 Jishang Huang1
Jishang Huang1 Guanglin Ji
Guanglin Ji Tiansheng Zheng
Tiansheng Zheng- 1Department of Orthopedics, First Affiliated Hospital of Gannan Medical University, Ganzhou, China
- 2College of Pharmacy, Gannan Medical University, Ganzhou, China
- 3Department of Orthopedics, Shangyou Hospital of traditional Chinese Medicine, Ganzhou, China
Endoplasmic reticulum (ER) is an important player in various intracellular signaling pathways that regulate cellular functions in many diseases. Intervertebral disc degeneration (IDD), an age-related degenerative disease, is one of the main clinical causes of low back pain. Although the pathological development of IDD is far from being fully elucidated, many studies have been shown that ER stress (ERS) is involved in IDD development and regulates various processes, such as inflammation, cellular senescence and apoptosis, excessive mechanical loading, metabolic disturbances, oxidative stress, calcium homeostasis imbalance, and extracellular matrix (ECM) dysregulation. This review summarizes the formation of ERS and the potential link between ERS and IDD development. ERS can be a promising new therapeutic target for the clinical management of IDD.
1. Introduction
In recent decades, low back pain has been regarded as the main cause of disability, which brings heavy mental pressure and economic burden on patients (1, 2). An epidemiological survey finds that the direct medical cost of low back pain is as high as $100 billion in the United States, without considering the indirect costs caused by the decline of patients’ labor capacity (3). It is well known that intervertebral disc degeneration (IDD) is one of the leading causes of low back pain, and the prevalence of IDD increases dramatically with aging. Studies have been shown that more than 90% of people over 50 have IDD (4). Although the etiology and mechanism of IDD are still being explored, a large number of studies indicate that the pathological development of IDD can be attributed to various factors, such as excessive mechanical load, insufficient nutritional supply, metabolic disorders, immune mechanism, and inflammation.
IDD is a pathological process in which the intervertebral disc (IVD) degenerates with aging, resulting from a complex interaction between environmental and genetic factors. Currently, many therapeutic strategies are available for the treatment of IDD, mainly including drug treatment and surgical therapy. However, most of these strategies have limitations (5). Early drug treatment can only slightly reduce patients’ suffering, and very few drugs are approved against IDD pathogenesis and for disc protection. Arthroplasty with artificial disc replacement for surgical treatment has been introduced with the notion that it may decrease the risk of adjacent segment disease (6). Although the degenerated IVD can be removed surgically, some patients after surgical therapy may suffer from long-term post-spine syndromes and lumbar pain due to the destruction of the original biological structure (7). In addition, a discectomy may accelerate the progress of disc degeneration (8).
The degenerative process of IVD, demonstrated by the pathological changes at the molecular, cellular, and tissue levels, is orchestrated by a variety of signaling pathways. Endoplasmic reticulum (ER) plays an important role in maintaining the normal physiological structure and exerting physiological functions in IVD. Endoplasmic reticulum stress (ERS) can be induced by abnormal accumulation of unfolded or misfolded proteins and be involved in the mechanism of musculoskeletal disorders, including IDD (9–12), osteoarthritis (13), osteoporosis (14), rheumatoid arthritis (15), and muscular dystrophy (16). Particularly, some studies have been shown that ERS is associated with the pathogenesis of IDD by mediating oxidative stress, calcium homeostasis imbalance, inflammatory responses, excessive mechanical loading, and metabolic disturbance. In this review, we will specifically focus on the activation of ERS and its role in the pathological development of IDD, as well as discussion on the potential of reducing ERS as a therapeutic target against IDD.
2. The pathological development of IDD
Anatomically, IVD is a cylindrical avascular connective tissue located between two adjacent vertebral bodies. It is composed of a central nucleus pulposus (NP), a peripheral annulus fibrosus (AF), and a cartilage endplate (CEP) and plays a vital role in maintaining normal spine height and physiological curvature and coordinating spine movement. The extracellular matrix (ECM) provides a microenvironment for IVD cells and it maintains a dynamic balance between synthesis and degradation under normal conditions and ensures the physiological functions of IVD. Proteoglycans and collagens are the main biological macromolecules in ECM. Once the molecular dysregulation and structural abnormalities in ECM occur, the degradation and loss of proteoglycan will be triggered and the earliest pathological changes are initiated during the pathological development of IDD. It has been demonstrated IDD development is influenced by various factors, such as excessive mechanical loading, hypoxia, metabolic disorders, immune dysregulation, and inflammation (17–23). The pathological changes include a progressive decrease in nutrient supply, alterations in ECM composition, and cell apoptosis in IVD. The imbalance between the anabolism and catabolism of IVD cells can be reflected by the changes in ECM components, including the decrease of proteoglycan content and the replacement of type II by type I collagen fiber. More importantly, studies have been shown that the degenerated IVD cells are often accompanied by abnormal gene and protein expression of matrix proteases, such as MMPs and a disintegrin and metalloproteinase with thrombospondin motifs (ADAMTSs), through the possible mechanisms in mediating oxidative stress, inflammatory responses, and acidic metabolites (24–26).
3. Activation of ERS contributes to IDD pathological development
3.1. ERS and the unfolded protein responses
ER is a three-dimensional tubular network structure composed of small tubes, vesicles, and flat sacs. As the largest organelle in eukaryotic cells, ER is involved in the folding of proteins, the post-translational modification of various proteins, and a reservoir for intracellular calcium ions (27). Almost all proteins on the endomembrane system are synthesized in ER membrane-bound ribosomes and followed by post-translational folding in the ER lumen (28). According to the metabolic needs, cells regulate ER’s functions to ensure the accuracy of protein folding. Although the folding activity in the ER is well maintained, misfolding of nascent peptide chains and defects in protein assembly are unavoidable. Occasionally, some generated residues are recognized by the transporters and polyubiquitinated in the cytoplasm by the proteases for degradation, which is called ER-associated degradation (ERAD) (29).
ERS occurs when the protein processing is blocked or the misfolded proteins accumulate in the ER (30). The stability of ER internal environment is a prerequisite for the realization of functions. When ER is overwhelmed, there will be a large accumulation of unfolded or misfolded proteins. When the accumulated proteins exceed the folding and degradation capacity of ER, ERS will be initiated. Many factors are involved in the induction of ERS, such as inflammatory responses, excessive mechanical loading, and metabolic disorders.
When ER is stressed, cells sense the accumulation of the unfolded or misfolded proteins and restore ER homeostasis through three signaling pathways (31). This regulation is called unfolded protein response (UPR) (32), which is an adaptive response against the accumulation of the unfolded or misfolded proteins. Cells relieve ERS by reducing protein synthesis, promoting protein degradation and efflux, and inducing the synthesis of molecular chaperones to enhance the protein folding capacity. Interestingly, UPR also increases the area of the ER membrane by increasing the expression of lipid metabolism-related genes, thereby expanding the ER capacity (33, 34). Ultimately, if ERS is overwhelmed or prolonged beyond the cell’s adaptive capacity, UPR will trigger cell apoptosis (31, 35).
The UPR regulates the folding processes of proteins through three ER-localized transmembrane proteins, protein kinase R (PKR)-like endoplasmic reticulum kinase (PERK), inositol-requiring enzyme-1α (IRE1α), and activating transcription factor 6 (ATF6) (Figure 1) (33, 36). Normally, the N-termini of these ER transmembrane proteins are tightly bound to the ER molecular chaperone GRP78/BiP to maintain their inactive forms. However, when the unfolded protein accumulates in the ER, GRP78/BiP is ectopically released, and phosphorylation of these ER transmembrane proteins results from a conformational change.
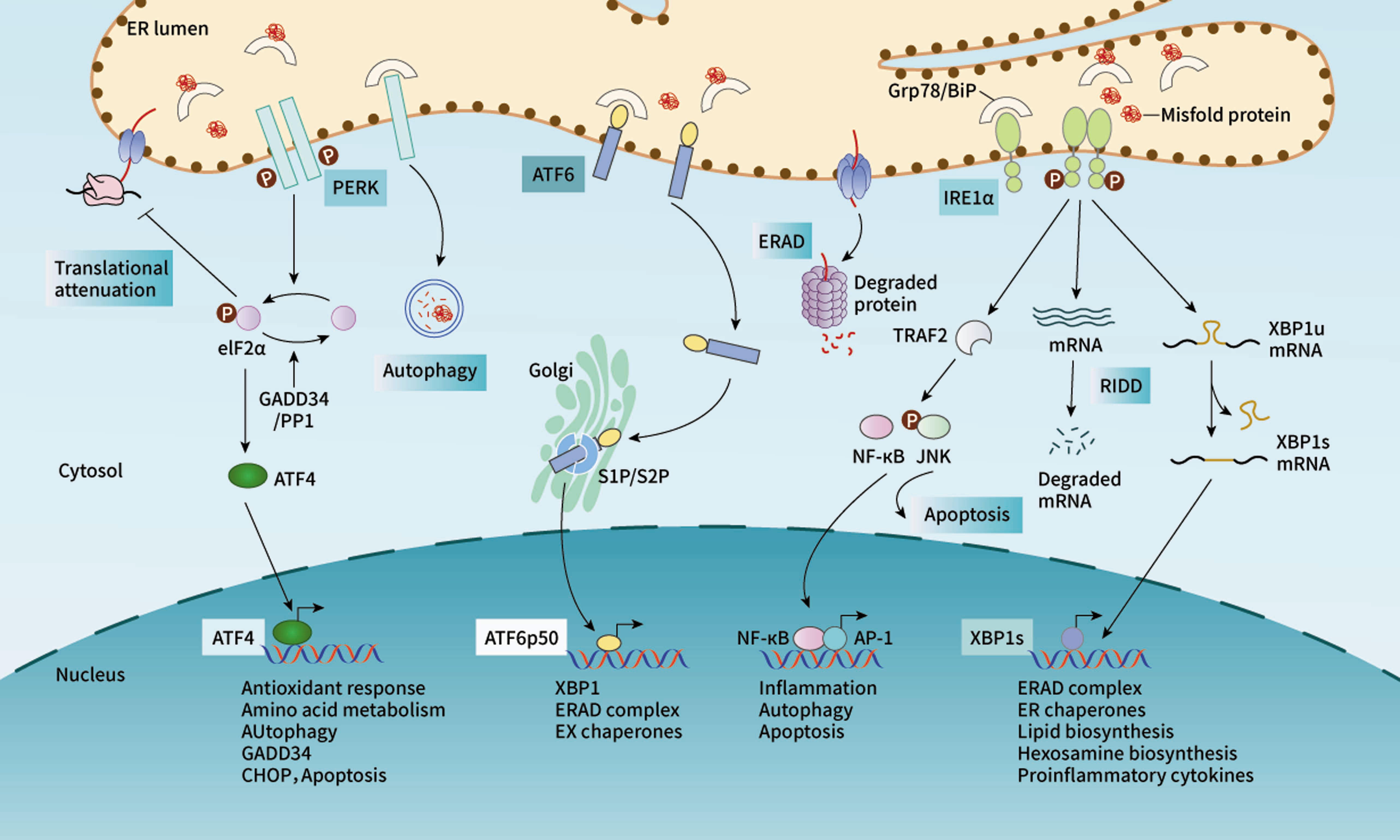
Figure 1 The UPR regulates the protein folding processes by activating three signaling pathways. Under stress, the release of GRP78 induces activation of PERK, which phosphorylates eIF2α and then upregulates the expression of ATF4. As a transcriptional factor, ATF4 enters the nucleus and mediates the expression of downstream factors. Similarly, IRE1α triggers the splicing activity to produce spliced XBP1, which is also a transcriptional factor in regulating the target gene expression. In addition, IRE1α may also initiate cell apoptosis by activating TRAF2/JNK signaling. ATF6 is transported to Golgi, where it is cleaved and activated.
PERK is an ER-resident Ser/Thr protein kinase (37) and its catalytic domain shares high homology with the kinases in the eukaryotic initiation factor 2α (eIF2α) family. Once GRP78 is released, PERK is phosphorylated and its kinase domain is activated (37, 38). Activated PERK phosphorylates the protein translation initiation factor eIF2α, which up regulates the expression of ATF4. Additionally, ATF4 may drive the expression of the downstream target gene CHOP and control the encoding of apoptosis-related genes (39). IRE1α is a bifunctional transmembrane kinase/endoribonuclease (RNase) to exhibit dual enzymatic activities. When GRP78 is released, IRE1α dimerizes and stimulates autophosphorylation, inducing a conformational change to activate the ribonuclease domain. IRE1α determines cell fate through unconventional splicing of mRNA and regulated IRE1α-dependent decay (RIDD). When the UPR occurs, the XBP1 mRNA becomes the direct target of the endonuclease action of IRE1α. The spliced XBP-1 can promote the expression of UPR target molecules containing ERS response elements, such as GRP78. IRE1α initiates RIDD for the UPR target genes selectively to enhance ERS intensity (40). Once the intensity reaches the threshold, RIDD initiates mitochondria-dependent apoptosis (41–46). In addition, activated IRE1α also induces the phosphorylation of c-Jun N-terminal kinase (JNK), which leads to cell apoptosis (42). Compared with PERK and IRE1α, ATF6 turns on a distinct protein activation mechanism after dissociation from GRP78. When the unfolded protein accumulates, GRP78 dissociates from ATF6 and loses its inhibitory effect on Golgi localization signal (GLS), leading to the translocation of ATF6 to the Golgi apparatus. The transmembrane fragment of ATF6 is cleaved by a resident protease at the juxtamembrane site of the Golgi apparatus (47). The fragment activated by ATF6 cleavage can enter the nucleus to upregulate the expression of UPR molecules, such as GRP78 and GRP94. Meanwhile, activated ATF6 can also promote the expression of XBP1 and CHOP and work together with ATF4 to activate the ERAD pathway, alleviating ERS (48, 49).
3.2. The role of ERS in the pathological changes of IVD cells
IVD is a high loading site for protein synthesis, and ER is more likely to withstand the pressure of protein synthesis and folding. Numerous studies have demonstrated that ERS plays an important role in IVD degeneration (11, 50, 51). It has been reported that the expression of ERS-related markers is significantly increased in a rat IDD model, which is constructed by surgically removing the sacrospinous muscle, spinous process, supraspinous ligament, interspinous ligament, and the posterolateral part of the bilateral facet joints of the lumbar spine (5, 52). Interestingly, when the UPR branch inhibitor GSK2606414 is added or the transcriptional expression of ERS pathway target proteins is knocked down by lentiviral transfection, the apoptosis rate of FBS starvation-treated AF cells is significantly reduced. Increased AF cell apoptosis can accelerate the progression of IDD, and GSK2606414 treatment may ameliorate IDD development (51). These studies strongly demonstrate the involvement of ERS in the development of IDD.
3.2.1. ERS induces cell apoptosis during IDD development
The epidemiological study shows that the proportion of middle-aged and elderly patients with IDD is significantly higher than that of young people (4). Through transmission electron microscopy and tissue section observation, the apoptosis of human degenerated NP cells and endplate chondrocytes also increased significantly (51). Another key pathogenic factor for the induction of IDD is cellular senescence. NP cells can be induced to senesce under a simulated acidic microenvironment in vitro, and activation of UPR signaling can compromise the process (26). Cell death triggered by activation of apoptotic signals in IVD cells is the main cause of cells loss and ECM degradation in IDD (Figure 2).
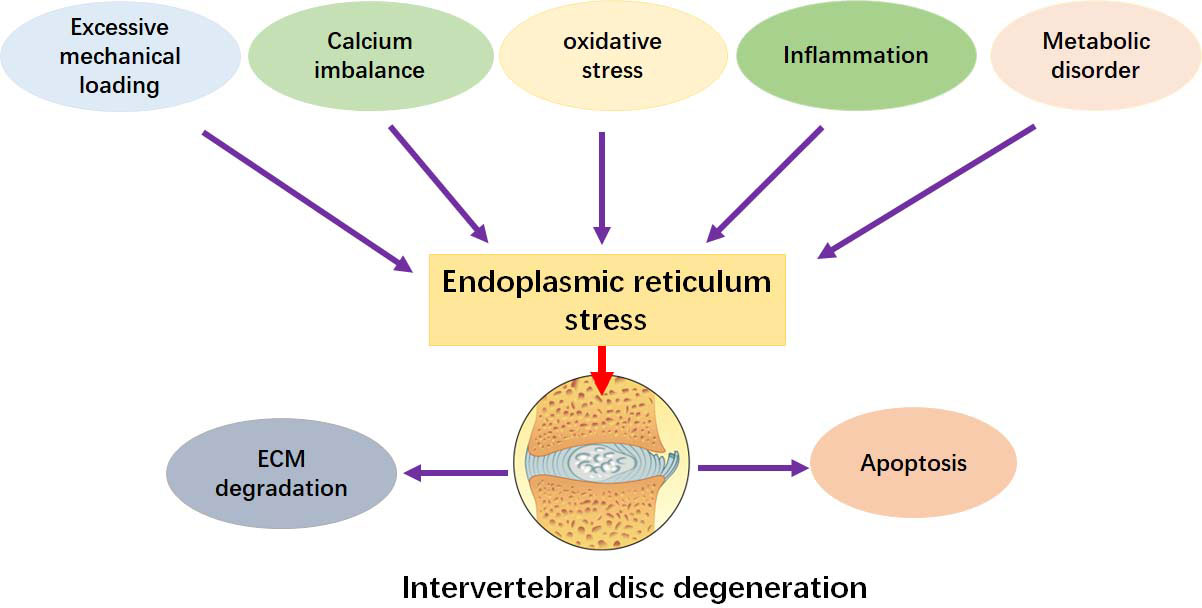
Figure 2 ERS promotes the development of IDD. ERS can be activated by inflammation, apoptosis, senescence, oxidative stress, mechanical pressure, calcium dyshomeostasis, and metabolic disorder. These factors can facilitate the pathological changes by stimulating ERS.
IDD-related apoptosis pathways include death receptor pathway, mitochondrial pathway, and ERS pathway. In the degenerated IVD tissues, the ERS levels and apoptosis rates increase, and ERS promotes NP cell apoptosis and IDD progression. Specifically, IRE1α inhibits the transcription of microRNA and initiates mitochondria-dependent IVD apoptosis (45). Inhibition of IRE1α activity significantly slows the degeneration of NP cells and the progression of IDD in vivo (53). Similarly, activation of PERK/eIF2α pathway at the early stage of the UPR can inhibit the apoptosis of NP cells by activating autophagy. However, CHOP acts as the main transcription factor linking ERS to apoptosis. The expression of CHOP and its downstream effectors caspase-12 and GADD34 in IVD cells can be significantly upregulated (54, 55). In one study, TNF-α promotes NP cells apoptosis by enhancing the expression of CHOP (55). Apoptosis activation is closely related to the expression of CHOP (56). Mild ERS has a protective effect on IVD (57, 58), and severe ERS may be related to cell senescence and apoptosis, which participate in the development of IDD. Collectively, ERS-induced cell senescence and apoptosis play an important role in the induction of IDD pathological features. However, it is still unclear what threshold of ERS will lead to the switch from protection to injury.
3.2.2. ERS affects ECM metabolism during IDD development
The constituents of ECM mainly include water, collagen, proteoglycan, elastin, glycosaminoglycan, and glycoprotein. Various elements cross-link into a coordinated functional network (59, 60), which is involved in the metabolism regulation, compression cushion, and microenvironmental homeostasis. ECM acts as a repository of ER-secreted growth factors and regulates the metabolism of IVD through the interaction of matrix components with various growth factors (61–63). Current evidence shows (53, 64–67) that a series of factors, including mechanical stress, inflammation, and oxidative stress, promotes the degradation of ECM. These may induce persistent overloading of ER or dysfunction of ER-resident molecular chaperones, contributing to the main pathological characteristics of IDD (68–70).
IDD development is associated with an ERS-mediated process of ECM metabolism that ultimately leads to pathological changes in the structures and functions of IVD (60, 71). Consistently, another study shows that more dilated and abundant ER and higher UPR target gene expression are observed in the degenerated NP cells isolated from IDD (12). Impaired anabolism of ECM has been associated with the development of IDD (53, 64, 72). Zhen Lin et al. reports that eicosapentaenoic acid can inhibit the mRNA and protein expression of ERS markers, such as GRP78, ATF4, and CHOP, and promote the synthesis of ECM components collagen II and aggrecan, indicating potential crosstalk between ERS and ECM metabolism in IDD (73). Previous studies identify that AGEs accumulation in IVD acts as an essential risk factor associating with cell apoptosis and impeding ECM metabolism via ERS (74). Furthermore, suppression of AGEs-induced metabolism impairment and cell apoptosis after ERS inhibition has been observed (74). Inflammation also impairs the metabolic activity of collagen and proteoglycans (53, 64).
Excessive activation of ECM catabolism promotes ERS-induced IDD progression (75). It has been demonstrated the presence of aggrecan catabolic products in both normal and degenerated human IVD tissues activates ERS with different levels (76). Consistently, the levels of biglycan and decorin, the degradation products of native glycanated forms, increase in rat NP cells with persistent activation of ERS (71, 72). Additionally, NP cells cultured under hypoxia show higher ERS and ADAMTSs expression, which contribute to ECM degradation (75, 76). Accumulation of ECM catabolic products may in turn lead to a stress-related response involving increased expression of Grp78 and protein disulfide isomerase, which further deteriorate the microenvironment of IVD (77–79). The imbalanced gene expression of MMPs and ADAMTSs is the important risk factor to disrupt ECM homeostasis in IDD (80, 81). Recent evidence shows that ERS acts as a key regulatory mechanism to regulate the expression of MMPs and their inhibitors (53, 72, 82). Specifically, cholesterol increases the gene and protein expression of MMPs and activates ERS in NP cells by stimulating the maturation of SREBP1 (72). Inhibition of IRE1 activity prevents the gene and protein expression of MMPs and ADAMTSs in IL-1β-treated NP cells (53).
Collectively, both metabolism balance and IVD structure can be negatively affected by ERS, as indicated by disorganized disc morphology, disruption of the lamellar collagen architecture, and reduction of collagen fibers and collagen (10, 51, 83, 84). However, the specific mechanisms of ERS in affecting IVD structure by interfering with metabolism still needs to be further studied.
3.3. The possible mechanisms of ERS in mediating IDD development
3.3.1. The interaction between ERS and oxidative stress during the development of IDD
It has been reported that the degeneration of IVD is associated with an increased level of oxidative stress (65). H2O2 can promote NP cell apoptosis by activating the ATF4/CHOP signaling pathway. Peroxynitrite, a potent oxidant, may cause cellular tyrosine nitrosylation, which has been considered a hallmark of ROS overproduction (85). Notably, the number of nitrotyrosine-positive cells in human NP tissues increases with the aggravation of IDD (86, 87). Furthermore, oxidative stress has been implicated in matrix degradation, inflammation (88), and the reduction of cell viability and functions in the diagnostic device microenvironment in vitro (89, 90). Specifically, excessive ROS modifies matrix protein expression, causing oxidative damage to ECM, nutrient metabolism dysregulation, and mechanical impairment in IVD (67, 91). ERS may produce oxidative stress through the formation of disulfide bonds, activation of NADPH oxidase 4, and ER flavoprotein, promoting the progression of IDD (92–94). Oxidative stress can be a linker between ERS and IDD pathological features (95, 96). Some potential mechanisms might be implicated in the regulation of ERS-related oxidative stress in IDD development. Firstly, increased levels of ROS trigger calcium release from the ER and increase the sensitivity of calcium channels on the ER membrane, thereby delivering positive feedback for enhancing ERS (97, 98). Secondly, ROS generation impairs the ubiquitin-proteasome pathway and impedes the degradation of unfolded proteins (99–101). Finally, ROS also enforces ERS by mediating the formation of disulfide bonds and affecting the folding of proteins (92).
3.3.2. The interaction between ERS and imbalanced calcium homeostasis during the development of IDD
Oxidative stress may contribute to the imbalance of calcium homeostasis. In addition, imbalance of calcium homeostasis and oxidative stress are the two factors involved in the progression of IDD (Figure 2). Ca2+ is an important messenger for intracellular signal transmission, and ER is responsible for intracellular Ca2+ storage. Under the condition of severe or prolonged ER dysfunction, Ca2+ is released from ER into mitochondria and subsequently triggers a series of apoptotic signal transduction pathways (102). This translocation is regulated by the IP3R-GRP75-VDAC1 signaling, and the imbalanced calcium homeostasis is involved in the development of IDD by initiating NP apoptosis (67). Advanced glycation end products (AGEs) have been reported to elevate intracellular calcium, deplete Ca2+ in the ER lumen, impair Ca2+ homeostasis, and promote NP apoptosis through ERS in a concentration- and time-dependent manner, exacerbating IDD development in rats (74).
3.3.3. The interaction between ERS and inflammation during the development of IDD
Oxidative stress can interact with inflammation reciprocally. Inflammation is also one of the important factors involved in the development of IDD (Figure 2) (57, 103). Several studies have been shown that the expression of TNF-α and IL-1β in the degenerative IVD is significantly higher than that in the non-degenerative IVD (64, 104). Proinflammatory cytokines IL-1β, IL-6, and TNF-α are the key factors to activate ERS signaling and promote IVD cell apoptosis, leading to the development of IDD. In addition, the expression levels of TNF-α and IL-1β are positively correlated with the severity of IDD (64, 105–108). In IL-1β-treated human NP cells, a decrease in the expression of type II collagen and proteoglycan and an increase in the expression of TNF-α (mRNA), IL-6 (mRNA), MMP-13 (mRNA), GRP78 (mRNA and protein), and CHOP (mRNA and protein) are observed (79). In addition, in human NP cells co-stimulated with 5 ng/ml TNF-α and 1 ng/ml IL-1 β for 12 hours, the proliferation and anabolism are inhibited and the expression of PERK and IRE1 is increased. Inversely, knockdown of PERK and IRE1 increases the anabolism of NP cells. However, the expression of ATF6 is not significantly changed (12). Another study shows that IL-1β activates the protein expression of ER stress markers GRP78 and CHOP in human primary IVD cells (109).
It has been shown that upregulation of ERS and UPR signaling may compromise TNF-α-induced apoptosis in rat NP cells at an early stage and promote cell proliferation (110). Later, it is reported that PERK/eIF2α activation-induced autophagy protects TNF-α-treated NP cells from apoptosis (111). Another study reports that TNF-α (5 and 10 ng/mL) does not induce ER stress, while IL-1β (5 and 10 ng/mL) activates GRP78 expression but does not affect calcium mobilization (24). This discrepancy might be attributed to the different cell types and the reagent concentrations. The dual effects of PERK and IRE1-α on IVD cells may be associated with different stimuli under certain circumstances (12, 112, 113).
Evidence shows that ERS may synergy with inflammation to exacerbate IDD progression. ERS can activate the NLRP3 inflammasome to induce inflammatory responses through oxidative stress, calcium dysregulation, and NF-κB activation (114, 115). NOD-like receptors may mediate ERS-induced inflammation, and ERS-inducing agents trigger the production of the proinflammatory cytokine IL-6 in a NOD1/2-dependent manner (116, 117). ERS increases the mRNA expression of TNF-α and IL-6 in a NF-κB pathway-dependent manner in rat AF cells (51). Translational inhibition of IκB by PERK and upregulation of p65 phosphorylation by IRE1/XBP1 signaling may promote nuclear translocation of p65, effectively activating NF-κB signaling. However, the regulatory link between the UPR pathways and NF-κB signaling still needs to be elucidated.
3.3.4. The interaction between ERS and excessive mechanical loading during the development of IDD
Excessive mechanical loading on IVD is another important factor in the development of IDD (66, 118) (Figure 2). Exposing AF cells to 18% deformation stress for 3 consecutive days can significantly increase the expression of caspase-12, caspase-3, and CHOP (119). Piezo1 is a mechanosensitive calcium channel, and it can be upregulated under the stimulation of mechanical tensile stress or shear stress. Excessive mechanical stress-activated Piezo1increases the senescence and apoptosis of NP cells, as well as the expression of GRP78 and CHOP (120). Cyclic stretching with a frequency of 0.5 Hz and an elongation rate of 20% has been reported to increase the expression of CHOP and GRP78 and induce apoptosis in rat AF cells (54, 56). Furthermore, the rate of apoptosis in CHOP shRNA-transfected AF cells was significantly reduced under the cyclic tensile stress (54). Excessive mechanical stress may transduce the signals into cells by mediating ERS, thereby regulating cell fate. In addition, some mechanosensitive channels, such as TRPV4, N-cadherin adhesions, and integrins, are involved in the regulation of IDD development. However, whether they are mediated by ERS is still unclear (121, 122).
3.3.5. The interaction between ERS and metabolic disorder during the development of IDD
Metabolic disorder can stimulate IDD development (Figure 2). Abnormal glucose metabolism has been demonstrated to be a risk factor (123). A study shows that people with diabetes have a higher rate of IDD than healthy persons (124). In addition, high glucose also inhibits cell anabolism (119) and promotes IVD cells apoptosis (125). In AF cells, high glucose stimulates ERS and increases the gene and protein expression of CHOP, ATF6, and GRP78, which are the important factors in ERS. In addition, the apoptosis of AF cells is also increased (126). Due to the high blood glucose level, the chemotaxis of leukocytes is decreased, and the outcome of IDD surgery is often worse (127). Interestingly, glucose deprivation also induces apoptosis in NP cells, and this action can be inhibited by autophagy-mediated p-eIF2α/ATF4 pathway (128). Hypercholesterolemia may be a potential risk factor for IDD development. Specifically, cholesterol induces pyroptosis and matrix degradation through mSREBP1-driven ERS in a rat model with hypercholesterolemia. Similarly, the cholesterol-lowering drug atorvastatin can counteract these adverse effects of cholesterol on IVD (72). It has been reported that high concentrations of lactate promote the degradation of type II collagen and participate in the apoptosis regulation by activating caspase-3 expression and mediating autophagy (82). A study finds that activation of the acid-sensitive ion channel ASIC1a can promote the apoptosis of NP cells by regulating ERS (129). Interestingly, UPR-induced autophagy exerts an anti-degeneration effect on acid-treated rat NP cells (26).
4. ERS may become the potential therapeutic target to prevent IDD development
At present, there is no effective clinical strategies to cure IDD, due to the limited understanding of the molecular mechanisms. ERS has been demonstrated to play a key role in the development of IDD, and it has become the potential target for the therapeutic management of IDD. In recent years, some scholars have proposed the term “promoter of protein homeostasis” to define some small molecules with “chemical chaperone” activity (130, 131). For example, 4-phenylbutyric acid (4-Phenylbutyric acid, 4-PBA) and tauroursodeoxycholic acid (TUDCA) have been shown to reduce ERS by enhancing the folding ability of proteins (132). 4-PBA, a modified fatty acid, inhibits ERS and alleviates the progression of IDD. Consistently, 4-PBA can inhibit tension-induced IDD development, as indicated by reduced apoptosis, ameliorated ERS, and decreased ROS generation in AF cells (119). 4-PBA can also inhibit high glucose-induced apoptosis by inhibiting ERS in AF cells (126). TUDCA, a hydrophilic bile acid, has been reported to inhibit ERS-induced catabolism and reduce excessive mechanical compression-stimulated apoptosis in NP cells (133, 134). However, due to the poor selectivity, these chaperones usually require high concentrations to be effective, and it still needs further investigation to make the chemical chaperone-based therapeutics available for IDD management.
Inhibition of ATF4 expression can alleviate TNF-α-induced ROS production and apoptosis in NP cells (135). In addition, starvation-mediated TUNEL-positive frequency and apoptotic protein expression in AF cells can be significantly inhibited by PERK inhibitors (51). Despite the marked therapeutic efficacy, the on-target toxicity PERK inhibitors still needs for further investigation (136). In addition, ISRIB, a potent eIF2α inhibitor, significantly suppresses PTEN-deficient and MYC-overexpressing prostate cancer progression, extending the survival of tumor-bearing mice (137, 138). One study shows that pro-inflammatory cytokine intervention significantly upregulates the expression of IRE1-α and PERK, but does not change the expression of ATF6 (12). B-I09 has been proven a safe and selective IRE1α RNase inhibitor suitable for in vivo use. A preclinical study shows that treatment with B-I09 suppresses leukemic growth in mouse models with chronic lymphocytic leukemia without causing systemic toxicity (139). An RNase inhibitor of IRE1α can alleviate ERS-induced inflammatory responses in a mouse model with lipid metabolism disorder (140). These suggest that IRE1α pharmacological inhibitors may ameliorate the pathological development of IDD.
The Bip inducer sodium 2-propylvalerate (valproate) has been approved for clinical use in neuronal diseases and been in trials to protect pancreatic beta cells from ERS-induced apoptosis (141). BiP has been shown safe and effective anti-inflammatory and immunomodulatory properties in patients with rheumatoid arthritis (142). In addition, it has been reported that estrogen exhibits a critical role in protecting against the development of IDD in ovariectomized rats (143, 144). In addition, estrogen exhibits regulatory activity against ERS in different tissues (145, 146). However, whether estrogen is involved in inhibiting the pathogenesis of IDD by mediating ERS signaling deserves further study.
It is important to find that effective natural compounds from traditional Chinese medicine (TCM) play a role in regulating ERS and UPR pathways. This makes it possible to explore these natural compounds for the treatment of IDD. Berberine, an isoquinoline alkaloid isolated from Coptis chinensis and Phellodendri (147), has been reported to inhibit ERS-induced apoptosis via the IRE1/JNK pathway in NP cells (148). Quercetin, a bioflavonoid widely used in the treatment of the respiratory and cardiovascular diseases, has been shown to inhibit ERS and reduce chondrocyte degeneration and apoptosis by activating SIRT1/AMPK signaling pathway (149). Curcumin can significantly attenuate ERS-related chondrocyte apoptosis and improve the progression of osteoarthritis in vivo (150). Furthermore, 5-hydroxymethylfurfural and salidroside also exhibit the protective effects against ERS in various cell lines (151–153).
Exosomes are vesicle-like structures containing complex RNAs and/or proteins, which are widely involved in cell proliferation, migration, and other processes. Recently, it has been shown that exosomes have certain potentials in IDD treatment. Mesenchymal stem cell-derived exosomes can inhibit the apoptosis and calcification of endplate chondrocytes, and the mechanism may be related to the regulation of miR-31-5p/ATF6/ERS pathway (154). In addition, human urinary stem cell-derived exosomes (USCs-exos) have been also shown to significantly ameliorate ERS and inhibit apoptosis through AKT and ERK signaling pathways in NP cells, leading to attenuation of IDD development (155). Unfortunately, the specific mechanism by which exosomes mediate ERS signaling for the treatment of IDD has not yet been established.
5. Future perspective
The pathogenesis of IDD is complex and diverse. ER plays an important role in maintaining the functions of IVD and participating in the synthesis, transport, secretion, and degradation of ECM-related proteins (59, 156, 157). Current studies have been shown (18, 20–23) that various pathological factors, such as excessive mechanical loading, insufficient nutrient supply, metabolic disorders, immune mechanisms, and inflammation, lead to the chronic activation of ERS and promote the progression of IDD. However, ERS under physiological conditions has a protective effect on IVD cells (26, 110). Thus, there are still many problems to be solved in these areas. The dynamics of ERS activation remain unclear. For example, how does the process of accumulation of unfolded and misfolded proteins in the ER lumen affect ERS signaling activation and transmission? Does it include the chronological sequence of the three branch pathways of the UPR in response to ERS and of their respective roles? Do the mechanisms by which chronic noxious stimuli and acute severe factors trigger ERS and their effects act on IDD progression in the same way? How to determine the dividing line between whether ERS regulates life activities is beneficial or harmful? And how the ERS under physiological stress is regulated is still unknown. Many studies have been confirmed the benefits of inhibiting ERS in IDD. Whether ERS inhibitors have certain side effects on IVD or other parts of the body still needs to be further explored.
6. Conclusion
IDD has become a global public health problem. It is important to study the pathogenesis of IDD and explore effective prevention and treatment. Various potential mechanisms, including inflammatory responses, excessive mechanical loading, metabolic disturbance, oxidative stress, calcium homeostasis imbalance, and nutritional deficiencies, are included in the development of IDD. ERS plays an important role in the pathogenesis of IDD. However, targeting ERS for the treatment of IDD remains challenging. In recent years, many studies have been carried out to explore the relationship between ERS and IDD and investigate the intervention of ERS signaling to improve IDD. Clues are being enriched in ERS-targeted therapy for disc degeneration.
Author contributions
TZ provided the idea of this paper. JH, QZ, QR, LL, and GJ collected the information and revised and finalized the paper. All authors approved the final paper.
Funding
This study was supported by Team Construction Projects from Gannan Medical University under Grant TS202002.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Maher C, Underwood M, Buchbinder R. Non-specific low back pain. Lancet (London England) (2017) 389(10070):736–47. doi: 10.1016/S0140-6736(16)30970-9
2. Cieza A, Causey K, Kamenov K, Hanson SW, Chatterji S, Vos T. Global estimates of the need for rehabilitation based on the global burden of disease study 2019: a systematic analysis for the global burden of disease study 2019. Lancet (London England) (2021) 396(10267):2006–17. doi: 10.1016/S0140-6736(20)32340-0
3. Katz JN. Lumbar disc disorders and low-back pain: socioeconomic factors and consequences. J Bone Joint Surg Am (2006) 88 Suppl 2:21–4. doi: 10.2106/00004623-200604002-00005
4. Teraguchi M, Yoshimura N, Hashizume H, Muraki S, Yamada H, Minamide A, et al. Prevalence and distribution of intervertebral disc degeneration over the entire spine in a population-based cohort: the Wakayama spine study. Osteoarthr Cartil (2014) 22(1):104–10. doi: 10.1016/j.joca.2013.10.019
5. Sharifi S, Bulstra SK, Grijpma DW, Kuijer R. Treatment of the degenerated intervertebral disc; closure, repair and regeneration of the annulus fibrosus. J Tissue Eng Regen Med (2015) 9(10):1120–32. doi: 10.1002/term.1866
6. Khalaf K, Nikkhoo M. Comparative biomechanical analyses of lower cervical spine post anterior fusion versus intervertebral disc arthroplasty: A geometrically patient-specific poroelastic finite element investigation. J Orthop Translat (2022) 36:33–43. doi: 10.1016/j.jot.2022.05.008
7. Palmer N, Guan Z, Chai NC. Spinal cord stimulation for failed back surgery syndrome – patient selection considerations. Trans Perioper Pain Med (2019) 6(3):81–90.
8. Senck S, Trieb K, Kastner J, Hofstaetter SG, Lugmayr H, Windisch G. Visualization of intervertebral disc degeneration in a cadaveric human lumbar spine using microcomputed tomography. J Anat (2020) 236(2):243–51. doi: 10.1111/joa.13105
9. Zhao C-Q, Zhang Y-H, Jiang S-D, Jiang L-S, Dai L-Y. Both endoplasmic reticulum and mitochondria are involved in disc cell apoptosis and intervertebral disc degeneration in rats. Age (Dordrecht Netherlands) (2010) 32(2):161–77. doi: 10.1007/s11357-009-9121-4
10. Liao Z, Luo R, Li G, Song Y, Zhan S, Zhao K, et al. Exosomes from mesenchymal stem cells modulate endoplasmic reticulum stress to protect against nucleus pulposus cell death and ameliorate intervertebral disc degeneration in vivo. Theranostics (2019) 9(14):4084–100. doi: 10.7150/thno.33638
11. Wang D, He X, Zheng C, Wang C, Peng P, Gao C, et al. Endoplasmic reticulum stress: An emerging therapeutic target for intervertebral disc degeneration. Front In Cell Dev Biol (2021) 9:819139. doi: 10.3389/fcell.2021.819139
12. Wen T, Xue P, Ying J, Cheng S, Liu Y, Ruan D. The role of unfolded protein response in human intervertebral disc degeneration: Perk and IRE1- as two potential therapeutic targets. Oxid Med Cell Longevity (2021) 2021:6492879. doi: 10.1155/2021/6492879
13. Hughes A, Oxford AE, Tawara K, Jorcyk CL, Oxford JT. Endoplasmic reticulum stress and unfolded protein response in cartilage pathophysiology; contributing factors to apoptosis and osteoarthritis. Int J Mol Sci (2017) 18(3):665. doi: 10.3390/ijms18030665
14. Li J, Yang S, Li X, Liu D, Wang Z, Guo J, et al. Role of endoplasmic reticulum stress in disuse osteoporosis. Bone (2017) 97:2–14. doi: 10.1016/j.bone.2016.12.009
15. Rahmati M, Moosavi MA, McDermott MF. ER stress: A therapeutic target in rheumatoid arthritis? Trends In Pharmacol Sci (2018) 39(7):610–23. doi: 10.1016/j.tips.2018.03.010
16. Ikezoe K, Nakamori M, Furuya H, Arahata H, Kanemoto S, Kimura T, et al. Endoplasmic reticulum stress in myotonic dystrophy type 1 muscle. Acta Neuropathol (2007) 114(5):527–35. doi: 10.1007/s00401-007-0267-9
17. Akmal M, Kesani A, Anand B, Singh A, Wiseman M, Goodship A. Effect of nicotine on spinal disc cells: a cellular mechanism for disc degeneration. Spine (2004) 29(5):568–75. doi: 10.1097/01.brs.0000101422.36419.d8
18. Huang Y-C, Urban JPG, Luk KDK. Intervertebral disc regeneration: do nutrients lead the way? Nat Rev Rheumatol (2014) 10(9):561–6. doi: 10.1038/nrrheum.2014.91
19. Hanaei S, Abdollahzade S, Khoshnevisan A, Kepler CK, Rezaei N. Genetic aspects of intervertebral disc degeneration. Rev In Neurosci (2015) 26(5):581–606. doi: 10.1515/revneuro-2014-0077
20. Vergroesen PPA, Kingma I, Emanuel KS, Hoogendoorn RJW, Welting TJ, van Royen BJ, et al. Mechanics and biology in intervertebral disc degeneration: a vicious circle. Osteoarthr Cartil (2015) 23(7):1057–70. doi: 10.1016/j.joca.2015.03.028
21. Wang F, Cai F, Shi R, Wang XH, Wu XT. Aging and age related stresses: a senescence mechanism of intervertebral disc degeneration. Osteoarthr Cartil (2016) 24(3):398–408. doi: 10.1016/j.joca.2015.09.019
22. Alpantaki K, Kampouroglou A, Koutserimpas C, Effraimidis G, Hadjipavlou A. Diabetes mellitus as a risk factor for intervertebral disc degeneration: a critical review. Eur Spine J (2019) 28(9):2129–44. doi: 10.1007/s00586-019-06029-7
23. Wang Y, Che M, Xin J, Zheng Z, Li J, Zhang S. The role of IL-1β and TNF-α in intervertebral disc degeneration. Biomed Pharmacother = Biomed Pharmacother (2020) 131:110660. doi: 10.1016/j.biopha.2020.110660
24. Krupkova O, Sadowska A, Kameda T, Hitzl W, Hausmann ON, Klasen J, et al. p38 MAPK facilitates crosstalk between endoplasmic reticulum stress and IL-6 release in the intervertebral disc. Front In Immunol (2018) 9:1706. doi: 10.3389/fimmu.2018.01706
25. Zhang X-B, Hu Y-C, Cheng P, Zhou H-Y, Chen X-Y, Wu D, et al. Targeted therapy for intervertebral disc degeneration: inhibiting apoptosis is a promising treatment strategy. Int J Med Sci (2021) 18(13):2799–813. doi: 10.7150/ijms.59171
26. Zhu L, Xie Z-Y, Jiang Z-L, Wang X-H, Shi H, Chen L, et al. Unfolded protein response alleviates acid-induced premature senescence by promoting autophagy in nucleus pulposus cells. Cell Biol Int (2022) 46(4):568–78. doi: 10.1002/cbin.11751
27. Schwarz DS, Blower MD. The endoplasmic reticulum: structure, function and response to cellular signaling. Cell Mol Life Sci CMLS (2016) 73(1):79–94. doi: 10.1007/s00018-015-2052-6
28. Lin JH, Walter P, Yen TSB. Endoplasmic reticulum stress in disease pathogenesis. Annu Rev Pathol (2008) 3:399–425. doi: 10.1146/annurev.pathmechdis.3.121806.151434
29. Hwang J, Qi L. Quality control in the endoplasmic reticulum: Crosstalk between ERAD and UPR pathways. Trends In Biochem Sci (2018) 43(8):593–605. doi: 10.1016/j.tibs.2018.06.005
30. Wang M, Kaufman RJ. Protein misfolding in the endoplasmic reticulum as a conduit to human disease. Nature (2016) 529(7586):326–35. doi: 10.1038/nature17041
31. Hetz C. The unfolded protein response: controlling cell fate decisions under ER stress and beyond. Nat Rev Mol Cell Biol (2012) 13(2):89–102. doi: 10.1038/nrm3270
32. Ron D, Walter P. Signal integration in the endoplasmic reticulum unfolded protein response. Nat Rev Mol Cell Biol (2007) 8(7):519–29. doi: 10.1038/nrm2199
33. Walter P, Ron D. The unfolded protein response: from stress pathway to homeostatic regulation. Sci (New York N.Y.) (2011) 334(6059):1081–6. doi: 10.1126/science.1209038
34. Volmer R, Ron D. Lipid-dependent regulation of the unfolded protein response. Curr Opin In Cell Biol (2015) 33:67–73. doi: 10.1016/j.ceb.2014.12.002
35. Lin JH, Li H, Yasumura D, Cohen HR, Zhang C, Panning B, et al. IRE1 signaling affects cell fate during the unfolded protein response. Sci (New York N.Y.) (2007) 318(5852):944–9. doi: 10.1126/science.1146361
36. Sonenberg N, Hinnebusch AG. Regulation of translation initiation in eukaryotes: mechanisms and biological targets. Cell (2009) 136(4):731–45. doi: 10.1016/j.cell.2009.01.042
37. Harding HP, Zhang Y, Ron D. Protein translation and folding are coupled by an endoplasmic-reticulum-resident kinase. Nature (1999) 397(6716):271–4. doi: 10.1038/16729
38. Harding HP, Zhang Y, Bertolotti A, Zeng H, Ron D. Perk is essential for translational regulation and cell survival during the unfolded protein response. Mol Cell (2000) 5(5):897–904. doi: 10.1016/s1097-2765(00)80330-5
39. Oyadomari S, Araki E, Mori M. Endoplasmic reticulum stress-mediated apoptosis in pancreatic beta-cells. Apoptosis an Int J On Programmed Cell Death (2002) 7(4):335–45. doi: 10.1023/a:1016175429877
40. Han D, Lerner AG, Vande Walle L, Upton J-P, Xu W, Hagen A, et al. IRE1alpha kinase activation modes control alternate endoribonuclease outputs to determine divergent cell fates. Cell (2009) 138(3):562–75. doi: 10.1016/j.cell.2009.07.017
41. Upton J-P, Wang L, Han D, Wang ES, Huskey NE, Lim L, et al. IRE1α cleaves select microRNAs during ER stress to derepress translation of proapoptotic caspase-2. Sci (New York N.Y.) (2012) 338(6108):818–22. doi: 10.1126/science.1226191
42. Chen Y, Brandizzi F. IRE1: ER stress sensor and cell fate executor. Trends In Cell Biol (2013) 23(11):547–55. doi: 10.1016/j.tcb.2013.06.005
43. Maurel M, Chevet E, Tavernier J, Gerlo S. Getting RIDD of RNA: IRE1 in cell fate regulation. Trends In Biochem Sci (2014) 39(5):245–54. doi: 10.1016/j.tibs.2014.02.008
44. Moore K, Hollien J. Ire1-mediated decay in mammalian cells relies on mRNA sequence, structure, and translational status. Mol Biol Cell (2015) 26(16):2873–84. doi: 10.1091/mbc.E15-02-0074
45. Chang T-K, Lawrence DA, Lu M, Tan J, Harnoss JM, Marsters SA, et al. Coordination between two branches of the unfolded protein response determines apoptotic cell fate. Mol Cell (2018) 71(4):629–36. doi: 10.1016/j.molcel.2018.06.038
46. Bashir S, Banday M, Qadri O, Bashir A, Hilal N, Nida IF, et al. The molecular mechanism and functional diversity of UPR signaling sensor IRE1. Life Sci (2021) 265:118740. doi: 10.1016/j.lfs.2020.118740
47. Schindler AJ, Schekman R. In vitro reconstitution of ER-stress induced ATF6 transport in COPII vesicles. Proc Natl Acad Sci United States America (2009) 106(42):17775–80. doi: 10.1073/pnas.0910342106
48. Shoulders MD, Ryno LM, Genereux JC, Moresco JJ, Tu PG, Wu C, et al. Stress-independent activation of XBP1s and/or ATF6 reveals three functionally diverse ER proteostasis environments. Cell Rep (2013) 3(4):1279–92. doi: 10.1016/j.celrep.2013.03.024
49. Bao Q, Zhao M, Chen L, Wang Y, Wu S, Wu W, et al. MicroRNA-297 promotes cardiomyocyte hypertrophy via targeting sigma-1 receptor. Life Sci (2017) 175:1–10. doi: 10.1016/j.lfs.2017.03.006
50. Xie Z-Y, Chen L, Wang F, Liu L, Zhang C, Wang K, et al. Endoplasmic reticulum stress is involved in nucleus pulposus degeneration and attenuates low pH-induced apoptosis of rat nucleus pulposus cells. DNA Cell Biol (2017) 36(8):627–37. doi: 10.1089/dna.2017.3736
51. Fujii T, Fujita N, Suzuki S, Tsuji T, Takaki T, Umezawa K, et al. The unfolded protein response mediated by PERK is casually related to the pathogenesis of intervertebral disc degeneration. J Orthop Res Off Publ Orthop Res Soc (2018) 36(5):1334–45. doi: 10.1002/jor.23787
52. Wang Y-J, Shi Q, Lu WW, Cheung KCM, Darowish M, Li T-F, et al. Cervical intervertebral disc degeneration induced by unbalanced dynamic and static forces: a novel in vivo rat model. Spine (2006) 31(14):1532–8. doi: 10.1097/01.brs.0000222019.84095.23
53. Kang H, Dong Y, Peng R, Liu H, Guo Q, Song K, et al. Inhibition of IRE1 suppresses the catabolic effect of IL-1β on nucleus pulposus cell and prevents intervertebral disc degeneration in vivo. Biochem Pharmacol (2022) 197:114932. doi: 10.1016/j.bcp.2022.114932
54. Zhang Y-H, Zhao C-Q, Jiang L-S, Dai L-Y. Lentiviral shRNA silencing of CHOP inhibits apoptosis induced by cyclic stretch in rat annular cells and attenuates disc degeneration in the rats. Apoptosis an Int J On Programmed Cell Death (2011) 16(6):594–605. doi: 10.1007/s10495-011-0596-y
55. Zhang J, Wang X, Liu H, Li Z, Chen F, Wang H, et al. TNF-α enhances apoptosis by promoting chop expression in nucleus pulposus cells: role of the MAPK and NF-κB pathways. J Orthop Res Off Publ Orthop Res Soc (2019) 37(3):697–705. doi: 10.1002/jor.24204
56. Zhang Y-H, Zhao C-Q, Jiang L-S, Dai L-Y. Cyclic stretch-induced apoptosis in rat annulus fibrosus cells is mediated in part by endoplasmic reticulum stress through nitric oxide production. Eur Spine J (2011) 20(8):1233–43. doi: 10.1007/s00586-011-1718-5
57. Oakes SA, Papa FR. The role of endoplasmic reticulum stress in human pathology. Annu Rev Pathol (2015) 10:173–94. doi: 10.1146/annurev-pathol-012513-104649
58. Oakes SA. Endoplasmic reticulum proteostasis: a key checkpoint in cancer. Am J Physiol Cell Physiol (2017) 312(2):C93–102. doi: 10.1152/ajpcell.00266.2016
59. Le Maitre CL, Pockert A, Buttle DJ, Freemont AJ, Hoyland JA. Matrix synthesis and degradation in human intervertebral disc degeneration. Biochem Soc Trans (2007) 35(Pt 4):652–5. doi: 10.1042/BST0350652
60. Raj PP. Intervertebral disc: anatomy-physiology-pathophysiology-treatment. Pain Pract (2008) 8(1):18–44. doi: 10.1111/j.1533-2500.2007.00171.x
61. Hynes RO. The extracellular matrix: not just pretty fibrils. Sci (New York N.Y.) (2009) 326(5957):1216–9. doi: 10.1126/science.1176009
62. Theocharis AD, Skandalis SS, Gialeli C, Karamanos NK. Extracellular matrix structure. Adv Drug Del Rev (2016) 97:4–27. doi: 10.1016/j.addr.2015.11.001
63. Alcorta-Sevillano N, Macías I, Infante A, Rodríguez CI. Deciphering the relevance of bone ECM signaling. Cells (2020) 9(12):2630. doi: 10.3390/cells9122630
64. Le Maitre CL, Hoyland JA, Freemont AJ. Catabolic cytokine expression in degenerate and herniated human intervertebral discs: IL-1beta and TNFalpha expression profile. Arthritis Res Ther (2007) 9(4):R77. doi: 10.1186/ar2275
65. Hou G, Lu H, Chen M, Yao H, Zhao H. Oxidative stress participates in age-related changes in rat lumbar intervertebral discs. Arch Gerontol Geriatr (2014) 59(3):665–9. doi: 10.1016/j.archger.2014.07.002
66. Desmoulin GT, Pradhan V, Milner TE. Mechanical aspects of intervertebral disc injury and implications on biomechanics. Spine (2020) 45(8):E457–64. doi: 10.1097/BRS.0000000000003291
67. Lin H, Peng Y, Li J, Wang Z, Chen S, Qing X, et al. Reactive oxygen species regulate endoplasmic reticulum stress and ER-mitochondrial Ca crosstalk to promote programmed necrosis of rat nucleus pulposus cells under compression. Oxid Med Cell Longevity (2021) 2021:8810698. doi: 10.1155/2021/8810698
68. Ishikawa Y, Holden P, Bächinger HP. Heat shock protein 47 and 65-kDa FK506-binding protein weakly but synergistically interact during collagen folding in the endoplasmic reticulum. J Biol Chem (2017) 292(42):17216–24. doi: 10.1074/jbc.M117.802298
69. Besio R, Garibaldi N, Leoni L, Cipolla L, Sabbioneda S, Biggiogera M, et al. Cellular stress due to impairment of collagen prolyl hydroxylation complex is rescued by the chaperone 4-phenylbutyrate. Dis Models Mech (2019) 12(6):dmm038521. doi: 10.1242/dmm.038521
70. Ito S, Nagata K. Roles of the endoplasmic reticulum-resident, collagen-specific molecular chaperone Hsp47 in vertebrate cells and human disease. J Biol Chem (2019) 294(6):2133–41. doi: 10.1074/jbc.TM118.002812
71. Zhang S, Liu W, Chen S, Wang B, Wang P, Hu B, et al. Extracellular matrix in intervertebral disc: basic and translational implications. Cell Tissue Res (2022) 390(1):1–22. doi: 10.1007/s00441-022-03662-5
72. Yan J, Li S, Zhang Y, Deng Z, Wu J, Huang Z, et al. Cholesterol induces pyroptosis and matrix degradation mSREBP1-driven endoplasmic reticulum stress in intervertebral disc degeneration. Front In Cell Dev Biol (2021) 9:803132. doi: 10.3389/fcell.2021.803132
73. Lin Z, Ni L, Teng C, Zhang Z, Wu L, Jin Y, et al. Eicosapentaenoic acid-induced autophagy attenuates intervertebral disc degeneration by suppressing endoplasmic reticulum stress, extracellular matrix degradation, and apoptosis. Front In Cell Dev Biol (2021) 9:745621. doi: 10.3389/fcell.2021.745621
74. Luo R, Song Y, Liao Z, Yin H, Zhan S, Wang K, et al. Impaired calcium homeostasis via advanced glycation end products promotes apoptosis through endoplasmic reticulum stress in human nucleus pulposus cells and exacerbates intervertebral disc degeneration in rats. FEBS J (2019) 286(21):4356–73. doi: 10.1111/febs.14972
75. Novais EJ, Choi H, Madhu V, Suyama K, Anjo SI, Manadas B, et al. Hypoxia and hypoxia-inducible factor-1α regulate endoplasmic reticulum stress in nucleus pulposus cells: Implications of endoplasmic reticulum stress for extracellular matrix secretion. Am J Pathol (2021) 191(3):487–502. doi: 10.1016/j.ajpath.2020.11.012
76. Patel KP, Sandy JD, Akeda K, Miyamoto K, Chujo T, An HS, et al. Aggrecanases and aggrecanase-generated fragments in the human intervertebral disc at early and advanced stages of disc degeneration. Spine (2007) 32(23):2596–603. doi: 10.1097/BRS.0b013e318158cb85
77. Domowicz MS, Pirok EW, Novak TE, Schwartz NB. Role of the c-terminal G3 domain in sorting and secretion of aggrecan core protein and ubiquitin-mediated degradation of accumulated mutant precursors. J Biol Chem (2000) 275(45):35098–105. doi: 10.1074/jbc.275.45.35098
78. Xu M, Stattin E-L, Shaw G, Heinegård D, Sullivan G, Wilmut I, et al. Chondrocytes derived from mesenchymal stromal cells and induced pluripotent cells of patients with familial osteochondritis dissecans exhibit an endoplasmic reticulum stress response and defective matrix assembly. Stem Cells Trans Med (2016) 5(9):1171–81. doi: 10.5966/sctm.2015-0384
79. Yang CH, Qi WL, Zhao CW, Cai WJ, Gong Q, Niu JY, et al. Parecoxib prevents nucleus pulposus cells apoptosis by suppressing endoplasmic reticulum stress. Eur Rev For Med Pharmacol Sci (2020) 24(21):11295–304. doi: 10.26355/eurrev_202011_23619
80. Vo NV, Hartman RA, Yurube T, Jacobs LJ, Sowa GA, Kang JD. Expression and regulation of metalloproteinases and their inhibitors in intervertebral disc aging and degeneration. Spine J (2013) 13(3):331–41. doi: 10.1016/j.spinee.2012.02.027
81. Wang W-J, Yu X-H, Wang C, Yang W, He W-S, Zhang S-J, et al. MMPs and ADAMTSs in intervertebral disc degeneration. Clin Chim Acta; Int J Clin Chem (2015) 448:238–46. doi: 10.1016/j.cca.2015.06.023
82. Wu W, Zhang X, Hu X, Wang X, Sun L, Zheng X, et al. Lactate down-regulates matrix systhesis and promotes apoptosis and autophagy in rat nucleus pulposus cells. J Orthop Res Off Publ Orthop Res Soc (2014) 32(2):253–61. doi: 10.1002/jor.22503
83. Gruber HE, Hanley EN. Observations on morphologic changes in the aging and degenerating human disc: secondary collagen alterations. BMC Musculoskelet Disord (2002) 3:9. doi: 10.1186/1471-2474-3-9
84. Chooi WH, Chan BP. Compression loading-induced stress responses in intervertebral disc cells encapsulated in 3D collagen constructs. Sci Rep (2016) 6:26449. doi: 10.1038/srep26449
85. Hogg N, Darley-Usmar VM, Wilson MT, Moncada S. Production of hydroxyl radicals from the simultaneous generation of superoxide and nitric oxide. Biochem J (1992) 281(Pt 2):419–24. doi: 10.1042/bj2810419
86. Poveda L, Hottiger M, Boos N, Wuertz K. Peroxynitrite induces gene expression in intervertebral disc cells. Spine (2009) 34(11):1127–33. doi: 10.1097/BRS.0b013e31819f2330
87. Suzuki S, Fujita N, Hosogane N, Watanabe K, Ishii K, Toyama Y, et al. Excessive reactive oxygen species are therapeutic targets for intervertebral disc degeneration. Arthritis Res Ther (2015) 17:316. doi: 10.1186/s13075-015-0834-8
88. Dimozi A, Mavrogonatou E, Sklirou A, Kletsas D. Oxidative stress inhibits the proliferation, induces premature senescence and promotes a catabolic phenotype in human nucleus pulposus intervertebral disc cells. Eur Cells Mater (2015) 30:89–102. doi: 10.22203/eCM.v030a07
89. Bhattacharyya S, Sinha K, Sil PC. Cytochrome P450s: mechanisms and biological implications in drug metabolism and its interaction with oxidative stress. Curr Drug Metab (2014) 15(7):719–42. doi: 10.2174/1389200215666141125121659
90. Ochoa CD, Wu RF, Terada LS. ROS signaling and ER stress in cardiovascular disease. Mol Aspects Med (2018) 63:18–29. doi: 10.1016/j.mam.2018.03.002
91. Feng C, Yang M, Lan M, Liu C, Zhang Y, Huang B, et al. ROS: Crucial intermediators in the pathogenesis of intervertebral disc degeneration. Oxid Med Cell Longevity (2017) 2017:5601593. doi: 10.1155/2017/5601593
92. Tu BP, Weissman JS. Oxidative protein folding in eukaryotes: mechanisms and consequences. J Cell Biol (2004) 164(3):341–6. doi: 10.1083/jcb.200311055
93. Santos CXC, Tanaka LY, Wosniak J, Laurindo FRM. Mechanisms and implications of reactive oxygen species generation during the unfolded protein response: roles of endoplasmic reticulum oxidoreductases, mitochondrial electron transport, and NADPH oxidase. Antioxid Redox Signaling (2009) 11(10):2409–27. doi: 10.1089/ARS.2009.2625
94. Eletto D, Chevet E, Argon Y, Appenzeller-Herzog C. Redox controls UPR to control redox. J Cell Sci (2014) 127(Pt 17):3649–58. doi: 10.1242/jcs.153643
95. Chen D, Xia D, Pan Z, Xu D, Zhou Y, Wu Y, et al. Metformin protects against apoptosis and senescence in nucleus pulposus cells and ameliorates disc degeneration in vivo. Cell Death Dis (2016) 7(10):e2441. doi: 10.1038/cddis.2016.334
96. Zeeshan HMA, Lee GH, Kim H-R, Chae H-J. Endoplasmic reticulum stress and associated ROS. Int J Mol Sci (2016) 17(3):327. doi: 10.3390/ijms17030327
97. Gerasimenko JV, Gerasimenko OV, Palejwala A, Tepikin AV, Petersen OH, Watson AJM. Menadione-induced apoptosis: roles of cytosolic Ca(2+) elevations and the mitochondrial permeability transition pore. J Cell Sci (2002) 115(Pt 3):485–97. doi: 10.1242/jcs.115.3.485
98. Li G, Mongillo M, Chin K-T, Harding H, Ron D, Marks AR, et al. Role of ERO1-alpha-mediated stimulation of inositol 1,4,5-triphosphate receptor activity in endoplasmic reticulum stress-induced apoptosis. J Cell Biol (2009) 186(6):783–92. doi: 10.1083/jcb.200904060
99. Bence NF, Sampat RM, Kopito RR. Impairment of the ubiquitin-proteasome system by protein aggregation. Sci (New York N.Y.) (2001) 292(5521):1552–5. doi: 10.1126/science.292.5521.1552
100. Schaffar G, Breuer P, Boteva R, Behrends C, Tzvetkov N, Strippel N, et al. Cellular toxicity of polyglutamine expansion proteins: mechanism of transcription factor deactivation. Mol Cell (2004) 15(1):95–105. doi: 10.1016/j.molcel.2004.06.029
101. Liu Y. Hydrogen peroxide induces nucleus pulposus cell apoptosis by ATF4/CHOP signaling pathway. Exp Ther Med (2020) 20(4):3244–52. doi: 10.3892/etm.2020.9052
102. Szabadkai G, Bianchi K, Várnai P, De Stefani D, Wieckowski MR, Cavagna D, et al. Chaperone-mediated coupling of endoplasmic reticulum and mitochondrial Ca2+ channels. J Cell Biol (2006) 175(6):901–11. doi: 10.1083/jcb.200608073
103. Cao SS, Luo KL, Shi L. Endoplasmic reticulum stress interacts with inflammation in human diseases. J Cell Physiol (2016) 231(2):288–94. doi: 10.1002/jcp.25098
104. Hiyama A, Hiraishi S, Sakai D, Mochida J. CCAAT/enhancer binding protein β regulates the expression of tumor necrosis factor-α in the nucleus pulposus cells. J Orthop Res (2016) 34(5):865–75. doi: 10.1002/jor.23085
105. Weiler C, Nerlich AG, Bachmeier BE, Boos N. Expression and distribution of tumor necrosis factor alpha in human lumbar intervertebral discs: a study in surgical specimen and autopsy controls. Spine (2005) 30(1):44–53. doi: 10.1097/01.brs.0000149186.63457.20
106. Bachmeier BE, Nerlich AG, Weiler C, Paesold G, Jochum M, Boos N. Analysis of tissue distribution of TNF-alpha, TNF-alpha-receptors, and the activating TNF-alpha-converting enzyme suggests activation of the TNF-alpha system in the aging intervertebral disc. Ann New York Acad Sci (2007) 1096:44–54. doi: 10.1196/annals.1397.069
107. Takada T, Nishida K, Maeno K, Kakutani K, Yurube T, Doita M, et al. Intervertebral disc and macrophage interaction induces mechanical hyperalgesia and cytokine production in a herniated disc model in rats. Arthritis Rheum (2012) 64(8):2601–10. doi: 10.1002/art.34456
108. Chen Z-H, Jin S-H, Wang M-Y, Jin X-L, Lv C, Deng Y-F, et al. Enhanced NLRP3, caspase-1, and IL- 1β levels in degenerate human intervertebral disc and their association with the grades of disc degeneration. Anatomical Rec (Hoboken N.J. 2007) (2015) 298(4):720–6. doi: 10.1002/ar.23059
109. Xu D, Jin H, Wen J, Chen J, Chen D, Cai N, et al. Hydrogen sulfide protects against endoplasmic reticulum stress and mitochondrial injury in nucleus pulposus cells and ameliorates intervertebral disc degeneration. Pharmacol Res (2017) 117:357–69. doi: 10.1016/j.phrs.2017.01.005
110. Chen L, Liu L, Xie Z-Y, Wang F, Sinkemani A, Zhang C, et al. Endoplasmic reticulum stress facilitates the survival and proliferation of nucleus pulposus cells in TNF-α stimulus by activating unfolded protein response. DNA Cell Biol (2018) 37(4):347–58. doi: 10.1089/dna.2017.4029
111. Chen L, Liu L, Xie Z-Y, Wang F, Zhu L, Zhang C, et al. Protein kinase RNA-like ER kinase/eukaryotic translation initiation factor 2α pathway attenuates tumor necrosis factor alpha-induced apoptosis in nucleus pulposus cells by activating autophagy. J Cell Physiol (2019) 234(7):11631–45. doi: 10.1002/jcp.27820
112. Karali E, Bellou S, Stellas D, Klinakis A, Murphy C, Fotsis T. VEGF signals through ATF6 and PERK to promote endothelial cell survival and angiogenesis in the absence of ER stress. Mol Cell (2014) 54(4):559–72. doi: 10.1016/j.molcel.2014.03.022
113. Duran-Aniotz C, Cornejo VH, Espinoza S, Ardiles ÁO, Medinas DB, Salazar C, et al. IRE1 signaling exacerbates alzheimer's disease pathogenesis. Acta Neuropathol (2017) 134(3):489–506. doi: 10.1007/s00401-017-1694-x
114. Jiang M, Wang H, Liu Z, Lin L, Wang L, Xie M, et al. Endoplasmic reticulum stress-dependent activation of iNOS/NO-NF-κB signaling and NLRP3 inflammasome contributes to endothelial inflammation and apoptosis associated with microgravity. FASEB J Off Publ Fed Am Soc For Exp Biol (2020) 34(8):10835–49. doi: 10.1096/fj.202000734R
115. Li W, Cao T, Luo C, Cai J, Zhou X, Xiao X, et al. Crosstalk between ER stress, NLRP3 inflammasome, and inflammation. Appl Microbiol Biotechnol (2020) 104(14):6129–40. doi: 10.1007/s00253-020-10614-y
116. Zhang K, Kaufman RJ. From endoplasmic-reticulum stress to the inflammatory response. Nature (2008) 454(7203):455–62. doi: 10.1038/nature07203
117. Keestra-Gounder AM, Byndloss MX, Seyffert N, Young BM, Chávez-Arroyo A, Tsai AY, et al. NOD1 and NOD2 signalling links ER stress with inflammation. Nature (2016) 532(7599):394–7. doi: 10.1038/nature17631
118. Adams MA, Freeman BJ, Morrison HP, Nelson IW, Dolan P. Mechanical initiation of intervertebral disc degeneration. Spine (2000) 25(13):1625–36. doi: 10.1097/00007632-200007010-00005
119. Liu Z, Zhang Z, Zhang A, Zhang F, Du W, Zhang Y, et al. Osteogenic protein-1 alleviates high glucose microenvironment-caused degenerative changes in nucleus pulposus cells. Biosci Rep (2019) 39(4):BSR20190170. doi: 10.1042/BSR20190170
120. Wang B, Ke W, Wang K, Li G, Ma L, Lu S, et al. Mechanosensitive ion channel Piezo1 activated by matrix stiffness regulates oxidative stress-induced senescence and apoptosis in human intervertebral disc degeneration. Oxid Med Cell Longevity (2021) 2021:8884922. doi: 10.1155/2021/8884922
121. Hwang PY, Jing L, Michael KW, Richardson WJ, Chen J, Setton LA. N-Cadherin-Mediated signaling regulates cell phenotype for nucleus pulposus cells of the intervertebral disc. Cell Mol Bioeng (2015) 8(1):51–62. doi: 10.1007/s12195-014-0373-4
122. Kim MK, Ramachandran R, Séguin CA. Spatiotemporal and functional characterisation of transient receptor potential vanilloid 4 (TRPV4) in the murine intervertebral disc. Eur Cells Mater (2021) 41:194–203. doi: 10.22203/eCM.v041a14
123. Zhang Z, Lin J, Nisar M, Chen T, Xu T, Zheng G, et al. The Sirt1/P53 axis in diabetic intervertebral disc degeneration pathogenesis and therapeutics. Oxid Med Cell Longevity (2019) 2019:7959573. doi: 10.1155/2019/7959573
124. Mobbs RJ, Newcombe RL, Chandran KN. Lumbar discectomy and the diabetic patient: incidence and outcome. J Clin Neurosci (2001) 8(1):10–3. doi: 10.1054/jocn.2000.0682
125. Wang W, Li P, Xu J, Wu X, Guo Z, Fan L, et al. Resveratrol attenuates high glucose-induced nucleus pulposus cell apoptosis and senescence through activating the ROS-mediated PI3K/Akt pathway. Biosci Rep (2018) 38(2):BSR20171454. doi: 10.1042/BSR20171454
126. Pang L, Yang K, Zhang Z. High-glucose environment accelerates annulus fibrosus cell apoptosis by regulating endoplasmic reticulum stress. Biosci Rep (2020) 40(7):BSR20200262. doi: 10.1042/BSR20200262
127. Sakellaridis N. The influence of diabetes mellitus on lumbar intervertebral disk herniation. Surg Neurol (2006) 66(2):152–4. doi: 10.1016/j.surneu.2006.01.019
128. Chang H, Cai F, Zhang Y, Xue M, Liu L, Yang A, et al. Early-stage autophagy protects nucleus pulposus cells from glucose deprivation-induced degeneration via the p-eIF2α/ATF4 pathway. Biomed Pharmacother = Biomed Pharmacother (2017) 89:529–35. doi: 10.1016/j.biopha.2017.02.074
129. Xie Z-Y, Chen L, Zhang C, Liu L, Wang F, Cai F, et al. Acid-sensing ion channel 1a regulates fate of rat nucleus pulposus cells in acid stimulus through endoplasmic reticulum stress. Biores Open Access (2018) 7(1):2–9. doi: 10.1089/biores.2017.0049
130. Kolb PS, Ayaub EA, Zhou W, Yum V, Dickhout JG, Ask K. The therapeutic effects of 4-phenylbutyric acid in maintaining proteostasis. Int J Biochem Cell Biol (2015) 61:45–52. doi: 10.1016/j.biocel.2015.01.015
131. Vega H, Agellon LB, Michalak M. The rise of proteostasis promoters. IUBMB Life (2016) 68(12):943–54. doi: 10.1002/iub.1576
132. Perlmutter DH. Chemical chaperones: a pharmacological strategy for disorders of protein folding and trafficking. Pediatr Res (2002) 52(6):832–6. doi: 10.1203/00006450-200212000-00004
133. Wang W, Qing X, Wang B, Ma K, Wei Y, Shao Z. Tauroursodeoxycholic acid protects nucleus pulposus cells from compression-induced apoptosis and necroptosis via inhibiting endoplasmic reticulum stress. Evidence-Based Complement Altern Med ECAM (2018) 2018:6719460. doi: 10.1155/2018/6719460
134. Yang L, Li Z, Ouyang Y. Taurine attenuates ER stress−associated apoptosis and catabolism in nucleus pulposus cells. Mol Med Rep (2022) 25(5):172. doi: 10.3892/mmr.2022.12688
135. Bao J, Qian Z, Liu L, Hong X, Che H, Wu X. Pharmacological disruption of phosphorylated eukaryotic initiation factor-2α/Activating transcription factor 4/Indian hedgehog protects intervertebral disc degeneration via reducing the reactive oxygen species and apoptosis of nucleus pulposus cells. Front In Cell Dev Biol (2021) 9:675486. doi: 10.3389/fcell.2021.675486
136. Atkins C, Liu Q, Minthorn E, Zhang S-Y, Figueroa DJ, Moss K, et al. Characterization of a novel PERK kinase inhibitor with antitumor and antiangiogenic activity. Cancer Res (2013) 73(6):1993–2002. doi: 10.1158/0008-5472.CAN-12-3109
137. Nguyen HG, Conn CS, Kye Y, Xue L, Forester CM, Cowan JE, et al. Development of a stress response therapy targeting aggressive prostate cancer. Sci Trans Med (2018) 10(439):eaar2036. doi: 10.1126/scitranslmed.aar2036
138. Chen X, Cubillos-Ruiz JR. Endoplasmic reticulum stress signals in the tumour and its microenvironment. Nat Rev Cancer (2021) 21(2):71–88. doi: 10.1038/s41568-020-00312-2
139. Tang C-HA, Ranatunga S, Kriss CL, Cubitt CL, Tao J, Pinilla-Ibarz JA, et al. Inhibition of ER stress-associated IRE-1/XBP-1 pathway reduces leukemic cell survival. J Clin Invest (2014) 124(6):2585–98. doi: 10.1172/JCI73448
140. Tufanli O, Telkoparan Akillilar P, Acosta-Alvear D, Kocaturk B, Onat UI, Hamid SM, et al. Targeting IRE1 with small molecules counteracts progression of atherosclerosis. Proc Natl Acad Sci U S A (2017) 114(8):E1395–404. doi: 10.1073/pnas.1621188114
141. Huang S, Zhu M, Wu W, Rashid A, Liang Y, Hou L, et al. Valproate pretreatment protects pancreatic β-cells from palmitate-induced ER stress and apoptosis by inhibiting glycogen synthase kinase-3β. J Biomed Sci (2014) 21:38. doi: 10.1186/1423-0127-21-38
142. Kirkham B, Chaabo K, Hall C, Garrood T, Mant T, Allen E, et al. Safety and patient response as indicated by biomarker changes to binding immunoglobulin protein in the phase I/IIA RAGULA clinical trial in rheumatoid arthritis. Rheumatol (Oxford England) (2016) 55(11):1993–2000. doi: 10.1093/rheumatology/kew287
143. Yang S, Zhang F, Ma J, Ding W. Intervertebral disc ageing and degeneration: The antiapoptotic effect of oestrogen. Ageing Res Rev (2020) 57:100978. doi: 10.1016/j.arr.2019.100978
144. Tian T, Wang H, Li Z, Yang S, Ding W. Intervertebral disc degeneration induced by needle puncture and ovariectomy: A rat coccygeal model. BioMed Res Int (2021) 2021:5510124. doi: 10.1155/2021/5510124
145. Li R, Wang Y, Chen P, Meng J, Zhang H. G-Protein-coupled estrogen receptor protects retinal ganglion cells via inhibiting endoplasmic reticulum stress under hyperoxia. J Cell Physiol (2021) 236(5):3780–8. doi: 10.1002/jcp.30149
146. Chen J, Liu Y, Pan D, Xu T, Luo Y, Wu W, et al. Estrogen inhibits endoplasmic reticulum stress and ameliorates myocardial ischemia/reperfusion injury in rats by upregulating SERCA2a. Cell Commun Signaling CCS (2022) 20(1):38. doi: 10.1186/s12964-022-00842-2
147. Imenshahidi M, Hosseinzadeh H. Berberis vulgaris and berberine: An update review. Phytother Res PTR (2016) 30(11):1745–64. doi: 10.1002/ptr.5693
148. Luo R, Liao Z, Song Y, Yin H, Zhan S, Li G, et al. Berberine ameliorates oxidative stress-induced apoptosis by modulating ER stress and autophagy in human nucleus pulposus cells. Life Sci (2019) 228:85–97. doi: 10.1016/j.lfs.2019.04.064
149. Feng K, Chen Z, Pengcheng L, Zhang S, Wang X. Quercetin attenuates oxidative stress-induced apoptosis via SIRT1/AMPK-mediated inhibition of ER stress in rat chondrocytes and prevents the progression of osteoarthritis in a rat model. J Cell Physiol (2019) 234(10):18192–205. doi: 10.1002/jcp.28452
150. Feng K, Ge Y, Chen Z, Li X, Liu Z, Li X, et al. Curcumin inhibits the PERK-eIF2-CHOP pathway through promoting SIRT1 expression in oxidative stress-induced rat chondrocytes and ameliorates osteoarthritis progression in a rat model. Oxid Med Cell Longevity (2019) 2019:8574386. doi: 10.1155/2019/8574386
151. Wu M-F, Li P-C, Chen C-C, Ye S-S, Chien C-T, Yu C-C. Cordyceps sobolifera extract ameliorates lipopolysaccharide-induced renal dysfunction in the rat. Am J Chin Med (2011) 39(3):523–35. doi: 10.1142/S0192415X11009007
152. Jiang Z-Q, Ma Y-X, Li M-H, Zhan X-Q, Zhang X, Wang M-Y. 5-hydroxymethylfurfural protects against ER stress-induced apoptosis in GalN/TNF-α-injured L02 hepatocytes through regulating the PERK-eIF2α signaling pathway. Chin J Nat Med (2015) 13(12):896–905. doi: 10.1016/S1875-5364(15)30095-9
153. Tao K, Wang B, Feng D, Zhang W, Lu F, Lai J, et al. Salidroside protects against 6-Hydroxydopamine-Induced cytotoxicity by attenuating ER stress. Neurosci Bull (2016) 32(1):61–9. doi: 10.1007/s12264-015-0001-x
154. Xie L, Chen Z, Liu M, Huang W, Zou F, Ma X, et al. MSC-derived exosomes protect vertebral endplate chondrocytes against apoptosis and calcification via the miR-31-5p/ATF6 axis. Mol Ther Nucleic Acids (2020) 22:601–14. doi: 10.1016/j.omtn.2020.09.026
155. Xiang H, Su W, Wu X, Chen W, Cong W, Yang S, et al. Exosomes derived from human urine-derived stem cells inhibit intervertebral disc degeneration by ameliorating endoplasmic reticulum stress. Oxid Med Cell Longevity (2020) 2020:6697577. doi: 10.1155/2020/6697577
156. Akintayo A, Stanley P. Roles for golgi glycans in oogenesis and spermatogenesis. Front In Cell Dev Biol (2019) 7:98. doi: 10.3389/fcell.2019.00098
Keywords: intervertebral disc degeneration, endoplasmic reticulum stress, apoptosis, PERK, IRE1α, ATF6
Citation: Huang J, Zhou Q, Ren Q, Luo L, Ji G and Zheng T (2023) Endoplasmic reticulum stress associates with the development of intervertebral disc degeneration. Front. Endocrinol. 13:1094394. doi: 10.3389/fendo.2022.1094394
Received: 10 November 2022; Accepted: 27 December 2022;
Published: 12 January 2023.
Edited by:
Yichuan Pang, Tongji University, ChinaReviewed by:
Esther Potier, Centre National de la Recherche Scientifique (CNRS), FranceFarhath Sultana, Icahn School of Medicine at Mount Sinai, United States
Copyright © 2023 Huang, Zhou, Ren, Luo, Ji and Zheng. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Tiansheng Zheng, emhlbmd0aWFuc2hlbmcyMDAyQDE2My5jb20=