 Marle Pretorius
Marle Pretorius Carol Huang
Carol Huang- Department of Biochemistry and Molecular Biology, Cumming School of Medicine, University of Calgary, Calgary, AB, Canada
During pregnancy, the mother develops insulin resistance to shunt nutrients to the growing fetus. As a result, the maternal islets of Langerhans undergo several changes to increase insulin secretion in order to maintain glucose homeostasis and prevent the development of gestational diabetes. These changes include an increase in β-cell proliferation and β-cell mass, upregulation of insulin synthesis and insulin content, enhanced cell-to-cell communication, and a lowering of the glucose threshold for insulin secretion, all of which resulting in an increase in glucose-stimulated insulin secretion. Emerging data suggests that a change in intracellular calcium dynamics occurs in the β-cell during pregnancy as part of the adaptive process. Influx of calcium into β-cells is crucial in the regulation of glucose-stimulated insulin secretion. Calcium fluxes into and out of the cytosol, endoplasmic reticulum, and mitochondria are also important in controlling β-cell function and survival. Here, we review calcium dynamics in islets in response to pregnancy-induced changes in hormones and signaling molecules, and how these changes may enhance insulin secretion to stave off gestational diabetes.
Introduction
It has long been known that during pregnancy, the maternal insulin demand increases due to the physiologic increase in insulin resistance (1). To accommodate this increased demand, pancreatic islets adapt through several mechanisms including increasing insulin synthesis and lowering the threshold for glucose-stimulated insulin secretion (GSIS) (2, 3), which has been demonstrated in both rodents and humans (4–10). Change in calcium dynamics within pancreatic islets, and specifically in various subcellular compartments of the pancreatic β-cell, can affect β-cell function such as insulin secretion and β-cell survival (11–13). This review will explore and summarize the current knowledge on calcium dynamics in pancreatic islets during pregnancy, and the implications in gestational diabetes mellitus (GDM).
Changes in Islets During Pregnancy
During pregnancy, the maternal pancreatic islets are placed under high demand for insulin production due to an increase in insulin resistance of maternal tissues (8, 14), a physiologic change that encourages the diversion of nutrient from the mother to the developing fetus. In order to accommodate for this increase in insulin demand, studies in rodent islets and β-cell lines have identified several mechanisms involved, including an increase in β-cell proliferation, β-cell size (7, 10, 15), insulin gene expression, insulin synthesis, and insulin content, as well as lowering the glucose threshold for insulin secretion (16, 17). An increase in β-cell mass and number has also been demonstrated in human pregnancy (9, 18). An increase in gap-junction coupling and islet vasculature density are also part of the adaptive mechanism (19, 20). In contrast, there is no significant change in α-cell number or size during pregnancy, and no significant change in the spatial organization of the islets, i.e. the majority of the β-cells form the core of the islet surrounded by α-cells in the periphery (15, 21). Gene ontology analysis of the islet transcriptome during pregnancy has identified enrichment of genes that regulate cell proliferation, apoptosis, response to stress, cell communication, cellular physiological processes such as proteolysis and vesicle trafficking, as well as cellular metabolic processes such as lipid metabolism and electron transport (22–25). Many of these adaptive responses are regulated by pregnancy hormones such as lactogens (15, 26), growth hormone (27–29), estrogen (30, 31), progesterone (20, 32), and other factors such as hepatic growth factors (33)and serotonin (24). Whether these or other pregnancy-associated factors regulate β-cell Ca2+ handling directly and contribute to the enhanced insulin secretion observed during pregnancy requires much more investigation, although indirect evidence suggests that they may participate in the regulation of Ca2+ dynamics in β-cells.
Estrogen (17 β-oestradiol or E2) level increases throughout pregnancy (34). It acts through the classic nuclear hormone estrogen receptor α and β isoforms (ERα and ERβ) as well as through the G protein-coupled estrogen receptor 1 (GPER1, or GPR30) in both rodents and humans islets (35). The 3 receptors have distinctive functions in β-cells. Activation of ERα regulates insulin synthesis and β-cell survival (31, 36). Activation of ERβ in mouse islets stimulates guanylyl cyclase A and rapidly increases cyclic GMP levels, leading to a reduction in KATP channel activity in the plasma membrane, an increase in calcium oscillation and cytosolic calcium ([Ca2+]c), and augments GSIS (37, 38). Mice treated with an ERβ agonist also demonstrated a higher β-cell mass and β-cell proliferation (38). GPER1 (aka GPR30) is a plasma membrane receptor that upon 17 β-estradiol binding, stimulates cGMP synthesis and activates protein kinase G (PKG), leading to closure of the KATP channel and increases frequency of Ca2+ oscillation and intracellular Ca2+ concentration, enhancing GSIS (35, 39–42). Martensson et al. found that in GPR30-/- mice, there is a defect in 1st-phase insulin secretion in vivo and when these islets were tested in vitro, E2-stimulated insulin secretion was completely abolished, suggesting its dominant role in regulating E2-mediated insulin secretion (42). GPER1 activation also protects β-cells against apoptosis (43). During pregnancy, estrogen receptor ERα and GPER expression are up regulated in rodent islets (44, 45). Recently, Ma et al. reported activation of the transient receptor potential ankyrin-repeat 1 (TRPA1) channels in INS-1 cells as well as rodent and human β-cells by estradiol metabolites (46). TRPA1 is a cation channel that is activated by a wide variety of exogenous irritants and inflammatory cytokines (47). It has been shown to regulate insulin secretion (48). In this study (46), estradiol metabolites (but not estradiol) induced strong inward current and a robust and sustained elevation in [Ca2+]c an increase that closely parallels their effect to enhance GSIS.
Serotonin has been identified as a key regulator of β-cell proliferation during pregnancy. Expression of serotonin, as well as tryptophan hydroxylase-1, the enzyme responsible for serotonin synthesis, was found to increase significantly in rodent islets during pregnancy (24). Inhibition of serotonin synthesis during pregnancy blocks β-cell proliferation, resulting in glucose intolerance. This was found to be downstream of lactogen and prolactin receptor (PRLR) signaling. In human islets, serotonin is secreted by β-cells and it exerts paracrine action on α-cells, inhibiting glucagon secretion (49). Activation of the serotonin receptor, 5-HT2B by a-methyl serotonin maleate salt has been shown to alter [Ca2+]c oscillation, causing an increase in both peak duration and distance between peaks in mouse islets, and an increase in insulin secretion from both human and mouse islets (50). Interestingly, exposure of β-cells in culture (MIN6 mouse insulinoma cells) to a selective serotonin reuptake inhibitor (SSRI) reduced ER calcium stores and inhibited ER calcium release and store-operated calcium entry activation (51). SSRIs can also inhibit insulin secretion by inhibiting mitochondrial complex I and II, decrease oxidative respiration, ATP generation and KATP channel activity, although this study did not measure [Ca2+]c (52). Whether the increase in endogenous serotonin in islets during pregnancy regulates intracellular Ca2+ dynamics and contributes to GSIS require further investigation.
The corticotropin-releasing hormone (CRH) family of peptides activates the cAMP/PKA signaling pathways, potentiate Ca2+ influx through the L-type Ca2+ channels and modulate insulin secretion in rat islets (53). CRH and its paralogs, urocortin 1 (Ucn1), Ucn2, and Ucn3 act through their cognate G-protein-coupled receptors, CRH receptor 1 (CRHR1) and CRH receptor 2 (CRHR2); both are expressed in pancreatic β-cells, such as the MIN6 mouse insulinoma cells and primary rodent islets (53–56). A recent study by Simpson et al. found that during mouse pregnancy, urocortin 2 (Ucn2) is up regulated and it acts through CRHR2 to regulate glucose homeostasis, most likely via its effect on insulin secretion (57), This conclusion was based on the observation that CRHR2 blockade had no effect on insulin sensitivity or β-cell proliferation while both in vitro and in vivo blockage of CRHR2 have been shown to attenuate GSIS in mice (58).
Hepatic growth factor (HGF) is another hormone that has been shown to be important in the regulation of β-cell adaptation to pregnancy (33). During pregnancy, there is an increase in HGF level in the serum and HGF expression in the β-cells. Transgenic mice with β-cell specific HGF overexpression had increased glucokinase expression, glucose transport, and insulin secretion (59). In kidney epithelial cells, HGF has been found to inhibit Ca2+ release from the ER while in hepatocytes, HGF activates the inositol-triphosphate-PLCγ pathway and causes a rapid rise in [Ca2+]c (60). Whether similar response to HGF occurs in β-cells has yet to be determined.
Lastly, many of the changes observed in the islets during pregnancy are due to actions of prolactin and placental lactogens, both signaling through the Prolactin Receptor (PRLR) (15, 26). Work by us and others have shown that PRLR deletion led to impaired glucose tolerance during mouse pregnancy, mainly by dampening pregnancy-induced β-cell proliferation. This results in a smaller β-cell mass, lower serum insulin levels, and reduced pancreatic insulin content. Interestingly, several transcriptome analyses have identified Leucine Rich Repeat Containing 55 (Lrrc55) (61), an auxiliary protein of big-potassium channels (62), as one of the most highly upregulated genes downstream of PRLR in the pancreatic islets during pregnancy (24, 63), and we found that Lrrc55 is a novel, pro-survival factor in β-cells, potentially by regulating calcium handling (25).
Calcium Dynamics in β-Cells
The electrically excitable pancreatic β-cells utilize the controlled flux of a few key ions, all of which coupled to calcium flux, to precisely regulate insulin release in response to high levels of blood glucose (Figure 1).
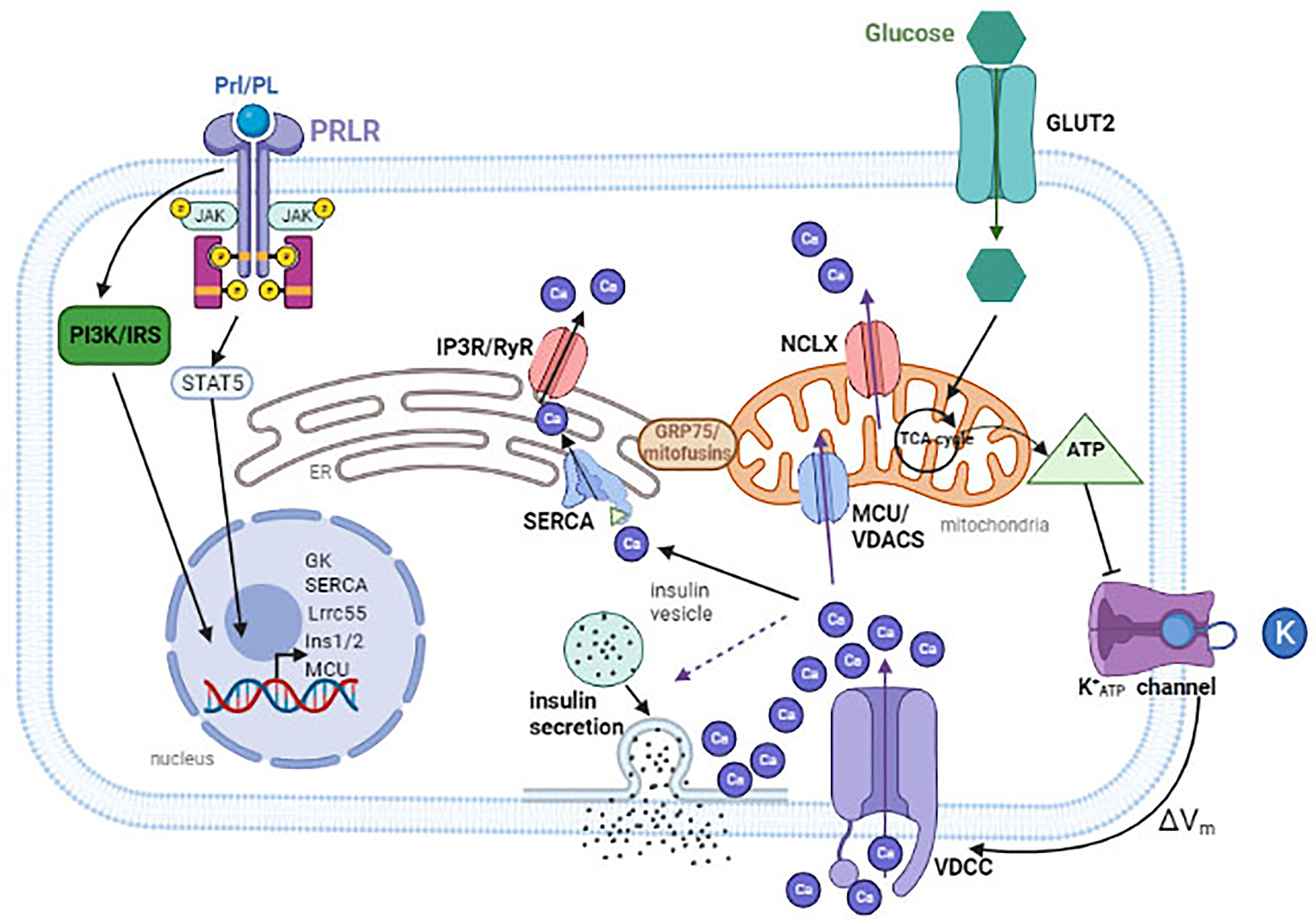
Figure 1 Glucose is transported into the cytoplasm through glucose transporters (GLUT2 in rodents). Once inside, glucose are metabolized in the mitochondria to generate ATP. An increase in ATP to ADP ratio will cause the closure of ATP-sensitive potassium channel (KATP), resulting in membrane depolarization. This leads to the opening of the L-type voltage-dependent Ca2+ channels (VDCC) and an influx of Ca2+. To maintain [Ca2+]m, Ca2+ enters through the MCU or the VDACs and escapes through the NCLX. Ca2+ is pumped into the ER through ATP-dependent SERCA, and is released back into the cytosol through the IP3R or RyR calcium channels. Calcium can also flow from the ER to the mitochondria. During pregnancy, placental hormones bind to PrlR, inducing a signal cascade that results in up regulation of several genes to increase insulin production and secretion. These include Lrrc55 and SERCA to maintain [Ca2+]ER, MCU to maintain [Ca2+]m, and INS1/2 to increase insulin expression. VDAC, voltage-dependent anion channels; MCU, mitochondrial Ca2+ uniporter; PRLR, prolactin receptor; Prl/PL, prolactin/placental lactogen. The dashed line arrow indicates that influx of calcium induces insulin secretion. Created with BioRender.com.
Glucose enters β-cells via the facilitated glucose transporter GLUT2 in rodents and GLUT1 and GLUT3 in humans. Upon phosphorylation by glucokinase, glucose-6-phosphate is metabolized by glycolysis and the TCA cycle to generate ATP. The increase in the ATP/ADP ratio results in closure of the KATP channels in the plasma membrane and a burst of action potentials, which leads to opening of the voltage-dependent Ca2+ channels (VDCC), allowing Ca2+ influx and an increase in [Ca2+]c, especially in submembrane areas near the Ca2+ channels. At moderate glucose concentration (7-15mM), the intermittent opening of VDCC causes [Ca2+]c oscillation while at high glucose concentration (>20mM), the continued opening of VDCC causes a sustained increase in [Ca2+]c (11). VDCC is not distributed uniformly throughout the plasma membrane, generating microdomains with high [Ca2+] (64). These subdomains of high [Ca2+]c are in close proximity to the voltage-gated Ca2+ channels and form hot spots for insulin granule to dock and fuse (65), a process that is facilitated by soluble N-ethylmaleimide-sensitive factor attachment protein receptors (SNAREs) and SNARE regulator proteins, such as syntaxin 1A (66), SNAP-25 (67–69) and synaptotagmins (70–73). Repolarization of the plasma membrane results from the rapid inactivation of VDCC and the opening of potassium channels (Kv2.1 voltage-dependent channels and large-conductance Ca2+-activated K+ channels, (BK) (11, 74, 75).
Limited data are available on pregnancy-induced changes in intracellular calcium dynamics. In a study on the effect of protein restriction on insulin secretion and intracellular calcium concentration, Marin et al. reported that in islets from control (not protein restricted) rats, on day-15 of pregnancy, glucose elicited a larger and earlier rise in [Ca2+]c in comparison to β-cells from non-pregnant rats. Interestingly, while this larger rise in [Ca2+]c did not result in a change in the total amount of Ca2+ influx during plasma membrane depolarization, it was accompanied by a more sustained and gradual insulin secretion profile. Islets from pregnant rats also expressed more SNAP-25, although its specific role on calcium dynamics was not explored (76). Vanzela et al. reported that islets from pregnant rats (15th or 16th day of pregnancy) had increased calcium oscillation and a higher level of expression of Cavα1.2 and SERCA2a in comparison to non-pregnant rats (77). In a model of cafeteria-diet (Caf) induced obesity, islets from Caf-exposed rats exhibited a blunted glucose-induced calcium increase and insulin secretion, both of which were reversed by pregnancy. The Caf-induced reduction in Cavα1.2 expression was also reversed by pregnancy. They concluded that pregnancy reversed the deleterious effects of Caf on islet function by restoring calcium handling, in part through the up regulation of Cavα1.2 and SERCA2a expression. Expression of the SNARE protein, synaptotagmin 4 (Syt4), has also been shown to be up regulated in islets isolated from rats on day 15 of pregnancy and in prolactin-treated islets in vitro (70). Syt4 regulates Ca2+ sensitivity in β-cells and its expression is increased by ~8-fold during β-cell maturation, leading to increased GSIS in mature islets in comparison to neonatal islets in mice (78). Syt4 is expressed in insulin vesicles, Golgi, and the ER, and potentially regulates general Ca2+ signaling in the ER of β-cells (78). Syn4 is expressed in human β-cells and it regulates insulin secretion in the human β-cell line EndoC-βH1 (78). Taken together, these changes in expression and activities of Ca2+ channels, SNARE proteins, and glucose metabolism may contribute to the changes in Ca2+ influx and insulin secretion in pregnancy.
ER Calcium in β-Cells
ER calcium concentrations are mainly determined by the sarco/endoplasmic reticulum Ca2+-ATPase (SERCA), which actively pumps calcium into the ER from the cytosol, as well as two receptor-type membrane proteins, the ryanodine receptor (RyR) and the Inositol Triphosphate Receptor (IP3R) (79–81), both of which releases calcium from the ER. Blocking IP3R, RyR, and combinations thereof, has been shown to rescue β-cells from ER calcium depletion caused by SERCA inhibitor (82). Luciani et al. found that in the mouse β-cell line MIN6, blocking IP3R appeared to have a more pronounced effect on β-cell survival than blocking RyR (82). Interestingly, Hara et al. found that in conditions associated with β-cell death, such as ER stress, oxidative stress, exposure to palmitate, chronic high glucose, and overexpression of mutated insulin, there were decreases in ER calcium levels (in INS-1 cells) and SERCA expression (in INS-1, human and mouse islets) (83). They hypothesize that genetic and environmental stressors cause β-cell stress, leading to a reduction in SERCA2b and an increase in IP3R expression, resulting in a reduction in ER calcium and an increase in [Ca2+]c leading to β-cell death. How ER stress causes a reduction in SERCA2b expression is currently unknown.
The increased insulin demand seen during pregnancy and the associated increase in protein synthesis can activate the unfolded protein response (UPR), which if unresolved, can lead to ER stress. Indeed, while we did not observe an increase in the number of apoptotic β-cells during mouse pregnancy (15), we found that expression of IRE1α and CHOP, components of the UPR pathways, are up regulated in mouse islets during early pregnancy (25). Anhe et al. reported that on d19 of pregnancy, SERCA2 expression was upregulated in rat islets. This increase may contribute to the increase in GSIS observed during pregnancy, as inhibition of SERCA2 activity by thapsigargin led to a reduction in 1st phase insulin secretion in isolated rat islets (84). Activation of prolactin receptor may also contribute to these pregnancy-associated changes, as levels of prolactin and its related hormones, placental lactogens, are high throughout pregnancy, and treatment of the rat islet cell line, RINm5F, with prolactin recapitulated the increase in SERCA2 expression observed during pregnancy (84). Interestingly, prolactin has been shown to up regulate SERCA2b expression, increase [Ca2+]ER, and stimulate cell proliferation in prostate cells. Conversely, SERCA2b knockdown reduced both [Ca2+]ER and cell proliferation (85). Whether primary β-cells would have a similar response to prolactin in terms of increasing [Ca2+]ER has not been examined.
Protein kinase R (PKR)-like endoplasmic reticulum kinase (PERK) (EIF2AK3) is an eIF2α kinase in the ER membrane, known to regulate β-cell development, function, and ER stress response (13, 86, 87). PERK inhibition lowered the glucose-induced rise in intracellular calcium and blunted GSIS in INS-1 cells as well as in rat and human islets (88). PERK regulates intracellular calcium levels by at least two mechanisms: first, it controls calcium influx into the cytosol by regulating store-operated Ca2+ channel (SOCC) activity (89); second, it stimulates SERCA-mediated calcium reuptake into the ER after [Ca2+]ER depletion or release via a calcineurin dependent pathway (88). During pregnancy, expression of PERK in the islets rises near the end of gestation (day 19 in mice) and peaks on day 1 of lactation, but promptly drops below pre-pregnancy level by day 2 of lactation (90). How and whether this increase in PERK expression contributes to pregnancy-associated changes in calcium dynamics in β-cells remains to be determined.
Mitochondrial Calcium Dynamics in β-Cells
Calcium influx into the mitochondria is important for facilitating GSIS from β-cells. Under basal condition, intramitochondrial calcium ([Ca2+]m) is low, comparable to that of [Ca2+]c (<100nM) (91). Calcium is transported into mitochondria through voltage-dependent anion channels (VDACs) in the outer mitochondrial membrane and the mitochondrial Ca2+ uniporter (MCU) (92) in the inner mitochondrial membrane, while calcium exits the mitochondria through the Na+/Ca2+ exchanger (NCLX) (93). Endoplasmic reticulum is another source of Ca2+ for mitochondria, and tethering molecules such as GRP75 and mitofusins are present in microdomains between ER and mitochondria to facilitate calcium flow from ER (which has basal calcium concentration of ~5mM) into the mitochondria (94, 95).
In mouse β-cells, Tarasov et al. demonstrated that [Ca2+]m follows the slow but not fast changes in [Ca2+]c, and [Ca2+]m is highly sensitive to calcium oscillation (91). At low glucose concentration, the small spikes in [Ca2+]c is not transmitted to the [Ca2+]m but as glucose levels rises, the increase in [Ca2+]c oscillation leads to an increase in [Ca2+]m. The rise in intramitochondrial calcium activates mitochondrial dehydrogenases and further stimulates ATP production, resulting in a biphasic phase increase in [ATP/ADP]c. Tarasov et al. speculated that the second phase of ATP production may be involved in mobilization of the reserve pool of insulin granule (91).
While changes in [Ca2+]m have not been directly determined in β-cells during pregnancy, it is tempting to speculate that the subtle changes in cytosolic calcium oscillation discussed above may influence [Ca2+]m, and explain in part the increase in insulin secretion observed during pregnancy. Expression of MCU increased throughout gestation in human placenta (96). Whether a similar change is present in pancreatic islets and the functional consequence to mitochondrial calcium handling is unknown.
Calcium and Gestational Diabetes
Gestational diabetes mellitus (GDM) is defined as diabetes diagnosed for the first time during pregnancy (97). While estimates differ depending on the populations studied, approximately 3-20% of pregnancies are complicated by GDM (97, 98). GDM is associated with a higher risk of maternal and neonatal adverse outcomes, such as pre-eclampsia, macrosomia, stillbirth, and neonatal hypoglycemia (99). Women who have developed GDM are at high risk of developing GDM in subsequent pregnancies as well as progressing to type 2 diabetes. Additionally, exposure of the offspring to hyperglycemia in utero significantly increases their risk of developing Type 2 diabetes later in life (14, 100, 101).
Many human studies have examined the relationship between serum calcium and vitamin D levels and the risk of diabetes (102–104). A large epidemiological study in >3400 US women found that periconceptional calcium intake was inversely associated with the risk of developing GDM (105). They hypothesized that the relationship between calcium intake and GDM risk may lie in the positive association between intracellular calcium ([Ca2+]i) and insulin secretion in β-cells; they also observed a U-shaped relationship between [Ca2+]i and insulin sensitivity in vascular smooth muscle and adipocytes (106). Calcium sensing receptor (CaSR) may potentially link serum calcium levels to β-cell function. CaSR is expressed in β-cells and it contributes to β-cell adhesion, coupling, and communication. CaSR has also been shown to inhibit basal and GSIS in human islets (107). Transgenic mice with gain-of-function mutation of CaSR have reduced islet mass and β-cell proliferation, as well as hypoinsulinemia and hyperglycemia (108). In human pregnancies complicated by GDM, expression of CaSR was found to be significantly reduced in the placenta, which may have contributed to the hypocalcemia observed in 16% of the newborns (109). Whether CaSR expression was also altered in islets of women with GDM could not be determined due to the inaccessibility of pancreatic tissue for analysis.
In support of the link between calcium dynamics and GDM risk, Goldstein et al. used an informatics-based approach to determine the association between GDM and/or type 2 diabetes, disease-associated SNPs, and the effects of a list of 129 active drugs in 9960 patients. They found that the use of a calcium channel blocker (CCB) such as nifedipine was associated with a reduction in serum glucose during glucose tolerance tests, and there was a strong association between genes targeted by CCBs and GDM risk (110). Mechanistically, treatment of mouse islets with CCB increased basal insulin secretion and reduced glucagon secretion (111) while blocking calcium entry was shown to protect β-cells and human islets against ER stress and apoptosis (112–114).
The level of circulating vitamin D (25-hydroxyvitamin D [25(OH)D]) during pregnancy has also been shown to inversely correlate with GDM risk (103, 115). In a meta-analysis, Wei et al. reported that low levels of circulating vitamin D increase the risk of GDM by 1.38-fold (116). Vitamin D levels may contribute to GDM via its putative effect on both insulin sensitivity and insulin secretion (117). Non-genomic signaling through vitamin D receptor has been shown to augment GSIS by increasing intracellular Ca2+ concentration, which was blocked by the CCB nitrendipine (118). Norman et al. found that pancreas from vitamin D deficient rats showed a 48% reduction in GSIS in an ex-vivo perfusion experiment (119). Moreover, in a study of 126 healthy human subjects, serum vitamin D levels were found to negatively correlate with 1st and 2nd phase insulin release during a hyperglycemic clamp, and an effect on β-cell function remains after correction for insulin sensitivity index (120).
Conclusion
In this review, we discussed the current knowledge of calcium dynamics in pancreatic islets during pregnancy. The hallmark of β-cell adaptation to pregnancy is an increase in insulin secretion, a process that is tightly regulated by intracellular calcium dynamics. Hormones and small molecules such as estrogen, corticotropin releasing hormone, serotonin, HGF, and placental lactogens potentially enhance insulin secretion and β-cell proliferation via regulating calcium dynamics. Furthermore, up regulation of GLUT2 and glucokinase during pregnancy allows more efficient glucose metabolism. Coupled with the increased expression of L-type Ca2+ channel, these changes allow β-cells to secrete insulin more efficiently. The finding that calcium channel blockers improve glycemia is very exciting and encouraging, as it points to abnormal intracellular calcium dynamics as a potential contributing factor to GDM but it also provides a safe and effective treatment for those with GDM. Currently, our understanding of how intracellular calcium dynamics changes as part of β-cell adaptation to the increased insulin demand of pregnancy is very limited and fragmented. A thorough understanding of this field would allow for design of more targeted therapy for GDM and prevent the vicious cycle of GDM begetting more GDM.
Author Contributions
MP and CH wrote the manuscript. Both authors contributed to the article and approved the submitted version.
Funding
This work was supported by funds from Natural Sciences and Engineering Research Council of Canada (RGPIN-2020-05247) to CH.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Kuhl C. Insulin Secretion and Insulin Resistance in Pregnancy and GDM. Implications for Diagnosis and Management. Diabetes (1991) 40 Suppl 2:18–24. doi: 10.2337/diab.40.2.S18
2. Sorenson RL, Johnson MG, Parsons JA, Sheridan JD. Decreased Glucose Stimulation Threshold, Enhanced Insulin Secretion, and Increased Beta Cell Coupling in Islets of Prolactin-Treated Rats. Pancreas (1987) 2:283–8. doi: 10.1097/00006676-198705000-00006
3. Mills JL, Jovanovic L, Knopp R, Aarons J, Conley M, Park E, et al. Physiological Reduction in Fasting Plasma Glucose Concentration in the First Trimester of Normal Pregnancy: The Diabetes in Early Pregnancy Study. Metabolism (1998) 47:1140–4. doi: 10.1016/S0026-0495(98)90290-6
4. Spellacy WN, Goetz FC, Greenberg BZ, Ells J. Plasma Insulin in Normal “Early” Pregnancy. Obstet Gynecol (1965) 25:862–5.
5. Spellacy WN, Goetz FC, Greenberg BZ, Ells J. Plasma Insulin in Normal Midpregnancy. Am J Obstet Gynecol (1965) 92:11–5. doi: 10.1016/0002-9378(65)90099-2
6. Spellacy WN, Goetz FC. Plasma Insulin in Normal Late Pregnancy. N Engl J Med (1963) 268:988–91. doi: 10.1056/NEJM196305022681805
7. Parsons JA, Brelje TC, Sorenson RL. Adaptation of Islets of Langerhans to Pregnancy: Increased Islet Cell Proliferation and Insulin Secretion Correlates With the Onset of Placental Lactogen Secretion. Endocrinology (1992) 130:1459–66. doi: 10.1210/endo.130.3.1537300
8. Sorenson RL, Brelje TC. Prolactin Receptors Are Critical to the Adaptation of Islets to Pregnancy. Endocrinology (2009) 150:1566–9. doi: 10.1210/en.2008-1710
9. Van Assche FA, Aerts L, De Prins F. A Morphological Study of the Endocrine Pancreas in Human Pregnancy. Br J Obstet Gynaecol (1978) 85:818–20. doi: 10.1111/j.1471-0528.1978.tb15835.x
10. Sorenson RL, Brelje TC. Adaptation of Islets of Langerhans to Pregnancy: Beta-Cell Growth, Enhanced Insulin Secretion and the Role of Lactogenic Hormones. Horm Metab Res (1997) 29:301–7. doi: 10.1055/s-2007-979040
11. Gilon P, Chae HY, Rutter GA, Ravier MA. Calcium Signaling in Pancreatic Beta-Cells in Health and in Type 2 Diabetes. Cell Calcium (2014) 56:340–61. doi: 10.1016/j.ceca.2014.09.001
12. Herchuelz A, Nguidjoe E, Jiang L, Pachera N. Beta-Cell Preservation and Regeneration in Diabetes by Modulation of Beta-Cell Ca(2)(+) Homeostasis. Diabetes Obes Metab (2012) 14 Suppl 3:136–42. doi: 10.1111/j.1463-1326.2012.01649.x
13. Cunha DA, Hekerman P, Ladriere L, Bazarra-Castro A, Ortis F, Wakeham MC, et al. Initiation and Execution of Lipotoxic ER Stress in Pancreatic Beta-Cells. J Cell Sci (2008) 121:2308–18. doi: 10.1242/jcs.026062
14. Buchanan TA, Xiang AH. Gestational Diabetes Mellitus. J Clin Invest (2005) 115:485–91. doi: 10.1172/JCI200524531
15. Huang C, Snider F, Cross JC. Prolactin Receptor Is Required for Normal Glucose Homeostasis and Modulation of Beta-Cell Mass During Pregnancy. Endocrinology (2009) 150:1618–26. doi: 10.1210/en.2008-1003
16. Green IC, Taylor KW. Effects of Pregnancy in the Rat on the Size and Insulin Secretory Response of the Islets of Langerhans. J Endocrinol (1972) 54:317–25. doi: 10.1677/joe.0.0540317
17. Weinhaus AJ, Stout LE, Sorenson RL. Glucokinase, Hexokinase, Glucose Transporter 2, and Glucose Metabolism in Islets During Pregnancy and Prolactin-Treated Islets In Vitro: Mechanisms for Long Term Up-Regulation of Islets. Endocrinology (1996) 137:1640–9. doi: 10.1210/endo.137.5.8612496
18. Butler AE, Cao-Minh L, Galasso R, Rizza RA, Corradin A, Cobelli C, et al. Adaptive Changes in Pancreatic Beta Cell Fractional Area and Beta Cell Turnover in Human Pregnancy. Diabetologia (2010) 53:2167–76. doi: 10.1007/s00125-010-1809-6
19. Sheridan JD, Anaya PA, Parsons JA, Sorenson RL. Increased Dye Coupling in Pancreatic Islets From Rats in Late-Term Pregnancy. Diabetes (1988) 37:908–11. doi: 10.2337/diab.37.7.908
20. Banerjee RR. Piecing Together the Puzzle of Pancreatic Islet Adaptation in Pregnancy. Ann N Y Acad Sci (2018) 1411:120–39. doi: 10.1111/nyas.13552
21. Brelje TC, Svensson AM, Stout LE, Bhagroo NV, Sorenson RL. An Immunohistochemical Approach to Monitor the Prolactin-Induced Activation of the JAK2/STAT5 Pathway in Pancreatic Islets of Langerhans. J Histochem Cytochem (2002) 50:365–83. doi: 10.1177/002215540205000308
22. Rieck S, White P, Schug J, Fox AJ, Smirnova O, Gao N, et al. The Transcriptional Response of the Islet to Pregnancy in Mice. Mol Endocrinol (2009) 23:1702–12. doi: 10.1210/me.2009-0144
23. Layden BT, Durai V, Newman MV, Marinelarena AM, Ahn CW, Feng G, et al. Regulation of Pancreatic Islet Gene Expression in Mouse Islets by Pregnancy. J Endocrinol (2010) 207:265–79. doi: 10.1677/JOE-10-0298
24. Kim H, Toyofuku Y, Lynn FC, Chak E, Uchida T, Mizukami H, et al. Serotonin Regulates Pancreatic Beta Cell Mass During Pregnancy. Nat Med (2010) 16:804–8. doi: 10.1038/nm.2173
25. Makkar G, Shrivastava V, Hlavay B, Pretorius M, Kyle BD, Braun AP, et al. Lrrc55 Is a Novel Prosurvival Factor in Pancreatic Islets. Am J Physiol Endocrinol Metab (2019) 317:E794–804. doi: 10.1152/ajpendo.00028.2019
26. Banerjee RR, Cyphert HA, Walker EM, Chakravarthy H, Peiris H, Gu X, et al. Gestational Diabetes Mellitus From Inactivation of Prolactin Receptor and Mafb in Islet Beta-Cells. Diabetes (2016) 65:2331–41. doi: 10.2337/db15-1527
27. Brelje TC, Scharp DW, Lacy PE, Ogren L, Talamantes F, Robertson M, et al. Effect of Homologous Placental Lactogens, Prolactins, and Growth Hormones on Islet B-Cell Division and Insulin Secretion in Rat, Mouse, and Human Islets: Implication for Placental Lactogen Regulation of Islet Function During Pregnancy. Endocrinology (1993) 132:879–87. doi: 10.1210/endo.132.2.8425500
28. Brelje TC, Stout LE, Bhagroo NV, Sorenson RL. Distinctive Roles for Prolactin and Growth Hormone in the Activation of Signal Transducer and Activator of Transcription 5 in Pancreatic Islets of Langerhans. Endocrinology (2004) 145:4162–75. doi: 10.1210/en.2004-0201
29. Fleenor D, Oden J, Kelly PA, Mohan S, Alliouachene S, Pende M, et al. Roles of the Lactogens and Somatogens in Perinatal and Postnatal Metabolism and Growth: Studies of a Novel Mouse Model Combining Lactogen Resistance and Growth Hormone Deficiency. Endocrinology (2005) 146:103–12. doi: 10.1210/en.2004-0744
30. Moldrup A, Petersen ED, Nielsen JH. Effects of Sex and Pregnancy Hormones on Growth Hormone and Prolactin Receptor Gene Expression in Insulin-Producing Cells. Endocrinology (1993) 133:1165–72. doi: 10.1210/endo.133.3.8365359
31. Wong WP, Tiano JP, Liu S, Hewitt SC, Le May C, Dalle S, et al. Extranuclear Estrogen Receptor-Alpha Stimulates Neurod1 Binding to the Insulin Promoter and Favors Insulin Synthesis. Proc Natl Acad Sci USA (2010) 107:13057–62. doi: 10.1073/pnas.0914501107
32. Sorenson RL, Brelje TC, Roth C. Effects of Steroid and Lactogenic Hormones on Islets of Langerhans: A New Hypothesis for the Role of Pregnancy Steroids in the Adaptation of Islets to Pregnancy. Endocrinology (1993) 133:2227–34. doi: 10.1210/endo.133.5.8404674
33. Demirci C, Ernst S, Alvarez-Perez JC, Rosa T, Valle S, Shridhar V, et al. Loss of HGF/C-Met Signaling in Pancreatic Beta-Cells Leads to Incomplete Maternal Beta-Cell Adaptation and Gestational Diabetes Mellitus. Diabetes (2012) 61:1143–52. doi: 10.2337/db11-1154
34. Nadal A, Alonso-Magdalena P, Soriano S, Ropero AB, Quesada I. The Role of Oestrogens in the Adaptation of Islets to Insulin Resistance. J Physiol (2009) 587:5031–7. doi: 10.1113/jphysiol.2009.177188
35. Kumar R, Balhuizen A, Amisten S, Lundquist I, Salehi A. Insulinotropic and Antidiabetic Effects of 17beta-Estradiol and the GPR30 Agonist G-1 on Human Pancreatic Islets. Endocrinology (2011) 152:2568–79. doi: 10.1210/en.2010-1361
36. Le May C, Chu K, Hu M, Ortega CS, Simpson ER, Korach KS, et al. Estrogens Protect Pancreatic Beta-Cells From Apoptosis and Prevent Insulin-Deficient Diabetes Mellitus in Mice. Proc Natl Acad Sci USA (2006) 103:9232–7. doi: 10.1073/pnas.0602956103
37. Soriano S, Ropero AB, Alonso-Magdalena P, Ripoll C, Quesada I, Gassner B, et al. Rapid Regulation of K(ATP) Channel Activity by 17{Beta}-Estradiol in Pancreatic {Beta}-Cells Involves the Estrogen Receptor {Beta} and the Atrial Natriuretic Peptide Receptor. Mol Endocrinol (2009) 23:1973–82. doi: 10.1210/me.2009-0287
38. Alonso-Magdalena P, Ropero AB, Garcia-Arevalo M, Soriano S, Quesada I, Muhammed SJ, et al. Antidiabetic Actions of an Estrogen Receptor Beta Selective Agonist. Diabetes (2013) 62:2015–25. doi: 10.2337/db12-1562
39. Nadal A, Rovira JM, Laribi O, Leon-quinto T, Andreu E, Ripoll C, et al. Rapid Insulinotropic Effect of 17beta-Estradiol via a Plasma Membrane Receptor. FASEB J (1998) 12:1341–8. doi: 10.1096/fasebj.12.13.1341
40. Nadal A, Ropero AB, Laribi O, Maillet M, Fuentes E, Soria B. Nongenomic Actions of Estrogens and Xenoestrogens by Binding at a Plasma Membrane Receptor Unrelated to Estrogen Receptor Alpha and Estrogen Receptor Beta. Proc Natl Acad Sci USA (2000) 97:11603–8. doi: 10.1073/pnas.97.21.11603
41. Ropero AB, Fuentes E, Rovira JM, Ripoll C, Soria B, Nadal A. Non-Genomic Actions of 17beta-Oestradiol in Mouse Pancreatic Beta-Cells Are Mediated by a Cgmp-Dependent Protein Kinase. J Physiol (1999) 521 Pt 2:397–407. doi: 10.1111/j.1469-7793.1999.00397.x
42. Martensson UE, Salehi SA, Windahl S, Gomez MF, Sward K, Daszkiewicz-Nilsson J, et al. Deletion of the G Protein-Coupled Receptor 30 Impairs Glucose Tolerance, Reduces Bone Growth, Increases Blood Pressure, and Eliminates Estradiol-Stimulated Insulin Release in Female Mice. Endocrinology (2009) 150:687–98. doi: 10.1210/en.2008-0623
43. Liu S, Le May C, Wong WP, Ward RD, Clegg DJ, Marcelli M, et al. Importance of Extranuclear Estrogen Receptor-Alpha and Membrane G Protein-Coupled Estrogen Receptor in Pancreatic Islet Survival. Diabetes (2009) 58:2292–302. doi: 10.2337/db09-0257
44. Shrivastava V, Lee M, Lee D, Pretorius M, Radford B, Makkar G, et al. Beta Cell Adaptation to Pregnancy Requires Prolactin Action on Both Beta and Non-Beta Cells. Sci Rep (2021) 11:10372. doi: 10.1038/s41598-021-89745-9
45. Jacovetti C, Abderrahmani A, Parnaud G, Jonas JC, Peyot ML, Cornu M, et al. Micrornas Contribute to Compensatory Beta Cell Expansion During Pregnancy and Obesity. J Clin Invest (2012) 122:3541–51. doi: 10.1172/JCI64151
46. Ma W, Chen X, Cerne R, Syed SK, Ficorilli JV, Cabrera O, et al. Catechol Estrogens Stimulate Insulin Secretion in Pancreatic Beta-Cells via Activation of the Transient Receptor Potential A1 (TRPA1) Channel. J Biol Chem (2019) 294:2935–46. doi: 10.1074/jbc.RA118.005504
47. Bautista DM, Pellegrino M, Tsunozaki M. TRPA1: A Gatekeeper for Inflammation. Annu Rev Physiol (2013) 75:181–200. doi: 10.1146/annurev-physiol-030212-183811
48. Cao DS, Zhong L, Hsieh TH, Abooj M, Bishnoi M, Hughes L, et al. Expression of Transient Receptor Potential Ankyrin 1 (TRPA1) and Its Role in Insulin Release From Rat Pancreatic Beta Cells. PloS One (2012) 7:e38005. doi: 10.1371/journal.pone.0038005
49. Almaca J, Molina J, Menegaz D, Pronin AN, Tamayo A, Slepak V, et al. Human Beta Cells Produce and Release Serotonin to Inhibit Glucagon Secretion From Alpha Cells. Cell Rep (2016) 17:3281–91. doi: 10.1016/j.celrep.2016.11.072
50. Bennet H, Mollet IG, Balhuizen A, Medina A, Nagorny C, Bagge A, et al. Serotonin (5-HT) Receptor 2b Activation Augments Glucose-Stimulated Insulin Secretion in Human and Mouse Islets of Langerhans. Diabetologia (2016) 59:744–54. doi: 10.1007/s00125-015-3847-6
51. Chang HY, Chen SL, Shen MR, Kung ML, Chuang LM, Chen YW. Selective Serotonin Reuptake Inhibitor, Fluoxetine, Impairs E-Cadherin-Mediated Cell Adhesion and Alters Calcium Homeostasis in Pancreatic Beta Cells. Sci Rep (2017) 7:3515. doi: 10.1038/s41598-017-03747-0
52. Elmorsy E, Al-Ghafari A, Helaly ANM, Hisab AS, Oehrle B, Smith PA. Editor’s Highlight: Therapeutic Concentrations of Antidepressants Inhibit Pancreatic Beta-Cell Function via Mitochondrial Complex Inhibition. Toxicol Sci (2017) 158:286–301. doi: 10.1093/toxsci/kfx090
53. Kanno T, Suga S, Nakano K, Kamimura N, Wakui M. Corticotropin-Releasing Factor Modulation of Ca2+ Influx in Rat Pancreatic Beta-Cells. Diabetes (1999) 48:1741–6. doi: 10.2337/diabetes.48.9.1741
54. Huising MO, van der Meulen T, Vaughan JM, Matsumoto M, Donaldson CJ, Park H, et al. CRFR1 Is Expressed on Pancreatic Beta Cells, Promotes Beta Cell Proliferation, and Potentiates Insulin Secretion in a Glucose-Dependent Manner. Proc Natl Acad Sci USA (2010) 107:912–7. doi: 10.1073/pnas.0913610107
55. Schmid J, Ludwig B, Schally AV, Steffen A, Ziegler CG, Block NL, et al. Modulation of Pancreatic Islets-Stress Axis by Hypothalamic Releasing Hormones and 11beta-Hydroxysteroid Dehydrogenase. Proc Natl Acad Sci USA (2011) 108:13722–7.
56. Amisten S, Salehi A, Rorsman P, Jones PM, Persaud SJ. An Atlas and Functional Analysis of G-Protein Coupled Receptors in Human Islets of Langerhans. Pharmacol Ther (2013) 139:359–91. doi: 10.1016/j.pharmthera.2013.05.004
57. Simpson SJS, Smith LIF, Jones PM, Bowe JE. UCN2: A New Candidate Influencing Pancreatic Beta-Cell Adaptations in Pregnancy. J Endocrinol (2020) 245:247–57. doi: 10.1530/JOE-19-0568
58. Li C, Chen P, Vaughan J, Lee KF, Vale W. Urocortin 3 Regulates Glucose-Stimulated Insulin Secretion and Energy Homeostasis. Proc Natl Acad Sci USA (2007) 104:4206–11. doi: 10.1073/pnas.0611641104
59. Garcia-Ocana A, Vasavada RC, Cebrian A, Reddy V, Takane KK, Lopez-Talavera JC, et al. Transgenic Overexpression of Hepatocyte Growth Factor in the Beta-Cell Markedly Improves Islet Function and Islet Transplant Outcomes in Mice. Diabetes (2001) 50:2752–62. doi: 10.2337/diabetes.50.12.2752
60. Baffy G, Yang L, Michalopoulos GK, Williamson JR. Hepatocyte Growth Factor Induces Calcium Mobilization and Inositol Phosphate Production in Rat Hepatocytes. J Cell Physiol (1992) 153:332–9. doi: 10.1002/jcp.1041530213
61. Yan J, Aldrich RW. BK Potassium Channel Modulation by Leucine-Rich Repeat-Containing Proteins. Proc Natl Acad Sci USA (2012) 109:7917–22. doi: 10.1073/pnas.1205435109
62. Zhang J, Yan J. Regulation of BK Channels by Auxiliary Gamma Subunits. Front Physiol (2014) 5:401. doi: 10.3389/fphys.2014.00401
63. Rieck S, Kaestner KH. Expansion of Beta-Cell Mass in Response to Pregnancy. Trends Endocrinol Metab (2010) 21:151–8. doi: 10.1016/j.tem.2009.11.001
64. Rutter GA, Tsuboi T, Ravier MA. Ca2+ Microdomains and the Control of Insulin Secretion. Cell Calcium (2006) 40:539–51. doi: 10.1016/j.ceca.2006.08.015
65. Ravier MA, Cheng-Xue R, Palmer AE, Henquin JC, Gilon P. Subplasmalemmal Ca(2+) Measurements in Mouse Pancreatic Beta Cells Support the Existence of an Amplifying Effect of Glucose on Insulin Secretion. Diabetologia (2010) 53:1947–57. doi: 10.1007/s00125-010-1775-z
66. Yang SN, Larsson O, Branstrom R, Bertorello AM, Leibiger B, Leibiger IB, et al. Syntaxin 1 Interacts With the L(D) Subtype of Voltage-Gated Ca(2+) Channels in Pancreatic Beta Cells. Proc Natl Acad Sci USA (1999) 96:10164–9. doi: 10.1073/pnas.96.18.10164
67. Ji J, Yang SN, Huang X, Li X, Sheu L, Diamant N, et al. Modulation of L-Type Ca(2+) Channels by Distinct Domains Within SNAP-25. Diabetes (2002) 51:1425–36. doi: 10.2337/diabetes.51.5.1425
68. Nagamatsu S, Nakamichi Y, Yamamura C, Matsushima S, Watanabe T, Ozawa S, et al. Decreased Expression of T-SNARE, Syntaxin 1, and SNAP-25 in Pancreatic Beta-Cells Is Involved in Impaired Insulin Secretion From Diabetic GK Rat Islets: Restoration of Decreased T-SNARE Proteins Improves Impaired Insulin Secretion. Diabetes (1999) 48:2367–73. doi: 10.2337/diabetes.48.12.2367
69. Zhuang GQ, Wu W, Liu F, Ma JL, Luo YX, Xiao ZX, et al. SNAP-25(1-180) Enhances Insulin Secretion by Blocking Kv2.1 Channels in Rat Pancreatic Islet Beta-Cells. Biochem Biophys Res Commun (2009) 379:812–6. doi: 10.1016/j.bbrc.2008.12.059
70. Cunha DA, Amaral ME, Carvalho CP, Collares-Buzato CB, Carneiro EM, Boschero AC. Increased Expression of SNARE Proteins and Synaptotagmin IV in Islets From Pregnant Rats and In Vitro Prolactin-Treated Neonatal Islets. Biol Res (2006) 39:555–66. doi: 10.4067/S0716-97602006000300016
71. Dolai S, Xie L, Zhu D, Liang T, Qin T, Xie H, et al. Synaptotagmin-7 Functions to Replenish Insulin Granules for Exocytosis in Human Islet Beta-Cells. Diabetes (2016) 65:1962–76. doi: 10.2337/db15-1436
72. Iezzi M, Eliasson L, Fukuda M, Wollheim CB. Adenovirus-Mediated Silencing of Synaptotagmin 9 Inhibits Ca2+-Dependent Insulin Secretion in Islets. FEBS Lett (2005) 579:5241–6. doi: 10.1016/j.febslet.2005.08.047
73. Iezzi M, Kouri G, Fukuda M, Wollheim CB, Synaptotagmin V. And Ix Isoforms Control Ca2+ -Dependent Insulin Exocytosis. J Cell Sci (2004) 117:3119–27. doi: 10.1242/jcs.01179
74. Houamed KM, Sweet IR, Satin LS. BK Channels Mediate a Novel Ionic Mechanism That Regulates Glucose-Dependent Electrical Activity and Insulin Secretion in Mouse Pancreatic Beta-Cells. J Physiol (2010) 588:3511–23. doi: 10.1113/jphysiol.2009.184341
75. Rorsman P, Braun M. Regulation of Insulin Secretion in Human Pancreatic Islets. Annu Rev Physiol (2013) 75:155–79. doi: 10.1146/annurev-physiol-030212-183754
76. Marin BK, de Lima Reis SR, de Fatima Silva Ramalho A, Lemes SF, Marin L, Vanzela EC, et al. Protein Restriction in Early Life Increases Intracellular Calcium and Insulin Secretion, But Does Not Alter Expression of SNARE Proteins During Pregnancy. Exp Physiol (2019) 104:1029–37. doi: 10.1113/EP087045
77. Vanzela EC, Ribeiro RA, de Oliveira CA, Rodrigues FB, Bonfleur ML, Carneiro EM, et al. Pregnancy Restores Insulin Secretion From Pancreatic Islets in Cafeteria Diet-Induced Obese Rats. Am J Physiol Regul Integr Comp Physiol (2010) 298:R320–8. doi: 10.1152/ajpregu.00256.2009
78. Huang C, Walker EM, Dadi PK, Hu R, Xu Y, Zhang W, et al. Synaptotagmin 4 Regulates Pancreatic Beta Cell Maturation by Modulating the Ca(2+) Sensitivity of Insulin Secretion Vesicles. Dev Cell (2018) 45:347–61.e5. doi: 10.1016/j.devcel.2018.03.013
79. Chen L, Koh DS, Hille B. Dynamics of Calcium Clearance in Mouse Pancreatic Beta-Cells. Diabetes (2003) 52:1723–31. doi: 10.2337/diabetes.52.7.1723
80. Johnson JD, Han Z, Otani K, Ye H, Zhang Y, Wu H, et al. Ryr2 and Calpain-10 Delineate a Novel Apoptosis Pathway in Pancreatic Islets. J Biol Chem (2004) 279:24794–802. doi: 10.1074/jbc.M401216200
81. Lee B, Jonas JC, Weir GC, Laychock SG. Glucose Regulates Expression of Inositol 1,4,5-Trisphosphate Receptor Isoforms in Isolated Rat Pancreatic Islets. Endocrinology (1999) 140:2173–82. doi: 10.1210/endo.140.5.6738
82. Luciani DS, Gwiazda KS, Yang TL, Kalynyak TB, Bychkivska Y, Frey MH, et al. Roles of IP3R and Ryr Ca2+ Channels in Endoplasmic Reticulum Stress and Beta-Cell Death. Diabetes (2009) 58:422–32. doi: 10.2337/db07-1762
83. Hara T, Mahadevan J, Kanekura K, Hara M, Lu S, Urano F. Calcium Efflux From the Endoplasmic Reticulum Leads to Beta-Cell Death. Endocrinology (2014) 155:758–68. doi: 10.1210/en.2013-1519
84. Anhe GF, Nogueira TC, Nicoletti-Carvalho JE, Lellis-Santos C, Barbosa HC, Cipolla-Neto J, et al. Signal Transducer and Activator of Transcription 3-Regulated Sarcoendoplasmic Reticulum Ca2+-Atpase 2 Expression by Prolactin and Glucocorticoids Is Involved in the Adaptation of Insulin Secretory Response During the Peripartum Period. J Endocrinol (2007) 195:17–27. doi: 10.1677/JOE-07-0010
85. Crepin A, Bidaux G, Vanden-Abeele F, Dewailly E, Goffin V, Prevarskaya N, et al. Prolactin Stimulates Prostate Cell Proliferation by Increasing Endoplasmic Reticulum Content Due to SERCA 2b Over-Expression. Biochem J (2007) 401:49–55. doi: 10.1042/BJ20060870
86. Harding HP, Zeng H, Zhang Y, Jungries R, Chung P, Plesken H, et al. Diabetes Mellitus and Exocrine Pancreatic Dysfunction in Perk-/- Mice Reveals a Role for Translational Control in Secretory Cell Survival. Mol Cell (2001) 7:1153–63. doi: 10.1016/S1097-2765(01)00264-7
87. Cardozo AK, Ortis F, Storling J, Feng YM, Rasschaert J, Tonnesen M, et al. Cytokines Downregulate the Sarcoendoplasmic Reticulum Pump Ca2+ Atpase 2b and Deplete Endoplasmic Reticulum Ca2+, Leading to Induction of Endoplasmic Reticulum Stress in Pancreatic Beta-Cells. Diabetes (2005) 54:452–61. doi: 10.2337/diabetes.54.2.452
88. Wang R, McGrath BC, Kopp RF, Roe MW, Tang X, Chen G, et al. Insulin Secretion and Ca2+ Dynamics in Beta-Cells Are Regulated by PERK (EIF2AK3) in Concert With Calcineurin. J Biol Chem (2013) 288:33824–36. doi: 10.1074/jbc.M113.503664
89. Sabourin J, Le Gal L, Saurwein L, Haefliger JA, Raddatz E, Allagnat F. Store-Operated Ca2+ Entry Mediated by Orai1 and TRPC1 Participates to Insulin Secretion in Rat Beta-Cells. J Biol Chem (2015) 290:30530–9. doi: 10.1074/jbc.M115.682583
90. Bromati CR, Lellis-Santos C, Yamanaka TS, Nogueira TC, Leonelli M, Caperuto LC, et al. UPR Induces Transient Burst of Apoptosis in Islets of Early Lactating Rats Through Reduced AKT Phosphorylation via ATF4/CHOP Stimulation of TRB3 Expression. Am J Physiol Regul Integr Comp Physiol (2011) 300:R92–100. doi: 10.1152/ajpregu.00169.2010
91. Tarasov AI, Griffiths EJ, Rutter GA. Regulation of ATP Production by Mitochondrial Ca(2+). Cell Calcium (2012) 52:28–35. doi: 10.1016/j.ceca.2012.03.003
92. Vishnu N, Hamilton A, Bagge A, Wernersson A, Cowan E, Barnard H, et al. Mitochondrial Clearance of Calcium Facilitated by MICU2 Controls Insulin Secretion. Mol Metab (2021) 51:101239. doi: 10.1016/j.molmet.2021.101239
93. Nita II, Hershfinkel M, Fishman D, Ozeri E, Rutter GA, Sensi SL, et al. The Mitochondrial Na+/Ca2+ Exchanger Upregulates Glucose Dependent Ca2+ Signalling Linked to Insulin Secretion. PloS One (2012) 7:e46649. doi: 10.1371/journal.pone.0046649
94. Filadi R, Greotti E, Turacchio G, Luini A, Pozzan T, Pizzo P. On the Role of Mitofusin 2 in Endoplasmic Reticulum-Mitochondria Tethering. Proc Natl Acad Sci USA (2017) 114:E2266–7. doi: 10.1073/pnas.1616040114
95. Filadi R, Theurey P, Pizzo P. The Endoplasmic Reticulum-Mitochondria Coupling in Health and Disease: Molecules, Functions and Significance. Cell Calcium (2017) 62:1–15. doi: 10.1016/j.ceca.2017.01.003
96. Vishnyakova PA, Tarasova NV, Volodina MA, Tsvirkun DV, Sukhanova IA, Kurchakova TA, et al. Gestation Age-Associated Dynamics of Mitochondrial Calcium Uniporter Subunits Expression in Feto-Maternal Complex at Term and Preterm Delivery. Sci Rep (2019) 9:5501. doi: 10.1038/s41598-019-41996-3
97. Diabetes Canada Clinical Practice Guidelines Expert C, Feig DS, Berger H, Donovan L, Godbout A, Kader T, et al. Diabetes and Pregnancy. Can J Diabetes (2018) 42 Suppl 1:S255–82. doi: 10.1016/j.jcjd.2017.10.038
98. Cheung NW, Byth K. Population Health Significance of Gestational Diabetes. Diabetes Care (2003) 26:2005–9. doi: 10.2337/diacare.26.7.2005
99. Damm P, Houshmand-Oeregaard A, Kelstrup L, Lauenborg J, Mathiesen ER, Clausen TD. Gestational Diabetes Mellitus and Long-Term Consequences for Mother and Offspring: A View From Denmark. Diabetologia (2016) 59:1396–9. doi: 10.1007/s00125-016-3985-5
100. Hjort L, Novakovic B, Grunnet LG, Maple-Brown L, Damm P, Desoye G, et al. Diabetes in Pregnancy and Epigenetic Mechanisms-How the First 9 Months From Conception Might Affect the Child’s Epigenome and Later Risk of Disease. Lancet Diabetes Endocrinol (2019) 7:796–806. doi: 10.1016/S2213-8587(19)30078-6
101. Portha B, Chavey A, Movassat J. Early-Life Origins of Type 2 Diabetes: Fetal Programming of the Beta-Cell Mass. Exp Diabetes Res (2011) 2011:105076. doi: 10.1155/2011/105076
102. Najjar L, Sutherland J, Zhou A, Hypponen E. Vitamin D and Type 1 Diabetes Risk: A Systematic Review and Meta-Analysis of Genetic Evidence. Nutrients (2021) 13(12):4260. doi: 10.3390/nu13124260
103. Liu S. The Role of Vitamin D Receptor Gene Polymorphisms in Gestational Diabetes Mellitus Susceptibility: A Meta-Analysis. Diabetol Metab Syndr (2021) 13:144. doi: 10.1186/s13098-021-00764-y
104. Zheng JS, Luan J, Sofianopoulou E, Sharp SJ, Day FR, Imamura F, et al. The Association Between Circulating 25-Hydroxyvitamin D Metabolites and Type 2 Diabetes in European Populations: A Meta-Analysis and Mendelian Randomisation Analysis. PloS Med (2020) 17:e1003394. doi: 10.1371/journal.pmed.1003394
105. Osorio-Yanez C, Qiu C, Gelaye B, Enquobahrie DA, Williams MA. Risk of Gestational Diabetes Mellitus in Relation to Maternal Dietary Calcium Intake. Public Health Nutr (2017) 20:1082–9. doi: 10.1017/S1368980016002974
106. Zemel MB. Nutritional and Endocrine Modulation of Intracellular Calcium: Implications in Obesity, Insulin Resistance and Hypertension. Mol Cell Biochem (1998) 188:129–36. doi: 10.1023/A:1006880708475
107. Squires PE, Jones PM, Younis MY, Hills CE. The Calcium-Sensing Receptor and Beta-Cell Function. Vitam Horm (2014) 95:249–67. doi: 10.1016/B978-0-12-800174-5.00010-7
108. Babinsky VN, Hannan FM, Ramracheya RD, Zhang Q, Nesbit MA, Hugill A, et al. Mutant Mice With Calcium-Sensing Receptor Activation Have Hyperglycemia That Is Rectified by Calcilytic Therapy. Endocrinology (2017) 158:2486–502. doi: 10.1210/en.2017-00111
109. Papadopoulou A, Gole E, Moutafi A, Sfikas C, Oehrl W, Samiotaki M, et al. Calcium Sensing Receptor in Pregnancies Complicated by Gestational Diabetes Mellitus. Placenta (2014) 35:632–8. doi: 10.1016/j.placenta.2014.05.001
110. Goldstein JA, Bastarache LA, Denny JC, Roden DM, Pulley JM, Aronoff DM. Calcium Channel Blockers as Drug Repurposing Candidates for Gestational Diabetes: Mining Large Scale Genomic and Electronic Health Records Data to Repurpose Medications. Pharmacol Res (2018) 130:44–51. doi: 10.1016/j.phrs.2018.02.013
111. Le Marchand SJ, Piston DW. Glucose Decouples Intracellular Ca2+ Activity From Glucagon Secretion in Mouse Pancreatic Islet Alpha-Cells. PloS One (2012) 7:e47084. doi: 10.1371/journal.pone.0047084
112. Wang Y, Gao L, Li Y, Chen H, Sun Z. Nifedipine Protects INS-1 Beta-Cell From High Glucose-Induced ER Stress and Apoptosis. Int J Mol Sci (2011) 12:7569–80. doi: 10.3390/ijms12117569
113. Ma Z, Moruzzi N, Catrina SB, Hals I, Oberholzer J, Grill V, et al. Preconditioning With Associated Blocking of Ca2+ Inflow Alleviates Hypoxia-Induced Damage to Pancreatic Beta-Cells. PloS One (2013) 8:e67498. doi: 10.1371/journal.pone.0067498
114. Xu G, Chen J, Jing G, Shalev A. Preventing Beta-Cell Loss and Diabetes With Calcium Channel Blockers. Diabetes (2012) 61:848–56. doi: 10.2337/db11-0955
115. Milajerdi A, Abbasi F, Mousavi SM, Esmaillzadeh A. Maternal Vitamin D Status and Risk of Gestational Diabetes Mellitus: A Systematic Review and Meta-Analysis of Prospective Cohort Studies. Clin Nutr (2021) 40:2576–86. doi: 10.1016/j.clnu.2021.03.037
116. Wei SQ, Qi HP, Luo ZC, Fraser WD. Maternal Vitamin D Status and Adverse Pregnancy Outcomes: A Systematic Review and Meta-Analysis. J Matern Fetal Neonatal Med (2013) 26:889–99. doi: 10.3109/14767058.2013.765849
117. Jain R, von Hurst PR, Stonehouse W, Love DR, Higgins CM, Coad J. Association of Vitamin D Receptor Gene Polymorphisms With Insulin Resistance and Response to Vitamin D. Metabolism (2012) 61:293–301. doi: 10.1016/j.metabol.2011.06.018
118. Kajikawa M, Ishida H, Fujimoto S, Mukai E, Nishimura M, Fujita J, et al. An Insulinotropic Effect of Vitamin D Analog With Increasing Intracellular Ca2+ Concentration in Pancreatic Beta-Cells Through Nongenomic Signal Transduction. Endocrinology (1999) 140:4706–12. doi: 10.1210/endo.140.10.7025
119. Norman AW, Frankel JB, Heldt AM, Grodsky GM. Vitamin D Deficiency Inhibits Pancreatic Secretion of Insulin. Science (1980) 209:823–5. doi: 10.1126/science.6250216
Keywords: pregnancy, beta-cell, calcium, insulin secretion, apoptosis, islets, endoplasmic reticulum
Citation: Pretorius M and Huang C (2022) Beta-Cell Adaptation to Pregnancy – Role of Calcium Dynamics. Front. Endocrinol. 13:853876. doi: 10.3389/fendo.2022.853876
Received: 13 January 2022; Accepted: 21 February 2022;
Published: 25 March 2022.
Edited by:
Simone Baltrusch, University Hospital Rostock, GermanyReviewed by:
Andrei I. Tarasov, Ulster University, United KingdomStephan Scherneck, Technische Universitat Braunschweig, Germany
Copyright © 2022 Pretorius and Huang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Carol Huang, Y3RodWFuZ0B1Y2FsZ2FyeS5jYQ==