). As previously described (10), the transfection concentration of a single plasmid was 2.5 μg/ml, and the concentration of each plasmid was 1.5 μg/ml at the cotransfection of the two plasmids.
After 48 h, the cells were lysed with mammalian protein extraction reagent (Thermo Fisher Scientific) containing protease inhibitor cocktail (Sigma-Aldrich). HEK293T/17-cell lysates were used to analyze the protein expression of LPL by Western blotting. We used Amicon® Ultra15 ultrafiltration centrifuge tubes (Millipore-Sigma, Burlington, MA, USA) to concentrate the medium 80 times. The concentrated medium was used for LPL activity analysis.
The Lipoprotein Lipase Activity Assay Kit (Fluorometric, Biovision) was used to determine LPL activity in the culture medium of HEK293T/17 cells transfected with LPL wild-type plasmids and plasmids containing LPL variants.
We boiled the cell lysates mixed with Laemmli buffer containing 2-mercaptoethanol for 5 min at 99°CC. Proteins were separated by sodium dodecyl sulfate–polyacrylamide gel electrophoresis (SDS-PAGE; 100 V, 90 min) and then transferred onto a nitrocellulose membrane (400 mA, 1 h). Membranes were incubated overnight with primary antibodies and washed three times for 10 min with 0.2% Tris-buffered saline Tween (TBST). Then, the membranes were incubated with secondary antibodies for 1 h and washed three times for 10 min with 0.2% TBST. Next, a 5-min incubation between chemiluminescent horseradish peroxidase (HRP) substrate (Millipore Corporation, Billerica, MA, USA) and membranes was conducted, and bands were visualized by a ChemiDoc XRS System (Bio-Rad, Hercules, CA, USA). The following antibodies were used: rabbit monoclonal anti-LPL antibody (Abcam Ab91606) and mouse monoclonal anti-GAPDH (glyceraldehyde-3-phosphatee dehydrogenase) antibody (Abcam Ab8245).
Statistical analysis was performed using the SPSS 24.0 software package (SPSS Inc., Chicago, IL, USA). The Kolmogorov–Smirnov test was used to determine the distribution of continuous variables. Continuous variables with a normal distribution are given as the mean ± standard deviation (SD) and were compared by the Mann–Whitney U test. The results were considered statistically significant when the p-value was <0.05.
Proband 1, female, was hospitalized with acute necrotizing pancreatitis and underwent abdominal surgery at the age of 24. At 25 years old, she was found to have high triglyceride (TG) levels (approximately 17 mmol/L). However, TG levels increased further after she was administered some lipid-lowering drugs (including fenofibrate, rosuvastatin, Chinese medicine, etc.). In recent years, TG levels fluctuated at 13.63–33.94 mmol/L, and TC levels fluctuated at 4.92–10.51 mmol/L after drug withdrawal (Figures 1A, B). At the age of 49, the proband presented symptoms of hyperglycemia with fasting plasma glucose at 15.22 mmol/L and developed vomiting and diarrhea after oral administration of Chinese medicine decoction. Abdominal computerized tomography (CT) scan (Figure 1C) showed that the pancreas was irregular in shape; no obvious abnormality was observed in other parts. After giving patient 1 the low-fat diet, insulin, and liver protection drugs, the last follow-up in June 2020 showed that the TG, TC, and fasting plasma glucose were 15.37, 9.43, and 6.01 mmol/L, respectively (Table 1). The family history of the patient showed that the patient’s parents and her sister were deceased and could not be tested. However, it was recalled that her sister had also suffered from recurrent pancreatitis (Figure 2A).
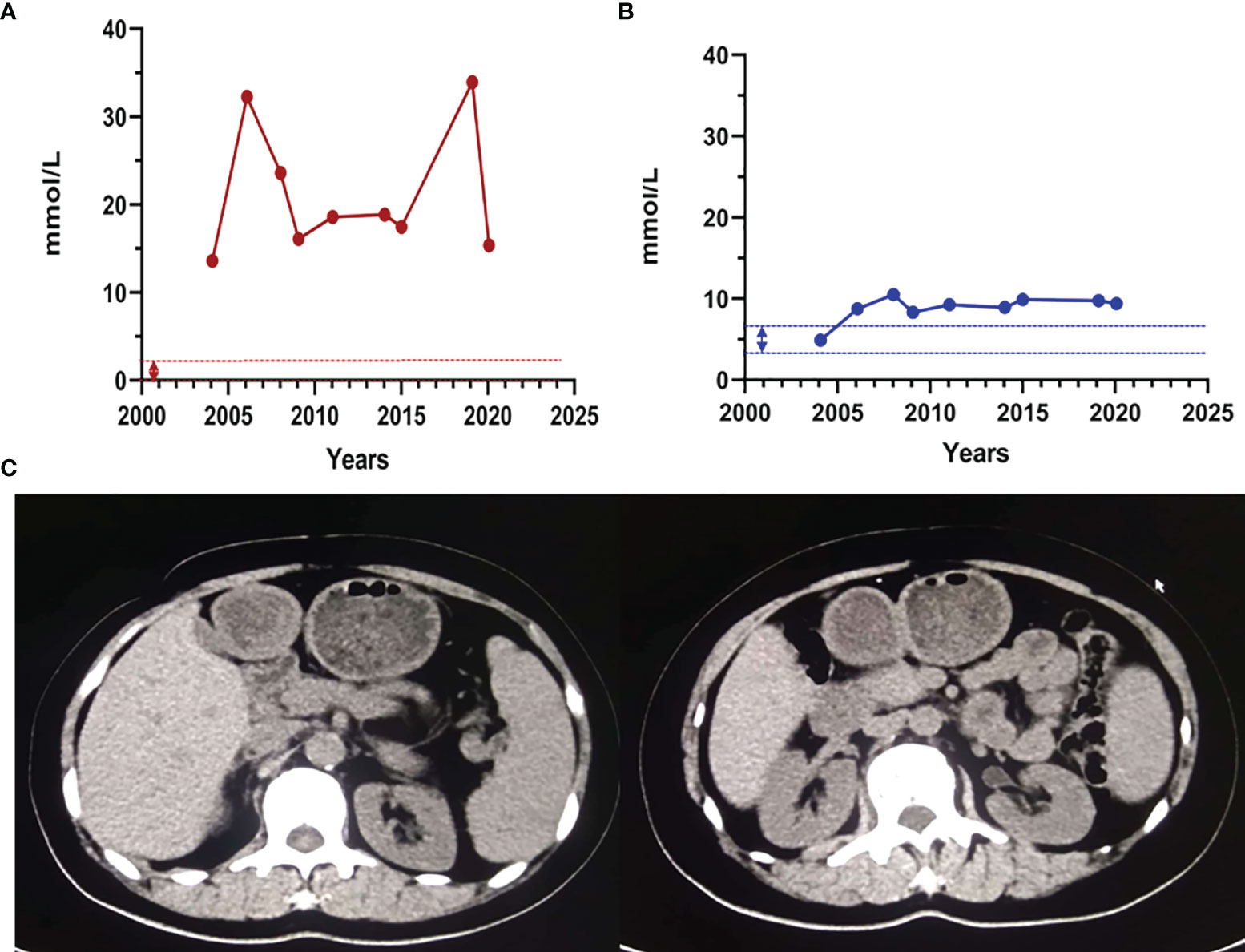
Figure 1 The clinical characteristics of proband 1. (A) The proband 1 triglyceride and total cholesterol levels. The dashed line indicates the normal reference range. Abbreviations: TG, triglycerides; TC, total cholesterol. Normal range: TG: 0.4–1.8 mmol/L; TC: 3.6–6.2 mmol/L. (B) Abdomen and lower limbs of patient 1. (C) Abdominal CT scan of patient 1. The pancreas was irregular in shape; the liver was normal in shape and size; no abnormal density shadow was observed in the parenchyma; no expansion of the bile ducts in and outside the liver was observed; the shape and size of the gallbladder were fine; no abnormal density shadows were found in the gallbladder; and the spleen was normal in shape and size.

Table 1 Clinical features and lipoprotein profile of the individuals screened for LPL variants.
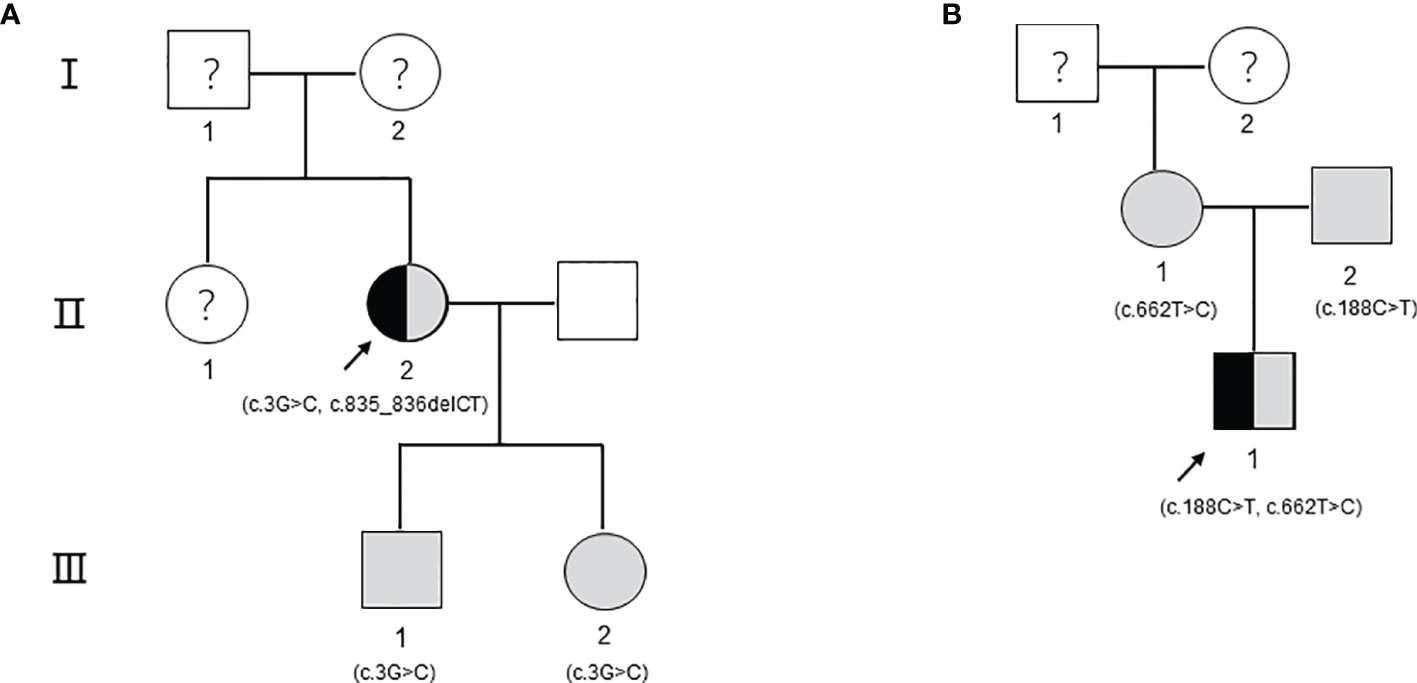
Figure 2 The pedigree including the patients. The arrow refers to the proband. Black indicates that the person has type I hyperlipoproteinemia. The shading indicates that the person carries gene mutations but has a healthy phenotype. Circles indicate female individuals reared, and squares indicate male individuals reared. The question mark means that it cannot be determined whether the individual carries a genetic mutation. (A) The pedigree of patient 1. (B) The pedigree of patient 2.
Proband 2 came to our hospital for treatment because his blood sample showed severe hyperlipemia during the physical examination at 9 months old. On the second day of admission, his TG level was 20.35 mmol/L, and his TC level was 8.89 mmol/L. A low-fat diet and 4 U of continuous intravenous infusion of insulin were ordered to lower blood lipids. After 3 days, the TG level was 22.77 mmol/L, and the TC was 7.60 mmol/L. After 7 days, the TG level was 12.32 mmol/L, and the TC level was 4.55 mmol/L. After 10 days, the proband was discharged, his blood lipids improved significantly, with TG at 10.25 mmol/L and TC at 4.93 mmol/L. After being discharged from the hospital, he continued to consume a low-fat diet without any special discomfort. The last follow-up conducted in June 2020 showed that the serum TG was 3.35 mmol/L, the TC was 2.72 mmol/L, and the fasting plasma glucose was 5.60 mmol/L (Table 1). The family history of the patient shows that the parents of patient 2 have thus far been healthy (Figure 2B).
To further identify disease-causing genes to facilitate diagnosis, we subsequently applied the WES technique for genetic analysis of the two pedigrees. According to HGMD, we found a novel compound variant of LPL (c.3G>C and c.835_836delCT, Figure 3A) in proband 1, which may cause truncation mutations (p. M1)?, and frameshift mutations (p. L279Vfs*3) leading to premature amino acid stop codes for protein synthesis, respectively. The p. M1? variant has been reported in a related clinical case, and its region is the translation initiation codon of this protein (11). The p. L279Vfs*3 variant is absent in the GnomAD, TOPMED, and ExAC databases. Notably, the son and daughter of proband 1 carry the p. M1? variant derived from the maternal line, as verified by Sanger sequencing, but their lipid levels and other phenotypes were normal.
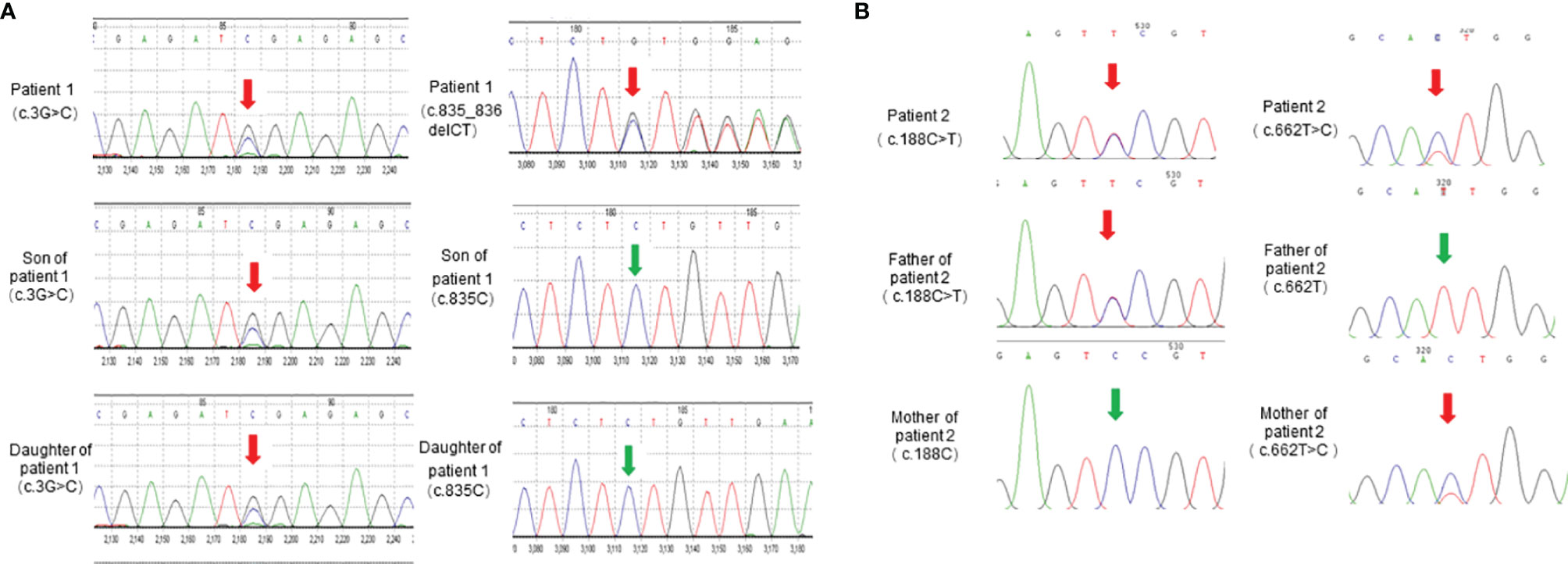
Figure 3 LPL gene sequencing diagram. (A) LPL gene (reference sequence NM_000237) sequencing diagram of patient 1 and her children. (B) LPL gene (reference sequence NM_000237) sequencing diagram of patient 2 and his parents.
We also identified proband 2 as carrying a novel compound variant of LPL (c.188C>T and c.662T>C, Figure 3B) that resulted in a change in the 63rd amino acid of the encoded protein from serine to phenylalanine (p. Ser63Phe) and a change in the 221st amino acid of the encoded protein from isoleucine to threonine (p. Ile221Thr), respectively. The p. Ser63Phe variant is absent in the GnomAD, TOPMED, and ExAC databases, while the p. Ile221Thr variant has been reported in three unrelated probands causing lipoprotein lipase deficiency (12). In addition, the parents of proband 2 each carried a variant. The Ser63Phe variant comes from the father, and the p. Ile221Thr variant comes from the mother, but they have thus far been healthy.
Accordingly, the postheparin LPL activity of patient 1 (0.46 ± 0.13 mU/ml) and patient 2 (0.75 ± 0.11 mU/ml) actually showed a decrease of 72.22 ± 9.46% (p<0.01) and 54.60 ± 9.03% (p<0.01), respectively, compared with the control (1.66 ± 0.09 mU/ml). The differences between the son and daughter of proband 1, the parents of proband 2, and the control seem to be negligible (Figure 4). Therefore, two novel compound heterozygous variants of LPL may cause changes in LPL enzyme activity in the patient’s plasma, leading to defects in triglyceride metabolism and resulting in extremely elevated serum triglycerides.
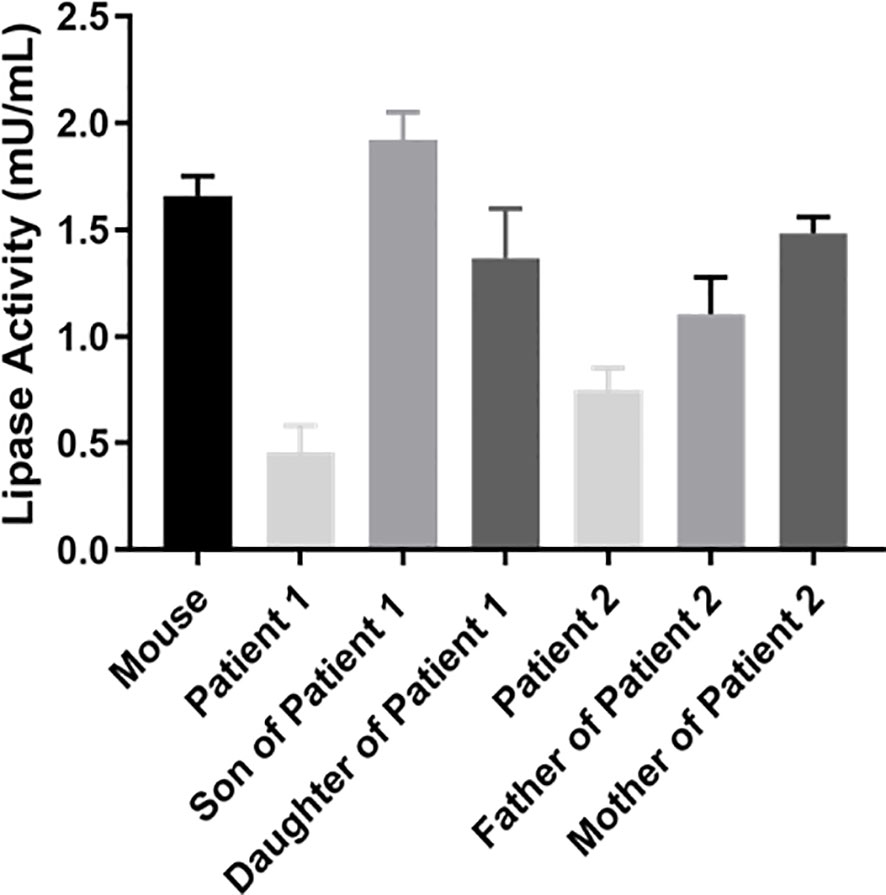
Figure 4 Analysis of LPL activity in plasma. Peripheral blood was collected at 10 min after heparin injection (60 IU/kg) to assay lipase activity by the enzyme-fluorescent method.
To understand the effect of the identified LPL variants at the protein level, we transiently transfected the plasmids containing pcDNA3.1-LPL-WT and pcDNA3.1-LPL-MU into HEK293T/17 cells. HEK293T/17 cells transfected with the p. M1? mutation, p. L279Vfs*3 mutation, or p. M1? and p. L279Vfs*3 mutation showed virtually no protein in the cell lysate (p<0.01, Figure 5). Compared with cells transfected with the plasmids containing the pcDNA3.1-LPL-WT, there was no significant difference in the amount of protein reduction produced by the cells transfected with the p. S63F mutation or the two variants (p. S63F and p. I221T), but protein synthesis was reduced by 48.33 ± 1.78% in cells transfected with the p. I221T variant (p<0.01, Figure 5).
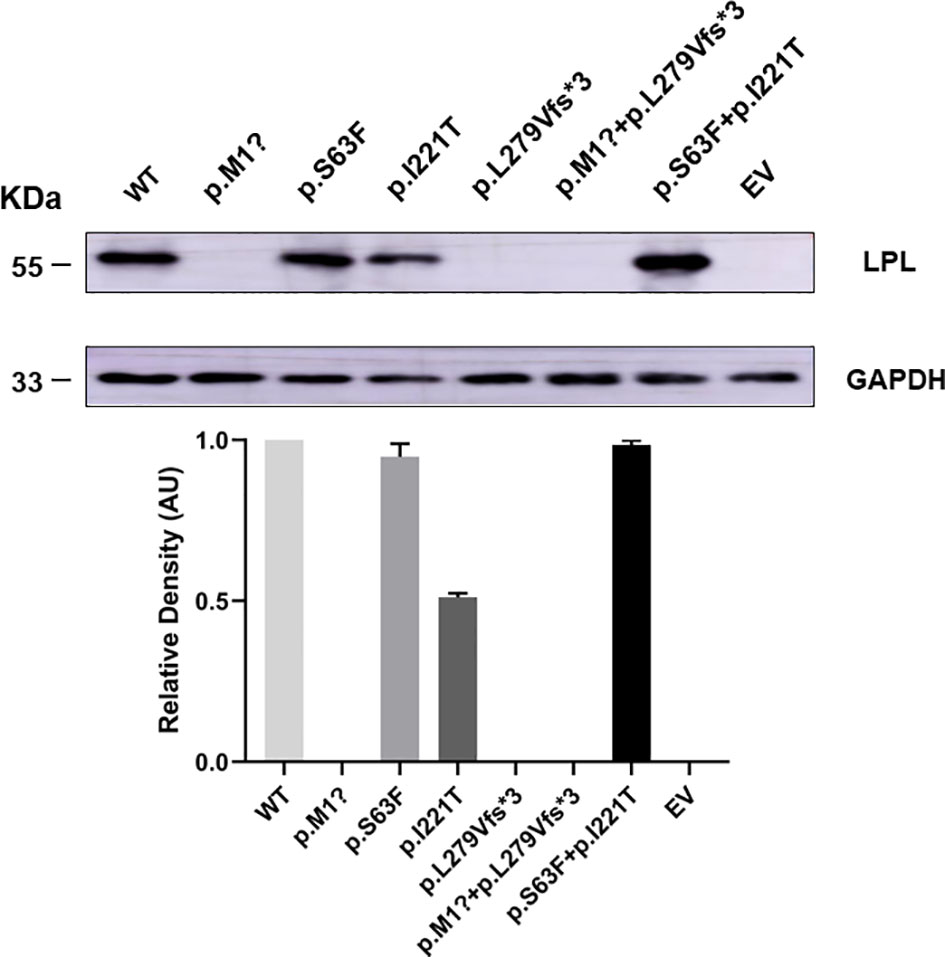
Figure 5 HEK 293T/17 cells transiently transfected showed a reduction in the production of LPL variants. HEK293T/17 cells transfected with the p. M1? mutation, p. L279Vfs*3 mutation, or p. M1? and p. L279Vfs*3 mutation showed virtually no protein in the cell lysate (p<0.01). Compared with cells transfected with the plasmids containing the pcDNA3.1-LPL-WT, there was no significant difference in the amount of protein reduction produced by the cells transfected with the p. S63F mutation or the two variants (p. S63F and p. I221T), but protein synthesis was reduced by 48.33 ± 1.78% in cells transfected with the p. I221T variant (p<0.01). Abbreviations: EV, empty vector; WT, wild type; LPL, lipoprotein lipase.
Next, we tested LPL activity in media of HEK 293T/17 cells. Wild-type LPL medium was used as a positive control. Except for the p. L279Vfs*3 mutation, LPL activity in the medium transfected with other plasmids containing pcDNA3.1-LPL-MU was significantly reduced compared with the LPL activity of the wild type. Specifically, cells transfected with the p. M1? or the two variants (p. M1? and p. L279Vfs*3) showed reductions in protein activity of approximately 55.93 ± 3.28% (p<0.01) or 59.84 ± 4.47% (p<0.01), while LPL activity was reduced by 62.76 ± 9.90% (p<0.01) or 63.25 ± 10.55% (p<0.01) in the media of HEK 293T/17 cells transfected with p. Ser63Phe or the two variants (p. Ser63Phe and Ser63Phe+Ile221Thr). The media of cells transfected with p. Ile221Thr demonstrated a maximum reduction in LPL activity of approximately 81.56 ± 4.35% (p<0.01, Figure 6).
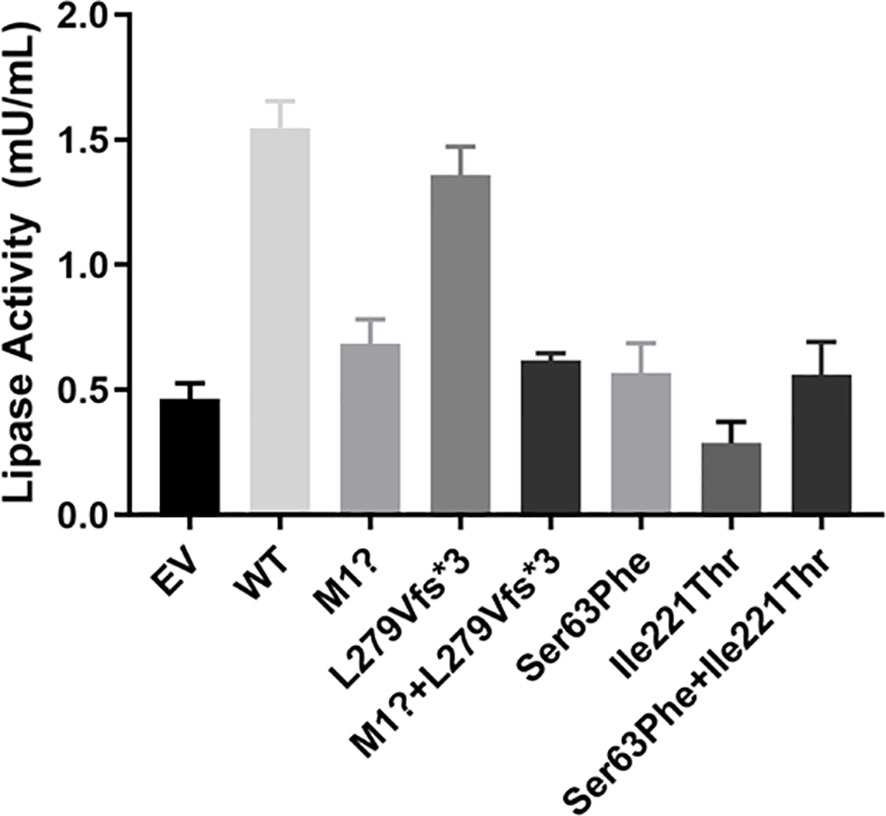
Figure 6 Functional analysis of LPL mutants in the medium. Except for the p. L279Vfs*3 mutation, LPL activity in the medium transfected with other plasmids containing pcDNA3.1-LPL-MU was significantly reduced compared with the wild type. Specifically, cells transfected with the p. M1? or the two variants (p. M1? and p. L279Vfs*3) showed reductions in protein activity of approximately 55.93 ± 3.28% (p<0.01) or 59.84 ± 4.47% (p<0.01), while LPL activity was reduced by 62.76 ± 9.90% (p<0.01) or 63.25 ± 10.55% (p<0.01) in the media of HEK 293T/17 cells transfected with p. Ser63Phe or the two variants (p. Ser63Phe and Ser63Phe+Ile221Thr). The media of cells transfected with p. Ile221Thr demonstrated a maximum reduction in LPL activity of approximately 81.56 ± 4.35% (p<0.01).
In the present study, we systematically identified and characterized two novel compound heterozygous variants of the LPL gene in patients with type I hyperlipoproteinemia. This harmful blood lipid metabolism disorder prompted us to solve this problem by determining the activity of LPL in plasma after heparin and analyzing the activity and quality defects of LPL in vitro. The functional characterization of these variants was in keeping with the postulated LPL mutant activity.
LPL is the rate-limiting enzyme in normal triglyceride metabolism, which plays a central role in the hydrolysis of triglycerides present in very-low-density lipoproteins and chylomicrons (13). Therefore, defective LPL can cause severe hypertriglyceridemia, such as type I hyperlipoproteinemia, a rare autosomal recessive genetic disease (14). In patients with hypertriglyceridemia, low plasma LPL activity and quality have been found (4, 15), and this phenomenon is usually caused by pathogenic LPL variants (16–18). The LPL protein consists of a large N-terminal domain (amino acid residues 1–315) and a small C-terminal domain (amino acid residues 316–448). The N-terminal domain contains the catalytic center, active site region, substrate-binding site, and heparin-binding site (19, 20). In addition to participating in the binding of heparin to the substrate, the C-terminal domain has been considered to be critical to the formation and stability of the LPL head and tail non-covalent homodimers (21, 22). Although LPL has always been considered to be active only as a homodimer, there are reports of biochemical data showing that LPL in complex with GPIHBP1 can be active as a monomer 1:1 complex (23).
Interestingly, we detected a novel compound variant of LPL (p. M1? and p. L279Vfs*3) in a Chinese patient with type I hyperlipoproteinemia. This patient exhibited severe hypertriglyceridemia and recurrent acute pancreatitis. In particular, the p. M1? variant, an initiation codon mutation, has previously been identified in an 18-year-old patient from China who also carried the previously reported heterozygous substitution of glutamic acid at residue 242 with lysine (p. Glu242Lys) (11). In vitro experiments found that the p. M1? had approximately 3% protein mass and 2% activity, whereas p. Glu242Lys had normal mass but undetectable activity; that is, the p. M1? variant rather than the heterozygous p. Glu242Lys variant is mainly responsible for the phenotypic expression of type I hyperlipoproteinemia in this patient. Here, our in vitro expression of both mutations separately or in combination displayed virtually no protein in the cell lysate. Except for the p. L279Vfs*3 mutation, the LPL activity in the medium transfected with p. M1? or both mutations in combination was significantly reduced compared with the LPL activity of the wild type. Therefore, we speculate that the p. M1? mutation will have a significant impact on the quality and activity of LPL, but p. L279Vfs*3 has little effect on the activity. What needs illustration is that the son and daughter also carry the p. M1? variant derived from proband 1, but their lipid levels and other phenotypes were normal. Compared with the control, the postheparin LPL activity in the plasma of the son and daughter was not different. Hence, what caused proband 1 to have severe hyperlipidemia? This requires more in-depth research to explore.
Proband 2 also carries a novel compound variant of LPL (p. Ser63Phe and p. Ile221Thr). The p. Ile221Thr variant has been reported in three unrelated probands causing lipoprotein lipase deficiency (12). The synthesis and secretion of a catalytically defective protein induced by the p. Ile221Thr mutation were confirmed through in vitro experiments in COS-1 cells. In accordance with the previous data, two new compound LPL variants of patient 2 in our study caused changes in the LPL enzyme activity in the plasma of the patient, whereas the differences in enzyme activity between the parents of proband 2 and the control seemed to be negligible. Curiously, the parents each carried a variant of proband 2. Sanger sequencing confirmed that the Ser63Phe variant comes from the father and the p. Ile221Thr variant comes from the mother, but the parents have thus far been healthy. Our in vitro experiments to understand the effect of the two LPL variants at the protein level indicated that the p. I221T variant can reduce approximately 48.33 ± 1.78% mass and approximately 81.56 ± 4.35% activity, and the LPL activity was reduced by 62.76 ± 9.90% or 63.25 ± 10.55% in the media of HEK 293T/17 cells transfected with p. Ser63Phe or the two variants (p. Ser63Phe and Ser63Phe+Ile221Thr).
Functional studies have potential benefits for the diagnosis and treatment of severe hypertriglyceridemia and can promote the mechanistic study of the occurrence of type I hyperlipoproteinemia. Ultimately, the findings may lead to the development of more effective drugs for the precise intervention for such patient rather than limited symptomatic treatment. In this study, differences in the activity of LPL would affect the corresponding clinical research, and interested researchers may summarize more cases and find some rules, such as mutations in one exon region having a greater impact on LPL activity, while mutations in another exon region have little impact on LPL activity.
This study has some limitations. For instance, in our study, we could not analyze whether the parents of patient 1 had hypertriglyceridemia because they had passed away. Second, we have not been able to conduct a more in-depth study on why the son and daughter of proband 1 and the parents of proband 2 carry a certain variant, but the phenotype is normal. Our patients did not have two harmful lifestyle factors, namely, severe obesity and severe tobacco abuse. Obesity is not only related to primary hypertriglyceridemia but also a risk factor for secondary hypertriglyceridemia (24). Tobacco can affect fat metabolism in the liver and increase TG levels due to defects in the lipolysis system (25). In short, it can basically rule out the possibility that our two probands had secondary hypertriglyceridemia.
To conclude, we have described two novel compound heterozygous variants of LPL in patients with type I hyperlipoproteinemia, showing that all these variants are pathogenic by disrupting LPL mass and the enzymatic activity of LPL. Although LPL variants are rare, WES is important to discover the monogenic and oligogenic genetic patterns in severe hyperlipoproteinemia, a challenge now predigested by the access to next-generation sequencing.
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found below: GenBank, 2561205.
The studies involving human participants were reviewed and approved by Ethics Committee of Shandong Provincial Hospital affiliated to Shandong University. Written informed consent to participate in this study was provided by the participants’ legal guardian/next of kin.
SW and YC designed the experiments, researched the data, and wrote the manuscript. YS contributed to the statistical analysis. LG and WZ contributed to the experiments and technical supporting. LF, XJ, XH, and QS to the data Interpretation and Technical Assistance. GL, JZ and CX conceived the study, analyzed the data, and wrote the manuscript. All authors read and approved the final manuscript.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
We are grateful to all study participants. We also thank all investigators for engagement and efforts in collecting and analyzing the data. This work was supported by grants from the National Natural Science Foundation (No. 81974124), special funds for Taishan Scholar Project (No. tsqn20161071), and the Natural Science Foundation of Shandong Province (ZR201911130757).
1. Hoffmann MM, Jacob S, Luft D, Schmülling RM, Rett K, März W, et al. Type I Hyperlipoproteinemia Due to a Novel Loss of Function Mutation of Lipoprotein Lipase, Cys(239)-->TRP, Associated with Recurrent Severe Pancreatitis. J Clin Endocrinol Metab (2000) 85(4):4795–8.
2. Evans D, Arzer J, Aberle J, Beil FU. Rare Variants in the Lipoprotein Lipase (LPL) Gene Are Common in Hypertriglyceridemia But Rare in Type III Hyperlipidemia. Atherosclerosis (2011) 214(2):386–90. doi: 10.1016/j.atherosclerosis.2010.11.026
3. Rodrigues R, Artieda M, Tejedor D, Martinez A, Konstantinova P, Petry H, et al. Pathogenic Classification of LPL Gene Variants Reported to be Associated With LPL Deficiency. J Clin Lipidol (2016) 10(2):394–409. doi: 10.1016/j.jacl.2015.12.015
4. Caddeo A, Mancina RM, Pirazzi C, Russo C, Sasidharan K, Sandstedt J, et al. Molecular Analysis of Three Known and One Novel LPL Variants in Patients With Type I Hyperlipoproteinemia. Nutrition Metabol Cardiovasc Dis: NMCD (2018) 28(2):158–64. doi: 10.1016/j.numecd.2017.11.003
5. Deeb SS, Peng RL. Structure of the Human Lipoprotein Lipase Gene. Biochemistry (1989) 28(10):4131–5. doi: 10.1021/bi00436a001
6. Wang H, Eckel RH. Lipoprotein Lipase: From Gene to Obesity. Am J Physiol Endocrinol Metab (2009) 297(2):E271–88. doi: 10.1152/ajpendo.90920.2008
7. Kirchgessner TG, Chuat JC, Heinzmann C, Etienne J, Guilhot S, Svenson K, et al. Organization of the Human Lipoprotein Lipase Gene and Evolution of the Lipase Gene Family. Proc Natl Acad Sci USA (1989) 86(24):9647–51. doi: 10.1073/pnas.86.24.9647
8. Gilbert B, Rouis M, Griglio S, de Lumley L, Laplaud P. Lipoprotein Lipase (LPL) Deficiency: A New Patient Homozygote for the Preponderant Mutation Gly188Glu in the Human LPL Gene and Review of Reported Mutations: 75% are Clustered in Exons 5 and 6. Ann Genet (2001) 44(1):25–32. doi: 10.1016/s0003-3995(01)01037-1
9. Brahm AJ, Hegele RA. Chylomicronaemia–current Diagnosis and Future Therapies. Nat Rev Endocrinol (2015) 11(6):352–62. doi: 10.1038/nrendo.2015.26
10. Cheng Y, Sun Y, Ji Y, Jiang D, Teng G, Zhou X, et al. Novel Compound Variants of the AR and MAP3K1 Genes are Related to the Clinical Heterogeneity of Androgen Insensitivity Syndrome. Biosci Rep (2020) 40(5):BSR20200616. doi: 10.1042/BSR20200616
11. Yu XH, Zhao TQ, Wang L, Liu ZP, Zhang CM, Chen R, et al. A Novel Substitution at the Translation Initiator Codon (ATG–>ATC) of the Lipoprotein Lipase Gene Is Mainly Responsible for Lipoprotein Lipase Deficiency in a Patient With Severe Hypertriglyceridemia and Recurrent Pancreatitis. Biochem Biophys Res Commun (2006) 341(1):82–7. doi: 10.1016/j.bbrc.2005.12.165
12. Henderson HE, Ma Y, Hassan MF, Monsalve MV, Marais AD, Winkler F, et al. Amino Acid Substitution (Ile194—Thr) in Exon 5 of the Lipoprotein Lipase Gene Causes Lipoprotein Lipase Deficiency in Three Unrelated Probands. Support Multicentric Origin J Clin Invest (1991) 87(6):2005–11. doi: 10.1172/JCI115229
13. Emmerich J, Beg OU, Peterson J, Previato L, Brunzell JD, Brewer HB Jr., et al. Human Lipoprotein Lipase. Analysis of the Catalytic Triad by Site-Directed Mutagenesis of Ser-132, Asp-156, and His-241. J Biol Chem (1992) 267(6):4161–5. doi: 10.1016/S0021-9258(19)50642-1
14. Rioja J, Ariza MJ, García-Casares N, Coca-Prieto I, Arrobas T, Muñiz-Grijalvo O, et al. Evaluation of the Chylomicron-TG to VLDL-TG Ratio for Type I Hyperlipoproteinemia Diagnostic. Eur J Clin Invest (2020) 50(12):e13345. doi: 10.1111/eci.13345
15. Hoffmann MM, Jacob S, Luft D, Schmülling RM, Rett K, März W, et al. Type I Hyperlipoproteinemia Due to a Novel Loss of Function Mutation of Lipoprotein Lipase, Cys(239)–>Trp, Associated With Recurrent Severe Pancreatitis. J Clin Endocrinol Metab (2000) 85(12):4795–8. doi: 10.1210/jcem.85.12.7069
16. Chen TZ, Xie SL, Jin R, Huang ZM. A Novel Lipoprotein Lipase Gene Missense Mutation in Chinese Patients With Severe Hypertriglyceridemia and Pancreatitis. Lipids Health Dis (2014) 13:52. doi: 10.1186/1476-511X-13-52
17. Khovidhunkit W, Charoen S, Kiateprungvej A, Chartyingcharoen P, Muanpetch S, Plengpanich W. Rare and Common Variants in LPL and APOA5 in Thai Subjects With Severe Hypertriglyceridemia: A Resequencing Approach. J Clin Lipidol (2016) 10(3):505–11.e1. doi: 10.1016/j.jacl.2015.11.007
18. Li X, Yang Q, Shi X, Chen W, Pu N, Li W, et al. Compound But non-Linked Heterozygous P.W14X and P.L279 V LPL Gene Mutations in a Chinese Patient With Long-Term Severe Hypertriglyceridemia and Recurrent Acute Pancreatitis. Lipids Health Dis (2018) 17(1):144. doi: 10.1186/s12944-018-0789-2
19. Plengpanich W, Kiateprungvej A, Charoen S, Khovidhunkit W. Clinical and Functional Studies of Two Novel Variants in the LPL Gene in Subjects With Severe Hypertriglyceridemia. Clin Chim Acta Int J Clin Chem (2018) 487:22–7. doi: 10.1016/j.cca.2018.08.041
20. Hata A, Ridinger DN, Sutherland S, Emi M, Shuhua Z, Myers RL, et al. Binding of Lipoprotein Lipase to Heparin. Identification of Five Critical Residues in Two Distinct Segments of the Amino-Terminal Domain. J Biol Chem (1993) 268(12):8447–57. doi: 10.1016/S0021-9258(18)52896-9
21. Kobayashi Y, Nakajima T, Inoue I. Molecular Modeling of the Dimeric Structure of Human Lipoprotein Lipase and Functional Studies of the Carboxyl-Terminal Domain. Eur J Biochem (2002) 269(18):4701–10. doi: 10.1046/j.1432-1033.2002.03179.x
22. Lookene A, Groot NB, Kastelein JJ, Olivecrona G, Bruin T. Mutation of Tryptophan Residues in Lipoprotein Lipase. Effects on Stability, Immunoreactivity, and Catalytic Properties. J Biol Chem (1997) 272(2):766–72. doi: 10.1074/jbc.272.2.766
23. Arora R, Nimonkar AV, Baird D, Wang C, Chiu CH, Horton PA, et al. Structure of Lipoprotein Lipase in Complex With GPIHBP1. Proc Natl Acad Sci USA (2019) 116(21):10360–5. doi: 10.1073/pnas.1820171116
24. Chen WW, Yang Q, Li XY, Shi XL, Pu N, Lu GT, et al. Identification of a Novel and Heterozygous LMF1 Nonsense Mutation in an Acute Pancreatitis Patient With Severe Hypertriglyceridemia, Severe Obesity and Heavy Smoking. Lipids Health Dis (2019) 18(1):68. doi: 10.1186/s12944-019-1012-9
Keywords: lipoprotein lipase (LPL), type 1, hypertriglyceridemia, variants, pedigree
Citation: Wang S, Cheng Y, Shi Y, Zhao W, Gao L, Fang L, Jin X, Han X, Sun Q, Li G, Zhao J and Xu C (2022) Identification and Characterization of Two Novel Compounds: Heterozygous Variants of Lipoprotein Lipase in Two Pedigrees With Type I Hyperlipoproteinemia. Front. Endocrinol. 13:874608. doi: 10.3389/fendo.2022.874608
Received: 12 February 2022; Accepted: 03 June 2022;
Published: 18 July 2022.
Edited by:
Anna Alisi, Bambino Gesù Children’s Hospital (IRCCS), ItalyReviewed by:
Naishi Li, Peking Union Medical College Hospital (CAMS), ChinaCopyright © 2022 Wang, Cheng, Shi, Zhao, Gao, Fang, Jin, Han, Sun, Li, Zhao and Xu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jiajun Zhao, amp6aGFvQHNkdS5lZHUuY24=; Guimei Li, bGlndWltZWkyMDEzQDEyNi5jb20=; Chao Xu, ZG9jdG9yeHVjaGFvQDE2My5jb20=
†These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.