 Allan Langlois
Allan Langlois Aurore Dumond
Aurore Dumond Karim Bouzakri
Karim Bouzakri
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Endocrinol. , 03 February 2022
Sec. Diabetes: Molecular Mechanisms
Volume 13 - 2022 | https://doi.org/10.3389/fendo.2022.836344
This article is part of the Research Topic Look who's Talking: Dialogues with Beta Cells View all 5 articles
The regulation of insulin secretion is under control of a complex inter-organ/cells crosstalk involving various metabolites and/or physical connections. In this review, we try to illustrate with current knowledge how β-cells communicate with other cell types and organs in physiological and pathological contexts. Moreover, this review will provide a better understanding of the microenvironment and of the context in which β-cells exist and how this can influence their survival and function. Recent studies showed that β-cell insulin secretion is regulated also by a direct and indirect inter-organ/inter-cellular communication involving various factors, illustrating the idea of “the hidden face of the iceberg”. Moreover, any disruption on the physiological communication between β-cells and other cells or organs can participate on diabetes onset. Therefore, for new anti-diabetic treatments’ development, it is necessary to consider the entire network of cells and organs involved in the regulation of β-cellular function and no longer just β-cell or pancreatic islet alone. In this context, we discuss here the intra-islet communication, the β-cell/skeletal muscle, β-cell/adipose tissue and β-cell/liver cross talk.
Maintaining glucose homeostasis requires pancreatic islets cells’ secretion of several hormones including insulin by β-cells, glucagon by α-cells, somatostatin by δ-cells and the pancreatic polypeptide (PP) by PP-cells. Glycaemia regulation is also allowed thanks to several insulin sensitive/responsive tissues like liver, adipose tissue and skeletal muscle (1, 2). Specifically, β-cells releasing insulin, a hypoglycaemic hormone, play a critical role in this physiological regulation. Indeed, defective insulin secretion is the cause of all forms of diabetes (3).
Diabetes in its two main forms is characterized by an absolute or relative insulin deficit. Several signals are thought to lead to impaired cell function and possibly a decrease in cell mass in type 2 diabetes (T2D), with autoimmune cell destruction underlying type 1 diabetes (T1D). More precisely, T1D results from the destruction of pancreatic β-cells that is mediated by the immune system. Multiple genetic and environmental factors found in variable combinations in individual patients are involved in the development of T1D. It was recently described that the two first auto-antibodies which initiate the autoimmune process are insulin autoantibodies or glutamic acid decarboxylase autoantibodies. Furthermore, the autoimmune response is affected by environmental factors such as nutrition etc… (4). T2D concerns the majority of diabetic patients (around 90–95%) and is due to the development of insulin resistance that can result in a progressive loss of β-cell insulin secretion (5).
Insulin resistance is defined as the inability of cells to respond normally to the insulin, leading to a decrease in glucose uptake in primary insulin-sensitive organs such as skeletal muscle and adipose tissue. Moreover, hepatic insulin resistance impairs suppression of glucose production by insulin in hepatocytes, participating in the chronic increase in glycaemia characteristic of diabetes (6, 7). In parallel with alterations in glucose metabolism, insulin resistance induces an accumulation of circulating free fatty acids due to an increase in adipose tissue lipolysis and hepatic de novo lipogenesis (8). This contributes to ectopic fat deposition in liver or skeletal muscle, which in turn exacerbate insulin resistance. Usually, it appears that insulin resistance onset involves multiple and complex pathways, with inter-linking and multidirectional effects (9). These mechanisms include inflammation, lipotoxicity, endothelial reticulum stress and mitochondrial dysfunction (10). Moreover, insulin resistance plays a larger role in T1D pathological process than it is generally recognized. Along with the increasing incidence of T1D, obesity and physical inactivity have steadily increased in children and adolescents. The role of insulin resistance in T1D has only recently been accepted.
Interestingly, the insulin resistance alone does not induce T2D. Indeed, healthy β-cells are able to compensate this insulin resistance by increasing in number and enhancing their secretory capacities (11). Thus, insulin resistance, in addition to impaired β-cell function, is a hallmark of T2D. Therefore, for a long time, β-cell was considered as the main target for the treatment of diabetes. However, it is now well established that optimal glycaemic regulation involves cross communication between several cell type, organs and tissues such as intra-islet cells, intestine L-cells, pancreas, liver, skeletal muscle, adipose tissue etc… (12–14). Moreover, for adaptation to nutritional and environmental conditions, this close conversation is essential. Therefore, to treat diabetes mellitus, it seems important to consider not only a cell type but all the organs involved in this glycaemic regulation. In addition, it is widely described that this inter-organ communication involves many metabolites, including myokines, adipokines, hepatokines, isletokines, extracellular vesicles (EV) etc… (12–14). To prove the importance of this communication network, numerous studies have shown that any disruption of it induces metabolic dysfunctions and metabolic syndromes such as obesity and diabetes (12–15).
In this review, we sought to illustrate with current knowledge how β-cells communicate with other cell types and organs in physiological and pathological contexts. Moreover, this review will provide a better understanding of the microenvironment and the context in which β-cells exist and how this can influence their survival and function. In this context, we discuss here the intra-islet communication, the β-cell/skeletal muscle, β-cell/adipose tissue and β-cell/liver cross talk.
In response to glucose elevation, β-cells secrete insulin through the canonical insulin pathway involving GLUT2 transporter, KATP channels, intra-cellular Ca 2+ increasing etc…. Conversely, at low levels, β-cells employ a negative feedback mechanism to help maintain the blood glucose within a safe range (16–18). However, it is clearly established that β-cell insulin secretion mechanism is even more complex and is regulated also by a close inter-organ/cells cross-talk involving various metabolites and/or physical connections. This communication is also found into the islet.
For decades, the challenge has been to determine how β-cells finely regulate their insulin secretion linked to glucose concentration and whether this resides in a collective response via a close communication between β-cells.
The arrangement of β-cells within pancreatic islets plays a critical role for insulin secretion through the generation of rhythmic activity. Indeed, Johnston NR et al. demonstrated that the islet functional architecture is composed of hub β-cells with pacemaker properties. Moreover, authors showed that silencing of hubs abolished coordinated islet responses to glucose, whereas specific stimulation restored communication patterns (19). Activation of these “Hub cells”, recently renamed “Leader cells” (20), may trigger [Ca 2+]i waves that diffuse to the other cells within the islets, called “Followers cells”. Then, the coordinated increases and decreases in [Ca 2+]i drive pulses of insulin secretion (21, 22). Interestingly, it was demonstrated that these “Hub cells” are targeted by pro-inflammatory and glucolipotoxic factors inducing β-cells dysfunction (19). Therefore, these “Hub cells” are essential for regulating synchronization of islet insulin secretion and any disruption of it could contribute to diabetes onset. Nevertheless, Satin LS et al. have recently reported that depolarization of “Hub cells” is sufficient to trigger electrical activity and [Ca 2+]i waves but it is difficult for one cell to supply enough current to repolarize the entire islet. Thus, authors suggest the involvement of diffusible factors released by specialized cells or groups of cells within islets like nitric oxide, carbon monoxide, GABA … but more studies are necessary to conclude (22).
It is widely described in the literature that Gap junctions provide one of the micro-anatomical bases for appropriate glucose-induced insulin release (23, 24). Indeed, β-cell is known to express gap junctions involved in cell coupling and in the exchange of ions and small metabolites between β-cells (24–26). Particularly, Speier S et al. highlighted an important role for Cx36-gap junctions in modulating stimulation threshold and kinetics of insulin release (24). Moreover, it was shown that coupling of β-cells improves insulin synthesis and secretion, while uncoupling leads to altered β-cell function (27). Very recently, in order to improve knowledge about the exact role and effects of β-cell/β-cell communication on glucose homeostasis, Boris P et al. developed a new study model. This consists on a dynamical network model in which N network nodes represent individual β-cells and network links represent couplings with k neighboring cells (18). Thanks to their study model, they demonstrated the existence of a glucose-induced transition in β-cell activity thanks to increasing coordination through gap-junctional signaling and paracrine interactions. In conclusion, insulin β-cell response to glucose stimulation is a collective and coordinated action involving Gap junctions.
Among elements involved in β-cell to β-cell communication, it is well established that EVs play a critical role and can influence β-cell function (28). Indeed, Javeed N et al. have recently showed that pro-inflammatory β-cell EVs induce a complete loss of insulin secretion in response to glucose and promote a pro-inflammatory islet transcriptome (29). More precisely, Guay C et al. demonstrated that these effects are mediated by miRNA derived-Evs, which are transferred to neighboring β-cells. Indeed, down-regulation of the miRNA-mediating silencing protein Ago2 in recipient cells prevent deleterious effect of non-coding RNAs (30).
In conclusion, EVs transfer is an important cell-to-cell communication mechanism regulating β-cell function and constitute targets of interest to develop therapeutic strategies.
α-cells, with β-cells, are among the two most abundant and essential endocrine cell type in the pancreatic islet for the maintenance of glycaemia balance. The role of α-cells is to release glucagon into the bloodstream in order to increase blood glucose levels in opposition to β-cells that secrete a hypoglycaemic hormone (31). Thus, they were considered for long time as functional antagonists but the function of these two cellular types is influenced by each other. Indeed, glucagon produced by the pancreatic α-cell stimulates β-cell function while insulin has an inhibitory effect on glucagon release (32). This cross communication between β and α-cells enables active regulation to maintain stable blood glucose concentration (15, 31, 33). For that, β-cells release some inhibitory factors of α-cellular function including insulin, Zn2+, ATP and γ-aminobutyric acid (31, 34). Conversely, α-cells produce, in addition of glucagon, factors which affect the regulation of β-cells activity. These can have both autocrine and paracrine signaling properties (15, 31).
Rodriguez-Diaz R et al. have recently shown that glucagon input increases insulin secretion also from the neighboring β-cell in human pancreatic islet (35). For that, authors transplanted human islets into the anterior chamber of the eye of diabetic nude mice. Once restoring normoglycaemia, they inhibited human glucagon receptors with a specific antagonist (L-168,049). Then, they showed that this treatment decreased insulin secretion from human islet grafts and increased glycaemia to pre-diabetic levels. Consequently, for the first time, it was demonstrated that insulin secretion has to be amplified by input from adjacent α-cells (35).
Furthermore, in intestinal L-cells, proglucagon, release by α-cells, is converted to GLP-1 by PC1/3 that is able to potentiate insulin secretion under conditions of elevated blood glucose concentration (36). Moreover, some studies suggest that GLP-1 may be also directly produced in the α-cell, also through PC1/3 expression, in order to increase insulin secretion (37–39). Recently, it was shown that this α- to β-cell communication and the subsequent enhancement of insulin secretion are lost in KO mice for proglucagon peptides in the α-cell. The same observation was done if the receptors for proglucagon peptides (GLP-1R and GCGR) are silenced (37, 40, 41). Therefore, β- to α-cell communication seems currently attributed to proglucagon products but further studies are necessary to clarify the exact contribution of glucagon and GLP-1 in this communication and the mechanisms involved.
Finally, Human α-cells secrete acetylcholine, which strongly potentiates the glucose-induced insulin secretion (42, 43). Indeed, human α-cells provide paracrine cholinergic input to surrounding endocrine cells, whose β-cells. Particularly, human α-cells express acetylcholine transporter, release acetylcholine in response to glucose concentration and amplify β-cell answer to increase glucose concentration (43).
In conclusion, a very close and complex relationship exists between the two cell types that is fundamental for setting the regulation of insulin secretion and thus for glucose homeostasis, involving the secretion of various metabolites. Moreover, α/β-cell communication is not only a direct but is also an indirect crosstalk involving other intermediate cell types as intestine. Consequently, any cell dysfunction can alter this communication between α- and β-cells and thus can affect insulin secretion in response to glucose elevation. Therefore, for the development of therapeutic strategies it is necessary to be not only interested in the communication between two cell types but also in the impact that other organs/cells could have on this communication. This increases sharply the task.
Other component of pancreatic islet cyto-architecture are the δ-cells. With α- et β-cells, these 3 cell types constitute the endocrine functional part of the islet which are finely connected to allow an adequate response to glycaemic variations (44–46). δ-cell is an important paracrine regulator of β and α-cell’s secretory activity by secreting somatostatin. More precisely, somatostatin is an inhibitor of both glucagon and insulin release and so, is an important regulator of glucose homeostasis (46). It acts through different isoforms of the somatostatin receptor (47, 48). Furthermore, the interaction between δ-cells and other intra-islet cells also becomes defective in diabetes. Indeed, this defect reduces paracrine feedback to β-cells to exacerbate hyperglycemia or enhanced inhibition of α-cells, disabling counter-regulation, to cause hypoglycemia (49).
δ-cell release somatostatin in response to high glucose and to local-acting signaling molecules secreted by islet cells, such as acetylcholine, glutamate, urocortin3 (Ucn3), ghrelin (48, 50–53). In addition, β- and δ-cells can regulate glucagon secretion by α-cells through gap junction communication (54).
Interestingly, Arrojo e Drigo R et al. have shown that δ-cell structure is composed of filopodia, which play a crucial role in β-cell function regulation. Indeed, authors observed that these filopodia are dynamic structures that contain a secretory machinery, enabling the δ-cell to reach a large number of β-cells within the islet (46). Furthermore, they showed that endogenous IGF-1/VEGF-A signaling modulates this. Therefore, any disruption in this regulation may contribute to early stages of beta cell failure and diabetes pathophysiology (46, 55).
In order to bring new knowledge on intra-islet cells communication, a recent study from Germany aimed to determine how α- and β-cells regulate somatostatin secretion (56). For that, authors studied in transgenic mice models the effects of varying glucose concentrations together with infusions of arginine, glucagon, insulin and somatostatin, as well as infusions of antagonists of insulin, somatostatin and GLP-1 receptors. Interestingly, they demonstrated that somatostatin and glucagon secretion are linked in a reciprocal feedback cycle with somatostatin inhibiting glucagon secretion at low and high glucose levels. Moreover, they observed that glucagon stimulates somatostatin secretion thanks to glucagon and GLP-1 receptors activation. Conversely, they showed that insulin or activation of its receptor did not affect somatostatin secretion. Therefore, glucagon pathway plays a crucial role on somatostatin secretion and could be an interesting therapeutic target to control somatostatin secretion as this hormone is a strong regulator of glucagon and insulin secretion.
Pancreatic islets are highly vascularized and composed of fenestrated capillaries. This dense and tortuous vascular network is essential to deliver quickly insulin in the bloodstream in response to glucose elevation. Indeed, islets receive 6–20% of the pancreas’s direct arterial blood flow, although they only represent 1–2% of the organ mass (57–59). Moreover, the connection between endothelial cells and endocrine cells enables proper gas and nutrition’s exchange and waste removal via the bloodstream.
The existence of cross-talk between β-cells and intra-islet endothelial cells which play a crucial role in β-cell development and function providing non-nutritional signals to islets, has been well established for decades (58, 60). For example, few years ago it was shown that endothelial cell signals regulate the expression of transcription factors in order to initiate dorsal pancreas development by selectively inducing the transcription factor Ptf1α (61). Moreover, using transgenic mice, Lammert E et al. discovered that vessels not only provide metabolic sustenance, but also provide inductive signals inducing insulin expression and islet hyperplasia (62). Among these signals, the main ones is the Vascular Endothelial Growth Factor A (VEGF-A) known to promote endothelial migration and proliferation after binding to its receptor (63). Thus, a defect in VEGF-A signaling can impairs β cell proliferation, insulin secretion and glucose homeostasis (58, 64). Furthermore, Hepatocyte Growth Factor (HGF) produced by intra-islets vessels, also regulates β-cell proliferation. Moreover, it is essential for β cell differentiation, function, and proliferation (58). Then, it was demonstrated that markers of endothelial cell function (E-selectin, Il6, endothelin-1 and endothelial nitric oxide synthase) are overexpressed in islet endothelial cells from diabetic db/db mice and the exposure of these molecules on pancreatic islets decreases insulin secretion during a glucose stimulation test (GSIS) and decreases insulin content. Thus, authors highlighted that in diabetes, islet endothelial cells have a dysfunctional phenotype, which may contribute to loss of β-cell function (65, 66). Finally, it is widely described that endothelial cells modulate β-cell function in mature islets by secreting basement membrane components that interact with specific receptors in β-cells (67–70). For example, Daniel B et al, showed the existence of an effective paracrine interaction between islets microcapillary endothelial cells and β-cells that modulates glucose-induced insulin secretion via the TPI-sulfonylurea receptor-KATP channel (SUR1-Kir6.2) complex attenuating interactions (70).
Interestingly, β-cells also release factors, which influence intra-islet endothelial cell function. Indeed, Figliolini et al. demonstrated that biologically active islet-derived EVs are able to shuttle anti-apoptotic and pro-angiogenic mRNAs and miRNAs into endothelial cells, which is a promising target to improve islet transplantation (71).
On this part, the role of intra-islet endothelial cells is not only to deliver quickly hormones into the bloodstream to regulate glucose homeostasis but is also to influence the function and survival of cells through a close communication with β-cells involving secreted molecules. In conclusion, this is a two-way communication and any endothelial and/or β-cell dysfunction will participate in diabetes onset.
Cellular plasticity in the pancreas also plays a crucial role in glycaemia homeostasis and it represents a strong illustration of pancreatic cell communication. Interestingly, maintaining normoglycaemia involves a certain degree of pancreatic islet’s biological adaptation. For example, an increased demand for insulin can lead to an increase in β-cell mass. Moreover, any changes in these adaptive mechanisms can lead to the incapacity in maintaining normoglycaemia and to the development of diabetes (72).
It is well described that pancreatic cells are able to transdifferentiate in functional β-cells to compensate insulin secretion impairment to maintain normoglycaemia. Indeed, it was demonstrated that pancreatic α-cells and δ-cells become insulin expressers upon ablation of insulin-secreting β-cells, promoting diabetes recovery (73). Interestingly, in young mice, δ-cells (but not α-cells) undergo spontaneous conversion into β-cells, proceeding through a dedifferentiated intermediate (74, 75). Moreover, it was recently shown that α-cells and PPY-producing γ–cells, obtained from deceased non-diabetic or diabetic human donors, can be lineage-traced and reprogrammed by the transcription factors Pdx1 and MafA to produce and secrete insulin in response to glucose (73, 76). Furthermore, Furuyama K et al. demonstrated that transplantation of human α-cells converted in β-cells in diabetic mice reverses diabetes and that the graft remains functional for 6 months (73). However, in diabetic patients it was observed a β-cell plasticity and a loss of β-cell identity. Thus, it is proposed that β-cells fail to maintain a fully differentiated glucose-responsive and drug-responsive state in diabetic individuals with poorly controlled and long-lasting periods of hyperglycemia (77). Then, it has been demonstrated that β-cell neogenesis occurs via transdifferentiation of acinar or ductal cells, differentiation of progenitors to β-cells in exocrine and endocrine tissue or by replication of preexisting β-cells (78, 79).
Finally, this concept of cellular plasticity is primordial to understand the importance of the role of pancreatic cell intercommunication in β-cell function and glycaemia regulation. Indeed, the loss of cell identity due to transdifferentiation process will disturb intra and inter pancreatic cell communication (described all along this chapter) essential for optimal β-cell function. Thus, it represents another way of interest for future diabetic patients’ treatments.
Skeletal muscle is the largest insulin-sensitive organ in the body, so it plays a major role in postprandial glucose homeostasis. Consequently, altered insulin action in skeletal muscle can lead to a pathological state of insulin resistance, in which normal concentrations of insulin induce an impaired biological response (80). Interestingly, Dr Bente Klarlund Pedersen was the first to suggest that skeletal muscle is as an endocrine organ. Indeed, she suggested that certain cytokines and other peptides, called “myokines”, were produced, expressed and released by muscle fibers (81). Moreover, several studies have shown that the contraction of skeletal muscles releases a selected panel of myokines, which can act hormonally both locally and in distant tissues. For example, these secreted myokines exert specific endocrine effects on visceral or median fat and have direct anti-inflammatory effects (82–84). Other myokines act locally within muscle via paracrine mechanisms, exerting their effects on signaling pathways involved in lipid metabolism (83).
Skeletal muscle tissue is composed of a heterogeneous population of muscle fibers ranging from slow contraction type I fibers to fast contraction type IIx/d fibers (85). Type I fibers will use lipids for their function, while type IIx/d fibers will mostly use glucose (86, 87). Recently, our laboratory showed that skeletal muscle cells can secrete different myokine profiles depending on their insulin sensitivity and fibrillar composition and can impact β-cell function and survival (12).
Our team showed that skeletal muscle with different insulin sensitivity can have a differential impact on pancreatic β cell function and survival (12). Moreover, our work demonstrated that myokines secreted by insulin-sensitive skeletal muscle increased pancreatic β-cell proliferation as well as glucose-induced insulin secretion. In contrast, myokines secreted by insulin-resistant skeletal muscle induce loss of insulin secretion and destruction of pancreatic β-cells. In conclusion, skeletal muscle secretome contains factors that can have either a positive (insulin-sensitive muscle) or a negative (insulin-resistant muscle) impact on pancreatic β-cells. For this review, we have chosen to discuss about cytokines and chemokines independently tested for their effects on pancreatic β cells.
For example, IL-1β alone can have a positive or a negative impact on β-cell function, survival and proliferation. Furthermore, these differential effects are depending on its concentration and on its duration of exhibition (88). In addition, we have also shown positive effects on β-cell survival and proliferation for low levels of IL-1β secreted by the cells themselves when cultured on a particular extracellular matrix (89, 90).
Moreover, we showed that TNF-α, at high concentration, decreases insulin secretion induced by glucose and stimulates apoptosis. However, as observed with IL-1β, at low concentration, TNF-α promotes β-cell proliferation and improves insulin secretion. Thus, this indicates the existence of bimodal effects for this cytokine (12).
Then, the interferon-gamma-inducible protein (IP-10), also called C-X-C motif chemokine ligand 10 (CXCL10), is increased in the secretome of insulin-resistant human skeletal muscle cells (12). In addition, its circulating level is increased in the serum of patients with type 1 and type 2 diabetes (91, 92). This has led several groups to study the impact of this chemokine on the endocrine pancreas. The expression of CXCL10 in the pancreas was first shown to accelerate the autoimmune process (93). Secondly, a direct impact of CXCL10 on the function and survival of pancreatic β cells was shown with induction of their apoptosis, altered insulin secretion and decrease in insulin mRNA.
Studies on the effects of IL-6 on β-cell function have shown mixed results, with some finding a negative effect (94) and others a positive effect on insulin production (95, 96). Furthermore, it appears that these effects are more complex than a direct effect. Indeed, IL-6 plays an essential role in a muscle-entero-pancreatic communication loop. During exercise IL-6 releases GLP-1 from L cells in the intestine and further improves β-cell function through local production of GLP-1 in α-cells, leading to better glycaemic control (97). Interestingly, high and acute secretion of IL-6, observed during exercise, have beneficial effects. However, chronic elevation of plasma IL-6 is associated with negative clinical parameters, including the development of type 2 diabetes (98).
A decade ago, it was shown that plasma follistatin is rapidly elevated during physical activity, peaks during the recovery phase and remains elevated for a few hours (99). The origin of exercise-induced follistatin appears to depend on the type of exercise performed. During resistance exercise such as weight training, follistatin mRNA expression increases in skeletal muscle tissue biopsies from women on hormone replacement therapy (100). The same observation was made in healthy young men after a session of strength training (101). However, recently we have shown that follistatin is also secreted by the liver in response to exercise. This follistatin secreted into the bloodstream can then target the pancreas and regulate the secretion of insulin and glucagon. The latter will subsequently target the liver and in turn regulate the secretion of follistatin (102). This example perfectly illustrates the complexity of the role played by the inter-organs communication on β-cell function and survival.
Another myokine has generated great interest in the treatment of diabetic patients, fractalkine, also known as CX3CL1. It is a CX3C chemokine expressed in various cell types such as skeletal muscle cells and pancreatic β-cells (103–105). Furthermore, knockdown of CX3CR1 (fractalkine receptor) in a mouse model was found to induce hyperglycaemia and to reduce nutrient-stimulated insulin secretion. In addition, the injection of fractalkine into C57BL/6N mice made it possible to potentiate their β-cell function, to increase plasma insulin levels and to improve their glucose tolerance (106). In another study conducted by our team, we showed that fractalkine protects human β-cells from the harmful effects of TNFα on the molecular mechanisms involved in the trafficking of insulin granules. We then highlighted that this myokine restored the phosphorylation and expression of key proteins involved in the insulin secretion pathway such as AKT, AS160, paxillin, IRS2 for example (105). In summary, all these data suggest that the Fractalkine-CX3CR1 axis could be a target of interest for treating diabetes and that fractalkine could be an interesting pharmacological candidate for treating diabetic patients.
Recently, we have highlighted a set of myokines secreted by glycolytic skeletal muscles. These myokines, such as osteoprotegerin (OPG), a member of the TNF receptor superfamily, are being evaluated therapeutically for replacement of pancreatic β-cells destroyed in T1D. Indeed, recent studies have shown that OPG enables the replication of human pancreatic beta cells by modulating the CREB and GSK3 pathways, by binding RANKL and thus, interfering with the RANKL/RANK antiproliferative interaction. Furthermore, it has been shown by plasma insulin assay and a glucose tolerance test that the glycemic balance is significantly improved in diabetic mice treated with 1.0 mg/g of mOPG-Fc (107). In addition, we recently demonstrated in a study carried out on primary cultures of human myotubes, that OPG is a specific myokine of the triceps and that it significantly decreases the apoptosis of pancreatic β-cells. Finally, we found that OPG counteracts both the negative effects of cytomix and TNFα on primary pancreatic β-cell proliferation and insulin secretion (108).
In conclusion, far from being an inert tissue in terms of inter-organ communication, skeletal muscle secretes myokines, which can affect the function of distant organs/tissues either favorably or unfavorably. Here, we have summarized the potential impact of myokines in the communication between skeletal muscle and endocrine pancreas. This is a new route of communication that we believe is further altered by the degree of insulin resistance in skeletal muscle. Finally, the identification of the skeletal muscle secretome may have important implications for understanding the decrease in the functional mass of β cells in diabetes and for developing an innovative therapy.
For decades, white adipose tissue has long been considered a mere storage tissue. In 1990s, it has been highlighted that adipose tissue is an endocrine tissue able to secrete heterogeneous bioactive factors including proteins (i.e. adipokines), lipids (i.e. lipokines) and extracellular vesicles (e.g. exosomes). Since the 1990s, the number of adipose tissue’s secretions has continuously increased and new factors are regularly identified. All of these secretions establish communications with a variety of organs and cells including the pancreas and particularly β-cells. A lot of original papers and reviews have already been published concerning the impact of the “classic” factors (e.g. leptin, adiponectin) (13). For these reasons, this review focuses on recently described adipose tissue’s secretions that crosstalk with β-cells.
Asprosin, identified as a novel adipokine in 2016, is a C-terminal product generated by the cleavage of a proprotein by activated protease furin, which generates mature fibrillin-1 and 140 amino-acid asprosin (109). Plasma asprosin level increases during starvation to stimulate hepatic gluconeogenesis and prevent hypoglycaemia. An increase is also observed in patients with obesity and T2D (110, 111). Interestingly, asprosin is negatively correlated with homeostasis model assessment for β-cells function (HOMA-β) (112) that indicates that asprosin may be involved in β-cells dysfunction during T1D. Indeed, it has been recently shown that treatment of MIN6 cells by asprosin increases caspase 3 activity as a marker of apoptosis and decreases cell viability (113). This is associated with an impairment of GSIS. Surprisingly, it has been shown that asprosin secretion is enhanced by irisin, an exercise-induced myokine (114) which raises the question of its beneficial or deleterious effect. So, it will be necessary to elucidate more precisely asprosin’s mechanisms. Then, this observation highlights the idea of a complex and indirect crosstalk between organs that can influence β-cells function. All of these results suggest asprosin as a potential therapeutic target for preserving pancreatic β-cells.
Furthermore, Adipsin or complement factor D was the first described adipokine and plays a central role in metabolism (115). This adipokine is also synthesized by the liver but adipose tissue remains the major source of its secretion. Adipsin stimulates the production of C3a, a component complement able to enhance insulin secretion by increasing the concentration of cytosolic Ca2+ in β-cells (116). Interestingly, adipsin expression in adipose tissue is decreased in T2D patients with β-cell failure compared to T2D patients (116). Recently, Gomez-Banoy et al. found that treatment of transplanted pancreatic islets with an adeno-associated virus (AAV) expressing adipsin preserves β-cell mass by inhibiting death in a model of T2D mice (117). Moreover, adipsin maintains β-cell transcription identity and so inhibits dedifferentiation observed during diabetes. Despite these attractive results, to date no study has investigated the role of adipsin in the context of T1D. This adipokine could be an attractive pharmacological candidate and this path deserves to be explored in the future.
Finally, Secreted frizzled-related protein 5 (Sfrp5) is an endogenous inhibitor of Wnt signaling pathway identified as a novel anti-inflammatory adipokine in 2010 (118). Typically, its adipose tissue mRNA expression is reduced in rodent models of obesity (118). Moreover, KO mice fed with a high fat diet develop systemic metabolic dysfunction, such as glucose intolerance or adipose tissue inflammation. In 2019, Carstensen-Kirberg et al. showed that treatment of INS-1E cells with Sfrp5 during 24h dose dependently increased GSIS (119). This is associated with a decrease in the phosphorylation levels of c-jun N terminal kinase (JNK), a deleterious pathway for β-cell function (120). In light of these first observations, Sfrp5 appeared to represent a potential target for diabetes mellitus and it seems necessary to deal with it in depth.
In addition to adipokines, adipose tissue is also able to secrete bioactive lipids called lipokines. By their nature, there factors are difficult to isolate and still few of them have been characterized. Among them, some have shown interesting impact on β-cells and more will certainly be identified in the years to come.
Palmitoleate was the first lipid identified as being released from adipose tissue and having metabolic effects on distant organs. It is mostly derived from de novo lipogenesis and acts as an insulin sensitizer in liver and skeletal muscle tissues (121). Concerning its impact in pancreatic β-cells, it seems to stimulate insulin secretion for high glucose levels (122) but mechanisms of action have not yet been elucidated. Now, further studies are needed to understand the link between palmitoleate and diabetes and to consider this factor as a biomarker or a possible treatment.
Then, Fatty acid esters of hydroxy fatty acids (FAHFA) are a novel class of lipids, which showed beneficial metabolic and anti-inflammatory effects (123). The most well characterized FAHFA species are palmitic acid esters of hydroxyl stearic acid (PAHSA) whose serum and adipose tissue levels are decreased in insulin-resistant subjects (124). In 2019, Syed I et al. showed beneficial impacts of PAHSA on β-cells. Indeed, chronic treatment of NOD mice delays the onset and reduces the incidence of diabetes (125), associated with an enhancement of GSIS. In accordance with that, PAHSA increases proliferation and attenuates cytokine-induced MIN6 cells death. Interestingly, these effects are mediated in part by GLP-1R, widely described as activator of insulin secretion (126). Taken together, these results highlight a new class of potential candidates for treatment of diabetes.
Besides white adipose tissue, brown adipose tissue (BAT) is also able to secrete proteins called batokines. By its ability to increase energy expenditure, targeting BAT to treat diabetes is a particularly attractive strategy. However, the secretome of this tissue remains poorly characterized and nowadays there is no evidence of a direct dialogue between BAT and pancreas during diabetes. The identification and characterization of batokines could open new areas of research and potentially future treatment options.
EV are lipid bilayer particles naturally secreted by cells into the extracellular space. They envelop and release intracellular molecules including proteins, microRNA and bioactive lipids to mediate cell-to-cell communication. Several studies have shown that EV proteins and microRNAs content were altered during diabetes (127, 128). In this way, Gesmundo I et al. showed that EV from human lean adipose tissue promote survival and insulin secretion of β-cells while EV from human obese adipose tissue alters these same settings (129). The authors have not investigated the cargo of the EV but it could be interesting to identify modulated factors in this context. Future studies of adipose tissue-pancreas crosstalk will take into account this important mode of communication.
The liver is composed of many different cell types: parenchymal cells representing 80% of the liver, including hepatocytes and bile duct cells, and non-parenchymal cells including Kupffer cells (liver macrophages), hepatic stellate cells and sinusoidal endothelial cells (14). For a long period, the secretory role of liver was mainly known for its regulation of coagulation and hemostasis, however the different cells composing the liver give a wide range of secreted proteins, thus its endocrine role in metabolic diseases has started to be described recently (14). Furthermore, its structure composed of sinusoids enable to deliver the liver secretome to the peripheral organs through the central veins and the inferior vena cava (14), making liver one of the largest metabolic organ (130). Indeed, the liver has also an impact on β-cells, principally by regulating the compensatory effect on islets induced in insulin-resistant state. Here, we focus on different liver secretory products that have an impact on β-cells and which represent a real therapeutic interest.
FGF21 was first identified in 2000 principally in the liver (131) but was first described as a metabolic regulator in 2005 (132), involved in lipid metabolism, by decreasing adipose tissue lipolysis, increasing fatty acid oxidation and reducing hepatic lipids. Secondly, it is involved in glucose metabolism by decreasing plasma glucose, which mainly explain its higher circulating expression in patients with obesity and/or hepatic steatosis as a compensatory effect (14, 133). In addition to its metabolic regulator role, FGF21 has been reported as a potential anti-diabetic therapy thanks to its ability to improve β-cell function and mass (132). This anti-diabetic role is explained by different mechanisms: decrease in plasma glucose and triglyceride levels which reduces β-cells’ glucolipotoxicity and activate AKT signaling pathway which improves insulin sensitivity (14, 134, 135).
SerpinB1 is a protease inhibitor mainly expressed in hepatocytes (136). SerpinB1 inhibits particularly the neutrophil elastase that interferes with β-cell proliferation by inhibiting the phosphorylation of proteins involved in insulin or IGF-1 signaling pathways, such as MAPK3 or GSK. Thus, SerpinB1 increases β-cells proliferation particularly in insulin-resistant state (137).
HGF is expressed in liver in response to ERK signaling and induce its signal through its tyrosine-kinase receptor Met (138). The treatment of β-cells by HGF increases the phosphorylation of IRS2, AKT and ERK (138). This increase is the response of MET activation by HGF, which induce the formation of a MET-Insulin receptor complex responsible of an increase in insulin signal (136, 138, 139).
Selenoprotein P is a secretory protein predominantly expressed by the liver (14, 140), which plays an important role in selenium metabolism (141). Selenoprotein P expression induces insulin resistance and impairs glucose metabolism in hepatocytes through the inhibition of AMPK activity (140, 141). Selenoprotein P high level is also involved in the reduction of β-cells and α-cell mass and in the re-arrangement of the position of β-cells and α-cells in the pancreas that might explain the pancreatic insulin level decrease observed when selenoprotein P is overexpressed (141).
miRNAs are small RNA sequences (around 20 nucleotides) that regulate gene expression by binding on mRNAs to suppress translation or induce their lysosomal degradation (14, 142). Thus, intracellular accumulation of miRNAs could specifically regulate metabolic functions such as insulin secretion of β-cells (14, 143).
For example, miR-7218-5p is expressed in hepatocellular EV and its expression is decreased in high fat diet rats EV (130). miR-7218-5p regulates β-cells’ (MIN6) proliferation as a compensatory effect in an insulin resistance state (130). This impact on β-cells’ proliferation is induced by miR-7218-5p’s regulation of Cd74 gene expression. Cd74 is a transmembrane protein involved in immunological processes but also in cell proliferation regulating ERK1/2 and AKT signaling pathway (130).
Finally, other miRNA secreted by the liver are known to impair insulin secretion in β-cells as miR-375, miR-9, miR-143 (14).
Previously, it has been shown that the liver exerts effects on β-cells through its secretory proteins or extracellular vesicles. However, liver can also have an impact on β-cells indirectly. Indeed, ERK pathway is over-expressed in liver of obese mice and increases β-cell proliferation (136). Imai J. et al. have highlighted that ERK over-expression in hepatocytes regulates β-cell proliferation through splanchnic nerves that deliver a signal to the brain which transfers this signal to the β-cells through vagal nerves (136, 144). The vagal nerves release different neural factors, as acetylcholine, adenylate cyclase activating polypeptide (PACAP), vasoactive intestinal polypeptide (VIP), that are going to activate β-cell proliferation through FoxM1 pathway upregulation, which is a critical factor for β-cell mass expansion (136).
Thus, liver also have indirect effect on β-cells through this liver-β-cells inter organ neuronal network involved in β-cell proliferation in insulin-resistant and obese state (136).
On another hand, liver is also involved in the regulation of glucagon, through a hepatic/α-cells axis (145). α-cells produce glucagon, which, by binding onto the hepatic glucagon receptor, induces hepatic glucose’s production through gluconeogenesis. However, in case of interrupted glucose signalling, a decrease of amino acid (AA) catabolism in the liver is observed increasing amino acid in the blood circulation (145, 146). AA’s increase is involved in α-cell proliferation particularly through glutamine, which is transported in α-cells through Slc38a5 (AA transporter) and, which increases mTOR expression involved in α-cell proliferation (145, 146). This glutamine-dependent α-cell proliferation re-establishes the production of glucagon and, so hepatic glucose’s production (147). Thus, this axis presents several target for diabetes’ therapies (146).
The gastrointestinal tract houses a complex population of microorganisms called gut microbiota. In physiological conditions, it offers many benefits to the host including nutrient and drug metabolism, immunomodulation or maintenance of the structure of gut integrity (148). Dysbiosis of the gut microbiome has been implicated in various diseases including diabetes. Recently, gene sequencing of fecal samples from healthy and diabetic people showed an increase in pro-inflammatory bacteria and a decrease in anti-inflammatory bacteria with diabetes (149). It is also interesting to point out a decline in bacteria that produce short chain fatty acid (SCFA), some of which have shown beneficial effects on the pancreas. Indeed, Pingitore et al. showed that long-term colonic propionate delivery improves β-cell function by potentiating insulin secretion in response to glucose and by protecting cells from cytokine- and palmitate-induced apoptosis (150). In accordance with that, it has been shown that acetate, another SCFA, is able to diminish the frequency of autoimmune T cells in the pancreas and so protect against T1D (151). Finally, intestinal lysozyme can also release Nod1 ligands from commensal bacteria and promote insulin granule transport in β-cell thanks to the recruitment of the protein Rab1a (152). In conclusion, all of these data highlight the importance of taking into account the gut microbiota in the regulation of β-cell function.
In this review, we sought to provide recent knowledge on how β-cell function is regulated. First of all, it is important to highlight that this regulation is more complex as some had thought. Indeed, it is largely demonstrated that β-cell insulin secretion is regulated also by a direct and indirect inter-organ/inter-cellular communication involving various factors, illustrating the idea of “the hidden face of the iceberg” (Figure 1). Then, this very close and complex relationship between all these cell types and organs is fundamental for setting the regulation of insulin secretion and thus for glucose homeostasis. Moreover, this review provides a better understanding of the microenvironment and the context in which β-cells exist. All of these recent studies showed how this communication can influence β-cell survival, function and that any disruption on this physiological communication can participate on diabetes onset. Therefore, for the development of new treatments for diabetic patients, it is necessary to consider the entire network of cells and organs involved in the regulation of β-cellular function and no longer than β-cell or pancreatic islet alone. Finally, many metabolites and signaling pathways, which improve insulin secretion, could be targeted to treat diabetes but further investigations are required to propose the best therapy.
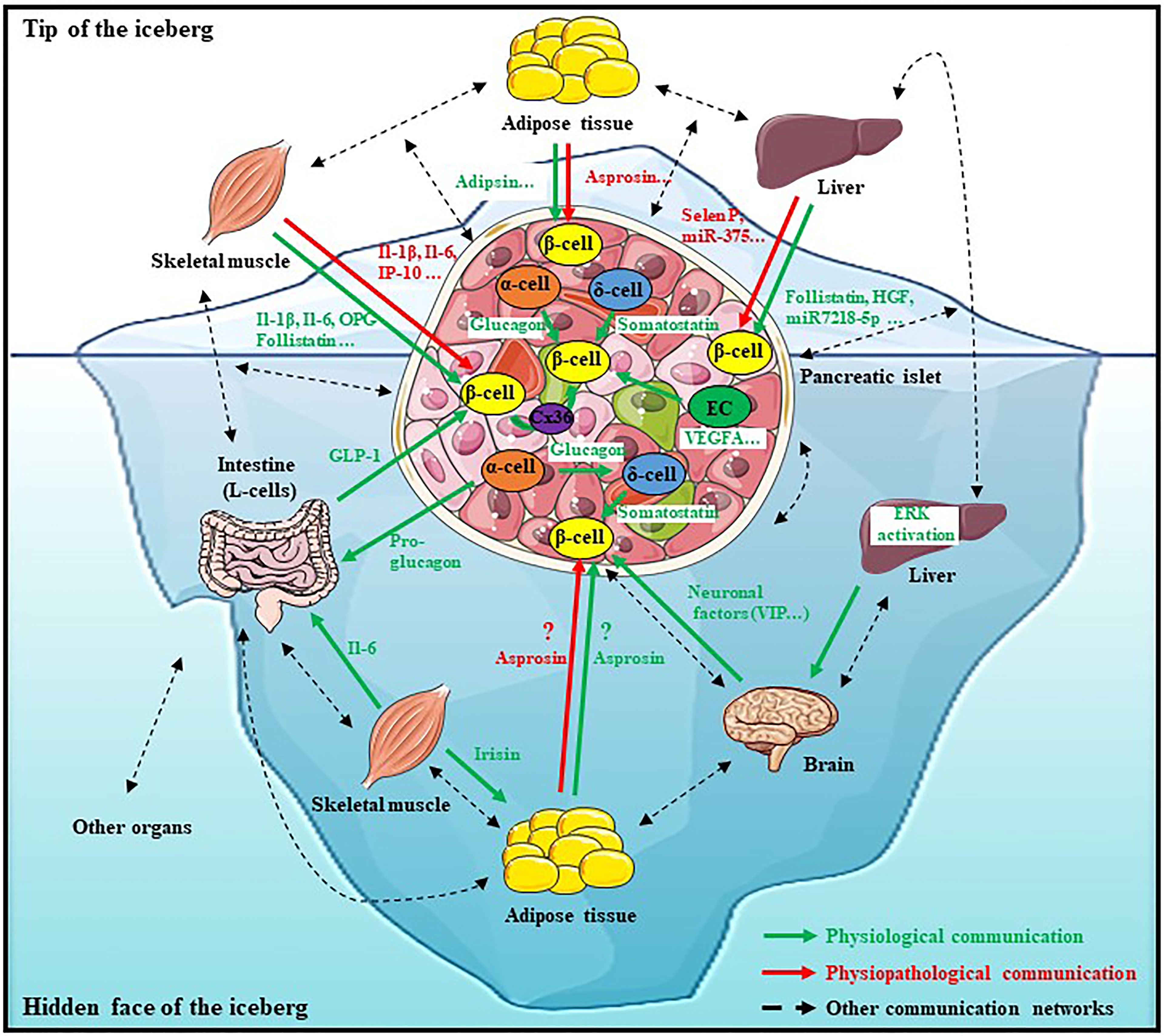
Figure 1 The iceberg of β-cell’s communication network. The physiological β-cellular function is finely regulated by a dense and complex communication network including direct and indirect interactions with various cell types and organs. This communication takes place thanks to various factors secreted from many organs such as the liver, adipose tissue, skeletal muscle, intestine or even the brain which constitute a strong union to allow a physiological β-cell function. However, this description is not exhaustive, this network is more complex and should also involve other organs (Dotted black arrows). Any disturbance in this communication network can lead to dysfunction of β-cell function and to metabolic diseases such as diabetes onset. Therefore, for new anti-diabetic treatments’ development, it is necessary to consider the entire network of cells and organs involved in the regulation of β-cellular function and no longer just the tip of the iceberg.
AL, AD, and JV wrote the review. MP and KB edit the text. KB designed and edited all the review. All authors contributed to the article and approved the submitted version.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Thanks to Christophe Lindley for proofreading the manuscript.

1. Shirakawa J, De Jesus DF, Kulkarni RN. Exploring Inter-Organ Crosstalk to Uncover Mechanisms That Regulate β-Cell Function and Mass. Eur J Clin Nutr (2017) 71(7):896−903. doi: 10.1038/ejcn.2017.13
2. Tanabe K, Amo-Shiinoki K, Hatanaka M, Tanizawa Y. Interorgan Crosstalk Contributing to β -Cell Dysfunction. J Diabetes Res (2017) 2017:1−8. doi: 10.1155/2017/3605178
3. Rutter GA, Pullen TJ, Hodson DJ, Martinez-Sanchez A. Pancreatic β-Cell Identity, Glucose Sensing and the Control of Insulin Secretion. Biochem J (2015) 466(2):203−18. doi: 10.1042/BJ20141384
4. Ilonen J, Lempainen J, Veijola R. The Heterogeneous Pathogenesis of Type 1 Diabetes Mellitus. Nat Rev Endocrinol (2019) 15(11):635−50. doi: 10.1038/s41574-019-0254-y
5. Artasensi A, Pedretti A, Vistoli G, Fumagalli L. Type 2 Diabetes Mellitus: A Review of Multi-Target Drugs. Molecules (2020) 25(8):1987. doi: 10.3390/molecules25081987
6. Meshkani R, Adeli K. Hepatic Insulin Resistance, Metabolic Syndrome and Cardiovascular Disease. Clin Biochem (2009) 42(13−14):1331−46. doi: 10.1016/j.clinbiochem.2009.05.018
7. Petersen MC, Shulman GI. Mechanisms of Insulin Action and Insulin Resistance. Physiol Rev (2018) 98(4):2133−223. doi: 10.1152/physrev.00063.2017
8. Sears B, Perry M. The Role of Fatty Acids in Insulin Resistance. Lipids Health Dis (2015) 14(1):121. doi: 10.1186/s12944-015-0123-1
9. Barber TM, Kyrou I, Randeva HS, Weickert MO. Mechanisms of Insulin Resistance at the Crossroad of Obesity With Associated Metabolic Abnormalities and Cognitive Dysfunction. IJMS (2021) 22(2):546. doi: 10.3390/ijms22020546
10. Silva Rosa SC, Nayak N, Caymo AM, Gordon JW. Mechanisms of Muscle Insulin Resistance and the Cross-Talk With Liver and Adipose Tissue. Physiol Rep (2020) 8(19):e14607. doi: 10.14814/phy2.14607
11. Kitamura T. The Role of FOXO1 in β-Cell Failure and Type 2 Diabetes Mellitus. Nat Rev Endocrinol (2013) 9(10):615−23. doi: 10.1038/nrendo.2013.157
12. Bouzakri K, Plomgaard P, Berney T, Donath MY, Pedersen BK, Halban PA. Bimodal Effect on Pancreatic β-Cells of Secretory Products From Normal or Insulin-Resistant Human Skeletal Muscle. Diabetes (2011) 60(4):1111−21. doi: 10.2337/db10-1178
13. Dunmore SJ, Brown JEP. The Role of Adipokines in β-Cell Failure of Type 2 Diabetes. J Endocrinol (2013) 216(1):T37−45. doi: 10.1530/JOE-12-0278
14. Watt MJ, Miotto PM, De Nardo W, Montgomery MK. The Liver as an Endocrine Organ-Linking NAFLD and Insulin Resistance. Endocr Rev (2019) 40(5):1367−93. doi: 10.1210/er.2019-00034
15. Rodriguez-Diaz R, Tamayo A, Hara M, Caicedo A. The Local Paracrine Actions of the Pancreatic α-Cell. Diabetes (2020) 69(4):550−8. doi: 10.2337/dbi19-0002
16. Prentki M, Matschinsky FM, Madiraju SRM. Metabolic Signaling in Fuel-Induced Insulin Secretion. Cell Metab (2013) 18(2):162−85. doi: 10.1016/j.cmet.2013.05.018
17. Maechler P, Wollheim CB. Mitochondrial Function in Normal and Diabetic Beta-Cells. Nature (2001) 414(6865):807−12. doi: 10.1038/414807a
18. Podobnik B, Korošak D, Skelin Klemen M, Stožer A, Dolenšek J, Slak Rupnik M, et al. β Cells Operate Collectively to Help Maintain Glucose Homeostasis. Biophys J (2020) 118(10):2588−95. doi: 10.1016/j.bpj.2020.04.005
19. Johnston NR, Mitchell RK, Haythorne E, Pessoa MP, Semplici F, Ferrer J, et al. Beta Cell Hubs Dictate Pancreatic Islet Responses to Glucose. Cell Metab (2016) 24(3):389−401. doi: 10.1016/j.cmet.2016.06.020
20. Salem V, Silva LD, Suba K, Georgiadou E, Neda Mousavy Gharavy S, Akhtar N, et al. Leader β-Cells Coordinate Ca2+ Dynamics Across Pancreatic Islets In Vivo. Nat Metab (2019) 1(6):615−29. doi: 10.1038/s42255-019-0075-2
21. Westacott MJ, Ludin NWF, Benninger RKP. Spatially Organized β-Cell Subpopulations Control Electrical Dynamics Across Islets of Langerhans. Biophys J (2017) 113(5):1093−108. doi: 10.1016/j.bpj.2017.07.021
22. Satin LS, Zhang Q, Rorsman P. “Take Me To Your Leader”: An Electrophysiological Appraisal of the Role of Hub Cells in Pancreatic Islets. Diabetes (2020) 69(5):830−6. doi: 10.2337/dbi19-0012
23. Pipeleers D, Veld PI, Maes E, Van De Winkel M. Glucose-Induced Insulin Release Depends on Functional Cooperation Between Islet Cells. Proc Natl Acad Sci USA (1982) 79(23):7322−5. doi: 10.1073/pnas.79.23.7322
24. Speier S, Gjinovci A, Charollais A, Meda P, Rupnik M. Cx36-Mediated Coupling Reduces -Cell Heterogeneity, Confines the Stimulating Glucose Concentration Range, and Affects Insulin Release Kinetics. Diabetes (2007) 56(4):1078−86. doi: 10.2337/db06-0232
25. Eddlestone GT, Gonçalves A, Bangham JA, Rojas E. Electrical Coupling Between Cells in Islets of Langerhans From Mouse. J Membr Biol (1984) 77(1):1−14. doi: 10.1007/BF01871095
26. Michaels RL, Sheridan JD. Islets of Langerhans: Dye Coupling Among Immunocytochemically Distinct Cell Types. Science (1981) 214(4522):801−3. doi: 10.1126/science.6117129
27. Meda P, Bosco D, Chanson M, Giordano E, Vallar L, Wollheim C, et al. Rapid and Reversible Secretion Changes During Uncoupling of Rat Insulin-Producing Cells. J Clin Invest (1990) 86(3):759−68. doi: 10.1172/JCI114772
28. Chidester S, Livinski AA, Fish AF, Joseph PV. The Role of Extracellular Vesicles in β-Cell Function and Viability: A Scoping Review. Front Endocrinol (2020) 11:375. doi: 10.3389/fendo.2020.00375
29. Javeed N, Her TK, Brown MR, Vanderboom P, Rakshit K, Egan AM, et al. Pro-Inflammatory β Cell Small Extracellular Vesicles Induce β Cell Failure Through Activation of the CXCL10/CXCR3 Axis in Diabetes. Cell Rep (2021) 36(8):109613. doi: 10.1016/j.celrep.2021.109613
30. Guay C, Menoud V, Rome S, Regazzi R. Horizontal Transfer of Exosomal microRNAs Transduce Apoptotic Signals Between Pancreatic Beta-Cells. Cell Commun Signal (2015) 13(1):17. doi: 10.1186/s12964-015-0097-7
31. Moede T, Leibiger IB, Berggren P-O. Alpha Cell Regulation of Beta Cell Function. Diabetologia (2020) 63(10):2064−75. doi: 10.1007/s00125-020-05196-3
32. Samols E, Marri G, Marks V. PROMOTION OF INSULIN SECRETION BY GLUCAGON. Lancet (1965) 2(7409):415−6. doi: 10.1016/S0140-6736(65)90761-0
33. Koeslag JH, Saunders PT, Terblanche E. A Reappraisal of the Blood Glucose Homeostat Which Comprehensively Explains the Type 2 Diabetes Mellitus-Syndrome X Complex. J Physiol (2003) 549(Pt 2):333−46. doi: 10.1113/jphysiol.2002.037895
34. Hughes JW, Ustione A, Lavagnino Z, Piston DW. Regulation of Islet Glucagon Secretion: Beyond Calcium. Diabetes Obes Metab (2018) 20(Suppl 2):127−36. doi: 10.1111/dom.13381
35. Rodriguez-Diaz R, Molano RD, Weitz JR, Abdulreda MH, Berman DM, Leibiger B, et al. Paracrine Interactions Within the Pancreatic Islet Determine the Glycemic Set Point. Cell Metab (2018) 27(3):549–558.e4. doi: 10.1016/j.cmet.2018.01.015
36. Kieffer TJ, Habener JF. The Glucagon-Like Peptides. Endocr Rev (1999) 20(6):876−913. doi: 10.1210/edrv.20.6.0385
37. Chambers AP, Sorrell JE, Haller A, Roelofs K, Hutch CR, Kim K-S, et al. The Role of Pancreatic Preproglucagon in Glucose Homeostasis in Mice. Cell Metab (2017) 25(4):927–934.e3. doi: 10.1016/j.cmet.2017.02.008
38. Campbell SA, Golec DP, Hubert M, Johnson J, Salamon N, Barr A, et al. Human Islets Contain a Subpopulation of Glucagon-Like Peptide-1 Secreting α Cells That Is Increased in Type 2 Diabetes. Mol Metab (2020) 39:101014. doi: 10.1016/j.molmet.2020.101014
39. El K, Capozzi ME, Campbell JE. Repositioning the Alpha Cell in Postprandial Metabolism. Endocrinology (2020) 161(11):bqaa169. doi: 10.1210/endocr/bqaa169
40. Finan B, Capozzi ME, Campbell JE. Repositioning Glucagon Action in the Physiology and Pharmacology of Diabetes. Diabetes (2020) 69(4):532−41. doi: 10.2337/dbi19-0004
41. Capozzi ME, DiMarchi RD, Tschöp MH, Finan B, Campbell JE. Targeting the Incretin/Glucagon System With Triagonists to Treat Diabetes. Endocr Rev (2018) 39(5):719−38. doi: 10.1210/er.2018-00117
42. Huypens P, Ling Z, Pipeleers D, Schuit F. Glucagon Receptors on Human Islet Cells Contribute to Glucose Competence of Insulin Release. Diabetologia (2000) 43(8):1012−9. doi: 10.1007/s001250051484
43. Rodriguez-Diaz R, Dando R, Jacques-Silva MC, Fachado A, Molina J, Abdulreda MH, et al. Alpha Cells Secrete Acetylcholine as a Non-Neuronal Paracrine Signal Priming Beta Cell Function in Humans. Nat Med (2011) 17(7):888−92. doi: 10.1038/nm.2371
44. Cabrera O, Berman DM, Kenyon NS, Ricordi C, Berggren P-O, Caicedo A. The Unique Cytoarchitecture of Human Pancreatic Islets Has Implications for Islet Cell Function. Proc Natl Acad Sci USA (2006) 103(7):2334−9. doi: 10.1073/pnas.0510790103
45. Brissova M, Fowler MJ, Nicholson WE, Chu A, Hirshberg B, Harlan DM, et al. Assessment of Human Pancreatic Islet Architecture and Composition by Laser Scanning Confocal Microscopy. J Histochem Cytochem (2005) 53(9):1087−97. doi: 10.1369/jhc.5C6684.2005
46. Arrojo e Drigo R, Jacob S, García-Prieto CF, Zheng X, Fukuda M, Nhu HTT, et al. Structural Basis for Delta Cell Paracrine Regulation in Pancreatic Islets. Nat Commun (2019) 10(1):3700. doi: 10.1038/s41467-019-11517-x
47. Adriaenssens AE, Svendsen B, Lam BYH, Yeo GSH, Holst JJ, Reimann F, et al. Transcriptomic Profiling of Pancreatic Alpha, Beta and Delta Cell Populations Identifies Delta Cells as a Principal Target for Ghrelin in Mouse Islets. Diabetologia (2016) 59(10):2156−65. doi: 10.1007/s00125-016-4033-1
48. DiGruccio MR, Mawla AM, Donaldson CJ, Noguchi GM, Vaughan J, Cowing-Zitron C, et al. Comprehensive Alpha, Beta and Delta Cell Transcriptomes Reveal That Ghrelin Selectively Activates Delta Cells and Promotes Somatostatin Release From Pancreatic Islets. Mol Metab (2016) 5(7):449−58. doi: 10.1016/j.molmet.2016.04.007
49. Gao R, Yang T, Zhang Q. δ-Cells: The Neighborhood Watch in the Islet Community. Biology (2021) 10(2):74. doi: 10.3390/biology10020074
50. Zhang Q, Bengtsson M, Partridge C, Salehi A, Braun M, Cox R, et al. R-Type Ca(2+)-Channel-Evoked CICR Regulates Glucose-Induced Somatostatin Secretion. Nat Cell Biol (2007) 9(4):453−60. doi: 10.1038/ncb1563
51. Molina J, Rodriguez-Diaz R, Fachado A, Jacques-Silva MC, Berggren P-O, Caicedo A. Control of Insulin Secretion by Cholinergic Signaling in the Human Pancreatic Islet. Diabetes (2014) 63(8):2714−26. doi: 10.2337/db13-1371
52. Muroyama A, Uehara S, Yatsushiro S, Echigo N, Morimoto R, Morita M, et al. A Novel Variant of Ionotropic Glutamate Receptor Regulates Somatostatin Secretion From Delta-Cells of Islets of Langerhans. Diabetes (2004) 53(7):1743−53. doi: 10.2337/diabetes.53.7.1743
53. van der Meulen T, Donaldson CJ, Cáceres E, Hunter AE, Cowing-Zitron C, Pound LD, et al. Urocortin3 Mediates Somatostatin-Dependent Negative Feedback Control of Insulin Secretion. Nat Med (2015) 21(7):769−76. doi: 10.1038/nm.3872
54. Briant LJB, Reinbothe TM, Spiliotis I, Miranda C, Rodriguez B, Rorsman P. δ-Cells and β-Cells Are Electrically Coupled and Regulate α-Cell Activity via Somatostatin. J Physiol (2018) 596(2):197−215. doi: 10.1113/JP274581
55. Unger RH, Orci L. Paracrinology of Islets and the Paracrinopathy of Diabetes. Proc Natl Acad Sci (2010) 107(37):16009−12. doi: 10.1073/pnas.1006639107
56. Svendsen B, Holst JJ. Paracrine Regulation of Somatostatin Secretion by Insulin and Glucagon in Mouse Pancreatic Islets. Diabetologia (2021) 64(1):142−51. doi: 10.1007/s00125-020-05288-0
57. Bonner-Weir S, Orci L. New Perspectives on the Microvasculature of the Islets of Langerhans in the Rat. Diabetes (1982) 31(10):883−9. doi: 10.2337/diabetes.31.10.883
58. Aamodt KI, Powers AC. Signals in the Pancreatic Islet Microenvironment Influence β-Cell Proliferation. Diabetes Obes Metab (2017) 19 Suppl 1:124−36. doi: 10.1111/dom.13031
59. Cao Z, Wang X. The Endocrine Role Between β Cells and Intra-Islet Endothelial Cells. Endocr J (2014) 61(7):647−54. doi: 10.1507/endocrj.ej14-0045
60. Nikolova G, Strilic B, Lammert E. The Vascular Niche and Its Basement Membrane. Trends Cell Biol (2007) 17(1):19−25. doi: 10.1016/j.tcb.2006.11.005
61. Yoshitomi H, Zaret KS. Endothelial Cell Interactions Initiate Dorsal Pancreas Development by Selectively Inducing the Transcription Factor Ptf1a. Development (2004) 131(4):807−17. doi: 10.1242/dev.00960
62. Lammert E, Cleaver O, Melton D. Induction of Pancreatic Differentiation by Signals From Blood Vessels. Science (2001) 294(5542):564−7. doi: 10.1126/science.1064344
63. Ferrara N, Gerber H-P, LeCouter J. The Biology of VEGF and Its Receptors. Nat Med (2003) 9(6):669−76. doi: 10.1038/nm0603-669
64. Reinert RB, Brissova M, Shostak A, Pan FC, Poffenberger G, Cai Q, et al. Vascular Endothelial Growth Factor-A and Islet Vascularization Are Necessary in Developing, But Not Adult, Pancreatic Islets. Diabetes (2013) 62(12):4154−64. doi: 10.2337/db13-0071
65. Hogan MF, Liu AW, Peters MJ, Willard JR, Rabbani Z, Bartholomew EC, et al. Markers of Islet Endothelial Dysfunction Occur in Male B6.BKS(D)-Leprdb/J Mice and May Contribute to Reduced Insulin Release. Endocrinology (2017) 158(2):293−303. doi: 10.1210/en.2016-1393
66. Hogan MF, Hull RL. The Islet Endothelial Cell: A Novel Contributor to Beta Cell Secretory Dysfunction in Diabetes. Diabetologia (2017) 60(6):952−9. doi: 10.1007/s00125-017-4272-9
67. Bosco D, Meda P, Halban PA, Rouiller DG. Importance of Cell-Matrix Interactions in Rat Islet Beta-Cell Secretion In Vitro: Role of Alpha6beta1 Integrin. Diabetes (2000) 49(2):233−43. doi: 10.2337/diabetes.49.2.233
68. Parnaud G, Hammar E, Rouiller DG, Armanet M, Halban PA, Bosco D. Blockade of Beta1 Integrin-Laminin-5 Interaction Affects Spreading and Insulin Secretion of Rat Beta-Cells Attached on Extracellular Matrix. Diabetes (2006) 55(5):1413−20. doi: 10.2337/db05-1388
69. Johansson A, Lau J, Sandberg M, Borg L a H, Magnusson PU, Carlsson P-O. Endothelial Cell Signalling Supports Pancreatic Beta Cell Function in the Rat. Diabetologia (2009) 52(11):2385−94. doi: 10.1007/s00125-009-1485-6
70. Daniel B, Livne A, Cohen G, Kahremany S, Sasson S. Endothelial Cell–Derived Triosephosphate Isomerase Attenuates Insulin Secretion From Pancreatic Beta Cells of Male Rats. Endocrinology (2021) 162(3):bqaa234. doi: 10.1210/endocr/bqaa234
71. Figliolini F, Cantaluppi V, De Lena M, Beltramo S, Romagnoli R, Salizzoni M, et al. Isolation, Characterization and Potential Role in Beta Cell-Endothelium Cross-Talk of Extracellular Vesicles Released From Human Pancreatic Islets. Abderrahmani A, Éditeur. PloS One (2014) 9(7):e102521. doi: 10.1371/journal.pone.0102521
72. Ilegems E, Berggren P-O. The Eye as a Transplantation Site to Monitor Pancreatic Islet Cell Plasticity. Front Endocrinol (2021) 12:652853. doi: 10.3389/fendo.2021.652853
73. Furuyama K, Chera S, van Gurp L, Oropeza D, Ghila L, Damond N, et al. Diabetes Relief in Mice by Glucose-Sensing Insulin-Secreting Human α-Cells. Nature (2019) 567(7746):43−8. doi: 10.1038/s41586-019-0942-8
74. Grimont A, Leach SD, Chandwani R. Uncertain Beginnings: Acinar and Ductal Cell Plasticity in the Development of Pancreatic Cancer. Cell Mol Gastroenterol Hepatol (2022) 13(2):369−82. doi: 10.1016/j.jcmgh.2021.07.014
75. Chera S, Baronnier D, Ghila L, Cigliola V, Jensen JN, Gu G, et al. Diabetes Recovery by Age-Dependent Conversion of Pancreatic δ-Cells Into Insulin Producers. Nature (2014) 514(7523):503−7. doi: 10.1038/nature13633
76. Thorel F, Népote V, Avril I, Kohno K, Desgraz R, Chera S, et al. Conversion of Adult Pancreatic α-Cells to β-Cells After Extreme β-Cell Loss. Nature (2010) 464(7292):1149−54. doi: 10.1038/nature08894
77. Remedi MS, Emfinger C. Pancreatic β-Cell Identity in Diabetes. Diabetes Obes Metab (2016) 18:110−6. doi: 10.1111/dom.12727
78. Kaya-Dagistanli F, Ozturk M. Transdifferentiation of Both Intra- and Extra-Islet Cells Into Beta Cells in Nicotinamide Treated Neonatal Diabetic Rats: An In Situ Hybridization and Double Immunohistochemical Study. Acta Histochemica (2020) 122(7):151612. doi: 10.1016/j.acthis.2020.151612
79. Kim H, Lee M. β-Cell Regeneration Through the Transdifferentiation of Pancreatic Cells: Pancreatic Progenitor Cells in the Pancreas. J Diabetes Investig (2016) 7(3):286−96. doi: 10.1111/jdi.12475
80. Zierath JR, Krook A, Wallberg-Henriksson H. Insulin Action and Insulin Resistance in Human Skeletal Muscle. Diabetologia (2000) 43(7):821−35. doi: 10.1007/s001250051457
81. Febbraio MA, Pedersen BK. Contraction-Induced Myokine Production and Release: Is Skeletal Muscle an Endocrine Organ? Exerc Sport Sci Rev (2005) 33(3):114−9. doi: 10.1097/00003677-200507000-00003
82. Pedersen BK, Akerström TCA, Nielsen AR, Fischer CP. Role of Myokines in Exercise and Metabolism. J Appl Physiol (2007) 103(3):1093−8. doi: 10.1152/japplphysiol.00080.2007
83. Brandt C, Pedersen BK. The Role of Exercise-Induced Myokines in Muscle Homeostasis and the Defense Against Chronic Diseases. J Biomedicine Biotechnol (2010) 2010:1−6. doi: 10.1155/2010/520258
84. Pedersen BK. Exercise-Induced Myokines and Their Role in Chronic Diseases. Brain Behav Immun (2011) 25(5):811−6. doi: 10.1016/j.bbi.2011.02.010
85. Bárány M. ATPase Activity of Myosin Correlated With Speed of Muscle Shortening. J Gen Physiol (1967) 50(6):197–218. doi: 10.1085/jgp.50.6.197
86. Andersen JL, Schiaffino S. Mismatch Between Myosin Heavy Chain mRNA and Protein Distribution in Human Skeletal Muscle Fibers. Am J Physiol (1997) 272(6 Pt 1):C1881–1889. doi: 10.1152/ajpcell.1997.272.6.C1881
87. Spamer C, Pette D. Activity Patterns of Phosphofructokinase, Glyceraldehydephosphate Dehydrogenase, Lactate Dehydrogenase and Malate Dehydrogenase in Microdissected Fast and Slow Fibres From Rabbit Psoas and Soleus Muscle. Histochemistry (1977) 52(3):201−16. doi: 10.1007/BF00495857
88. Maedler K, Schumann DM, Sauter N, Ellingsgaard H, Bosco D, Baertschiger R, et al. Low Concentration of Interleukin-1beta Induces FLICE-Inhibitory Protein-Mediated Beta-Cell Proliferation in Human Pancreatic Islets. Diabetes (2006) 55(10):2713−22. doi: 10.2337/db05-1430.
89. Ribaux P, Ehses JA, Lin-Marq N, Carrozzino F, Böni-Schnetzler M, Hammar E, et al. Induction of CXCL1 by Extracellular Matrix and Autocrine Enhancement by Interleukin-1 in Rat Pancreatic Beta-Cells. Endocrinology (2007) 148(11):5582−90. doi: 10.1210/en.2007-0325
90. Parnaud G, Hammar E, Ribaux P, Donath MY, Berney T, Halban PA. Signaling Pathways Implicated in the Stimulation of Beta-Cell Proliferation by Extracellular Matrix. Mol Endocrinol (2009) 23(8):1264−71. doi: 10.1210/me.2009-0008
91. Nicoletti F, Conget I, Di Mauro M, Di Marco R, Mazzarino MC, Bendtzen K, et al. Serum Concentrations of the Interferon-Gamma-Inducible Chemokine IP-10/CXCL10 Are Augmented in Both Newly Diagnosed Type I Diabetes Mellitus Patients and Subjects at Risk of Developing the Disease. Diabetologia (2002) 45(8):1107−10. doi: 10.1007/s00125-002-0879-5
92. Xu H, Nakayama K, Ogawa S, Sugiura A, Kato T, Sato T, et al. Elevated Plasma Concentration of IP-10 in Patients With Type 2 Diabetes Mellitus. Nihon Jinzo Gakkai Shi (2005) 47(5):524−30.
93. Rhode A, Pauza ME, Barral AM, Rodrigo E, Oldstone MBA, von Herrath MG, et al. Islet-Specific Expression of CXCL10 Causes Spontaneous Islet Infiltration and Accelerates Diabetes Development. J Immunol (2005) 175(6):3516−24. doi: 10.4049/jimmunol.175.6.3516
94. Ellingsgaard H, Ehses JA, Hammar EB, Van Lommel L, Quintens R, Martens G, et al. Interleukin-6 Regulates Pancreatic Alpha-Cell Mass Expansion. Proc Natl Acad Sci USA (2008) 105(35):13163−8. doi: 10.1073/pnas.0801059105
95. Suzuki T, Imai J, Yamada T, Ishigaki Y, Kaneko K, Uno K, et al. Interleukin-6 Enhances Glucose-Stimulated Insulin Secretion From Pancreatic -Cells: Potential Involvement of the PLC-IP3-Dependent Pathway. Diabetes (2011) 60(2):537−47. doi: 10.2337/db10-0796
96. Handschin C, Choi CS, Chin S, Kim S, Kawamori D, Kurpad AJ, et al. Abnormal Glucose Homeostasis in Skeletal Muscle–Specific PGC-1α Knockout Mice Reveals Skeletal Muscle–Pancreatic β Cell Crosstalk. J Clin Invest (2007) 117(11):3463−74. doi: 10.1172/JCI31785
97. Ellingsgaard H, Hauselmann I, Schuler B, Habib AM, Baggio LL, Meier DT, et al. Interleukin-6 Enhances Insulin Secretion by Increasing Glucagon-Like Peptide-1 Secretion From L Cells and Alpha Cells. Nat Med (2011) 17(11):1481−9. doi: 10.1038/nm.2513
98. Pedersen M, Bruunsgaard H, Weis N, Hendel HW, Andreassen BU, Eldrup E, et al. Circulating Levels of TNF-Alpha and IL-6-Relation to Truncal Fat Mass and Muscle Mass in Healthy Elderly Individuals and in Patients With Type-2 Diabetes. Mech Ageing Dev (2003) 124(4):495−502. doi: 10.1016/S0047-6374(03)00027-7
99. Hansen J, Brandt C, Nielsen AR, Hojman P, Whitham M, Febbraio MA, et al. Exercise Induces a Marked Increase in Plasma Follistatin: Evidence That Follistatin Is a Contraction-Induced Hepatokine. Endocrinology (2011) 152(1):164−71. doi: 10.1210/en.2010-0868
100. Dieli-Conwright CM, Spektor TM, Rice JC, Sattler FR, Schroeder ET. Hormone Therapy and Maximal Eccentric Exercise Alters Myostatin-Related Gene Expression in Postmenopausal Women. J Strength Cond Res (2012) 26(5):1374−82. doi: 10.1519/JSC.0b013e318251083f
101. Laurentino GC, Ugrinowitsch C, Roschel H, Aoki MS, Soares AG, Neves M, et al. Strength Training With Blood Flow Restriction Diminishes Myostatin Gene Expression. Med Sci Sports Exerc (2012) 44(3):406−12. doi: 10.1249/MSS.0b013e318233b4bc
102. Hansen JS, Pedersen BK, Xu G, Lehmann R, Weigert C, Plomgaard P. Exercise-Induced Secretion of FGF21 and Follistatin Are Blocked by Pancreatic Clamp and Impaired in Type 2 Diabetes. J Clin Endocrinol Metab (2016) 101(7):2816−25. doi: 10.1210/jc.2016-1681
103. Scheler M, Irmler M, Lehr S, Hartwig S, Staiger H, Al-Hasani H, et al. Cytokine Response of Primary Human Myotubes in an In Vitro Exercise Model. Am J Physiol Cell Physiol (2013) 305(8):C877–886. doi: 10.1152/ajpcell.00043.2013
104. Pedersen BK, Febbraio MA. Muscles, Exercise and Obesity: Skeletal Muscle as a Secretory Organ. Nat Rev Endocrinol (2012) 8(8):457−65. doi: 10.1038/nrendo.2012.49
105. Rutti S, Arous C, Schvartz D, Timper K, Sanchez J-C, Dermitzakis E, et al. Fractalkine (CX3CL1), a New Factor Protecting β-Cells Against Tnfα. Mol Metab (2014) 3(7):731−41. doi: 10.1016/j.molmet.2014.07.007
106. Lee YS, Morinaga H, Kim JJ, Lagakos W, Taylor S, Keshwani M, et al. The Fractalkine/CX3CR1 System Regulates β Cell Function and Insulin Secretion. Cell (2013) 153(2):413−25. doi: 10.1016/j.cell.2013.03.001
107. Kondegowda NG, Fenutria R, Pollack IR, Orthofer M, Garcia-Ocaña A, Penninger JM, et al. Osteoprotegerin and Denosumab Stimulate Human Beta Cell Proliferation Through Inhibition of the Receptor Activator of NF-κb Ligand Pathway. Cell Metab (2015) 22(1):77−85. doi: 10.1016/j.cmet.2015.05.021
108. Rutti S, Dusaulcy R, Hansen JS, Howald C, Dermitzakis ET, Pedersen BK, et al. Angiogenin and Osteoprotegerin are Type II Muscle Specific Myokines Protecting Pancreatic Beta-Cells Against Proinflammatory Cytokines. Sci Rep (2018) 8(1):10072. doi: 10.1038/s41598-018-28117-2
109. Romere C, Duerrschmid C, Bournat J, Constable P, Jain M, Xia F, et al. Asprosin, a Fasting-Induced Glucogenic Protein Hormone. Cell (2016) 165(3):566−79. doi: 10.1016/j.cell.2016.02.063
110. Wang C-Y, Lin T-A, Liu K-H, Liao C-H, Liu Y-Y, Wu VC-C, et al. Serum Asprosin Levels and Bariatric Surgery Outcomes in Obese Adults. Int J Obes (2019) 43(5):1019−25. doi: 10.1038/s41366-018-0248-1
111. Zhang L, Chen C, Zhou N, Fu Y, Cheng X. Circulating Asprosin Concentrations Are Increased in Type 2 Diabetes Mellitus and Independently Associated With Fasting Glucose and Triglyceride. Clinica Chimica Acta (2019) 489:183−8. doi: 10.1016/j.cca.2017.10.034
112. Wang Y, Qu H, Xiong X, Qiu Y, Liao Y, Chen Y, et al. Plasma Asprosin Concentrations Are Increased in Individuals With Glucose Dysregulation and Correlated With Insulin Resistance and First-Phase Insulin Secretion. Mediators Inflammation (2018) 2018:1−7. doi: 10.1155/2018/9471583
113. Lee T, Yun S, Jeong JH, Jung TW. Asprosin Impairs Insulin Secretion in Response to Glucose and Viability Through TLR4/JNK-Mediated Inflammation. Mol Cell Endocrinol (2019) 486:96−104. doi: 10.1016/j.mce.2019.03.001
114. Ozcan S, Ulker N, Bulmus O, Yardimci A, Ozcan M, Canpolat S. The Modulatory Effects of Irisin on Asprosin, Leptin, Glucose Levels and Lipid Profile in Healthy and Obese Male and Female Rats. Arch Physiol Biochem (2020). doi: 10.1080/13813455.2020.1722706
115. Cook KS, Min HY, Johnson D, Chaplinsky RJ, Flier JS, Hunt CR, et al. Adipsin: A Circulating Serine Protease Homolog Secreted by Adipose Tissue and Sciatic Nerve. Science (1987) 237(4813):402−5. doi: 10.1126/science.3299705
116. Lo JC, Ljubicic S, Leibiger B, Kern M, Leibiger IB, Moede T, et al. Adipsin Is an Adipokine That Improves β Cell Function in Diabetes. Cell (2014) 158(1):41−53. doi: 10.1016/j.cell.2014.06.005
117. Gómez-Banoy N, Guseh JS, Li G, Rubio-Navarro A, Chen T, Poirier B, et al. Adipsin Preserves Beta Cells in Diabetic Mice and Associates With Protection From Type 2 Diabetes in Humans. Nat Med (2019) 25(11):1739−47. doi: 10.1038/s41591-019-0610-4
118. Ouchi N, Higuchi A, Ohashi K, Oshima Y, Gokce N, Shibata R, et al. Sfrp5 Is an Anti-Inflammatory Adipokine That Modulates Metabolic Dysfunction in Obesity. Science (2010) 329(5990):454−7. doi: 10.1126/science.1188280
119. Carstensen-Kirberg M, Röhrig K, Niersmann C, Ouwens DM, Belgardt BF, Roden M, et al. Sfrp5 Increases Glucose-Stimulated Insulin Secretion in the Rat Pancreatic Beta Cell Line INS-1e. Fürnsinn C, Éditeur. PloS One (2019) 14(3):e0213650. doi: 10.1371/journal.pone.0213650
120. Lanuza-Masdeu J, Arévalo MI, Vila C, Barberà A, Gomis R, Caelles C. In Vivo JNK Activation in Pancreatic B-Cells Leads to Glucose Intolerance Caused by Insulin Resistance in Pancreas. Diabetes (2013) 62(7):2308–17. doi: 10.2337/db12-1097
121. Cao H, Gerhold K, Mayers JR, Wiest MM, Watkins SM, Hotamisligil GS. Identification of a Lipokine, a Lipid Hormone Linking Adipose Tissue to Systemic Metabolism. Cell (2008) 134(6):933−44. doi: 10.1016/j.cell.2008.07.048
122. Gravena C, Mathias P, Ashcroft S. Acute Effects of Fatty Acids on Insulin Secretion From Rat and Human Islets of Langerhans. J Endocrinol (2002) 173(1):73−80. doi: 10.1677/joe.0.1730073
123. Brejchova K, Balas L, Paluchova V, Brezinova M, Durand T, Kuda O. Understanding FAHFAs: From Structure to Metabolic Regulation. Prog Lipid Res (2020) 79:101053. doi: 10.1016/j.plipres.2020.101053
124. Yore MM, Syed I, Moraes-Vieira PM, Zhang T, Herman MA, Homan EA, et al. Discovery of a Class of Endogenous Mammalian Lipids With Anti-Diabetic and Anti-Inflammatory Effects. Cell (2014) 159(2):318−32. doi: 10.1016/j.cell.2014.09.035
125. Syed I, Rubin de Celis MF, Mohan JF, Moraes-Vieira PM, Vijayakumar A, Nelson AT, et al. PAHSAs Attenuate Immune Responses and Promote β Cell Survival in Autoimmune Diabetic Mice. J Clin Invest (2019) 129(9):3717−31. doi: 10.1172/JCI122445
126. Meloni AR, DeYoung MB, Lowe C, Parkes DG. GLP -1 Receptor Activated Insulin Secretion From Pancreatic β-Cells: Mechanism and Glucose Dependence. Diabetes Obes Metab (2013) 15(1):15−27. doi: 10.1111/j.1463-1326.2012.01663.x
127. Freeman DW, Noren Hooten N, Eitan E, Green J, Mode NA, Bodogai M, et al. Altered Extracellular Vesicle Concentration, Cargo, and Function in Diabetes. Diabetes (2018) 67(11):2377−88. doi: 10.2337/db17-1308
128. Gao X, Salomon C, Freeman DJ. Extracellular Vesicles From Adipose Tissue—A Potential Role in Obesity and Type 2 Diabetes? Front Endocrinol (2017) 8:202. doi: 10.3389/fendo.2017.00202
129. Gesmundo I, Pardini B, Gargantini E, Gamba G, Birolo G, Fanciulli A, et al. Adipocyte-Derived Extracellular Vesicles Regulate Survival and Function of Pancreatic β Cells. JCI Insight 8 Mars (2021) 6(5):e141962. doi: 10.1172/jci.insight.141962
130. Fu Q, Li Y, Jiang H, Shen Z, Gao R, He Y, et al. Hepatocytes Derived Extracellular Vesicles From High-Fat Diet Induced Obese Mice Modulate Genes Expression and Proliferation of Islet β Cells. Biochem Biophys Res Commun (2019) 516(4):1159−66. doi: 10.1016/j.bbrc.2019.06.124
131. Staiger H, Keuper M, Berti L, Hrabě de Angelis M, Häring H-U. Fibroblast Growth Factor 21—Metabolic Role in Mice and Men. Endocrine Rev (2017) 38(5):468−88. doi: 10.1210/er.2017-00016
132. Phan P, Saikia BB, Sonnaila S, Agrawal S, Alraawi Z, Kumar TKS, et al. The Saga of Endocrine FGFs. Cells (2021) 10(9):2418. doi: 10.3390/cells10092418
133. Xie T, So WY, Li XY, Leung PS. Fibroblast Growth Factor 21 Protects Against Lipotoxicity-Induced Pancreatic β-Cell Dysfunction via Regulation of AMPK Signaling and Lipid Metabolism. Clin Sci (Lond) (2019) 133(19):2029−44. doi: 10.1042/CS20190093
134. Wente W, Efanov AM, Brenner M, Kharitonenkov A, Köster A, Sandusky GE, et al. Fibroblast Growth Factor-21 Improves Pancreatic β-Cell Function and Survival by Activation of Extracellular Signal–Regulated Kinase 1/2 and Akt Signaling Pathways. Diabetes (2006) 55(9):2470−8. doi: 10.2337/db05-1435
135. Yan J, Nie Y, Cao J, Luo M, Yan M, Chen Z, et al. The Roles and Pharmacological Effects of FGF21 in Preventing Aging-Associated Metabolic Diseases. Front Cardiovasc Med (2021) 8:655575. doi: 10.3389/fcvm.2021.655575
136. Imai J. Regulation of Compensatory β-Cell Proliferation by Inter-Organ Networks From the Liver to Pancreatic β-Cells. Endocr J (2018) 65(7):677–84. doi: 10.1507/endocrj.EJ18-0241
137. El Ouaamari A, Dirice E, Gedeon N, Hu J, Zhou J-Y, Shirakawa J, et al. SerpinB1 Promotes Pancreatic β Cell Proliferation. Cell Metab (2016) 23(1):194−205. doi: 10.1016/j.cmet.2015.12.001
138. Araújo TG, Oliveira AG, Carvalho BM, Guadagnini D, Protzek AOP, Carvalheira JBC, et al. Hepatocyte Growth Factor Plays a Key Role in Insulin Resistance-Associated Compensatory Mechanisms. Endocrinology (2012) 153(12):5760−9. doi: 10.1210/en.2012-1496
139. Zhou D, Mou X, Liu K, Liu W, Xu Y, Zhou D. In silico Prediction and Validation of Potential Therapeutic Genes in Pancreatic Β−Cells Associated With Type 2 Diabetes. Exp Ther Med (2020) 20(5):1−1. doi: 10.3892/etm.2020.9188
140. Misu H, Takamura T, Takayama H, Hayashi H, Matsuzawa-Nagata N, Kurita S, et al. A Liver-Derived Secretory Protein, Selenoprotein P, Causes Insulin Resistance. Cell Metab (2010) 12(5):483−95. doi: 10.1016/j.cmet.2010.09.015
141. Saito Y. Selenoprotein P as a Significant Regulator of Pancreatic β Cell Function. J Biochem (2020) 167(2):119−24. doi: 10.1093/jb/mvz061
142. Guo H, Ingolia NT, Weissman JS, Bartel DP. Mammalian microRNAs Predominantly Act to Decrease Target mRNA Levels. Nature (2010) 466(7308):835−40. doi: 10.1038/nature09267
143. Hashimoto N, Tanaka T. Role of miRNAs in the Pathogenesis and Susceptibility of Diabetes Mellitus. J Hum Genet (2017) 62(2):141−50. doi: 10.1038/jhg.2016.150
144. Katagiri H, Imai J, Oka Y. Neural Relay From the Liver Induces Proliferation of Pancreatic β Cells: A Path to Regenerative Medicine Using the Self-Renewal Capabilities. Communicative Integr Biol (2009) 2(5):425−7. doi: 10.4161/cib.2.5.9053
145. Dean ED, Li M, Prasad N, Wisniewski SN, Von Deylen A, Spaeth J, et al. Interrupted Glucagon Signaling Reveals Hepatic α Cell Axis and Role for L-Glutamine in α Cell Proliferation. Cell Metab (2017) 25(6):1362–1373.e5. doi: 10.1016/j.cmet.2017.05.011
146. Dean ED. A Primary Role for α-Cells as Amino Acid Sensors. Diabetes (2020) 69(4):542−9. doi: 10.2337/dbi19-0021
147. Rhyu J, Yu R. Newly Discovered Endocrine Functions of the Liver. WJH (2021) 13(11):1611−28. doi: 10.4254/wjh.v13.i11.1611
148. Jandhyala SM. Role of the Normal Gut Microbiota. WJG (2015) 21(29):8787. doi: 10.3748/wjg.v21.i29.8787
149. Das T, Jayasudha R, Chakravarthy S, Prashanthi GS, Bhargava A, Tyagi M, et al. Alterations in the Gut Bacterial Microbiome in People With Type 2 Diabetes Mellitus and Diabetic Retinopathy. Sci Rep (2021) 11(1):2738. doi: 10.1038/s41598-021-82538-0
150. Pingitore A, Chambers ES, Hill T, Maldonado IR, Liu B, Bewick G, et al. The Diet-Derived Short Chain Fatty Acid Propionate Improves Beta-Cell Function in Humans and Stimulates Insulin Secretion From Human Islets In Vitro: PINGITORE Et al. Diabetes Obes Metab (2017) 19(2):257−65. doi: 10.1111/dom.12811
151. Mariño E, Richards JL, McLeod KH, Stanley D, Yap YA, Knight J, et al. Gut Microbial Metabolites Limit the Frequency of Autoimmune T Cells and Protect Against Type 1 Diabetes. Nat Immunol (2017) 18(5):552−62. doi: 10.1038/ni.3713
Keywords: Beta cell (B-cell), insulin secretion, islets, skeletal muscle, adipose tissue, Liver
Citation: Langlois A, Dumond A, Vion J, Pinget M and Bouzakri K (2022) Crosstalk Communications Between Islets Cells and Insulin Target Tissue: The Hidden Face of Iceberg. Front. Endocrinol. 13:836344. doi: 10.3389/fendo.2022.836344
Received: 15 December 2021; Accepted: 06 January 2022;
Published: 03 February 2022.
Edited by:
Anabel Rojas, Andalusian Center of Molecular Biology and Regenerative Medicine (CSIC), SpainReviewed by:
Valentine Comaills, Andalusian Center of Molecular Biology and Regenerative Medicine (CSIC), SpainCopyright © 2022 Langlois, Dumond, Vion, Pinget and Bouzakri. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Karim Bouzakri, ay5ib3V6YWtyaUBjZWVkLWRpYWJldGUub3Jn
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.