
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Endocrinol. , 28 April 2022
Sec. Reproduction
Volume 13 - 2022 | https://doi.org/10.3389/fendo.2022.829153
 Tingting Meng1,2,3,4,5†
Tingting Meng1,2,3,4,5† Wenzhe Zhang1,2,3,4,5†
Wenzhe Zhang1,2,3,4,5† Rongrong Zhang1,2,3,4,5
Rongrong Zhang1,2,3,4,5 Jie Li1,2,3,4,5Yuan Gao1,2,3,4,5Yingying Qin1,2,3,4,5
Jie Li1,2,3,4,5Yuan Gao1,2,3,4,5Yingying Qin1,2,3,4,5 Xue Jiao1,2,3,4,5,6*
Xue Jiao1,2,3,4,5,6*Objective: To characterize the status of ovarian reserve and ART outcomes in BPES women and provide informative reference for clinical diagnosis and treatment.
Methods: Twenty-one women with BPES were screened for mutations in the FOXL2 gene and underwent assisted reproductive technology (ART) treatment. Indicators for ovarian reserve and ART outcomes were compared between patients with and without FOXL2 mutations. Additionally, ART outcomes were compared among patients with different subtypes of FOXL2 mutations.
Results: A total of 13 distinct heterozygous variants in the FOXL2 gene were identified in 80.95% of BPES women, including 4 novel mutations with plausible pathogenicity (c.173_175dup, c.481C>T, c.576del and c.675_714del). Compared to non-mutation group, patients with FOXL2 mutations had elevated levels of FSH (P=0.007), decreased AMH levels (P=0.012) and less AFC (P=0.015). They also had worse ART outcomes with large amount of Gn dosage (P=0.008), fewer oocytes (P=0.001), Day3 good quality embryos (P=0.001) and good quality blastocysts (P=0.037), and a higher cancellation rate (P=0.272). High heterogeneity of ART outcomes existed in BPES patients with different FOXL2 mutation types.
Conclusions: BPES patients with FOXL2 mutations had diminished ovarian reserve and adverse ART outcomes. The genotype-reproductive phenotype correlations were highly heterogeneous and cannot be generalized. Genetic counseling for fertility planning and preimplantation or prenatal genetic diagnosis to reduce offspring inheritance are recommended.
Blepharophimosis-ptosis-epicanthus inversus syndrome (BPES, OMIM #110100) is a rare autosomal dominant genetic disorder with an estimated prevalence of 1/50,000 (1). It is characterized by narrow horizontal palpebral fissures, ptosis, epicanthus inversus and telecanthus. In addition to isolated ophthalmic features (type II BPES), BPES patients may also present with female infertility or ovarian dysfunction associated with premature ovarian insufficiency (POI), classified as type I BPES (2).
Several pathogenic or candidate genes of BPES have been found, including FOXL2, UBE3B, KAT6 and ITGB5 (3, 4). Approximately 70% of BPES is contributed by heterozygous variations in FOXL2 gene (5, 6). This single-exon gene encodes a forkhead transcription factor highly conserved among species and predominantly expressed in developing eyelid mesenchyme and fetal and adult ovaries. To date, more than 270 variants in the FOXL2 gene have been reported to be associated with BPES. Of these, intragenic mutations accounted for 80% and could be subdivided into frameshift mutations (44%), in-frame changes (33%), nonsense (12%) and missense mutations (11%) (5, 7). However, a genotype-phenotype correlation remains elusive given that ovarian function is largely unavailable. A tendency was observed that truncated mutations before the polyalanine tract were preferentially associated with BPES type I, whereas polyalanine expansion variants frequently led to BPES type II (5). Nonetheless, the phenotype associated with missense substitutions and with mutations leading to a truncated or extended protein with an intact forkhead domain and polyalanine tract is not clearly predictable. Furthermore, high heterogeneity existed in BPES patients, either with interfamilial or intrafamilial genetic or phenotypic variability (8). Therefore, the correlation between the FOXL2 genotype and BPES reproductive phenotype warrants further investigation in larger cohorts.
FOXL2 is involved in fetal development as well as maintenance of the mature ovary. In the postnatal ovary, it supports follicular growth (9). FOXL2 deficiency in mice led to atresia of the oocytes with no maturation of secondary follicles. Mutations in the FOXL2 gene may affect the proliferation, differentiation and steroidogenesis of granulosa cells by inhibiting the transcription of aromatase, P450scc and Cyclin D2, leading to oocyte atresia and progressive follicular depletion in the absence of functional granulosa cells (10–12). Female patients carrying FOXL2 mutations represent a challenge for genetic counseling in terms of both fertility planning and offspring inheritance. However, the reproductive characteristics in reproductive-aged BPES women were only reported by case reports, including at most 3 cases (13–15). Few studies have previously summarized the clinical, hormonal, and ovarian profiles in BPES women with FOXL2 mutations, let alone the commonly accepted fertility recommendation and practical intervention for successful pregnancy and healthy offspring.
In the current study, we characterized the ovarian reserve status and assisted reproductive technology (ART) outcomes and further evaluated the genotype-reproductive phenotype correlations in female BPES patients with FOXL2 mutations, which will provide informative references for the clinical diagnosis, fertility counseling and treatment of BPES patients.
A total of 21 Han Chinese women with a clinical diagnosis of BPES were recruited from the Reproductive Hospital Affiliated to Shandong University between April 2018 and March 2021. The typical features of eyelid deformities included bilateral dysplasia of the eyelids with narrow horizontal palpebral fissures (blepharophimosis), droopy upper lids reducing the vertical palpebral aperture (ptosis), bilateral skin fold arising from the medial lower eyelid ascending to the upper lid (epicanthus inversus), and an increased distance between the medial canthi (telecanthus). The diagnostic criteria of POI included oligo/amenorrhea for at least 4 months, and elevated FSH level >25 IU/L (on two occasions >4 weeks apart). Subjects with bilateral fallopian tube obstruction, a history of ovarian surgery, chemo- or radiotherapy, and male factor infertility were excluded. The standardized evaluation consisted of clinical history, physical examination, reproductive characteristics, and genetic and iatrogenic factors. The study was approved by the Institutional Review Board of Center for Reproductive Medicine, Shandong University ([2018]- NO.34). Written informed consents were obtained from all participants.
Peripheral blood was sampled on day 2-4 of menstrual cycle. Endocrine hormones follicle-stimulating hormone (FSH), luteinizing hormone (LH), estradiol (E2), and testosterone (T) were detected through chemiluminescence immunoassay (Roche Diagnostics), and anti-Müllerian hormone (AMH) was detected by enzyme-linked immunosorbent assay (Kangrun Biotech). The intra- and inter-assay coefficients of variation were <10% and <15%, respectively. Transvaginal ultrasonography was routinely conducted. Antral follicle count (AFC) was defined as the number of bilateral follicles (2-10 mm in diameter) in early follicular phase.
All patients underwent karyotype analysis and exon sequencing of the FOXL2 gene. Karyotype analysis was performed on GTG-banded metaphase chromosomes prepared from peripheral lymphocyte cultures using a standard protocol that generated 400-450 band resolutions, as detailed in our previous study (16). Chromosome polymorphisms were recorded but classified as normal. For the mutation screening of the FOXL2 gene, genomic DNA was extracted from the peripheral blood samples using QIAamp DNA Blood Kits (Qiagen). The entire coding sequence and its splice site junctions of FOXL2 gene were amplified by the polymerase chain reaction (PCR) with overlapping sets of primers designed by Primer Premier 5.0 software: FOXL2-1F (5’-GCAGTCTGGCTTCCTCAACAA-3′), FOXL2-1R (5’-AGGGGACAAAGAGGAGCGAC-3′), FOXL2-2F (5’-CTGCGAAGACATGTTCGAGAAG-3′), and FOXL2-2R (5’-GGACAAAGAGGAGCGACAGG-3′). The PCR products were first analyzed by agarose gel electrophoresis, purified by polyethylene glycol precipitation and then sequenced on an ABI 3730XL DNA Analyzer (Applied Biosystems, Foster City, CA) with the BigDye Terminator Cycle Sequencing Kit (Applied Biosystems, Carlsbad, CA, USA). The results were analyzed using Sequencer version 4.9 software, and the variants identified were compared from the gnomAD, ExAC and 1000 Genomes database. All novel variants were validated by three independent PCRs and bidirectional sequencing. The nomenclature of the mutations identified was based on Human Genome Variation Society guidelines. Amino acid conservation was analyzed by multiple sequence alignment by ClustalW software, and the plausible pathogenic effect of these mutations was classified according to American College of Medical Genetics and Genomics/Association for Molecular Pathology (ACMG/AMP) guidelines and analyzed by PolyPhen-2 and Mutation Taster.
Patients enrolled in the present study received a standardized ovarian stimulation regimen, including gonadotropin releasing hormone (GnRH) agonist long protocol, GnRH agonist short protocol, and GnRH antagonist protocol. The starting dosage was determined by age, ovarian reserve, body mass index (BMI), and previous response to COH. When at least one dominant follicle reached 18 mm in diameter, human chorionic gonadotropin (hCG) at a dose of 6,000-10,000 IU was used for oocyte maturation triggering. Oocyte retrieval was transvaginally performed 36-38 h after hCG injection. Oocytes were fertilized through either conventional insemination or intracytoplasmic sperm injection (ICSI), depending on semen parameters. In vitro maturation (IVM) was performed for immature oocytes, including those at either the germinal vesicle (GV) or metaphase I (MI) stage. The IVM culture medium was prepared and balanced overnight one day in advance, and then 10 ng/ml epidermal growth factor (EGF) (Sigma, USA), 75 U/L FSH (Serono, Switzerland) and 75 U/L LH (Serono, Switzerland) were added 4 h before immature oocytes were available. Then the number of metaphase II (MII) stage oocytes with the extrusion of the first polar body was counted. For patients requiring granulosa cell coculture, the cumulus granulosa cells were isolated from the cumulus-oocyte complexes (COCs) by digestion with 80 IU/mL hyaluronidase, cut into small pieces at 500 μm with the microscope, and then washed in the IVM medium. After washing, the cells were added for immature oocyte culture in an incubator at 37°C in 6% CO2.
Fertilization was assessed 17-19 h after insemination and defined by the presence of two pronuclei (2PN) or two polar bodies (2PB). Embryo development and quality were assessed 68-72 h (day 3) after insemination based on the number of blastomeres, blastomere symmetry, percentage of fragmentation, and quality of cytoplasm. Day 3 good quality embryo was defined as a minimum of seven blastomeres on day 3 according to the criteria established by the Istanbul Consensus Workshop on Embryo Assessment (17). Day 5/6 embryos were graded using the Gardner scoring system with respect to the inner cell mass and trophectoderm, in which blastocysts equal to or greater than grade 4 BC were defined as good quality blastocysts (18, 19).
SPSS 23.0 (SPSS Inc., Chicago, IL) was used for data analysis. The Shapiro-wilk test was used for normality of distribution. Continuous variables in normality distribution were expressed as mean ± standard deviation and compared by Student’s t-test or one-way analysis of variance. Continuous variables that were not normally distributed were presented as median (quartile interval) and compared by nonparametric test. Categorical variables were presented as percentage (n/N) and compared by chi-square or Fisher’s exact test. A two-tailed value of P<0.05 was considered statistically significant.
A total of 21 participants were recruited with an average age of 30 years old (29.91 ± 4.27 yrs). They showed cardinal features of eyelid deformities from birth with variable severity, including shortened palpebral fissures, drooping eyelids and a tiny skin fold arising from the lower eyelid and an increased distance between the medial canthi. Ten patients (47.62%) had a family history of BPES. All showed infertility (76.2% in primary infertility) with an average duration of 5 years (5.17 ± 3.58 yrs), except for one fertile fetus with 3 fetuses positive for maternal FOXL2 mutations (patient #12). Menstruation irregularity was found in 57.14% of cases, with one presenting with secondary amenorrhea. Seventeen patients had at least one or more abnormal hormone profile(s), with either elevated FSH (>10 IU/L) and/or decreased AMH (<1.2 ng/mL). However, none of them reached the diagnostic criteria for POI. All patients had normal 46, XX karyotype.
Among 21 women with BPES, 17 (80.95%) cases carried mutations in the FOXL2 gene. A total of 13 distinct heterozygous variants were identified, including 2 in-frame duplications, 3 missense variants, 4 nonsense and 4 frameshift variants (Table 1 and Figure 1). These variant sites were highly conserved among species. Of note, four novel mutations were also identified in 4 patients: c.173_175dup (p.Ser58dup), truncated mutations c.481C>T (p.Gln161*), c.576del (p.Lys193Serfs*78) and c.675_714del (p.Ala226Leufs*32) (5, 20–29). None was found in the gnomAD, ExAC, or 1000 Genomes databases or previously reported in the literature. All were predicted to be potentially pathogenic by PolyPhen-2 or Mutation Taster and classified as pathogenic (P) or likely pathogenic (LP) according to ACMG/AMP guidelines.
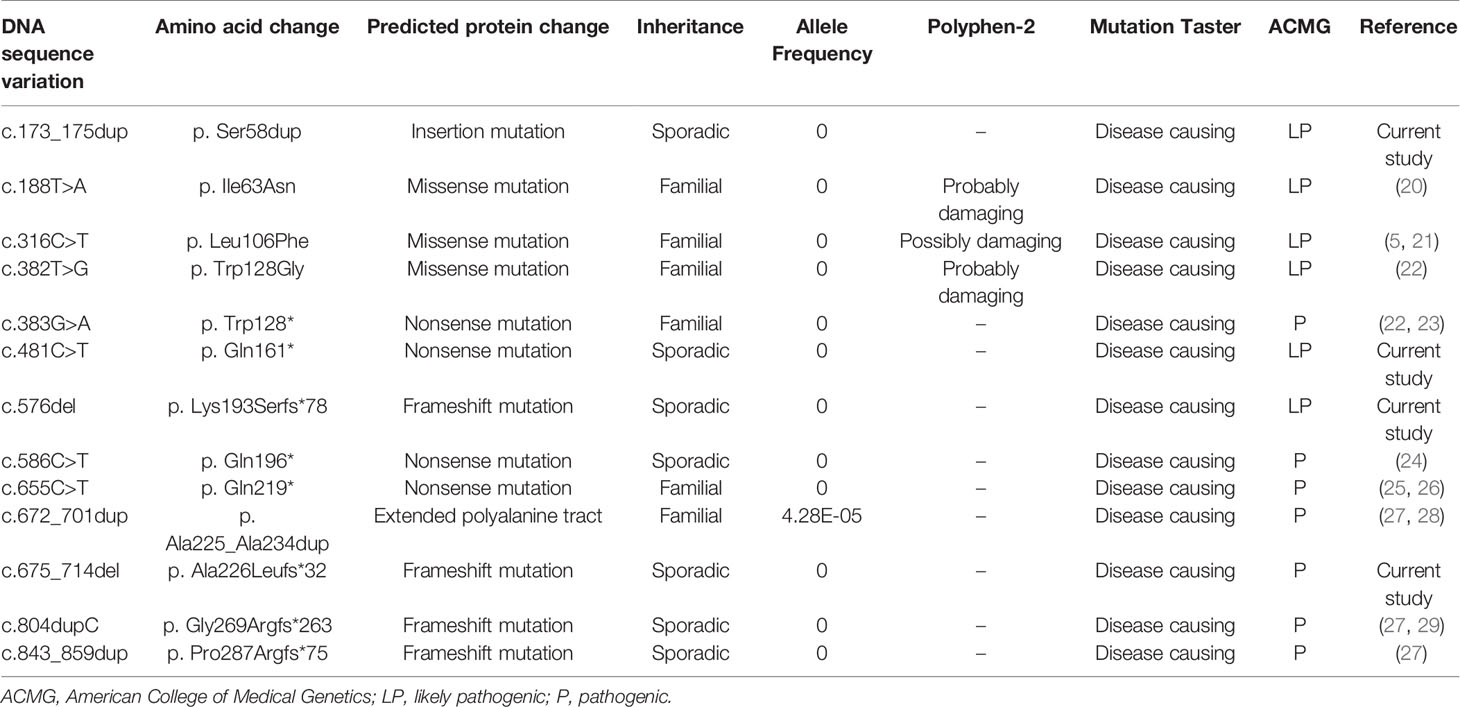
Table 1 The FOXL2 mutations identified in the current study.
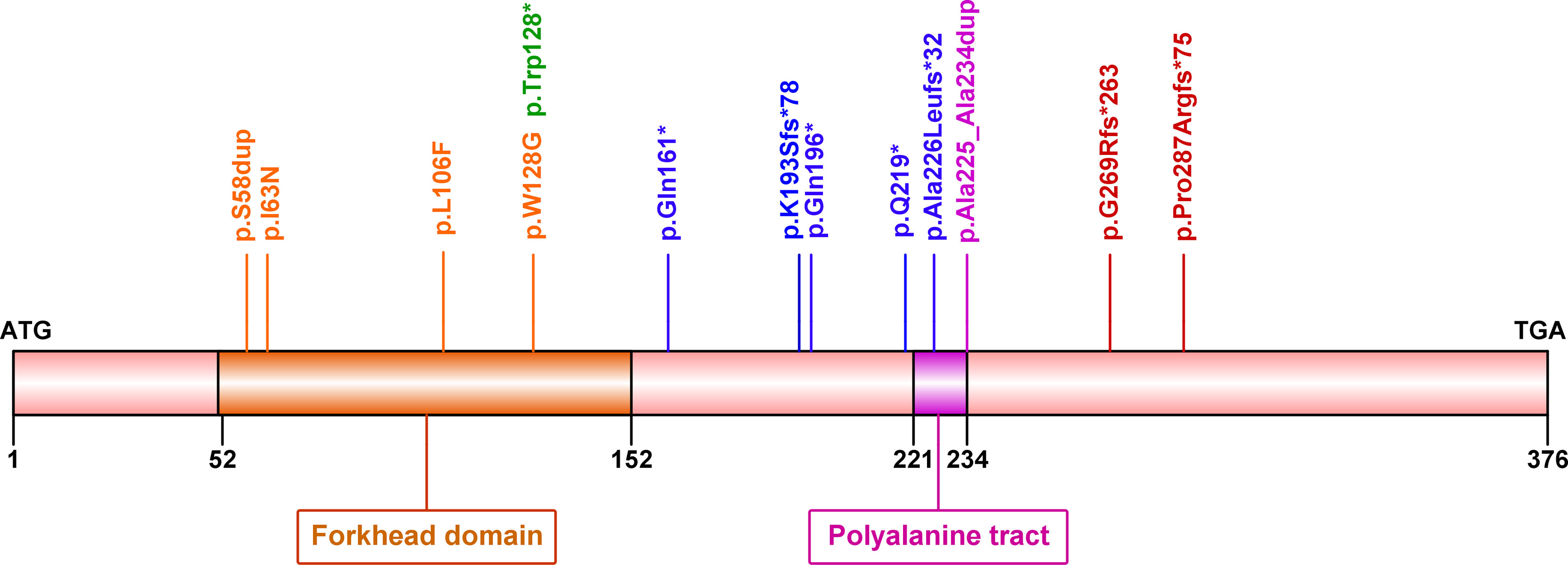
Figure 1 Diagram showing forkhead box L2 (FOXL2) protein structure and mutations identified in the current study. Human FOXL2 encodes a 376 amino acid protein, containing a DNA-binding forkhead domain (amino acid 52–152) and a polyalanine tract (amino acid 221–234).
The status of ovarian reserve was compared between BPES patients with and without FOXL2 gene mutations (Table 2). There was no significant difference in age at diagnosis, BMI or duration of infertility between the two groups (P>0.05). The levels of FSH (17.08 ± 12.74 IU/L vs. 6.66 ± 1.86 IU/L, P=0.007) and LH (11.00 ± 5.76 IU/L vs. 3.23 ± 1.56 IU/L, P=0.017) were significantly higher in patients with FOXL2 mutations than in those without mutations. Consistently, patients with FOXL2 mutations also had significantly decreased AMH concentration (0.460 [0.170, 0.858] ng/mL vs. 2.295 [1.120, 4.518] ng/mL, P=0.012) and AFC (8.18 ± 5.13 vs. 15.25 ± 1.71, P=0.015). In addition, we found that 76.47% of patients in the mutation group had irregular menstruation or amenorrhea, while regular menstrual cycles were reported in all four patients without FOXL2 mutations (P=0.012).

Table 2 The ovarian reserve in BPES patients with and without FOXL2 mutations.
For these 21 women with BPES, 17 ART cycles in total were performed, among which 7 were cancelled due to poor ovarian response (POR). The cycle characteristics and reproductive outcomes of ART were shown in Table 3. The gonadotropin (Gn) starting dosage (229.17 ± 54.18 IU vs. 172.50 ± 22.36 IU, P=0.042), Gn duration (15.08 ± 4.54 days vs. 10.00 ± 1.73 days, P=0.014) and total Gn dosage (4926.25 ± 2073.73 IU vs. 2002.50 ± 648.22 IU, P=0.008) in the mutation group were significantly higher than those in the nonmutation group. Furthermore, patients with FOXL2 mutations had significantly lower level of E2 on trigger day (1553.63 ± 1067.67 pg/ml vs. 2768.00 ± 1039.91 pg/ml, P=0.048) and decreased number of follicles above 14 mm in diameter on the trigger day (4.83 ± 3.97 vs. 14.60 ± 6.19, P=0.001), as well as a significantly lower number of retrieved oocytes (2.75 ± 1.91 vs. 12.40 ± 6.19, P=0.001) than those without mutations.
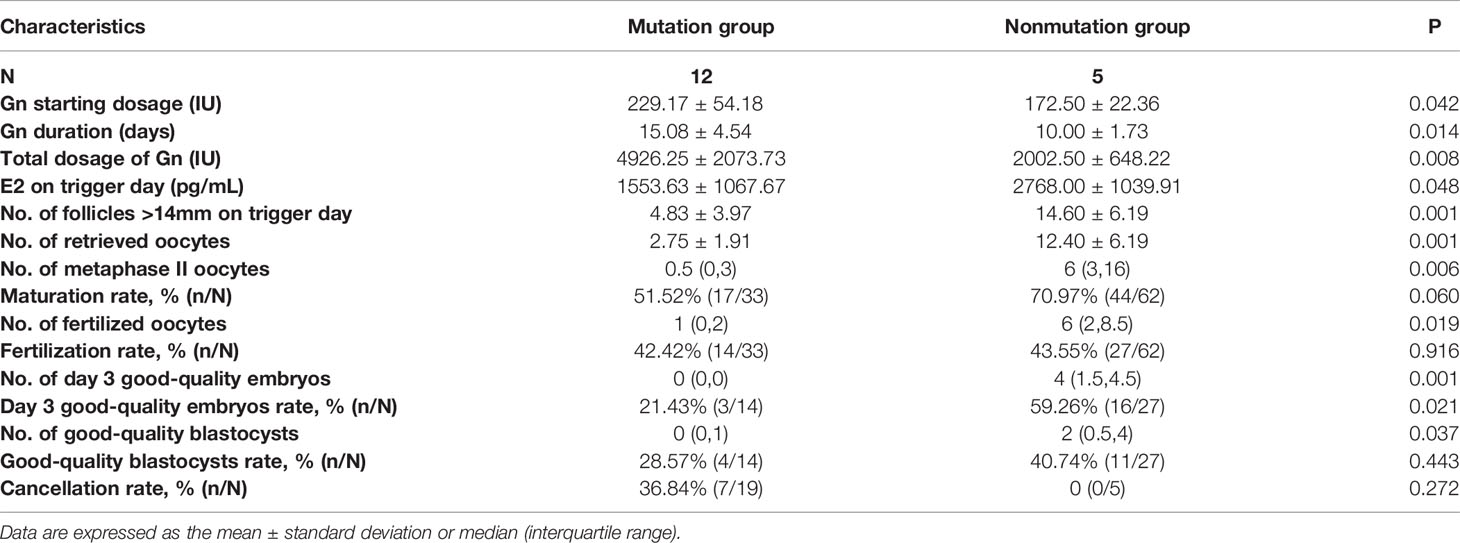
Table 3 The cycle characteristics and reproductive outcomes of ART in BPES patients with and without FOXL2 mutations.
For reproductive outcomes related to oocyte quality, fertilization and embryo quality, we found that the number of MII oocytes (0.5 [0, 3] vs. 6 [3, 16], P=0.006) and fertilized oocytes (1 [0, 2] vs. 6 [2,8.5], P=0.019) in the mutation group were significantly decreased compared with those in nonmutation group, as well as the number of day 3 good quality embryos (0 [0,0] vs. 4 [1.5, 4.5], P=0.001) and good quality blastocysts (0 [0,1] vs. 2 [0.5, 4], P=0.037). Consistently, the maturation rate and day 3 good-quality embryo rate were also decreased in patients with FOXL2 mutations. Additionally, cancelled cycles due to poor ovarian response were present only in the mutation group, although the difference in cycle cancellation rate did not reach statistical significance (36.84% [7/19] vs. 0 [0/5], P=0.272).
To identify a possible genotype-reproductive phenotype correlation, the identified FOXL2 mutations were further classified into five groups according to their location and the predicted effect on protein. Groups A to C present truncated proteins, with a partial forkhead domain (A), with a complete forkhead and without complete polyalanine domain (B), and with a complete forkhead and polyalanine domain (C). Group D comprised duplication mutations leading to elongated polyalanine tracts, and group E contained missense and single amino acid insertion mutations in the forkhead domain. (Figure 2 and Table 4).
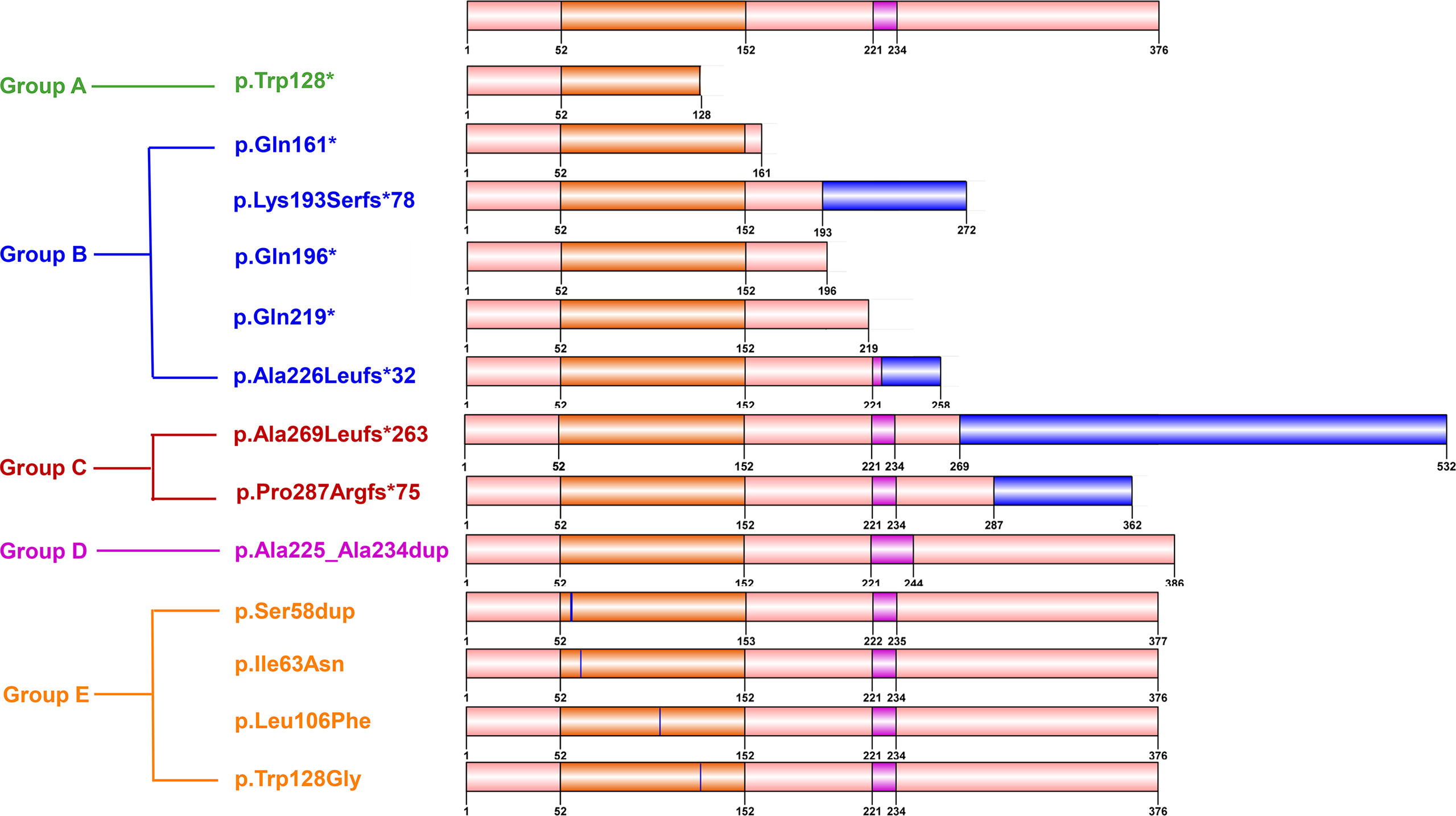
Figure 2 Diagram showing five groups with different types of FOXL2 mutations. Groups A to C: truncated proteins, with partial forkhead domain (Group A), with complete forkhead and without complete polyalanine tract (Group B), and with complete forkhead and polyalanine domain (Group C). Group D: duplication mutations leading to elongated polyalanine tract, group E: missense and single amino acid insertion mutations in forkhead domain.

Table 4 The ART characteristics and outcomes in BPES patients with different types of FOXL2 mutations.
In group A, the patient #1 and #2 were siblings. They both showed primary infertility (~ 4 yrs) and diminished ovarian reserve (DOR) with elevated FSH (>10 IU/L) and decreased AMH (<1.2 ng/ml). They experienced 4 ART cycles in total. Undergoing one ART cycle, patient #1 had 3 oocytes retrieved but failed pregnancy with one good-quality blastocyst transferred. Among the 3 ART cycles for patient #2, only one high-quality blastocyst was obtained, and a baby girl with a maternal FOXL2 mutation was born after frozen embryo transfer.
Group B consisted of 5 infertile patients with DOR (FSH >10 IU/L or AMH <1.2 ng/ml). A total of 8 ART cycles were performed, of which 6 were cancelled due to POR. Two high-quality embryos were obtained for patient #5 within 2 cycles, but no pregnancy was achieved after fresh embryo transfer.
Group C included 2 women with primary infertility and the worst ovarian reserve status (patient #8 and #9). Both of them had FSH >15 IU/L, AMH <1.2 ng/ml and AFC <5 simultaneously. They underwent at least 2 COH cycles without dominant follicles available in other hospitals, and hence receiving oocyte donation was recommended.
Three patients were included in group D. Patients #10 and #11 were infertile with DOR (FSH >15 IU/L and AMH <1.2 ng/ml), turned out without available embryo with single oocyte retrieved or ART cycle canceled due to POR. Patient #12 had normal fertility but multiple offspring with FOXL2 mutations. She had 4 natural pregnancies, including 1 BPES daughter and 3 abortions due to positivity for FOXL2 mutations in the fetus by prenatal diagnosis. Consequently, she asked for fertility counseling and underwent 2 cycles of preimplantation genetic test (PGT) in our center. In the first cycle, 3 immature oocytes were retrieved and matured in vitro by coculturing with granulosa cells but subsequently degenerated after fertilization. In the second cycle, 4 oocytes were retrieved and matured in vitro with granulosa cells from normal donors, which resulted in 2 high-quality blastocysts. However, neither was transferred owing to carrying maternal pathogenic mutations indicated by PGT.
There were 5 patients in group E, of which patients #15 and #17 were sisters. All 5 patients had relatively normal ovarian reserve indicated by FSH and AMH, and two of them (patient #13 and #15) experienced natural pregnancies. Of these, 3 patients (patient #14, #15 and #16) underwent 3 ART cycles, except for #17 with donor eggs. For patient #15, no oocytes were retrieved except 3 empty mucus masses within one cycle. In the ART cycle of patients #14 and #16, the oocytes retrieved with poor oocyte-corona-cumulus complexes remained immature or degenerated after IVM, and consequently no transferable embryos were available.
In summary, among 17 BPES patients with FOXL2 mutations, 4 had transferable embryos available but clinical pregnancy with maternal FOXL2 mutation was achieved in one case.
BPES is a rare genetic disorder characterized by eyelid manifestation in the presence or absence of infertility or ovary dysfunction, with FOXL2 as the dominant pathogenic gene. Currently, the genotype-reproductive phenotype correlation remains elusive. In the current study, we identified 13 distinct FOXL2 heterozygous variants in 6 BPES families and 7 sporadic cases, of which 4 were novel with potentially pathogenic effects. The ovarian reserve in BPES patients with FOXL2 gene mutations was significantly more impaired than that in those without mutations. Furthermore, BPES patients with FOXL2 mutations had worse ART outcomes, characterized by a large Gn dosage, fewer oocytes and high-quality embryos, and a higher cycle cancellation rate. High heterogeneity existed in the ART outcomes of BPES patients with distinct mutation types. We did not discover more grievous reproductive impairments related to FOXL2 mutations with more severe deleterious functional effects. Furthermore, for women with BPES with FOXL2 mutations, PGT is recommended to reduce the risk of offspring inheritance.
Since the BPES classification was first proposed in 1983, an increasing number of studies on FOXL2 mutations and functional experiments have been reported (2). The majority focused on ophthalmology or included mainly children and male patients, and reproductive characteristics have seldom been comprehensively evaluated in reproductive-aged women except for some case reports (15, 25, 30, 31). Here, we are the first to characterize fertility, ovarian reserve status and ART outcomes in BPES women with FOXL2 mutations. We found that the ovarian reserve, quality of oocytes and embryos, and reproductive outcomes were significantly compromised in BPES women positive for FOXL2 mutations compared to those without mutations, although they all presented with infertility. In the adult ovary, FOXL2 is predominately expressed in the undifferentiated granulosa cells of the growing follicles and affects granulosa cell proliferation, differentiation and steroidogenesis by inhibiting the activity of steroidogenic acute regulatory (StAR) protein, aromatase, P450scc and Cyclin D2 (9–11, 32). Ablation of FOXL2 could result in female infertility with follicles blocked between the primordial and primary stages and subsequent follicle atresia. Therefore, the impairment of reproductive phenotypes of our patients could be contributed by the functional deficiency of FOXL2 in ovarian development and functional maintenance.
Notably, unlike the severe ovarian phenotype in FOXL2 knockout mice, patients with heterozygous FOXL2 mutations usually undergo complete follicle development and normal menarche, followed by oligomenorrhea and amenorrhea due to accelerated depletion of the follicle pool and subsequent ovarian dysfunction or even POI. The BPES patients with FOXL2 mutations in our study were infertile due to ovarian dysfunction with variable severity, but none exhibited POI until now. Inappropriate luteinization of graafian follicles might be the major pathophysiological mechanism involved. Alternatively, the patients are still younger than 40 years old and they might suffer from the more severe phenotype of POI later. In addition, FOXL2 could also be expressed in the adult pituitary and essential for FSHB transcription in the pituitary and for maintaining the FSH levels required (33, 34). Defects in FSH secretion resulting from the FOXL2 mutations might partially explain the low FSH levels in most BPES patients (35). Given that, the classification of BPES should not merely rely on the presence of abnormal FSH levels but focus on whether there is intact ovarian function or fertility.
The establishment of a genotype-phenotype correlation is of great importance for fertility prediction and timely intervention for reproductive-aged women. To date, it remains a great challenge to distinguish BPES types I and II and ovarian dysfunction with varied severity based on different FOXL2 mutations. It is widely believed that truncations before the polyalanine tract, inducing loss of the C-terminal region acting as a transcriptional inhibitory domain, are preferentially associated with BPES type I (5, 36–38). Consistently, five truncations p. Trp128*, p. Gln161*, p. Lys193Serfs*78, p. Gln196* and p. Ala226Leufs*32 were first reported to be related to BPES type I in our study. Interestingly, we also found truncated mutations of p. Gly269Argfs and p. Pro287Argfs*75 after the polyalanine tract, which have been strongly associated with BPES type I (6, 29, 39, 40). Polyalanine expansion mutations, acting as hypomorphic alleles and retaining partial transactivation effects, are commonly considered to lead to BPES type II (5, 36, 41). However, the mutation p. Ala225_Ala234dup, which has previously been reported in patients with BPES type II, was detected for the first time in our patient with BPES type I. The ovarian phenotype might relate to the abnormal nuclear aggregation and cytoplasmic mislocalization of mutated FOXL2 proteins (42, 43). Missense mutations were considered particularly associated with typical palpebral malformations independent of ovarian phenotypes. Intriguingly, the 3 missense mutations previously reported in BPES type II were also found in our type I patients. These mutations have been shown to have subcellular mislocalization and adverse impacts on the transactivation of CYP19A1, CCND2, and StAR by in-vitro functional experiments (7, 20–22). Therefore, the genotype-phenotype BPES classification correlation cannot be generalized. In addition, high phenotypic variability existed within FOXL2 mutations of the same domain and even within the same mutation, although with the presence of ovarian dysfunction. In our study, although with the same c.672_701dup mutation, patient #12 was fertile with normal ovarian reserve, while patients #10 and #11 exhibited infertility (>5 yrs duration) and a DOR phenotype, both of whom had FSH >15 IU/L, AMH <1.2 ng/ml and AFC ≤5 simultaneously. Of note, when we classified different FOXL2 mutations found in our BPES type I patients from severe to mild predicted functional impairment (from group A to E), we unfortunately did not find more severe reproductive phenotypes associated with more serious functional defects. Taken together, the clinical findings of our patient confirm phenotypic overlap and question a clear-cut prediction of female fertility based on FOXL2 molecular defects only. The wide clinical variability observed indicates that the reproductive phenotype of FOXL2 mutations may depend not only on the causal mutation but also on other contributing factors, including unknown modifier genes, epigenetics and/or environmental factors. Of note, although our study is the largest study describing the ART outcomes in BPES women currently, the sample size is small and additional studies in larger cohorts are required to establish clear-cut genotype-phenotype correlations.
The genetic counseling approach should be routinely established in clinics for BPES patients. First, FOXL2 mutation testing in BPES women of childbearing age should be handled with caution, given that genotype-phenotype correlation cannot be generalized. Nonetheless, based on our data, BPES patients with FOXL2 gene mutations had reduced fertility and decreased ovarian reserve, and they might have a higher risk for POI than mutation-negative BPES patients. Risk evaluation and surveillance and effective and timely fertility intervention are recommended before POI finally occurs. Furthermore, BPES has a high familial heritability with an inheritance risk of 50%. In our study, patient #12 had multiple offspring or pregnancies with FOXL2 mutations, including 1 BPES daughter, 3 abortions indicated by prenatal diagnosis, and 2 PGT embryos, which brought her great physical and psychological trauma. Therefore, prenatal genetic counseling for offspring health is also particularly important and highly recommended for BPES patients with FOXL2 mutations. For adolescent girls, delayed development of secondary sexual characteristics and menarche should be considered. For reproductive-aged women, ovarian function should be monitored, and fertility planning is recommended for future risk of POI. For those with natural pregnancy, prenatal diagnosis through amniotic fluid or fetal umbilical cord blood should be performed, whereas for those with infertility, ART treatment, PGT for normal and high-quality embryo screening, or egg donation are recommended.
Of note, BPES patients with FOXL2 mutations frequently suffer from failure of IVF pregnancy due to poor oocyte quality with compromised developmental potential, even after IVM. The hypothesis is that the inherent functional defects of granulosa cells due to FOXL2 mutations might not provide an optimal microenvironment for the growth and maturation of oocytes. Currently, there is no consensus or recommendation to improve reproductive success for BPES patients with FOXL2 mutations. We assume that remedial supplementation or coculturing of granulosa cells with immature oocytes during IVM treatment would improve oocyte quality by simulating or improving the microenvironment of oocyte maturation, and establishing communication with oocytes through gap junctions (44). For patient #12, two blastocysts were obtained after coculturing granulosa cells from normal donors. These preliminary data to some extent supported our hypothesis and might provide a practical intervention to improve reproductive outcomes for BPES patients with FOXL2 mutations. However, research on granulosa cell coculture is still in its infancy. Further exploration of coculturing the immature oocytes with granulosa cells from normal donors in larger cohorts is warranted.
In conclusion, our study is the first to summarize the reproductive characteristics of women of reproductive age with BPES and demonstrates that these patients with FOXL2 mutations have decreased ovarian reserve and fertility, as well as poor ART outcomes. The genotype-reproductive phenotype correlations were highly heterogeneous and cannot be generalized. Genetic counseling for fertility planning and preimplantation or prenatal genetic diagnosis to reduce offspring inheritance is recommended.
The original contributions presented in the study are included in the article/supplementary material. Further inquiries can be directed to the corresponding author.
The studies involving human participants were reviewed and approved by Institutional Review Board of Center for Reproductive Medicine, Shandong University. The patients/participants provided their written informed consent to participate in this study.
XJ and YQ contributed to the study concept and design. TM, WZ, RZ, JL, and YG contributed to acquisition of data. TM, WZ, and XJ contributed to analysis and interpretation of data, and manuscript writing. All authors contributed to reviewing and approval of the final version of this work.
This work was supported by the National Key Research & Developmental Program of China (2018YFC1003803), the National Natural Science Foundation of China (82171651, 82125014, 81971352), and the Young Scholars Program of Shandong University.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
We thank all the patients who participated in the study.
1. Bertini V, Valetto A, Baldinotti F, Azzarà A, Cambi F, Toschi B, et al. FOXL2Blepharophimosis, Ptosis, Epicanthus Inversus Syndrome: New Report With a 197-Kb Deletion Upstream of and Review of the Literature. Mol Syndromol (2019) 10(3):147–53. doi: 10.1159/000497092
2. Zlotogora J, Sagi M, Cohen T. The Blepharophimosis, Ptosis, and Epicanthus Inversus Syndrome: Delineation of Two Types. Am J Hum Genet (1983) 35(5):1020–7.
3. Yu HC, Geiger EA, Medne L, Zackai EH, Shaikh TH. An Individual With Blepharophimosis-Ptosis-Epicanthus Inversus Syndrome (BPES) and Additional Features Expands the Phenotype Associated With Mutations in KAT6B. Am J Med Genet A (2014) 164A(4):950-7. doi: 10.1002/ajmg.a.36379
4. Cheng T, Yuan X, Yuan S, Zhu J, Tang S, Zhang Y. ITGB5 Mutation Discovered in a Chinese Family With Blepharophimosis-Ptosis-Epicanthus Inversus Syndrome. Open Life Sci (2021) 16(1):1268–77. doi: 10.1515/biol-2021-0129
5. De Baere E, Beysen D, Oley C, Lorenz B, Cocquet J, De Sutter P, et al. FOXL2 and BPES: Mutational Hotspots, Phenotypic Variability, and Revision of the Genotype-Phenotype Correlation. Am J Hum Genet (2003) 72(2):478–87. doi: 10.1086/346118
6. Bunyan DJ, Thomas NS. Screening of a Large Cohort of Blepharophimosis, Ptosis, and Epicanthus Inversus Syndrome Patients Reveals a Very Strong Paternal Inheritance Bias and a Wide Spectrum of Novel FOXL2 Mutations. Eur J Med Genet (2019) 62(7):103668. doi: 10.1016/j.ejmg.2019.05.007
7. Beysen D, De Paepe A, De Baere E. FOXL2 Mutations and Genomic Rearrangements in BPES. Hum Mutat (2009) 30(2):158–69. doi: 10.1002/humu.20807
8. Fan J, Zhou Y, Huang X, Zhang L, Yao Y, Song X, et al. The Combination of Polyalanine Expansion Mutation and a Novel Missense Substitution in Transcription Factor FOXL2 Leads to Different Ovarian Phenotypes in Blepharophimosis-Ptosis-Epicanthus Inversus Syndrome (BPES) Patients. Hum Reprod (2012) 27(11):3347–57. doi: 10.1093/humrep/des306
9. Caburet S, Georges A, L'Hôte D, Todeschini AL, Benayoun BA, Veitia RA. The Transcription Factor FOXL2: At the Crossroads of Ovarian Physiology and Pathology. Mol Cell Endocrinol (2012) 356:55–64. doi: 10.1016/j.mce.2011.06.019
10. Pisarska MD, Barlow G, Kuo FT. Minireview: Roles of the Forkhead Transcription Factor FOXL2 in Granulosa Cell Biology and Pathology. Endocrinology (2011) 152(4):1199–208. doi: 10.1210/en.2010-1041
11. Bentsi-Barnes IK, Kuo FT, Barlow GM, Pisarska MD. Human Forkhead L2 Represses Key Genes in Granulosa Cell Differentiation Including Aromatase, P450scc, and Cyclin D2. Fertil Steril (2010) 94(1):353–6. doi: 10.1016/j.fertnstert.2009.09.050
12. Nicol B, Rodriguez K, Yao. Aberrant HH. And Constitutive Expression of FOXL2 Impairs Ovarian Development and Functions in Mice. Biol Reprod (2020) 103(5):966–77. doi: 10.1093/biolre/ioaa146
13. Yu Y, Ji M, Xu W, Zhang L, Qi M, Shu J. Confrontment and Solution to Gonadotropin Resistance and Low Oocyte Retrieval in In Vitro Fertilization for Type I BPES: A Case Series With Review of Literature. J Ovarian Res (2021) 14(1):143. doi: 10.1186/s13048-021-00900-2
14. Bonus ML, Pothast R, Lamb JD, Feinberg EC, Bernardi LA. Planned Oocyte Cryopreservation in Women With Blepharophimosis-Ptosis-Epicanthus Inversus Syndrome: A Case Series. F&S Rep (2021) 2(3):332–7. doi: 10.1016/j.xfre.2021.05.006
15. Nuovo S, Passeri M, Di Benedetto E, Calanchini M, Meldolesi I, Di Giacomo MC, et al. Characterization of Endocrine Features and Genotype-Phenotypes Correlations in Blepharophimosis-Ptosis-Epicanthus Inversus Syndrome Type 1. J Endocrinol Invest (2016) 39(2):227–33. doi: 10.1007/s40618-015-0334-3
16. Jiao X, Qin C, Li J, Qin Y, Gao X, Zhang B, et al. Cytogenetic Analysis of 531 Chinese Women With Premature Ovarian Failure. Hum Reprod (2012) 27(7):2201–7. doi: 10.1093/humrep/des104
17. Alpha Scientists in Reproductive Medicine and ESHRE Special Interest Group of Embryology. The Istanbul Consensus Workshop on Embryo Assessment: Proceedings of an Expert Meeting. Hum Reprod (2011) 26(6):1270–83. doi: 10.1093/humrep/der037
18. Gardner DK, Balaban B. Assessment of Human Embryo Development Using Morphological Criteria in an Era of Time-Lapse, Algorithms and 'OMICS': Is Looking Good Still Important? Mol Hum Reprod (2016) 22(10):704–18. doi: 10.1093/molehr/gaw057
19. Gardner DK, Schoolcraft WB. Culture and Transfer of Human Blastocysts. Curr Opin Obstet Gynecol (1999) 11(3):307–11. doi: 10.1097/00001703-199906000-00013
20. Hu J, Ke H, Luo W, Yang Y, Liu H, Li G, et al. A Novel FOXL2 Mutation in Two Infertile Patients With Blepharophimosis-Ptosis-Epicanthus Inversus Syndrome. J Assist Reprod Genet (2020) 37(1):223–9. doi: 10.1007/s10815-019-01651-2
21. Beysen D, Moumné L, Veitia R, Peters H, Leroy BP, De Paepe A, et al. Missense Mutations in the Forkhead Domain of FOXL2 Lead to Subcellular Mislocalization, Protein Aggregation and Impaired Transactivation. Hum Mol Genet (2008) 17(13):2030–8. doi: 10.1093/hmg/ddn100
22. Li F, Chen H, Wang Y, Yang J, Zhou Y, Song X, et al. Functional Studies of Novel FOXL2 Variants in Chinese Families With Blepharophimosis-Ptosis-Epicanthus Inversus Syndrome. Front Genet (2021) 12:616112. doi: 10.3389/fgene.2021.616112
23. Lin WD, Chou IC, Lee NC, Wang CH, Hwu WL, Lin SP, et al. FOXL2 Mutations in Taiwanese Patients With Blepharophimosis, Ptosis, Epicanthus Inversus Syndrome. Clin Chem Lab Med (2010) 48(4):485–8. doi: 10.1515/cclm.2010.100
24. Udar N, Yellore V, Chalukya M, Yelchits S, Silva-Garcia R, Small K. Comparative Analysis of the FOXL2 Gene and Characterization of Mutations in BPES Patients. Hum Mutat (2003) 22(3):222–8. doi: 10.1002/humu.10251
25. Beysen D, De Jaegere S, Amor D, Bouchard P, Christin-Maitre S, Fellous M, et al. Identification of 34 Novel and 56 Known FOXL2 Mutations in Patients With Blepharophimosis Syndrome. Hum Mutat (2008) 29(11):E205–19. doi: 10.1002/humu.20819
26. Méjécase C, Nigam C, Moosajee M, Bladen JC. The Genetic and Clinical Features of FOXL2-Related Blepharophimosis, Ptosis and Epicanthus Inversus Syndrome. Gene (2021) 12(3):364. doi: 10.3390/genes12030364
27. Kaur I, Hussain A, Naik MN, Murthy R, Honavar SG. Mutation Spectrum of Fork-Head Transcriptional Factor Gene (FOXL2) in Indian Blepharophimosis Ptosis Epicanthus Inversus Syndrome (BPES) Patients. Br J Ophthalmol (2011) 95(6):881–6. doi: 10.1136/bjo.2009.177972
28. De Baere E, Dixon MJ, Small KW, Jabs EW, Leroy BP, Devriendt K, et al. Spectrum of FOXL2 Gene Mutations in Blepharophimosis-Ptosis-Epicanthus Inversus (BPES) Families Demonstrates a Genotype–Phenotype Correlation. Hum Mol Genet (2001) 10(15):1591–600. doi: 10.1093/hmg/10.15.1591
29. Shen J, Qu D, Gao Y, Sun F, Xie J, Sun X, et al. Genetic Etiologic Analysis in 74 Chinese Han Women With Idiopathic Premature Ovarian Insufficiency by Combined Molecular Genetic Testing. J Assist Reprod Genet (2021) 38(4):965–78. doi: 10.1007/s10815-021-02083-7
30. Grzechocińska B, Warzecha D, Wypchło M, Ploski R, Wielgoś M. Premature Ovarian Insufficiency as a Variable Feature of Blepharophimosis, Ptosis, and Epicanthus Inversus Syndrome Associated With C.223C > T P.(Leu75Phe) FOXL2 Mutation: A Case Report. BMC Med Genet (2019) 20(1):132. doi: 10.1186/s12881-019-0865-0
31. Corrêa FJ, Tavares AB, Pereira RW, Abrão MS. A New FOXL2 Gene Mutation in a Woman With Premature Ovarian Failure and Sporadic Blepharophimosis-Ptosis-Epicanthus Inversus Syndrome. Fertil Steril (2010) 93(3):1006.e3–6. doi: 10.1016/j.fertnstert.2009.08.034
32. Kuo FT, Bentsi-Barnes IK, Barlow GM, Pisarska MD. Mutant Forkhead L2 (FOXL2) Proteins Associated With Premature Ovarian Failure (POF) Dimerize With Wild-Type FOXL2, Leading to Altered Regulation of Genes Associated With Granulosa Cell Differentiation. Endocrinology (2011) 152(10):3917–29. doi: 10.1210/en.2010-0989
33. Lamba P, Fortin J, Tran S, Wang Y, Bernard DJ. A Novel Role for the Forkhead Transcription Factor FOXL2 in Activin A-Regulated Follicle-Stimulating Hormone Beta Subunit Transcription. Mol Endocrinol (2009) 23(7):1001–13. doi: 10.1210/me.2008-0324
34. Ongaro L, Schang G, Zhou Z, Kumar TR, Treier M, Deng CX, et al. Human Follicle-Stimulating Hormone ß Subunit Expression Depends on FOXL2 and SMAD4. Endocrinology (2020) 161(5):bqaa045. doi: 10.1210/endocr/bqaa045
35. Justice NJ, Blount AL, Pelosi E, Schlessinger D, Vale W, Bilezikjian LM. Impaired FSHbeta Expression in the Pituitaries of Foxl2 Mutant Animals. Mol Endocrinol (2011) 25(8):1404–15. doi: 10.1210/me.2011-0093
36. Crisponi L, Deiana M, Loi A, Chiappe F, Uda M, Amati P, et al. The Putative Forkhead Transcription Factor FOXL2 is Mutated in Blepharophimosis/Ptosis/Epicanthus Inversus Syndrome. Nat Genet (2001) 27(2):159–66. doi: 10.1038/84781
37. Li F, Chai P, Fan J, Wang X, Lu W, Li J, et al. A Novel FOXL2 Mutation Implying Blepharophimosis-Ptosis-Epicanthus Inversus Syndrome Type I. Cell Physiol Biochem (2018) 45(1):203–11. doi: 10.1159/000486358
38. Yang XW, He WB, Gong F, Li W, Li XR, Zhong CG, et al. Novel FOXL2 Mutations Cause Blepharophimosis-Ptosis-Epicanthus Inversus Syndrome With Premature Ovarian Insufficiency. Mol Genet Genomic Med (2018) 6(2):261–7. doi: 10.1002/mgg3.366
39. Chacón-Camacho OF, Salgado-Medina A, Alcaraz-Lares N, López-Moreno D, Barragán-Arévalo T, Nava-Castañeda A, et al. Clinical Characterization and Identification of Five Novel FOXL2 Pathogenic Variants in a Cohort of 12 Mexican Subjects With the Syndrome of Blepharophimosis-Ptosis-Epicanthus Inversus. Gene (2019) 706:62–8. doi: 10.1016/j.gene.2019.04.073
40. Krepelova A, Simandlova M, Vlckova M, Kuthan P, Vincent AL, Liskova P. Analysis of FOXL2 Detects Three Novel Mutations and an Atypical Phenotype of Blepharophimosis-Ptosis-Epicanthus Inversus Syndrome. Clin Exp Ophthalmol (2016) 44(9):757–62. doi: 10.1111/ceo.12783
41. Benayoun BA, Caburet S, Dipietromaria A, Bailly-Bechet M, Batista F, Fellous M, et al. The Identification and Characterization of a FOXL2 Response Element Provides Insights Into the Pathogenesis of Mutant Alleles. Hum Mol Genet (2008) 17(20):3118–27. doi: 10.1093/hmg/ddn209
42. Moumné L, Dipietromaria A, Batista F, Kocer A, Fellous M, Pailhoux E, et al. Differential Aggregation and Functional Impairment Induced by Polyalanine Expansions in FOXL2, a Transcription Factor Involved in Cranio-Facial and Ovarian Development. Hum Mol Genet (2008) 17(7):1010–9. doi: 10.1093/hmg/ddm373
43. Nallathambi J, Moumné L, De Baere E, Beysen D, Usha K, Sundaresan P, et al. A Novel Polyalanine Expansion in FOXL2: The First Evidence for a Recessive Form of the Blepharophimosis Syndrome (BPES) Associated With Ovarian Dysfunction. Hum Genet (2007) 121(1):107–12. doi: 10.1007/s00439-006-0276-0
Keywords: blepharophimosis-ptosis-epicanthus inversus syndrome (BPES), FOXL2, infertility, ovarian reserve, assisted reproductive technology (ART)
Citation: Meng T, Zhang W, Zhang R, Li J, Gao Y, Qin Y and Jiao X (2022) Ovarian Reserve and ART Outcomes in Blepharophimosis-Ptosis-Epicanthus Inversus Syndrome Patients With FOXL2 Mutations. Front. Endocrinol. 13:829153. doi: 10.3389/fendo.2022.829153
Received: 05 December 2021; Accepted: 30 March 2022;
Published: 28 April 2022.
Edited by:
Tom Kelsey, University of St Andrews, United KingdomReviewed by:
Laura Alessandra Favetta, University of Guelph, CanadaCopyright © 2022 Meng, Zhang, Zhang, Li, Gao, Qin and Jiao. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Xue Jiao, amlhb3h1ZUBzZHUuZWR1LmNu
†These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.