
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Endocrinol. , 29 March 2022
Sec. Diabetes: Molecular Mechanisms
Volume 13 - 2022 | https://doi.org/10.3389/fendo.2022.853822
This article is part of the Research Topic Cellular Dysfunction and Cell Death in Diabetes (etio)Pathology: Novel Insights into Molecular Mechanisms and Therapeutic Targeting View all 6 articles
 Xi-Ding Yang1,2
Xi-Ding Yang1,2 Yong-Yu Yang1,3*
Yong-Yu Yang1,3*The global diabetes epidemic and its complications are increasing, thereby posing a major threat to public health. A comprehensive understanding of diabetes mellitus (DM) and its complications is necessary for the development of effective treatments. Ferroptosis is a newly identified form of programmed cell death caused by the production of reactive oxygen species and an imbalance in iron homeostasis. Increasing evidence suggests that ferroptosis plays a pivotal role in the pathogenesis of diabetes and diabetes-related complications. In this review, we summarize the potential impact and regulatory mechanisms of ferroptosis on diabetes and its complications, as well as inhibitors of ferroptosis in diabetes and diabetic complications. Therefore, understanding the regulatory mechanisms of ferroptosis and developing drugs or agents that target ferroptosis may provide new treatment strategies for patients with diabetes.
According to the International Diabetes Federation, 536.6 million adults are affected by diabetes worldwide (2021), and the total number of adults with diabetes is predicted to rise to 783.2 million by 2045 (1). The World Health Organization proposes that diabetes will be the seventh leading cause of death by 2030 (2). There are two major forms of diabetes: type 1 and type 2 diabetes mellitus (T2DM). Type 1 diabetes mellitus (T1DM) is caused by insufficient insulin secretion, and T2DM results from insulin resistance (3). Intervention studies have confirmed that hyperglycemia is a major cause of diabetic complications (4). Long-term hyperglycemia damages multiple organs, including the kidneys, cardiovascular system, nerves, bones, and eyes, ultimately resulting in severe complications (5). Major diabetic complications include diabetic kidney disease (DKD), retinopathy (DR), neuropathy (DNP), osteoporosis (DOP), and cardiomyopathy (DCM). With increasing trends in diabetes worldwide, mortality and associated costs owing to diabetes and diabetes-related complications are a major global public health concern (6). It is generally believed that intensifying glucose control reduces the risk of diabetic complications and ameliorates them (7). However, the occurrence and progression of diabetic complications cannot be prevented in some patients (8).
As diabetes progresses, persistent hyperglycemia increases the levels of inflammatory factors, advanced glycation end products (AGEs), reactive oxygen species (ROS), free fatty acids (FFAs), triacylglycerol, and diacylglycerol in the heart, retina, kidneys, and nervous system (9–12). These factors individually and/or synergistically result in cellular death, including apoptosis, autophagy, and necroptosis (9, 10, 13). Diabetes complications are believed to be a consequence of cellular death (14–16). For example, increasing evidence demonstrates that death of pancreatic B cells is the main cause of insufficient insulin secretion in diabetes (17). Similarly, cell death is associated with diabetes complications such as DKD (14), DR (18), DN (19), DCM (15), and DOP (20).
Ferroptosis is a recently recognized form of programmed cell death that is genetically, biochemically, and morphologically distinct from apoptosis, necroptosis, autophagy, and other types of cell death (21, 22). In recent years, ferroptosis has been found to be involved in diabetes and its multiple complications (23–32). Thus, pharmacological modulation of ferroptosis has emerged as a promising therapeutic strategy for diabetes and various diabetic complications. In this study, we provide an update on the contribution of ferroptosis to the pathogenesis of diabetes and its complications. Potential therapeutic drugs or compounds targeting the ferroptosis pathway for diabetic complications are also discussed.
In 2003, a new compound, erastin, was found to induce a new way of cell death (33). This mechanism of cell death can be inhibited by iron-chelating agents (34). In 2012, Dixon first named this cell death pattern as ferroptosis (35). Ferroptosis differs from apoptosis, necroptosis, and autophagy in terms of cell morphological characteristics and functions (21, 22). Ferroptosis does not exhibit the typical morphological characteristics of necrosis, such as swelling of the cytoplasm and organelles and rupture of the cell membrane (36). It also does not result in morphological changes similar to typical apoptosis, including cell shrinkage, chromatin condensation, and the formation of apoptotic bodies (37). Ferroptosis does not form a classical closed bilayer membrane structure, which is the main difference between ferroptosis and autophagy (38).
Morphologically, ferroptosis is mainly characterized by the shrinkage of mitochondria with increased membrane density, as well as the decrease or disappearance of mitochondrial cristae (39). Biochemically, ferroptosis is characterized by iron-dependent lipid peroxidation (35, 40). Cysteine deficiency and glutathione (GSH) synthesis inhibition contribute to ferroptosis (22, 41), while iron-chelating agents and lipophilic antioxidants can prevent ferroptosis (42). The pathways in the ferroptosis cascade can be roughly divided into three major categories: GSH/glutathione peroxidase 4 (Gpx4) pathway, iron metabolism, and lipid metabolism (Figure 1). Currently, the molecular mechanisms underlying ferroptosis remain elusive. To date, multiple proteins, such as voltage dependent anion channel 2/3, p53, nuclear receptor coactivator 4 (NCOA4), cysteinyl-tRNA synthetase (CARS), mitogen-activated protein kinase (MAPK), nicotinamide adenine dinucleotide phosphate oxidase (NOX), and other proteins, can positively regulate ferroptosis (43–48). In addition, some genes are involved in the negative regulation of ferroptosis, including Gpx4, nuclear factor erythroid 2-related factor 2 (Nrf2), heat shock protein beta-1(HSPB1), and system Xc- (xCT), which protect cells against ferroptosis (49–52).
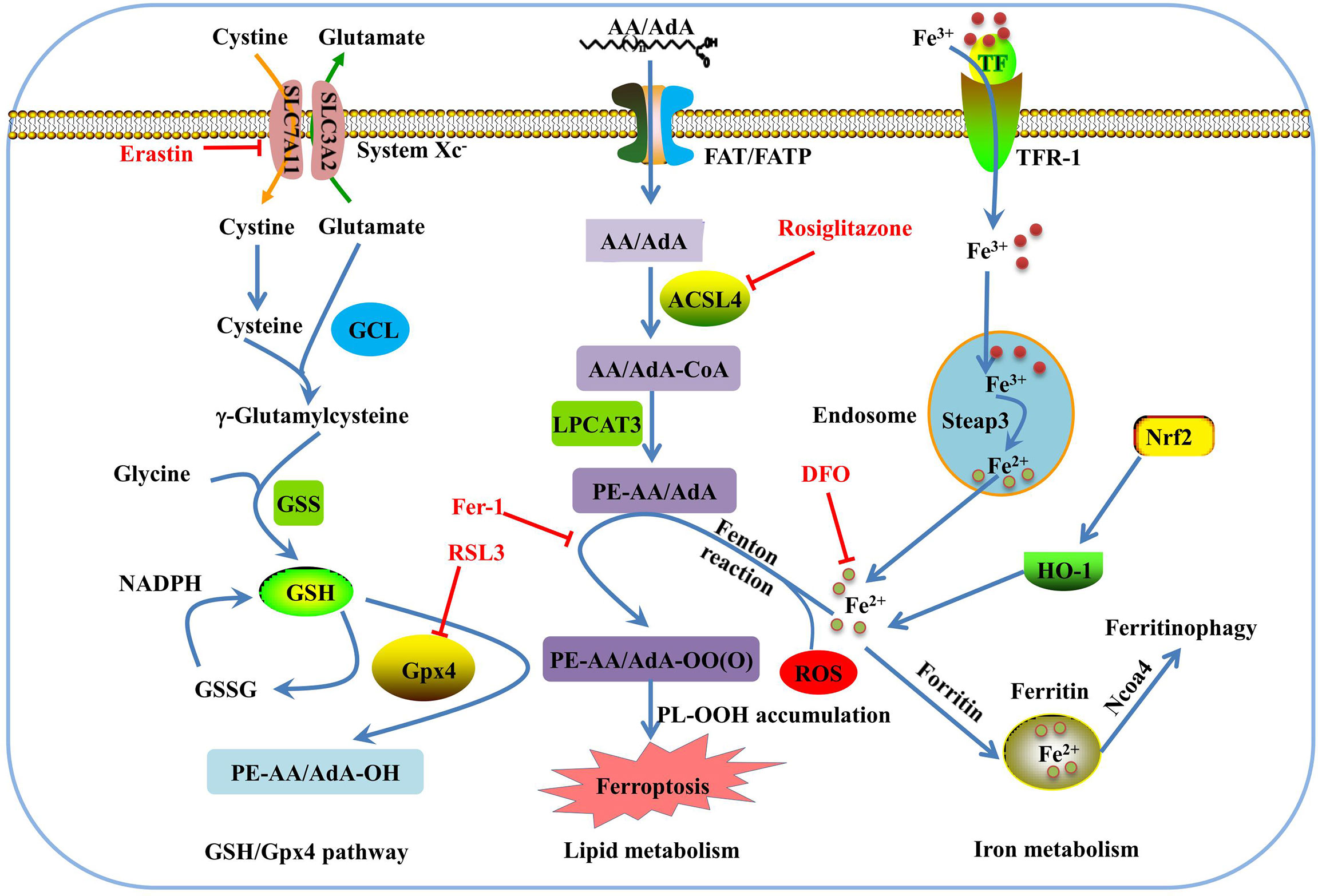
Figure 1 Overview of the ferroptosis pathway. GSH is synthesized from glutamate, cysteine, and glycine, and catalyzed by glutamate cysteine ligase (GCL) and glutathione synthetase (GSS). GPX4 converts GSH to glutathione oxidized (GSSG) to inhibit lipid ROS production. Erasin and RSL3 trigger ferroptosis by inhibiting of system Xc- and Gpx4. In lipid metabolism pathway, phosphatidylethanolamine-adrenic acid/arachidonic acid (PE-AA/AdA) is synthesized in two steps catalyzed by Acyl-CoA synthetase long-chain family member 4 (ACSL4) and lysophosphatidylcholine acyltransferase 3 (LPCAT3). Rosiglitazone is the strongest inhibitor of ACSL4 that alleviates ferroptosis. Transferrin receptor 1 (TFR-1), transferrin (TF), ferritin, and Ncoa4 are involved in iron metabolism and are closely associated with ferroptosis. Deferoxamine (DFO) and ferrostatin-1(Fer-1) inhibit ferroptosis by regulating iron level and lipid oxidation, respectively.
Ferroptosis is involved in various human diseases, such as acute kidney injury (53), cancers (54–56), Parkinson’s disease (PD) (57), and other neurodegenerative diseases (58). As a new mechanism of cell death, the discovery of ferroptosis and the mechanisms involved in its regulation may help to develop novel treatments for several diseases. Recent studies have documented significant effects of ferroptosis on the development and progression of T1DM (59); T2DM (60, 61); gestational diabetes mellitus (60–62); and diabetes complications such as DKD (27), DCM (29), DR (63, 64), diabetes-induced endothelial dysfunction (32), cognitive dysfunction (31), wound healing (65), diabetic atherosclerosis (66), and DOP (67). The molecular mechanisms of ferroptosis in diabetic complications involve multiple proteins, including acyl-CoA synthetase long chain family member 4 (ACSL4), high-mobility group box-1 (HMGB1), hypoxia-inducible factor-1α (HIF-1α), heme oxygenase-1 (HO-1), tripartite motif containing 46 (TRIM46), circ-PSEN1, NCOA4 (68), and Nox2, and negative genes, Gpx4, Nrf2, xCT, adenosine monophosphate-activated protein kinase, heat shock factor 1 (HSF1), and nutrient-deprivation autophagy factor-1 (NAF-1) (69) (Figure 2). The key molecular mechanisms underlying ferroptosis in diabetes and its complications are discussed in detail below.
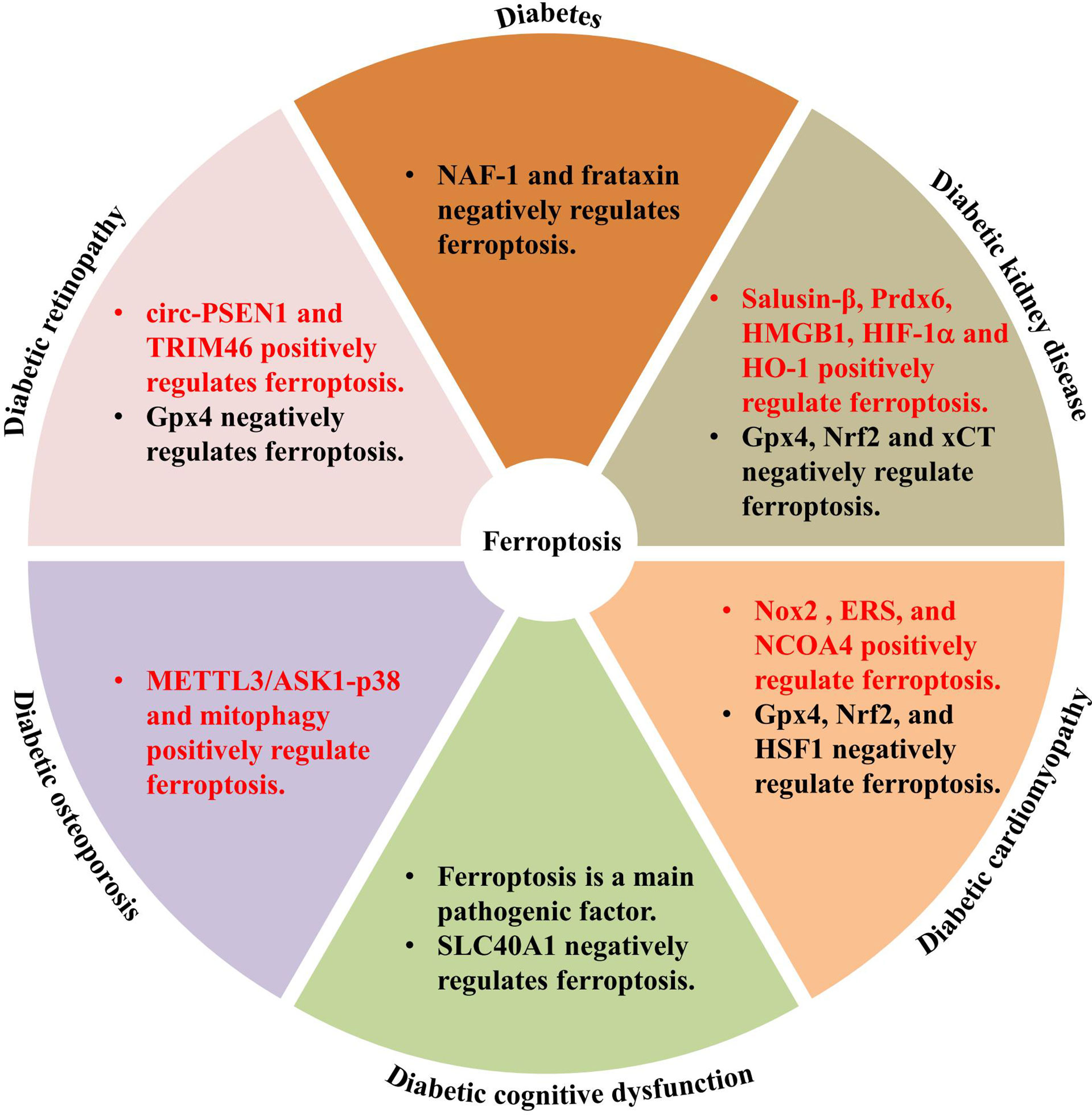
Figure 2 Genes involved in ferroptosis regulation in diabetes and diabetic complications.
Excessive iron stores have been demonstrated to be associated with the development of T2DM, and ferritin levels are increased in T2DM (70) and gestational diabetes mellitus patients (71). The activity of islet cells is closely associated with diabetes, and insufficient insulin secretion caused by pancreatic β-cell failure contributes to hyperglycemia. Increased cellular iron modulates the expression of genes involved in β-cell function and causes pancreatic β-cell dysfunction (72). Erastin, a small potent molecule capable of selectively inhibiting the Xc-cystine/glutamate antiporter required for GSH biosynthesis, induces ferroptosis (73) and affects the growth and function of human pancreatic islet-like cell clusters (74). NAF-1, a member of the [2Fe-2S] NEET protein family, regulates mitochondrial iron levels (75). Suppressed expression of NAF-1 in INS-1E pancreatic β-cells results in the appearance of ferroptosis-like features characterized by enhanced lipid peroxidation, decreased expression of Gpx4, and enhanced expression of tTfR. Fer-1 treatment of INS-1E NAF-1(−) cells significantly reduced ferroptosis and improved cell growth (69).
Acrolein and arsenic are common environmental pollutants that threaten public health (76, 77). Growing evidence has revealed that chronic arsenic exposure is a high-risk factor for T2DM (78, 79). Arsenic induced ferroptosis in vitro in a dose-dependent manner. Ferroptosis occurs in animal models of arsenic-induced pancreatic dysfunction (61). The ferroptotic effect of arsenic on mouse insulinoma cells depends on regulation of the mitochondrial ROS-autophagy-lysosomal pathway (61). In mouse pancreatic β-cell MIN6 cells, acrolein induces ferroptosis and insulin secretion dysfunction via the endoplasmic reticulum (ER) stress-related PERK pathway (77). These data indicate that ferroptosis plays an important role in maintaining homeostasis in the pancreatic islet cells.
In addition to their involvement in the regulation of islet cell death and dysfunction, ferroptosis-related proteins modulate diabetes-related metabolic phenotypes. Friedreich’s ataxia (FRDA) is a neurodegenerative disease caused by a defect in mitochondrial frataxin (FXN), a key regulator of ferroptosis, and patients with FRDA are predisposed to diabetes (80). FXN knock-in/knock-out (KIKO) mice exhibited hyperlipidemia, reduced energy expenditure and insulin sensitivity, and reduced thermogenic activities of brown adipose tissue. Moreover, the mRNA levels of the iron modulators (Ncoa4 and solute carrier family 39 member 14) were upregulated in brown adipocyte precursors of KIKO mice. Increased susceptibility of brown adipocyte precursors to ferroptosis could be one of the causes of brown adipose tissue (BAT) dysfunction (81). These data suggest that ferroptosis and its related proteins are involved in disorders of glucose and lipid metabolism. However, little is known about the exact mechanisms by which iron dysmetabolism affects metabolic phenotypes in vivo, and further studies are needed.
DKD is a major complication of diabetes and is characterized by proteinuria and decreased glomerular filtration rate (82). Accumulating evidence has indicated that hyperglycemia-stimulated oxidative stress, AGE production, inflammation, and fibrosis are all involved in the pathogenesis of DKD (83–85). Iron homeostasis is essential for normal functioning of renal cells (86). In patients with DKD, the indicators of ferroptosis, including the release of serum ferritin and lactate dehydrogenase, are upregulated (24). In kidney biopsy samples from patients with DKD, xCT and Gpx4 mRNA expression was also reduced compared to that in control samples (26). A low-iron diet or iron chelator can delay the progression of DKD in diabetic rats (87). Recent studies have demonstrated that ferroptosis is involved in the development of DKD and may be a new approach to explore the progression of DKD (23–26, 87).
Animal experiments showed that iron content was increased in the kidney tissue of streptozotocin (STZ)-induced DKD mice and diabetic (db/db) DKD mice, especially in the renal tubules. The ACSL4 inhibitor rosiglitazone improves kidney function in DKD mice and reduces the lipid peroxidation product and iron content, and these effects are correlated with attenuated ferroptosis (23). Renal tubular injury is a critical factor in DKD. High glucose (HG) triggered iron overload, antioxidant capability reduction, massive ROS production, and lipid peroxidation in renal tubular cell death (23, 88). Salusin−β, a bioactive peptide of 20 amino acids, participates in HG−induced HK−2 cell ferroptosis in an Nrf−2−dependent manner (88). Feng et al. found that ferroptosis might damage renal tubules in diabetic models via the HIF-1α/HO-1 pathway (25). Another study revealed that inhibition of ferroptosis by upregulating Nrf2 could delay the progression of DKD (27). Transforming growth factor-β1 (TGF-β1) is an important factor that leads to renal fibrosis, and renal tubular cell death induced by TGF-β1 is known to be involved in DKD (89). GSH concentration and xCT and Gpx4 expression were significantly decreased, and lipid peroxidation was enhanced in TGF-β1-stimulated renal tubular cells. These changes were significantly ameliorated by Fer-1 (26).
Mesangial cells, specialized smooth muscle cells, are distributed between the capillary loops of glomerular capillaries, and their injury is a basic pathological change in DN (90). High mobility group box 1 (HMGB1) has been reported to be a damage-associated molecular pattern molecule, and its levels are elevated in patients with DKD. Further results identified HMGB1 as having a regulatory role in the ferroptosis of mesangial cells. Suppression of HMGB1 restores cellular proliferation, prevents ROS and lactate dehydrogenase (LDH) generation, decreases ACSL4, and increases Gpx4 levels in mesangial cells (24). Ferroptosis is also involved in podocyte injury in diabetic conditions. Podocytes are an important component of the glomerular filtration barrier (GFB), and podocyte damage is considered one of the main mechanisms leading to GFB injury (91). An in vitro study demonstrated that HG triggered ferroptosis in mouse glomerular podocyte MPCs, and these effects were mediated by peroxiredoxin 6 (Prdx6), a new member of antioxidant enzymes (92). Taken together, the above studies demonstrate that ferroptosis is closely related to the pathogenesis of DKD and promotes DKD through a variety of mechanisms in podocytes, mesangial cells, and tubule cells. An in-depth study of the pathological mechanism of ferroptosis would help in the timely and reasonable prevention and treatment of DKD by targeted ferroptosis.
DCM, characterized by diastolic and systolic dysfunction, left ventricular hypertrophy, myocyte hypertrophy, and fibrosis (93), is a major cause of heart failure in patients with diabetes. Increasing evidence suggests that ferroptosis is involved in the pathogenesis of cardiomyocyte injury (28, 29). Wang et al. first found that diabetes can cause more severe myocardial ischemia-reperfusion injury (I/RI) by promoting ferroptosis in myocardial cells, as well as two other forms of programmed cell death, including apoptosis and pyroptosis. However, it is not known which type of cell death has the dominant function in DCM (28). However, what is certain is that cardiomyocytes of diabetic myocardial I/RI rats were injured accompanied by increased ferroptosis level. Inhibition of ferroptosis by Fer can alleviate this injury. ERS aggravates hypoxia/reoxygenation (H/R)-induced cardiomyocyte injury and inhibition of ERS alleviates ferroptosis (29). Ferritinophagy is a newly identified selective autophagy pathway that is mediated by NCOA4. NCOA4 mediates ferritin degradation in the autophagosome and causes the release of iron ions from ferritin, resulting in ferroptosis (94). Knockdown of NCOA4 reduced ferritinophagy and ferroptosis in a high-glucose hypoxia reoxygenation model (68).
Excessive saturated fatty acids, such as palmitic acid (PA), resulting from lipid metabolism in patients with diabetes, have been shown to play a role in cardiomyocyte death and in the development of DCM (95). It is widely accepted that apoptosis and necroptosis contribute greatly to PA-induced myocardial injury (96–98). Ferroptosis is also known to be involved in PA-induced myocardial injury. Ferroptosis inhibitors significantly reduced cell death in H9c2 cardiomyoblasts exposed to PA (30). HSF1 is an important defender of ferroptosis in cardiomyocytes. HSF1 negatively regulates PA-induced cell death, and HSF1-/- mice treated with PA exhibited more serious ferroptosis by decreasing SLC40A1, FTH1, and Gpx4 expression and increasing TFRC expression in the heart (30). Nrf2, a transcription factor controlling the expression of many ferroptosis-related genes such as Gpx4, is generally considered to have an inhibitory effect on ferroptosis (99). An interesting study demonstrated that Nrf2 has detrimental effects on the heart by promoting ferroptosis during myocardial autophagy deficiency (100). However, targeting Nrf2 and its related targets remains a viable approach to prevent or treat DCM by regulating ferroptosis. In summary, these findings suggest that ferroptosis is involved in the pathogenesis of DCM. At present, the mechanism of ferroptosis in DCM remains poorly understood, and areas related to ferroptosis and cardiomyocyte function need to be further explored.
Diabetes is a risk factor for neurodegenerative disorders such as Alzheimer’s disease (AD), PD, Huntington’s disease, amyotrophic lateral sclerosis, and FRDA (101). Ferroptosis is involved in the development of neurodegenerative diseases (102). In some neurodegenerative diseases, iron is redistributed in specific regions of the central and peripheral nervous systems; for example, iron content is significantly increased in the hypothalamus of patients with AD (103) and in the dopaminergic neurons of the substantia nigra in PD (104). AD is characterized by a loss of neurons in the frontal lobe, amygdala, and hippocampus. Hao et al. (31) reported that ferroptosis is the main pathogenic factor in diabetes-induced cognitive dysfunction. Among the ferroptosis signaling pathway genes, the SLC40A1 gene (ferroportin) was significantly downregulated in the diabetic rat hippocampus (31). Hyperglycemia or hypoglycemia can cause nerve cell injury, resulting in cognitive dysfunction. Another study showed that ferroptosis occurs in the hippocampus of db/db mice, and liraglutide, a drug approved for the treatment of obesity and diabetes, reduces ferroptosis by suppressing oxidative stress and iron overload, improving hippocampal neuronal and synaptic plasticity, thereby restoring cognitive impairment (105). In diabetic rats, but not in control rats, an iron chelator, deferoxamine, improved sensorimotor and cognitive outcomes of rats subjected to thromboembolic middle cerebral artery occlusion (106). These data indicate that the inhibition of ferroptosis may prevent brain injury and provide a novel disease-modifying therapeutic strategy for the prevention of cognitive impairment in diabetes.
DR is one of the most devastating complications of diabetes, affecting millions of working-age adults worldwide (107). DR is a leading cause of global vision loss. Retinal microvasculopathy, inflammation, and retinal neurodegeneration are the major causes of DR. Hyperglycemia causes pericyte damage, endothelial cell dysfunction, and basement membrane thickening in retinal vessels, leading to disruption of the blood-retinal barrier (108). Dysfunction and increased permeability of the retinal capillary endothelial cells (RCECs) are essential features of DR progression. HG inhibits human RCEC growth and induces ferroptosis, which can be reversed by Fer-1. TRIM46 mediated Gpx4 ubiquitination pathway is involved in HG-induced ferroptosis in human RCECs (63). In addition to microvascular changes, retinal neurodegeneration or loss of the retinal pigment epithelium (RPE) also contribute to diabetic retinal damage early in DR, and HG has detrimental effects on RPE cells by producing ROS and decreasing the Gpxs expression (109). An in vitro study in ARPE-19 cells showed that circular RNAs (circRNAs) play a regulatory role in HG-induced ferroptosis via competing endogenous RNAs to regulate target microRNAs (miRNAs). Circ-PSNE1 was significantly upregulated in DR patients and in HG-treated ARPE-19 cells, and downregulation of circ-PSEN1 reduced cell death, decreased MDA and Fe2+ content, and increased the expression of Gpx4 and solute carrier family 7 member 11 (SLC7A11). Furthermore, Zhu, et al. demonstrated that circ-PSEN1 mitigates HG-induced ferroptosis via the miR-200b-3p/cofilin-2 axis (64). These studies suggest that ferroptosis plays an important role in damage to RCECs and retinal pigment epithelium under hyperglycemic conditions. However, whether ferroptosis is involved in HG-triggered pericyte loss remains unclear and requires further investigation.
Recently, osteoporosis has been recognized as a complication of diabetes, and DOP has a serious impact on the quality of life of the elderly. A decrease in bone mineral density (BMD) is a characteristic of patients with T1DM. Conversely, most studies have demonstrated that BMD does not decrease in patients with T2DM (110). Although seemingly paradoxical, the fracture risks in patients with T1DM and T2DM are similar (111). Although BMD is variable in T1DM and T2DM, prolonged hyperglycemia promotes changes in bone metabolism and destroys bone micro-architecture through multiple mechanisms, such as production of AGEs, inflammation, calcium and phosphorus metabolism disorders, and oxidative stress (112). DOP has also been reported to be associated with ferroptosis, and iron overload can promote osteoclast differentiation and bone resorption through ROS production (113). HG induces ferroptosis, and Fer-1 inhibits the downregulation of the expression of osteogenesis-associated proteins, osteoprotegerin (OPG) and osteocalcin (OCN), in MC3T3-E1 cells, suggesting that ferroptosis is involved in osteogenic ability in the presence of HG (20). Recent studies have shown that Fer-1 and DFO block ferroptosis induced by HG and palmitic acid (HGPA) in MC3T3-E1 cells. The mechanism of high glucose and high fat (HGHF)-induced ferroptosis in osteoblasts is related to the activation of the METTL3/ASK1-p38 signaling pathway (114). Morphological changes of mitochondrial are the important hallmark of ferroptosis, and studies have shown that mitochondria play an important role in the regulation of ferroptosis via regulation of energetic metabolism, iron metabolism, and other pathways (115, 116). Mitochondrial ferritin (FtMt), an iron-storage protein, can intercept excessive iron ions to decrease ROS levels in mice and is involved in osteoblastic ferroptosis in type 2 DOP. Overexpression of FtMt reduced osteoblastic ferroptosis and improved osteoblast function under HG conditions, whereas knockdown of FtMt decreased osteogenic function and induced mitophagy through the ROS/PINK1/Parkin pathway (67). These studies indicate that the occurrence of T2DOP is correlated with iron homeostasis imbalance and ferroptosis in osteoblasts. However, the detailed mechanism requires further investigation.
With an in-depth study on the mechanism of ferroptosis, many specific inhibitors of ferroptosis have been identified, such as Fer-1, deferoxamine, liproxstatin-1, mitoquinone, vitamin E, and zileuton (38, 49). Ferroptosis inhibitors in diabetes and its complications have also been well studied. Some compounds that specifically inhibit iron death have not yet entered clinical stages. However, studies have shown that some drugs in the market inhibit ferroptosis and are beneficial for diabetes and its complications. Rosiglitazone is the strongest inhibitor of ACSL4 (117) and ACSL4 is an essential component of ferroptosis (118). Rosiglitazone ameliorates DKD by maintaining kidney function and inhibiting the production of pro-inflammatory cytokines by inhibiting ACSL4 and blocking renal tubular cell ferroptosis (23). Fenofibrate is a third-generation fibric acid derivative widely used to treat patients with atherogenic dyslipidemia (119). Many studies have shown that fenofibrate can improve diabetes complications, but this effect does not depend on its ability to improve lipid levels (120). Fenofibrate delays the progression of DKD by inhibiting diabetes-related ferroptosis via upregulation of Nrf2 (120). Metformin is a commonly used anti-diabetic agent (121). Metformin attenuates hyperlipidemia/PA-induced vascular calcification through anti-ferroptosis effects (122). It is possible that metformin may also exert its anti-diabetic effects through this pathway, but further studies are needed to confirm this hypothesis.
Some compounds from natural products have been developed to protect cells against ferroptosis. Quercetin is one of the most widely distributed natural polyphenolic flavonoids in the plant kingdom (123). It has been reported to have potential anti-diabetic effects both in vivo and in vitro (124, 125). Epidemiological investigations have demonstrated that quercetin can reduce the risk of T2DM (124–127). Li et al. (60) investigated the mechanisms by which quercetin protects against T2DM and found that quercetin exerts beneficial effects on T2DM by inhibiting pancreatic iron deposition and pancreatic beta cell ferroptosis. Resveratrol, a natural polyphenol, inhibits acrolein-induced ferroptosis in MIN6 cells (77). Germacrone, one of the main bioactive components extracted from Curcuma zedoaria Roscoe, has been reported to have numerous pharmacological activities, including anti-inflammatory, anti-tumor, and anti-fibrotic effects (128, 129). Jin et al. found that germacrone combined with exogenous mmu_circRNA_0000309 facilitated DKD treatment by inactivating ferroptosis-dependent mitochondrial injury and podocyte apoptosis (130). Cryptochlorogenic acid was the main active component in the mulberry leaf extract. The extracts of mulberry leaf have been confirmed to improve inflammation and insulin resistance (131). Cryptochlorogenic acid exerts excellent anti-diabetic effects by inhibiting ferroptosis by activating cystine/xCT/Gpx4/Nrf2 and inhibiting NCOA4 in diabetes (132). These natural products have potential anti-ferroptosis properties; however, the targets of these compounds need to be further verified. Furthermore, randomized clinical trials of these anti-ferroptotic compounds for diabetic complications need to be conducted.
In this review, we summarize the role and possible mechanism of ferroptosis in diabetes and its complications. To date, studies have demonstrated that ferroptosis plays an important role in diabetes and its complications, and drugs or agents that target ferroptosis may provide new treatment strategies for patients with diabetes. Morphologically, ferroptotis-experiencing cells, including HK-2, MC3T3, and hFOB 1.19 cells, exhibited reduced mitochondrial volume and cristae, and increased mitochondrial membrane density in diabetes (Table 1). These changes are consistent with the mitochondrial phenotypic changes induced by erastin. Moreover, HG and other risk factors (FFA or H/R) could induce ferroptosis in target cells through the following three categories: iron metabolism, antioxidant system, and lipid oxidation pathway. These defects may predispose cells to ferroptosis in diabetes (Figure 3).
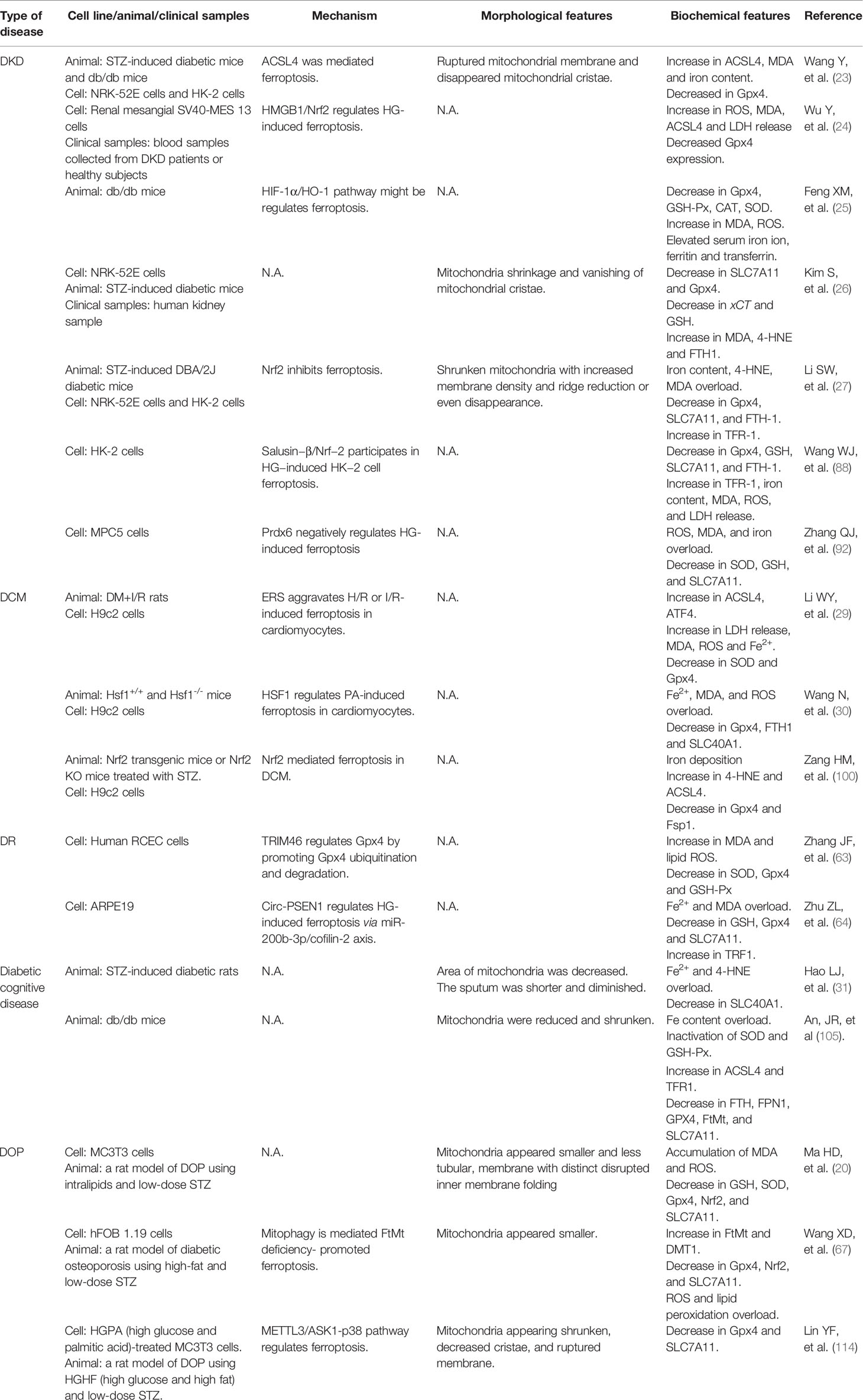
Table 1 The main regulatory mechanism, morphological and biochemical feature of ferroptosis in diabetes and related complications.
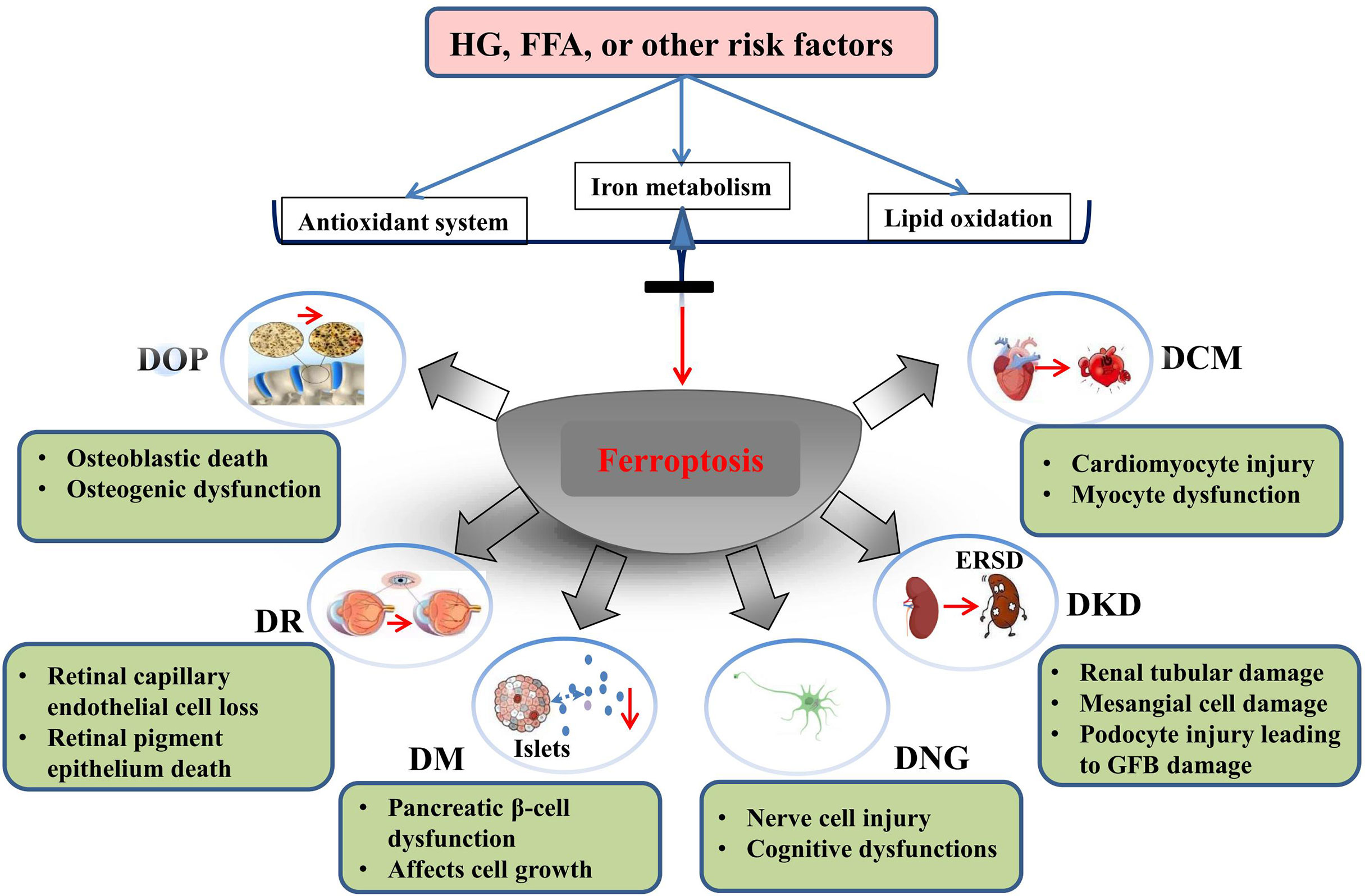
Figure 3 Ferroptosis is involved in the development of diabetes and related complications. HG and other risk factors (FFA or H/R) could induce ferroptosis by interfering with the balance of ion metabolism, reduction system, and lipid oxidation. Ferroptosis is involved in cell death of target organ and dysfunction in diabetes and its metabolic diseases.
Further studies are needed to understand the regulatory mechanisms of ferroptosis in diabetes and its related complications. At present, there is a lack of knowledge regarding potential biomarkers that can be used to specifically diagnose ferroptosis in clinical settings. Although proteins related to iron metabolism, such as ferritin, are elevated in the serum of patients with diabetes and related complications, their specificity cannot meet the requirements of clinical diagnosis. Therefore, identification of biomarkers for ferroptosis will benefit the treatment of diabetes and its complications. Techniques for detecting ferroptosis in vivo should be developed in the future. In addition, the identification and development of specific inhibitors for ferroptosis is crucial, and much work remains to be done before specific inhibitors can be used clinically. In some cases, at least two or more types of programmed cell death may occur because of diabetic complications. Therefore, targeting of apoptosis, autophagy, necrosis, or ferroptosis alone may not achieve the desired effects, and multi-target therapeutics may be effective for treating complex diseases in the future.
X-DY wrote the manuscript. Y-YY contributions to design of the work and revised the work. All authors contributed to the article and approved the submitted version.
This study was supported by the National Natural Science Foundation (No. 81603171), Hunan Provincial Natural Scientific Foundation (No. 2018JJ3743), and Hunan Traditional Chinese Medicine Science and Technology Project (No. 2021061).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Sun H, Saeedi P, Karuranga S, Pinkepank M, Ogurtsova K, Duncan BB, et al. IDF Diabetes Atlas: Global, Regional and Country-Level Diabetes Prevalence Estimates for 2021 and Projections for 2045. Diabetes Res Clin Pract (2022) 183:109119. doi: 10.1016/j.diabres.2021.109119
2. Emami-Riedmaier A, Schaeffeler E, Nies AT, Morike K, Schwab M. Stratified Medicine for the Use of Antidiabetic Medication in Treatment of Type II Diabetes and Cancer: Where do We Go From Here? J Intern Med (2015) 277:235–47. doi: 10.1111/joim.12330
3. Antonelli A, Ferrari SM, Giuggioli D, Di Domenicantonio A, Ruffilli I, Corrado A, et al. Hepatitis C Virus Infection and Type 1 and Type 2 Diabetes Mellitus. World J Diabetes (2014) 5:586–600. doi: 10.4239/wjd.v5.i5.586
4. Gruden G, Barutta F, Kunos G, Pacher P. Role of the Endocannabinoid System in Diabetes and Diabetic Complications. Br J Pharmacol (2016) 173:1116–27. doi: 10.1111/bph.13226
5. Forbes JM, Cooper ME. Mechanisms of Diabetic Complications. Physiol Rev (2013) 93:137–88. doi: 10.1152/physrev.00045.2011
6. van Dieren S, Beulens JW, van der Schouw YT, Grobbee DE, Neal B. The Global Burden of Diabetes and its Complications: An Emerging Pandemic. Eur J Cardiovasc Prev Rehabil (2010) 17 Suppl 1:S3–8. doi: 10.1097/01.hjr.0000368191.86614.5a
7. Diabetes Control and Complications Trial Research Group, Nathan DM, Genuth S, Lachin J, Cleary P, Crofford O, et al. The Effect of Intensive Treatment of Diabetes on the Development and Progression of Long-Term Complications in Insulin-Dependent Diabetes Mellitus. N Engl J Med (1993) 329:977–86. doi: 10.1056/NEJM199309303291401
8. Eid S, Sas KM, Abcouwer SF, Feldman EL, Gardner TW, Pennathur S, et al. New Insights Into the Mechanisms of Diabetic Complications: Role of Lipids and Lipid Metabolism. Diabetologia (2019) 62:1539–49. doi: 10.1007/s00125-019-4959-1
9. Volpe CMO, Villar-Delfino PH, Dos Anjos PMF, Nogueira-Machado JA. Cellular Death, Reactive Oxygen Species (ROS) and Diabetic Complications. Cell Death Dis (2018) 9:119. doi: 10.1038/s41419-017-0135-z
10. Kang Q, Yang C. Oxidative Stress and Diabetic Retinopathy: Molecular Mechanisms, Pathogenetic Role and Therapeutic Implications. Redox Biol (2020) 37:101799. doi: 10.1016/j.redox.2020.101799
11. Opazo-Rios L, Mas S, Marin-Royo G, Mezzano S, Gomez-Guerrero C, Moreno JA, et al. Lipotoxicity and Diabetic Nephropathy: Novel Mechanistic Insights and Therapeutic Opportunities. Int J Mol Sci (2020) 21:2632. doi: 10.3390/ijms21072632
12. Borradaile NM, Schaffer JE. Lipotoxicity in the Heart. Curr Hypertens Rep (2005) 7:412–7. doi: 10.1007/s11906-005-0035-y
13. Clarke M, Dodson PM. PKC Inhibition and Diabetic Microvascular Complications. Best Pract Res Clin Endocrinol Metab (2007) 21:573–86. doi: 10.1016/j.beem.2007.09.007
14. Balasescu E, Ion DA, Cioplea M, Zurac S. Caspases, Cell Death and Diabetic Nephropathy. Rom J Intern Med (2015) 53:296–303. doi: 10.1515/rjim-2015-0038
15. Cai L, Kang YJ. Cell Death and Diabetic Cardiomyopathy. Cardiovasc Toxicol (2003) 3:219–28. doi: 10.1385/ct:3:3:219
16. Khan ZA, Chakrabarti S. Cellular Signaling and Potential New Treatment Targets in Diabetic Retinopathy. Exp Diabetes Res (2007) 2007:31867. doi: 10.1155/2007/31867
17. Sramek J, Nemcova-Furstova V, Kovar J. Molecular Mechanisms of Apoptosis Induction and its Regulation by Fatty Acids in Pancreatic Beta-Cells. Int J Mol Sci (2021) 22:4285. doi: 10.3390/ijms22084285
18. Kowluru RA. Diabetic Retinopathy: Mitochondrial Dysfunction and Retinal Capillary Cell Death. Antioxid Redox Signal (2005) 7:1581–87. doi: 10.1089/ars.2005.7.1581
19. Vincent AM, Brownlee M, Russell JW. Oxidative Stress and Programmed Cell Death in Diabetic Neuropathy. Ann N Y Acad Sci (2002) 959:368–83. doi: 10.1111/j.1749-6632.2002.tb02108.x
20. Ma H, Wang X, Zhang W, Li H, Zhao W, Sun J, et al. Melatonin Suppresses Ferroptosis Induced by High Glucose via Activation of the Nrf2/HO-1 Signaling Pathway in Type 2 Diabetic Osteoporosis. Oxid Med Cell Longev (2020) 2020:9067610. doi: 10.1155/2020/9067610
21. Cao JY, Dixon SJ. Mechanisms of Ferroptosis. Cell Mol Life Sci (2016) 73:2195–209. doi: 10.1007/s00018-016-2194-1
22. Stockwell BR, Friedmann Angeli JP, Bayir H, Bush AI, Conrad M, Dixon SJ, et al. Ferroptosis: A Regulated Cell Death Nexus Linking Metabolism, Redox Biology, and Disease. Cell (2017) 171:273–85. doi: 10.1016/j.cell.2017.09.021
23. Wang Y, Bi R, Quan F, Cao Q, Lin Y, Yue C, et al. Ferroptosis Involves in Renal Tubular Cell Death in Diabetic Nephropathy. Eur J Pharmacol (2020) 888:173574. doi: 10.1016/j.ejphar.2020.173574
24. Wu Y, Zhao Y, Yang HZ, Wang YJ, Chen Y. HMGB1 Regulates Ferroptosis Through Nrf2 Pathway in Mesangial Cells in Response to High Glucose. Biosci Rep (2021) 41:BSR20202924. doi: 10.1042/BSR20202924
25. Feng X, Wang S, Sun Z, Dong H, Yu H, Huang M, et al. Ferroptosis Enhanced Diabetic Renal Tubular Injury via HIF-1alpha/HO-1 Pathway in Db/Db Mice. Front Endocrinol (Lausanne) (2021) 12:626390. doi: 10.3389/fendo.2021.626390
26. Kim S, Kang SW, Joo J, Han SH, Shin H, Nam BY, et al. Characterization of Ferroptosis in Kidney Tubular Cell Death Under Diabetic Conditions. Cell Death Dis (2021) 12:160. doi: 10.1038/s41419-021-03452-x
27. Li S, Zheng L, Zhang J, Liu X, Wu Z. Inhibition of Ferroptosis by Up-Regulating Nrf2 Delayed the Progression of Diabetic Nephropathy. Free Radic Biol Med (2021) 162:435–49. doi: 10.1016/j.freeradbiomed.2020.10.323
28. Wang C, Zhu L, Yuan W, Sun L, Xia Z, Zhang Z, et al. Diabetes Aggravates Myocardial Ischaemia Reperfusion Injury via Activating Nox2-Related Programmed Cell Death in an AMPK-Dependent Manner. J Cell Mol Med (2020) 24:6670–79. doi: 10.1111/jcmm.15318
29. Li W, Li W, Leng Y, Xiong Y, Xia Z. Ferroptosis is Involved in Diabetes Myocardial Ischemia/Reperfusion Injury Through Endoplasmic Reticulum Stress. DNA Cell Biol (2020) 39:210–25. doi: 10.1089/dna.2019.5097
30. Wang N, Ma H, Li J, Meng C, Zou J, Wang H, et al. HSF1 Functions as a Key Defender Against Palmitic Acid-Induced Ferroptosis in Cardiomyocytes. J Mol Cell Cardiol (2021) 150:65–76. doi: 10.1016/j.yjmcc.2020.10.010
31. Hao L, Mi J, Song L, Guo Y, Li Y, Yin Y, et al. SLC40A1 Mediates Ferroptosis and Cognitive Dysfunction in Type 1 Diabetes. Neuroscience (2021) 463:216–26. doi: 10.1016/j.neuroscience.2021.03.009
32. Luo EF, Li HX, Qin YH, Qiao Y, Yan GL, Yao YY, et al. Role of Ferroptosis in the Process of Diabetes-Induced Endothelial Dysfunction. World J Diabetes (2021) 12:124–37. doi: 10.4239/wjd.v12.i2.124
33. Dolma S, Lessnick SL, Hahn WC, Stockwell BR. Identification of Genotype-Selective Antitumor Agents Using Synthetic Lethal Chemical Screening in Engineered Human Tumor Cells. Cancer Cell (2003) 3:285–96. doi: 10.1016/s1535-6108(03)00050-3
34. Yang WS, Stockwell BR. Synthetic Lethal Screening Identifies Compounds Activating Iron-Dependent, Nonapoptotic Cell Death in Oncogenic-RAS-Harboring Cancer Cells. Chem Biol (2008) 15:234–45. doi: 10.1016/j.chembiol.2008.02.010
35. Dixon SJ, Lemberg KM, Lamprecht MR, Skouta R, Zaitsev EM, Gleason CE, et al. Ferroptosis: An Iron-Dependent Form of Nonapoptotic Cell Death. Cell (2012) 149:1060–72. doi: 10.1016/j.cell.2012.03.042
36. Yu H, Guo P, Xie X, Wang Y, Chen G. Ferroptosis, a New Form of Cell Death, and its Relationships With Tumourous Diseases. J Cell Mol Med (2017) 21:648–57. doi: 10.1111/jcmm.13008
37. Zhang P, Chen L, Zhao Q, Du X, Bi M, Li Y, et al. Ferroptosis was More Initial in Cell Death Caused by Iron Overload and its Underlying Mechanism in Parkinson's Disease. Free Radic Biol Med (2020) 152:227–34. doi: 10.1016/j.freeradbiomed.2020.03.015
38. Li J, Cao F, Yin HL, Huang ZJ, Lin ZT, Mao N, et al. Ferroptosis: Past, Present and Future. Cell Death Dis (2020) 11:88. doi: 10.1038/s41419-020-2298-2
39. Li Z, Chen L, Chen C, Zhou Y, Hu D, Yang J, et al. Targeting Ferroptosis in Breast Cancer. biomark Res (2020) 8:58. doi: 10.1186/s40364-020-00230-3
40. Yang WS, SriRamaratnam R, Welsch ME, Shimada K, Skouta R, Viswanathan VS, et al. Regulation of Ferroptotic Cancer Cell Death by GPX4. Cell (2014) 156:317–31. doi: 10.1016/j.cell.2013.12.010
41. Wang H, Li J, Follett PL, Zhang Y, Cotanche DA, Jensen FE, et al. 12-Lipoxygenase Plays a Key Role in Cell Death Caused by Glutathione Depletion and Arachidonic Acid in Rat Oligodendrocytes. Eur J Neurosci (2004) 20:2049–58. doi: 10.1111/j.1460-9568.2004.03650.x
42. Wang X, Wang Z, Cao J, Dong Y, Chen Y. Ferroptosis Mechanisms Involved in Hippocampal-Related Diseases. Int J Mol Sci (2021) 22:9902. doi: 10.3390/ijms22189902
43. Lemasters JJ. Evolution of Voltage-Dependent Anion Channel Function: From Molecular Sieve to Governator to Actuator of Ferroptosis. Front Oncol (2017) 7:303. doi: 10.3389/fonc.2017.00303
44. Kang R, Kroemer G, Tang D. The Tumor Suppressor Protein P53 and the Ferroptosis Network. Free Radic Biol Med (2019) 133:162–68. doi: 10.1016/j.freeradbiomed.2018.05.074
45. Santana-Codina N, Mancias JD. The Role of NCOA4-Mediated Ferritinophagy in Health and Disease. Pharm (Basel) (2018) 11:114. doi: 10.3390/ph11040114
46. Hayano M, Yang WS, Corn CK, Pagano NC, Stockwell BR. Loss of cysteinyl-tRNA Synthetase (CARS) Induces the Transsulfuration Pathway and Inhibits Ferroptosis Induced by Cystine Deprivation. Cell Death Differ (2016) 23:270–8. doi: 10.1038/cdd.2015.93
47. Gao H, Bai Y, Jia Y, Zhao Y, Kang R, Tang D, et al. Ferroptosis is a Lysosomal Cell Death Process. Biochem Biophys Res Commun (2018) 503:1550–56. doi: 10.1016/j.bbrc.2018.07.078
48. Kuang F, Liu J, Tang D, Kang R. Oxidative Damage and Antioxidant Defense in Ferroptosis. Front Cell Dev Biol (2020) 8:586578. doi: 10.3389/fcell.2020.586578
49. Ju J, Song YN, Wang K. Mechanism of Ferroptosis: A Potential Target for Cardiovascular Diseases Treatment. Aging Dis (2021) 12:261–76. doi: 10.14336/AD.2020.0323
50. Seibt TM, Proneth B, Conrad M. Role of GPX4 in Ferroptosis and its Pharmacological Implication. Free Radic Biol Med (2019) 133:144–52. doi: 10.1016/j.freeradbiomed.2018.09.014
51. Sun X, Ou Z, Xie M, Kang R, Fan Y, Niu X, et al. HSPB1 as a Novel Regulator of Ferroptotic Cancer Cell Death. Oncogene (2015) 34:5617–25. doi: 10.1038/onc.2015.32
52. Dodson M, Castro-Portuguez R, Zhang DD. NRF2 Plays a Critical Role in Mitigating Lipid Peroxidation and Ferroptosis. Redox Biol (2019) 23:101107. doi: 10.1016/j.redox.2019.101107
53. Martin-Sanchez D, Ruiz-Andres O, Poveda J, Carrasco S, Cannata-Ortiz P, Sanchez-Nino MD, et al. Ferroptosis, But Not Necroptosis, is Important in Nephrotoxic Folic Acid-Induced AKI. J Am Soc Nephrol (2017) 28:218–29. doi: 10.1681/ASN.2015121376
54. Shen Z, Liu T, Li Y, Lau J, Yang Z, Fan W, et al. Fenton-Reaction-Acceleratable Magnetic Nanoparticles for Ferroptosis Therapy of Orthotopic Brain Tumors. ACS Nano (2018) 12:11355–65. doi: 10.1021/acsnano.8b06201
55. Sun X, Ou Z, Chen R, Niu X, Chen D, Kang R, et al. Activation of the P62-Keap1-NRF2 Pathway Protects Against Ferroptosis in Hepatocellular Carcinoma Cells. Hepatology (2016) 63:173–84. doi: 10.1002/hep.28251
56. Gentric G, Kieffer Y, Mieulet V, Goundiam O, Bonneau C, Nemati F, et al. PML-Regulated Mitochondrial Metabolism Enhances Chemosensitivity in Human Ovarian Cancers. Cell Metab (2019) 29:156–73 e10. doi: 10.1016/j.cmet.2018.09.002
57. Do Van B, Gouel F, Jonneaux A, Timmerman K, Gele P, Petrault M, et al. Ferroptosis, a Newly Characterized Form of Cell Death in Parkinson's Disease That is Regulated by PKC. Neurobiol Dis (2016) 94:169–78. doi: 10.1016/j.nbd.2016.05.011
58. Hambright WS, Fonseca RS, Chen L, Na R, Ran Q. Ablation of Ferroptosis Regulator Glutathione Peroxidase 4 in Forebrain Neurons Promotes Cognitive Impairment and Neurodegeneration. Redox Biol (2017) 12:8–17. doi: 10.1016/j.redox.2017.01.021
59. Krummel B, Plotz T, Jorns A, Lenzen S, Mehmeti I. The Central Role of Glutathione Peroxidase 4 in the Regulation of Ferroptosis and its Implications for Pro-Inflammatory Cytokine-Mediated Beta-Cell Death. Biochim Biophys Acta Mol Basis Dis (2021) 1867:166114. doi: 10.1016/j.bbadis.2021.166114
60. Li D, Jiang C, Mei G, Zhao Y, Chen L, Liu J, et al. Quercetin Alleviates Ferroptosis of Pancreatic Beta Cells in Type 2 Diabetes. Nutrients (2020) 12:2954. doi: 10.3390/nu12102954
61. Wei S, Qiu T, Yao X, Wang N, Jiang L, Jia X, et al. Arsenic Induces Pancreatic Dysfunction and Ferroptosis via Mitochondrial ROS-Autophagy-Lysosomal Pathway. J Hazard Mater (2020) 384:121390. doi: 10.1016/j.jhazmat.2019.121390
62. Han D, Jiang L, Gu X, Huang S, Pang J, Wu Y, et al. SIRT3 Deficiency is Resistant to Autophagy-Dependent Ferroptosis by Inhibiting the AMPK/mTOR Pathway and Promoting GPX4 Levels. J Cell Physiol (2020) 235:8839–51. doi: 10.1002/jcp.29727
63. Zhang J, Qiu Q, Wang H, Chen C, Luo D. TRIM46 Contributes to High Glucose-Induced Ferroptosis and Cell Growth Inhibition in Human Retinal Capillary Endothelial Cells by Facilitating GPX4 Ubiquitination. Exp Cell Res (2021) 407:112800. doi: 10.1016/j.yexcr.2021.112800
64. Zhu Z, Duan P, Song H, Zhou R, Chen T. Downregulation of Circular RNA PSEN1 Ameliorates Ferroptosis of the High Glucose Treated Retinal Pigment Epithelial Cells via miR-200b-3p/Cofilin-2 Axis. Bioengineered (2021) 12:12555–67. doi: 10.1080/21655979.2021.2010369
65. Li S, Li Y, Wu Z, Wu Z, Fang H. Diabetic Ferroptosis Plays an Important Role in Triggering on Inflammation in Diabetic Wound. Am J Physiol Endocrinol Metab (2021) 321:E509–E20. doi: 10.1152/ajpendo.00042.2021
66. Meng Z, Liang H, Zhao J, Gao J, Liu C, Ma X, et al. HMOX1 Upregulation Promotes Ferroptosis in Diabetic Atherosclerosis. Life Sci (2021) 284:119935. doi: 10.1016/j.lfs.2021.119935
67. Wang X, Ma H, Sun J, Zheng T, Zhao P, Li H, et al. Mitochondrial Ferritin Deficiency Promotes Osteoblastic Ferroptosis via Mitophagy in Type 2 Diabetic Osteoporosis. Biol Trace Elem Res (2022) 200:298–307. doi: 10.1007/s12011-021-02627-z
68. Li W, Li W, Wang Y, Leng Y, Xia Z. Inhibition of DNMT-1 Alleviates Ferroptosis Through NCOA4 Mediated Ferritinophagy During Diabetes Myocardial Ischemia/Reperfusion Injury. Cell Death Discov (2021) 7:267. doi: 10.1038/s41420-021-00656-0
69. Karmi O, Sohn YS, Marjault HB, Israeli T, Leibowitz G, Ioannidis K, et al. A Combined Drug Treatment That Reduces Mitochondrial Iron and Reactive Oxygen Levels Recovers Insulin Secretion in NAF-1-Deficient Pancreatic Cells. Antioxid (Basel) (2021) 10:1160. doi: 10.3390/antiox10081160
70. Liu J, Li Q, Yang Y, Ma L. Iron Metabolism and Type 2 Diabetes Mellitus: A Meta-Analysis and Systematic Review. J Diabetes Investig (2020) 11:946–55. doi: 10.1111/jdi.13216
71. Gautam S, Alam F, Moin S, Noor N, Arif SH. Role of Ferritin and Oxidative Stress Index in Gestational Diabetes Mellitus. J Diabetes Metab Disord (2021) 20:1615–19. doi: 10.1007/s40200-021-00911-2
72. Hamad M, Mohammed AK, Hachim MY, Mukhopadhy D, Khalique A, Laham A, et al. Heme Oxygenase-1 (HMOX-1) and Inhibitor of Differentiation Proteins (ID1, ID3) are Key Response Mechanisms Against Iron-Overload in Pancreatic β-Cells. Mol Cell Endocrinol (2021) 538:111462. doi: 10.1016/j.mce.2021.111462
73. Linkermann A, Skouta R, Himmerkus N, Mulay SR, Dewitz C, De Zen F, et al. Synchronized Renal Tubular Cell Death Involves Ferroptosis. Proc Natl Acad Sci USA (2014) 111:16836–41. doi: 10.1073/pnas.1415518111
74. Li XY, Leung PS. Erastin-Induced Ferroptosis is a Regulator for the Growth and Function of Human Pancreatic Islet-Like Cell Clusters. Cell Regener (2020) 9:16. doi: 10.1186/s13619-020-00055-3
75. Mittler R, Darash-Yahana M, Sohn YS, Bai F, Song L, Cabantchik IZ, et al. NEET Proteins: A New Link Between Iron Metabolism, Reactive Oxygen Species, and Cancer. Antioxid Redox Signal (2019) 30:1083–95. doi: 10.1089/ars.2018.7502
76. Hsu LI, Hsieh FI, Wang YH, Lai TS, Wu MM, Chen CJ, et al. Arsenic Exposure From Drinking Water and the Incidence of CKD in Low to Moderate Exposed Areas of Taiwan: A 14-Year Prospective Study. Am J Kidney Dis (2017) 70:787–97. doi: 10.1053/j.ajkd.2017.06.012
77. Zhang X, Jiang L, Chen H, Wei S, Yao K, Sun X, et al. Resveratrol Protected Acrolein-Induced Ferroptosis and Insulin Secretion Dysfunction via ER-Stress- Related PERK Pathway in MIN6 Cells. Toxicology (2022) 465:153048. doi: 10.1016/j.tox.2021.153048
78. Grau-Perez M, Kuo CC, Spratlen M, Thayer KA, Mendez MA, Hamman RF, et al. The Association of Arsenic Exposure and Metabolism With Type 1 and Type 2 Diabetes in Youth: The SEARCH Case-Control Study. Diabetes Care (2017) 40:46–53. doi: 10.2337/dc16-0810
79. Eick SM, Ferreccio C, Acevedo J, Castriota F, Cordero JF, Roh T, et al. Socioeconomic Status and the Association Between Arsenic Exposure and Type 2 Diabetes. Environ Res (2019) 172:578–85. doi: 10.1016/j.envres.2019.03.013
80. Du J, Zhou Y, Li Y, Xia J, Chen Y, Chen S, et al. Identification of Frataxin as a Regulator of Ferroptosis. Redox Biol (2020) 32:101483. doi: 10.1016/j.redox.2020.101483
81. Turchi R, Tortolici F, Guidobaldi G, Iacovelli F, Falconi M, Rufini S, et al. Frataxin Deficiency Induces Lipid Accumulation and Affects Thermogenesis in Brown Adipose Tissue. Cell Death Dis (2020) 11:51. doi: 10.1038/s41419-020-2253-2
82. Alicic RZ, Rooney MT, Tuttle KR. Diabetic Kidney Disease: Challenges, Progress, and Possibilities. Clin J Am Soc Nephrol (2017) 12:2032–45. doi: 10.2215/CJN.11491116
83. Kashihara N, Kidokoro K, Kanda E. Renoprotective Effects of Sodium-Glucose Cotransporter-2 Inhibitors and Underlying Mechanisms. Curr Opin Nephrol Hypertens (2020) 29:112–18. doi: 10.1097/MNH.0000000000000561
84. Wu XQ, Zhang DD, Wang YN, Tan YQ, Yu XY, Zhao YY. AGE/RAGE in Diabetic Kidney Disease and Ageing Kidney. Free Radic Biol Med (2021) 171:260–71. doi: 10.1016/j.freeradbiomed.2021.05.025
85. Chen J, Luo SF, Yuan X, Wang M, Yu HJ, Zhang Z, et al. Diabetic Kidney Disease-Predisposing Proinflammatory and Profibrotic Genes Identified by Weighted Gene Co-Expression Network Analysis (WGCNA). J Cell Biochem (2022) 123:481–92. doi: 10.1002/jcb.30195
86. van Swelm RPL, Wetzels JFM, Swinkels DW. The Multifaceted Role of Iron in Renal Health and Disease. Nat Rev Nephrol (2020) 16:77–98. doi: 10.1038/s41581-019-0197-5
87. Matsumoto M, Sasaki N, Tsujino T, Akahori H, Naito Y, Masuyama T. Iron Restriction Prevents Diabetic Nephropathy in Otsuka Long-Evans Tokushima Fatty Rat. Ren Fail (2013) 35:1156–62. doi: 10.3109/0886022X.2013.819729
88. Wang WJ, Jiang X, Gao CC, Chen ZW. Salusin−β Participates in High Glucose−Induced HK−2 Cell Ferroptosis in a Nrf−2−dependent Manner. Mol Med Rep (2021) 24:674. doi: 10.3892/mmr.2021.12313
89. Dai C, Yang J, Liu Y. Transforming Growth Factor-Beta1 Potentiates Renal Tubular Epithelial Cell Death by a Mechanism Independent of Smad Signaling. J Biol Chem (2003) 278:12537–45. doi: 10.1074/jbc.M300777200
90. Yang YY, Deng RR, Chen Z, Yao LY, Yang XD, Xiang DX. Piperazine Ferulate Attenuates High Glucose−Induced Mesangial Cell Injury via the Regulation of P66(Shc). Mol Med Rep (2021) 23:374. doi: 10.3892/mmr.2021.12013
91. Daehn IS, Duffield JS. The Glomerular Filtration Barrier: A Structural Target for Novel Kidney Therapies. Nat Rev Drug Discov (2021) 20:770–88. doi: 10.1038/s41573-021-00242-0
92. Zhang Q, Hu Y, Hu JE, Ding Y, Shen Y, Xu H, et al. Sp1-Mediated Upregulation of Prdx6 Expression Prevents Podocyte Injury in Diabetic Nephropathy via Mitigation of Oxidative Stress and Ferroptosis. Life Sci (2021) 278:119529. doi: 10.1016/j.lfs.2021.119529
93. Wang J, Song Y, Wang Q, Kralik PM, Epstein PN. Causes and Characteristics of Diabetic Cardiomyopathy. Rev Diabetes Stud (2006) 3:108–17. doi: 10.1900/RDS.2006.3.108
94. Santana-Codina N, Gikandi A, Mancias JD. The Role of NCOA4-Mediated Ferritinophagy in Ferroptosis. Adv Exp Med Biol (2021) 1301:41–57. doi: 10.1007/978-3-030-62026-4_4
95. Wu KM, Hsu YM, Ying MC, Tsai FJ, Tsai CH, Chung JG, et al. High-Density Lipoprotein Ameliorates Palmitic Acid-Induced Lipotoxicity and Oxidative Dysfunction in H9c2 Cardiomyoblast Cells via ROS Suppression. Nutr Metab (Lond) (2019) 16:36. doi: 10.1186/s12986-019-0356-5
96. Zhao M, Lu L, Lei S, Chai H, Wu S, Tang X, et al. Inhibition of Receptor Interacting Protein Kinases Attenuates Cardiomyocyte Hypertrophy Induced by Palmitic Acid. Oxid Med Cell Longev (2016) 2016:1451676. doi: 10.1155/2016/1451676
97. Park M, Sabetski A, Kwan Chan Y, Turdi S, Sweeney G. Palmitate Induces ER Stress and Autophagy in H9c2 Cells: Implications for Apoptosis and Adiponectin Resistance. J Cell Physiol (2015) 230:630–9. doi: 10.1002/jcp.24781
98. Zeng X, Zhu M, Liu X, Chen X, Yuan Y, Li L, et al. Oleic Acid Ameliorates Palmitic Acid Induced Hepatocellular Lipotoxicity by Inhibition of ER Stress and Pyroptosis. Nutr Metab (Lond) (2020) 17:11. doi: 10.1186/s12986-020-0434-8
99. Jenkins T, Gouge J. Nrf2 in Cancer, Detoxifying Enzymes and Cell Death Programs. Antioxid (Basel) (2021) 10:1030. doi: 10.3390/antiox10071030
100. Zang H, Wu W, Qi L, Tan W, Nagarkatti P, Nagarkatti M, et al. Autophagy Inhibition Enables Nrf2 to Exaggerate the Progression of Diabetic Cardiomyopathy in Mice. Diabetes (2020) 69:2720–34. doi: 10.2337/db19-1176
101. Morsi M, Maher A, Aboelmagd O, Johar D, Bernstein L. A Shared Comparison of Diabetes Mellitus and Neurodegenerative Disorders. J Cell Biochem (2018) 119:1249–56. doi: 10.1002/jcb.26261
102. Devos D, Moreau C, Devedjian JC, Kluza J, Petrault M, Laloux C, et al. Targeting Chelatable Iron as a Therapeutic Modality in Parkinson's Disease. Antioxid Redox Signal (2014) 21:195–210. doi: 10.1089/ars.2013.5593
103. Raven EP, Lu PH, Tishler TA, Heydari P, Bartzokis G. Increased Iron Levels and Decreased Tissue Integrity in Hippocampus of Alzheimer's Disease Detected In Vivo With Magnetic Resonance Imaging. J Alzheimers Dis (2013) 37:127–36. doi: 10.3233/jad-130209
104. Ayton S, Lei P. Nigral Iron Elevation is an Invariable Feature of Parkinson's Disease and is a Sufficient Cause of Neurodegeneration. BioMed Res Int (2014) 2014:581256. doi: 10.1155/2014/581256
105. An JR, Su JN, Sun GY, Wang QF, Fan YD, Jiang N, et al. Liraglutide Alleviates Cognitive Deficit in Db/Db Mice: Involvement in Oxidative Stress, Iron Overload, and Ferroptosis. Neurochem Res (2021) 47:279–94. doi: 10.1007/s11064-021-03442-7
106. Abdul Y, Li W, Ward R, Abdelsaid M, Hafez S, Dong G, et al. Deferoxamine Treatment Prevents Post-Stroke Vasoregression and Neurovascular Unit Remodeling Leading to Improved Functional Outcomes in Type 2 Male Diabetic Rats: Role of Endothelial Ferroptosis. Transl Stroke Res (2021) 12:615–30. doi: 10.1007/s12975-020-00844-7
107. Cheloni R, Gandolfi SA, Signorelli C, Odone A. Global Prevalence of Diabetic Retinopathy: Protocol for a Systematic Review and Meta-Analysis. BMJ Open (2019) 9:e022188. doi: 10.1136/bmjopen-2018-022188
108. Wang W, Lo ACY. Diabetic Retinopathy: Pathophysiology and Treatments. Int J Mol Sci (2018) 19:1816. doi: 10.3390/ijms19061816
109. Gonzalez de Vega R, Garcia M, Fernandez-Sanchez ML, Gonzalez-Iglesias H, Sanz-Medel A. Protective Effect of Selenium Supplementation Following Oxidative Stress Mediated by Glucose on Retinal Pigment Epithelium. Metallomics (2018) 10:83–92. doi: 10.1039/c7mt00209b
110. Valderrabano RJ, Linares MI. Diabetes Mellitus and Bone Health: Epidemiology, Etiology and Implications for Fracture Risk Stratification. Clin Diabetes Endocrinol (2018) 4:9. doi: 10.1186/s40842-018-0060-9
111. Leidig-Bruckner G, Grobholz S, Bruckner T, Scheidt-Nave C, Nawroth P, Schneider JG. Prevalence and Determinants of Osteoporosis in Patients With Type 1 and Type 2 Diabetes Mellitus. BMC Endocr Disord (2014) 14:33. doi: 10.1186/1472-6823-14-33
112. Mohsin S, Baniyas MM, AlDarmaki RS, Tekes K, Kalasz H, Adeghate EA. An Update on Therapies for the Treatment of Diabetes-Induced Osteoporosis. Expert Opin Biol Ther (2019) 19:937–48. doi: 10.1080/14712598.2019.1618266
113. Jia P, Xu YJ, Zhang ZL, Li K, Li B, Zhang W, et al. Ferric Ion Could Facilitate Osteoclast Differentiation and Bone Resorption Through the Production of Reactive Oxygen Species. J Orthop Res (2012) 30:1843–52. doi: 10.1002/jor.22133
114. Lin Y, Shen X, Ke Y, Lan C, Chen X, Liang B, et al. Activation of Osteoblast Ferroptosis via the METTL3/ASK1-P38 Signaling Pathway in High Glucose and High Fat (HGHF)-Induced Diabetic Bone Loss. FASEB J (2022) 36:e22147. doi: 10.1096/fj.202101610R
115. Otasevic V, Vucetic M, Grigorov I, Martinovic V, Stancic A. Ferroptosis in Different Pathological Contexts Seen Through the Eyes of Mitochondria. Oxid Med Cell Longev (2021) 2021:5537330. doi: 10.1155/2021/5537330
116. Battaglia AM, Chirillo R, Aversa I, Sacco A, Costanzo F, Biamonte F. Ferroptosis and Cancer: Mitochondria Meet the "Iron Maiden" Cell Death. Cells (2020) 9:1505. doi: 10.3390/cells9061505
117. Kim JH, Lewin TM, Coleman RA. Expression and Characterization of Recombinant Rat Acyl-CoA Synthetases 1, 4, and 5. Selective Inhibition by Triacsin C and Thiazolidinediones. J Biol Chem (2001) 276:24667–73. doi: 10.1074/jbc.M010793200
118. Yuan H, Li X, Zhang X, Kang R, Tang D. Identification of ACSL4 as a Biomarker and Contributor of Ferroptosis. Biochem Biophys Res Commun (2016) 478:1338–43. doi: 10.1016/j.bbrc.2016.08.124
119. McKeage K, Keating GM. Fenofibrate: A Review of its Use in Dyslipidaemia. Drugs (2011) 71:1917–46. doi: 10.2165/11208090-000000000-00000
120. Cheng Y, Zhang X, Ma F, Sun W, Wang W, Yu J, et al. The Role of Akt2 in the Protective Effect of Fenofibrate Against Diabetic Nephropathy. Int J Biol Sci (2020) 16:553–67. doi: 10.7150/ijbs.40643
122. Ma WQ, Sun XJ, Zhu Y, Liu NF. Metformin Attenuates Hyperlipidaemia-Associated Vascular Calcification Through Anti-Ferroptotic Effects. Free Radic Biol Med (2021) 165:229–42. doi: 10.1016/j.freeradbiomed.2021.01.033
123. Zhang J, Ning L, Wang J. Dietary Quercetin Attenuates Depressive-Like Behaviors by Inhibiting Astrocyte Reactivation in Response to Stress. Biochem Biophys Res Commun (2020) 533:1338–46. doi: 10.1016/j.bbrc.2020.10.016
124. Wang Z, Zhai D, Zhang D, Bai L, Yao R, Yu J, et al. Quercetin Decreases Insulin Resistance in a Polycystic Ovary Syndrome Rat Model by Improving Inflammatory Microenvironment. Reprod Sci (2017) 24:682–90. doi: 10.1177/1933719116667218
125. Bardy G, Virsolvy A, Quignard JF, Ravier MA, Bertrand G, Dalle S, et al. Quercetin Induces Insulin Secretion by Direct Activation of L-Type Calcium Channels in Pancreatic Beta Cells. Br J Pharmacol (2013) 169:1102–13. doi: 10.1111/bph.12194
126. Eitah HE, Maklad YA, Abdelkader NF, Gamal El Din AA, Badawi MA, Kenawy SA. Modulating Impacts of Quercetin/Sitagliptin Combination on Streptozotocin-Induced Diabetes Mellitus in Rats. Toxicol Appl Pharmacol (2019) 365:30–40. doi: 10.1016/j.taap.2018.12.011
127. Yao Z, Gu Y, Zhang Q, Liu L, Meng G, Wu H, et al. Estimated Daily Quercetin Intake and Association With the Prevalence of Type 2 Diabetes Mellitus in Chinese Adults. Eur J Nutr (2019) 58:819–30. doi: 10.1007/s00394-018-1713-2
128. Li Z, Wang Z, Dong F, Shi W, Dai W, Zhao J, et al. Germacrone Attenuates Hepatic Stellate Cells Activation and Liver Fibrosis via Regulating Multiple Signaling Pathways. Front Pharmacol (2021) 12:745561. doi: 10.3389/fphar.2021.745561
129. Riaz A, Rasul A, Kanwal N, Hussain G, Shah MA, Sarfraz I, et al. Germacrone: A Potent Secondary Metabolite With Therapeutic Potential in Metabolic Diseases, Cancer and Viral Infections. Curr Drug Metab (2020) 21:1079–90. doi: 10.2174/1389200221999200728144801
130. Jin J, Wang Y, Zheng D, Liang M, He Q. A Novel Identified Circular RNA, Mmu_Mmu_circRNA_0000309 Involves in Germacrone-Mediated the Improvement of Diabetic Nephropathy Through Regulating Ferroptosis by Targeting miR-188-3p/GPX4 Signaling Axis. Antioxid Redox Signal (2021). doi: 10.1089/ars.2021.0063
131. Tian S, Wang M, Liu C, Zhao H, Zhao B. Mulberry Leaf Reduces Inflammation and Insulin Resistance in Type 2 Diabetic Mice by TLRs and Insulin Signalling Pathway. BMC Complement Altern Med (2019) 19:326. doi: 10.1186/s12906-019-2742-y
Keywords: ferroptosis, diabetes, diabetic complications, diabetic kidney disease, iron
Citation: Yang X-D and Yang Y-Y (2022) Ferroptosis as a Novel Therapeutic Target for Diabetes and Its Complications. Front. Endocrinol. 13:853822. doi: 10.3389/fendo.2022.853822
Received: 13 January 2022; Accepted: 02 March 2022;
Published: 29 March 2022.
Edited by:
Ana Stancic, University of Belgrade, SerbiaReviewed by:
Isha Sharma, Northwestern University, United StatesCopyright © 2022 Yang and Yang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Yong-Yu Yang, eW9uZ3l1eWFuZ0Bjc3UuZWR1LmNu
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.