 Roxana Marino
Roxana Marino Angélica Moresco
Angélica Moresco Natalia Perez Garrido
Natalia Perez Garrido Pablo Ramirez
Pablo Ramirez Alicia Belgorosky
Alicia Belgorosky
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
MINI REVIEW article
Front. Endocrinol. , 25 February 2022
Sec. Cellular Endocrinology
Volume 13 - 2022 | https://doi.org/10.3389/fendo.2022.803226
This article is part of the Research Topic Molecular-Genetic Causes Underlying Primary Adrenal Insufficiency: Current Insights into Diagnosis and Treatments View all 11 articles
Congenital adrenal hyperplasia (CAH) secondary to 21-hydroxylase deficiency is an autosomal recessive disorder. The 21-hydroxylase enzyme P450c21 is encoded by the CYP21A2 gene located on chromosome 6p21.33 within the HLA major histocompatibility complex. This locus also contains the CYP21A1P, a non-functional pseudogene, that is highly homologous to the CYP21A2 gene. Other duplicated genes are C4A and C4B, that encode two isoforms of complement factor C4, the RP1 gene that encodes a serine/threonine protein kinase, and the TNXB gene that, encodes the extracellular matrix glycoprotein tenascin-X (TNX). TNX plays a role in collagen deposition by dermal fibroblasts and is expressed in the dermis of the skin and the connective tissue of the heart and skeletal muscle. During meiosis, misalignment may occur producing large gene deletions or gene conversion events resulting in chimeric genes. Chimeric recombination may occur between TNXB and TNXA. Three TNXA/TNXB chimeras have been described that differ in the junction site (CH1 to CH3) and result in a contiguous CYP21A2 and TNXB gene deletion, causing CAH-X syndrome. TNXB deficiency is associated with Ehlers Danlos syndrome (EDS). EDS comprises a clinically and genetically heterogeneous group of connective tissue disorders. As molecular analysis of the TNXB gene is challenging, the TNX-deficient type EDS is probably underdiagnosed. In this minireview, we will address the different strategies of molecular analysis of the TNXB-gene, as well as copy number variations and genetic status of TNXB in different cohorts. Furthermore, clinical features of EDS and clinical recommendations for long-term follow-up are discussed.
Congenital adrenal hyperplasia (CAH) comprises a group of autosomal recessive enzymatic disorders, caused by a deficiency of one of the enzymes required for cortisol biosynthesis in the adrenal cortex. CAH is mostly associated with pathogenic variants in the 21-hydroxylase (CYP21A2) gene (1, 2). Residual enzyme activity defines the clinical severity of the disease. The most common form of CAH is 21-hydroxylase deficiency (21-OHD) accounting for 95% of cases. Prevalence of the most severe or classic forms is 1:16,000 live births in the Caucasian population, while the non-classic or late-onset form is the most common, with a prevalence between 1:1000-1:500 live births depending on ethnicity and geographic area (1–3).
The CYP21A2 gene is located on the long arm of chromosome 6, within the major human histocompatibility complex (HLA), a region with a complex gene organization (4–7) (Figure 1). There is a nonfunctional pseudogene (CYP21A1P), located approximately 30 kb from the CYP21A2 gene. Both the functional gene and the pseudogene comprise ten exons that share a 98% nucleotide sequence identity (4–7). The close proximity and high level of homology between the functional and the non-functional gene facilitates misalignment resulting in recombination events that frequently produce large CYP21A2 gene deletions and conversions as well as point pathogenic variants in the CYP21A2 gene. The locus, is one of the most complex in the human genome since it contains three other genes, RP1 also called STK19 that encodes a serine/threonine protein kinase, C4A and C4B that encode two isoforms of complement factor C4, and TNXB that encodes an extracellular matrix glycoprotein tenascin-X (TNX), as well as two pseudogenes, RP2 and TNXA, that together constitute a 30-kb genetic unit called the RCCX module (4–7). The genetic diversity of the RCCX module is highly attributable to nonallelic homologous recombination (NAHR). Unequal crossover during meiosis generates large structural rearrangements and copy number changes, whereas gene conversion mediates relatively short sequence transfers (8).
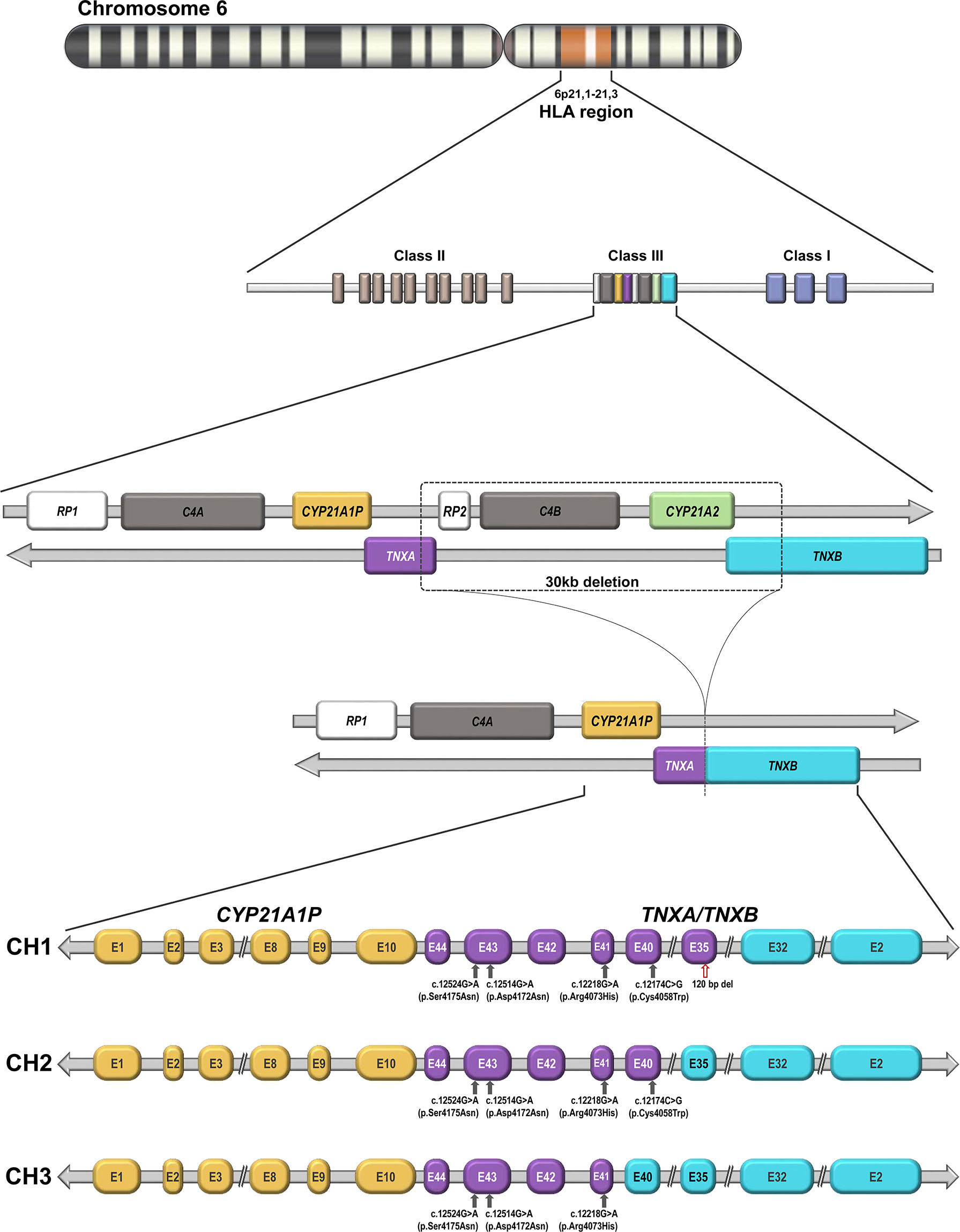
Figure 1 Genomic organization of the CYP21 locus on chromosome 6p21.1-21.3: The functional CYP21A2 gene and its non-functional CYP21A1P pseudogene are arranged in tandem repeat with the two C4 genes that encode factor four of the complement system, the serine–threonine nuclear protein kinase active gene RP1 (STK19) and a truncated pseudogene RP2 (STK19P). The functional TNXB and the TNXA pseudogene are located on the complement strand. Schematic representation of a 30-kb deletion as a result of unequal crossover during meiosis and the formation of the three described TNXA/TNXB chimeras (CH1, CH2 and CH3), which differ in the junction site. CH1 is characterized by a 120-bp deletion in exon and intron 35 carried over from TNXA pseudogene sequences. CH2 lacks this deletion but contains a pseudogene-derived variant c.12174C>G (p.Cys4058Trp) in TNXB exon 40. CH3 is characterized by the presence of any variant of a cluster of three pseudogene-derived variants (exon 41: c.12218G>A, p.Arg4073His; exon 43: c.12514G>A, p.Asp4172Asn and c.12524G>A, p.Ser4175Asn). These three pseudogene-derived variants do not always co-segregate in the three chimeras. TNXB gene exons are represented in cyan and TNXA gene exons in purple. The red arrow indicates a 120-bp deletion in exon 35 and grey arrows indicate different pseudogene-derived variants. CH1: TNXA/TNXB chimera 1. CH2: TNXA/TNXB chimera 2. CH3: TNXA/TNXB chimera 3.
The TNXA and TNXB genes lie on the opposite strand of DNA from the other genes of the cluster and therefore have the opposite transcriptional direction. The last exon of TNXA and TNXB partially overlap the 3′ untranslated region of exon 10 in CYP21A1P and CYP21A2, respectively (9). TNXB is a large gene composed of 44 exons spanning 68.2 kb, whereas TNXA is a truncated gene of 4.5kb, homologous to exons 32 to 44 of TNXB.
As mentioned above, chimeric CYP21A1P/CYP21A2 genes are caused by homologous recombination between CYP21A2 and its pseudogene CYP21A1P as a result of unequal crossover and are found in 20-25% of alleles in CAH due to 21-OHD. To date, nine different CYP21A1P/CYP21A2 genes have been characterized (10).
The unequal crossover may, in some cases, produce TNXA/TNXB chimeras from which the CYP21A2 gene is completely removed (Figure 1). At present three different TNXA/TNXB chimeras have been described- CH1, CH2 and CH3-that differ in the junction site. CH1 is characterized by a 120-bp deletion in exon and intron 35 carried over from TNXA pseudogene sequences leading to haploinsufficiency (11). CH2 lacks this deletion but contains a pseudogene-derived variant- c.12174C>G (p.Cys4058Trp)- in TNXB exon 40 producing the loss of a critical disulfide bond in the tertiary structure of the TNX C-terminal fibrinogen-like domain (12). CH3 is characterized by the presence of any variant of a cluster of three pseudogene-derived variants -exon 41: c.12218G>A, (p.Arg4073His); exon 43: c.12514G>A (p.Asp4172Asn) and c.12524G>A (p.Ser4175Asn)-. The cluster of these three pseudogene-derived variants may differ in the haplotypes found in the three chimeras and they do not always co-segregate. Modeling and energy calculations suggest that the p.Arg4073His variant is detrimental to proper TNX folding while the remaining variants in the cluster did not significantly affect the folding energies in the models (13) . In addition, some CH1 haplotypes that harbor the 120-bp deletion in exon 35 but lack the p.Cys4058Trp variant in exon 40 have been found. This may be explained by the fact that the allele frequency of pseudogene derived-variants is not 100 percent. Since CH2 and CH3 produce altered proteins rather than reducing TNX expression, they are associated with a dominant-negative effect. The TNX protein belongs to a family of evolutionarily conserved large glycoproteins of the extracellular matrix. It plays a role in collagen deposition by dermal fibroblasts and is expressed in the dermis of the skin and in the connective tissue of the heart and skeletal muscle. TNXB deficiency leads to Ehlers-Danlos Syndrome (EDS) and up to 10% of CAH patients also have EDS, an entity called CAH-X. EDS comprises a clinically and genetically heterogeneous group of connective tissue disorders characterized by joint hypermobility, skin hyperextensibility, and tissue fragility (14).
As molecular analysis of the TNXB gene is challenging, the TNX-deficient type EDS is probably underdiagnosed. In this minireview, we will address the different strategies of molecular analysis of the TNXB gene, as well as copy number variations and genetic status of TNXB in different cohorts. Furthermore, clinical features of EDS and clinical recommendations for long-term follow-up are discussed.
The first report of TNX deficiency was a description of a patient with CAH and EDS (15). In 2001, Schalkwijk et al. reported a subtype of EDS that is now known as classic-like EDS (clEDS) (16). clEDS is an autosomal recessive form of EDS and is caused by a deficiency of TNX encoded by the TNXB gene. The authors evaluated 151 patients with the classic hypermobility or vascular types of EDS, together with 168 patients with other conditions (psoriasis and rheumatoid arthritis) and 21 healthy individuals for the presence of TNX and tenascin-C by enzyme-linked immunosorbent assay. The patients were tested for the 30-kb deletion leading to a TNXA/TNXB chimeric gene by allele-specific PCR and for other point mutations by sequencing the coding sequence of the TNXB gene. Four of five TNX–deficient patients were identified to have homozygous TNXB mutations. Subsequently, the same authors reported an association between haploinsufficiency of the TNXB gene and the hypermobility-type EDS (hEDS) in 20 heterozygous family members; however, generalized joint hypermobility (GJH) was observed in only nine female patients (17).
The first evaluation of the potential clinical implications of TNXB heterozygosity in CAH patients was reported by Merke et al. in a large prospective observational study from the National Institutes of Health (NIH) (11). One hundred ninety-three unrelated patients with CAH were evaluated clinically for manifestations of EDS and genetically for TNXB mutations. DNA was analyzed for the presence of a contiguous gene deletion syndrome caused by deletion of CYP21A2 and its flanking gene TNXB by PCR multiplex ligation-dependent probe amplification (MLPA) and confirmed by Southern blot. This deletion generated a TNXA/TNXB chimera characterized by a 120-bp deletion in exon 35 of the TNXB gene, which is replaced by TNXA sequences. In addition, TNXB sequencing was performed in a group of patients with one or more joint or skin manifestations. Heterozygosity for the TNXB deletion was observed in 7% of CAH patients, who were considered to have CAH-X syndrome. Here, the association between the hypermobility phenotype and TNXB-gene haploinsufficiency was established. In 2015, the same authors identified a pseudogene-derived variant- c.12174C>G (p.C4058W)- representing a novel TNXA/TNXB chimera that did not involve a 120-bp deletion in exon 35 in seven families with CAH-X (12). Interestingly, this variant did not affect the protein expression of tenascin in dermal fibroblasts and for this missense variant a dominant-negative mechanism was proposed, which is different from the haploinsufficiency caused by the above-described chimera. Of 246 CAH probands screened, 14 carried previously described TNXA/TNXB chimeras (CH1) while seven unrelated patients carried the novel TNXB variant (CH2) resulting in a prevalence of CAH-X of 8.5% (12). The same authors later reported three patients with biallelic CAH-X and identified a novel dominant-negative chimera (CH3) characterized by any of three TNXB variants [exon 41: c.12218G>A (p.Arg4073His); 191 exon 43: c.12514G>A (p.Asp4172Asn) and c.12524G>A (p.Ser4175Asn)] (Figure 1). This study presented evidence for disrupted TNX function, since by computational data the p.Arg4073His variant was predicted to reduce protein-folding energy by interfering with a cation-pi interaction between p.Arg4073 and p.Phe4080 (13).
Molecular analysis of the TNXB gene is challenging due to the presence of the pseudogene, which makes next-generation sequencing highly complicated in these cases. In 2019, Lao et al. reported a high-throughput CAH-X screening method based on allele-specific PCR to assess the copy numbers of TNXB exons 35 and 40. The method is compatible with either quantitative PCR or droplet digital PCR and allows detection of CH1 and CH2. Using this methodology, the authors found a 15.6% prevalence of CAH-X, which was higher than previously estimated. The prevalence was especially high (29.2%) in subjects with a 30-kb deletion genotype (18).
In 2020, Gao et al. assessed the prevalence of the chimeric TNXA/TNXB gene and clinical symptoms in a Chinese cohort of 424 21-OHD patients. MLPA analysis and Sanger sequencing was performed to genetically identify the CAH-X syndrome. In this cohort, 14% of the patients with 21-OHD were found to have a chimeric TNXA/TNXB gene (19). Finally, also in 2020, our group reported the molecular TNXB-gene status and clinical evaluation of the EDS phenotype in a cohort of 337 Argentine 21-OHD patients to assess the prevalence of this condition in our population. TNXB gene analysis was performed in 66 unrelated CAH patients that were carriers of the 30-kb CYP21A2 gene deletion. A molecular strategy based on MLPA and Sanger sequencing analysis was developed for the detection of the three previously described TNXA/TNXB chimeras (20). The overall prevalence of CAH-X in 21-OHD patients in our cohort was 14%, which was similar to that previously found in the large cohort from the NIH and in the Chinese population (15% and 14%, respectively). In our population of 21-OHD patients carrying the 30-kb CYP21A2 gene deletion in which the junction site was downstream exon 7 both in the homozygous or the heterozygous state, the incidence of TNXA/TNXB chimeras was 73% (48/66), similar to the prevalence of 62.8% (59/94) found in the Chinese population. On the other hand, in the NIH cohort a prevalence of 29.2% (21/72) was reported. The reason for the lower prevalence found in the latter study is that the authors reported the presence of TNXA/TNXB chimeras in CAH patients that were carriers of all types of 30-kb CYP21A2 deletions described.
In addition to TNXA/TNXB chimeras, pathogenic variants in the TNXB gene have been described as a less frequent cause of TNX deficiency. Pathogenic variants were detected in the coding region of the EGF-like repeats, the fibronectin type III domain or C-terminal domain structurally related to fibrinogen of the TNX protein. Moreover, recently a splice donor site variant has been described as a cause of the hEDS type (21). Finally, the variable prevalence of CAH-X reported in the different cohorts might be related to the molecular strategies used.
CAH-X patients are reported to have a wide range of connective tissue abnormalities, including generalized joint hypermobility, subluxations, chronic arthralgias, soft or velvety skin, mild skin hyperextensibility, and variable systemic manifestations. The severity of the phenotype may be correlated with the dosage of the dominant alleles, as monoallelic CAH-X is associated with hEDS, the mildest and most common EDS type, and biallelic CAH-X with the more severe clEDS subtype.
Biallelic CAH-X patients resemble the clEDS type, with extreme joint laxity, with or without recurrent joint dislocations, and skin hyperextensibility with a velvety skin texture and absence of atrophic scarring. Easily bruisable skin and soft-tissue injuries as well as organ prolapse, pes planus, piezogenic papules, chronic pain, arthralgias, and cardiac abnormalities have been described. Thus far, 12 patients with biallelic CAH-X syndrome have been reported (13, 15, 16, 19, 20); however, clinical information is not available for all cases. Our group reported four biallelic CAH-X patients (two with a CH1/CH1 and two with a CH1/CH2 combination) (20). Both CH1/CH2 patients had a more severe EDS phenotype, with greater skin involvement. Nevertheless, the low number of homozygous patients reported to date limits the possibility to draw robust conclusions based on these data. In addition, in the latter patients cardiac defects were detected; one had an atrial septal and the other a mild pulmonary valve defect. None of the patients developed either atrophic scarring, organ prolapse, or any other complications during the 3 years of follow-up; however, these observations are limited by the young age of our patients and the short-term follow-up. Chen et al. reported three biallelic patients of 14, 19, and 29 years of age, all displaying unique combinations of TNXB variants in both alleles; one of them was homozygous for CH2/CH2, the other was a CH2/CH3 compound, and the third a CH2/CH1 compound (13). All of them had skin hyperextensibility and significant joint hypermobility. Joint laxity was extreme and two patients had a history of joint dislocations, chronic arthralgias, and chronic tendinitis and/or bursitis. Unlike our findings, the authors reported widened atrophic scarring, rectal prolapse, severe gastroesophageal reflux, high palate, and elongated uvula in all three biallelic patients. Mild ventricular enlargement was detected in two patients. Currently, the limited number of cases reported and the heterogeneous combination of TNXB variants they display make it difficult to establish a certain genotype/phenotype correlation.
Long-term follow-up is needed to specifically evaluate quality of life in CAH-X patients. On the other hand, clEDS patients (without CAH) are affected by soft-tissue fragility and long-term complications (13, 15, 16, 19, 20).
Most patients with the monoallelic form of CAH-X syndrome present with the clinical spectrum of hEDS with variable expression at different stages of life, predominantly characterized by GJH, mild skin hyperextensibility, and soft velvety skin, without abnormal scarring. Related musculoskeletal complications, such as recurrent joint dislocations, pes planus, and chronic arthralgias, have been reported. Other associated features include functional gastrointestinal alterations and cardiac disorders. Although this type of EDS may cause chronic pain and reduced quality of life, life-threatening complications are uncommon (11, 12, 15, 17, 19, 20).
The underlying chimera translates into different degrees of hEDS phenotypes. Compared to TNXB haploinsufficiency caused by CAH-X CH1, a dominant negative effect related to CAH-X CH2 causes a more severe phenotype with increased joint and skin manifestations (12, 20). Gastrointestinal disorders, such as chronic gastroesophageal reflux and irritable bowel syndrome, hernias, and organ prolapse, are also more frequently reported in patients that are heterozygous for CAH-X CH2 than in those with CAH-X CH1. Data on the phenotype associated with the less frequent CAH-X CH3 are scarce (12, 19, 20, 22–24). Recently, Gao et al. reported the presence of CH3 in 11 patients; however, clinical information was available for only one of them, who had joint hypermobility and poor wound healing (19). In our cohort, CH3 was only found in one monoallelic patient who was not available for clinical evaluation (20).
Although the exact role of CAH-related hormonal factors and/or chronic glucocorticoid treatment in connective tissue dysplasia is not yet completely understood, it has been shown that CAH-X patients are consistently more severely affected than patients with homozygous or heterozygous TNX-deficient-type EDS without CAH (11, 12, 16, 19, 25).
Furthermore, a phenotype that is varied and usually milder than that of monoallelic CAH-X patients has been reported in the relatives of CAH-X patients, who were carriers of one CAH-X allele, but not affected by CAH. Some carriers were observed to have less joint, heart, or gastrointestinal symptoms, while others were asymptomatic (12, 13, 17, 19, 20). On the other hand, connective tissue dysplasia has been described in CAH patients without demonstrated TNXB deficiency (11, 12).
Recently, Lao et al. reported a novel cause of CAH-X syndrome, not associated with pathogenic TNXA/TNXB chimeras but due to a TNXB splice donor site variant (21). As currently TNXB testing remains challenging, at least in the routine diagnostic approach, the diagnosis of CAH-X still relies on clinical evaluation.
Connective tissue dysplasia should be evaluated in all CAH patients, especially in those harboring a deletion in the CYP21A2 gene. Screening for GJH and other soft tissue features presents age-related difficulties and should therefore be adapted to age, systematically evaluated, and retrospectively asked for. The Beighton Score (shown in Figure 2) remains the most objective assessment tool to measure GJH (22, 26).
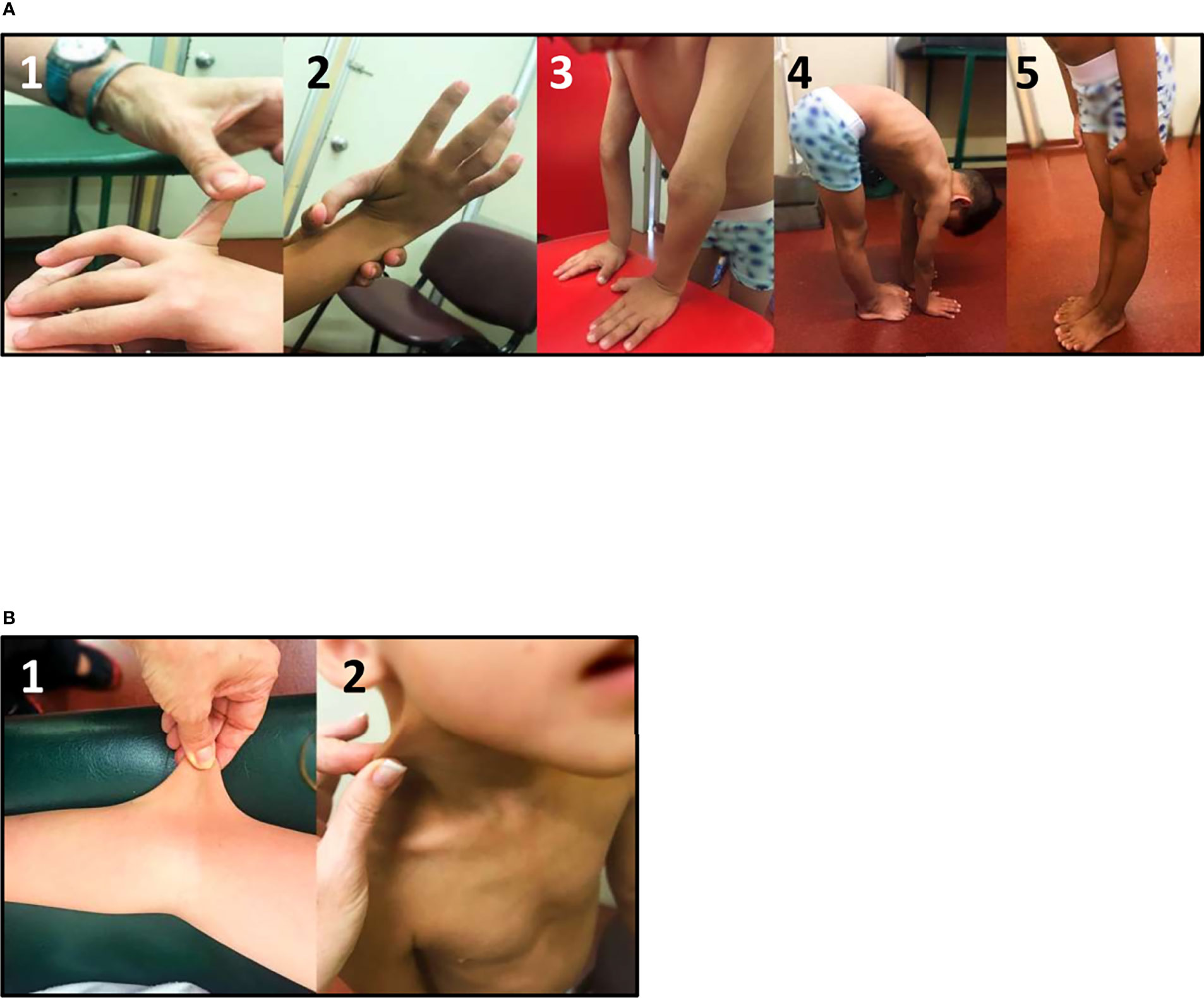
Figure 2 Clinical evaluation of CAH-X Syndrome patients: (A) Generalized joint hypermobility Beighton score: 1) fifth finger extension test, 2) wrist flexion thumb abduction test, 3) elbow extension test, 4) trunk and hip flexion test, and 5) knee extension test. (B) Skin extensibility can be measured by lifting the cutaneous and subcutaneous layers of the skin and is considered hyperextensible if it can be stretched over 1.5 cm at the forearm and the dorsum of hands, and 3 cm for neck, elbow, and knees. Photographs shown belong to a biallelic CAH-X patient (compound heterozygous for CH1 and CH2).
Knowledge of the TNXB status in CAH will not only offer patients a better understanding of their symptomatology with the burden of the diagnosis of a second genetic disease, but also assures specific clinical management with a focus on preventing musculoskeletal manifestations and complications. Cardiovascular alterations may be underreported in CAH-X patients, probably related to the difficulties patients without cardiac symptoms may have to access specific diagnostic studies, such as cardiac magnetic resonance imaging (MRI), outside the context of a research study protocol, as observed in different series. A variable prevalence of cardiac abnormalities was found in different studies using different diagnostic methods.
Currently, no specific medical or genetic therapies are available for CAH-X patients. Management consists of interdisciplinary medical care, rehabilitation, and monitoring of major and organ-specific complications (24, 27, 28). Further TNXB and CAH-X studies are necessary to define detailed surveillance guidelines for these and other long-term complications and to develop prevention strategies.
Different studies on the CYP21A2 gene have improved our knowledge on TNXB-related disorders. The TNXB gene encodes an extracellular matrix glycoprotein named TNX. EDS may be due to TNXB deficiency and up to 10% of 21-OHD CAH patients also have CAH-X. Chimeric recombination of the TNXB and TNXA genes may occur, and three TNXA/TNXB chimeras that differ in the junction site (CH1 to CH3) resulting in a contiguous CYP21A2 and TNXB gene deletion, named CAH-X syndrome, have been described. Molecular studies are the gold standard to assess the presence of TNXA/TNXB chimeras. On the other hand, molecular analysis of the TNXB gene is challenging due to the presence of a pseudogene and next generation sequencing is highly complicated in these cases. For this reason, among others, TNX-deficient type EDS may be underdiagnosed. The variable prevalence of CAH-X reported in different cohorts may be related to the molecular strategies applied. Systematic study of TNXB status in individuals with a previous diagnosis of CAH and carriers of the complete 30-kb deletion of CYP21A2 is highly recommended. Moreover, molecular genetic testing of CAH-21OHD should include TNXA/TNXB chimera analysis (29).
EDS comprises a clinically and genetically heterogeneous group of connective tissue disorders characterized by joint hypermobility, skin hyperextensibility, and tissue fragility as well as cardiovascular alterations. Cardiac disorders, in particular heart valve abnormalities, may be underdiagnosed, probably because of the young age of the majority of reported CAH-X patients and EDS-related cardiac abnormalities may appear with aging. In addition, it is unlikely that patients without heart symptoms are routinely checked for cardiac abnormalities with echocardiogram and/or cardiac MRI and currently only data from patients participating in specific CAH-X research studies are available. Severity of the phenotype may be correlated with the dosage of the dominant alleles, as monoallelic CAH-X patients have the mildest and most common EDS type and biallelic CAH-X patients the more severe clEDS subtype. CAH-X patients are consistently more severely affected than patients with homozygous or heterozygous TNX-deficient type EDS without CAH; however, the impact of the hormonal milieu on TNX and its role in connective-tissue pathophysiology is still poorly understood. In this line, carriers of one CAH-X allele who are not affected with CAH have a varied and milder EDS phenotype than monoallelic CAH-X patients.
Once the diagnosis of CAH-X has been established, it is advisable to guarantee long-term follow-up of these patients by medical specialists with a focus on preventing musculoskeletal manifestations and complications.
Finally, in order to prevent long-term musculoskeletal disorders, timely diagnosis of CAH-X is important and physical therapy for joint instability is recommended. In addition, molecular characterization of CAH-X is relevant for genetic counseling.
Written informed consent was obtained from the participant’s legal guardians for the publication of any identifiable material in this study.
All authors contributed equally to design of the manuscript. RM, AM, and AB wrote the first draft of the manuscript. All authors contributed to manuscript revision, read, and approved the submitted version.
Fondo Nacional de Ciencia y Tecnología, Argentina, Director of the Grant, Award number: 2016, 0028.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Speiser PW, White PC. Congenital Adrenal Hyperplasia. N Engl J Med (2003) 349(8):776–88. doi: 10.1056/NEJMra021561
2. Speiser PW, Arlt W, Auchus RJ, Baskin LS, Conway GS, Merke DP, et al. Congenital Adrenal Hyperplasia Due to Steroid 21-Hydroxylase Deficiency: An Endocrine Society Clinical Practice Guideline. J Clin Endocrinol Metab (2018) 103(11):4043–88. doi: 10.1210/jc.2018-01865
3. Witchel SF, Azziz R. Nonclassic Congenital Adrenal Hyperplasia. Int J Pediatr Endocrinol (2010) 2010:1–11. doi: 10.1155/2010/625105
4. Higashi Y, Yoshioka H, Yamane M, Gotoh O, Fujii-Kuriyama Y. Complete Nucleotide Sequence of Two Steroid 21-Hydroxylase Genes Tandemly Arranged in Human Chromosome: A Pseudogene and a Genuine Gene. Proc Natl Acad Sci USA (1986) 387 83(9):2841–5. doi: 10.1073/pnas.83.9.2841
5. Yang Z, Mendoza AR, Welch TR, Zipf WB, Yu CY. Modular Variations of the Human Major Histocompatibility Complex Class III Genes for Serine/Threonine Kinase RP, Complement Component C4, Steroid 21-Hydroxylase CYP21, and Tenascin TNX (the RCCX Module). A Mechanism for Gene Deletions and Disease Associations. J Biol Chem (1999) 274(17):12147–56. doi: 10.1074/jbc.274.17.12147
6. Pignatelli D, Carvalho BL, Palmeiro A, Barros A, Guerreiro SG and Macut D. The Complexities in Genotyping of Congenital Adrenal Hyperplasia: 21-Hydroxylase 395 Deficiency. Front Endocrinol (2019) 10:432. doi: 10.3389/fendo.2019.00432
7. Blanchong CA, Zhou B, Rupert KL, Chung EK, Jones KN, Sotos JF, et al. Deficiencies of Human Complement Component C4A and C4B and Heterozygosity in Length Variants of RP-C4-CYP21-TNX (RCCX) Modules in Caucasians. The Load of RCCX Genetic Diversity on Major Histocompatibility Complex-Associated Disease. J Exp Med (2000) 191(12):2183–96. doi: 10.1084/jem.191.12.2183
8. Carrozza C, Foca L, De Paolis E, Concolino P. Genes and Pseudogenes: Complexity of the RCCX Locus and Disease. Front Endocrinol (Lausanne) (2021) 12:709758:709758. doi: 10.3389/fendo.2021.709758
9. Morel Y, Bristow J, Gitelman SE, Miller WL. Transcript Encoded on the Opposite Strand of the Human Steroid 21-Hydroxylase Complement Component C4 Gene Locus. Proc Natl Acad Sci U.S.A. (1989) 86(17):6582–6. doi: 10.1073/pnas.86.17.6582
10. Chen W, Xu Z, Sullivan A, Finkielstain GP, Van Ryzin C, Merke DP, et al. Junction Site Analysis of Chimeric CYP21A1P/CYP21A2 Genes in 21-Hydroxylase Deficiency. Clin Chem (2012) 58(2):421–30. doi: 10.1373/clinchem.2011.174037
11. Merke DP, Chen W, Morissette R, Xu Z, Van Ryzin C, Sachdev V, et al. Tenascin- X Haploinsufficiency Associated With Ehlers-Danlos Syndrome in Patients With Congenital Adrenal Hyperplasia. J Clin Endocrinol Metab (2013) 98(2):E379–87. doi: 10.1210/jc.2012-3148
12. Morissette R, Chen W, Perritt AF, Dreiling JL, Arai AE, Sachdev V, et al. Broadening the Spectrum of Ehlers Danlos Syndrome in Patients With Congenital Adrenal Hyperplasia. J Clin Endocrinol Metab (2015) 100(8):E1143–52. doi: 10.1210/jc.2015-2232
13. Chen W, Perritt A, Morissette R, Dreiling J, Bohn M, Mallappa A, et al. Ehlers- Danlos Syndrome Caused by Biallelic TNXB Variants in Patients With Congenital Adrenal Hyperplasia. Brief Report. Hum Mutat (2016) 37(9):893–7. doi: 10.1002/humu.23028
14. Bristow J, Carey W, Egging D, Schalkwijk J. Tenascin-X, Collagen, Elastin, and the Ehlers-Danlos Syndrome. Am J Med Genet C Semin Med Genet (2005) 139C(1):24–30. doi: 10.1002/ajmg.c.30071
15. Burch GH, Gong Y, Liu W, Dettman RW, Curry CJ, Smith L, et al. Tenascin-X Deficiency is Associated With Ehlers-Danlos Syndrome. Nat Genet (1997) 17(1):104–8. doi: 10.1038/ng0997-104
16. Schalkwijk J, Zweers MC, Steijlen PM, Dean WB, Taylor G, van Vlijmen IM, et al. A Recessive Form of the Ehlers-Danlos Syndrome Caused by Tenascin-X Deficiency. N Engl J Med (2001) 345(16):1167–75. doi: 10.1056/NEJMoa002939
17. Zweers MC, Bristow J, Steijlen PM, Dean WB, Hamel BC, Otero M, et al. Haploinsufficiency of TNXB is Associated With Hypermobility Type of Ehlers-Danlos Syndrome. Am J Hum Genet (2003) 73(1):214–7. doi: 10.1086/376564
18. Lao Q, Brookner B, Merke DP. High-Throughput Screening for CYP21A1P- TNXA/TNXB Chimeric Genes Responsible for Ehlers-Danlos Syndrome in Patients With Congenital Adrenal Hyperplasia. J Mol Diagn (2019) 21(5):924–31. doi: 10.1016/j.jmoldx.2019.06.001
19. Gao Y, Lu L, Yu B, Mao J, Wang X, Nie M, et al. The Prevalence of the Chimeric TNXA/TNXB Gene and Clinical Symptoms of Ehlers-Danlos Syndrome With 21- Hydroxylase Deficiency. Clin Endocrinol Metab (2020) 105(7):dgaa199. doi: 10.1210/clinem/dgaa199
20. Marino R, Perez Garrido N, Ramirez P, Notaristéfano G, Moresco A, Touzon MS, et al. Ehlers-Danlos Syndrome: Molecular and Clinical Characterization of TNXA/TNXB Chimeras in Congenital Adrenal Hyperplasia. J Clin Endocrinol Metab (2021) 106(7):e2789–802. doi: 10.1210/clinem/dgab033
21. Lao Q, Mallappa A, Rueda Faucz F, Joyal E, Veeraraghavan P, Chen W, et al. A TNXB Splice Donor Site Variant as a Cause of Hypermobility Type Ehlers-Danlos Syndrome in Patients With Congenital Adrenal Hyperplasia. Mol Genet Genomic Med (2021) 9:e1556. doi: 10.1002/mgg3.1556
22. Malfait F, Francomano C, Byers P, Belmont J, Berglund B, Black J, et al. The 2017 International Classification of the Ehlers–Danlos Syndromes. Am J Med Genet Part C Semin Med Genet (2017) 175C:8–26. doi: 10.1002/ajmg.c.31552
23. Forghani I. Updates in Clinical and Genetics Aspects of Hypermobile Ehlers Danlos Syndrome. Balkan Med J (2019) 36(1):12–6. doi: 10.4274/balkanmedj.2018.1113
24. Brady AF, Demirdas S, Fournel-Gigleux S, Ghali N, Giunta C, Kapferer-Seebacher I, et al. The Ehlers-Danlos Syndromes, Rare Types. Am J Med Genet C Semin Med Genet (2017) 175(1):70–115. doi: 10.1002/ajmg.c.31550
25. Demirdas S, Dulfer E, Robert L, Kempers M, van Beek D, Micha D, et al. Recognizing the Tenascin-X Deficient Type of Ehlers-Danlos Syndrome: A Cross-Sectional Study in 17 Patients. Clin Genet (2017) 91(3):411–25. doi: 10.1111/cge.12853
26. Beighton P, De Paepe A, Steinmann B, Tsipouras P, Wenstrup RJ. Ehlers-Danlos Syndromes: Revised Nosology, Villefranche, 1997. Ehlers-Danlos National Foundation (USA) and Ehlers-Danlos Support Group (Uk). Am J Med Genet (1998) 77(1):31–7. doi: 10.1002/(sici)1096-8628(19980428)77:1<31::aid-ajmg8>3.0.co;2-o
27. Bowen JM, Sobey GJ, Burrows NP, Colombi M, Lavallee ME, Malfait F, et al. Ehlers-Danlos Syndrome, Classical Type. Am J Med Genet C Semin Med Genet (2017) 175(1):27–39. doi: 10.1002/ajmg.c.31548
28. Engelbert RH, Juul-Kristensen B, Pacey V, de Wandele I, Smeenk S, Woinarosky N, et al. The Evidence-Based Rationale for Physical Therapy Treatment of Children, Adolescents, and Adults Diagnosed With Joint Hypermobility Syndrome/Hypermobile Ehlers Danlos Syndrome. Am J Med Genet C Semin Med Genet (2017) 175(1):158–67. doi: 10.1002/ajmg.c.31545
Keywords: congenital adrenal hyperplasia, CAH-X, CYP21A2, TNXB, Ehlers-Danlos Syndrome
Citation: Marino R, Moresco A, Perez Garrido N, Ramirez P and Belgorosky A (2022) Congenital Adrenal Hyperplasia and Ehlers-Danlos Syndrome. Front. Endocrinol. 13:803226. doi: 10.3389/fendo.2022.803226
Received: 27 October 2021; Accepted: 31 January 2022;
Published: 25 February 2022.
Edited by:
Liliana Dain, Centro Nacional de Genética Médica, ArgentinaReviewed by:
Tony Yuen, Icahn School of Medicine at Mount Sinai, United StatesCopyright © 2022 Marino, Moresco, Perez Garrido, Ramirez and Belgorosky. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Alicia Belgorosky, YWJlbGdvMTIzNDVAZ21haWwuY29t
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.