
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Endocrinol., 30 March 2022
Sec. Cancer Endocrinology
Volume 13 - 2022 | https://doi.org/10.3389/fendo.2022.841118
This article is part of the Research TopicInsights in Cancer Endocrinology: 2021View all 14 articles
 Giuseppe Giuffrida1
Giuseppe Giuffrida1 Valeria D’Argenio2,3
Valeria D’Argenio2,3 Francesco Ferraù1,4*Vito Alessandro Lasorsa3,5Francesca Polito6Federica Aliquò1Marta Ragonese1Oana Ruxandra Cotta4Ylenia Alessi4Rosaria Oteri6
Francesco Ferraù1,4*Vito Alessandro Lasorsa3,5Francesca Polito6Federica Aliquò1Marta Ragonese1Oana Ruxandra Cotta4Ylenia Alessi4Rosaria Oteri6 Federica Di Maggio3,5
Federica Di Maggio3,5 Alessio Asmundo7
Alessio Asmundo7 Petronilla Daniela Romeo1
Petronilla Daniela Romeo1 Federica Spagnolo4
Federica Spagnolo4 Lucio Pastore3,5
Lucio Pastore3,5 Filippo Flavio Angileri7
Filippo Flavio Angileri7 Mario Capasso3,5
Mario Capasso3,5 Salvatore Cannavò1,4†
Salvatore Cannavò1,4† M’Hammed Aguennouz6†
M’Hammed Aguennouz6†Pituitary adenomas (PAs), usually benign lesions, can sometimes present with “aggressive” features (rapid growth, local invasiveness, scarce response to conventional treatments). Despite the fact that a few genetic alterations have been associated to this clinical behavior, the role of epigenetic modifications, mainly methylation and miRNAs activity, is now opening new frontiers in this field. We evaluated the methylation profile of 21 PA (11 GH-omas, 10 nonfunctioning tumors—NFPAs) samples from TNS surgery and 5 normal pituitaries, collected at our neurosurgery between 2015 and 2017. DNA was extracted and sequenced, selecting 184,841 target regions. Moreover, methylation profiles were correlated with demographic, radiological, and clinicopathological features. NFPAs showed higher methylation levels vs. GH-omas, with 178 differentially methylated regions (DMRs) mainly consisting of noncoding and intronic sequences, and mostly localized in the open sea regions. We also found three hypermethylated genes (C7orf50, GNG7, and BAHCC1) involved in tumorigenesis processes and potentially influencing pituitary tumor pathophysiology. Among the clinicopathological features, only the maximum diameter resulted significantly higher in NFPAs. Our data provide further evidence of the complex epigenetic background of pituitary tumors. In line with the current literature, we confirmed a significant prevalence of hypermethylation in NFPAs vs. GH-omas, whose pathophysiological consequence is yet to be defined.
Pituitary adenomas (PAs) are distinguished by the presence of hormonal secretion and/or the expression of cell line-specific growth factors (1, 2). Although the presence of distant metastases is linked to the definition of pituitary carcinomas, even PAs can show an aggressive biological behavior, being characterized by local invasion, rapid proliferation, and scarce response to conventional treatments in up to 45% of cases (3, 4). In this context, there is a growing amount of data about PA (epi)genetic features predicting their behavior and/or their treatment response/relapse. In terms of genetics, for example, germinal mutations of the AIP (aryl hydrocarbon receptor-interacting protein) gene are associated to the development of familial isolated pituitary adenomas (FIPA), with early onset, higher aggressiveness, and resistance to somatostatin analogs (SSAs) (5). Similarly, the mutations involving the MEN1 oncosuppressor, linked to the homonymous syndrome, are associated with PAs in 15–50% of affected patients and a higher frequency of macroadenomas, that in 1/3 of cases are more invasive than non-MEN1 tumors (6). On the other hand, there is some evidence about somatic changes in sporadic pituitary tumors. These mutations can consist of sequence changes, qualitative alterations of chromosomes, or modification in their copy numbers, but they are often aspecific and infrequent, suggesting an additional oncogenic contribution from nonmutational factors (7, 8). Epigenetic modifications, which take place without altering the DNA sequences, comprehend both the alterations in mRNA transcription (nucleotides methylation, histones acetylation) and the different expression of long noncoding mRNAs (lncmRNAs) and, as also recently described by our group, microRNAs (miRNAs) (9). Methylation, that is, the apposition of methyl groups on DNA chains by specific enzymes—the DNA methyl-n-transferases (DMNTs)—is a physiological mechanism acting to silence specific genes in order to regulate their expression (8). Many DMNT isoforms are known, but DMNT1 and 3A are overexpressed in more aggressive PTs, with the DMNT1 more frequently found in macroadenomas (10). On the contrary, it seems that this DMNT hyperactivity would lead to hypomethylation of other DNA regions, which consequently result to being overtranscripted, as already observed in tumorigenesis processes (7). In such a context, the search for epigenetic changes can be crucial in order to identify potential predictors of clinical behavior and/or treatment response, as well as targets for tailored therapies (8). For example, in the case of GH-secreting PAs causing acromegaly, the presence of parameters predicting treatment response would be useful to avoid potentially inefficacious therapies that could have an impact on other conditions like glucose metabolism, or to guide drug dosing (11–14). Furthermore, even environmental factors, especially pollutants with endocrine disrupting activities, which have been increasingly demonstrated to have a role in PA pathophysiology, could have an impact on tumor epigenetic profile and molecular features, and consequently on their biological behavior (15–18).
This study aimed to assess the methylation status, as compared to normal pituitary tissues, of nonfunctioning pituitary adenomas (NFPAs) and GH-omas, and to correlate the methylation status of NFPAs and GH-omas with their epidemiological and clinicopathological features.
Twenty-one PA samples (11 GH-omas, 10 NFPAs) were collected by the Neurosurgery Unit of Messina University Hospital between 2015 and 2017. All patients gave their written informed consent to the study. Demographic information, including sex, age, and clinical data, of the enrolled patients are summarized in Table 1. Five nontumor pituitary tissue samples were collected through an autopsy of subjects who died due to non-endocrine causes. The research protocol was approved by the local ethics committee. For DNA methylation analyses (see below), genomic DNA was extracted from each collected tissue using the QIAamp DNA mini kit (Qiagen), according to the manufacturer’s instructions.
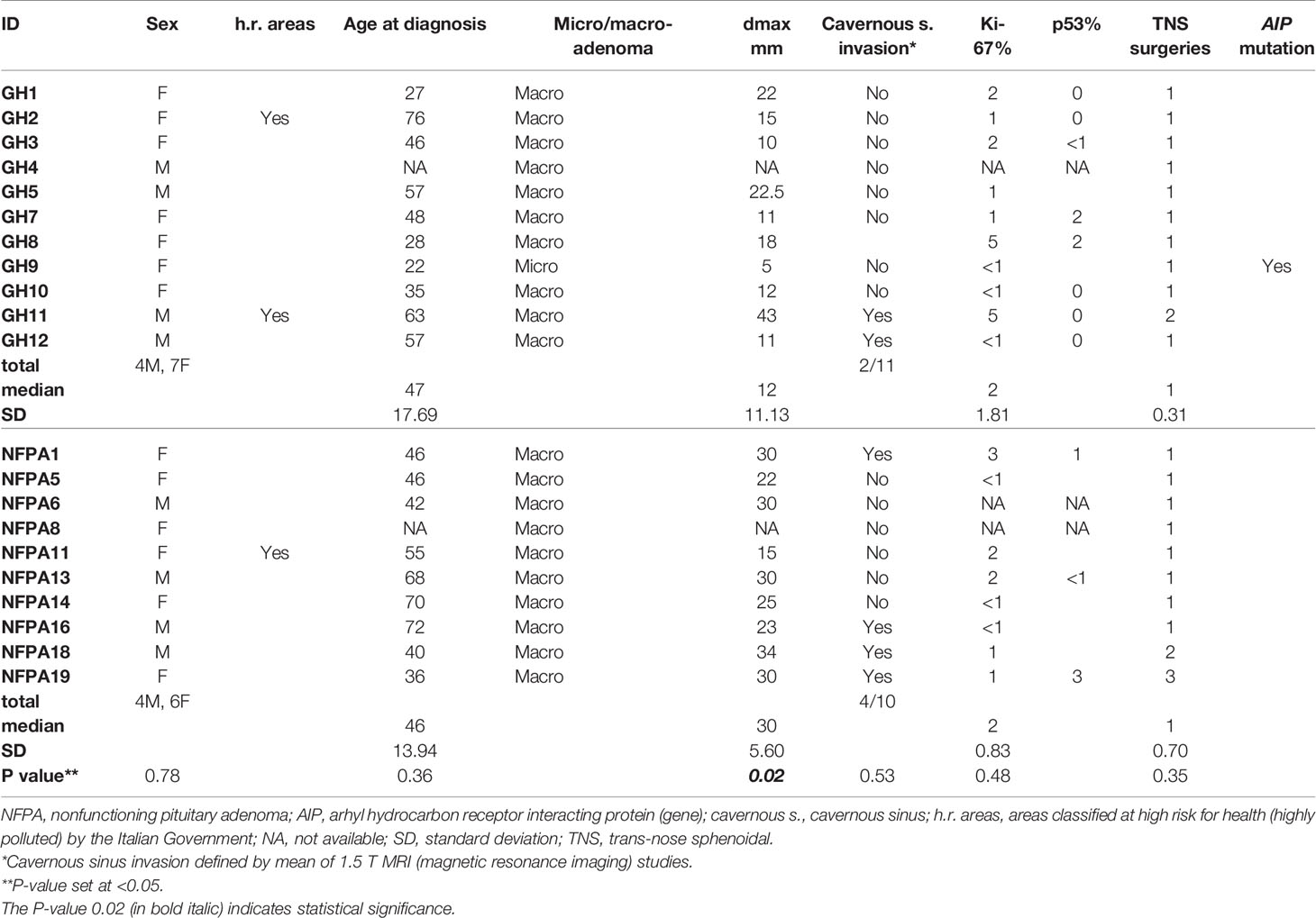
Table 1 Demographic, radiological, and clinicopathological features of the studied cohort of patients.
The whole-genome DNA methylation profiling of 11 GH-omas, 10 NFPAs, and 5 normal pituitaries was carried out with the TruSeq Methyl Capture EPIC library preparation protocol followed by next-generation sequencing (NGS) (Illumina). Genomic DNAs underwent picogreen quantification on the Qubit fluorimetric system (dsDNA HS assay, Life Technologies) in order to obtain 1,000 ng of DNA/sample for subsequent library preparation. Libraries were carried out following the manufacturer’s instructions. In detail, 1,000 ng of each sample was sonicated (Covaris M220 System) to obtain small DNA fragments (average size 150–200 bp) as assessed by the Tape Station quality check system (High Sensitivity D1000, Agilent). After end repair and adapter ligation, these DNA fragments were enriched by hybridization with specific capture probes. The enriched fragments were bisulfite converted and amplified. These obtained libraries were checked for quantity (Qubit, dsDNA HS assay, Life Technologies) and quality (Tape Station, High Sensitivity D1000, Agilent) before sequencing. NGS was carried out on the Illumina HiSeq1500 System. Up to 12 different DNA libraries, each univocally identified by a specific barcode or index, were pooled in equimolar amounts and sequenced in 4 different lanes in order to avoid analytical biases.
A multistep bioinformatic pipeline was used to analyze the obtained sequencing data. First, sequencing reads quality check was carried out using the FASTQC software. For the sequence alignment and downstream quantification steps, we used the “QuasR” (version 1.22.1), an R-Bioconductor package installed on R (version 3.5.0) (19). The QuasR package integrates the functionality of several R packages for genomic intervals and alignment files manipulation and external software [e.g., Bowtie (20)] for the real sequence alignment. Sequence mapping was carried out using a BS pre-processed reference genome version (version GRCh37/hg19) that was generated exploiting the “QuasR” functions. The tool was run with default parameters. PCR duplicated reads were removed during the alignment. Subsequently, to quantify methylated and unmethylated cytosines in each sample, we used the function qMeth of the QuasR package. We considered a total of 437,792 genomic regions (mean length of 245 bp; from 2 to 8,131 bp) covered in the manifest file of the TruSeq Methyl Capture EPIC Library Prep kit (Illumina). In this step, the tool collapses the information of individual cytosines by query region. Finally, the methylation fraction of each target region for each sample was obtained as the ratio between methylated reads and the total number of aligned reads and ranged between 0 (un-methylated) and 1 (totally methylated). The tools were run with default parameters.
For the differential methylation analysis, we considered GH-oma and NFPAs samples. To improve the consistency of the results, we kept all the target regions (n=184,841) that were covered in all the GH-oma and NFPAs samples and calculated the methylation fold enrichment (as Log2) between NFPAs and GH-omas mean methylation. Statistical significance was calculated with t-test, and P-values were corrected for multiple testing with the Bonferroni method. Significant results were considered if the Bonferroni adjusted P value was less than 0.05 and if the Log2 fold change was above or below 0.5. Functional annotation, to get distances from the nearest genes and other genomic information, of differentially methylated regions was performed with the ANNOVAR software.
The statistical evaluation of demographic and clinicopathological parameters was performed by means of t-test and chi-square test (with Yates’ correction), and significance was set at a P value less than 0.05.
Sequencing reads quality evaluation returned good-quality paired-end reads of length between 35 and 101 bp. The percentage of reads with quality scores above 20 (Q20) and above 30 (Q30) was 98.91 and 94.42, respectively (Supplementary Figure 1A). The mean base quality was 36.20 (Supplementary Figure 1B). The overall mapping rate ranged between 71.3% and 78.95%, as reported in Supplementary Figure 1C.
The methylation quantification step was performed by restricting the analysis to the genomic regions covered in the TruSeq Methyl Capture EPIC manifest file provided by Illumina. A total of 437,792 regions with a mean length of 245 bp (from 2 to 8,131 bp), being equivalent to 301,525 non-CpGs, 61,703 CpG islands, 18,707 N_shelf, 19,038 N_shore, 19,201 S_shelf, and 17,618 S_shore regions, were analyzed. The general positioning of these sequences in the human genome is summarized in Figure 1A.
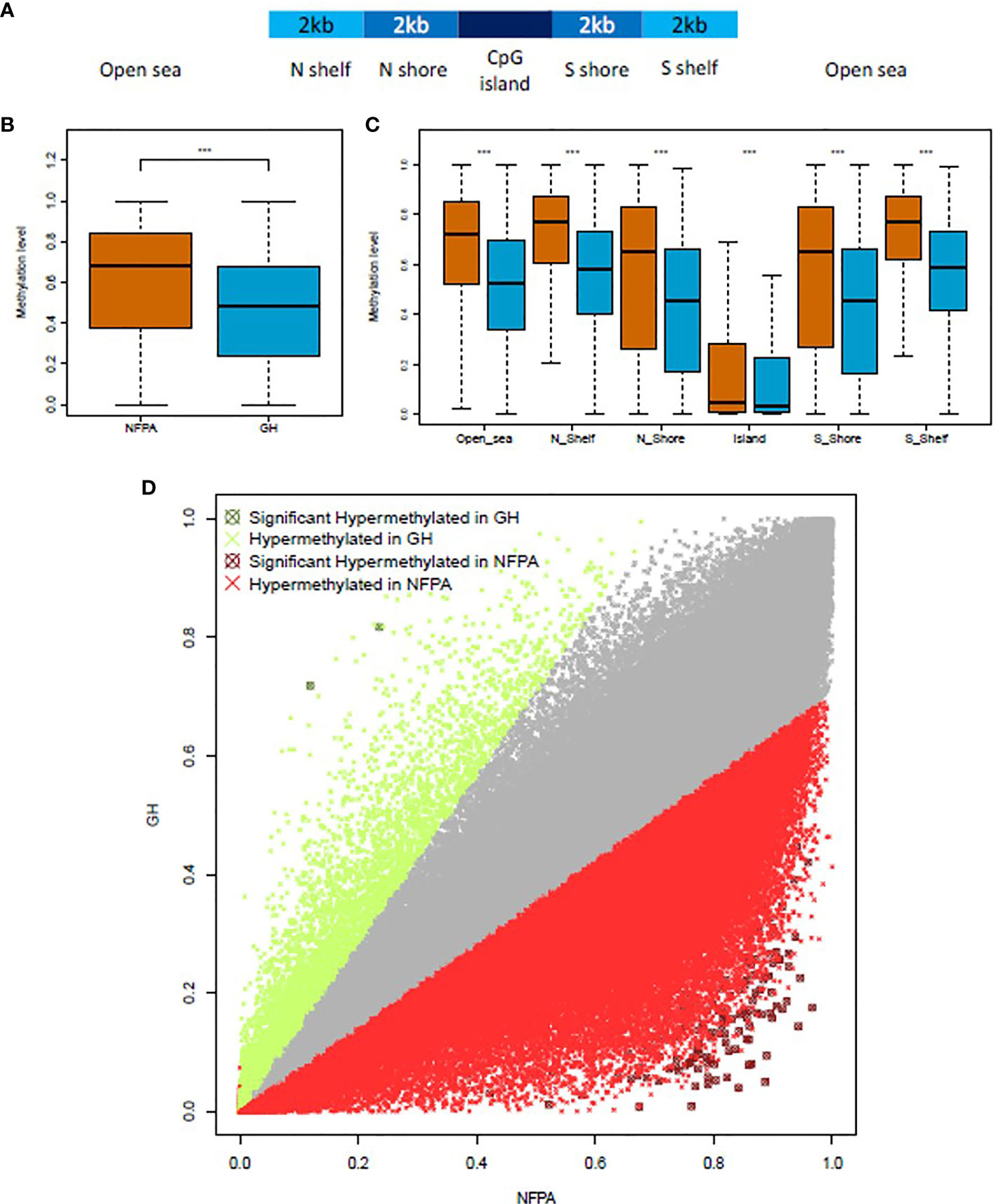
Figure 1 NFPAs are hypermethylated when compared to GH-secreting tumors. (A) Schematic representation of the CpG-related region annotation. (B) Boxplot showing the global level of methylation in nonfunctioning pituitary adenomas (NFPAs—brown) and GH-secreting (blue) tumors. (C) Methylation levels in CpG-related regions. (D) The scatterplot compares the methylation levels of 184,841 regions in NFPAs (x-axis) and GH-omas (y-axis). Data points in gray did not pass the Log2 fold change cutoffs. Points in red or green passed the Log2 fold change cutoffs. Points in dark red or dark green were significantly hypermethylated in NFPAs or GH, respectively. Mann–Whitney test was used in (B, C) t-test was used in (D). P < 0.0001 (***).
For the differential methylation analysis, we compared GH-oma and NFPAs samples. To improve the consistency of the results, we kept all the target regions (n=184,841) that were covered in all the GH-oma and NFPAs samples. As reported in Figure 1B and Supplementary Figure 1D, globally, NFPAs showed higher methylation levels (median=0.68) compared to GH-secreting pituitary tumors (median=0.48) (P<2.2x10-16; Mann–Whitney test). Moreover, we evaluated the methylation levels of CpG-related regions and found that NFPAs were hypermethylated, as compared to GH-secreting pituitary tumors. In particular, we found hypermethylation in Open sea (median=0.7210 vs median=0.5236, respectively); in N Shelfs (median=0.7707 vs median=0.5786, respectively); in N Shores (median=0.6534 vs median=0.4517, respectively); in Islands (median=0.04584 vs median=0.030993, respectively); in S Shores (median=0.6526 vs median=0.4569, respectively), and in S Shelfs (median=0.7748 vs median=0.5855, respectively) (Figure 1C; P<2.2x10-16; Mann–Whitney test).
Next, we calculated the methylation fold enrichment (as Log2) between NFPA and GH-oma samples to identify the differentially methylated regions (DMRs).
NFPAs showed a distinct methylation profile as compared to GH-omas. In particular, we obtained 178 target regions that were differentially methylated (corrected P-value ≤0.05; Log2 Fold Change ± 0.5) between the two tumor types (Figure 1D and Supplementary Table 1). Of note, only two regions resulted significantly hypermethylated in GH-omas compared to NFPAs (Figure 1D).
We classified as H-DMRs (high differentially methylated regions) those regions having Log2 FC above 2 or below -2. This category counted a total of 111 DMRs (62.36%), one of which was hypomethylated in NFPAs as compared to GH-oma. DMRs with Log2 FC values between 2 and -2 were deemed as L-DMRs (low differentially methylated regions). This class included a total of 67 DMRs (37.64%), one of which was hypermethylated in NFPAs as compared to GH-oma.
Subsequently, we functionally annotated the list of DMRs and found that the majority of them mapped in noncoding regions (92.7%), of which 64.85% were intronic sequences (Figure 2A). Moreover, we found that DMRs within the coding regions were significantly hypermethylated in NFPAs (median beta value=0.83 and 0.25 for NFPAs and GH-oma, respectively; P = 1.92x10-07). Accordingly, also noncoding sequences were significantly hypermethylated in NFPAs (median beta value=0.81 and 0.16 for NFPAs and GH-oma, respectively; P = 4.38x10-53) (Figure 2B, and Supplementary Table 1).
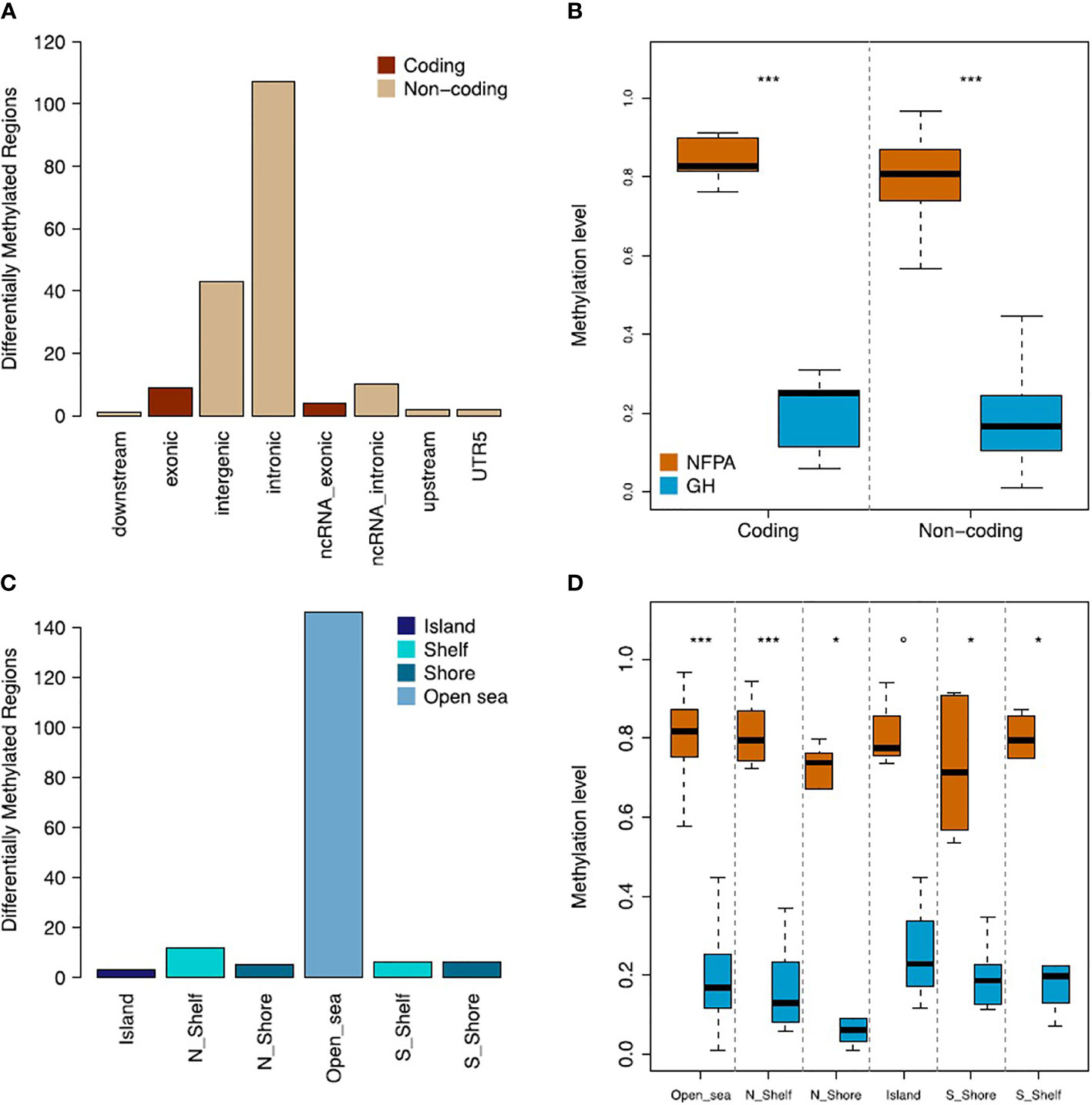
Figure 2 NFPAs and GH tumors are differentially methylated. (A) Barplot showing the gene annotation of the 178 DMRs. (B) Boxplot reporting the comparison of methylation levels of coding and noncoding regions between nonfunctioning pituitary adenomas (NFPAs) and GH tumors. (C) Barplot showing the CpG-related annotation of the 178 DMRs. (D) Boxplot reporting the comparison of methylation levels of CpG-related regions between NFPAs and GH tumors. Mann–Whitney test was used in (B, D) P = 0.1 (°), P < 0.01 (*), P < 0.0001 (***).
The CpG-centric annotation of DMRs (see Figure 1A) highlighted that the large majority of DMRs were annotated as Open sea (82.02%) (Figure 2C). These DMRs were significantly hypermethylated in NFPAs (median beta value=0.82 and 0.17 for NFPAs and GH-oma, respectively; P = 4.60x10-47). Overall, as reported in Figures 2B, D, we observed generalized hypermethylation in NFPAs as compared to GH-oma (P < 4.38x10-53).
To assess if the DMR-related genes were involved in specific pathways, we conducted a Gene Ontology Biological Process Enrichment Analysis using the web app ShinyGO (v0.741) (PMID: 31882993) and set the FDR cutoff to 0.05. Of note, among the significantly enriched GO terms, we found biological processes related to cell and neuron development (Figure 3 and Supplementary Table 2).
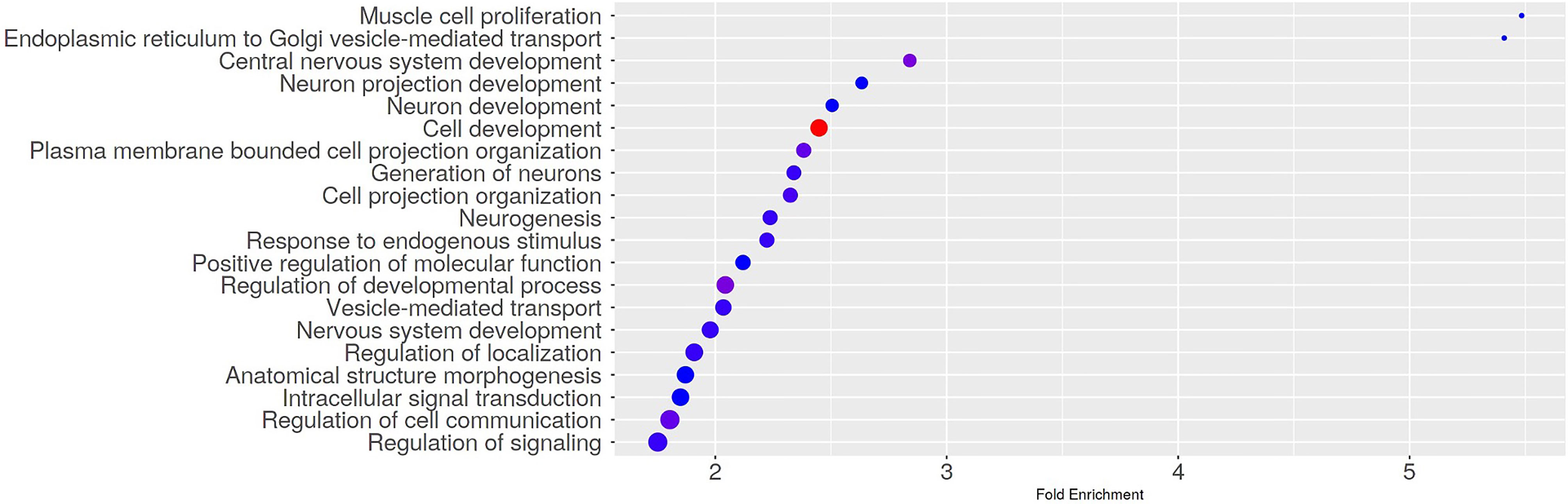
Figure 3 Gene Ontology Biological Process Enrichment results. The dotplot shows the top 20 significantly enriched GO terms. The Gene Ontology Biological Process Enrichment Analysis was run using the web app ShinyGO (v0.741) with the FDR cutoff set to 0.05.
With regard to the correlation between methylation profile and demographic (including the degree of pollution of the residence area) or clinicopathological features of pituitary tumors, no statistically significant differences were observed between GH-omas and NFPAs, except for the maximum tumor diameter (Table 1), which resulted significantly higher in the latter group (median ± SD: 30 ± 5.6 vs 12 ± 11.13 mm; P = 0.02). Of note, 4 out of 10 (40%) patients with NFPAs presented with a neuroradiologically documented invasion of cavernous sinus vs 2 out of 11 (18.2%) in the GH-oma group, but this difference was not statistically significant (Table 1).
Finally, we found three hypermethylated genes (C7orf50, GNG7, and BAHCC1), involved in tumorigenesis processes, whose role could be related to pituitary tumor pathophysiology.
The role of epigenetic modifications, especially methylation, has increased its importance in the genetic background of sporadic pituitary tumors in the last few years. In fact, only a few somatic mutations with significant effects are known, such as GNAS alterations (this gene codifies for the α stimulatory subunit of G proteins) in GH-omas or USP8 mutations in ACTH-omas causing EGFR overexpression and promoting corticotroph cells growth and ACTH hypersecretion (21). Also, our group recently demonstrated a novel somatic deletion in exon 10 of the AHR (aryl hydrocarbon receptor) gene in patients affected by GH-omas, whose role could be related to an altered AHR/AIP pathway favoring tumorigenesis (22). On the other hand, in pituitary tumors, it has been observed that methylation is preferentially concentrated in the so-called CpG islands, sequences of about 500 bp strictly connected to promoter regions, leading to the silencing of genes often involved in cell cycle regulation (23). Of note, a lot of oncosuppressors can be found among these genes, as for the couple CDKN2A/Rb1, whose deregulation can cause altered apoptosis regulation (8). In fact, methylation of CDNK2A leads to a reduced expression of p16, which, in turn, determines pRb phosphorylation and cell cycle progression through the activation of E2F transcription factors (24). However, the hardest challenge in the assessment of pituitary tumor methylation profile is to find a consistent “signature,” potentially useful as biological/prognostic/therapeutic marker. As it emerges from many studies—also confirmed by our findings—NFPAs tend to present with a higher degree of methylation compared to GH-omas, although some invasive NFPAs can even be characterized by hypomethylation (7, 23, 25, 26). Besides, NFPAs more frequently harbor CDKN2A/p16 alterations, inversely from what were observed in GH-secreting pituitary tumors, which often do not express pRb (8). CDKN2A methylation has been related to the pituitary tumor volume, grade, and patients’ age, with higher methylation levels in macroadenomas (8). Furthermore, p27 hypermethylation was found in ACTH-omas, while EML2, HOXB1, and RHOD epigenetic modifications have been reported in NFPAs, GH-omas, and PRL-omas, respectively (7, 8). Gu et al. demonstrated that methylation would lead to the downregulation of genes like GALNT9, CDH1, and CDH13 (E-cadherin and H-cadherin, respectively), involved into cellular adhesion processes, and potentially linked to the development of invasiveness (26). The same study observed that DMRs were located not only in CpG islands but also in the gene body in 40% of the cases (26). Accordingly, in our study, only 3 DMRs were found in known CpG islands, while the remaining alterations were found in genome open sea regions. Other genome elements prone to methylation are lncRNAs, RNA fragments of about 200 nucleotides functionally similar to the respective coding RNAs (27). In this regard, the downregulation following the hypermethylation of MEG3, which interacts with p53 and acts as oncosuppressor, has been found in gonadotropinomas (8, 27, 28). Another lncRNA, called C5orf66-AS1, regulates several genes, including PAQR7, a progesterone receptor that causes a progesterone A-B receptor-independent reduction in GnRH, whose expression has been found to have a role in progression and invasion of null-cell pituitary adenomas (24, 29).
With regard to our findings, DMR analysis revealed a prevalence of methylation of noncoding sequences, including lncRNAs (Figure 2A). Most of the methylation profile alterations were localized in open-sea regions more than involving promoters, with an inverse trend if compared to the literature (Figure 2C). Anyway, the prevalence of hypermethylation in NFPAs vs GH-omas has been confirmed (Figures 2B, D). Interestingly, we found hypermethylation of 3 known CpG islands belonging to genes thought to have a role in tumorigenesis processes: C7orf50, GNG7, and BAHCC1.
C7orf50 is a ubiquitarian gene whose product is implicated in the assembling of ribosomal RNA to the nucleus, even if part of its sequences could also originate some regulatory miRNAs. Its full function is still unknown, although some evidence suggests it could bind the Sp1 transcriptional factor, which has several regulatory functions (i.e., cell cycle, apoptosis, etc.) including an interaction with AHR favoring the ubiquitination and consequent degradation of the estrogenic receptor α (ERα) in murine breast and uterine cancer (30).
GNG7 is a gene located on chromosome 19 codifying for the γ7 subunit of guanin-binding G proteins, which is involved in contact-mediated cell growth blockade and acts as an oncosuppressor (31), whose promoter methylation has been found in many cases of head/neck cancer and associated with higher tumor volume and lesser metastatic potential (31). Similarly, Xu et al. observed methylation-mediated, reduced expression of GNG7 in renal clear cell carcinoma. In this case, methylation, not present in normal tissue, led to the impairment of the mTOR1 signaling pathway and was linked to a higher stadium/grade of the disease and a reduced overall survival (32).
BAHCC1 is a chromatin transcriptional silencer, implied into cell replication and transcriptional regulation mechanisms. Amplifications and deletions of this gene would make it potentially part of aberrant cell regeneration processes linked to the development of liver cancer, according to still-not-well-known mechanisms, but possibly due to downstream alterations in the signaling pathways (33). However, there are few data about epigenetic modifications of BAHCC1, although an experimental study by Gitik et al. found an increase in its methylation in the dorsal hippocampus of mice treated with nicotine before adolescence, in an animal model correlating substance abuse in the age of adolescence (or alcohol exposure in utero) with addiction. These chromatin modifications were linked to the development of cognitive deficits in the adult age, otherwise preventable by the simultaneous administration of choline (34).
With regards to the correlation between methylation profile and clinicopathological features, the higher maximum diameter of NFPAs could hypothetically be linked to a higher proliferative potential in this subtype of pituitary tumors. The same could apply to the higher frequency of cavernous sinus invasion—although not statistically significant—in the NFPA group. These findings are in line with the study by Gu et al. in which hypermethylation altered the expression profile of cell adhesion proteins (26). Finally, no relationship was observed between the methylation status and the degree of pollution of the residence area of our patients, but the number of pituitary tumors evaluated in this study is very small. Furthermore, the hypermethylation of C7orf50, a gene interacting with AHR, should be investigated in larger cohorts of patients. In fact, better defining such an interaction could add new information to the complex role played by AHR, which along the years we demonstrated to significantly influence morphology, secretion, and therapeutic response in GH-omas (16–18, 35).
In conclusion, our data provide further evidence on the complexity of the epigenetic background of pituitary tumors. We found a significant prevalence of hypermethylation in NFPAs, as compared to GH-omas, whose pathophysiological consequence is yet to be defined. Further studies are needed to clarify the role and relevance of C7orf50, GNG7, and BAHCC1 genes—which have been found to be methylated—in pituitary tumor biology, oncogenesis, and clinical expression.
The data presented in the study are available on the European Nucleotide Archive (ENA) repository. Accession nr: PRJEB50807.
The studies involving human participants were reviewed and approved by the Ethics Committee of The Province of Messina. The patients/participants provided their written informed consent to participate in this study.
GG and FF wrote the paper and organized research data. GG, MR, OC, FS, and YA contributed to the gathering of clinical data. FP, FAl, PR, RO, and M'HA (Messina) and VD’A, FM, LP, VL, and MC (Naples) performed DNA sequencing and methylome analysis. FAn provided samples from patients by means of TNS surgery, while AA provided healthy pituitary samples from autopsies. FF, SC, M'HA, and MC conceived the entire study and revised the final paper version. All authors contributed to the article and approved the submitted version.
This work was supported by the following grants of the Italian government: Ricerca Finalizzata 2013: “Role of environment-gene interaction in etiology and promotion of pituitary tumours” (code: RF-2013-02356201); Programma di Ricerca di Interesse Nazionale 2015: “Epidemiological determinants, molecular mechanisms and clinical criteria of treatment outcome and resistance in pituitary disease syndromes” (code: PRIN-2015-2015ZHKFTA); and Progetto Rilevante di Interesse Nazionale 2017: “Identification of new biomarkers and clinical determinants for management improvement of patients with pituitary tumor related syndromes” (code: PRIN 2017S55RXB).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fendo.2022.841118/full#supplementary-material
Supplementary Figure 1 | Evaluation of sequencing, mapping, and methylation analysis data. (A) Boxplot reporting the percentage of reads with quality scores above 20 (Q20) and above 30 (Q30). (B) Boxplot reporting the mean base quality of sequencing reads. (C) Summary of the mapping rates. (D) Boxplot of the global methylation level of the 437,792 regions analyzed.
1. Melmed S. Pituitary-Tumor Endocrinopaties. N Engl J Med (2020) 382:937–50. doi: 10.1056/NEJMra1810772
2. Trouillas J, Jaffrain-Rea ML, Vasiljevic A, Raverot G, Roncaroli F, Villa C. How to Classify Pituitary Neuroendocrine Tumors (PitNETs) in 2020. Cancers (2020) 12:514. doi: 10.3390/cancers12020514
3. Raverot G, Burman P, McCormack A, Heaney A, Petersenn S, Popovic V, et al. Clinical Practice Guidelines for the Management of Aggressive Pituitary Tumors and Carcinomas. Eur J Endocrinol (2018) 178:G1–24. doi: 10.1530/EJE-17-0796
4. Giuffrida G, Ferraù F, Laudicella R, Cotta OR, Messina E, Granata F, et al. Peptide Receptor Radionuclide Therapy for Aggressive Pituitary Tumors: A Monocentric Experience. Endocr Connect (2019) 8:528–35. doi: 10.1530/EC-19-0065
5. Korbonits M, Kumar AV. AIP Familial Isolated Pituitary Adenomas. In: Adam MP, Ardinger HH, Pagon RA, Wallace SE, Bean LJH, Stephens K, Amemiya A, editors. GeneReviews®. Seattle (WA: University of Washington, Seattle (2012). p. 1993–2020. updated 2020 Apr 16.
6. Thakker RV, Newey PG, Walls GV, Bilezikian J, Dralle H, Ebeling PR, et al. Clinical Practice Guidelines for Multiple Endocrine Neoplasia Type 1 (Men1). J Clin Endocrinol Metab (2012) 97:2990–3011. doi: 10.1210/jc.2012-1230
7. Hauser BM, Lau A, Gupta S, Bi WL, Dunn IF. The Epigenomics of Pituitary Adenoma. Front Endocrinol (2019) 10:290. doi: 10.3389/fendo.2019.00290
8. Pease M, Ling C, Mack WJ, Wang K, Zada G. The Role of Epigenetic Modification in Tumorigenesis and Progression of Pituitary Adenomas: A Systematic Review of the Literature. PloS One (2013) 8:e82619. doi: 10.1371/journal.pone.0082619
9. Vicchio TM, Aliquò F, Ruggeri RM, Ragonese M, Giuffrida G, Cotta OR, et al. MicroRNAs Expression in Pituitary Tumors: Differences Related to Functional Status, Pathological Features, and Clinical Behavior. J Endocrinol Invest (2020) 43:947–58. doi: 10.1007/s40618-019-01178-4
10. Ma HS, Wang EL, Xu WF, Yamada S, Yoshimoto K, Qian ZR, et al. Overexpression of DNA (Cytosine-5)-Methyltransferase 1 (DNMT1) and DNA (Cytosine-5)-Methyltransferase 3A (DNMT3A) Is Associated With Aggressive Behavior and Hypermethylation of Tumor Suppressor Genes in Human Pituitary Adenomas. Med Sci Monit (2018) 24:4841–50. doi: 10.12659/MSM.910608
11. Cozzolino A, Feola T, Simonelli I, Puliani G, Pozza C, Giannetta E, et al. Somatostatin Analogs and Glucose Metabolism in Acromegaly: A Meta-Analysis of Prospective Interventional Studies. J Clin Endocrinol Metab (2018) 41(5):575–81. doi: 10.1210/jc.2017-02566
12. Feola T, Cozzolino A, Simonelli I, Sbardella E, Pozza C, Giannetta E, et al. Pegvisomant Improves Glucose Metabolism in Acromegaly: A Meta-Analysis of Prospective Interventional Studies. J Clin Endocrinol Metab (2019) 104(7):2892–902. doi: 10.1210/jc.2018-02281
13. Cozzolino A, Feola T, Simonelli I, Puliani G, Hasenmajer V, Minnetti M, et al. Metabolic Complications in Acromegaly After Neurosurgery: A Meta-Analysis. Eur J Endocrinol (2020) 183(6):597–606. doi: 10.1530/EJE-20-0497
14. Ragonese M, Grottoli S, Maffei P, Alibrandi A, Ambrosio MR, Arnaldi G, et al. How to Improve Effectiveness of Pegvisomant Treatment in Acromegalic Patients. J Endocrinol Invest (2018) 41:575–81. doi: 10.1007/s40618-017-0773-0
15. Diamanti-Kandarakis E, Bourguignon JP, Giudice LC, Hauser R, Prins GS, Soto AS, et al. Endocrine-Disrupting Chemicals: An Endocrine Society Scientific Statement. Endocrine Rev (2009) 30:293–342. doi: 10.1210/er.2009-0002
16. Cannavò S, Ferraù F, Ragonese M, Curtò L, Torre ML, Magistri M, et al. Increased Prevalence of Acromegaly in a Highly Polluted Area. Eur J Endocrinol (2010) 163:509–13. doi: 10.1530/EJE-10-0465
17. Cannavò S, Ferrau F, Ragonese M, Romeo PD, Torre ML, Puglisi S, et al. Increased Frequency of the Rs2066853 Variant of Aryl Hydrocarbon Receptor Gene in Patients With Acromegaly. Clin Endocrinol (2014) 81(2):249–53. doi: 10.1111/cen.12424
18. Cannavò S, Ragonese M, Puglisi S, Romeo PD, Torre ML, Alibrandi A, et al. Acromegaly is More Severe in Patients With AHR or AIP Gene Variants Living in Highly Polluted Areas. J Clin Endocr Metab (2016) 101:1872–9. doi: 10.1210/jc.2015-4191
19. Gaidatzis D, Lerch A, Hahne F, Stadler MB. QuasR: Quantification and Annotation of Short Reads in R. Bioinformatics (2015) 31:1130–2. doi: 10.1093/bioinformatics/btu781
20. Langmead B, Salzberg SL. Fast Gapped-Read Alignment With Bowtie 2. Nat Methods (2012) 9(4):357–9. doi: 10.1038/nmeth.1923
21. Nadhamuni VS, Korbonits M. Novel Insights Into Pituitary Tumorigenesis: Genetic and Epigenetic Mechanisms. Endocr Rev (2020) 41:bnaa006. doi: 10.1210/endrev/bnaa006
22. Re A, Ferraù F, Cafiero C, Spagnolo F, Barresi V, Romeo DP, et al. Somatic Deletion in Exon 10 of Aryl Hydrocarbon Receptor Gene in Human GH-Secreting Pituitary Tumors. Front Endocrinol (2020) 12(11):591039. doi: 10.3389/fendo.2020.591039
23. Duong CV, Emes RD, Wessely F, Yacub-Usman K, Clayton RN, Farrell WE. Quantitative, Genome-Wide Analysis of the DNA Methylome in Sporadic Pituitary Adenomas. Endocr Relat Cancer (2012) 19:805–16. doi: 10.1530/ERC-12-0251
24. Chang M, Yang C, Bao X, Wang R. Genetic and Epigenetic Causes of Pituitary Adenomas. Front Endocrinol (2021) 11:596554. doi: 10.3389/fendo.2020.596554
25. Ling C, Pease M, Shi L, Punj V, Shiroishi MS, Commins D, et al. A Pilot Genome-Scale Profiling of DNA Methylation in Sporadic Pituitary Macroadenomas: Association With Tumor Invasion and Histopathological Subtype. PloS One (2014) 9:e96178. doi: 10.1371/journal.pone.0096178
26. Gu Y, Zhou X, Hu F, Yu Y, Xie T, Huang Y, et al. Differential DNA Methylome Profiling of Nonfunctioning Pituitary Adenomas Suggesting Tumour Invasion is Correlated With Cell Adhesion. J Neurooncol (2016) 129:23–31. doi: 10.1007/s11060-016-2139-4
27. Beylerli O, Gareev I, Pavlov V, Chen X, Zhao S. The Role of Long Noncoding RNAs in the Biology of Pituitary Adenomas. World Neurosurg (2020) 137:252–6. doi: 10.1016/j.wneu.2019.10.137
28. Gejman R, Batista DL, Zhong Y, Zhou Y, Zhang X, Swearingen B, et al. Selective Loss of MEG3 Expression and Intergenic Differentially Methylated Region Hypermethylation in the MEG3/DLK1 Locus in Human Clinically Nonfunctioning Pituitary Adenomas. J Clin Endocrinol Metab (2008) 93:4119–25. doi: 10.1210/jc.2007-2633
29. Yu G, Li C, Xie W, Wang Z, Gao H, Cao L, et al. Long non-Coding RNA C5orf66-AS1 Is Downregulated in Pituitary Null Cell Adenomas and is Associated With Their Invasiveness. Oncol Rep (2017) 38:1140–8. doi: 10.3892/or.2017.5739
30. Wormke M, Stoner M, Saville B, Walker K, Abdelrahim M, Burghardt R, et al. The Aryl Hydrocarbon Receptor Mediates Degradation of Estrogen Receptor Alpha Through Activation of Proteasomes. Mol Cell Biol (2003) 23(6):1843–55. doi: 10.1128/MCB.23.6.1843-1855.2003
31. Hartmann S, Szaumkessel M, Salaverria I, Simon R, Sauter G, Kiwerska K, et al. Loss of Protein Expression and Recurrent DNA Hypermethylation of the GNG7 Gene in Squamous Cell Carcinoma of the Head and Neck. J Appl Genet (2012) 53:167–74. doi: 10.1007/s13353-011-0079-4
32. Xu S, Zhang H, Liu T, Chen Y, He D, Li L. G Protein γ Subunit 7 Loss Contributes to Progression of Clear Cell Renal Cell Carcinoma. J Cell Physiol (2019) 234:20002–12. doi: 10.1002/jcp.28597
33. Nalesnik MA, Tseng G, Ding Y, Xiang GS, Zheng Z, Yu YP, et al. 2012 Gene Deletions and Amplifications in Human Hepatocellular Carcinomas. Am J Pathol (2012) 180:1496–508. doi: 10.1016/j.ajpath.2011.12.021
34. Gitik M, Holliday ED, Leung M, Yuan Q, Logue SF, Tikkanen R, et al. Choline Ameliorates Adult Learning Deficits and Reverses Epigenetic Modification of Chromatin Remodeling Factors Related to Adolescent Nicotine Exposure. Neurobiol Learn Mem (2018) 155:239–48. doi: 10.1016/j.nlm.2018.08.009
Keywords: GH-OMAs, methylation, pituitary adenomas, NFPAs, pituitary tumors
Citation: Giuffrida G, D’Argenio V, Ferraù F, Lasorsa VA, Polito F, Aliquò F, Ragonese M, Cotta OR, Alessi Y, Oteri R, Di Maggio F, Asmundo A, Romeo PD, Spagnolo F, Pastore L, Angileri FF, Capasso M, Cannavò S and Aguennouz M’H (2022) Methylome Analysis in Nonfunctioning and GH-Secreting Pituitary Adenomas. Front. Endocrinol. 13:841118. doi: 10.3389/fendo.2022.841118
Received: 21 December 2021; Accepted: 21 February 2022;
Published: 30 March 2022.
Edited by:
Antonino Belfiore, University of Catania, ItalyReviewed by:
Elisa Giannetta, Sapienza University of Rome, ItalyCopyright © 2022 Giuffrida, D’Argenio, Ferraù, Lasorsa, Polito, Aliquò, Ragonese, Cotta, Alessi, Oteri, Di Maggio, Asmundo, Romeo, Spagnolo, Pastore, Angileri, Capasso, Cannavò and Aguennouz. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Francesco Ferraù, ZmZlcnJhdUB1bmltZS5pdA==
†These authors share senior authorship
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.