 Simona Alexandra Iacob
Simona Alexandra Iacob Diana Gabriela Iacob
Diana Gabriela Iacob- 1Department of Infectious Diseases, Carol Davila University of Medicine and Pharmacy, Bucharest, Romania
- 2Department of Infectious Diseases, National Institute of Infectious Diseases “Prof. Dr. Matei Bals”, Bucharest, Romania
- 3Department of Infectious Diseases, Emergency University Hospital, Bucharest, Romania
Non-alcoholic fatty liver disease (NAFLD) is strongly associated with the metabolic syndrome and is one of the most prevalent comorbidities in HIV and HBV infected patients. HIV plays an early and direct role in the development of metabolic syndrome by disrupting the mechanism of adipogenesis and synthesis of adipokines. Adipokines, molecules that regulate the lipid metabolism, also contribute to the progression of NAFLD either directly or via hepatic organokines (hepatokines). Most hepatokines play a direct role in lipid homeostasis and liver inflammation but their role in the evolution of NAFLD is not well defined. The role of HBV in the pathogenesis of NAFLD is controversial. HBV has been previously associated with a decreased level of triglycerides and with a protective role against the development of steatosis and metabolic syndrome. At the same time HBV displays a high fibrogenetic and oncogenetic potential. In the HIV/HBV co-infection, the metabolic changes are initiated by mitochondrial dysfunction as well as by the fatty overload of the liver, two interconnected mechanisms. The evolution of NAFLD is further perpetuated by the inflammatory response to these viral agents and by the variable toxicity of the antiretroviral therapy. The current article discusses the pathogenic changes and the contribution of the hepatokine/adipokine axis in the development of NAFLD as well as the implications of HIV and HBV infection in the breakdown of the hepatokine/adipokine axis and NAFLD progression.
1 Introduction
Non-alcoholic fatty liver disease (NAFLD) encompasses a spectrum of pathological changes induced by the accumulation of fat in the liver parenchyma, in the absence of alcohol consumption. According to the literature, NAFLD is encountered in 25% of the general population and represents the most common liver-related disease, especially in developed countries (1, 2). From a histopathological perspective NAFLD progresses from the simple accumulation of fat (fatty liver or steatosis) to liver inflammation (non-alcoholic steatohepatitis, NASH) and to liver fibrosis, potentially leading to hepatocellular carcinoma (HCC) (3). The deposition of fat in the liver is a reversible process, while NASH is an aggressive form of NAFLD leading to cirrhotic transformation and carcinogenesis.
As much as 27-44% of patients with NAFLD display NASH on liver biopsy and 30-42% of these have been shown to progress to liver fibrosis depending on various factors such as age, gender, geographical location or other comorbidities (4–7). However, the high prevalence of NAFLD and its close relation with the metabolic syndrome (MetS) (8) increase the risk of mortality and morbidity during NAFLD (8). The outcome of NAFLD is further aggravated by HIV and HBV infections, as a result of the intrahepatic inflammatory response and metabolic imbalances triggered by both viruses (9, 10). The pathological changes behind these events consist in the ability of viruses to simultaneously control key enzymes needed for viral replication and transcription factors involved in metabolic processes.
The HIV/HBV viral replication, as well as the inflammatory response and fatty deposition within the liver contribute to hepatic mitochondrial toxicity, one of the main mechanisms responsible for the development of NAFLD. Additionally, the antiretroviral (ARV) drugs induces a spectrum of metabolic abnormalities strongly associated with NAFLD known as HIV/ART–associated lipodystrophy syndrome (HALS). The limited ability of the liver to coordinate all these events through the hepatokine/adipokine network enables the progression of liver lesions and aggravates the ensuing metabolic imbalance.
Data related to the evolution of the hepatokine/adipokine axis in HIV and HBV infected patients with NAFLD are scarce. Currently no review has previously approached the mechanisms through which the hepatokine/adipokine axis controls the liver impairment induced by HIV/HBV. The article sums up available data on the immune and metabolic implications of the hepatokine/adipokine axis in HIV/HBV-infected patients with NAFLD. On the long term, the modulation of hepatokine/adipokine axis represents an important direction for research and could play a significant therapeutic benefit towards the attenuation or prevention of NAFLD
The article is structured in two parts. The first part discusses the cellular mechanisms and the contribution of the hepatokine/adipokine axis in the development of NAFLD, whereas the second part presents the implications of HIV/HBV infections on these mechanisms.
2 Cellular Mechanisms Involved in the Development of NAFLD and the Role of the Oxidative Stress
The liver holds a central role in metabolic homeostasis given its key function in the fat and glucose metabolism, namely in fat absorption and fatty acid (FA) metabolism, in the conversion of glucose to glycogen and vice versa and in the regulation of insulin signals from liver receptors. Hence, the resulting liver-related lesions during NAFLD are closely linked with additional metabolic changes and particularly with the progression of the MetS. NAFLD could favor the development of type 2 diabetes (T2D), insulin resistance (IR), atherogenic dyslipidemia and obesity, all inducing MetS (11) while at the same time, these conditions are risk factors for the evolution of NAFLD (12, 13). In this respect, both hyperinsulinemia and the excessive accumulation of triglycerides (TGs) in the adipose tissue interfere with liver lipogenesis and de novo lipogenesis (DNL) and contribute to the onset of steatosis, the first stage of NAFLD (14). Fatty deposition within the liver cells gradually induces mitochondrial oxidative damage and generates oxidative stress (OxS), a fundamental cellular process in the development of NAFLD (15).
OxS represents a cellular imbalance between free radicals (reactive oxygen species, ROS) and antioxidants accompanied by the reduction of the oxidative capacity and antioxidant response in the mitochondria, and subsequently in the endoplasmic reticulum. The OxS response involves the activation of numerous transcription factors which upregulate the inflammatory response and modulate the glycolipid metabolism, further favoring the occurrence of NASH (16). In moderate amounts, ROS play a key role as signalling molecules that control the immune response and protect against invasive pathogens (17). However, the overproduction of ROS in the liver parenchyma generates an inflammatory response that triggers cell necrosis and subsequently favors liver fibrosis (18). ROS also initiate a process of oxidative damage of the polyunsaturated FA belonging to the cell membranes through lipid peroxidation and further release toxic intermediate products, namely the lipid hydroperoxides. The accumulation and continuous conversion of lipid hydroperoxides to alkoxyl and peroxyl radicals aggravates all the more the oxidative damage of cells and membranes and facilitates the release of ROS and mitochondrial toxicity in a vicious circle (19). The ensuing mitochondrial toxicity further contributes to liver inflammation and fibrosis (20, 21). In addition, hydroperoxides diffuse across cell membranes and serve as pancreatic signalling molecules to influence glucose-stimulated insulin secretion and to suspend the pancreatic glycemic control (22). All these aspects highlight the close link between OxS, liver inflammation and glycolipid metabolism.
3 Hepatokine/Adipokine Axis in NAFLD Pathogenesis
3.1 Overview
Currently, there is ample evidence that NAFLD is a multifactorial disorder dependent on metabolic, genetic, environmental, toxic and infectious factors in different combinations (“multiple-hits” theory) (23). NAFLD develops as a stress response to these factors. Recently, the pathogenesis of NAFLD has been associated with the release of “organokines”, peptides that are synthesized within the liver or in various tissues. Notably, these organokines play an active role in the regulation of metabolic and inflammatory processes and connect numerous tissues/organs, particularly the liver and the adipose tissue.
Organkines are secreted in various physiological or pathological conditions predominantly in the liver (“hepatokines”), adipose tissue (“adipokines”) or muscles (“myokines”).Various authors have described the functions and features of these structures, their connections and their involvement in NAFLD (24–27). Some of these bioactive peptides are currently studied as therapeutic targets or biomarkers of NAFLD severity (28). However, the number and specific functions of these regulatory peptides have not been elucidated and their role in the pathogenesis of NAFLD remains unclear. Currently, the adipose tissue is considered the largest endocrine organ, producing over 700 adipokines of which only a few have been characterized. Two of these adipokines, namely adiponectin and leptin have been validated by clinical and histological studies in NAFLD (26, 29). Additionally, hepatocytes secrete more than 500 proteins of which only a small number have been studied and very few proteins such as fetuins A or FGF21 have been clearly correlated with distinct manifestations of MetS or NAFLD (30).
3.2 Cellular Mechanisms Activated by Hepatokines and Adipokines
Hepatokines and adipokines carry multiple cellular functions, regulating various transcriptional factors, receptors or key enzymes connected to metabolic, immune, antiviral, or antitumor processes. Hence, these organokines control the transcription factors that interfere in the regulation of insulin or lipid signalling (31–33) (e.g. carbohydrate response element binding protein- ChREBP or sterol regulatory element-binding transcription factor 1-SREBP-1c), as well as in hepatic inflammatory response, fibrogenesis or carcinogenesis (34–37) (e.g., nuclear factor kappa-light-chain-enhancer of activated B cells-NF-kB or members of the signal transducer and activator of transcription-STAT family). Adipokines and hepatokines can also activate specific receptors (e.g AdipoR1/R2, ChemR, LepRb) (38–41) or metabolic receptors (e.g. the peroxisome proliferator-activated receptors-PPAR family) (33, 42–47). Additionally, certain organokines (adiponectin, resisitin, fetuin A) could display a competitive binding to the cellular receptors TLR4/CD14, which transduce the bacterial lipopolysaccharide (LPS) signal into inflammatory signals (48–51). Some organokines such as adiponectin and fibroblast growth factor 21 (FGF21) impair the metabolic activity of c-Jun N-terminal kinases (JNK). Also leptin, ghrelin, adiponectin or FGF21 dysregulate the signalling pathways of mammalian target of rapamycin (mTOR1), a protein kinase that coordinates lipid homeostasis and cellular growth but also, liver inflammation and carcinogenesis (52). Furthermore, the contribution of organokines in liver pathology is highly intricate as a result of the numerous synergistic or antagonistic interactions that are established between them. The main hepatokines and adipokines which contribute to NAFLD and their mechanism of action is presented in the Table 1. The interactions of these organokines are presented in the Table 2.
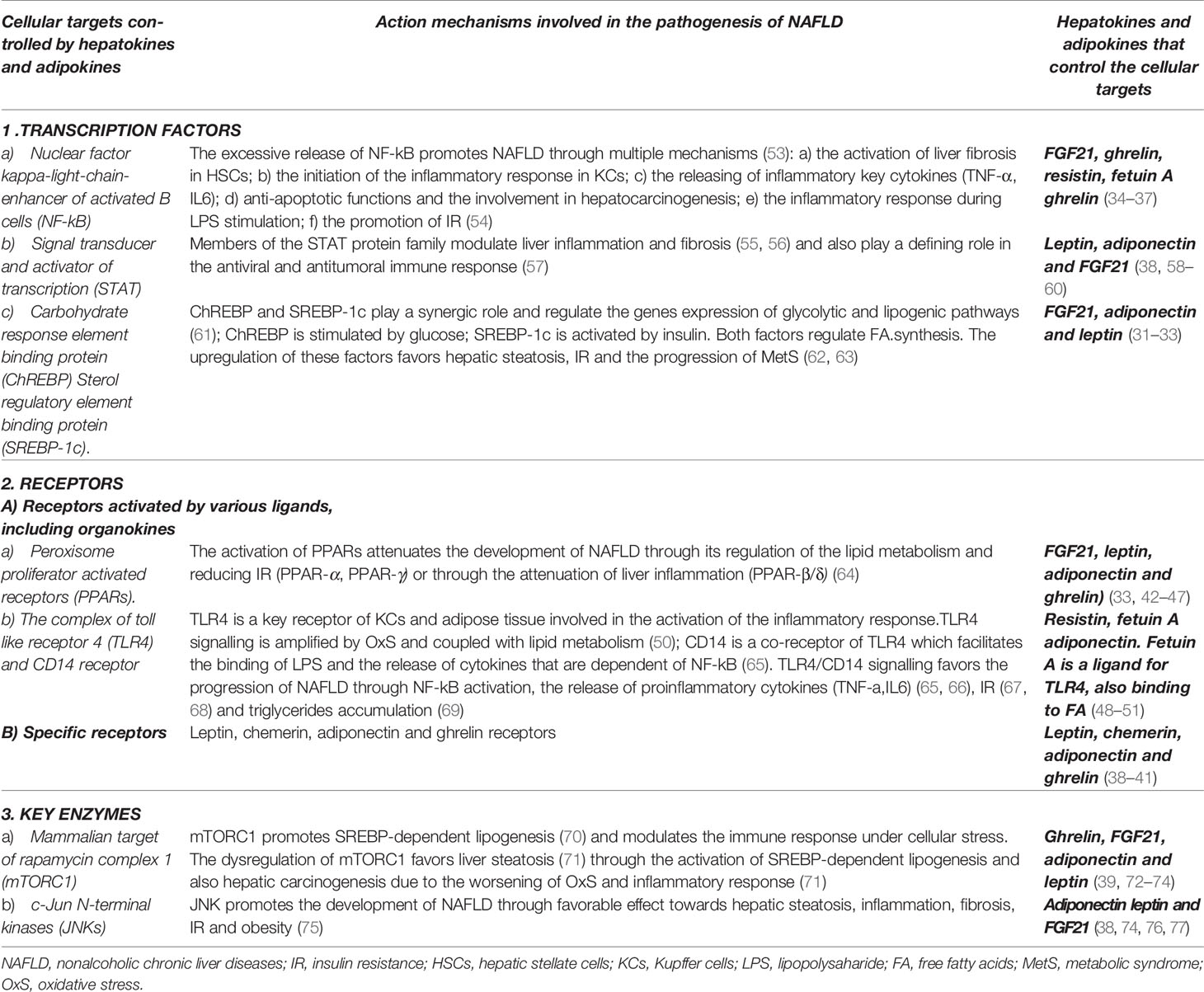
Table 1 Cellular targets regulated by adipokines and hepatokines with relevance in the pathogenesis of non-alcoholic fatty liver disease and metabolic syndrome.

Table 2 Correlations between various hepatokines and adipokines with a possible role in the pathogenesis of non-alcoholic fatty liver disease.
3.3 NAFLD Pathogenic Pathways Regulated by Hepatokines and Adipokines
Hepatokines and adipokines act as intercellular signals which modulate the liver lipogenic, inflammatory and fibrogenic pathways (25, 89). Lipogenic pathways are modulated by hepatokines and adipokines through their specific control over the FA flux, DNL and insulin signals. These carbohydrate-dependent processes are regulated through the activation of PPAR-α/γ receptors (64) as well as by transcription factors and enzymes involved in the lipid homeostasis (e.g. mTOR/SREBP1c and ChREBP) (25, 90). A predominant lipogenic effect can be observed in the case of resistin, ghrelin, fetuin A, chemerin and selenoprotein P (SeP). Inflammatory and fibrogenic pathways are modulated mainly through activation/inhibition of proinflammatory transcription factors (e.g NF-κB expression or STAT-3), thus modifying the profile of the released cytokines (53, 57, 75). Leptin and resistin are significantly associated with liver inflammation and leptin, resistin, chemerin, fetuin A and SeP with a profibrotic effect. The main adipokines and hepatokines presented in this article along with their metabolic, inflammatory and fibrogenic pathways are depicted in Table 3 and their potential effect in NAFLD are represented in Table 4 and Figure 1. As shown in Figure 1, visfatin chemerin, resisitin and SeP aggravate NAFLD through multiple pathways including the activation of receptors (TLR4/CD14), the regulation of transcription factors ((NF-κB) and the release of inflammatory or profibrotic cytokines (TNF-α/IL-6, respectively TGF-β) (98, 99, 104, 123, 124, 126, 132, 150, 154–156). These pathways involved in the control of liver inflammation are mediated by Kupffer cells (KCs) and hepatic stellate cells, (HSCs) and are associated with the exacerbation of inflammation, adipogenesis and finally, with the development of MetS (24, 99, 150, 157–161). By comparison, adiponectin and FGF21 display a hepatoprotective activity (34, 76, 111, 162–164) and attenuate the MetS (24). Various organokines play dual roles. For example, leptin reverses steatosis through SREBP/mTOR inhibition, while also promoting liver inflammation and fibrosis in non-parenchymal cells (79, 96, 165). Additionally, leptin promotes adipogenesis and the immune response and favors MetS (95) Other examples include ghrelin (39, 108) and fetuin A (51, 118, 166) which concurrently aggravate hepatic steatosis and prevent liver fibrosis. Similarly, most adipokines and hepatokines display contradictory roles regarding the progression of NAFLD (27, 167) (Tables 3 and 4). In this regard, resistin, a pro-inflammatory adipokine acting via NF-κB/TLR4 mediated pathway and overexpressed in NASH (98) can still promote an anti-inflammatory response in the presence of LPS (100, 137, 168). Fetuin A, a hepatokine which induces MetS has been associated with a controversial role in liver fibrosis (119, 120) similar to chemerin in liver inflammation (86, 124). The level of visfatin, a proinflammatory and anti-steatosis adipokine (148) has been correlated with exacerbation as well as protection of liver inflammation (150) while it lacked a correlation with NAFLD in certain studies (146, 151). SeP, a hepatokine shown to display a protective antioxidant role in all stages of NAFLD, with a therapeutic benefit (132), has nevertheless been incriminated in liver fibrosis and IR (129). All of these conflicting aspects underline the need to clarify the roles of hepatokines and adipokines in the evolution of NAFLD and in particular, the need for studies with comparable designs and entry criteria regarding the patient’s metabolic status, age and NAFLD staging (29).
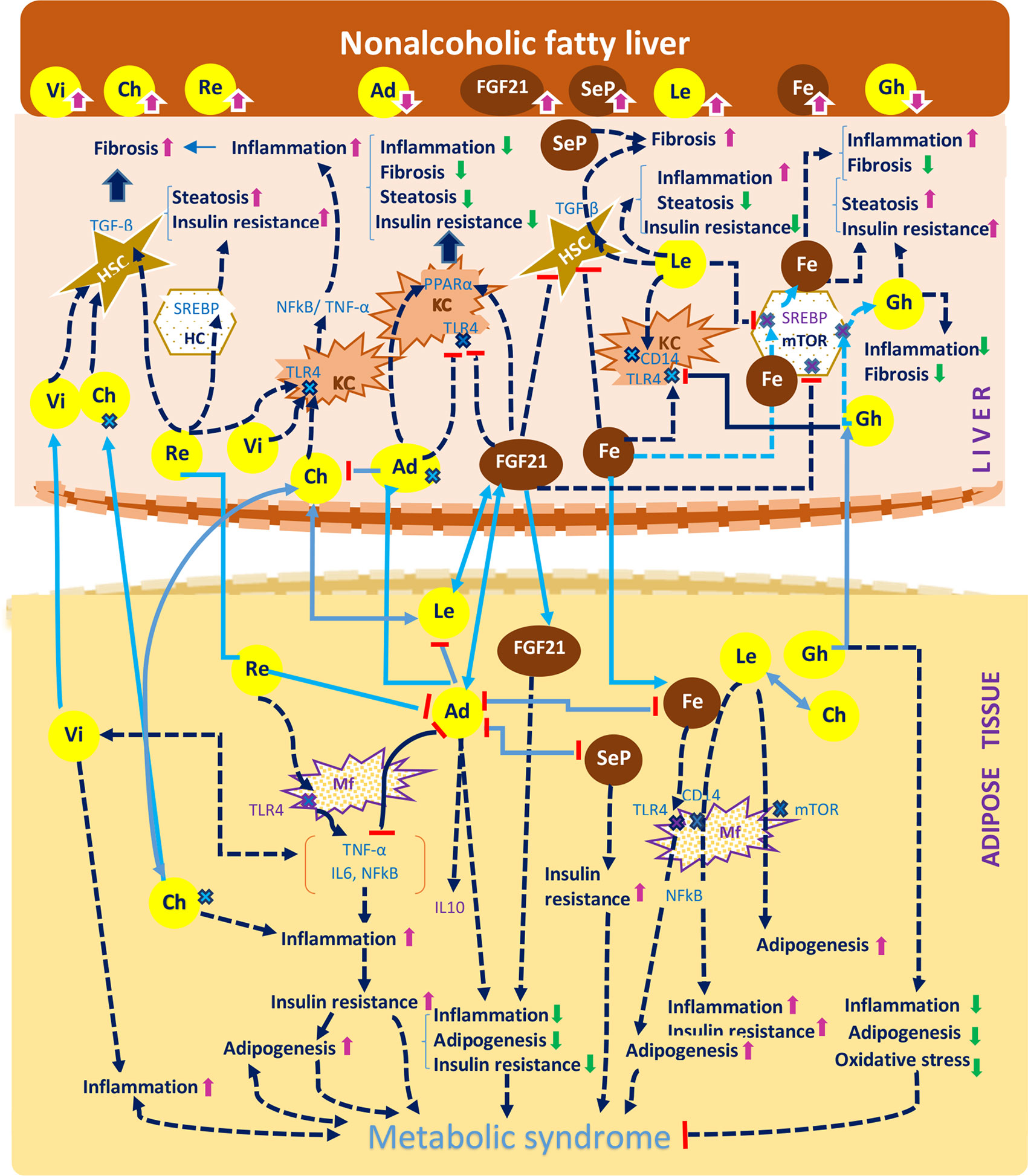
Figure 1 A concise representation of the hepatokines and adipokines presented in the article and their implications in the pathogenesis of non-alcoholic fatty liver disease according to the current studies. The diagram indicates the effect of hepatokines (FGF21, selenoprotein P, fetuin A, chemerin) and of the adipokines (visfatin, resistin, adiponectin, ghrelin), against the mechanisms that drive the pathogenesis of NAFLD, namely steatosis, inflammation, fibrosis and insulin resistance and also against the development of the metabolic syndrome, namely adipogenesis, inflammation and insulin resistance. The diagram depicts the following processes: a. The effects of organokines on liver inflammation: visfatin, chemerin, leptin, resistin, fetuin A activate the NF-kB and TNF-α/IL6 pathway and induce a proinflammatory effect mediated by Kuppfer cells (KCs). The previous organokines exhibit high concentrations in NAFLD. By comparison, adiponectin, FGF21 and ghrelin exert an anti-inflammatory effect. b. The effects of organokines on liver fibrosis: visfatin, chemerin, leptin, selenoprotein P mediate the release of TGF-β in hepatic stelatte cells (HSCs), while adiponectin and FGF21 exert an antifibrotic effect. c. The additive effect of organokines against the evolution of the metabolic syndrome: visfatin, chemerin and leptin promote the metabolic syndrome through their proinflammatory and proadipogenic effect, as well as through their role in the aggravation of insulin resistance. On the other hand, adiponectin and FGF21 play a protective role against the metabolic syndrome. Organokines can stimulate each other (e.g: adiponectin and leptin with FGF21) or inhibit each other (e.g: fetuin A, leptin, selenoprotein P, resistin with adiponectin). Organokines synthesized predominantly in the liver are presented in brown and those synthesized predominantly in the adipose tissue are presented in yellow. The correlations between these different organokines are shown in blue. The serum concentrations of these organokines in NAFLD (high or low) are represented by arrows. Vi, visfatin; Re, resistin; Fe, fetuin A; Le, leptin; Ch, chemerin; Ad, adiponectin; Gh, ghrelin; SeP, selenoprotein; Re, resistin; Mf, macrophage; HC, hepatic cell;  receptor.
receptor.
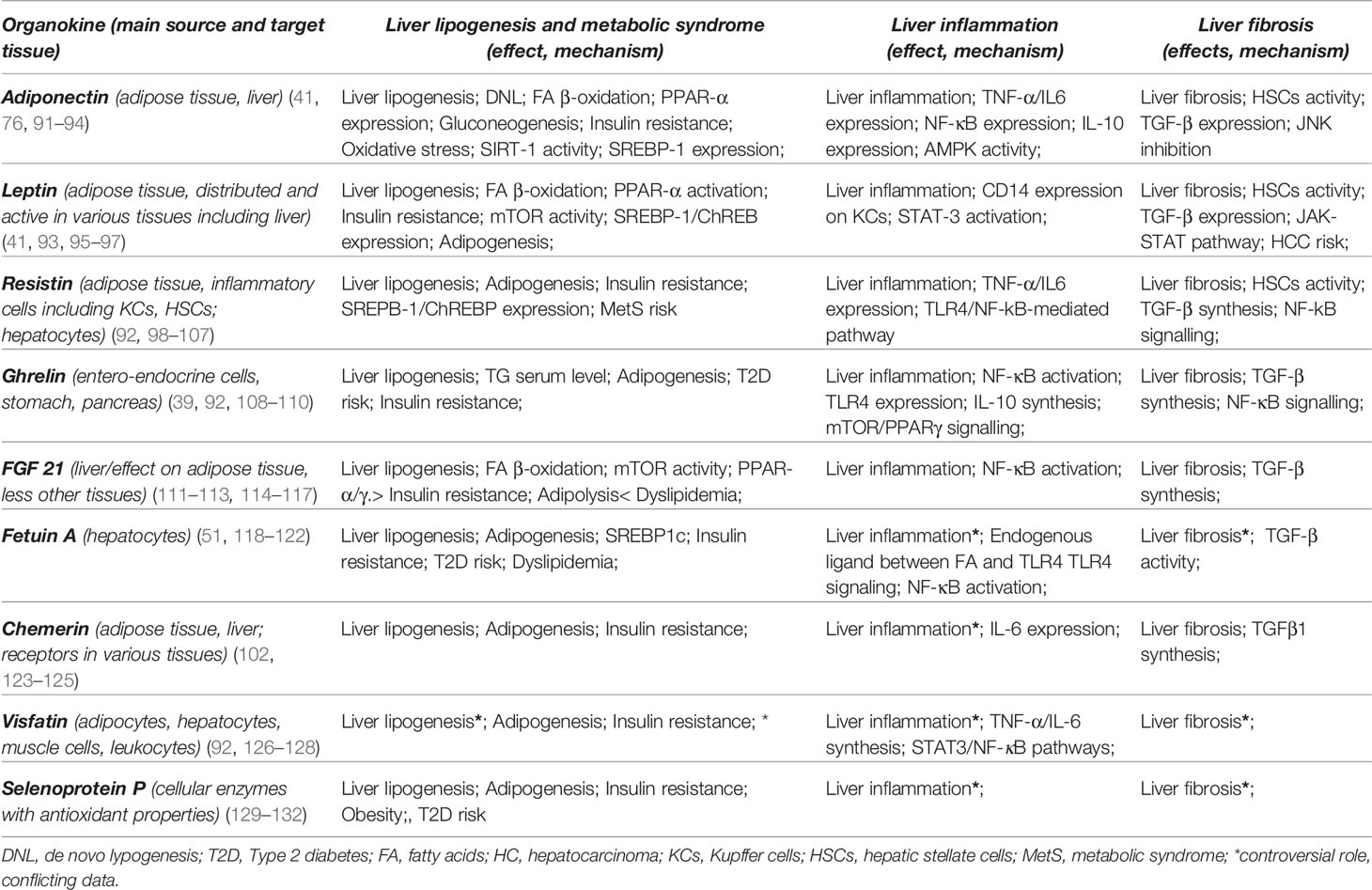
Table 3 The main hepatokines and adipokines and their mechanisms in the development of non-alcoholic fatty liver disease.

Table 4 Potential effects of the main hepatokines and adipokines on non-alcoholic fatty liver disease pathogenesis (29, 30, 92, 102).
4 HIV and HBV Role in NAFLD Pathogenesis and in the Breakdown of Hepatokine/Adipokine Axis
HIV/HBVco-infection promotes hepatic injuries during NAFLD through several mechanisms triggered by their persistent replication, enhanced inflammatory response and metabolic interference. The toxicity of antiretroviral therapy (ART) can also contribute to NAFLD. Hence, the prevalence of NAFLD can amount to 50-65% of HIV infected patients, depending cumulative risk exposure to metabolic factors, drug toxicities, age, disease duration or ARV regimen (169, 170). By comparison, the prevalence of NAFLD in HBV-infected patients is estimated at 31.4% (171) while HIV/HBV co-infected patients also display a lower prevalence of only 30% (172). On the other hand HIV/HBV co-infected patients show a higher risk of liver fibrosis of 37-40% (173) and exhibit a more rapid progression to liver cirrhosis and HCC compared with HIV mono-infected patients (173, 174). The pathogenic differences between the two viruses explain their different influence on the progression of NAFLD (174). We present below the pathogenic mechanisms associated with the progression to NAFLD in HIV and HBV infection along with the role of antiretroviral treatment (ART) and the ensuing disruption of the hepatokine/adipokine axis.
4.1 HIV-Associated NAFLD Mechanisms
HIV promotes NAFLD through multiple mechanisms, as follows: a) the generation of an inflammatory response within the liver; b) the metabolic changes induced in the adipose tissue; c) the disruption of the hepatokine/adipokine axis which maintains the immune and metabolic balance. These mechanisms will be presented below.
4.1.1 HIV- Associated Liver Inflammation
HIV promotes liver inflammation through direct mechanisms or indirectly, following the breakdown of the intestinal immune response during HIV enteropathy.
4.1.1.1 HIV and liver inflammation
HIV was detected in the mitochondria of non-parenchymal cells (175), while the infection of hepatocytes was observed only experimentally (176). HIV replication in HSCs and KCs induces mitochondrial toxicity accompanied by inflammation and fibrosis (177). At the same time, HIV releases numerous profibrogenic, proinflammatory and proapoptotic signals in the liver parenchyma (178) which contribute to ROS generation (179), metabolic activation of CD4+T cells and immune stimulation (180). Consequently, HIV induces the cellular death by apoptosis or pyroptosis along with hepatocytes necrosis (181). Additionally, the activation of hepatic T and B lymphocytes during HIV infection and their ensuing apoptosis aggravates the inflammatory response and accelerates the fibrogenesis induced by HSCs (182) (Figure 2).

Figure 2 A brief representation of metabolic and inflammatory mechanisms generated by HIV and HBV infections and their interference with the hepatokine/adipokine axis during the progression of non-alcoholic fatty liver disease. The figure shows: a. The effects of HIV on parenchymal liver cells: activation of CCR5 receptors of hepatocytes; the release of reactive oxygen species (ROS) and their subsequent effect on metabolic alterations inducing hepatic steatosis and non-alcoholic steatohepatitis (NASH). ROS excess also promotes lipid peroxidation and hepatocyte necrosis, which in turn aggravate liver inflammation and fibrosis either directly, through proinflammatory cytokines (TNF-α, IL6) and profibrogenic cytokines (TGF-β) or indirectly, through the ensuing proinflammatory and profibrotic response. b. The effects of HIV on non-parenchymal liver cells: both Kuppfer cells (KCs) and hepatic stellate cells (HSCs) can be regulated by HIV directly and indirectly via endotoxins (LPS), leading to the progression of the inflammatory response and fibrosis. c. The effects of HIV on adipose tissue cells: HIV ensures the transformation of these cells in cells with pro-inflammatory properties (adipocyte and preadipocyte cells, Th1/Th17-CD4ly lymphocytes and macrophages-MΦ1); HIV also promotes a disproportionate release of hepatokines and adipokines which in turn lead to liver inflammation, adipogenesis, insulin resistance and ultimately to HALS. d. The actions of HBV on hepatocytes: the induction of ROS with metabolic consequences; the changes in the concentrations belonging to hepatokines that promote fibrogenesis and hepatocellular carcinoma (HCC); the decreasing concentration of the protective hepatokine FGF21; the activation of mTOR, a metabolic receptor, stimulated by HBV protein x (HBx) e. The effect of antiretrovirals (ARVs) on the adipose tissue: ARVs favor the release of adipokines with lipogenetic role (e.g. resistin) and the reduction of anti-adipogenic adipokines (e.g. adiponectin, and leptin) f. The impact of HIV and ARVs on the occurrence of a specific metabolic syndrome, namely HIV/ARV associated lypodistrophy syndrome (HALS). HALS arises as a result of HIV-associated inflammatory changes and ARV-related impact on adipogenesis and is mediated by multiple mechanisms (adipocytes hypertrophy or atrophy, the evolution of the metabolic syndrome and the imbalance of hepatokine adipokine axis). Organokines synthesized predominantly in the liver are presented in brown and those synthesized predominantly in the adipose tissue are presented in yellow. The aggravating actions for the liver and adipose tissue are shown in pink. HC, hepatic cells; KC, Kupfer cells; HSC, hepatic stellate cells; ARV, antiretrovirals; Mϕ, macrophage; ROS, reactive oxygen species; HBx, hepatitis B virus X protein; Vis, visfatin; Re, resistin; Fe, fetuin; Le, leptin; Ch, chemerin; Ad, adiponectin; Gh, ghrelin; SeP, selenoprotein; Re, resistin; Th, T helper lymphocyte; LPS, endotoxin;  receptor;
receptor;  apoptotic cells; TLR4, Toll-like receptor 4.
apoptotic cells; TLR4, Toll-like receptor 4.
4.1.1.2 HIV enteropathy and inflammatory consequences
In the early stages of HIV infection the viral invasion of the gastrointestinal system weakens the intestinal mucosa through the depletion of CD4+T lymphocytes, thereby promoting liver injury through the microbial translocation of intestinal germs or immunogenic molecules such as LPS, a proinflammatory Gram negative endotoxin. The latter activate KCs through TLR4/CD14 signalling and induce proinflammatory cytokines (TNF-α, IL-1β and IL-6) and profibrogenic mediators (TGF-β) (183). These mechanisms further contribute to neutrophil recruitment and HSCs activation, aggravating liver fibrosis (184). Notably, the inflammatory enteropathy persists and worsens during HIV infection despite ART.
4.1.2 Metabolic Changes Induced by HIV and ART
4.1.2.1 HIV metabolic alterations
HIV promotes NAFLD mostly through its impact on the adipose tissue, an HIV reservoir and a promoter of metabolic alterations in this infection. HIV infects the resident immune cells of the adipose tissue and induces an extensive inflammatory response which leads to the activation of macrophages and pre-adipocytes with macrophage-like properties (185). The release of proinflammatory and profibrotic cytokines change over time the morphology and distribution of the adipose tissue and contributes to the development of HALS, a specific form of lipodystrophy. HALS is characterized by central fat gain along with the loss of adipose tissue in the periphery and weight loss. The metabolic disturbance leads to MetS and to a high cardiovascular risk (186) but is initially independent of ART (187). Later, HALS becomes a complication of ART.
4.1.2.2 ART-metabolic alterations
ART has been a major step towards improving the lifespan of HIV patients. It is undeniable that ART has saved millions of patients who have been given a chance at a normal life. ARV drug classes target various steps of the HIV replication cycle, namely preventing HIV cell entry (Entry inhibitors-EIs), blocking HIV reverse transcriptase (Nucleoside Reverse Transcriptase Inhibitors-NRTIs and Non-Nucleoside Reverse Transcriptase Inhibitors-NNRTIs), impeding protein synthesis (Protease Inhibitors-PIs) and inhibiting the integration of HIV DNA into host DNA (Integrase Strand Transfer Inhibitors-INSTIs). Unfortunately, ART is not completely effective against proviral HIV DNA (188–190) so that it does not provide the eradication of HIV and consequently ART lifelong administration is required. The sustained administration of ART is accompanied by multiple side effects including metabolic changes and HALS (191). The risk of HALS is associated with the duration and type of ART regimen and persists even after ART interruption (192).The exposure to NRTIs is particularly associated with peripheral lipoatrophy while PIs are frequently correlated with visceral lipohypertrophy (193, 194).
The metabolic changes occurring during ART are commonly associated with the cellular toxicity. Hence most ARVs have been shown to induce cumulative cellular toxicity irrespective of the ARV class and independent of HIV stimulation (195, 196). Of these, the most studied and the most aggressive type of cellular toxicity is represented by the mitochondrial toxicity (197) induced by NRTIs, NNRTIs, PIs and by some INSTI representatives (198, 199). This may occur shortly after ART starting and may be perpetuated depending on various risk factors (197, 199–201).
ART induces mitochondrial dysfunction by disrupting the oxidative phosphorylation and ATP synthesis as well as through the excessive release of ROS (196). The OxS generated by ART induces apoptosis, inflammation and fibrosis of the adipose tissue and of liver parenchymal cells. Both mitochondrial dysfunction and the metabolic changes play a decisive role in the development of HALS (194, 202) and favor liver steatosis and NAFLD development (194) as can be seen in the Figure 2. The degree of liver toxicity becomes apparent after 6-8 months of NRTI treatment (203) and differs depending on the type of NRTI and its persistence within the cell (200, 201). MetS also develops as a consequence of ART-mitochondrial toxicity (202, 204) and of visceral adipose tissue imbalance induced by hepatokines/adipokines disequilibrium (205). In turn, MetS favors lypodistrophy, increases the cardiovascular risk and contributes to liver steatosis (186, 191, 206) regardless of virological parameters (169).
In conclusion, the metabolic abnormalities arising during HIV and ART promote HALS, MetS and liver steatosis (205) and further lead to the development of NAFLD (207). It is estimated that each year of NRTI treatment may add an 11% risk to NAFLD (206). However the progression of NAFLD to liver fibrosis is a rare and lengthy process (208). The metabolic and inflammatory mechanisms generated by HIV and ART are depicted in the Figure 2.
4.1.3 Hepatokine/Adipokine Axis Breakdown in HIV-Specific NAFLD
In its attempt to restore the metabolic balance, and to reduce the HIV-lipodystrophy and IR, the liver regulates the metabolism of the adipose tissue by activating the hepatokines and adipokines circuit. Unfortunately, there is limited data regarding the hepatokines/adipokines roles in HIV patients. Currently, the most documented organokines involved in body fat distribution are leptin and adiponectin, two adipokines mainly secreted by adipocytes. Available data on adiponectin indicate that ART and in particular the treatment with IPs (ritonavir), lowers the levels of adiponectin and leptin especially at the beginning of therapy and favors the progression of lipodystrophy, steatosis and IR (195, 209, 210). In turn, the development of lipodystrophy lowers the concentration of these two adipokines independent of the ARV agent (211–214). Hence, an effective treatment against lipodystrophy could normalize the level of these two adipokines and could additionally lower the risk of steatosis. Recent studies indicate that the reduction of the serum adiponectin and leptin levels could be a consequence of SIRT1-depleted adipocytes found during lipodistrophy (215, 216). SIRT1, a member of the enzyme family of sirtuins, modulates the cellular metabolism, as well as HIV transcription (217). SIRT1 downregulates DNL and gluconeogenesis improving the lipid and carbohydrate metabolism, and protecting the liver from steatosis (218). The inhibition of SIRT1 along with the decrease of adiponectin and leptin expression during HIV infection as well as during PIs treatment could represent one of the steps required for the development of HIV-associated NAFLD (219). On the other hand, experimental data suggest that HIV could promote the release of adiponectin (211). Therefore HIV suppression during ART may lower the levels of adiponectin which would partly explain the metabolic changes associated within HALS (211). In this regard, HIV-infected patients starting ART may still develop MetS and NAFLD and may progress to HCC eventually (91, 220). Leptin is a liporegulatory adipokine which can display a protective role on pancreatic beta cells and liver steatosis but also increase the risk of inflammation in non-HIV infected patients (221, 222). As a result hipoleptinemia is linked to a lower risk of inflammation, fibrogenesis and carcinogenesis (96, 134, 223). On the other hand, in HIV-infected patients, the leptin secretion is closely related to the body fat mass, so that leptin deficiency prevails in cases with lipoatrophy, while hyperleptinemia accompanies patients with lipohypertrophy (212, 224). The treatment of obesity with rosiglitazone, an inhibitor of the leptin release could potentially reduce the hyperleptinemia in HIV infected patients improving metabolic parametric and the cardiovascular risk (225). The administration of leptin in hipoleptinemia could also contribute towards a metabolic balance but this type of therapy is not yet available in HIV patients. Human recombinant leptin (metrelin) is the only organokine that has been approved in the treatment of non-HIV associated lipodystrophy. Certain studies have suggested that two months of metreleptin treatment can also improve lipodistrophy and the metabolic syndrome including in patients undergoing ART (226, 227). However, in HIV patients the proinflammatory and profibrotic effect of leptin remains unknown. Therefore leptin treatment seems to be hazardous especially that serum leptin levels may be associated with leptin resistance, severe forms of NAFLD and HCC risk (135). Recombinant leptin has also been studied in NASH non-HIV patients as part of a clinical trial (ClinicalTrials.gov Identifier: NCT00596934), but it has not been studied in HIV/NAFLD patients. Nevertheless, there are still insufficient data to recommend this therapy in NAFLD patients.
Adiponectin has also been studied in both cell cultures and mice. Hence, the supplementation with adiponectin alleviates the metabolic syndrome, IR and steatosis and could antagonize the oncogenic effects of leptin against the liver (220, 228, 229). Additionally, adiponectin agonists have been used as an experimental antifibrotic therapy in liver diseases (230). It is considered that hypolipidemic therapies such as statins and antidiabetic medications thiazolidinediones or metformin could upregulate the level of adiponectin and induce a hepatoprotective effect without side effects (231, 232). However, there are conflicting data on the use of these drugs in HIV (212, 233–235) and the development of resistance to leptin and adiponectin limits their activity even in the presence of high serum levels (212, 236).
Data on other hepatokines and adipokines in HIV infected patients is summarized below.
SeP, a hepatokine with antioxidant properties during HIV activation and ROS release (237, 238) has been shown to display low levels in HIV patients. On the other hand, non-HIV patients with NAFLD display high serum concentrations of SeP, which are associated with the metabolic disturbances and NAFLD progression (129). FGF21, a hepatokine with a key metabolic role in glucose and lipid homeostasis is also expressed in HIV patients. The detection of a high FGF21 serum level in these patients was associated with MetS, lipodistrophy, and severe steatosis (239–241). Given the strong correlations between FGF21 and metabolic parameters, FGF21 has been suggested as a possible prognostic marker to monitor HALS and its therapy as well as the ART-associated cardiovascular risk (242). HIV infected patients also show high serum levels of resistin, an adipokine correlated with IR, lipoatrophy and NAFLD (243). In addition, it has been observed that genetic variants of resistin could play a role in the progression of HALS (244). HIV lipodistrophy also appears to be promoted by another regulator of adipogenesis, namely chemerin, an immunomodulatory adipokine, also with proinflamatory activity in NAFLD (88). Experimental data suggested that CMKLR1/ChemR23, a specific chemerin receptor could be used by HIV as a minor coreceptor but this remains to be confirmed (245). Visfatin, an adipokine released by various cells within the adipose and liver tissue interacts with a C-C chemokine receptor type 5 (CCR5), which is an HIV coreceptor but the result of this interaction is presently unknown (246, 247). Another organokine, namely ghrelin, has shown discordant results in HIV lipodystrophic patients (248, 249) The serum level of ghrelin has been concordant with the serum level of TGs on a small group of patients. Nevertheless, the administration of ghrelin was associated with the attenuation of liver inflammation in mouse models (250).
Overall, data on the direct impact of hepatokines and adipokines during HIV infection on ART is scarce and their role in the progression towards NAFLD remains unclear. The disruption of the hepatokine/adipokine axis during HIV infection and the ensuing implications for NAFLD progression are shown in the Figure 2 and Table 5.

Table 5 The main hepatokines and adipokines discussed in the article and their implications in HIV and HBV infection.
4.2 HBV-Associated NAFLD
HBV is a hepatotropic DNA virus and a major cause of cirrhosis and HCC in NAFLD patients. It is transmitted through the same pathways as HIV and infects about 10% of these patients (268). The HBV co-infection with an immunosuppressive virus such as HIV further reduces the ability of the immune response to recognize and eliminate HBV-infected cells. Thus HIV infection promotes HBV replication along with the synthesis of proinflammatory and profibrotic cytokines. In turn, HBV facilitates the ongoing HIV viral replication and impacts the recovery of CD4+T cells during ART (268–274). Furthermore, the use of NRTIs as a treatment in HIV/HBV patients could aggravate the mitochondrial dysfunction, inflammatory response (275) and favor additional metabolic complications. As a result, HIV-HBV co-infected patients display a more rapid progression towards liver cirrhosis and HCC (268, 272). Nevertheless, HBV appears to attenuate various metabolic effects induced by HIV and ART, so that the prevalence of NAFLD in HIV/HBV patients is lower than in HIV or HBV monoinfected patients (169, 170, 172). The mechanisms by which HBV influences the pathogenesis of NAFLD are depicted below.
4.2.1 HBV-Induced Hepatic Inflammatory Response
HBV is not a cytopathic virus; it generates 3 types of antigens that interfere with the immune response, preventing the recognition and elimination of virally infected hepatocytes. Of these, HBsAg is a modulator of the innate immune response which contributes to the suppression of inflammatory cytokines, the aggravation of liver fibrosis and the risk of malignant transformation (276, 277). HBcAg induces the secretion of IL18, a potent proinflammatory cytokine engaged in hepatocytes pyroptosis, one of the profibrotic mechanisms in HBV infection (278). HBeAg, the third viral antigen inhibits NF-κB pathway and ROS production in KCs and consequently suppresses NLRP3 inflammasomes and the inflammatory response (279). The inhibition of the inflammatory response allows HBV to escape the immune response and to induce a persistent infection along with HBV DNA insertion into the host genome, which further adds to the risk of oncogenesis.
4.2.2 HBV-Induced Metabolic Dysfunction
HBV has been viewed as a ‘‘metabolic virus’’ with multiple metabolic interferences (280). Still the correlation between HBV and the metabolic changes in the pathogenesis of NAFLD remains unclear. The metabolic dysfunctions are initiated as the same time with viral replication due to the activation of some transcription factors and nuclear receptors (281). These regulatory proteins once activated in the early stages of HBV transcription increase the expression of key enzymes involved in the control of metabolic pathways such as lipolysis, gluconeogenesis and cholesterol synthesis (281, 282). The activation of metabolic-related transcription factors is related to the overexpression of hepatitis B virus X protein (HBx), a regulatory protein involved in FA metabolism, steatosis and adipogenesis (282, 283). In addition to its metabolic contribution Hbx is a transcriptional activator mediating both NF-kB/TNF-α inflammatory signalling and cellular apoptosis, further contributing to DNA damage and liver carcinogenesis. Hence, HBx targets multiple mechanisms involved in the progress of NAFLD (284–286). However despite experimental evidence, the correlations between HBV and hepatic steatosis remain inconstant contradictory or even negative (284, 287, 288).
Studies on HBV infected patients have indicated that MetS appears to evolve independently of the HBV viral infection (289, 290). However, the presence of the metabolic changes once triggered (obesity, dyslipidaemia, diabetes) and the persistence of these changes supports the ongoing HBV viral replication. In turn, viral replication promotes hepatic steatosis and fibrosis, both of which contribute to the continuous progression of the metabolic dysfunction (280, 291–296). The duration of the MetS and of HBV infection may also influence the onset of steatosis (297).
In conclusion, HBV mainly promotes hepatic steatosis as well as liver fibrosis and carcinogenesis, yet it is less associated with liver inflammatory changes (Figure 2). By comparison, HIV induces early inflammation and irreversible metabolic changes further boosted by ART, so that during HIV and HBV coinfection both viruses play additive roles in the evolution of NAFLD (10, 298). It should be noted that both viruses also change the cellular metabolism through the conversion of glucose to lactate and further perpetuates the lactate synthesis in the presence of oxygen similar to the cancer cells (Warburg effect) (299). In this respect, HBV has been significantly linked to carcinogenesis, while HIV has been shown to strongly modulate the immune response and MetS.
4.2.3 Hepatokine/Adipokine Axis Breakdown in HBV Patients With NAFLD
There are few data on the hepatokine/adipokine axis in HBV infected patients. Studies on HBV infected patients have shown links between elevated levels of some hepatokines and insulin inhibition (e.g fetuin), between hepatokines, gluconeogenesis and the inflammatory response (fetuin A, leptin, resisitin, visfatin), between hepatokines and steatosis (leptin) as well as between hepatokines and HCC evolution (ghrelin, FGF21, fetuin A) (Table 2).
The hepatokines best documented in HBV infected patients include fetuin A and FGF21.
Fetuin A is a hepatokine predominantly released by the liver and correlated with the development of both MetS and NAFLD. The serum level of fetuin A is significantly higher in NAFLD but decreases in liver failure along with the extension of hepatic necrosis. A low level of fetuin A in HBV infected patients has been proposed as a marker of liver damage, as well as a predictive factor for poor prognosis (300). However, the highest fetuin A serum levels have been recorded in HBV patients with HCC and cirrhosis (261) while fetuin A levels in non-HBV patients has not been associated with fibrotic changes (118). Furthermore experimental data on HBV infected patients has shown that fetuin A attenuates the pro-inflammatory response induced by LPS administration (300). Thus, a low fetuin A secretion which follows hepatocyte necrosis explains the ensuing hepatic inflammatory response and the development of NASH, while fetuin A supplementation could alleviate the inflammatory response (301).
Another hepatokine that reflects the presence of liver damage is FGF21 secreted mainly in the liver but active especially in the adipose tissue. FGF21 is considered a stress hepatokine (302) with antioxidant and anti-inflammatory potential (303, 304). FGF21 secretion is reduced in chronic HBV infection, particularly in cirrhosis and the administration of FGF21 in HBV infected patients could improve liver inflammation and fibrosis (34, 304, 305). However FGF21 serum levels may increase in liver injuries and a very high level of FGF21 has been associated with HCC risk possibly as a protective response against the carcinogenesis process (306). Moreover, a high FGF21 serum level could be considered a marker of severity in patients with chronic HBV (260). A moderately elevated level has also been observed in NAFLD in non-HBV patients (307) in which case the FGF21 concentration has been correlated with metabolic improvement especially in the insulin response (308). Overall, the increased secretion of FGF21 arises as a compensatory response to OxS, and therefore a high level has a limited efficiency in patients with chronic HBV.
Certain adipokines with a fibrogenetic and neoplastic potential display high serum levels in HBV infected patients but their levels decrease during treatment, along with HBV viremia (30). Such is the case of leptin, a proinflammatory adipokine produced in HSCs and correlated with NAFLD fibrosis, which decreases during lamivudine HBV treatment (309).
Adiponectin is a metabolic, anti-inflammatory and antifibrotic adipokine. Experimental data indicate an improvement in steatosis, inflammation and liver fibrosis after adiponectin administration (228). Patients with chronic HBV infection display a high level of adiponectin and this level decreases during interferon therapy along with HBV viral load (253, 310). In these patients the level of adiponectin correlates with various stages of liver injury from liver inflammation and fibrosis to HCC (67, 311). Current reports indicate that adiponectin may promote HBV polymerase activity and HBV DNA replication, while in turn HBV replication induces the expression of adiponectin (312). This observation could partly explain the correlation above, as well as the hypothesis that adiponectin may play a role in the progression of HBV liver injury (252). The level of adiponectin differs in patients with HBV compared with non-HBV/NAFLD patients where, for example, the level of adiponectin decreases with the development of MetS and NASH yet may increase in patients with cirrhosis regardless of metabolic factors (133, 252, 313). In other studies, however, liver histology indicates a correlation between adiponectin and steatosis but not between adiponectin and viral factors (311).
Regarding other hepatokines and adipokines, chemerin is a multifunctional hepatokine with antioncogenic properties against HCC metastases, yet with reduced activity in HBV-HCC tissues (314). The low concentrations of chemerin in HCC-HBV infected patients have been associated with a favorable prognosis, thus suggesting a potential therapeutic role for this hepatokine. Nevertheless, the intratumoral level of chemerin was variable across different studies and experimental data on the matter are contradictory (262, 314). Resistin is another adipokine which regulates the development of obesity and IR and which has been correlated with the severity of liver damage in HBV infection (258, 315).
The imbalance of the hepatokine/adipokine axis in HBV infected patients and its implications in the progression of NAFLD are shown in Figure 2 and Table 5. In most cases the serum levels of adipokines released in HBV infected patients resemble the levels reported in other non-HBV infected patients with NAFLD, as seen in the cases of visfatin, leptin and resisitin. Regarding HIV infected patients, the serum levels of hepatokines or adipokines could be comparable to those detected in HBV as reported for SeP, or could exhibit a marked difference as for certain adipokines (leptin, adiponectin) or hepatokines (FGF21). These differences could be related to one of the following factors, namely the type of tissue damage (the lipodystrophy and adipose tissue damage predominate in HIV while the liver necrosis predominates in HBV), the incidence of liver fibrosis (more common in HBV), the degree of liver inflammation (more pronounced in HIV), the genetic polymorphisms of certain organokines (316) or the level of viral replication (211).
5 Conclusion
The liver contains a wide variety of cells through which it plays a complex role in the glycolipid metabolism, drug excretion and immune response. In this context, the metabolic changes, the treatments with hepatic metabolized drugs and the extent of the immune response have a direct impact on the structure and function of the liver parenchyma. One of the consequences of the immunometabolic imbalance is the development of NAFLD which gradually evolves from fatty liver and insulin resistance to inflammation, fibrosis and even to hepatic carcinogenesis. NAFLD in HIV/HBV co-infected patients could have a high severity due to the potential of the two viruses to replicate and to induce an inflammatory response. In addition both HIV and HBV play an important role in disrupting the glycolipid metabolism and liver communication through the imbalance of the hepatokines and adipokines. The two viruses act complementary and play an additive role in the emergence and progression of NAFLD. At the same time, ART aggravates the metabolic context induced by the two viruses, through the toxic effect on mitochondria and the development of lipodistrophy.
The HIV/HBV correlations with the breakdown of the hepatokine/adipokine axis during the progression of NAFLD are complex and largely unknown. Additional studies with comparable characteristics are needed to better formulate the assumptions of this axis and to further validate the potential therapeutic applications of hepatokines and adipokines.
Author Contributions
SI and DI contributed equally to the acquisition, analysis, and critical revision of the article. The authors had given their consent for the publication and agreed to be responsible for the accuracy and integrity of the article.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Cotter TG, Rinella M. Nonalcoholic Fatty Liver Disease 2020: The State of the Disease. Gastroenterology (2020) 158(7):1851–64. doi: 10.1053/J.GASTRO.2020.01.052
2. Younossi ZM, Koenig AB, Abdelatif D, Fazel Y, Henry L, Wymer M. Global Epidemiology of Nonalcoholic Fatty Liver Disease-Meta-Analytic Assessment of Prevalence, Incidence, and Outcomes. Hepatology (2016) 64(1):73–84. doi: 10.1002/hep.28431
3. Hassan K, Bhalla V, El RME, A-Kader HH. Nonalcoholic Fatty Liver Disease: A Comprehensive Review of a Growing Epidemic. World J Gastroenterol (2014) 20(34):12082. doi: 10.3748/wjg.v20.i34.12082
4. McPherson S, Hardy T, Henderson E, Burt AD, Day CP, Anstee QM. Evidence of NAFLD Progression From Steatosis to Fibrosing-Steatohepatitis Using Paired Biopsies: Implications for Prognosis and Clinical Management. J Hepatol (2015) 62(5):1148–55. doi: 10.1016/j.jhep.2014.11.034
5. Kleiner DE, Brunt EM, Wilson LA, Behling C, Guy C, Contos M, et al. Association of Histologic Disease Activity With Progression of Nonalcoholic Fatty Liver Disease. JAMA Netw Open (2019) 2(10):e1912565. doi: 10.1001/jamanetworkopen.2019.12565
6. Reddy YK, Marella HK, Jiang Y, Ganguli S, Snell P, Podila PSB, et al. Natural History of Non-Alcoholic Fatty Liver Disease: A Study With Paired Liver Biopsies. J Clin Exp Hepatol (2020) 10(3):245–54. doi: 10.1016/j.jceh.2019.07.002
7. Perumpail BJ, Khan MA, Yoo ER, Cholankeril G, Kim D, Ahmed A. Clinical Epidemiology and Disease Burden of Nonalcoholic Fatty Liver Disease. World J Gastroenterol (2017) 23(47):8263–76. doi: 10.3748/wjg.v23.i47.8263
8. Younossi ZM, Stepanova M, Afendy M, Fang Y, Younossi Y, Mir H, et al. Changes in the Prevalence of the Most Common Causes of Chronic Liver Diseases in the United States From 1988 to 2008. Clin Gastroenterol Hepatol (2011) 9(6):524–30.e1. doi: 10.1016/j.cgh.2011.03.020
9. Singh KP, Crane M, Audsley J, Avihingsanon A, Sasadeusz J, Lewin SR. HIV-Hepatitis B Virus Coinfection: Epidemiology, Pathogenesis, and Treatment. AIDS (2017) 31(15):2035–52. doi: 10.1097/QAD.0000000000001574
10. Amponsah-Dacosta E, Tamandjou Tchuem C, Anderson M. Chronic Hepatitis B-Associated Liver Disease in the Context of Human Immunodeficiency Virus Co-Infection and Underlying Metabolic Syndrome. World J Virol (2020) 9(5):54–66. doi: 10.5501/wjv.v9.i5.54
11. Huang PL. A Comprehensive Definition for Metabolic Syndrome. Dis Model Mech (2009) 2(5–6):231–7. doi: 10.1242/dmm.001180
12. Hamaguchi M, Kojima T, Takeda N, Nakagawa T, Taniguchi H, Fujii K, et al. The Metabolic Syndrome as a Predictor of Nonalcoholic Fatty Liver Disease. Ann Intern Med (2005) 143(10):722–8. doi: 10.7326/0003-4819-143-10-200511150-00009
13. Lonardo A, Ballestri S, Marchesini G, Angulo P, Loria P. Nonalcoholic Fatty Liver Disease: A Precursor of the Metabolic Syndrome. Dig Liver Dis (2015) 47(3):181–90. doi: 10.1016/j.dld.2014.09.020
14. Kawano Y, Cohen DE. Mechanisms of Hepatic Triglyceride Accumulation in Non-Alcoholic Fatty Liver Disease. J Gastroenterol (2013) 48(4):434–41. doi: 10.1007/s00535-013-0758-5
15. Spahis S, Delvin E, Borys J-M, Levy E. Oxidative Stress as a Critical Factor in Nonalcoholic Fatty Liver Disease Pathogenesis. Antioxid Redox Signal (2017) 26(10):519–41. doi: 10.1089/ars.2016.6776
16. Serviddio G, Bellanti F, Vendemiale G. Free Radical Biology for Medicine: Learning From Nonalcoholic Fatty Liver Disease. Free Radic Biol Med (2013) 65:952–68. doi: 10.1016/J.FREERADBIOMED.2013.08.174
17. Dröge W. Free Radicals in the Physiological Control of Cell Function. Physiol Rev (2002) 82(1):47–95. doi: 10.1152/physrev.00018.2001
18. Jaeschke H. Reactive Oxygen and Mechanisms of Inflammatory Liver Injury: Present Concepts. J Gastroenterol Hepatol (2011) 26(Suppl 1):173–9. doi: 10.1111/j.1440-1746.2010.06592.x
19. Morita M, Ishida N, Uchiyama K, Yamaguchi K, Itoh Y, Shichiri M, et al. Fatty Liver Induced by Free Radicals and Lipid Peroxidation. Free Radic Res (2012) 46(6):758–65. doi: 10.3109/10715762.2012.677840
20. Shimizu I, Shimamoto N, Saiki K, Furujo M, Osaw K. Lipid Peroxidation in Hepatic Fibrosis. In: Lipid Peroxidation. Rijeka: InTech (2012). doi: 10.5772/46180
21. Poli G, Parola M. Oxidative Damage and Fibrogenesis. Free Radic Biol Med (1997) 22(1–2):287–305. doi: 10.1016/s0891-5849(96)00327-9
22. Cohen G, Riahi Y, Shamni O, Guichardant M, Chatgilialoglu C, Ferreri C, et al. Role of Lipid Peroxidation and PPAR-δ in Amplifying Glucose-Stimulated Insulin Secretion. Diabetes (2011) 60(11):2830–42. doi: 10.2337/db11-0347
23. Buzzetti E, Pinzani M, Tsochatzis EA. The Multiple-Hit Pathogenesis of Non-Alcoholic Fatty Liver Disease (NAFLD). Metabolism (2016) 65(8):1038–48. doi: 10.1016/j.metabol.2015.12.012
24. Jensen-Cody SO, Potthoff MJ. Hepatokines and Metabolism: Deciphering Communication From the Liver. Mol Metab (2021) 44:101138. doi: 10.1016/j.molmet.2020.101138
25. de Oliveira Dos Santos AR, de Oliveira Zanuso B, Miola VFB, Barbalho SM, Santos Bueno PC, Flato UAP, et al. Adipokines, Myokines, and Hepatokines: Crosstalk and Metabolic Repercussions. Int J Mol Sci (2021) 22(5):2639. doi: 10.3390/ijms22052639
26. Boutari C, Perakakis N, Mantzoros CS. Association of Adipokines With Development and Progression of Nonalcoholic Fatty Liver Disease. Endocrinol Metab (Seoul Korea) (2018) 33(1):33–43. doi: 10.3803/EnM.2018.33.1.33
27. Lebensztejn DM, Flisiak-Jackiewicz M, Białokoz-Kalinowska I, Bobrus-Chociej A, Kowalska I. Hepatokines and Non-Alcoholic Fatty Liver Disease. Acta Biochim Pol (2016) 63(3):459–67. doi: 10.18388/abp.2015_1252
28. Marques V, Afonso MB, Bierig N, Duarte-Ramos F, Santos-Laso Á, Jimenez-Agüero R, et al. Adiponectin, Leptin, and IGF-1 Are Useful Diagnostic and Stratification Biomarkers of NAFLD. Front Med (2021) 8:683250. doi: 10.3389/fmed.2021.683250
29. Polyzos SA, Kountouras J, Mantzoros CS. Adipokines in Nonalcoholic Fatty Liver Disease. Metabolism (2016) 65(8):1062–79. doi: 10.1016/j.metabol.2015.11.006
30. Kucukoglu O, Sowa J-P, Mazzolini GD, Syn W-K, Canbay A. Hepatokines and Adipokines in NASH-Related Hepatocellular Carcinoma. J Hepatol (2021) 74(2):442–57. doi: 10.1016/j.jhep.2020.10.030
31. Awazawa M, Ueki K, Inabe K, Yamauchi T, Kaneko K, Okazaki Y, et al. Adiponectin Suppresses Hepatic SREBP1c Expression in an AdipoR1/LKB1/AMPK Dependent Pathway. Biochem Biophys Res Commun (2009) 382(1):51–6. doi: 10.1016/j.bbrc.2009.02.131
32. Kakuma T, Lee Y, Higa M, Wang ZW, Pan W, Shimomura I, et al. Leptin, Troglitazone, and the Expression of Sterol Regulatory Element Binding Proteins in Liver and Pancreatic Islets. Proc Natl Acad Sci USA (2000) 97(15):8536–41. doi: 10.1073/pnas.97.15.8536
33. Iroz A, Montagner A, Benhamed F, Levavasseur F, Polizzi A, Anthony E, et al. A Specific ChREBP and Pparα Cross-Talk Is Required for the Glucose-Mediated FGF21 Response. Cell Rep (2017) 21(2):403–16. doi: 10.1016/J.CELREP.2017.09.065
34. Xu P, Zhang Y, Liu Y, Yuan Q, Song L, Liu M, et al. Fibroblast Growth Factor 21 Attenuates Hepatic Fibrogenesis Through TGF-β/Smad2/3 and NF-κb Signaling Pathways. Toxicol Appl Pharmacol (2016) 290:43–53. doi: 10.1016/J.TAAP.2015.11.012
35. Cheng J, Zhang L, Dai W, Mao Y, Li S, Wang J, et al. Ghrelin Ameliorates Intestinal Barrier Dysfunction in Experimental Colitis by Inhibiting the Activation of Nuclear Factor-Kappa B. Biochem Biophys Res Commun (2015) 458(1):140–7. doi: 10.1016/j.bbrc.2015.01.083
36. Wen F, Xia Q, Zhang H, Shia H, Rajesh A, Wu Y, et al. Resistin Activates P65 Pathway and Reduces Glycogen Content Through Keratin 8. Int J Endocrinol (2020) 2020:1–11. doi: 10.1155/2020/9767926
37. Dasgupta S, Bhattacharya S, Biswas A, Majumdar SS, Mukhopadhyay S, Ray S, et al. NF-kappaB Mediates Lipid-Induced Fetuin-A Expression in Hepatocytes That Impairs Adipocyte Function Effecting Insulin Resistance. Biochem J (2010) 429(3):451–62. doi: 10.1042/BJ20100330
38. Frühbeck G. Intracellular Signalling Pathways Activated by Leptin. Biochem J (2006) 393(Pt 1):7–20. doi: 10.1042/BJ20051578
39. Li Z, Xu G, Qin Y, Zhang C, Tang H, Yin Y, et al. Ghrelin Promotes Hepatic Lipogenesis by Activation of mTOR-Pparγ Signaling Pathway. Proc Natl Acad Sci USA (2014) 111(36):13163–8. doi: 10.1073/pnas.1411571111
40. Kadowaki T, Yamauchi T. Adiponectin and Adiponectin Receptors. Endocr Rev (2005) 26(3):439–51. doi: 10.1210/er.2005-0005
41. Stern JH, Rutkowski JM, Scherer PE. Adiponectin, Leptin, and Fatty Acids in the Maintenance of Metabolic Homeostasis Through Adipose Tissue Crosstalk. Cell Metab (2016) 23(5):770–84. doi: 10.1016/J.CMET.2016.04.011
42. Giby VG, Ajith TA. Role of Adipokines and Peroxisome Proliferator-Activated Receptors in Nonalcoholic Fatty Liver Disease. World J Hepatol (2014) 6(8):570–9. doi: 10.4254/wjh.v6.i8.570
43. Ishtiaq SM, Rashid H, Hussain Z, Arshad MI, Khan JA. Adiponectin and PPAR: A Setup for Intricate Crosstalk Between Obesity and Non-Alcoholic Fatty Liver Disease. Rev Endocr Metab Disord (2019) 20(3):253–61. doi: 10.1007/s11154-019-09510-2
44. Dutchak PA, Katafuchi T, Bookout AL, Choi JH, Yu RT, Mangelsdorf DJ, et al. Fibroblast Growth Factor-21 Regulates Pparγ Activity and the Antidiabetic Actions of Thiazolidinediones. Cell (2012) 148(3):556–67. doi: 10.1016/j.cell.2011.11.062
45. Muise ES, Azzolina B, Kuo DW, El-Sherbeini M, Tan Y, Yuan X, et al. Adipose Fibroblast Growth Factor 21 Is Up-Regulated by Peroxisome Proliferator-Activated Receptor Gamma and Altered Metabolic States. Mol Pharmacol (2008) 74(2):403–12. doi: 10.1124/mol.108.044826
46. Gälman C, Lundåsen T, Kharitonenkov A, Bina HA, Eriksson M, Hafström I, et al. The Circulating Metabolic Regulator FGF21 Is Induced by Prolonged Fasting and PPARalpha Activation in Man. Cell Metab (2008) 8(2):169–74. doi: 10.1016/j.cmet.2008.06.014
47. Ge L, Li Q, Wang M, Ouyang J, Li X, Xing MMQ. Nanosilver Particles in Medical Applications: Synthesis, Performance, and Toxicity. Int J Nanomed (2014) 9:2399–407. doi: 10.2147/IJN.S55015
48. Dziegielewska KM, Andersen NA, Saunders NR. Modification of Macrophage Response to Lipopolysaccharide by Fetuin. Immunol Lett (1998) 60(1):31–5. doi: 10.1016/s0165-2478(97)00126-0
49. Tarkowski A, Bjersing J, Shestakov A, Bokarewa MI. Resistin Competes With Lipopolysaccharide for Binding to Toll-Like Receptor 4. J Cell Mol Med (2010) 14(6B):1419–31. doi: 10.1111/j.1582-4934.2009.00899.x
50. Baffy G. Kupffer Cells in Non-Alcoholic Fatty Liver Disease: The Emerging View. J Hepatol (2009) 51(1):212–23. doi: 10.1016/J.JHEP.2009.03.008
51. Pal D, Dasgupta S, Kundu R, Maitra S, Das G, Mukhopadhyay S, et al. Fetuin-A Acts as an Endogenous Ligand of TLR4 to Promote Lipid-Induced Insulin Resistance. Nat Med (2012) 18(8):1279–85. doi: 10.1038/nm.2851
52. Chen H. Nutrient Mtorc1 Signaling Contributes to Hepatic Lipid Metabolism in the Pathogenesis of Non-Alcoholic Fatty Liver Disease. Liver Res (2020) 4(1):15–22. doi: 10.1016/J.LIVRES.2020.02.004
53. Luedde T, Schwabe RF. NF-κb in the Liver–Linking Injury, Fibrosis and Hepatocellular Carcinoma. Nat Rev Gastroenterol Hepatol (2011) 8(2):108–18. doi: 10.1038/nrgastro.2010.213
54. Liu T, Zhang L, Joo D, Sun S-C. NF-κb Signaling in Inflammation. Signal Transduct Target Ther (2017) 2(1):17023. doi: 10.1038/sigtrans.2017.23
55. Zhao J, Qi Y-F, Yu Y-R. STAT3: A Key Regulator in Liver Fibrosis. Ann Hepatol (2021) 21:100224. doi: 10.1016/J.AOHEP.2020.06.010
56. Jeong WI, Park O, Radaeva S, Gao B. STAT1 Inhibits Liver Fibrosis in Mice by Inhibiting Stellate Cell Proliferation and Stimulating NK Cell Cytotoxicity. Hepatology (2006) 44(6):1441–51. doi: 10.1002/HEP.21419
57. Khodarev NN, Roizman B, Weichselbaum RR. Molecular Pathways Molecular Pathways: Interferon/Stat1 Pathway: Role in the Tumor Resistance to Genotoxic Stress and Aggressive Growth. Clin Cancer Res (2012) 18(11):3015–21. doi: 10.1158/1078-0432.CCR-11-3225
58. Miyazaki T, Bub JD, Uzuki M, Iwamoto Y. Adiponectin Activates C-Jun NH2-Terminal Kinase and Inhibits Signal Transducer and Activator of Transcription 3. Biochem Biophys Res Commun (2005) 333(1):79–87. doi: 10.1016/j.bbrc.2005.05.076
59. Shu R-Z, Zhang F, Wang F, Feng D-C, Li X-H, Ren W-H, et al. Adiponectin Deficiency Impairs Liver Regeneration Through Attenuating STAT3 Phosphorylation in Mice. Lab Investig (2009) 89(9):1043–52. doi: 10.1038/labinvest.2009.63
60. Opoku YK, Liu Z, Afrifa J, Kumi AK, Liu H, Ghartey-Kwansah G, et al. Fibroblast Growth Factor-21 Ameliorates Hepatic Encephalopathy by Activating the STAT3-SOCS3 Pathway to Inhibit Activated Hepatic Stellate Cells. EXCLI J (2020) 19:567–81. doi: 10.17179/excli2020-1287
61. Linden AG, Li S, Choi HY, Fang F, Fukasawa M, Uyeda K, et al. Interplay Between ChREBP and SREBP-1c Coordinates Postprandial Glycolysis and Lipogenesis in Livers of Mice. J Lipid Res (2018) 59(3):475–87. doi: 10.1194/jlr.M081836
62. Xu X, So J-S, Park J-G, Lee A-H. Transcriptional Control of Hepatic Lipid Metabolism by SREBP and ChREBP. Semin Liver Dis (2013) 33(4):301–11. doi: 10.1055/s-0033-1358523
63. Moslehi A, Hamidi-Zad Z. Role of SREBPs in Liver Diseases: A Mini-Review. J Clin Transl Hepatol (2018) 6(3):332–8. doi: 10.14218/JCTH.2017.00061
64. Wang N, Kong R, Luo H, Xu X, Lu J. Peroxisome Proliferator-Activated Receptors Associated With Nonalcoholic Fatty Liver Disease. PPAR Res (2017) 2017:1–8. doi: 10.1155/2017/6561701
65. Ogawa Y, Imajo K, Yoneda M, Kessoku T, Tomeno W, Shinohara Y, et al. Soluble CD14 Levels Reflect Liver Inflammation in Patients With Nonalcoholic Steatohepatitis. PloS One (2013) 8(6):e65211. doi: 10.1371/journal.pone.0065211
66. Miura K, Ohnishi H. Role of Gut Microbiota and Toll-Like Receptors in Nonalcoholic Fatty Liver Disease. World J Gastroenterol (2014) 20(23):7381. doi: 10.3748/wjg.v20.i23.7381
67. Liu C-J, Chen P-J, Lai M-Y, Liu C-H, Chen C-L, Kao J-H, et al. High Serum Adiponectin Correlates With Advanced Liver Disease in Patients With Chronic Hepatitis B Virus Infection. Hepatol Int (2009) 3(2):364–70. doi: 10.1007/s12072-008-9111-0
68. Fernández-Real JM, Pérez del Pulgar S, Luche E, Moreno-Navarrete JM, Waget A, Serino M, et al. CD14 Modulates Inflammation-Driven Insulin Resistance. Diabetes (2011) 60(8):2179–86. doi: 10.2337/db10-1210
69. Ferreira DF, Fiamoncini J, Prist IH, Ariga SK, de Souza HP, de Lima TM. Novel Role of TLR4 in NAFLD Development: Modulation of Metabolic Enzymes Expression. Biochim Biophys Acta (2015) 1851(10):1353–9. doi: 10.1016/j.bbalip.2015.07.002
70. Han J, Wang Y. Mtorc1 Signaling in Hepatic Lipid Metabolism. Protein Cell (2018) 9(2):145–51. doi: 10.1007/s13238-017-0409-3
71. Cho C-S, Kowalsky AH, Lee JH. Pathological Consequences of Hepatic Mtorc1 Dysregulation. Genes (Basel) (2020) 11(8):896. doi: 10.3390/genes11080896
72. Maya-Monteiro CM, Bozza PT. Leptin and mTOR: Partners in Metabolism and Inflammation. Cell Cycle (2008) 7(12):1713–7. doi: 10.4161/cc.7.12.6157
73. Minard AY, Tan S-X, Yang P, Fazakerley DJ, Domanova W, Parker BL, et al. Mtorc1 Is a Major Regulatory Node in the FGF21 Signaling Network in Adipocytes. Cell Rep (2016) 17(1):29–36. doi: 10.1016/j.celrep.2016.08.086
74. Saxena NK, Fu PP, Nagalingam A, Wang J, Handy J, Cohen C, et al. Adiponectin Modulates C-Jun N-Terminal Kinase and Mammalian Target of Rapamycin and Inhibits Hepatocellular Carcinoma. Gastroenterology (2010) 139(5):1762–73. doi: 10.1053/j.gastro.2010.07.001.
75. Czaja MJ. JNK Regulation of Hepatic Manifestations of the Metabolic Syndrome. Trends Endocrinol Metab (2010) 21(12):707–13. doi: 10.1016/j.tem.2010.08.010
76. Gamberi T, Magherini F, Modesti A, Fiaschi T. Adiponectin Signaling Pathways in Liver Diseases. Biomedicines (2018) 6(2):52. doi: 10.3390/biomedicines6020052
77. Lee KJ, Jang YO, Cha S-K, Kim MY, Park K-S, Eom YW, et al. Expression of Fibroblast Growth Factor 21 and β-Klotho Regulates Hepatic Fibrosis Through the Nuclear Factor-κb and C-Jun N-Terminal Kinase Pathways. Gut Liver (2018) 12(4):449–56. doi: 10.5009/gnl17443
78. Hennige AM, Staiger H, Wicke C, Machicao F, Fritsche A, Häring H-U, et al. Fetuin-A Induces Cytokine Expression and Suppresses Adiponectin Production. PloS One (2008) 3(3):e1765. doi: 10.1371/journal.pone.0001765
79. Martínez-Uña M, López-Mancheño Y, Diéguez C, Fernández-Rojo MA, Novelle MG. Unraveling the Role of Leptin in Liver Function and its Relationship With Liver Diseases. Int J Mol Sci (2020) 21(24):9368. doi: 10.3390/ijms21249368
80. Lin Z, Tian H, Lam KSL, Lin S, Hoo RCL, Konishi M, et al. Adiponectin Mediates the Metabolic Effects of FGF21 on Glucose Homeostasis and Insulin Sensitivity in Mice. Cell Metab (2013) 17(5):779–89. doi: 10.1016/j.cmet.2013.04.005
81. Holland WL, Adams AC, Brozinick JT, Bui HH, Miyauchi Y, Kusminski CM, et al. An FGF21-Adiponectin-Ceramide Axis Controls Energy Expenditure and Insulin Action in Mice. Cell Metab (2013) 17(5):790–7. doi: 10.1016/j.cmet.2013.03.019
82. Berti L, Irmler M, Zdichavsky M, Meile T, Böhm A, Stefan N, et al. Fibroblast Growth Factor 21 Is Elevated in Metabolically Unhealthy Obesity and Affects Lipid Deposition, Adipogenesis, and Adipokine Secretion of Human Abdominal Subcutaneous Adipocytes. Mol Metab (2015) 4(7):519–27. doi: 10.1016/j.molmet.2015.04.002
83. Asrih M, Veyrat-Durebex C, Poher A-L, Lyautey J, Rohner-Jeanrenaud F, Jornayvaz FR. Leptin as a Potential Regulator of FGF21. Cell Physiol Biochem (2016) 38(3):1218–25. doi: 10.1159/000443070
84. Misu H, Ishikura K, Kurita S, Takeshita Y, Ota T, Saito Y, et al. Inverse Correlation Between Serum Levels of Selenoprotein P and Adiponectin in Patients With Type 2 Diabetes. Luque RM, Editor. PloS One (2012) 7(4):e34952. doi: 10.1371/journal.pone.0034952
85. Jiang L-L, Li L, Hong X-F, Li Y-M, Zhang B-L. Patients With Nonalcoholic Fatty Liver Disease Display Increased Serum Resistin Levels and Decreased Adiponectin Levels. Eur J Gastroenterol Hepatol (2009) 21(6):662–6. doi: 10.1097/MEG.0b013e328317f4b5
86. Pohl R, Haberl EM, Rein-Fischboeck L, Zimny S, Neumann M, Aslanidis C, et al. Hepatic Chemerin mRNA Expression Is Reduced in Human Nonalcoholic Steatohepatitis. Eur J Clin Invest (2017) 47(1):7–18. doi: 10.1111/eci.12695
87. Wanninger J, Bauer S, Eisinger K, Weiss TS, Walter R, Hellerbrand C, et al. Adiponectin Upregulates Hepatocyte CMKLR1 Which Is Reduced in Human Fatty Liver. Mol Cell Endocrinol (2012) 349(2):248–54. doi: 10.1016/j.mce.2011.10.032
88. Sell H, Divoux A, Poitou C, Basdevant A, Bouillot J-L, Bedossa P, et al. Chemerin Correlates With Markers for Fatty Liver in Morbidly Obese Patients and Strongly Decreases After Weight Loss Induced by Bariatric Surgery. J Clin Endocrinol Metab (2010) 95(6):2892–6. doi: 10.1210/jc.2009-2374
89. Watt MJ, Miotto PM, De Nardo W, Montgomery MK. The Liver as an Endocrine Organ-Linking NAFLD and Insulin Resistance. Insulin Resist (2019) 40(5):1367–93. doi: 10.1210/er.2019-00034
90. Sanders FWB, Griffin JL. De Novo Lipogenesis in the Liver in Health and Disease: More Than Just a Shunting Yard for Glucose. Biol Rev Camb Philos Soc (2016) 91(2):452–68. doi: 10.1111/brv.12178
91. Wang ZV, Scherer PE. Adiponectin, the Past Two Decades. J Mol Cell Biol (2016) 8(2):93–100. doi: 10.1093/jmcb/mjw011
92. Stojsavljević S, Palčić MG, Jukić LV, Duvnjak LS, Duvnjak M. Adipokines and Proinflammatory Cytokines, the Key Mediators in the Pathogenesis of Nonalcoholic Fatty Liver Disease. World J Gastroenterol (2014) 20(48):18070. doi: 10.3748/wjg.v20.i48.18070
93. Saxena NK, Anania FA. Adipocytokines and Hepatic Fibrosis. Trends Endocrinol Metab (2015) 26(3):153–61. doi: 10.1016/j.tem.2015.01.002
94. Musso G, Gambino R, Biroli G, Carello M, Fagà E, Pacini G, et al. Hypoadiponectinemia Predicts the Severity of Hepatic Fibrosis and Pancreatic Beta-Cell Dysfunction in Nondiabetic Nonobese Patients With Nonalcoholic Steatohepatitis. Am J Gastroenterol (2005) 100(11):2438–46. doi: 10.1111/j.1572-0241.2005.00297.x
95. Palhinha L, Liechocki S, Hottz ED, Pereira JA da S, de Almeida CJ, Moraes-Vieira PMM, et al. Leptin Induces Proadipogenic and Proinflammatory Signaling in Adipocytes. Front Endocrinol (Lausanne) (2019) 10:841. doi: 10.3389/fendo.2019.00841
96. Ikejima K, Honda H, Yoshikawa M, Hirose M, Kitamura T, Takei Y, et al. Leptin Augments Inflammatory and Profibrogenic Responses in the Murine Liver Induced by Hepatotoxic Chemicals. Hepatology (2001) 34(2):288–97. doi: 10.1053/jhep.2001.26518
97. Polyzos SA, Kountouras J, Zavos C, Deretzi G. The Potential Adverse Role of Leptin Resistance in Nonalcoholic Fatty Liver Disease: A Hypothesis Based on Critical Review of the Literature. J Clin Gastroenterol (2011) 45(1):50–4. doi: 10.1097/MCG.0b013e3181ec5c66
98. Shen C, Zhao C-Y, Wang W, Wang Y-D, Sun H, Cao W, et al. The Relationship Between Hepatic Resistin Overexpression and Inflammation in Patients With Nonalcoholic Steatohepatitis. BMC Gastroenterol (2014) 14(1):39. doi: 10.1186/1471-230X-14-39
99. Tripathi D, Kant S, Pandey S, Ehtesham NZ. Resistin in Metabolism, Inflammation, and Disease. FEBS J (2020) 287(15):3141–9. doi: 10.1111/febs.15322
100. Han D, Chen J, Liu S, Zhang Z, Zhao Z, Jin W, et al. Serum Resistin Levels in Adult Patients With Nonalcoholic Fatty Liver Disease: A Systematic Review and Meta-Analysis. J Clin Transl Hepatol (2021) 9(4):484–93. doi: 10.14218/JCTH.2021.00018
101. Bertolani C, Sancho-Bru P, Failli P, Bataller R, Aleffi S, DeFranco R, et al. Resistin as an Intrahepatic Cytokine: Overexpression During Chronic Injury and Induction of Proinflammatory Actions in Hepatic Stellate Cells. Am J Pathol (2006) 169(6):2042–53. doi: 10.2353/ajpath.2006.060081
102. Bekaert M, Verhelst X, Geerts A, Lapauw B, Calders P. Association of Recently Described Adipokines With Liver Histology in Biopsy-Proven Non-Alcoholic Fatty Liver Disease: A Systematic Review. Obes Rev (2016) 17(1):68–80. doi: 10.1111/obr.12333
103. Li Y, Yang Q, Cai D, Guo H, Fang J, Cui H, et al. Resistin, a Novel Host Defense Peptide of Innate Immunity. Front Immunol (2021) 12:699807. doi: 10.3389/fimmu.2021.699807
104. Silswal N, Singh AK, Aruna B, Mukhopadhyay S, Ghosh S, Ehtesham NZ. Human Resistin Stimulates the Pro-Inflammatory Cytokines TNF-α and IL-12 in Macrophages by NF-κb-Dependent Pathway. Biochem Biophys Res Commun (2005) 334(4):1092–101. doi: 10.1016/J.BBRC.2005.06.202
105. Costandi J, Melone M, Zhao A, Rashid S. Human Resistin Stimulates Hepatic Overproduction of Atherogenic ApoB-Containing Lipoprotein Particles by Enhancing ApoB Stability and Impairing Intracellular Insulin Signaling. Circ Res (2011) 108(6):727–42. doi: 10.1161/CIRCRESAHA.110.238949
106. Sheng CH, Di J, Jin Y, Zhang YC, Wu M, Sun Y, et al. Resistin Is Expressed in Human Hepatocytes and Induces Insulin Resistance. Endocrine (2008) 33(2):135–43. doi: 10.1007/s12020-008-9065-y
107. Huang X, Yang Z. Resistin’s, Obesity and Insulin Resistance: The Continuing Disconnect Between Rodents and Humans. J Endocrinol Invest (2016) 39(6):607–15. doi: 10.1007/s40618-015-0408-2
108. Mao Y, Zhang S, Yu F, Li H, Guo C, Fan X. Ghrelin Attenuates Liver Fibrosis Through Regulation of TGF-β1 Expression and Autophagy. Int J Mol Sci (2015) 16(9):21911–30. doi: 10.3390/ijms160921911
109. Delhanty PJD, van der Lely AJ. Ghrelin and Glucose Homeostasis. Peptides (2011) 32(11):2309–18. doi: 10.1016/j.peptides.2011.03.001
110. Waseem T, Duxbury M, Ito H, Ashley SW, Robinson MK. Exogenous Ghrelin Modulates Release of Pro-Inflammatory and Anti-Inflammatory Cytokines in LPS-Stimulated Macrophages Through Distinct Signaling Pathways. Surgery (2008) 143(3):334–42. doi: 10.1016/j.surg.2007.09.039
111. Keinicke H, Sun G, Mentzel CMJ, Fredholm M, John LM, Andersen B, et al. FGF21 Regulates Hepatic Metabolic Pathways to Improve Steatosis and Inflammation. Endocr Connect (2020) 9(8):755–68. doi: 10.1530/EC-20-0152
112. Li H, Fang Q, Gao F, Fan J, Zhou J, Wang X, et al. Fibroblast Growth Factor 21 Levels Are Increased in Nonalcoholic Fatty Liver Disease Patients and Are Correlated With Hepatic Triglyceride. J Hepatol (2010) 53(5):934–40. doi: 10.1016/j.jhep.2010.05.018
113. Vernia S, Cavanagh-Kyros J, Garcia-Haro L, Sabio G, Barrett T, Jung DY, et al. The Pparα-FGF21 Hormone Axis Contributes to Metabolic Regulation by the Hepatic JNK Signaling Pathway. Cell Metab (2014) 20(3):512–25. doi: 10.1016/j.cmet.2014.06.010
114. Lin W, Zhang T, Zhou Y, Zheng J, Lin Z. Advances in Biological Functions and Clinical Studies of FGF21. Diabetes Metab Syndr Obes (2021) 14:3281–90. doi: 10.2147/DMSO.S317096
115. Yan H, Xia M, Chang X, Xu Q, Bian H, Zeng M, et al. Circulating Fibroblast Growth Factor 21 Levels Are Closely Associated With Hepatic Fat Content: A Cross-Sectional Study. Xu A, Editor. PloS One (2011) 6(9):e24895. doi: 10.1371/journal.pone.0024895
116. Fisher FM, Maratos-Flier E. Understanding the Physiology of FGF21. Annu Rev Physiol (2016) 78(1):223–41. doi: 10.1146/annurev-physiol-021115-105339
117. Tillman EJ, Rolph T. FGF21: An Emerging Therapeutic Target for Non-Alcoholic Steatohepatitis and Related Metabolic Diseases. Front Endocrinol (Lausanne) (2020) 11:601290. doi: 10.3389/fendo.2020.601290
118. Sato M, Kamada Y, Takeda Y, Kida S, Ohara Y, Fujii H, et al. Fetuin-A Negatively Correlates With Liver and Vascular Fibrosis in Nonalcoholic Fatty Liver Disease Subjects. Liver Int (2015) 35(3):925–35. doi: 10.1111/liv.12478
119. Yilmaz Y, Yonal O, Kurt R, Ari F, Oral AY, Celikel CA, et al. Serum Fetuin a/α2hs-Glycoprotein Levels in Patients With Non-Alcoholic Fatty Liver Disease: Relation With Liver Fibrosis. Ann Clin Biochem (2010) 47(Pt 6):549–53. doi: 10.1258/acb.2010.010169
120. Haukeland JW, Dahl TB, Yndestad A, Gladhaug IP, Løberg EM, Haaland T, et al. Fetuin A in Nonalcoholic Fatty Liver Disease: In Vivo and In Vitro Studies. Eur J Endocrinol (2012) 166(3):503–10. doi: 10.1530/EJE-11-0864
121. Liu S, Xiao J, Zhao Z, Wang M, Wang Y, Xin Y. Systematic Review and Meta-Analysis of Circulating Fetuin-A Levels in Nonalcoholic Fatty Liver Disease. J Clin Transl Hepatol (2021) 9(1):3–14. doi: 10.14218/JCTH.2020.00081
122. Li Z, Lin M, Liu C, Wang D, Shi X, Chen Z, et al. Fetuin-B Links Nonalcoholic Fatty Liver Disease to Type 2 Diabetes via Inducing Insulin Resistance: Association and Path Analyses. Cytokine (2018) 108:145–50. doi: 10.1016/j.cyto.2018.03.023
123. Kukla M, Zwirska-Korczala K, Hartleb M, Waluga M, Chwist A, Kajor M, et al. Serum Chemerin and Vaspin in Non-Alcoholic Fatty Liver Disease. Scand J Gastroenterol (2010) 45(2):235–42. doi: 10.3109/00365520903443852
124. Döcke S, Lock JF, Birkenfeld AL, Hoppe S, Lieske S, Rieger A, et al. Elevated Hepatic Chemerin mRNA Expression in Human Non-Alcoholic Fatty Liver Disease. Eur J Endocrinol (2013) 169(5):547–57. doi: 10.1530/EJE-13-0112
125. Buechler C. Chemerin in Liver Diseases. Chemerin Liver Dis Endocrinol Metab Synd (2014) 3(4):144. doi: 10.4172/2161-1017.1000144
126. Heo YJ, Choi S-E, Jeon JY, Han SJ, Kim DJ, Kang Y, et al. Visfatin Induces Inflammation and Insulin Resistance via the NF-κb and STAT3 Signaling Pathways in Hepatocytes. J Diabetes Res (2019) 2019:4021623. doi: 10.1155/2019/4021623
127. Ismaiel A, Leucuta D-C, Popa S-L, Dumitrascu DL. Serum Visfatin Levels in Nonalcoholic Fatty Liver Disease and Liver Fibrosis: Systematic Review and Meta-Analysis. J Clin Med (2021) 10(14):3029. doi: 10.3390/jcm10143029
128. Kukla M, Ciupińska-Kajor M, Kajor M, Wylezol M, Żwirska-Korczala K, Hartleb M, et al. Liver Visfatin Expression in Morbidly Obese Patients With Nonalcoholic Fatty Liver Disease Undergoing Bariatric Surgery. Pol J Pathol (2010) 61(3):147–53.
129. Caviglia GP, Rosso C, Armandi A, Gaggini M, Carli F, Abate ML, et al. Interplay Between Oxidative Stress and Metabolic Derangements in Non-Alcoholic Fatty Liver Disease: The Role of Selenoprotein P. Int J Mol Sci (2020) 21(22):8838. doi: 10.3390/ijms21228838
130. Reeves MA, Hoffmann PR. The Human Selenoproteome: Recent Insights Into Functions and Regulation. Cell Mol Life Sci (2009) 66(15):2457–78. doi: 10.1007/s00018-009-0032-4
131. Chen Y, He X, Chen X, Li Y, Ke Y. SeP Is Elevated in NAFLD and Participates in NAFLD Pathogenesis Through AMPK/ACC Pathway. J Cell Physiol (2021) 236(5):3800–7. doi: 10.1002/JCP.30121
132. Polyzos SA, Kountouras J, Goulas A, Duntas L. Selenium and Selenoprotein P in Nonalcoholic Fatty Liver Disease. Hormones (Athens) (2020) 19(1):61–72. doi: 10.1007/s42000-019-00127-3
133. Polyzos SA, Toulis KA, Goulis DG, Zavos C, Kountouras J. Serum Total Adiponectin in Nonalcoholic Fatty Liver Disease: A Systematic Review and Meta-Analysis. Metabolism (2011) 60(3):313–26. doi: 10.1016/j.metabol.2010.09.003
134. Saxena NK, Sharma D, Ding X, Lin S, Marra F, Merlin D, et al. Concomitant Activation of the JAK/STAT, PI3K/AKT, and ERK Signaling Is Involved in Leptin-Mediated Promotion of Invasion and Migration of Hepatocellular Carcinoma Cells. Cancer Res (2007) 67(6):2497–507. doi: 10.1158/0008-5472.CAN-06-3075
135. Polyzos SA, Aronis KN, Kountouras J, Raptis DD, Vasiloglou MF, Mantzoros CS. Circulating Leptin in Non-Alcoholic Fatty Liver Disease: A Systematic Review and Meta-Analysis. Diabetologia (2016) 59(1):30–43. doi: 10.1007/s00125-015-3769-3
136. Reitman ML. Leptin in the Liver: A Toxic or Beneficial Mix? Cell Metab (2012) 16(1):1–2. doi: 10.1016/j.cmet.2012.06.009
137. Wong VW, Hui AY, Tsang SW, Chan JL, Tse AM, Chan K, et al. Metabolic and Adipokine Profile of Chinese Patients With Nonalcoholic Fatty Liver Disease. Clin Gastroenterol Hepatol (2006) 4(9):1154–61. doi: 10.1016/j.cgh.2006.06.011
138. Machado MV, Coutinho J, Carepa F, Costa A, Proença H, Cortez-Pinto H. How Adiponectin, Leptin, and Ghrelin Orchestrate Together and Correlate With the Severity of Nonalcoholic Fatty Liver Disease. Eur J Gastroenterol Hepatol (2012) 24(10):1166–72. doi: 10.1097/MEG.0b013e32835609b0
139. Chopin LK, Seim I, Walpole CM, Herington AC. The Ghrelin Axis-Does it Have an Appetite for Cancer Progression? Endocr Rev (2012) 33(6):849–91. doi: 10.1210/er.2011-1007
140. Marchesini G, Pagotto U, Bugianesi E, De Iasio R, Manini R, Vanni E, et al. Low Ghrelin Concentrations in Nonalcoholic Fatty Liver Disease Are Related to Insulin Resistance. J Clin Endocrinol Metab (2003) 88(12):5674–9. doi: 10.1210/jc.2003-031094
141. Zheng Q, Martin RC, Shi X, Pandit H, Yu Y, Liu X, et al. Lack of FGF21 Promotes NASH-HCC Transition via hepatocyte-TLR4-IL-17A signaling. Theranostics (2020) 10(22):9923–36. doi: 10.7150/thno.45988
142. Goralski KB, Jackson AE, McKeown BT, Sinal CJ. More Than an Adipokine: The Complex Roles of Chemerin Signaling in Cancer. Int J Mol Sci (2019) 20(19):4778. doi: 10.3390/ijms20194778
143. Horn P, von Loeffelholz C, Forkert F, Stengel S, Reuken P, Aschenbach R, et al. Low Circulating Chemerin Levels Correlate With Hepatic Dysfunction and Increased Mortality in Decompensated Liver Cirrhosis. Sci Rep (2018) 8(1):9242. doi: 10.1038/s41598-018-27543-6
144. Polyzos SA, Kountouras J, Romiopoulos I, Polymerou V. Serum Visfatin in Nonalcoholic Fatty Liver Disease. Ann Hepatol 13(1):150–1. doi: 10.1016/S1665-2681(19)30917-2
145. Ninomiya S, Shimizu M, Imai K, Takai K, Shiraki M, Hara T, et al. Possible Role of Visfatin in Hepatoma Progression and the Effects of Branched-Chain Amino Acids on Visfatin-Induced Proliferation in Human Hepatoma Cells. Cancer Prev Res (Phila) (2011) 4(12):2092–100. doi: 10.1158/1940-6207.CAPR-11-0340
146. Genc H, Dogru T, Kara M, Tapan S, Ercin CN, Acikel C, et al. Association of Plasma Visfatin With Hepatic and Systemic Inflammation in Nonalcoholic Fatty Liver Disease. Ann Hepatol (2013) 12(4):380–7. doi: 10.1016/S1665-2681(19)31338-9
147. Akbal E, Koçak E, Taş A, Yüksel E, Köklü S. Visfatin Levels in Nonalcoholic Fatty Liver Disease. J Clin Lab Anal (2012) 26(2):115–9. doi: 10.1002/jcla.21491
148. Wang L-F, Wang X-N, Huang C-C, Hu L, Xiao Y-F, Guan X-H, et al. Inhibition of NAMPT Aggravates High Fat Diet-Induced Hepatic Steatosis in Mice Through Regulating Sirt1/Ampkα/SREBP1 Signaling Pathway. Lipids Health Dis (2017) 16(1):82. doi: 10.1186/s12944-017-0464-z
149. Johannsen K, Flechtner-Mors M, Kratzer W, Koenig W, Boehm BO, Schmidberger J, et al. Association Between Visfatin and Hepatic Steatosis in the General Population During Long-Term Follow-Up. Horm Metab Res (2019) 51(9):602–7. doi: 10.1055/a-0897-8565
150. Aller R, de Luis DA, Izaola O, Sagrado MG, Conde R, Velasco MC, et al. Influence of Visfatin on Histopathological Changes of Non-Alcoholic Fatty Liver Disease. Dig Dis Sci (2009) 54(8):1772–7. doi: 10.1007/s10620-008-0539-9
151. Qiu Y, Wang S-F, Yu C, Chen Q, Jiang R, Pei L, et al. Association of Circulating Adipsin, Visfatin, and Adiponectin With Nonalcoholic Fatty Liver Disease in Adults: A Case-Control Study. Ann Nutr Metab (2019) 74(1):44–52. doi: 10.1159/000495215
152. de Boer JF, Bahr MJ, Böker KHW, Manns MP, Tietge UJF. Plasma Levels of PBEF/Nampt/visfatin Are Decreased in Patients With Liver Cirrhosis. Am J Physiol Gastrointest Liver Physiol (2009) 296(2):G196–201. doi: 10.1152/ajpgi.00029.2008
153. Wang J, Shen P, Liao S, Duan L, Zhu D, Chen J, et al. Selenoprotein P Inhibits Cell Proliferation and ROX Production in HCC Cells. Ogunwobi O, Editor. PloS One (2020) 15(7):e0236491. doi: 10.1371/journal.pone.0236491
154. Lehrke M, Becker A, Greif M, Stark R, Laubender RP, von Ziegler F, et al. Chemerin Is Associated With Markers of Inflammation and Components of the Metabolic Syndrome But Does Not Predict Coronary Atherosclerosis. Eur J Endocrinol (2009) 161(2):339–44. doi: 10.1530/EJE-09-0380
155. Spirk M, Zimny S, Neumann M, McMullen N, Sinal CJ, Buechler C. Chemerin-156 Is the Active Isoform in Human Hepatic Stellate Cells. Int J Mol Sci (2020) 21(20):7555. doi: 10.3390/ijms21207555
156. Liang N-L, Men R, Zhu Y, Yuan C, Wei Y, Liu X, et al. Visfatin: An Adipokine Activator of Rat Hepatic Stellate Cells. Mol Med Rep (2015) 11(2):1073–8. doi: 10.3892/mmr.2014.2795
157. Curat CA, Wegner V, Sengenès C, Miranville A, Tonus C, Busse R, et al. Macrophages in Human Visceral Adipose Tissue: Increased Accumulation in Obesity and a Source of Resistin and Visfatin. Diabetologia (2006) 49(4):744–7. doi: 10.1007/s00125-006-0173-z
158. Filippatos TD, Derdemezis CS, Gazi IF, Lagos K, Kiortsis DN, Tselepis AD, et al. Increased Plasma Visfatin Levels in Subjects With the Metabolic Syndrome. Eur J Clin Invest (2007) 38(1):71–2. doi: 10.1111/j.1365-2362.2007.01904.x
159. Yang SJ, Hwang SY, Choi HY, Yoo HJ, Seo JA, Kim SG, et al. Serum Selenoprotein P Levels in Patients With Type 2 Diabetes and Prediabetes: Implications for Insulin Resistance, Inflammation, and Atherosclerosis. J Clin Endocrinol Metab (2011) 96(8):E1325–9. doi: 10.1210/jc.2011-0620
160. Helfer G, Wu Q-F. Chemerin: A Multifaceted Adipokine Involved in Metabolic Disorders. J Endocrinol (2018) 238(2):R79–94. doi: 10.1530/JOE-18-0174
161. Bozaoglu K, Bolton K, McMillan J, Zimmet P, Jowett J, Collier G, et al. Chemerin Is a Novel Adipokine Associated With Obesity and Metabolic Syndrome. Endocrinology (2007) 148(10):4687–94. doi: 10.1210/en.2007-0175
162. Yu Y, He J, Li S, Song L, Guo X, Yao W, et al. Fibroblast Growth Factor 21 (FGF21) Inhibits Macrophage-Mediated Inflammation by Activating Nrf2 and Suppressing the NF-κb Signaling Pathway. Int Immunopharmacol (2016) 38:144–52. doi: 10.1016/j.intimp.2016.05.026
163. Liu Y, Zhao C, Xiao J, Liu L, Zhang M, Wang C, et al. Fibroblast Growth Factor 21 Deficiency Exacerbates Chronic Alcohol-Induced Hepatic Steatosis and Injury. Sci Rep (2016) 6:31026. doi: 10.1038/srep31026
164. Liu J, Xu Y, Hu Y, Wang G. The Role of Fibroblast Growth Factor 21 in the Pathogenesis of Non-Alcoholic Fatty Liver Disease and Implications for Therapy. Metabolism (2015) 64(3):380–90. doi: 10.1016/j.metabol.2014.11.009
165. Lee Y, Wang M, Kakuma T. … ZW-J of B, 2001 Undefined. Liporegulation in Diet-Induced Obesity: The Antisteatotic Role of Hyperleptinemia. J Biol Chem (2001) 276(8):5629–35. doi: 10.1074/jbc.M008553200
166. Peter A, Kovarova M, Staiger H, Machann J, Schick F, Königsrainer A, et al. The Hepatokines Fetuin-A and Fetuin-B Are Upregulated in the State of Hepatic Steatosis and may Differently Impact on Glucose Homeostasis in Humans. Am J Physiol Endocrinol Metab (2018) 314(3):E266–73. doi: 10.1152/ajpendo.00262.2017
167. Dogru T, Kirik A, Gurel H, Rizvi AA, Rizzo M, Sonmez A. The Evolving Role of Fetuin-A in Nonalcoholic Fatty Liver Disease: An Overview From Liver to the Heart. Int J Mol Sci (2021) 22(12):6627. doi: 10.3390/ijms22126627
168. Jang JC, Li J, Gambini L, Batugedara HM, Sati S, Lazar MA, et al. Human Resistin Protects Against Endotoxic Shock by Blocking LPS-TLR4 Interaction. Proc Natl Acad Sci USA (2017) 114(48):E10399–408. doi: 10.1073/pnas.1716015114
169. Maurice JB, Patel A, Scott AJ, Patel K, Thursz M, Lemoine M. Prevalence and Risk Factors of Nonalcoholic Fatty Liver Disease in HIV-Monoinfection. AIDS (2017) 31(11):1621–32. doi: 10.1097/QAD.0000000000001504
170. Sterling RK, Smith PG, Brunt EM. Hepatic Steatosis in Human Immunodeficiency Virus: A Prospective Study in Patients Without Viral Hepatitis, Diabetes, or Alcohol Abuse. J Clin Gastroenterol (2013) 47(2):182–7. doi: 10.1097/MCG.0b013e318264181d
171. Khalili M, Kleiner DE, King WC, Sterling RK, Ghany MG, Chung RT, et al. Hepatic Steatosis and Steatohepatitis in a Large North American Cohort of Adults With Chronic Hepatitis B. Am J Gastroenterol (2021) 116(8):1686–97. doi: 10.14309/ajg.0000000000001257
172. Khalili M, King WC, Kleiner DE, Jain MK, Chung RT, Sulkowski M, et al. Fatty Liver Disease in a Prospective North American Cohort of Adults With Human Immunodeficiency Virus and Hepatitis B Virus Coinfection. Clin Infect Dis (2021) 73(9):e3275–85. doi: 10.1093/cid/ciaa1303
173. Sterling RK, Wahed AS, King WC, Kleiner DE, Khalili M, Sulkowski M, et al. Spectrum of Liver Disease in Hepatitis B Virus (HBV) Patients Co-Infected With Human Immunodeficiency Virus (HIV): Results of the HBV-HIV Cohort Study. Am J Gastroenterol (2019) 114(5):746–57. doi: 10.1038/s41395-018-0409-9
174. Klein MB, Althoff KN, Jing Y, Lau B, Kitahata M, Lo Re V, et al. Risk of End-Stage Liver Disease in HIV-Viral Hepatitis Coinfected Persons in North America From the Early to Modern Antiretroviral Therapy Eras. Clin Infect Dis (2016) 63(9):ciw531. doi: 10.1093/cid/ciw531
175. Somasundaran M, Zapp ML, Beattie LK, Pang L, Byron KS, Bassell GJ, et al. Localization of HIV RNA in Mitochondria of Infected Cells: Potential Role in Cytopathogenicity. J Cell Biol (1994) 126(6):1353–60. doi: 10.1083/jcb.126.6.1353
176. Cao YZ, Dieterich D, Thomas PA, Huang YX, Mirabile M, Ho DD. Identification and Quantitation of HIV-1 in the Liver of Patients With AIDS. AIDS (1992) 6(1):65–70. doi: 10.1097/00002030-199201000-00008
177. Zhang L, Bansal MB. Role of Kupffer Cells in Driving Hepatic Inflammation and Fibrosis in HIV Infection. Front Immunol (2020) 11:1086. doi: 10.3389/fimmu.2020.01086
178. Vlahakis SR, Villasis-Keever A, Gomez TS, Bren GD, Paya CV. Human Immunodeficiency Virus-Induced Apoptosis of Human Hepatocytes via CXCR4. J Infect Dis (2003) 188(10):1455–60. doi: 10.1086/379738
179. Lin W, Wu G, Li S, Weinberg EM, Kumthip K, Peng LF, et al. HIV and HCV Cooperatively Promote Hepatic Fibrogenesis via Induction of Reactive Oxygen Species and NFkappaB. J Biol Chem (2011) 286(4):2665–74. doi: 10.1074/jbc.M110.168286
180. Previte DM, O’Connor EC, Novak EA, Martins CP, Mollen KP, Piganelli JD. Reactive Oxygen Species Are Required for Driving Efficient and Sustained Aerobic Glycolysis During CD4+ T Cell Activation. PloS One (2017) 12(4):e0175549. doi: 10.1371/journal.pone.0175549
181. Gruevska A, Moragrega ÁB, Cossarizza A, Esplugues JV, Blas-García A, Apostolova N. Apoptosis of Hepatocytes: Relevance for HIV-Infected Patients Under Treatment. Cells (2021) 10(2):410. doi: 10.3390/cells10020410
182. Her Z, Tan JHL, Lim Y-S, Tan SY, Chan XY, Tan WWS, et al. CD4+ T Cells Mediate the Development of Liver Fibrosis in High Fat Diet-Induced NAFLD in Humanized Mice. Front Immunol (2020) 11:580968. doi: 10.3389/fimmu.2020.580968
183. Seki E, De Minicis S, Österreicher CH, Kluwe J, Osawa Y, Brenner DA, et al. TLR4 Enhances TGF-β Signaling and Hepatic Fibrosis. Nat Med (2007) 13(11):1324–32. doi: 10.1038/nm1663
184. Mosoian A, Zhang L, Hong F, Cunyat F, Rahman A, Bhalla R, et al. Frontline Science: HIV Infection of Kupffer Cells Results in an Amplified Proinflammatory Response to LPS. J Leukoc Biol (2017) 101(5):1083–90. doi: 10.1189/jlb.3HI0516-242R
185. Damouche A, Lazure T, Avettand-Fènoël V, Huot N, Dejucq-Rainsford N, Satie A-P, et al. Adipose Tissue Is a Neglected Viral Reservoir and an Inflammatory Site During Chronic HIV and SIV Infection. PloS Pathog (2015) 11(9):e1005153. doi: 10.1371/journal.ppat.1005153
186. Chen D, Misra A, Garg A. Clinical Review 153: Lipodystrophy in Human Immunodeficiency Virus-Infected Patients. J Clin Endocrinol Metab (2002) 87(11):4845–56. doi: 10.1210/jc.2002-020794
187. de Waal R, Cohen K, Maartens G. Systematic Review of Antiretroviral-Associated Lipodystrophy: Lipoatrophy, But Not Central Fat Gain, Is an Antiretroviral Adverse Drug Reaction. PloS One (2013) 8(5):e63623. doi: 10.1371/journal.pone.0063623
188. Avettand-Fènoël V, Hocqueloux L, Ghosn J, Cheret A, Frange P, Melard A, et al. Total HIV-1 DNA, a Marker of Viral Reservoir Dynamics With Clinical Implications. Clin Microbiol Rev (2016) 29(4):859–80. doi: 10.1128/CMR.00015-16
189. Viard J-P, Burgard M, Hubert J-B, Aaron L, Rabian C, Pertuiset N, et al. Impact of 5 Years of Maximally Successful Highly Active Antiretroviral Therapy on CD4 Cell Count and HIV-1 DNA Level. AIDS (2004) 18(1):45–9. doi: 10.1097/00002030-200401020-00005
190. Garrigue I, Pellegrin I, Hoen B, Dumon B, Harzic M, Schrive MH, et al. Cell-Associated HIV-1-DNA Quantitation After Highly Active Antiretroviral Therapy-Treated Primary Infection in Patients With Persistently Undetectable Plasma HIV-1 RNA. AIDS (2000) 14(18):2851–5. doi: 10.1097/00002030-200012220-00006
191. Finkelstein JL, Gala P, Rochford R, Glesby MJ, Mehta S. HIV/AIDS and Lipodystrophy: Implications for Clinical Management in Resource-Limited Settings. J Int AIDS Soc (2015) 18(1):19033. doi: 10.7448/IAS.18.1.19033
192. Calvo M, Martinez E. Update on Metabolic Issues in HIV Patients. Curr Opin HIV AIDS (2014) 9(4):332–9. doi: 10.1097/COH.0000000000000075
193. Haubrich RH, Riddler SA, DiRienzo AG, Komarow L, Powderly WG, Klingman K, et al. Metabolic Outcomes in a Randomized Trial of Nucleoside, Nonnucleoside and Protease Inhibitor-Sparing Regimens for Initial HIV Treatment. AIDS (2009) 23(9):1109–18. doi: 10.1097/QAD.0b013e32832b4377
194. Pérez-Matute P, Pérez-Martínez L, Blanco JR, Oteo JA. Role of Mitochondria in HIV Infection and Associated Metabolic Disorders: Focus on Nonalcoholic Fatty Liver Disease and Lipodystrophy Syndrome. Oxid Med Cell Longev (2013) 2013:493413. doi: 10.1155/2013/493413
195. Lagathu C, Bastard J-P, Auclair M, Maachi M, Kornprobst M, Capeau J, et al. Antiretroviral Drugs With Adverse Effects on Adipocyte Lipid Metabolism and Survival Alter the Expression and Secretion of Proinflammatory Cytokines and Adiponectin In Vitro. Antivir Ther (2004) 9(6):911–20.
196. Nolan D, Hammond E, Martin A, Taylor L, Herrmann S, McKinnon E, et al. Mitochondrial DNA Depletion and Morphologic Changes in Adipocytes Associated With Nucleoside Reverse Transcriptase Inhibitor Therapy. AIDS (2003) 17(9):1329–38. doi: 10.1097/00002030-200306130-00007
197. Margolis AM, Heverling H, Pham PA, Stolbach A. A Review of the Toxicity of HIV Medications. J Med Toxicol (2014) 10(1):26–39. doi: 10.1007/s13181-013-0325-8
198. Pinti M, Salomoni P, Cossarizza A. Anti-HIV Drugs and the Mitochondria. Biochim Biophys Acta (2006) 1757(5–6):700–7. doi: 10.1016/j.bbabio.2006.05.001
199. Sulkowski MS, Thomas DL, Mehta SH, Chaisson RE, Moore RD. Hepatotoxicity Associated With Nevirapine or Efavirenz-Containing Antiretroviral Therapy: Role of Hepatitis C and B Infections. Hepatology (2002) 35(1):182–9. doi: 10.1053/jhep.2002.30319
200. Johnson AA, Ray AS, Hanes J, Suo Z, Colacino JM, Anderson KS, et al. Toxicity of Antiviral Nucleoside Analogs and the Human Mitochondrial DNA Polymerase. J Biol Chem (2001) 276(44):40847–57. doi: 10.1074/jbc.M106743200
201. Bischoff J, Gu W, Schwarze-Zander C, Boesecke C, Wasmuth J-C, van Bremen K, et al. Stratifying the Risk of NAFLD in Patients With HIV Under Combination Antiretroviral Therapy (cART). EClinicalMedicine (2021) 40:101116. doi: 10.1016/j.eclinm.2021.101116
202. Duro M, Manso MC, Barreira S, Rebelo I, Medeiros R, Almeida C. Metabolic Syndrome in Human Immunodeficiency Virus-Infected Patients. Int J STD AIDS (2018) 29(11):1089–97. doi: 10.1177/0956462418775188
203. Boothby M, McGee KC, Tomlinson JW, Gathercole LL, McTernan PG, Shojaee-Moradie F, et al. Adipocyte Differentiation, Mitochondrial Gene Expression and Fat Distribution: Differences Between Zidovudine and Tenofovir After 6 Months. Antivir Ther (2009) 14(8):1089–100. doi: 10.3851/IMP1457
204. Carr A, Samaras K, Thorisdottir A, Kaufmann GR, Chisholm DJ, Cooper DA. Diagnosis, Prediction, and Natural Course of HIV-1 Protease-Inhibitor-Associated Lipodystrophy, Hyperlipidaemia, and Diabetes Mellitus: Acohort Study. Lancet (1999) 353(9170):2093–9. doi: 10.1016/S0140-6736(98)08468-2
205. Palios J, Kadoglou NPE, Lampropoulos S. The Pathophysiology of HIV-/HAART-Related Metabolic Syndrome Leading to Cardiovascular Disorders: The Emerging Role of Adipokines. Exp Diabetes Res (2012) 2012:103063. doi: 10.1155/2012/103063
206. Guaraldi G, Squillace N, Stentarelli C, Orlando G, D’Amico R, Ligabue G, et al. Nonalcoholic Fatty Liver Disease in HIV-Infected Patients Referred to a Metabolic Clinic: Prevalence, Characteristics, and Predictors. Clin Infect Dis (2008) 47(2):250–7. doi: 10.1086/589294
207. Xu H, Barnes GT, Yang Q, Tan G, Yang D, Chou CJ, et al. Chronic Inflammation in Fat Plays a Crucial Role in the Development of Obesity-Related Insulin Resistance. J Clin Invest (2003) 112(12):1821–30. doi: 10.1172/JCI19451
208. Bakasis A-D, Androutsakos T. Liver Fibrosis During Antiretroviral Treatment in HIV-Infected Individuals. Truth or Tale? Cells (2021) 10(5). doi: 10.3390/cells10051212
209. Gelpi M, Knudsen AD, Larsen KB, Mocroft A, Lebech A-M, Lindegaard B, et al. Long-Lasting Alterations in Adipose Tissue Density and Adiponectin Production in People Living With HIV After Thymidine Analogues Exposure. BMC Infect Dis (2019) 19(1):708. doi: 10.1186/s12879-019-4347-y
210. Addy CL, Gavrila A, Tsiodras S, Brodovicz K, Karchmer AW, Mantzoros CS. Hypoadiponectinemia Is Associated With Insulin Resistance, Hypertriglyceridemia, and Fat Redistribution in Human Immunodeficiency Virus-Infected Patients Treated With Highly Active Antiretroviral Therapy. J Clin Endocrinol Metab (2003) 88(2):627–36. doi: 10.1210/jc.2002-020795
211. Sankale J-L, Tong Q, Hadigan C, Tan G, Grinspoon S, Kanki P, et al. Regulation of Adiponectin in Adipocytes Upon Exposure to HIV-1. HIV Med (2006) 7(4):268–74. doi: 10.1111/j.1468-1293.2006.00372.x
212. Tsiodras S, Mantzoros C. Leptin and Adiponectin in the HIV Associated Metabolic Syndrome: Physiologic and Therapeutic Implications. Am J Infect Dis (2006) 2(3):141–52. doi: 10.3844/ajidsp.2006.141.152
213. Kim RJ, Wilson CG, Wabitsch M, Lazar MA, Steppan CM. HIV Protease Inhibitor-Specific Alterations in Human Adipocyte Differentiation and Metabolism. Obes (Silver Spring) (2006) 14(6):994–1002. doi: 10.1038/oby.2006.114
214. Lagathu C, Eustace B, Prot M, Frantz D, Gu Y, Bastard J-P, et al. Some HIV Antiretrovirals Increase Oxidative Stress and Alter Chemokine, Cytokine or Adiponectin Production in Human Adipocytes and Macrophages. Antivir Ther (2007) 12:489–500. doi: 10.1177/135965350701200407
215. Majeed Y, Halabi N, Madani AY, Engelke R, Bhagwat AM, Abdesselem H, et al. SIRT1 Promotes Lipid Metabolism and Mitochondrial Biogenesis in Adipocytes and Coordinates Adipogenesis by Targeting Key Enzymatic Pathways. Sci Rep (2021) 11(1):8177. doi: 10.1038/s41598-021-87759-x
216. Mariani S, Di Giorgio MR, Rossi E, Tozzi R, Contini S, Bauleo L, et al. Blood SIRT1 Shows a Coherent Association With Leptin and Adiponectin in Relation to the Degree and Distribution of Adiposity: A Study in Obesity, Normal Weight and Anorexia Nervosa. Nutrients (2020) 12(11):3506. doi: 10.3390/nu12113506
217. Pinzone MR, Cacopardo B, Condorelli F, Di Rosa M, Nunnari G. Sirtuin-1 and HIV-1: An Overview. Curr Drug Targets (2013) 14(6):648–52. doi: 10.2174/1389450111314060005
218. Nassir F, Ibdah JA. Sirtuins and Nonalcoholic Fatty Liver Disease. World J Gastroenterol (2016) 22(46):10084–92. doi: 10.3748/wjg.v22.i46.10084
219. Jurkowska K, Szymańska B, Knysz B, Kuźniarski A, Piwowar A. Sirtuins as Interesting Players in the Course of HIV Infection and Comorbidities. Cells (2021) 10(10):2739. doi: 10.3390/cells10102739
220. Sharma D, Wang J, Fu PP, Sharma S, Nagalingam A, Mells J, et al. Adiponectin Antagonizes the Oncogenic Actions of Leptin in Hepatocellular Carcinogenesis. Hepatology (2010) 52(5):1713–22. doi: 10.1002/hep.23892
221. Unger RH. Lipotoxic Diseases. Annu Rev Med (2002) 53:319–36. doi: 10.1146/annurev.med.53.082901.104057
222. Unger RH, Zhou YT. Lipotoxicity of Beta-Cells in Obesity and in Other Causes of Fatty Acid Spillover. Diabetes (2001) 50(Suppl;1):S118–21. doi: 10.2337/diabetes.50.2007.s118
223. Otero M, Lago R, Gomez R, Dieguez C, Lago F, Gómez-Reino J, et al. Towards a Pro-Inflammatory and Immunomodulatory Emerging Role of Leptin. Rheumatol (Oxf) (2006) 45(8):944–50. doi: 10.1093/rheumatology/kel157
224. Kosmiski L, Kuritzkes D, Lichtenstein K, Eckel R. Adipocyte-Derived Hormone Levels in HIV Lipodystrophy. Antivir Ther (2003) 8:9–15. doi: 10.1177/135965350300800102
225. Kovacic JC, Martin A, Carey D, Wand H, Mallon PW, Feneley MP, et al. Influence of Rosiglitazone on Flow-Mediated Dilation and Other Markers of Cardiovascular Risk in HIV-Infected Patients With Lipoatrophy. Antivir Ther (2005) 10(1):135–43. doi: 10.1177/135965350501000113
226. Lee JH, Chan JL, Sourlas E, Raptopoulos V, Mantzoros CS. Recombinant Methionyl Human Leptin Therapy in Replacement Doses Improves Insulin Resistance and Metabolic Profile in Patients With Lipoatrophy and Metabolic Syndrome Induced by the Highly Active Antiretroviral Therapy. J Clin Endocrinol Metab (2006) 91(7):2605–11. doi: 10.1210/jc.2005-1545
227. Mulligan K, Khatami H, Schwarz J-M, Sakkas GK, DePaoli AM, Tai VW, et al. The Effects of Recombinant Human Leptin on Visceral Fat, Dyslipidemia, and Insulin Resistance in Patients With Human Immunodeficiency Virus-Associated Lipoatrophy and Hypoleptinemia. J Clin Endocrinol Metab (2009) 94(4):1137–44. doi: 10.1210/jc.2008-1588
228. Xu A, Wang Y, Keshaw H, Xu LY, Lam KSL, Cooper GJS. The Fat-Derived Hormone Adiponectin Alleviates Alcoholic and Nonalcoholic Fatty Liver Diseases in Mice. J Clin Invest (2003) 112(1):91–100. doi: 10.1172/JCI17797
229. Berg AH, Combs TP, Du X, Brownlee M, Scherer PE. The Adipocyte-Secreted Protein Acrp30 Enhances Hepatic Insulin Action. Nat Med (2001) 7(8):947–53. doi: 10.1038/90992
230. Kumar P, Smith T, Rahman K, Thorn NE, Anania FA. Adiponectin Agonist ADP355 Attenuates CCl4-Induced Liver Fibrosis in Mice. PloS One (2014) 9(10):e110405. doi: 10.1371/journal.pone.0110405
231. Chruściel P, Sahebkar A, Rembek-Wieliczko M, Serban M-C, Ursoniu S, Mikhailidis DP, et al. Impact of Statin Therapy on Plasma Adiponectin Concentrations: A Systematic Review and Meta-Analysis of 43 Randomized Controlled Trial Arms. Atherosclerosis (2016) 253:194–208. doi: 10.1016/j.atherosclerosis.2016.07.897
232. Polyzos SA, Mantzoros CS. Adiponectin as a Target for the Treatment of Nonalcoholic Steatohepatitis With Thiazolidinediones: A Systematic Review. Metabolism (2016) 65(9):1297–306. doi: 10.1016/j.metabol.2016.05.013
233. Sutinen J, Häkkinen A-M, Westerbacka J, Seppälä-Lindroos A, Vehkavaara S, Halavaara J, et al. Rosiglitazone in the Treatment of Haart-Associated Lipodystrophy – A Randomized Double-Blind Placebo-Controlled Study. Antivir Ther (2003) 8(3):199–207. doi: 10.1177/135965350300800303
234. Mulligan K, Yang Y, Wininger DA, Koletar SL, Parker RA, Alston-Smith BL, et al. Effects of Metformin and Rosiglitazone in HIV-Infected Patients With Hyperinsulinemia and Elevated Waist/Hip Ratio. AIDS (2007) 21(1):47–57. doi: 10.1097/QAD.0b013e328011220e
235. Carr A, Workman C, Carey D, Rogers G, Martin A, Baker D, et al. No Effect of Rosiglitazone for Treatment of HIV-1 Lipoatrophy: Randomised, Double-Blind, Placebo-Controlled Trial. Lancet (Lond Engl) (2004) 363(9407):429–38. doi: 10.1016/S0140-6736(04)15489-5
236. Knight ZA, Hannan KS, Greenberg ML, Friedman JM. Hyperleptinemia Is Required for the Development of Leptin Resistance. PloS One (2010) 5(6):e11376. doi: 10.1371/journal.pone.0011376
237. Guillin OM, Vindry C, Ohlmann T, Chavatte L. Selenium, Selenoproteins and Viral Infection. Nutrients (2019) 11(9):1–33. doi: 10.3390/nu11092101
238. Dworkin BM. Selenium Deficiency in HIV Infection and the Acquired Immunodeficiency Syndrome (AIDS). Chem Biol Interact (1994) 91(2–3):181–6. doi: 10.1016/0009-2797(94)90038-8
239. Domingo P, Gallego-Escuredo JM, Domingo JC, Gutiérrez M, Mateo MG, Fernández I, et al. Serum FGF21 Levels Are Elevated in Association With Lipodystrophy, Insulin Resistance and Biomarkers of Liver Injury in HIV-1-Infected Patients. AIDS (2010) 24(17):2629–37. doi: 10.1097/QAD.0b013e3283400088
240. Eckard AR, Hughes HY, Hagood NL, O’Riordan MA, Labbato D, Kosco JC, et al. Fibroblast Growth Factor 21 Is Elevated in HIV and Associated With Interleukin-6. JAIDS J Acquir Immune Defic Syndr (2020) 83(5):e30–3. doi: 10.1097/QAI.0000000000002285
241. Urraza-Robledo AI, Giralt M, González-Galarza FF, Villarroya F, Miranda Pérez AA, Ruiz Flores P, et al. FGF21 Serum Levels Are Related to Insulin Resistance, Metabolic Changes and Obesity in Mexican People Living With HIV (PLWH). PloS One (2021) 16(5):e0252144. doi: 10.1371/journal.pone.0252144
242. Benedini S, Luzi L. Lipodystrophy HIV-Related and FGF21: A New Marker to Follow the Progression of Lipodystrophy? J Transl Intern Med (2016) 4(4):150–4. doi: 10.1515/jtim-2016-0026
243. Kamin D, Hadigan C, Lehrke M, Mazza S, Lazar MA, Grinspoon S. Resistin Levels in Human Immunodeficiency Virus-Infected Patients With Lipoatrophy Decrease in Response to Rosiglitazone. J Clin Endocrinol Metab (2005) 90(6):3423–6. doi: 10.1210/jc.2005-0287
244. Ranade K, Geese WJ, Noor M, Flint O, Tebas P, Mulligan K, et al. Genetic Analysis Implicates Resistin in HIV Lipodystrophy. AIDS (2008) 22(13):1561–8. doi: 10.1097/QAD.0b013e32830a9886
245. Mårtensson UEA, Maria Fenyö E, Olde B, Owman C. Characterization of the Human Chemerin Receptor – ChemR23/CMKLR1 – as Co-Receptor for Human and Simian Immunodeficiency Virus Infection, and Identification of Virus-Binding Receptor Domains. Virology (2006) 355(1):6–17. doi: 10.1016/J.VIROL.2006.07.010
246. Torretta S, Colombo G, Travelli C, Boumya S, Lim D, Genazzani AA, et al. The Cytokine Nicotinamide Phosphoribosyltransferase (eNAMPT; PBEF; Visfatin) Acts as a Natural Antagonist of C-C Chemokine Receptor Type 5 (CCR5). Cells (2020) 9(2):496. doi: 10.3390/cells9020496
247. Schindler K, Haider D, Wolzt M, Rieger A, Gmeinhart B, Luger A, et al. Impact of Antiretroviral Therapy on Visfatin and Retinol-Binding Protein 4 in HIV-Infected Subjects. Eur J Clin Invest (2006) 36(9):640–6. doi: 10.1111/j.1365-2362.2006.01699.x
248. Freitas P, Carvalho D, Santos AC, Madureira AJ, Martinez E, Pereira J, et al. Adipokines, Hormones Related to Body Composition, and Insulin Resistance in HIV Fat Redistribution Syndrome. BMC Infect Dis (2014) 14(1):347. doi: 10.1186/1471-2334-14-347
249. Koutkia P, Meininger G, Canavan B, Breu J, Grinspoon S. Metabolic Regulation of Growth Hormone by Free Fatty Acids, Somatostatin, and Ghrelin in HIV-Lipodystrophy. Am J Physiol Endocrinol Metab (2004) 286(2):E296–303. doi: 10.1152/ajpendo.00335.2003
250. Mao Y, Cheng J, Yu F, Li H, Guo C, Fan X. Ghrelin Attenuated Lipotoxicity via Autophagy Induction and Nuclear Factor-κb Inhibition. Cell Physiol Biochem (2015) 37(2):563–76. doi: 10.1159/000430377
251. Falasca K, Manigrasso MR, Racciatti D, Zingariello P, Dalessandro M, Ucciferri C, et al. Associations Between Hypertriglyceridemia and Serum Ghrelin, Adiponectin, and IL-18 Levels in HIV-Infected Patients. Ann Clin Lab Sci (2006) 36(1):59–66.
252. Tietge UJF, Böker KHW, Manns MP, Bahr MJ. Elevated Circulating Adiponectin Levels in Liver Cirrhosis Are Associated With Reduced Liver Function and Altered Hepatic Hemodynamics. Am J Physiol Endocrinol Metab (2004) 287(1):E82–9. doi: 10.1152/ajpendo.00494.2003
253. Chiang C-H, Lai J-S, Hung S-H, Lee L-T, Sheu J-C, Huang K-C. Serum Adiponectin Levels Are Associated With Hepatitis B Viral Load in Overweight to Obese Hepatitis B Virus Carriers. Obesity (2013) 21(2):291–6. doi: 10.1002/oby.20000
254. Nagy GS, Tsiodras S, Martin LD, Avihingsanon A, Gavrila A, Hsu WC, et al. Human Immunodeficiency Virus Type 1-Related Lipoatrophy and Lipohypertrophy Are Associated With Serum Concentrations of Leptin. Clin Infect Dis (2003) 36(6):795–802. doi: 10.1086/367859
255. Sinha U, Sinharay K, Sengupta N, Mukhopadhyay P. Benefits of Leptin Therapy in HIV Patients. Indian J Endocrinol Metab (2012) 16(Suppl 3):S637–43. doi: 10.4103/2230-8210.105583
256. Zhang Q-Y, Xu X, Luo M, Xue J-J, Li Y-L. Serum Leptin Levels in Patients With Hepatitis B: A Meta-Analysis. In: Proceedings of the 2017 2nd International Conference on Biological Sciences and Technology (BST 2017). Paris, France: Atlantis Press (2018). doi: 10.2991/bst-17.2018.1
257. Ataseven H, Bahcecioglu IH, Kuzu N, Yalniz M, Celebi S, Erensoy A, et al. The Levels of Ghrelin, Leptin, TNF-α, and IL-6 in Liver Cirrhosis and Hepatocellular Carcinoma Due to HBV and HDV Infection. Mediators Inflamm (2006) 2006:1–6. doi: 10.1155/MI/2006/78380
258. Meng Z, Zhang Y, Wei Z, Liu P, Kang J, Zhang Y, et al. High Serum Resistin Associates With Intrahepatic Inflammation and Necrosis: An Index of Disease Severity for Patients With Chronic HBV Infection. BMC Gastroenterol (2017) 17(1):6. doi: 10.1186/s12876-016-0558-5
259. Ataseven H, Bahcecioglu IH, Kuzu N, Yalniz M, Celebi S, Erensoy A, et al. The Levels of Ghrelin, Leptin, TNF-Alpha, and IL-6 in Liver Cirrhosis and Hepatocellular Carcinoma Due to HBV and HDV Infection. Mediators Inflamm (2006) 2006(4):78380. doi: 10.1155/MI/2006/78380
260. Wu L, Pan Q, Wu G, Qian L, Zhang J, Zhang L, et al. Diverse Changes of Circulating Fibroblast Growth Factor 21 Levels in Hepatitis B Virus-Related Diseases. Sci Rep (2017) 7(1):16482. doi: 10.1038/s41598-017-16312-6
261. Li L, Gu X, Fang M, Ji J, Yi C, Gao C. The Diagnostic Value of Serum Fucosylated Fetuin A in Hepatitis B Virus-Related Liver Diseases. Clin Chem Lab Med (2016) 54(4):693–701. doi: 10.1515/cclm-2015-0307
262. Li J-J, Yin H-K, Guan D-X, Zhao J-S, Feng Y-X, Deng Y-Z, et al. Chemerin Suppresses Hepatocellular Carcinoma Metastasis Through CMKLR1-PTEN-Akt Axis. Br J Cancer (2018) 118(10):1337–48. doi: 10.1038/s41416-018-0077-y
263. Tsai I-T, Wang C-P, Yu T-H, Lu Y-C, Lin C-W, Lu L-F, et al. Circulating Visfatin Level Is Associated With Hepatocellular Carcinoma in Chronic Hepatitis B or C Virus Infection. Cytokine (2017) 90:54–9. doi: 10.1016/j.cyto.2016.10.007
264. Baum MK, Shor-Posner G, Lai S, Zhang G, Lai H, Fletcher MA, et al. High Risk of HIV-Related Mortality Is Associated With Selenium Deficiency. J Acquir Immune Defic Syndr Hum Retrovirol (1997) 15(5):370–4. doi: 10.1097/00042560-199708150-00007
265. Gladyshev VN, Stadtman TC, Hatfield DL, Jeang KT. Levels of Major Selenoproteins in T Cells Decrease During HIV Infection and Low Molecular Mass Selenium Compounds Increase. Proc Natl Acad Sci USA (1999) 96(3):835–9. doi: 10.1073/pnas.96.3.835
266. Campa A, Shor-Posner G, Indacochea F, Zhang G, Lai H, Asthana D, et al. Mortality Risk in Selenium-Deficient HIV-Positive Children. J Acquir Immune Defic Syndr Hum Retrovirol (1999) 20(5):508–13. doi: 10.1097/00042560-199904150-00015
267. Yi Y-S, Park SG, Byeon SM, Kwon Y-G, Jung G. Hepatitis B Virus X Protein Induces TNF-Alpha Expression via Down-Regulation of Selenoprotein P in Human Hepatoma Cell Line, HepG2. Biochim Biophys Acta (2003) 1638(3):249–56. doi: 10.1016/s0925-4439(03)00090-5
268. Cheng Z, Lin P, Cheng N. HBV/HIV Coinfection: Impact on the Development and Clinical Treatment of Liver Diseases. Front Med (2021) 8:713981. doi: 10.3389/fmed.2021.713981
269. Iser DM, Avihingsanon A, Wisedopas N, Thompson AJ, Boyd A, Matthews GV, et al. Increased Intrahepatic Apoptosis But Reduced Immune Activation in HIV-HBV Co-Infected Patients With Advanced Immunosuppression. AIDS (2011) 25(2):197–205. doi: 10.1097/QAD.0b013e3283410ccb
270. Colin JF, Cazals-Hatem D, Loriot MA, Martinot-Peignoux M, Pham BN, Auperin A, et al. Influence of Human Immunodeficiency Virus Infection on Chronic Hepatitis B in Homosexual Men. Hepatology (1999) 29(4):1306–10. doi: 10.1002/hep.510290447
271. Parvez MK. HBV and HIV Co-Infection: Impact on Liver Pathobiology and Therapeutic Approaches. World J Hepatol (2015) 7(1):121. doi: 10.4254/wjh.v7.i1.121
272. Thio CL. Hepatitis B. And Human Immunodeficiency Virus Coinfection. Hepatology (2009) 49(S5):S138–45. doi: 10.1002/hep.22883
273. Wandeler G, Gsponer T, Bihl F, Bernasconi E, Cavassini M, Kovari H, et al. Hepatitis B Virus Infection Is Associated With Impaired Immunological Recovery During Antiretroviral Therapy in the Swiss HIV Cohort Study. J Infect Dis (2013) 208(9):1454–8. doi: 10.1093/infdis/jit351
274. Malagnino V, Teti E, Compagno M, Coppola L, Salpini R, Svicher V, et al. HBcAb Positivity Is a Risk Factor for an Increased Detectability of HIV RNA After Switching to a Two-Drug Regimen Lamivudine-Based (2DR-3TC-Based) Treatment: Analysis of a Multicenter Italian Cohort. Microorganisms (2021) 9(2):396. doi: 10.3390/microorganisms9020396
275. Gürtler LG. Effect of Antiretroviral HIV Therapy on Hepatitis B Virus Replication and Pathogenicity. Intervirology (2014) 57(3–4):212–7. doi: 10.1159/000360942
276. Hong M-Z, Huang W-Q, Min F, Xu J-C, Lin Z, Fang K-N, et al. Enhanced HBsAg Synthesis Correlates With Increased Severity of Fibrosis in Chronic Hepatitis B Patients. Lin W, Editor. PloS One (2014) 9(1):e87344. doi: 10.1371/journal.pone.0087344
277. Li Y-W, Yang F-C, Lu H-Q, Zhang J-S. Hepatocellular Carcinoma and Hepatitis B Surface Protein. World J Gastroenterol (2016) 22(6):1943–52. doi: 10.3748/wjg.v22.i6.1943
278. Gaul S, Leszczynska A, Alegre F, Kaufmann B, Johnson CD, Adams LA, et al. Hepatocyte Pyroptosis and Release of Inflammasome Particles Induce Stellate Cell Activation and Liver Fibrosis. J Hepatol (2021) 74(1):156–67. doi: 10.1016/J.JHEP.2020.07.041
279. Yu X, Lan P, Hou X, Han Q, Lu N, Li T, et al. HBV Inhibits LPS-Induced NLRP3 Inflammasome Activation and IL-1β Production via Suppressing the NF-κb Pathway and ROS Production. J Hepatol (2017) 66(4):693–702. doi: 10.1016/J.JHEP.2016.12.018
280. Zhang J, Ling N, Lei Y, Peng M, Hu P, Chen M. Multifaceted Interaction Between Hepatitis B Virus Infection and Lipid Metabolism in Hepatocytes: A Potential Target of Antiviral Therapy for Chronic Hepatitis B. Front Microbiol (2021) 12:636897. doi: 10.3389/fmicb.2021.636897
281. Bar-Yishay I, Shaul Y, Shlomai A. Hepatocyte Metabolic Signalling Pathways and Regulation of Hepatitis B Virus Expression. Liver Int (2011) 31(3):282–90. doi: 10.1111/j.1478-3231.2010.02423.x
282. Shi Y-X, Huang C-J, Yang Z-G. Impact of Hepatitis B Virus Infection on Hepatic Metabolic Signaling Pathway. World J Gastroenterol (2016) 22(36):8161–7. doi: 10.3748/wjg.v22.i36.8161
283. Kim KH, Shin H-J, Kim K, Choi HM, Rhee SH, Moon H-B, et al. Hepatitis B Virus X Protein Induces Hepatic Steatosis via Transcriptional Activation of SREBP1 and PPARgamma. Gastroenterology (2007) 132(5):1955–67. doi: 10.1053/j.gastro.2007.03.039
284. Wang B, Li W, Fang H, Zhou H. Hepatitis B Virus Infection Is Not Associated With Fatty Liver Disease: Evidence From a Cohort Study and Functional Analysis. Mol Med Rep (2018) 19(1):320–6. doi: 10.3892/mmr.2018.9619
285. Waris G, Huh KW, Siddiqui A. Mitochondrially Associated Hepatitis B Virus X Protein Constitutively Activates Transcription Factors STAT-3 and NF-Kappa B via Oxidative Stress. Mol Cell Biol (2001) 21(22):7721–30. doi: 10.1128/MCB.21.22.7721-7730.2001
286. Becker SA, Lee TH, Butel JS, Slagle BL. Hepatitis B Virus X Protein Interferes With Cellular DNA Repair. J Virol (1998) 72(1):266–72. doi: 10.1128/JVI.72.1.266-272.1998
287. Lin C, Huang X, Liu H, Wang Y. Interactions of Hepatitis B Virus Infection With Nonalcoholic Fatty Liver Disease: Possible Mechanisms and Clinical Impact. Dig Dis Sci (2015) 60(12):3513–24. doi: 10.1007/s10620-015-3772-z
288. Machado MV, Oliveira AG, Cortez-Pinto H. Hepatic Steatosis in Hepatitis B Virus Infected Patients: Meta-Analysis of Risk Factors and Comparison With Hepatitis C Infected Patients. J Gastroenterol Hepatol (2011) 26(9):1361–7. doi: 10.1111/j.1440-1746.2011.06801.x
289. Chiang C-H, Yang H-I, Jen C-L, Lu S-N, Wang L-Y, You S-L, et al. Association Between Obesity, Hypertriglyceridemia and Low Hepatitis B Viral Load. Int J Obes (Lond) (2013) 37(3):410–5. doi: 10.1038/ijo.2012.63
290. Wang C-H, Chen C-J, Lee M-H, Yang H-I, Hsiao CK. Chronic Hepatitis B Infection and Risk of Atherosclerosis-Related Mortality: A 17-Year Follow-Up Study Based on 22,472 Residents in Taiwan. Atherosclerosis (2010) 211(2):624–9. doi: 10.1016/j.atherosclerosis.2010.03.008
291. Zhu L, Jiang J, Zhai X, Baecker A, Peng H, Qian J, et al. Hepatitis B Virus Infection and Risk of Non-Alcoholic Fatty Liver Disease: A Population-Based Cohort Study. Liver Int (2019) 39(1):70–80. doi: 10.1111/liv.13933
292. Shi J, Fan J, Wu R, Gao X, Zhang L, Wang H, et al. Prevalence and Risk Factors of Hepatic Steatosis and its Impact on Liver Injury in Chinese Patients With Chronic Hepatitis B Infection. J Gastroenterol Hepatol (2008) 23(9):1419–25. doi: 10.1111/j.1440-1746.2008.05531.x
293. Wong GL-H, Wong VW-S, Choi PC-L, Chan AW-H, Chim AM-L, Yiu KK-L, et al. Metabolic Syndrome Increases the Risk of Liver Cirrhosis in Chronic Hepatitis B. Gut (2009) 58(1):111–7. doi: 10.1136/gut.2008.157735
294. Hsieh P-S, Hsieh Y-J. Impact of Liver Diseases on the Development of Type 2 Diabetes Mellitus. World J Gastroenterol (2011) 17(48):5240–5. doi: 10.3748/wjg.v17.i48.5240
295. Jarcuska P, Drazilova S, Fedacko J, Pella D, Janicko M. Association Between Hepatitis B and Metabolic Syndrome: Current State of the Art. World J Gastroenterol (2016) 22(1):155–64. doi: 10.3748/wjg.v22.i1.155
296. Wang C-C, Tseng T-C, Kao J-H. Hepatitis B Virus Infection and Metabolic Syndrome: Fact or Fiction? J Gastroenterol Hepatol (2015) 30(1):14–20. doi: 10.1111/jgh.12700
297. Rastogi A, Sakhuja P, Kumar A, Hissar S, Jain A, Gondal R, et al. Steatosis in Chronic Hepatitis B: Prevalence and Correlation With Biochemical, Histologic, Viral, and Metabolic Parameters. Indian J Pathol Microbiol (2011) 54(3):454–9. doi: 10.4103/0377-4929.85074
298. Maponga TG, Andersson MI, van Rensburg CJ, Arends JE, Taljaard J, Preiser W, et al. HBV and HIV Viral Load But Not Microbial Translocation or Immune Activation Are Associated With Liver Fibrosis Among Patients in South Africa. BMC Infect Dis (2018) 18(1):214. doi: 10.1186/s12879-018-3115-8
299. Proal AD V-EM.. Pathogens Hijack Host Cell Metabolism: Intracellular Infection as a Driver of the Warburg Effect in Cancer and Other Chronic Inflammatory Conditions. Immunometabolism (2021) 3(1):e210003. doi: 10.20900/immunometab20210003
300. Dai X-H, Zhang P, Xiao M-F, Zhou R-R, Zhang B-X, Hu G-S, et al. Protective Role of α2hs-Glycoprotein in HBV-Associated Liver Failure. Int J Mol Sci (2011) 12:3846–56. doi: 10.3390/ijms12063846
301. Zhang P, Shen H, Huang J, Wang H, Zhang B, Zhou R, et al. Intraperitoneal Administration of Fetuin-A Attenuates D-Galactosamine/Lipopolysaccharide-Induced Liver Failure in Mouse. Dig Dis Sci (2014) 59(8):1789–97. doi: 10.1007/s10620-014-3071-0
302. Schaap FG, Kremer AE, Lamers WH, Jansen PLM, Gaemers IC. Fibroblast Growth Factor 21 Is Induced by Endoplasmic Reticulum Stress. Biochimie (2013) 95(4):692–9. doi: 10.1016/j.biochi.2012.10.019
303. Woo YC, Xu A, Wang Y, Lam KSL. Fibroblast Growth Factor 21 as an Emerging Metabolic Regulator: Clinical Perspectives. Clin Endocrinol (Oxf) (2013) 78(4):489–96. doi: 10.1111/cen.12095
304. Luo Y, Ye S, Chen X, Gong F, Lu W, Li X. Rush to the Fire: FGF21 Extinguishes Metabolic Stress, Metaflammation and Tissue Damage. Cytokine Growth Factor Rev (2017) 38:59–65. doi: 10.1016/J.CYTOGFR.2017.08.001
305. Zhang J, Li J, Ma J, Wang H, Yi Y. Human Fibroblast Growth Factor-21 Serves as a Predictor and Prognostic Factor in Patients With Hepatitis B Cirrhosis Combined With Adrenal Insufficiency. Exp Ther Med (2018) 15(4):3189–96. doi: 10.3892/etm.2018.5840
306. Yang C, Lu W, Lin T, You P, Ye M, Huang Y, et al. Activation of Liver FGF21 in Hepatocarcinogenesis and During Hepatic Stress. BMC Gastroenterol (2013) 13:67. doi: 10.1186/1471-230X-13-67
307. Li H, Dong K, Fang Q, Hou X, Zhou M, Bao Y, et al. High Serum Level of Fibroblast Growth Factor 21 Is an Independent Predictor of Non-Alcoholic Fatty Liver Disease: A 3-Year Prospective Study in China. J Hepatol (2013) 58(3):557–63. doi: 10.1016/j.jhep.2012.10.029
308. Emanuelli B, Vienberg SG, Smyth G, Cheng C, Stanford KI, Arumugam M, et al. Interplay Between FGF21 and Insulin Action in the Liver Regulates Metabolism. J Clin Invest (2014) 124(2):515–27. doi: 10.1172/JCI67353
309. Manolakopoulos S, Bethanis S, Liapi C, Stripeli F, Sklavos P, Margeli A, et al. An Assessment of Serum Leptin Levels in Patients With Chronic Viral Hepatitis: A Prospective Study. BMC Gastroenterol (2007) 7:17. doi: 10.1186/1471-230X-7-17
310. Hui C-K, Zhang H-Y, Lee NP, Chan W, Yueng Y-H, Leung K-W, et al. Serum Adiponectin Is Increased in Advancing Liver Fibrosis and Declines With Reduction in Fibrosis in Chronic Hepatitis B. J Hepatol (2007) 47(2):191–202. doi: 10.1016/j.jhep.2007.02.023
311. Wu D, Li H, Xiang G, Zhang L, Li L, Cao Y, et al. Adiponectin and its Receptors in Chronic Hepatitis B Patients With Steatosis in China. Hepat Mon (2013) 13(4):e6065. doi: 10.5812/hepatmon.6065
312. Yoon S, Jung J, Kim T, Park S, Chwae Y-J, Shin H-J, et al. Adiponectin, a Downstream Target Gene of Peroxisome Proliferator-Activated Receptor γ, Controls Hepatitis B Virus Replication. Virology (2011) 409(2):290–8. doi: 10.1016/j.virol.2010.10.024
313. Abenavoli L, Luigiano C, Guzzi PH, Milic N, Morace C, Stelitano L, et al. Serum Adipokine Levels in Overweight Patients and Their Relationship With Non-Alcoholic Fatty Liver Disease. Panminerva Med (2014) 56(2):189–93.
314. Haberl EM, Feder S, Pohl R, Rein-Fischboeck L, Dürholz K, Eichelberger L, et al. Chemerin Is Induced in Non-Alcoholic Fatty Liver Disease and Hepatitis B-Related Hepatocellular Carcinoma. Cancers (Basel) (2020) 12(10):2967. doi: 10.3390/cancers12102967
315. Tsochatzis E, Papatheodoridis GV, Hadziyannis E, Georgiou A, Kafiri G, Tiniakos DG, et al. Serum Adipokine Levels in Chronic Liver Diseases: Association of Resistin Levels With Fibrosis Severity. Scand J Gastroenterol (2008) 43(9):1128–36. doi: 10.1080/00365520802085387
Keywords: non-alcoholic fatty liver disease, hepatitis B virus, HIV, hepatokines, adipokines, oxidative stress, metabolic syndrome, antiretroviral treatment
Citation: Iacob SA and Iacob DG (2022) Non-Alcoholic Fatty Liver Disease in HIV/HBV Patients – a Metabolic Imbalance Aggravated by Antiretroviral Therapy and Perpetuated by the Hepatokine/Adipokine Axis Breakdown. Front. Endocrinol. 13:814209. doi: 10.3389/fendo.2022.814209
Received: 12 November 2021; Accepted: 10 January 2022;
Published: 09 March 2022.
Edited by:
Mikiko Watanabe, Sapienza University of Rome, ItalyReviewed by:
Zheng Li, Jiangsu Normal University, ChinaEvanthia Tourkochristou, University of Patras, Greece
Copyright © 2022 Iacob and Iacob. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Diana Gabriela Iacob, ZGlhbmFnaWFjb2JAZ21haWwuY29t
†These authors have contributed equally to this work