
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Endocrinol. , 06 April 2022
Sec. Obesity
Volume 13 - 2022 | https://doi.org/10.3389/fendo.2022.846880
This article is part of the Research Topic Hypothalamic Obesity: Today and Future View all 10 articles
 Paul Dimitri1,2*
Paul Dimitri1,2*The hypothalamus is the centre of neuroendocrine regulation of energy homeostasis and appetite. Maldevelopment of, or damage to, the key hypothalamic nuclei disrupts the coordinated balance between energy intake and expenditure leading, to rapid and excessive weight gain. Hypothalamic obesity is compounded by a disruption of the hypothalamic-pituitary axis, sleep disruption, visual compromise, and neurological and vascular sequalae. Amongst suprasellar tumors, craniopharyngioma is the most common cause of acquired hypothalamic obesity, either directly or following surgical or radiotherapeutic intervention. At present, therapy is limited to strategies to manage obesity but with a modest and variable impact. Current approaches include optimizing pituitary hormone replacement, calorie restriction, increased energy expenditure through physical activity, behavioral interventions, pharmacotherapy and bariatric surgery. Current pharmacotherapeutic approaches include stimulants that increase energy consumption, anti-diabetic agents, hypothalamic–pituitary substitution therapy, octreotide, and methionine aminopeptidase 2 (MetAP2) inhibitors. Some pharmacological studies of hypothalamic obesity report weight loss or stabilization but reported intervention periods are short, and others report no effect. The impact of bariatric surgery on weight loss in hypothalamic obesity again is variable. Novel or combined approaches to manage hypothalamic obesity are thus required to achieve credible and sustained weight loss. Identifying etiological factors contributing hypothalamic obesity may lead to multi-faceted interventions targeting hyperphagia, insulin resistance, decreased energy expenditure, sleep disturbance, hypopituitarism and psychosocial morbidity. Placebo-controlled trials using current single, or combination therapies are required to determine the impact of therapeutic agents. A well-defined approach to defining the location of hypothalamic damage may support the use of future targeted therapies. Intranasal oxytocin is currently being investigated as an anorexogenic agent. Novel agents including those targeting pro-opimelanocortin-C and AgRP/NPY expressing neurons and the MC4 receptor may result in better outcomes. This article discusses the current challenges in the management of hypothalamic obesity in children and young people and future therapeutic approaches to increasing weight loss and quality of life in these patients.
The potential for a lesion in the hypothalamic-pituitary region to result in obesity is not a recent discovery. Over 180 years ago, von Mohr published a case of rapid onset obesity in a case of a pituitary tumour (1), and in 1939, Nevin published a report relating to the sequalae of hypothalamic-pituitary tumours which included ‘adiposity that may precede the depression of sexual function’ (2). At around the same time, the concept that the hypothalamus was in some way associated with fat metabolism was emerging (3), and only 4 years later, Brobeck demonstrated that rats with lesions in the hypothalamic ventromedial nucleus (VMN) showed voracious hunger (4); 10 years later, the central role of the hypothalamus in energy regulation was first proposed (5). It is over the last few decades that our knowledge of the hypothalamus as a key regulator energy regulation and appetite has significantly increased.
Nuclei in the mediobasal hypothalamus play a fundamental role in energy balance through the modulation of appetite and food consumption, regulation of fat storage and energy expenditure. By responding to circulating signals from peripheral energy stores the mediobasal hypothalamus adjusts energy consumption to ensure that body weight and in particular body fat remain stable (6, 7). The arcuate nucleus (ARC) within the mediobasal hypothalamus is considered the hypothalamic area that primarily senses metabolic signals from the periphery via the systemic circulation or the CSF, facilitated by its adjacency with the median eminence, and the third ventricle. There are two distinct populations of neurones within the (ARC) that balance food intake, through the production of the orexigenic peptides (appetite promoting) neuropeptide Y (NPY) and agouti-related peptide (AgRP), and the anorexigenic peptides (appetite suppressing) proopiomelanocortin (POMC), and cocaine- and amphetamine-regulated transcript (CART). These neurones respond to peripheral metabolic hormones, including leptin, insulin, ghrelin and nutrients. POMC neurons project to second-order neurons in the hypothalamic paraventricular nucleus (PVN), the dorsomedial hypothalamus (DMH), the lateral hypothalamus (LH) and the ventromedial hypothalamus (VMH) (8).
Peripheral signalling from leptin and insulin suppresses appetite through the activation of POMC and CART (9, 10), leading to an increase in the anorexigenic neuropeptide α-melanocyte-stimulating hormone (α-MSH) that binds to the melanocortin-3 and -4 receptors (MC3R and MC4R) in the hypothalamic PVN and LH, reducing food intake and increased energy expenditure (11). This is mediated by efferent neuronal projections from the PVN and LH that synapse in extrahypothalamic brain regions within the locus coeruleus, leading to activation of the sympathetic nervous system, and regulation of parasympathetic tone via the vagus nerve through the dorsal motor nucleus of the vagus (DMV) resulting in an increase in energy expenditure and net catabolic effect. Insulin secretion is regulated by parasympathetic vagal-efferent signalling by promoting depolarization of pancreatic β-cells via M3-muscarinc receptors (12, 13). Vagus nerve–mediated acetylcholine also increases intracellular phospholipases within β-cells, increasing intracellular release of calcium, inducing insulin vesicular exocytosis and insulin secretion (14, 15). Parasympathetic nervous system innervation to the pancreas is also associated with increased β-cell proliferation and mass (16), suggesting that the insulin hypersecretion secondary to hypothalamic damage is likely to result from increased β-cell proliferation and mass as well as insulin hypersecretion. Early studies demonstrate that ablation of the VMH in rats results in the insulin hypersecretion which can be obviated by pancreatic vagotomy (17, 18). Thus, damage to the VMH and LH results in hyperinsulinemia and a net reduction in lipolysis and fat mass accumulation. In the energy replete state, anorexigenic pathways within the hypothalamus activated by increased insulin and leptin stimulate downstream sympathetic efferent signalling resulting in increased brown adipose tissue thermogenesis and energy expenditure in part by raising resting heart rate and mean arterial pressure (19). As beta-adrenergic signalling also promotes lipolysis (20) and limits insulin secretion (21), abrogation of sympathetic outflow from the hypothalamus secondary to hypothalamic damage will result in disinhibited insulin secretion, a reduction in lipolysis with fat mass accumulation and reduced energy expenditure.
During fasting or energy deficit, neurons situated in the ARC stimulate feeding when they are activated by hormones such as ghrelin (12, 13) through the influence of NPY, AgRP and the neurotransmitter GABA on the PVN (5). AgRP downregulates the production of MC3R and MC4R, thereby preventing the anorexigenic effect of α-MSH on second-order neurones (22). AgRP/NPY neurons also directly inhibit POMC neurons via GABA at the level of the ARC (23). GABA release from AgRP/NPY projections to extrahypothalamic neurons, in the parabrachial nucleus, also plays a role in the stimulation of food intake (24). As well as stimulating feeding, activation of NPY results in energy conservation by reducing the metabolic activity of brown adipose tissue in a manner paradoxical to that seen with regulation of thermogenesis by POMC, by downregulation of sympathetic outflow from the locus coeruleus (25).
Centrally, POMC and AgRP/NPY neurons express receptors for insulin and leptin, indicating that these hormones play a key role in energy homeostasis and food intake. The adipocyte derived hormone leptin circulates at plasma levels directly correlated to adiposity (26) and plays a key role in energy homeostasis as a negative feedback regulator of adiposity by limiting energy intake and supporting energy expenditure thus preventing weight gain (27). Thus, during periods of starvation during which time fat mass is reduced, leptin is reduced in-turn promoting increased food intake and fat accumulation (28); conversely disruption of leptin signalling promotes hyperphagia and rapid weight gain (29). In the mediobasal hypothalamus, leptin activates POMC whilst directly inhibiting AgRP and NPY neurons with a net effect of increasing energy expenditure and decreasing food intake (30). In addition to this, in the dorsomedial hypothalamus, leptin promotes increased energy expenditure through activation of brown adipose tissue which results in a reduction in body weight that is independent of food intake (31).
Insulin is secreted from pancreatic β-cells upon nutrient ingestion and plays an important role in the peripheral regulation of energy and glucose homeostasis by peripheral glucose metabolism through the suppression hepatic glucose production via direct action on hepatic insulin receptors. Centrally, disruption of insulin receptors in the hypothalamus causes obesity, insulin resistance, hyperphagia, and hyperleptinemia in mice (32) and when insulin was injected into the cerebral ventricles of baboons in a study over 40 years ago, food intake and endogenous glucose production were both suppressed (9) underpinning the central role of insulin in energy homeostasis. The centrally mediated action of insulin has since been extensively reviewed in the last few years. At the level of the hypothalamus, insulin acts to suppress food intake, promote peripheral lipogenesis, inhibit hepatic glucose production and promote brown adipose tissue thermogenesis. These centrally mediated actions of insulin are fundamentally mediated through the excitation of POMC neurons and the concomitant suppression of AgRP and NPY neurons (33–35). Hypothalamic pathways regulating energy expenditure and feeding are detailed in Figure 1.
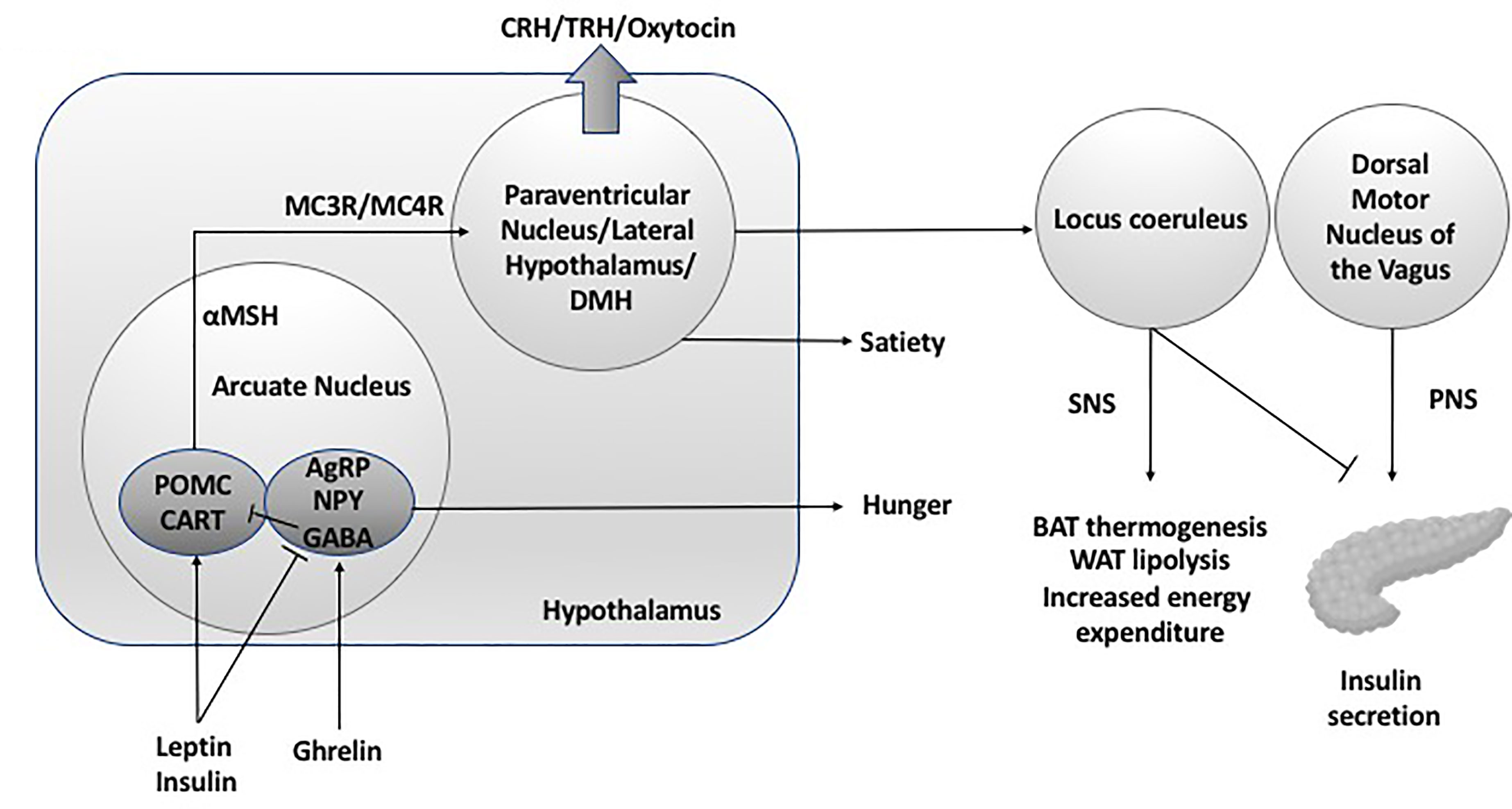
Figure 1 Hypothalamic neuroendocrine regulation of energy expenditure and feeding. DMH, dorsal medial hypothalamus; SNS, sympathetic nervous system; PNS, parasympathetic nervous system; POMC, pro-opiomelanocortin C; CART, cocaine and amphetamine related transcript; AgRP, Agouti related peptide; NPY, neuropeptide Y; GABA, gamma aminobutyric acid; MC3R, melanocortin 3 receptor; MC4R, melanocortin 4 receptor; BAT, brown adipose tissue; WAT, white adipose tissue; CRH, corticotroph releasing hormone; TRH, thyrotroph releasing hormone.
Given the fundamental role of the hypothalamus in energy homeostasis and appetite regulation, it follows that damage to the hypothalamus results in dysregulation of satiety and energy expenditure, leading to hyperphagia and rapid weight gain, reduced sympathetic tonicity and insulin hypersecretion. Thus, this provides multiple target areas for pharmacotherapeutic intervention to reduce weight gain and fat mass in patients with hypothalamic obesity.
Obesity is a well-recognized and common complication of hypothalamic damage either as a result of tumour invasion of, or treatment to, the hypothalamic regions critical to energy regulation. Imaging studies have demonstrated a direct correlation between the extent of hypothalamic damage and presentation of obesity (36, 37). In patients with craniopharyngioma, hypothalamic obesity occurs in up to 50% of patients (38–41) and up to twenty percent of patients with craniopharyngioma are obese at diagnosis (39–41), and this weight gain is irrespective of pituitary deficiency secondary to damage to the hypothalamic-pituitary axis. Higher preoperative BMI, radical tumour resection, larger preoperative tumour size, hypothalamic tumour invasion, adamantinomatous subtype, and familial predisposition to obesity are cited as factors that increase the risk of hypothalamic obesity (37, 42, 43). Rapid weight gain usually occurs within the first 3 years and often within the first year following surgical intervention, with surgical intervention increasing the prevalence of obesity in this patient group (38, 43).
Leptin and insulin levels are higher in patients with hypothalamic obesity suggesting that the normal mechanism by which leptin and insulin mediate satiety and play a central role in energy homeostasis is impaired, leading to uncontrolled and rapid gain in fat mass secondary to impaired satiety and disruption of energy homeostasis (44, 45). Assessment of dietary intake, energy expenditure and basal metabolic rate (BMR) in patients with craniopharyngioma report similar food intake to healthy controls, but instead demonstrate that reduced physical activity, energy expenditure and BMR may be central to rapid weight gain (45–47). Decrease in energy expenditure and basal metabolic rate is likely to be associated with a reduction in sympathetic tone as a direct result of hypothalamic damage. Evidence supporting this theory includes a reduction in the excretion of the urinary catecholamine metabolites homovanillic acid and vanillylmandelic acid that directly correlates with BMI (48), and a reduction in the 24-hour urinary excretion of adrenaline that is independent of adrenal insufficiency from pituitary damage (49).
In addition to the role that the hypothalamus plays in energy regulation, patients with hypothalamic obesity suffer with other co-morbidities that include endocrine dysfunction, sleep disturbance, visual dysfunction and neurological sequalae, underlying the fundamental role that the hypothalamus plays in physiological control, and the resultant damage caused by suprasellar tumours due to the intimate co-location of the hypothalamus with other structures such as that optic chiasm and cavernous sinus. The pituitary gland is dependent on hypothalamic signals that are frequently disrupted from hypothalamic damage, that affects secretion of growth hormone, gonadotropins, adrenocorticotrophic hormone (ACTH) and thyroid stimulating hormone (TSH). At the time of diagnosis up to 90% of patients with craniopharyngioma are reported to have at least one pituitary hormone deficiency (39, 40, 50). Thus, correction of pituitary hormone deficiency is key to the management of patients with suprasellar tumours. In particular, deficiency in growth hormone, and hypothyroidism secondary to TSH deficiency from the pituitary or thyrotropin releasing hormone from the hypothalamus (TRH), can compound excessive weight gain if left untreated, and treatment of these hormone deficiencies results in reduction in weight (51). Moreover, change in behaviour secondary to estrogen or testosterone deficiency due to hypogonadotrophic hypogonadism can lead to low mood and result in a reduction in physical activity thus compounding weight gain.
The hypothalamus is also a central regulator of sleep and arousal. The impact of hypothalamic lesions leading to sleep disruption was reported nearly 100 years ago (52). Hypothalamic damage results in disturbances in sleep-wake regulation with alterations in the circadian rhythm, sleep fragmentation, and increased daytime somnolence (53, 54). Polysomnography in children with craniopharyngioma demonstrates sleep patterns consistent hypersomnia and secondary narcolepsy (55, 56). This can be compounded by obstructive sleep apnoea secondary to obesity, leading to daytime somnolence secondary to poor sleep quality at night (57). Given that sleep is considered to be a period of energy conservation, hypersomnia in patients with hypothalamic damage can result in a reduction in energy expenditure (58). Concomitantly, although sleep disruption results in a rise in energy expenditure, energy consumption exceeds this rise resulting in a net weight gain (59). This is part is due to appetite dysregulation secondary to an increase in ghrelin and reduction in leptin (60), poor diet quality, disruption in the timing of eating, and a change in eating behaviours that promotes intake of higher calorific foods and emotional eating (61).
Due to the intimate co-location of the hypothalamus, pituitary and optic chiasm, visual disturbance from suprasellar tumours is relatively common at presentation, with visual disturbance identified in more than 50% of patients who present with craniopharyngioma (39). Moreover, due to the co-location of the hypothalamus to structures within the cavernous sinus, craniopharyngiomas can lead to other neurological sequalae including epilepsy, cranial nerve dysfunction and cerebrovascular events which increase in frequency with larger tumours (62). Any of these deficits either in isolation or in combination has the potential to impact on the ability to partake in physical activity, and thus increase the risk of obesity.
Craniopharyngiomas present a surgical challenge because of their proximity to the optic nerves optic, pituitary, hypothalamus, circle of Willis, brain stem, and temporal lobes. To minimize the risk of hypothalamic obesity, treatment strategies have become more conservative in an attempt to preserve normal hypothalamic function. Gross total resection results in higher rates of hypothalamic obesity, panhypopitutarism and limited improvement in 5-year progression-free survival (PFS) (63). Gross total resection is now used much less commonly and total tumour resection should be limited to tumours of less than 4 cm that can be easily removed without sequalae. The aim of surgical intervention should be to relieve tumour-associated compression symptoms, preserve or improve vision and hypothalamo-pituitary function, and minimize tumor recurrence through adjunct radiotherapy (64). Subtotal resection in isolation is associated with significantly inferior 5-year PFS compared with conservative surgery followed by adjuvant radiation (65). The approach of using subtotal resection followed by radiotherapy is of particular importance in patients with lesions within the hypothalamus as they have decreased 10-year overall survival and a significant risk of poor psychosocial and quality of life (66). More recently, surgical and radiotherapeutic approaches have evolved in an attempt to minimise treatment related damage. Current techniques include fractionated three-dimensional conformal radiotherapy, intensity modulated radiotherapy (IMRT), fractionated stereotactic radiotherapy (FSRT) and proton beam therapy. IMRT is an advanced mode of high-precision radiotherapy that delivers precise radiation doses to conform more precisely to the three-dimensional shape of a tumour by modulating the intensity of the radiation beam in multiple small volumes, thus limiting radiation scatter to surrounding unaffected structures by up to 45% (67). Proton beam therapy is now used preferentially in the treatment of craniopharyngiomas due to the ability to limit the distribution and intensity of the radiation dose, in-turn reducing the risk of complications and secondary cancer, whilst improving target conformity (68, 69), with some case series reporting a 100% 5-year control rate (70–72). Extension beyond tumour margins is now limited to 5mm to prevent the extension of radiation to normal surrounding structures (73). Collectively, these modern approaches to tumour radiation result in tumour control between 80-90% (74). Hypothalamic obesity has been reported in only 25% of children receiving IMRT or PBT (75), but in general hypothalamic obesity is under-reported and case-series are most-commonly retrospective. Prospective studies are required to determine whether a more conservative surgical approach together with newer radiotherapy modalities can reduce the incidence of post-interventional hypothalamic obesity.
The recent advances in our understanding of the centrally mediated pathways relevant to energy and appetite regulation have resulted in a targeted pharmacological approach in an attempt to bypass damaged hypothalamic pathways.
Damage to the VMH, PVN and LH results in a rise in vagal tone resulting in insulin hypersecretion, promoting energy deposition into the adipocyte and increased fat mass. The anorexigenic effect of insulin via afferent hypothalamic pathways may either be blunted or ablated by hypothalamic damage (9, 10). Pragmatically, carbohydrate restriction would appear to be the simplest way to reduce insulin secretion, although evidence to this approach is generally lacking and in general food intake does not appear to be significantly altered in hypothalamic obesity (45–47). Diazoxide is an inhibitor of glucose-stimulated insulin release and has been successfully used to lower insulin secretion in children with congenital hyperinsulinaemia. Diazoxide therapy in patients with hypothalamic obesity has however, been unsuccessful. In a study of 40 children, 18 of whom were randomized to a treatment arm using diazoxide (4mg/kg/day), there was no difference in weight when compared with the placebo group. More worryingly, 3 patients developed diabetes mellitus and plasma glucose was significantly higher in the treatment group (76).
In contrast, the combination of metformin and diazoxide has shown slightly more promising results in slowing weight gain (albeit not leading to weight loss). Metformin improves insulin sensitivity and decreases hepatic gluconeogenesis and intestinal glucose absorption. In a prospective open-label 6-month pilot treatment trial in 9 obese subjects with craniopharyngioma the administration of diazoxide (2 mg/kg/day) and metformin (1000 mg twice daily) resulted in an overall reduction in weight gain over a 6-month period compared with the weight gain observed over the 6-month period prior to the intervention. However, 2 patients were withdrawn due to vomiting and peripheral edema. This study is notably limited by the small number of participants and the lack of a comparator group, by instead assuming that weight gain would be uniformly similar during the pre-treatment and treatment phases (77).
Combination therapy of metformin and fenofibrate in hypothalamic obesity secondary to craniopharyngioma has again yielded disappointing results. Fenofibrate activates peroxisome proliferator-activated receptors alpha (PPAR-alpha), which upregulates lipoprotein lipase, induces high-density lipoprotein synthesis, and decreases liver production of apolipoprotein C and improves insulin sensitivity (78). Despite no significant improvement in weight gain, the fenofibrate/metformin combination therapy resulted in a reduction in dyslipidemia and improved insulin sensitivity in the treatment group (79).
Octreotide is a somatostatin analogue that improves insulin sensitivity (80). An initial pilot study in 1999 in eight paediatric patients demonstrated a reduction in body mass index that correlated with reduced insulin secretion and plasma leptin. However, a subsequent double-blind study enrolled 20 patients with hypothalamic obesity (13 subjects had craniopharyngioma, 4 subjects had hypothalamic astrocytoma, 1 had pineal germinoma, and 2 subjects had acute lymphoblastic leukaemia and received 24 Gy of cranial irradiation). Two patients were discontinued, one on octreatide who had recurrence of their craniopharyngioma, and the other on placebo developed diabetic hyperosmolar nonketotic coma. The 9 patients who were treated with octreotide showed a statistically significant decrease in BMI (i.e., –0.2 ± 0.2 on treatment versus +2.2 ± 0.5 on placebo) and improvement in insulin secretion that correlated with quality of life. Complications included diarrhea and abdominal discomfort in all those treated with octreotide which eventually subsided, and 4 patients developed gallbladder sludge or true cholelithiasis on ultrasound. Two patients developed impaired glucose tolerance, and 1 patient developed type 2 diabetes during the open-label extension, although this latter patient already exhibited acanthosis nigricans prior to treatment (81). A subsequent phase 4 study of octreotide depot compared with saline control in paediatric hypothalamic obesity patients led by Novartis (ClinicalTrials.gov Identifier: NCT00171613) commenced in February 2005 with patients receiving sandostatin (octreotide) LAR (SAS-LAR) depot for 6 months. After 6 months of treatment with SAS-LAR or placebo, no difference in change from baseline in BMI was seen between treatment group (0.1 kg/m2 in the SAS-LAR-treated group versus 0.0 kg/m2 in the placebo-treated group, p=0.74) (82). No further trials using octreotide therapy have been published since.
Given the evidence demonstrating a reduction in energy expenditure and BMR in patients with hypothalamic obesity (45–47), therapies that increase energy expenditure have been trialled to reduce BMI. CNS stimulants such as dextroamphetamine (83), sibutramine (84, 85) and a combination of caffeine and ephedrine (86) have been shown to reduce appetite and promote weight loss, albeit that sibutramine has since been withdrawn due to concerns over cardiovascular complications (84).
The rationale for using ephedrine in the treatment of hypothalamic obesity is based upon the reduction in sympathetic tone seen in these patients. Ephedrine is a sympathomimetic amine that activates adrenergic receptors, increasing heart rate and blood pressure, improving energy expenditure and increasing brown adipose tissue activity (87, 88). Ephedrine activates adrenergic α and β-receptors as well as inhibiting noradrenaline reuptake, and increasing the release of noradrenaline from vesicles in nerve cells. Caffeine affects peripheral metabolism through alterations in sympathetic nervous system activity (89) and by influencing peripheral metabolic targets directly through inhibition of cAMP phosphodiesterase or adenosine receptors or by activation of AMP-kinase (90). Three patients treated with a combination of caffeine and ephedrine showed an initial 8-18% reduction in weight, with 2 out of 3 showing sustained weight loss for 2 and 6 years respectively, and the other returning to the baseline weight (91). However, a meta-analysis of ephedrine with and without caffeine for weight loss and improving athletic performance demonstrated a 2.2 to 3.6 -fold increase in the odds of psychiatric, autonomic, or gastrointestinal symptoms and heart palpitations (86), thus underpinning the need to conduct a randomised placebo-controlled trial of caffeine and ephedrine in patients with hypothalamic obesity to monitor for treatment effect and complications.
The centrally mediated appetite suppressant side effect seen in patients with ADHD and narcolepsy treated with dextroamphetamine (92) suggests that a similar effect may be observed in patients with hypothalamic obesity. Dextroamphetamine mediates central anorexigenic control by direct modification of cerebral satiety signalling through stimulation of cocaine-amphetamine regulated transcript (CART) production in the ventromedial hypothalamus. Dextroamphetamine is also a potent sympathomimetic agent leading to an increase in α- and β-adrenergic tone (93), which may counteract the reduce sympathetic tone observed in hypothalamic obesity patients. Increased activity of the mesolimbic reward pathway and elevated extracellular dopamine in the nucleus accumbens following administration of dextroamphetamine may also rectify the compulsion to eat excessively (94). This action may be consistent with functional magnetic resonance imaging studies that demonstrate a trend towards higher activation in cerebral regions associated with hedonic appetite including the insula, nucleus accumbens, and medial orbitofrontal cortex observed in craniopharyngioma patients with hypothalamic obesity (95). In a study of 12 patients with hypothalamic obesity treated with low dose dextroamphetamine (5mg twice daily), 10 of the patients either lost weight or demonstrated weight stabilization and daytime somnolence improved in 11 patients (96). A similar effect of weight stabilization was observed in the first year in a case series of 7 patients treated with an upward dose titration regimen of dextroamphetamine commencing at 5 mg per day up to 20mg per day, although the results of the second year of dextroamphetamine treatment were heterogenous with several patients continuing to reduce weight, while others demonstrated significant gains in BMI z-score (97). In an earlier study of 5 children with hypothalamic obesity secondary to surgical intervention for craniopharyngioma, weight gain stabilized and significant improvements were observed in attention and activity following dextroamphetamine treatment (98). Longer-term studies are required to determine whether weight loss or stabilization is sustained by using dextroamphetamine in the treatment of hypothalamic obesity, to understand the mechanism by which dextroamphetamine is exerting an effect (e.g. as a sympathomimetic agent) and the potential for longer-term side effects.
In a small number (n=3), tri-iodothyroine monotherapy (T3) has also been trialled in patients with hypothalamic obesity, with a sustained reduction in weight observed over a 2-year period and with improvement in daytime somnolence (99). Only one additional study of T3 supplementation has been conducted since in a patient with hypothalamic obesity, by which levothyroxine therapy was switched to T3 monotherapy, with results demonstrating no change in sympathetic and metabolic BAT activity, energy expenditure, or BMI during T3 treatment (100). Thus, to date, only 4 patients have undergone treatment with T3 monotherapy with conflicting results. Given the ubiquitous impact on metabolic function mediated by thyroid hormones (101), further work is needed in this area to elucidate whether a select group of patients with hypothalamic obesity may respond with weight loss to T3 monotherapy.
An alternative approach to appetite regulation in patients with established hypothalamic obesity is to target areas of the brain that control satiety that are not impacted by hypothalamic damage. The quantity of food consumed is regulated by the nucleus tractus solitarus (NTS) located in the dorsomedial medulla and is controlled by gut mediated vagal afferents influenced by gut peptides including GLP1 and CCK (102, 103). Leptin appears to potentiate this effect by directly and indirectly enhancing the response of the NTS to gut peptides and leptin is increased in patients with hypothalamic obesity (6, 27, 104, 105). GLP1 receptor analogues (GLP1A) may therefore potentiate NTS sensitivity to GLP1 thus reducing the frequency and quantity of food consumed, leading to weight loss. In a rat model recapitulating the key features of hypothalamic obesity, the use of the GLP1A exendin-4 resulted in a significant reduction in food intake and weight compared to those treated with saline (106). However, results in adult studies to date have been variable with one adult study demonstrating sustained weight loss over 6-51 months in 9 patients with hypothalamic obesity and type-2 diabetes following treatment with a GLP1A (107), with a further 8 patients treated with twice daily exanetide for 1 year showing no significant weight loss, but no weight gain (108). The first study of children given 2 mg exenatide weekly for a 12-month period again showed no significant impact on weight or BMI, albeit one patient demonstrated a BMI SDS reduction of -0.33 after 12 months (109). In contrast, a recent randomized, multicentre, double-blind, placebo-controlled trial was conducted in 10- to 25-year-olds with hypothalamic injury following intracranial tumour and hypothalamic obesity. Participants were randomised to once-weekly subcutaneous injections of exenatide 2 mg or placebo for 36 weeks. Whilst there was no significant reduction in BMI in the treatment group compared to controls, there was a reduction in total body fat mass as measured by DXA in and stabilization of waist circumference in 50% of the treatment group, compared to an increase in waist circumference and fat mass in controls. Exanetide was generally well tolerated with the majority of side effects being related to gastrointestinal disturbance (110). The limited impact in response to GLP1A therapy on food consumption and weight loss in patients with hypothalamic obesity, may reflect a disruption of pathways involved in the complex interaction between the hypothalamus and other areas of the brain involved in hunger, satiety and hedonic appetite, that are disrupted and thus unresponsive to therapies that are aimed at targeting areas of the brain distant from the hypothalamus. Moreover, a select group of patients with limited hypothalamic damage may respond better to GLP1A, whilst others with more extensive hypothalamic damage fail to respond to the same therapy. However, contrary to this theory, stratification by MRI prior to enrolment into a trial of GLP1A to assess the degree of hypothalamic damage using an integrative hypothalamic lesion score (HLS), demonstrated that patients with more extensive hypothalamic damage with involvement of the mammillary body show greater reductions in adiposity following GLP1A treatment. The authors speculated that disruption of hypothalamic pathways involved in appetite and energy homeostasis may result in alterations in other pathways such as GLP1-mediated signalling in the brainstem, which remain intact in patients with hypothalamic obesity (111). The relevance of the mammillary body damage in this study is poorly understood in relation to GLP1A responsiveness and suggests that the mammillary body damage may act as a proxy for more extensive hypothalamic damage, or that the mammillary body has a functional role in feeding behaviour via interaction with the hippocampus (112). Future studies in patients treated with exanetide may thus benefit from additional stratification based on the degree of hypothalamic damage.
Hypothalamic PVN neurons secrete neuropeptides that have a net catabolic action, including corticotrophin-releasing hormone, thyrotropin-releasing hormone, somatostatin, vasopressin and oxytocin. In the hypothalamic PVN, oxytocin and other neurons tonically inhibit feeding and, during energy deficit, are inhibited by orexigenic input from the ARC, thereby stimulating feeding (11), leading the concept that oxytocin supplementation may promote weight loss in hypothalamic obesity. In 2018, administration of intranasal oxytocin was given for 10 weeks, followed by a combination of intranasal oxytocin and naloxone for 38 weeks in a 13-year-old male with confirmed hypothalamic obesity and hyperphagia following resection of a craniopharyngioma. The observed reduction in BMI and hyperphagia following oxytocin monotherapy has paved the way for future trials (113). Oxytocin is a neuropeptide produced in the hypothalamic PVN and supraoptic nucleus (SON) and modulates food intake and behaviour. The anorexigenic effect of oxytocin results from the extension of oxytocin neurons from the PVN and SON to other distal brain targets involved with appetite regulation (114). For example, the medial NTS receives oxytocinergic input that potentiates the effect of afferent gastrointestinal inputs resulting in a reduction in food intake (115). Damage to the hypothalamus from craniopharyngiomas and other tumours will result in disruption of POMC activation of PVH magnocellular oxytocin neurons and the release of oxytocin via α-melanocyte stimulating hormone and will thus abrogate the anorexigenic effect of oxytocin (116, 117). Moreover, VMH oxytocin administration has been shown to increase short-term energy expenditure and spontaneous physical activity (118, 119). Thus, oxytocin replacement may stimulate anorexigenic pathways that have been downregulated through hypothalamic damage and may play a role in upregulating pathways involved in energy expenditure. In patients with Prader-Willi syndrome (PWS), a syndrome associated with congenital hypothalamic damage, hypothalamic obesity and hyperphagia, intranasal oxytocin leads to a reduction in appetite drive, and improvements in socialization, anxiety, and repetitive behaviours. However, further, long-term studies with a larger population of participants are required to determine whether these effects are sustained (120, 121). Results from a placebo-controlled trial using Syntocinon (synthetic oxytocin) administered three times a day with meals in patients aged 10-21 years with hypothalamic obesity are awaited (ClinicalTrials.gov Identifier: NCT02849743).
Methionine Aminopeptidase Inhibitors are enzymes responsible for the removal of methionine from the amino-terminus of newly synthesized proteins (122). Increased expression of METAP2 gene is associated with various forms of cancer (123), and thus methionine aminopeptidase 2 inhibitors (MetAP2) were initially developed for treatment of cancer by preventing tumour proliferation (124). However, MetAP2 therapy also resulted in weight loss with increased adiponectin and decreased leptin, leading to decreased lipogenesis, increased fat oxidation, and increased lipolysis (125). The anti-obesity efficacy of MetAP2 inhibitors has been demonstrated in animal models of obesity and in humans at low doses that do not affect angiogenesis (126, 127). The precise mechanism for the anti-obesity effect of MetAP2 inhibitors is not clear but the ability of MetAP2 to suppress activity of extracellular signal regulated kinases 1 and 2 (ERK1/2) represents one of the key mechanisms for the observed anti-obesity effect (128). In the hypothalamic obesity mouse model, administration of the methionine aminopeptidase 2-inhibitor beloranib resulted in a reduction in body weight primarily related to a 30% reduction in food intake with improved glucose tolerance, reduced plasma insulin, and increased circulating levels of α-melanocyte stimulating hormone (129). A subsequent randomized, double-blind, placebo-controlled trial of 26-week beloranib therapy in patients with PWS demonstrated a reduction in food-seeking behavior and hyperphagia as well as weight loss secondary to an overall 85% reduction in fat mass, but the trial was stopped early due to an increase in serious thromboembolic events (2 fatal events of pulmonary embolism and 2 events of deep vein thrombosis) and mental health issues compared with placebo (130). In a Phase 2a, double-blind, placebo-controlled study of 14 adults with hypothalamic obesity, randomized to receive beloranib 1.8 mg or placebo subcutaneously twice weekly for 4 weeks with an optional 4-week open-label extension, patients in the treatment group lost weight that was sustained during the 4-week extension. No major adverse events were reported but the trial was short in duration (131). In the future, specific MetAP2 inhibitors developed to reduce the risk of thromboembolism may hold promise in the treatment of hypothalamic obesity.
Tesomet is an investigational fixed-dose combination therapy of tesofensine (a triple monoamine reuptake inhibitor) and metoprolol (a beta-1 selective blocker). Tesofensine acts on the brain to block reabsorption of three monoamine neurotransmitters - serotonin, noradrenaline and dopamine (132, 133), leading to a reduction in food cravings and subsequent reduction in weight. Tesofensine was originally developed to treat Alzheimers and Parkinson’s disease, but it was found to be ineffective and the unintended side effect of weight loss was observed in multiple clinical trials (134, 135). A phase II study of tesofensine in obese, nondiabetic subjects in 2008 demonstrated weight loss that was associated with increased doses of tesofensine, and beneficial effects on blood triglycerides, cholesterol, insulin and HbA1c were observed. Tesofensine has been combined with metoprolol to counteract previously observed side effects of tachycardia and hypertension at higher doses (136) thought to be related to an increase in sympathetic activity, which in fact may be of benefit to counteract the reduced sympathetic tone in patients with hypothalamic obesity (137). An ongoing phase 2 trial to evaluate the safety and efficacy of Tesomet in subjects 16 years of age or older with hypothalamic obesity is due to complete in May 2024 (ClinicalTrials.gov Identifier: NCT05147415).
Setmelanotide is a selective MC4 receptor agonist licensed for 6 years and older with obesity due to pro-opiomelanocortin deficiency, proprotein subtilisin/kexin type 1 (PCSK1) deficiency, and leptin receptor (LEPR) deficiency confirmed by genetic testing and is currently being trialled in other rare genetic disorders associated with obesity including Bardet-Biedl Syndrome and Alström Syndrome (138, 139). Setmelanotide stimulates MC4 receptors in the PVN and LH to activate anorexigenic pathways (140, 141). First year results of setmelanotide in patients with Bardet-Bield syndrome are promising, with weight loss and appetite suppression reported with no major adverse events (141). Results are awaited from a phase 2, open-label 20-week study to evaluate the safety and efficacy of Setmelanotide in subjects with hypothalamic obesity (ClinicalTrials.gov Identifier: NCT04725240), although drug response may be minimal due to potential damage of the PVN in these patients. A summary of the pharmacological interventions for hypothalamic obesity is provided in Table 1, and Figure 2 provides a schematic representation of the selective sites of targeted pharmacological intervention to suppress appetite and promote weight loss in patients with hypothalamic obesity.
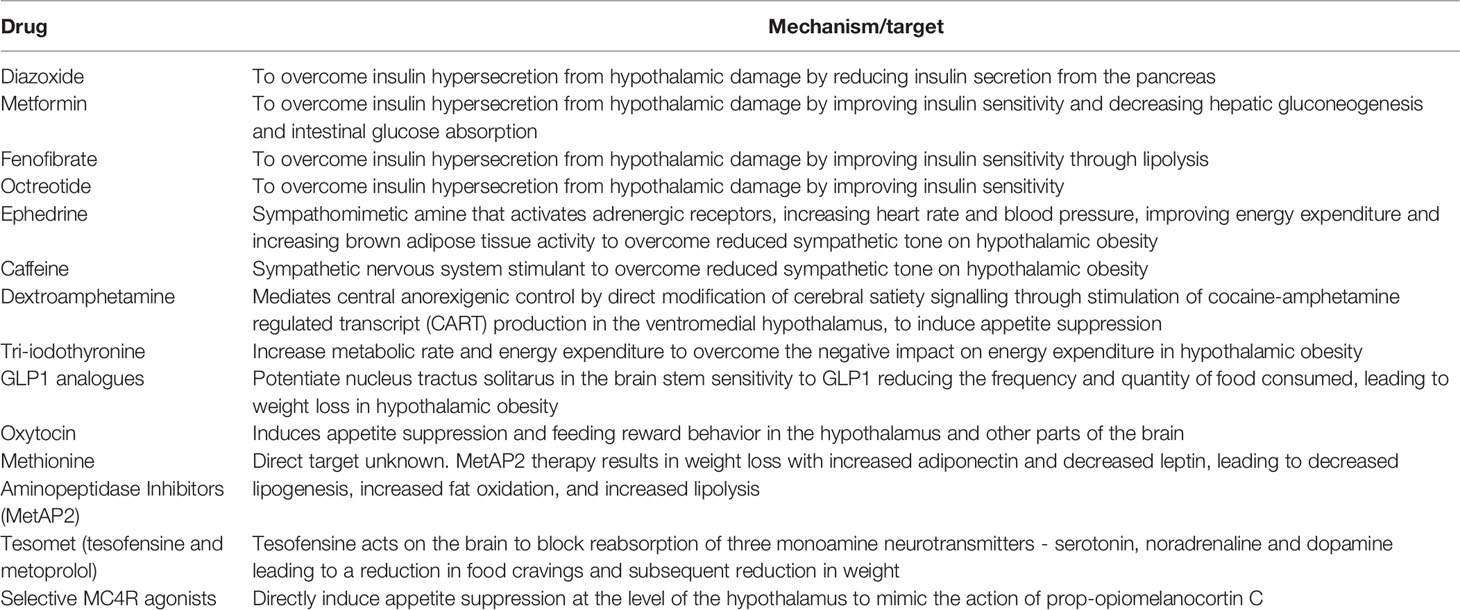
Table 1 Targeted action of pharmacological therapies in hypothalamic obesity.
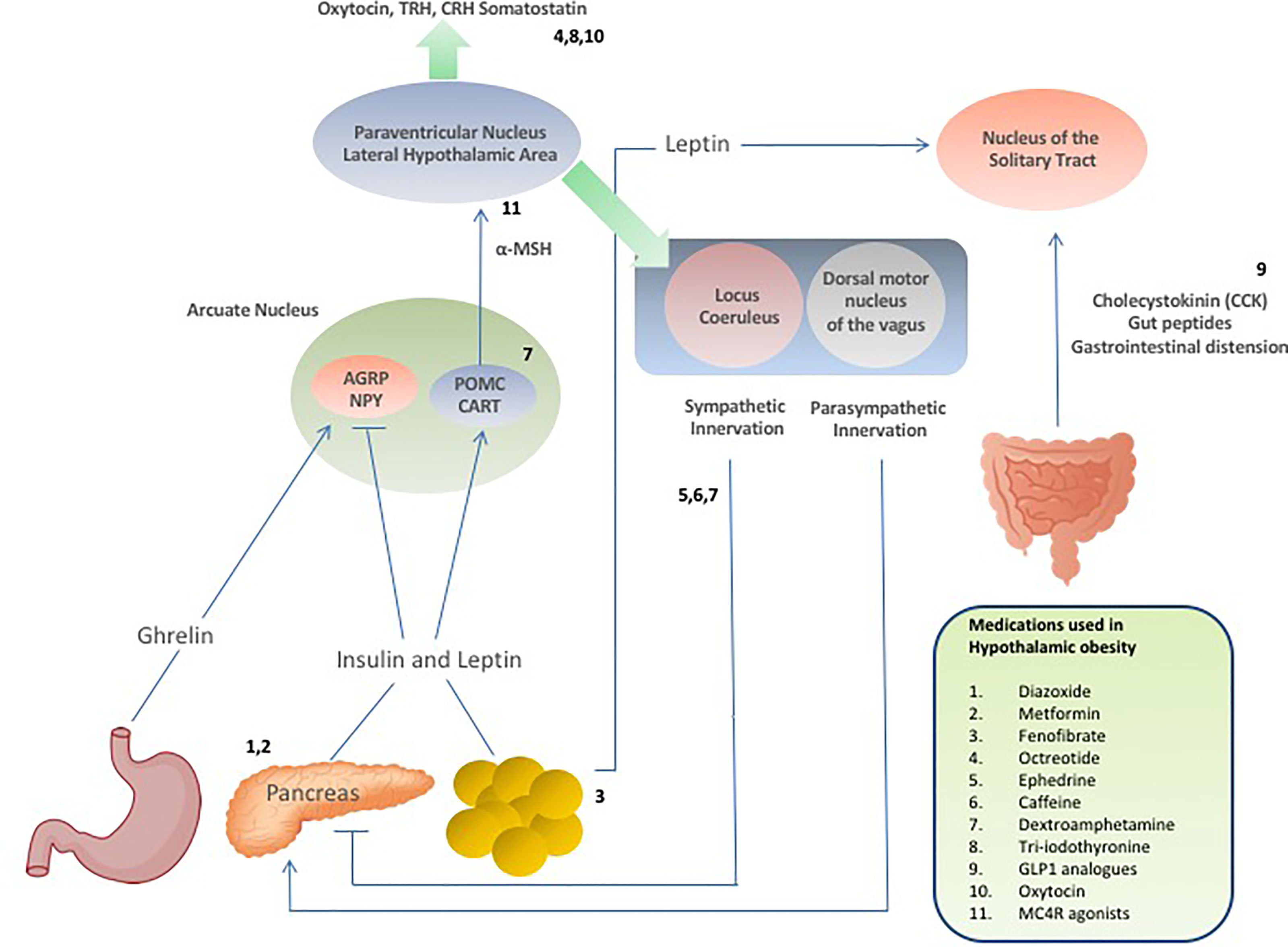
Figure 2 Schematic demonstrating hormonal and neuropeptide cerebral regulation of energy homeostasis and appetite and the selective sites of targeted drug action to suppress appetite and promote weight loss in patients with hypothalamic obesity.
A number of bariatric surgical approaches have been trialled in patients with hypothalamic obesity including Roux-en-Y Gastric Bypass (RYGB), Laproscopic Gastric Banding (LAGB) and sleeve gastrectomy (SG). In a number of patients, weight loss is relatively short-lived with patients returning to pre-surgical weights in studies with the longest follow-up duration, potentially reflecting the multi-faceted challenges of hypothalamic obesity that cannot be rectified by surgery, including reduced BMR and energy expenditure, low mood and increased daytime somnolence (142). The majority of studies reporting on bariatric surgery in patients with hypothalamic obesity include small number of patients. The largest study cohort reported includes 9 patients who underwent either LAGB (n = 6), SG (n = 4), and/or gastric bypass (n = 2). Two of these patients received SG after initial LAGB surgery, and one patient underwent gastric bypass after LAGB. No weight change occurred after LAGB or SG, but two patients who underwent gastric bypass surgery experienced weight loss after a mean follow-up period of 3 years (143). In a systematic review, van Iersel et al. report on 10 studies of bariatric surgery in patients with hypothalamic obesity. Of the studies that report on responders and non-responders to intervention, 19 of 24 patients experienced weight loss after bariatric surgery. The percentage of responders was 0.0% (0 of 4) for LAGB, 100.0% (6 of 6) for SG, and 92.9% (13 of 14) for gastric bypass surgery, although two patients regained weight after SG and RYGB (142).
To reduce insulin hypersecretion, truncal vagotomy has been reported in one patient resulting in 30 kg weight loss and reduced insulin secretion with delay in gastric emptying and foul smelling eructations as a side-effect (144). Other surgical approaches include the implantation of deep brain stimulation electrodes in a 19-year-old female to stimulate the nucleus accumbens, an approach that has been previously used in the treatment of morbid obesity (145, 146). DBS stimulation to the patient’s nucleus accumbens resulted in a sustained weight reduction and improved symptoms of hyperphagia after 14 months (147).
The challenge in achieving weight loss, modification in eating behaviour and improvement in energy expenditure in children and adults with acquired hypothalamic obesity reflects the complexity of hypothalamic control of energy homeostasis and feeding. Whist the hypothalamus is central to energy regulation and feeding, neuronal pathways to other parts of the brain are complex and extensive. In the last decade our understanding of central neuronal circuits that regulate feeding and satiety has led to the development of novel therapies capable of targeting afferent or efferent neuronal pathways that improve energy expenditure, modify insulin secretion and responsiveness, and upregulate anorexigenic and down regulate orexigenic neuropeptide pathways (148). The varied response to therapies in patients with hypothalamic obesity across studies may relate to the extent of hypothalamic damage, and future clinical trials will require enough patients to allow stratification to determine treatment response according to the location and extent of hypothalamic damage. Trials of this magnitude will require multinational collaboration to enroll enough patients to achieve relevant stratification. As our understanding of neuronal circuits controlling appetite and energy homeostasis outside of the hypothalamus increases, therapies should be developed that aim to circumvent damaged neuronal pathways, to target areas of the brain to modify eating behaviour and energy expenditure to promote weight loss. Given the thermogenic potential of brown adipose tissue, stimulating brown adipose tissue by novel targeted therapies may yield benefit in improving energy expenditure in patients with hypothalamic obesity. Trials using combinations of pharmacotherapeutic agents may yield more promising results but need to be considered carefully in the context of drug interactions and potential side effects. Combination therapies should potentially target one of the 6 domains of therapy recently proposed - hyperphagia, decreased energy expenditure, hyperinsulinemia, hypopituitarism, sleep disturbances, and psychosocial disorders (142). Bariatric surgical intervention may hold some promise in the treatment of hypothalamic obesity, but study numbers are small, and weight loss is potentially short-lived. The role of deep brain stimulation may also merit further consideration. Future trials should also consider a multidisciplinary approach by which novel therapies are combined with specific dietary intervention and exercise regimes (142, 149, 150). Ultimately, there is a significant need to act early to limit or reverse the rapid weight gain in patients with hypothalamic obesity to prevent the multiple associated physical and psychological co-morbidities that lead to early mortality in this patient population.
The author confirms being the sole contributor of this work and has approved it for publication.
The author declares that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. von Mohr U. Hypertrophie Der Hypophysis Cerebri Und Dadurch Bedingter Druck Auf Die Hirngrundfläche, Insbesondere Auf Die Sehnerven, Das Chiasma Desselben Und Den Linksseitigen Hirnschenkel. Wochenschr Ges Heilkd (1840) 6:556–71.
2. Nevin S. Hypothalamic and Pituitary Tumours. Postgrad Med J (1939) 15(163):210–9. doi: 10.1136/pgmj.15.163.210
4. Brobeck JR, Tepperman J, Long CNH. Experimental Hypothalamic Hyperphagia in the Albino Rat. Yale J Biol Med (1943) 15:831–53.
5. Kennedy G. The Role of Depot Fat in the Hypothalamic Control of Food Intake in the Rat. Proc R Soc Lond B (1953) 140:578–92. doi: 10.1098/rspb.1953.0009
6. Morton GJ, Meek TH, Schwartz MW. Neurobiology of Food Intake in Health and Disease. Nat Rev Neurosci (2015) 15:367–78. doi: 10.1038/nrn3745
7. Gao Q, Horvath TL. Neurobiology of Feeding and Energy Expenditure. Annu Rev Neurosci (2007) 30:367–39. doi: 10.1146/annurev.neuro.30.051606.094324
8. Timper K, Brüning JC. Hypothalamic Circuits Regulating Appetite and Energy Homeostasis: Pathways to Obesity. Dis Models Mech (2017) 10(6):679–89. doi: 10.1242/dmm.026609
9. Woods SC, Lotter EC, McKay LD, Porte D Jr. Chronic Intracerebroventricular Infusion of Insulin Reduces Food Intake and Body Weight of Baboons. Nature (1979) 282:503–5. doi: 10.1038/282503a0
10. Brüning JC, Gautam D, Burks DJ, Gillette J, Schubert M, Orban PC, et al. Role of Brain Insulin Receptor in Control of Body Weight and Reproduction. Science (2000) 289(5487):2122–5. doi: 10.1126/science.289.5487.2122
11. Roh E, Song D, Kim MS. Emerging Role of the Brain in the Homeostatic Regulation of Energy and Glucose Metabolism. Exp Mol Med (2016) 48:e216. doi: 10.1038/emm.2016.4
12. Miura Y, Gilon P, Henquin JC. Muscarinic Stimulation Increases Na+ Entry in Pancreatic B-Cells by a Mechanism Other Than the Emptying of Intracellular Ca2+ Pools. Biochem Biophys Res Commun (1996) 224(1):67–73. doi: 10.1006/bbrc.1996.0985
13. Rohner-Jeanrenaud F, Jeanrenaud B. Consequences of Ventromedial Hypothalamic Lesions Upon Insulin and Glucagon Secretion by Subsequently Isolated Perfused Pancreases in the Rat. J Clin Invest (1980) 65(4):902–10. doi: 10.1172/JCI109744
14. Arbuzova A, Murray D, McLaughlin S. MARCKS, Membranes, and Calmodulin: Kinetics of Their Interaction. Biochim Biophys Acta (1998) 1376(3):369–79. doi: 10.1016/S0304-4157(98)00011-2
15. Blondel O, Bell GI, Moody M, Miller RJ, Gibbons SJ. Creation of an Inositol 1,4,5-Trisphosphate-Sensitive Ca2+ Store in Secretory Granules of Insulin-Producing Cells. J Biol Chem (1994) 269(44):27167–70. doi: 10.1016/S0021-9258(18)46962-1
16. Lausier J, Diaz WC, Roskens V, LaRock K, Herzer K, Fong CG, et al. Vagal Control of Pancreatic ß-Cell Proliferation. Am J Physiol Endocrinol Metab (2010) 299(5):E786–93. doi: 10.1152/ajpendo.00202.2010
17. Berthoud HR, Jeanrenaud B. Acute Hyperinsulinemia and its Reversal by Vagotomy After Lesions of the Ventromedial Hypothalamus in Anesthetized Rats. Endocrinology (1979) 105(1):146–51. doi: 10.1210/endo-105-1-146
18. Inoue S, Bray GA. The Effects of Subdiaphragmatic Vagotomy in Rats With Ventromedial Hypothalamic Obesity. Endocrinology (1977) 100(1):108–14. doi: 10.1210/endo-100-1-108
19. Pandit R, Beerens S, Adan RAH. Role of Leptin in Energy Expenditure: The Hypothalamic Perspective. Am J Physiol Regul Integr Comp Physiol (2017) 312(6):R938–47. doi: 10.1152/ajpregu.00045.2016
20. Collins S. β-Adrenoceptor Signaling Networks in Adipocytes for Recruiting Stored Fat and Energy Expenditure. Front Endocrinol (Lausanne) (2012) 2:102. doi: 10.3389/fendo.2011.00102
21. Kiba T. Relationships Between the Autonomic Nervous System and the Pancreas Including Regulation of Regeneration and Apoptosis: Recent Developments. Pancreas (2004) 29(2):e51–8. doi: 10.1097/00006676-200408000-00019
22. Ollmann MM, Wilson BD, Yang YK, Kerns JA, Chen Y, GantzI, et al. Antagonism of Central Melanocortin Receptors In Vitro and In Vivo by Agouti-Related Protein. Science (1997) 278:135–8. doi: 10.1126/science.278.5335.135
23. Cowley MA, Smart JL, Rubinstein M, Cerdan MG, Diano S, Horvath TL. Leptin Activates Anorexigenic POMC Neurons Through a Neural Network in the Arcuate Nucleus. Nature (2001) 411:480–4. doi: 10.1038/35078085
24. Wu Q, Boyle MP, Palmiter RD. Loss of GABAergic Signaling by AgRP Neurons to the Parabrachial Nucleus Leads to Starvation. Cell (2009) 137:1225–34. doi: 10.1016/j.cell.2009.04.022
25. Shi YC, Lau J, Lin Z, Zhang H, Zhai L, Sperk G, et al. Arcuate NPY Controls Sympathetic Output and BAT Function via a Relay of Tyrosine Hydroxylase Neurons in the PVN. Cell Metab (2013) 17(2):236–48. doi: 10.1016/j.cmet.2013.01.006
26. Considine RV, Sinha MK, Heiman ML, Kriauciunas A, Stephens TW, Nyce MR, et al. Serum Immunoreactive-Leptin Concentrations in Normal-Weight and Obese Humans. N Engl J Med (1996) 334:292–29. doi: 10.1056/NEJM199602013340503
27. Morton GJ, Cummings DE, Baskin DG, Barsh GS, Schwartz MW. Central Nervous System Control of Food Intake and Body Weight. Nature (2006) 443:289–95. doi: 10.1038/nature05026
28. Cohen P, Zhao C, Cai X, Montez JM, Rohani SC, Feinstein P, et al. Selective Deletion of Leptin Receptor in Neurons Leads to Obesity. J Clin Invest (2001) 108:1113–21. doi: 10.1172/JCI200113914
29. Farooqi IS, Jebb SA, Langmack G, Lawrence E, Cheetham CH, Prentice AM, et al. Effects of Recombinant Leptin Therapy in a Child With Congenital Leptin Deficiency. N Engl J Med (1999) 341(12):879–84. doi: 10.1056/NEJM199909163411204
30. Sohn JW, Harris LE, Berglund ED, Liu T, Vong L, Lowell BB, et al. Melanocortin 4 Receptors Reciprocally Regulate Sympathetic and Parasympathetic Preganglionic Neurons. Cell (2013) 152:612–9. doi: 10.1016/j.cell.2012.12.022
31. Rezai-Zadeh K, Yu S, Jiang Y, Laque A, Schwartzenburg C, Morrison CD, et al. Leptin Receptor Neurons in the Dorsomedial Hypothalamus are Key Regulators of Energy Expenditure and Body Weight, But Not Food Intake. Mol Metab (2014) 3:681–93. doi: 10.1016/j.molmet.2014.07.008
32. Obici S, Feng Z, Karkanias G, Baskin DG, Rossetti L. Decreasing Hypothalamic Insulin Receptors Causes Hyperphagia and Insulin Resistance in Rats. Nat Neurosci (2002) 5(6):566–72. doi: 10.1038/nn0602-861
33. Ono H. Molecular Mechanisms of Hypothalamic Insulin Resistance. Int J Mol Sci (2019) 20(6):1317. doi: 10.3390/ijms20061317
34. Dodd GT, Tiganis T. Insulin Action in the Brain: Roles in Energy and Glucose Homeostasis. J Neuroendocrinol (2017) 29:(10). doi: 10.1111/jne.12513
35. Scherer T, Sakamoto K, Buettner C. Brain Insulin Signalling in Metabolic Homeostasis and Disease. Nat Rev Endocrinol (2021) 17(8):468–83. doi: 10.1038/s41574-021-00498-x
36. De Vile CJ, Grant DB, Hayward RD, Kendall BE, Neville BGR, Stanhope R. Obesity in Childhood Craniopharyngioma: Relation to Post-Operative Hypothalamic Damage Shown by Magnetic Resonance Imaging. J Clin Endocrinol Metab (1996) 81:2734–7. doi: 10.1210/jcem.81.7.8675604
37. Müller HL, Gebhardt U, Teske C, Faldum A, Zwiener I, Warmuth-Metz M, et al. Study Committee of KRANIOPHARYNGEOM 2000. Post-Operative Hypothalamic Lesions and Obesity in Childhood Craniopharyngioma: Results of the Multinational Prospective Trial KRANIOPHARYNGEOM 2000 After 3-Year Follow-Up. Eur J Endocrinol (2011) 165(1):17–24. doi: 10.1530/EJE-11-0158
38. Müller HL, Bueb K, Bartels U, Roth C, Harz K, Graf N, et al. Obesity After Childhood Craniopharyngioma–German Multicenter Study on Pre-Operative Risk Factors and Quality of Life. Klin Padiatr (2001) 213(4):244–9. doi: 10.1055/s-2001-16855
39. Muller HL. Childhood Craniopharyngioma. Recent Advances in Diagnosis, Treatment and Follow-Up. Horm Res (2008) 69(4):193–202. doi: 10.1159/000113019
40. Hoffman HJ, De Silva M, Humphreys RP, Drake JM, Smith ML, Blaser SI. Aggressive Surgical Management of Craniopharyngiomas in Children (1992). J Neurosurg (1992) 76:47–52. doi: 10.3171/jns.1992.76.1.0047
41. Ahmet A, Blaser S, Stephens D, Guger S, Rutkas JT, Hamilton J. Weight Gain in Craniopharyngioma–A Model for Hypothalamic Obesity. J Pediatr Endocrinol Metab (2006) 19:121–7. doi: 10.1515/JPEM.2006.19.2.121
42. Wu W, Sun Q, Zhu X, Xiang B, Zhang Q, Miao Q, et al. Risk Factors for Hypothalamic Obesity in Patients With Adult-Onset Craniopharyngioma: A Consecutive Series of 120 Cases. Front Endocrinol (Lausanne) (2021) 12:694213. doi: 10.3389/fendo.2021.694213
43. Van Iersel L, Meijneke RWH, Schouten-van Meeteren AYN, Reneman L, de Win MM, van Trotsenburg ASP, et al. The Development of Hypothalamic Obesity in Craniopharyngioma Patients: A Risk Factor Analysis in a Well-Defined Cohort. Pediatr Blood Cancer (2018) 65(5):e26911. doi: 10.1002/pbc.26911
44. Roth C, Wilken B, Hanefeld F, Schröter W, Leonhardt U. Hyperphagia in Children With Craniopharyngioma Is Associated With Hyperleptinemia and a Failure in the Down Regulation of Appetite. Eur J Endocrinol (1998) 138:89–91. doi: 10.1530/eje.0.1380089
45. Holmer H, Pozarek G, Wirfält E, Popovic V, Ekman B, Björk J, et al. Reduced Energy Expenditure and Impaired Feeding-Related Signals But Not High Energy Intake Reinforces Hypothalamic Obesity in Adults With Childhood Onset Craniopharyngioma. J Clin Endocrinol Metab (2010) 95(12):5395–402. doi: 10.1210/jc.2010-0993
46. Harz KJ, Müller HL, Waldeck E, Pudel V, Roth C. Obesity in Patients With Craniopharyngioma: Assessment of Food Intake and Movement Counts Indicating Physical Activity. J Clin Endocrinol Metab (2003) 88(11):5227–31. doi: 10.1210/jc.2002-021797
47. Shaikh MG, Grundy RG, Kirk JM. Reductions in Basal Metabolic Rate and Physical Activity Contribute to Hypothalamic Obesity. J Clin Endocrinol Metab (2008) 93:2588–93. doi: 10.1210/jc.2007-2672
48. Roth CL, Hunneman DH, Gebhardt U, Stoffel-Wagner B, Reinehr T, Müller HL. Reduced Sympathetic Metabolites in Urine of Obese Patients With Craniopharyngioma. Pediatr Res (2007) 61(4):496–501. doi: 10.1203/pdr.0b013e3180332cd6
49. Coutant R, Maurey H, Rouleau S, Mathieu E, Mercier P, Limal JM, et al. Defect in Epinephrine Production in Children With Craniopharyngioma: Functional or Organic Origin? J Clin Endocrinol Metab (2003) 88(12):5969–75. doi: 10.1210/jc.2003-030552
50. Caldarelli M, Massimi L, Tamburrini G, Cappa M, Di Rocco C. Long-Term Results of the Surgical Treatment of Craniopharyngioma: The Experience at the Policlinico Gemelli, Catholic University, Rome. Child’s Nervous System (2005) 21(8):747–57. doi: 10.1007/s00381-005-1186-5
51. Hoffman AR, Kuntze JE, Baptista J, Baum HB, Baumann GP, Biller BM, et al. Growth Hormone (GH) Replacement Therapy in Adult-Onset GH Deficiency: Effects on Body Composition in Men and Women in a Double-Blind, Randomized, Placebo-Controlled Trial. J Clin Endocrinol Metab (2004) 89(5):2048–56. doi: 10.1210/jc.2003-030346
52. Fulton JF, Bailey P. Tumors in the Region of the Third Ventricle: Their Diagnosis and Relation to Pathological Sleep. J Nervous Ment Dis (1929) 69:145–64. doi: 10.1097/00005053-192902000-00003
53. Cordani R, Veneruso M, Napoli F, Milanaccio C, Verrico A, Consales A, et al. Sleep Disturbances in Craniopharyngioma: A Challenging Diagnosis. J Neurol (2021) 268(11):4362–9. doi: 10.1007/s00415-021-10794-1
54. Müller HL, Handwerker G, Wollny B, Faldum A, Sörensen N. Melatonin Secretion and Increased Daytime Sleepiness in Childhood Craniopharyngioma Patients. J Clin Endocrinol Metab (2002) 87:3993–6. doi: 10.1210/jcem.87.8.8751
55. Müller HL, Müller-Stover S, Gebhardt U, Kolb R, Sörensen N, Handwerker G. Secondary Narcolepsy may be a Causative Factor of Increased Daytime Sleepiness in Obese Childhood Craniopharyngioma Patients. J Pediatr Endocrinol Metab 19(suppl (2006) 1):423–9. doi: 10.1055/s-2006-974095
56. Müller HL. Increased Daytime Sleepiness in Patients With Childhood Craniopharyngioma and Hypothalamic Tumor Involvement: Review of the Literature and Perspectives. Int J Endocrinol (2010) 519607. doi: 10.1155/2010/519607
57. O’Gorman CS, Simoneau-Roy J, Pencharz P, MacFarlane J, MacLusky I, Narang I, et al. Sleep-Disordered Breathing Is Increased in Obese Adolescents With Craniopharyngioma Compared With Obese Controls. J Clin Endocrinol Metab (2010) 95:2211–8. doi: 10.1210/jc.2009-2003
58. Jung CM, Melanson EL, Frydendall EJ, Perreault L, Eckel RH, Wright KP. Energy Expenditure During Sleep, Sleep Deprivation and Sleep Following Sleep Deprivation in Adult Humans. J Physiol 1;589(Pt (2011) 1):235–44. doi: 10.1113/jphysiol.2010.197517
59. Markwald RR, Melanson EL, Smith MR, Higgins J, Perreault L, Eckel RH, et al. Impact of Insufficient Sleep on Total Daily Energy Expenditure, Food Intake, and Weight Gain. Proc Natl Acad Sci USA (2013) 110(14):5695–700. doi: 10.1073/pnas.1216951110
60. Taheri S, Lin L, Austin D, Young T, Mignot E. Short Sleep Duration is Associated With Reduced Leptin, Elevated Ghrelin, and Increased Body Mass Index. PloS Med (2004) 1:210–7. doi: 10.1371/journal.pmed.0010062
61. Miller AL, Lumeng JC, LeBourgeois MK. Sleep Patterns and Obesity in Childhood. Curr Opin Endocrinol Diabetes Obes (2015) 22(1):41–7. doi: 10.1097/MED.0000000000000125
62. Poretti A, Grotzer MA, Ribi K, Schönle E, Boltshauser E. Outcome of Craniopharyngioma in Children: Long-Term Complications and Quality of Life. Dev Med Child Neurol. 2004;46:220–229, Elliott RE, Wisoff JH. Surgical Management of Giant Pediatric craniopharyngiomas. J Neurosurg Pediatr (2010) 46:403–16.
63. Schoenfeld A, Pekmezci M, Barnes MJ, Tihan T, Gupta N, Lamborn KR, et al. The Superiority of Conservative Resection and Adjuvant Radiation for Craniopharyngiomas. J Neurooncol (2012) 108(1):133–9. doi: 10.1007/s11060-012-0806-7
64. Spoudeas HA, Saran F, Pizer B. A Multimodality Approach to the Treatment of Craniopharyngiomas Avoiding Hypothalamic Morbidity: A UK Perspective. J Pediatr Endocrinol Metab (2006) Suppl 1:447–51.
65. Zacharia BE, Bruce SS, Goldstein H, Malone HR, Neugut AI, Bruce JN. Incidence, Treatment and Survival of Patients With Craniopharyngioma in the Surveillance, Epidemiology and End Results Program. Neuro Oncol (2012) 14(8):1070–8. doi: 10.1093/neuonc/nos142
66. Sterkenburg AS, Hoffmann A, Gebhardt U, Warmuth-Metz M, Daubenbüchel AM, Müller HL. Survival, Hypothalamic Obesity, and Neuropsychological/Psychosocial Status After Childhood-Onset Craniopharyngioma: Newly Reported Long-Term Outcomes. Neuro Oncol (2015) 17(7):1029–38. doi: 10.1093/neuonc/nov044
67. Beltran C, Naik M, Merchant TE. Dosimetric Effect of Target Expansion and Setup Uncertainty During Radiation Therapy in Pediatric Craniopharyngioma. Radiother Oncol (2010) 97(3):399–403. doi: 10.1016/j.radonc.2010.10.017
68. Boehling NS, Grosshans DR, Bluett JB, Palmer MT, Song X, Amos RA, et al. Dosimetric Comparison of Three-Dimensional Conformal Proton Radiotherapy, Intensity-Modulated Proton Therapy, and Intensity-Modulated Radiotherapy for Treatment of Pediatric Craniopharyngiomas. Int J Radiat Oncol Biol Phys (2012) 82(2):643–52. doi: 10.1016/j.ijrobp.2010.11.027
69. Beltran C, Roca M, Merchant TE. On the Benefits and Risks of Proton Therapy in Pediatric Craniopharyngioma. Int J Radiat Oncol Biol Phys Feb 1 (2012) 82(2):e281–7. doi: 10.1016/j.ijrobp.2011.01.005
70. Winkfield KM, Linsenmeier C, Yeap BY, Tarbell NJ, Yock TI. Proton Radiotherapy for Childhood Craniopharyngioma: Initial Clinical Outcomes. Int J Radiat Oncol Biol Phys (2008) 72(1):S496. doi: 10.1016/j.ijrobp.2008.06.1453
71. Chang AL, Fitzek MM, Kruter LE, Roach K, Thornton AF. Outcomes of Pediatric Craniopharyngioma Treated With Proton Radiation Therapy. Int J Radiat Oncol Biol Phys (2009) 75(3):S513. doi: 10.1016/j.ijrobp.2009.07.1170
72. Indelicato DJ, Rotondo R, Flampouri S, Kowalchik K, Sandler E, Aldana P, et al. Proton Therapy for Craniopharyngioma: Early Clinical Outcomes. Int J Radiat Oncol Biol Phys (2012) 84(3):S634. doi: 10.1016/j.ijrobp.2012.07.1692
73. Merchant TE, Hua CH, Sabin ND, Ezell SE, Madey MA, Wu S, et al. Necrosis, Vasculopathy, and Neurological Complications After Proton Therapy for Childhood Craniopharyngioma: Results From a Prospective Trial and a Photon Cohort Comparison. Int J Rad Biol Oncol Phys (2016) 96(2):S12–121. doi: 10.1016/j.ijrobp.2016.06.294
74. Kortmann RD. Different Approaches in Radiation Therapy of Craniopharyngioma. Front Endocrinol (Lausanne) (2011) 2:100. doi: 10.3389/fendo.2011.00100
75. Bishop AJ, Greenfield B, Mahajan A, Paulino AC, Okcu MF, Allen PK, et al. Proton Beam Therapy Versus Conformal Photon Radiation Therapy for Childhood Craniopharyngioma: Multi-Institutional Analysis of Outcomes, Cyst Dynamics, and Toxicity. Int J Radiat Oncol Biol Phys (2014) 90(2):354–61. doi: 10.1016/j.ijrobp.2014.05.051
76. Brauner R, Serreau R, Souberbielle JC, Pouillot M, Grouazel S, Recasens C, et al. Diazoxide in Children With Obesity After Hypothalamic-Pituitary Lesions: A Randomized, Placebo-Controlled Trial. J Clin Endocrinol Metab (2016) 101(12):4825–33. doi: 10.1210/jc.2016-2126
77. Hamilton JK, Conwell LS, Syme C, Ahmet A, Jeffery A, Daneman D. Hypothalamic Obesity Following Craniopharyngioma Surgery: Results of a Pilot Trial of Combined Diazoxide and Metformin Therapy. Int J Pediatr Endocrinol (2011) 1):417949. doi: 10.1155/2011/417949
78. Ramakrishnan SK, Russo L, Ghanem SS, Patel PR, Oyarce AM, Heinrich G, et al. Fenofibrate Decreases Insulin Clearance and Insulin Secretion to Maintain Insulin Sensitivity. J Biol Chem (2016) 291(46):23915–24. doi: 10.1074/jbc.M116.745778
79. Kalina MA, Wilczek M, Kalina-Faska B, Skała-Zamorowska E, Mandera M, Małecka Tendera E. Carbohydrate-Lipid Profile and Use of Metformin With Micronized Fenofibrate in Reducing Metabolic Consequences of Craniopharyngioma Treatment in Children: Single Institution Experience. J Pediatr Endocrinol Metab (2015) 28(1-2):45–51. doi: 10.1515/jpem-2014-0425
80. Orskov L, Møller N, Bak JF, Pørksen N, Schmitz O. Effects of the Somatostatin Analog, Octreotide, on Glucose Metabolism and Insulin Sensitivity in Insulin-Dependent Diabetes Mellitus. Metabolism (1996) 45(2)::211–7. doi: 10.1016/S0026-0495(96)90056-6
81. Lustig RH, Hinds PS, Ringwald-Smith K, Christensen RK, Kaste SC, Schreiber RE, et al. Octreotide Therapy of Pediatric Hypothalamic Obesity: A Double-Blind, Placebo-Controlled Trial. J Clin Endocrinol Metab (2003) 88(6):2586–92. doi: 10.1210/jc.2002-030003
82. Craig E. Supplemental NDA 21-008/S-018. Clinical Review. Available at: https://www.fda.gov/media/115272/download.
83. Silverstone JT, Stunkard AJ. The Anorectic Effect of Dexamphetamine Sulphate. Br J Pharmacol Chemother (1968) 33(3):513–22. doi: 10.1111/j.1476-5381.1968.tb00499.x
84. Mead E, Atkinson G, Richter B, Metzendorf MI, Baur L, Finer N, et al. Drug Interventions for the Treatment of Obesity in Children and Adolescents. Cochrane Database Syst Rev (2016) 11(11):CD012436. doi: 10.1002/14651858.CD012436
85. Danielsson P, Janson A, Norgren S, Marcus C. Impact Sibutramine Therapy in Children With Hypothalamic Obesity or Obesity With Aggravating Syndromes. J Clin Endocrinol Metab (2007) 92(11):4101–6. doi: 10.1210/jc.2007-0826
86. Shekelle PG, Hardy ML, Morton SC, Maglione M, Mojica WA, Suttorp MJ, et al. Efficacy and Safety of Ephedra and Ephedrine for Weight Loss and Athletic Performance: A Meta-Analysis. JAMA (2003) 289(12):1537–45. doi: 10.1001/jama.289.12.1537
87. Jimenez-Munoz CM, López M, Albericio F, Makowski K. Targeting Energy Expenditure-Drugs for Obesity Treatment. Pharmaceut (Basel) (2021) 14(5):435. doi: 10.3390/ph14050435
88. Loh R, Kingwell B, Carey A. Human Brown Adipose Tissue as a Target for Obesity Management; Beyond Cold-Induced Thermogenesis. Obes Rev (2017) 18:1227–42. doi: 10.1111/obr.12584
89. Dulloo AG, Seydoux J, Girardier L. Paraxanthine (Metabolite of Caffeine) Mimics Caffeine’s Interaction With Sympathetic Control of Thermogenesis. Am J Physiol (1994) 267(5 Pt 1):E801–804. doi: 10.1152/ajpendo.1994.267.5.E801
90. Clark KS, Coleman C, Shelton R, Heemstra LA, Novak CM. Caffeine Enhances Activity Thermogenesis and Energy Expenditure in Rats. Clin Exp Pharmacol Physiol (2019) 46(5):475–82. doi: 10.1111/1440-1681.13065
91. Greenway FL, Bray GA. Treatment of Hypothalamic Obesity With Caffeine and Ephedrine. Endocr Pract (2008) 14(6):697–703. doi: 10.4158/EP.14.6.697
92. Faraone SV, Biederman J, Morley CP, Spencer TJ. Effect of Stimulants on Height and Weight: A Review of the Literature. J Am Acad Child Adolesc Psychiatry (2008) 47((9):994–1009. doi: 10.1097/CHI.0b013e31817e0ea7
93. Seiden LS, Sabol KE, Ricaurte GA. Amphetamine: Effects on Catecholamine Systems and Behavior. Annu Rev Pharmacol Toxicol (1993) 33:639–77. doi: 10.1146/annurev.pa.33.040193.003231
94. Rowley HL, Butler SA, Prow MR, Dykes SG, Aspley S, Kilpatrick IC, et al. Comparison of the Effects of Sibutramine and Other Weight-Modifying Drugs on Extracellular Dopamine in the Nucleus Accumbens of Freely Moving Rats. Synapse (2000) 38(2):167–76. doi: 10.1002/1098-2396(200011)38:2<167::AID-SYN8>3.0.CO;2-W
95. Roth CL, Aylward E, Liang O, Kleinhans NM, Pauley G, Schur EA. Functional Neuroimaging in Craniopharyngioma: A Useful Tool to Better Understand Hypothalamic Obesity? Obes Facts (2012) 5(2):243–53. doi: 10.1159/000338695
96. Ismail D, O’Connell MA, Zacharin MR. Dexamphetamine Use for Management of Obesity and Hypersomnolence Following Hypothalamic Injury. J Pediatr Endocrinol Metab (2006) 19(2):129–34. doi: 10.1515/JPEM.2006.19.2.129
97. Denzer C, Denzer F, Lennerz BS, Vollbach H, Lustig RH, Wabitsch M. Treatment of Hypothalamic Obesity With Dextroamphetamine: A Case Series. Obes Facts (2019) 12(1):91–102. doi: 10.1159/000495851
98. Mason PW, Krawiecki N, Meacham LR. The Use of Dextroamphetamine to Treat Obesity and Hyperphagia in Children Treated for Craniopharyngioma. Arch Pediatr Adolesc Med (2002) 156(9):887–92. doi: 10.1001/archpedi.156.9.887
99. Fernandes JK, Klein MJ, Ater JL, Kuttesch JF, Vassilopoulou-Sellin R. Triiodothyronine Supplementation for Hypothalamic Obesity. Metabolism (2002) 51(11):1381–3. doi: 10.1053/meta.2002.35591
100. van Santen HM, Schouten-Meeteren AY, Serlie M, Meijneke RW, van Trotsenburg AS, Verberne H, et al. Effects of T3 Treatment on Brown Adipose Tissue and Energy Expenditure in a Patient With Craniopharyngioma and Hypothalamic Obesity. J Pediatr Endocrinol Metab (2015) 28(1-2):53–7. doi: 10.1515/jpem-2014-0337
101. Yavuz S, Salgado Nunez Del Prado S, Celi FS. Thyroid Hormone Action and Energy Expenditure. J Endocr Soc (2019) 3(7):1345–56. doi: 10.1210/js.2018-00423
102. Cawthon CR, de la Serre CB. The Critical Role of CCK in the Regulation of Food Intake and Diet-Induced Obesity. Peptides (2021) 138:170492. doi: 10.1016/j.peptides.2020.170492
103. Zanchi D, Depoorter A, Egloff L, Haller S, Mählmann L, Lang UE, et al. The Impact of Gut Hormones on the Neural Circuit of Appetite and Satiety: A Systematic Review. Neurosci Biobehav Rev (2017) 80:457–75. doi: 10.1016/j.neubiorev.2017.06.013
104. Barrachina MD, Martinez V, Wang L, Wei JY, Tache Y. Synergistic Interaction Between Leptin and Cholecystokinin to Reduce Short-Term Food Intake in Lean Mice. Proc Natl Acad Sci USA (1997) 94:10455–60. doi: 10.1073/pnas.94.19.10455
105. McMinn JE, Sindelar DK, Havel PJ, Schwartz MW. Leptin Deficiency Induced by Fasting Impairs the Satiety Response to Cholecystokinin. Endocrinology (2000) 141:4442–8. doi: 10.1210/endo.141.12.7815
106. Elfers CT, Simmons JH, Roth CL. Glucagon-Like Peptide-1 Agonist Exendin-4 Leads to Reduction of Weight and Caloric Intake in a Rat Model of Hypothalamic Obesity. Horm Res Paediatr (2012) 78(1):47–53. doi: 10.1159/000338464
107. Zoicas F, Droste M, Mayr B, Buchfelder M, Schöfl C. GLP-1 Analogues as a New Treatment Option for Hypothalamic Obesity in Adults: Report of Nine Cases. Eur J Endocrinol (2013) 168(5):699–706. doi: 10.1530/EJE-12-0997
108. Lomenick JP, Buchowski MS, Shoemaker AH. A 52-Week Pilot Study of the Effects of Exenatide on Body Weight in Patients With Hypothalamic Obesity. Obes (Silver Spring) (2016) 24(6):1222–5. doi: 10.1002/oby.21493
109. van Schaik J, Begijn DGA, van Iersel L, Vergeer Y, Hoving EW, Peeters B, et al. Experiences With Glucagon-Like Peptide-1 Receptor Agonist in Children With Acquired Hypothalamic Obesity. Obes Facts (2020) 13(4):361–70. doi: 10.1159/000509302
110. Roth CL, Perez FA, Whitlock KB, Elfers C, Yanovski JA, Shoemaker AH, et al. A Phase 3 Randomized Clinical Trial Using a Once-Weekly Glucagon-Like Peptide-1 Receptor Agonist in Adolescents and Young Adults With Hypothalamic Obesity. Diabetes Obes Metab (2021) 23(2):363–73. doi: 10.1111/dom.14224
111. Perez FA, Elfers C, Yanovski JA, Shoemaker AH, Abuzzahab MJ, Roth CL. MRI Measures of Hypothalamic Injury are Associated With Glucagon-Like Peptide-1 Receptor Agonist Treatment Response in People With Hypothalamic Obesity. Diabetes Obes Metab (2021) 23(7):1532–41. doi: 10.1111/dom.14366
112. Kanoski SE, Grill HJ. Hippocampus Contributions to Food Intake Control: Mnemonic, Neuroanatomical, and Endocrine Mechanisms. Biol Psychiat (2017) 81(9):748–56. doi: 10.1016/j.biopsych.2015.09.011
113. Hsu EA, Miller JL, Perez FA, Roth CL. Oxytocin and Naltrexone Successfully Treat Hypothalamic Obesity in a Boy Post-Craniopharyngioma Resection. J Clin Endocrinol Metab (2018) 103(2):370–5. doi: 10.1210/jc.2017-02080
114. Liu CM, Spaulding MO, Rea JJ, Noble EE, Kanoski SE. Oxytocin and Food Intake Control: Neural, Behavioral, and Signaling Mechanisms. Int J Mol Sci (2021) 22(19):10859. doi: 10.3390/ijms221910859
115. Ong ZY, Alhadeff AL, Grill HJ. Medial Nucleus Tractus Solitarius Oxytocin Receptor Signaling and Food Intake Control: The Role of Gastrointestinal Satiation Signal Processing. Am J Physiol Regul Integr Comp Physiol (2015) 308:R800–6. doi: 10.1152/ajpregu.00534.2014
116. Caquineau C, Leng G, Guan XM, Jiang M, van der Ploeg L, Douglas AJ. Effects of Alpha-Melanocyte-Stimulating Hormone on Magnocellular Oxytocin Neurones and Their Activation at Intromission in Male Rats. J Neuroendocrinol (2006) 18:685–91. doi: 10.1111/j.1365-2826.2006.01465.x
117. Maejima Y, Yokota S, Nishimori K, Shimomura K. The Anorexigenic Neural Pathways of Oxytocin and Their Clinical Implication. Neuroendocrinology (2018) 107:91–104. doi: 10.1159/000489263
118. Noble EE, Billington CJ, Kotz CM, Wang C. Oxytocin in the Ventromedial Hypothalamic Nucleus Reduces Feeding and Acutely Increases Energy Expenditure. Am J Physiol Regul Integr Comp Physiol (2014) 307:R737–45. doi: 10.1152/ajpregu.00118.2014
119. Blevins JE, Graham JL, Morton GJ, Bales KL, Schwartz MW, Baskin DG, et al. Chronic Oxytocin Administration Inhibits Food Intake, Increases Energy Expenditure, and Produces Weight Loss in Fructose-Fed Obese Rhesus Monkeys. Am J Physiol Regul Integr Comp Physiol (2015) 308(5):R431–8. doi: 10.1152/ajpregu.00441.2014
120. Miller JL, Tamura R, Butler MG, Kimonis V, Sulsona C, Gold JA, et al. Oxytocin Treatment in Children With Prader-Willi Syndrome: A Double-Blind, Placebo-Controlled, Crossover Study. Am J Med Genet A (2017) 173(5):1243–50. doi: 10.1002/ajmg.a.38160
121. Damen L, Grootjen LN, Juriaans AF, Donze SH, Huisman TM, Visser JA, et al. Oxytocin in Young Children With Prader-Willi Syndrome: Results of a Randomized, Double-Blind, Placebo-Controlled, Crossover Trial Investigating 3 Months of Oxytocin. Clin Endocrinol (Oxf) (2021) 94(5):774–85. doi: 10.1111/cen.14387
122. Solbiati J, Chapman-Smith A, Miller JL, Miller CG, Cronan JE. Processing of the N Termini of Nascent Polypeptide Chains Requires Deformylation Prior to Methionine Removal. J Mol Biol (1999) 290(3):607–14. doi: 10.1006/jmbi.1999.2913
123. Selvakumar P, Lakshmikuttyamma A, Das U, Pati HN, Dimmock JR, Sharma RK. NC2213: A Novel Methionine Aminopeptidase 2 Inhibitor in Human Colon Cancer HT29 Cells. Mol Cancer (2009) 8:65. doi: 10.1186/1476-4598-8-65
124. Heinrich T, Seenisamy J, Becker F, Blume B, Bomke J, Dietz M, et al. Identification of Methionine Aminopeptidase-2 (MetAP-2) Inhibitor M8891: A Clinical Compound for the Treatment of Cancer. J Med Chem (2019) 62(24):11119–34. doi: 10.1021/acs.jmedchem.9b01070
125. Joharapurkar AA, Dhanesha NA, Jain MR. Inhibition of the Methionine Aminopeptidase 2 Enzyme for the Treatment of Obesity. Diabetes Metab Syndr Obes (2014) 7:73–84. doi: 10.2147/DMSO.S56924
126. White HM, Acton AJ, Considine RV. The Angiogenic Inhibitor TNP-470 Decreases Caloric Intake and Weight Gain in High-Fat Fed Mice. Obes (Silver Spring) (2012) 20(10):2003–9. doi: 10.1038/oby.2012.87
127. Hughes TE, Kim DD, Marjason J, Proietto J, Whitehead JP, Vath JE. Ascending Dose-Controlled Trial of Beloranib, a Novel Obesity Treatment for Safety, Tolerability, and Weight Loss in Obese Women. Obes (Silver Spring) (2013) 21(9):1782–8. doi: 10.1002/oby.20356
128. Datta B, Majumdar A, Datta R, Balusu R. Treatment of Cells With the Angiogenic Inhibitor Fumagillin Results in Increased Stability of Eukaryotic Initiation Factor 2-Associated Glycoprotein, P67, and Reduced Phosphorylation of Extracellular Signal-Regulated Kinases. Biochemistry (2004) 43(46):14821–31. doi: 10.1021/bi049172p
129. Elfers CT, Roth CL. Robust Reductions of Excess Weight and Hyperphagia by Beloranib in Rat Models of Genetic and Hypothalamic Obesity. Endocrinology (2017) 158(1):41–55. doi: 10.1210/en.2016-1665
130. McCandless SE, Yanovski JA, Miller J, Fu C, Bird LM, Salehi P, et al. Effects of MetAP2 Inhibition on Hyperphagia and Body Weight in Prader-Willi Syndrome: A Randomized, Double-Blind, Placebo-Controlled Trial. Diabetes Obes Metab (2017) 19(12):1751–61. doi: 10.1111/dom.13021
131. Shoemaker A, Proietto J, Abuzzahab MJ, Markovic T, Malloy J, Kim DD. A Randomized, Placebo-Controlled Trial of Beloranib for the Treatment of Hypothalamic Injury-Associated Obesity. Diabetes Obes Metab (2017) 19(8):1165–70. doi: 10.1111/dom.12928
132. Lehr T, Staab A, Tillmann C, Nielsen EO, Trommeshauser D, Schaefer HG, et al. Contribution of the Active Metabolite M1 to the Pharmacological Activity of Tesofensine In Vivo: A Pharmacokinetic- Pharmacodynamic Modelling Approach. Br J Pharmacol (2008) 153(1):164–74. doi: 10.1038/sj.bjp.0707539
133. Hansen HH, Jensen MM, Overgaard A, Weikop P, Mikkelsen JD. Tesofensine Induces Appetite Suppression and Weight Loss With Reversal of Low Forebrain Dopamine Levels in the Diet-Induced Obese Rat. Pharmacol Biochem Behav (2013) 110:265–71. doi: 10.1016/j.pbb.2013.07.018
134. Hauser RA, Salin L, Juhel N, Konyago VL. Randomized Trial of the Triple Monoamine Reuptake Inhibitor NS 2330 (Tesofensine) in Early Parkinson’s Disease. Mov Disord (2007) 22(3):359–65. doi: 10.1002/mds.21258
135. Astrup A, Meier DH, Mikkelsen BO, Villumsen JS, Larsen TM. Weight Loss Produced by Tesofensine in Patients With Parkinson’s or Alzheimer’s Disease. Obes (Silver Spring) (2008) 16(6):1363–9. doi: 10.1038/oby.2008.56
136. Astrup A, Madsbad S, Breum L, Jensen TJ, Kroustrup JP, Larsen TM. Effect of Tesofensine on Bodyweight Loss, Body Composition, and Quality of Life in Obese Patients: A Randomised, Double-Blind, Placebo-Controlled Trial. Lancet (2008) 372(9653):1906–13. doi: 10.1016/S0140-6736(08)61525-1
137. Bentzen BH, Grunnet M, Hyveled-Nielsen L, Sundgreen C, Lassen JB, Hansen HH. Antihypertensive Treatment Preserves Appetite Suppression While Preventing Cardiovascular Adverse Effects of Tesofensine in Rats. Obes (Silver Spring) (2013) 21(5):985–92. doi: 10.1002/oby.20122
138. Kim GW, Lin JE, Blomain ES, Waldman SA. Antiobesity Pharmacotherapy: New Drugs and Emerging Targets. Clin Pharmacol Ther (2014) 95(1):53–66. doi: 10.1038/clpt.2013.204
139. Haws RM, Gordon G, Han JC, Yanovski JA, Yuan G, Stewart MW. The Efficacy and Safety of Setmelanotide in Individuals With Bardet-Biedl Syndrome or Alström Syndrome: Phase 3 Trial Design. Contemp Clin Trials Commun (2021) 22:100780. doi: 10.1016/j.conctc.2021.100780
140. Markham A. Setmelanotide: First Approval. Drugs (2021) 81(3):397–403. doi: 10.1007/s40265-021-01470-9
141. Haws R, Brady S, Davis E, Fletty K, Yuan G, Gordon G, et al. Effect of Setmelanotide, a Melanocortin-4 Receptor Agonist, on Obesity in Bardet-Biedl Syndrome. Diabetes Obes Metab (2020) 22(11):2133–40. doi: 10.1111/dom.14133
142. van Iersel L, Brokke KE, Adan RAH, Bulthuis LCM, van den Akker ELT, van Santen HM. Pathophysiology and Individualized Treatment of Hypothalamic Obesity Following Craniopharyngioma and Other Suprasellar Tumors: A Systematic Review. Endocr Rev (2019) 40(1):193–235. doi: 10.1210/er.2018-00017
143. Weismann D, Pelka T, Bender G, Jurowich C, Fassnacht M, Thalheimer A, et al. Bariatric Surgery for Morbid Obesity in Craniopharyngioma. Clin Endocrinol (Oxf) (2013) 78(3):385–90. doi: 10.1111/j.1365-2265.2012.04409.x
144. Smith DK, Sarfeh J, Howard L. Truncal Vagotomy in Hypothalamic Obesity. Lancet (1983) 1(8337):1330–1. doi: 10.1016/S0140-6736(83)92437-6
145. Pisapia JM, Halpern CH, Williams NN, Wadden TA, Baltuch GH, Stein SC. Deep Brain Stimulation Compared With Bariatric Surgery for the Treatment of Morbid Obesity: A Decision Analysis Study. Neurosurg Focus (2010) 29(2):E15. doi: 10.3171/2010.5.FOCUS10109
146. Oterdoom DLM, van Dijk G, Verhagen MHP, Jiawan VCR, Drost G, Emous M, et al. Therapeutic Potential of Deep Brain Stimulation of the Nucleus Accumbens in Morbid Obesity. Neurosurg Focus (2018) 45(2):E10. doi: 10.3171/2018.4.FOCUS18148
147. Harat M, Rudaś M, Zieliński P, Birska J, Sokal P. Nucleus Accumbens Stimulation in Pathological Obesity. Neurol Neurochir Pol (2016) 50(3):207–10. doi: 10.1016/j.pjnns.2016.01.014
148. Coulter AA, Rebello CJ, Greenway FL. Centrally Acting Agents for Obesity: Past, Present, and Future. Drugs (2018) 78(11):1113–32. doi: 10.1007/s40265-018-0946-y
149. Tessaris D, Matarazzo P, Tuli G, Tuscano A, Rabbone I, Spinardi A, et al. Multidisciplinary Approach for Hypothalamic Obesity in Children and Adolescents: A Preliminary Study. Children (Basel) (2021) 8(7):531. doi: 10.3390/children8070531
Keywords: hypothalamic obesity, craniopharyngioma, suprasellar tumors, hypothalamus, insulin, GLP1, oxytocin, methionine aminopeptidase inhibitors
Citation: Dimitri P (2022) Treatment of Acquired Hypothalamic Obesity: Now and the Future. Front. Endocrinol. 13:846880. doi: 10.3389/fendo.2022.846880
Received: 31 December 2021; Accepted: 07 March 2022;
Published: 06 April 2022.
Edited by:
Hanneke M. Van Santen, Wilhelmina Children’s Hospital, NetherlandsReviewed by:
Eva Erfurth, Lund University, SwedenCopyright © 2022 Dimitri. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Paul Dimitri, cGF1bC5kaW1pdHJpQG5ocy5uZXQ=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.