
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Endocrinol., 15 December 2022
Sec. Clinical Diabetes
Volume 13 - 2022 | https://doi.org/10.3389/fendo.2022.1092431
This article is part of the Research TopicThe Role of Diabetes in the Pathophysiology and Prognosis of Ischemic StrokeView all 13 articles
 Peng-Fei Ding1†
Peng-Fei Ding1† Hua-Sheng Zhang1,2†Jie Wang1†
Hua-Sheng Zhang1,2†Jie Wang1† Yong-Yue Gao1,2
Yong-Yue Gao1,2 Jian-Nan Mao1,2
Jian-Nan Mao1,2 Chun-Hua Hang1,2
Chun-Hua Hang1,2 Wei Li1,2*
Wei Li1,2*The pathological condition of insulin resistance prevents the neuroprotective effects of insulin. Numerous studies have demonstrated that insulin resistance, as an independent risk factor for ischemic stroke, accelerates the formation of thrombosis and promotes the development of atherosclerosis, both of which are major mechanisms of ischemic stroke. Additionally, insulin resistance negatively affects the prognosis of patients with ischemic stroke regardless of whether the patient has diabetes, but the mechanisms are not well studied. We explored the association between insulin resistance and the primary mechanisms of brain injury in ischemic stroke (inflammation, oxidative stress, and neuronal damage), looking for potential causes of poor prognosis in patients with ischemic stroke due to insulin resistance. Furthermore, we summarize insulin resistance therapeutic approaches to propose new therapeutic directions for clinically improving prognosis in patients with ischemic stroke.
Stroke incidence and patient prognosis have not changed significantly over the past few decades, despite significant advances in clinical interventions aimed at reducing stroke risk factors like hypertension, smoking, and diabetes. Stroke remains the second-leading cause of disability and death globally (1). Since energy metabolism is a prerequisite for life activity, many studies have examined disorders of energy metabolism in brain tissue, particularly insulin. Insulin protects brain tissue development by preventing ischemia, oxidative stress, and apoptosis-induced brain tissue damage, regulating cholesterol metabolism in neurons and astrocytes. Insulin also can effectively alleviate cognitive dysfunction caused by Alzheimer’s disease (2). Insulin resistance (IR) has long been linked to ischemic stroke, which makes up 87% of strokes and is increasing (3). IR, which is present in the majority of type II diabetes (T2D) patients, promotes the development of ischemic stroke and is associated with a poor prognosis (4, 5). One of the best-known effects of IR is the presence of hyperglycemia, which may negatively affect brain function through various mechanisms (6). This review evaluates studies on IR as an independent risk factor for ischemic stroke, with a particular focus on the mechanisms and therapy associated with IR and ischemic stroke. Firstly, we discuss the relationship between IR and the risk factors for ischemic stroke (hypertension, hyperlipidemia, etc.). Secondly, we discuss the potential causes of IR leading to poor outcomes in ischemic stroke patients. Lastly, we synthesize the research on IR inhibitors and ischemic stroke currently available and look ahead to a day when reducing IR may be a useful strategy for both preventing and treating ischemic stroke.
Insulin is a well-known hormone secreted by pancreatic β-cells, which regulates peripheral glucose metabolism. Insulin signaling from the central nervous system (CNS) regulates energy balance via complex mechanisms (7). The concentration of insulin in cerebrospinal fluid is correlated with the concentration in circulating plasma. Although it is still debated whether insulin can be produced in the CNS, circulating insulin can enter brain tissue via the (BBB) (8).
Neurons are the ultimate beneficiaries of brain tissue glucose uptake, consuming 85% of the energy needed by brain tissue (9). Brain tissue accounts for only about 2% of an adult’s body weight but consumes about 20% of total body energy, and glucose metabolism produces even more than 95% of the ATP required by brain tissue, making glucose metabolism in brain tissue especially important (10). Glucose transporter 3 (GLUT3) and glucose transporter 4 (GLUT4) are found on the neuronal cell membrane, with GLUT4 serving as a critical determinant of glucose homeostasis and being highly dependent on insulin (11, 12). Under insulin stimulation, GLUT4 translocation from the cytoplasm to the cell membrane in neurons in the hippocampus and cortex has a glucose-promoting transport effect (13). Insulin not only maintains the balance of energy metabolism in brain tissue but also stimulates neurite outgrowth, modulates catecholamine release and uptake, regulates the expression and localization of N-methyl-D-aspartate (NMDA), α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid (AMPA) and γ-aminobutyric acid (GABA) receptors, and modulates synaptic plasticity to enhance neuronal survival by inhibiting apoptosis (14).
Insulin needs to bind to insulin receptors (INSR) on plasma membranes to exert its known biological effects. Insulin receptor, which consists of two extracellular α subunits and β subunits, are found in the brain and on most cells. In brain tissue, insulin receptors are mainly distributed in the hypothalamus, olfactory bulb, hippocampus, striatum, cerebral cortex, and cerebellum (14). Insulin first binds to the extracellular α subunits of insulin receptors, which induces the autophosphorylation of intracellular β subunits (15). INSR has two isoforms, A and B. Evidence shows that the brain only expresses the shorter form INSR-A rather than the full-length INSR-B, which differs from many peripheral tissues (16). The most classic INSR scaffold is the insulin receptor substrate (IRS) family, which has six isoforms (IRS1-6). IRS1 and IRS2 are assumed to mediate most of the metabolic effects of INSR activation (17). In brain tissue, insulin acts through IRS1, and IRS2 is strongly related to the activity of insulin-like growth factor 1(IGF-1) (16). By phosphorylating several IRS tyrosine residues, INSR attracts downstream signaling effectors to relay and enhance insulin responses. Tyrosine-phosphorylated IRS proteins active phosphoinositide 3-kinase (PI3K) and then phosphorylate AKT to exert the known physiological effects of insulin (Figure 1) (17).
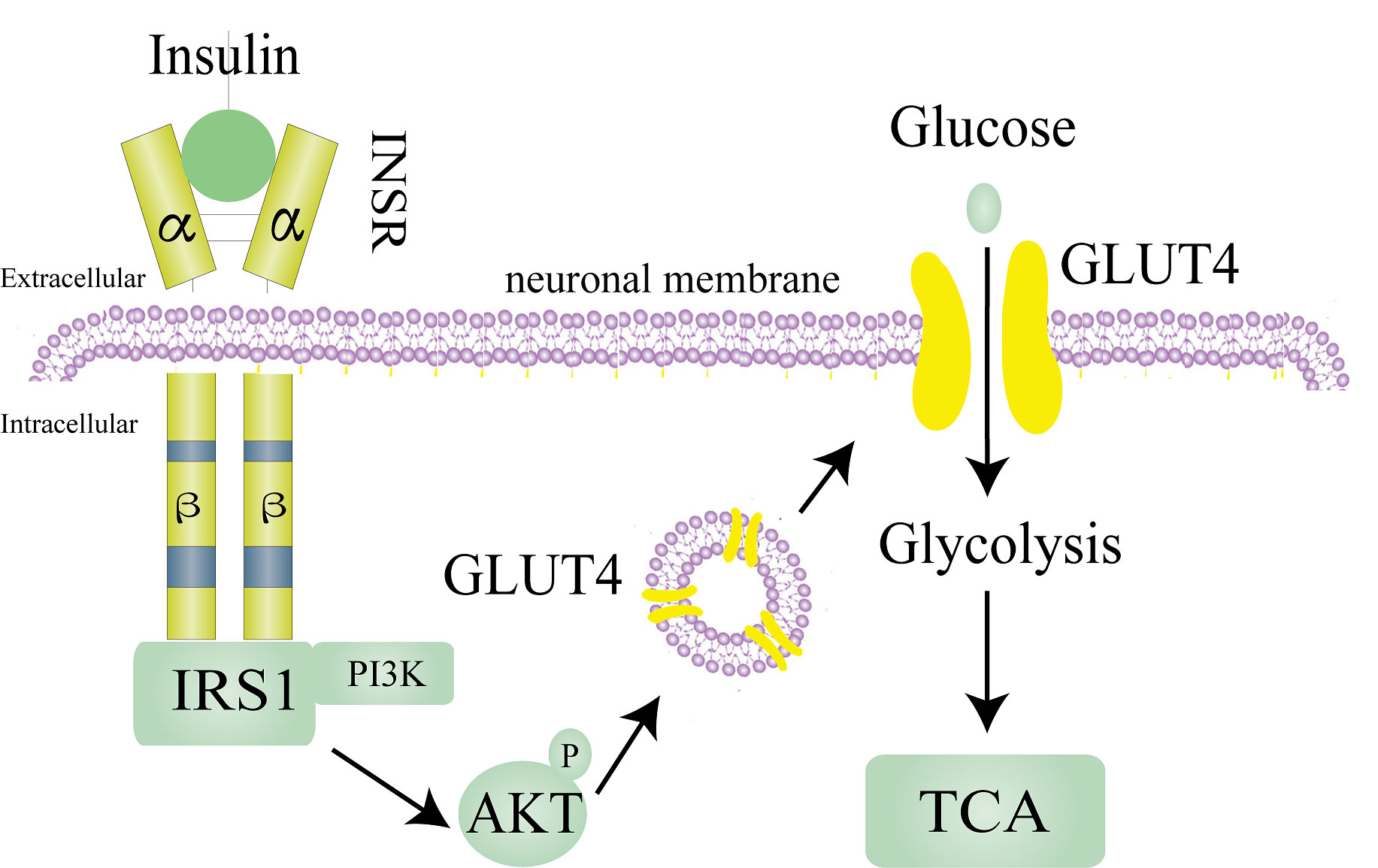
Figure 1 Normal insulin pathway in the neuron.
IR, as the name implies, is the failure of tissues to the normal response to insulin stimulation (18). IR appears earlier in brain tissue than in the periphery, suggesting that brain tissue is more vulnerable to IR, particularly in brain diseases such as ischemic stroke (14). There are four clinically accepted criteria for detecting the presence of IR in patients: (1) the gold standard assessment, homeostasis model assessment of IR (HOMA-IR); (2) oral glucose tolerance tests (OGTT); (3) C-peptide release test; and(4) triglyceride glucose (TyG) index, each with advantages and disadvantages (Table 1) (19–22). In addition, the TyG index combined with obesity indices, including body mass index (BMI), waist circumference (WC), and waist height ratio (WHtR) were found to be more accurate than the TyG index alone. TyG-BMI had the best ability to detect IR and the best consistency with HOMA-IR among them (23). However, TyG-BMI, TyG-WC, and TyG-WHtR share the same advantages and disadvantages as the TyG index. At the molecular level, while abnormalities in INSR number, receptor kinase activity, and various receptors for insulin action can all lead to IR production in tissues but abnormal serine phosphorylation of IRS blocking the PI3K/AKT pathway is thought to be the main cause of IR (17, 24). As a result, the ratio of serine phosphorylation to total phosphorylated IRS can be used as a marker of IR in either the brain or peripheral tissues, with a higher percentage indicating increased IR (25).
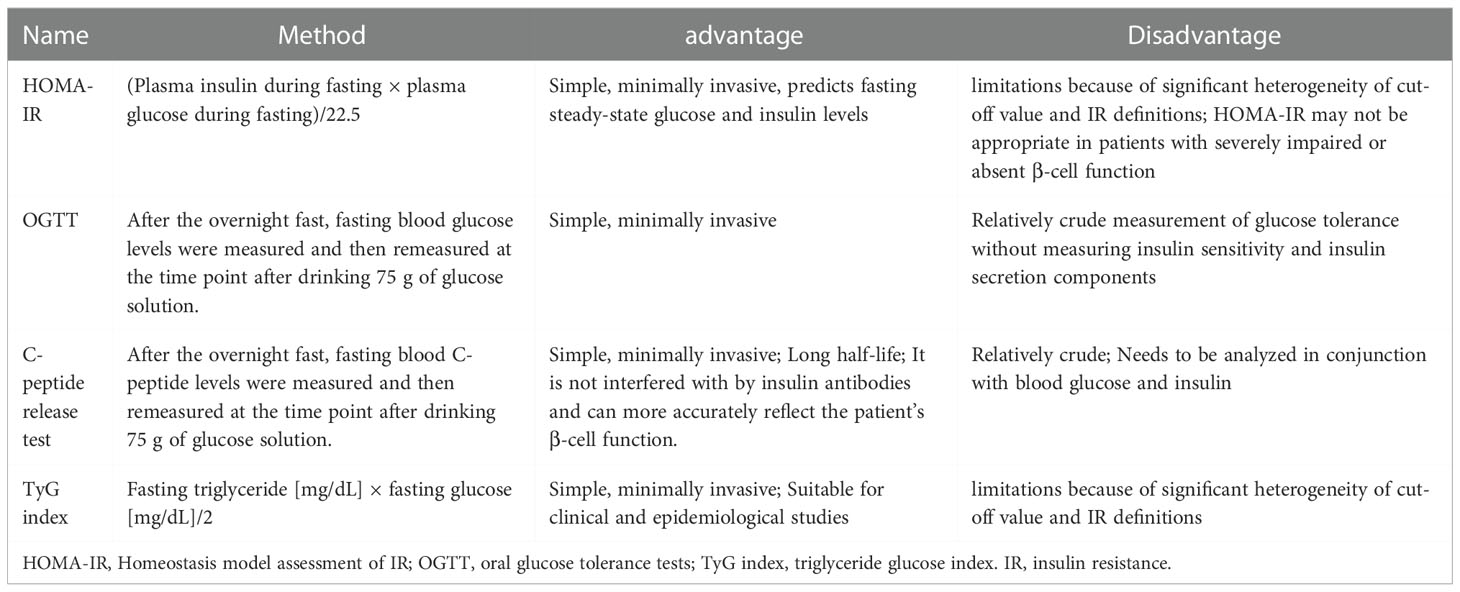
Table 1 Four normal methods for assessment of IR in clinical.
Ischemic stroke mechanisms are classified as embolism, large vessel disease, and small vessel disease (26). In up to two-thirds of acute stroke patients, abnormalities in glucose regulation (of which diabetes is a manifestation) are observed, and because ischemic stroke accounts for 87% of strokes, there have been numerous studies on T2D and ischemic stroke (3, 27). According to research, ischemic stroke and T2D share many causative factors, including IR and IR-associated syndrome. An earlier study found that 50% of 72 nondiabetic patients with transient ischemic attack (TIA) or ischemic stroke had significant IR (28). And in non-diabetic ischemic stroke patients, the value of HOMA-IR ≥2. 5, suggesting the presence of IR, in more than 20% of cases (29). Numerous studies show that IR is a risk factor for ischemic stroke and can lead to the incidence of ischemic stroke (30). Based on past reviews and contemporary research, this section will explain the relationship between IR and the two main causes of ischemic stroke—embolism and atherosclerosis—and how it influences the occurrence and progression of ischemic stroke (Figure 2).
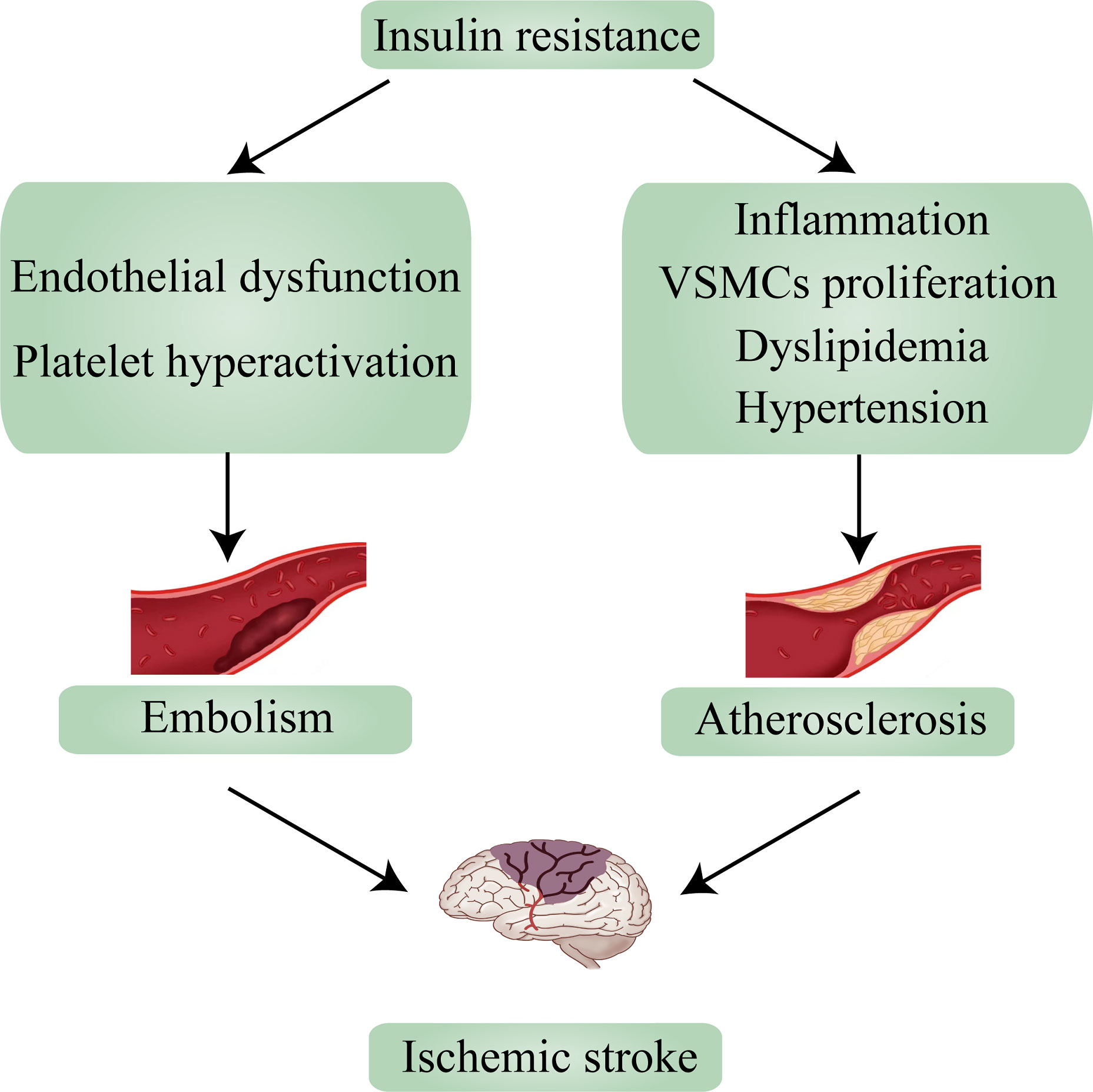
Figure 2 Insulin resistance leads to ischemic stroke via embolism and atherosclerosis. VSMCs, vascular smooth muscle cells.
Ischemic stroke is most frequently caused by embolism. Most embolisms are blood clots that arise from the heart due to heart disease (cardiogenic embolism). Atrial fibrillation, heart valve disease, and myocardial infarction or cardiomyopathy brought on by excessive blood pressure are common cardiac disorders that cause stroke (26). Thrombosis is commonly considered to be a pathological hemostatic deviation caused by coagulation and platelet activation. The formation of an intravascular thrombus (clot) and vascular occlusion constitute thrombosis. Ischemic stroke occurs when dislodged clots move and block cerebral blood vessels (31). Interventions using antiplatelet agents to prevent platelet activation and thrombosis are known to reduce the incidence and severity of ischemic strokes. The metabolic environment of T2D includes IR, hyperglycemia, excessive release of free fatty acids, and other metabolic abnormalities affecting blood vessel walls as a result of a variety of events such as endothelial dysfunction and platelet hyperactivation. Compared to non-diabetes, T2D had a two-to four-fold higher risk of recurrent atherosclerotic thrombotic events and vascular complications. The activation of these events causes even more vasoconstriction and promotes thrombosis (32). IR, the key role of T2D, has also attracted the interest of researchers. A rising number of studies are focusing on the connection between IR and thrombosis. Researchers found that IR can impair endothelial cell function and enhance platelet adhesion, activation, and aggregation, resulting in the formation of thrombosis (33). Endothelial cells serve several functions, including pro- and anti-coagulation, which are balanced under normal conditions. When endothelial cell function is impaired, the risk of thrombosis increases (34). In IR, the PI3K pathway is impaired, resulting in decreased NO (vasodilator) production, whereas the MAPK pathway is activated, leading to increased ET-1 (vasoconstrictor) production and ultimately endothelial dysfunction (35). Excessive free fatty acids (FFA) release caused by IR and excess FFA may cause lipotoxicity, which leads to increased expression of coagulation tissue factors (e.g., PAI-1) via the same mechanism as glucotoxicity, resulting in a prothrombotic state (36). Furthermore, FFA can set off a vicious cycle by activating several pathways which increases the percentage of IRS1 serine phosphorylation (37). Chronic hyperglycemia and decreased serum paraoxonase/arylesterase 1 (PON-1) activity may impair high-density lipoprotein’s (HDL) anti-inflammatory capacity. Eventually, serum tumor necrosis factor-α (TNF-α), interleukin-6 (IL-6), and C-reactive protein (CRP), which respond to inflammatory levels, were eventually elevated, indicating a link between the inflammatory response caused by IR and endothelial dysfunction (38). In addition, IR, assessed by HOMA-IR, was associated with endothelial dysfunction in patients with chest pain and non-diabetic patients with normal myocardial perfusion (39).
Several studies have shown that IR promotes vascular occlusion and cardiovascular disease (CVD) development by influencing platelet adhesion and aggregation (32). Platelets’ major function in the body is to support primary hemostasis and intravascular blood flow. Platelet adhesion, aggregation, and release are the three steps in the platelet activation process. Adhesion to the subendothelial extracellular matrix occurs through the initial interaction of the matrix with specific receptors on the platelet, including the GP1b/V/IX complex bound to the Von Willebrand factor and the GPVI and αIIβ1 receptors bound to the collagen component of the extracellular matrix on the platelet surface. Firm adhesion leads to initial clot or thrombus formation, and activated platelets bound within the thrombus will begin to take up new platelets from the circulation via platelet-platelet interactions mediated by integrin receptor αIIbβ3. Platelets express two purinergic receptors, P2Y1 and P2Y12, which have been shown to play an important role in platelet activation, one of which (P2Y12) is a target for antiplatelet therapy (40). Treatment with clopidogrel and aspirin improves prognosis in patients with cerebral infarction without increasing the risk of moderate to severe bleeding (41). The effect of hyperinsulinemia on platelets is complex and varies between insulin-resistant patients and healthy individuals. In healthy individuals, insulin reduces platelet aggregation and the release of pro-aggregation agents by facilitating the transfer of magnesium to platelets (such as thromboxane B2) (42). Insulin naturally inhibits platelet hyperactivity. It makes platelets more sensitive to prostacyclin (PGI2) and increases endothelial cell production of PGI2 and nitric oxide (NO), allowing platelets to maintain normal function. Platelet hyperactivation favors macrovascular and microvascular events when IR occurs, which may explain why platelets adhere to the vascular endothelium more frequently in T2D patients than in healthy individuals (43). In addition, hyperglycemia may activate platelets through miR-144 and miR-223 to downregulate IRS1 and upregulate P2Y12 expression in the platelets via IRS1/PI3K/AKT pathways (44).
Atherosclerosis is another common underlying cause of ischemic stroke (26). Atherosclerosis is a chronic inflammatory disease of large and medium-sized arteries that can lead to ischemic heart disease, stroke, and peripheral vascular disease collectively referred to as CVD (45). Increasing evidence has demonstrated that atherosclerosis is a major cause of ischemic stroke (46). Furthermore, there is strong evidence that IR or metabolic syndrome caused by IR contributes to the pathogenesis of ischemic stroke by promoting the formation of atherosclerotic and advanced plaques in the progression of atherosclerosis (47). According to epidemiological studies, hyperinsulinemia is an independent risk factor for atherosclerosis. Hyperinsulinemia caused by IR can exacerbate atherosclerosis by promoting vascular inflammation, vascular smooth muscle cells (VSMCs) development, a pathological cholesterol profile, hypertension, and immune cell recruitment to the endothelium (48). The relationship between inflammation and T2D can be traced back to the 1950s. At the molecular level, IR is promoted when macrophage polarization shifts from an alternative M2 (anti-inflammatory) activation state maintained by STAT6 and PPARs to a classical M1 (pro-inflammatory) activation state driven by NF-B, AP1. In short, inflammation promotes the development of IR (49). In contrast, researchers gave a 4-hour insulin intervention to healthy volunteers with normal glucose tolerance and no history of diabetes, took biopsies of their lateral femoral muscles, and discovered noticeably elevated levels of related inflammatory genes (50). We are aware of a mutual relationship between IR and inflammation. With both protective and pathogenic roles, monocyte-macrophage lineage cells and VSMCs are two key participants in the atherosclerotic process. Proliferation of VSMCs promotes plaque growth while also forming the atheroma’s fibrous cap (51). Hyperinsulinemia, which is caused by IR, is a potential growth factor that promotes its growth through the MAPK pathway, which catalyzes the phosphorylation of transcription factors that stimulate VSMCs growth, proliferation, and differentiation (52). It has been demonstrated that IR-related inflammation can also promote the development of VSMCs (53). C-peptide, produced by the cleavage of proinsulin in the β-cell. It is secreted equimolarly with the other cleavage product, insulin (54). C-peptide study extends to our knowledge of the mechanisms that promote VSMCs proliferation in hyperinsulinemia conditions (55). PI3K/AKT and ERK1/2-MAPK are thought to be critical signaling pathways controlling VSMCs. Through the activation of the protein tyrosine kinase Src, which can function as an intermediary in signaling networks that link G-protein-coupled receptors with downstream signaling cascades like the PI3K/Akt and the Ras/MAPK pathway, C-peptide can stimulate the growth of VSMCs (56, 57).
Atherosclerosis develops as a result of a disturbed cholesterol homeostasis balance (58). Numerous studies have shown that IR leads to disorders of lipid metabolism (59). In comparison to the normal metabolic state, insulin resistance promotes excess de novo lipogenesis as well as the production and secretion of very-low-density lipoprotein (VLDL) (60). Additionally, IR can accelerate the production of connective tissue in blood vessel walls and the aggregation of LDL cholesterol into arterial smooth muscle, both of which directly accelerate the development of atherosclerosis and, eventually, the occurrence of an ischemic stroke (47).
Hypertension is frequently regarded as a major cause of hemorrhagic stroke, as well as a risk factor for ischemic stroke (3). IR and hypertension are both independent risk factors for CVD, and a growing number of studies are beginning to recognize the link between these two diseases that promote the formation and progression of atherosclerosis, which together lead to CVD (61). Despite the fact that a short period of insulin stimulation did not significantly increase blood pressure in non-diabetic subjects, the researchers acknowledge that IR is directly correlated with the severity of hypertension (62). In a subsequent study, the researchers prolonged the insulin intervention and discovered that chronic insulin administration can significantly raise the blood pressure of lean rats (63). Leptin is crucial for controlling body weight, plays a role in the development of the IR syndrome, and is associated with cardiovascular disease. Increased sympathetic activity mediated by leptin may cause short- and long-term alterations in blood pressure through central and peripheral effects, which may help to explain how IR affects blood pressure (64). Although studies in humans appear controversial, the role of insulin in promoting antidiuretic effects and stimulating sympathetic nervous system activation has been confirmed as a potential mechanism by which insulin may elevate blood pressure (65). Furthermore, animal studies have shown that long-term insulin administration accelerates the development of atherosclerosis (47).
Stroke prevention and treatment are divided into two parts. Primary prevention includes modifying one’s lifestyle and treating risk factors such as hypertension, diabetes mellitus, etc. Secondary prevention includes surgical intervention and treatment of IR, etc. (66). In clinical studies, researchers have found that IR is independently associated with poor functional outcomes after acute ischemic stroke, regardless of whether the patient has T2D (5, 67). However, at the mechanistic level, it has not been well studied how IR negatively affects the prognosis of patients with ischemic stroke. Starting with the potential damage caused by IR, we summarize the potential mechanisms by which IR contributes to the poor prognosis of ischemic stroke patients in this section.
The inflammatory response is believed to play a crucial part in the cerebral damage caused by an ischemic stroke (68). Microglia are important immune cells in brain tissue. Brain microglia are activated in response to ischemia. On the one hand, activated microglia secrete pro-inflammatory factors such as TNF-α and interleukin-1β (IL-1β), which cause cellular damage; on the other hand, activated microglia have phagocytic and major histocompatibility complex (MHC) class II-restricted antigen-presenting properties, which help remove dead tissue and debris after ischemia. Furthermore, activated microglia can promote the production of neurotrophic growth factors such as brain-derived neurotrophic factor (BDNF) (69, 70). This is related to the state of microglia polarization. Microglia polarization refers to the development of a classically activated (M1, pro-inflammatory) or alternatively activated (M2, anti-inflammatory) phenotype of activated microglia (71). M1 and M2 are in a dynamic equilibrium under normal physiological conditions, but in ischemic stroke, M2 is converted to M1 (72). Severe ischemic injury accompanied by a pro-inflammatory environment can produce and release large amounts of inflammatory factors causing ischemic damage to brain tissue, among which IL-6 and TNF-α can also lead to an increase in neutrophils in circulating dead brain tissue. Increased neutrophil counts not only correlate with infarct size but also can disrupt the BBB (73). Obesity can induce activation of IKKβ, leading to nuclear translocation of NF-κB and, as a result, the production of various inflammatory markers and potential mediators, and obesity can also co-promote phosphorylation of IRS1 at serine sites (ser302 and ser307) through JNK activation, which together leads to the development of IR (74). A majority of studies have found that inflammation plays a significant role in the development of IR, some other research has also shown that IR actually promotes the development of an inflammatory response. Both sides agree that inflammation and IR feed off each other in a vicious cycle (49). In both diet-induced obesity and genetically (mTORC2-knockout) induced adipose-specific IR, researchers discovered that IR causes local accumulation of pro-inflammation macrophages. IR produces the chemokine monocyte chemoattractant protein 1 (MCP1), which recruits monocytes and activates pro-inflammatory macrophages (75). Researchers discovered that blocking glucose oxidative metabolism not only prevented macrophage polarization to the M2 phenotype but also drove macrophages into the M1 type. Simultaneously, forcing an M1 macrophage to undergo oxidative metabolism boosts the M2 phenotype (76, 77). IR prevents glucose from entering neurons for oxidative phosphorylation, which explains the potential cause of IR-induced inflammation in terms of macrophage metabolic reprogramming.
Oxidative stress and inflammation are two key points in ischemic stroke, and there has been considerable evidence in recent years that oxidative stress, associated with the overproduction of reactive oxygen species (ROS), is the underlying mechanism of brain injury in stroke (78). In ischemic stroke, excessive ROS production disrupts the balance of oxidant and antioxidant composition, resulting in oxidative stress (79). ROS are naturally occurring small molecule by products of oxygen metabolism that include superoxide anion radicals (O2-), hydrogen peroxide (H2O2), and hydroxyl radicals (-OH). The most reactive substance, -OH, can damage polyunsaturated fatty acids, causing loss of biofilm integrity, proteins, and enzymes, as well as affecting nucleic acids’ functional qualities and producing mutations that eventually result in cellular senescence or death (80). ROS are beneficial in certain physiological processes such as cell signaling, induction of pro-mitotic responses, immune defense, cellular senescence, apoptosis, and breakdown of toxic compounds. However, most studies indicate that ROS causes cellular damage and impaired function during biological stress (81). ROS can react with lipids to form peroxides, which are then degraded to aldehydes (e.g., hydroxynonenal) that are toxic to neurons and white matter in the brain, induce apoptosis, and are significantly associated with focal ischemia in rats (82). Increased ROS production after transient global ischemia upregulates p53 upregulated apoptosis regulator (PUMA) and Bcl-2 and Bax in neurons, and PUMA, along with anti-apoptotic Bcl-2 or pro-apoptotic Bax, plays an important role in ischemic neuronal death (83, 84). The effects of IR and oxidative stress also appear to be reciprocal. Oxidative stress-induced oxidative damage markers such as malondialdehyde (MDA), advanced glycation end products (AGEs), and 8‐hydroxy‐2′‐deoxyguanosine (8‐OH‐dG) were found to reduce insulin sensitivity in skeletal muscle cells and adipocytes (85). Some investigators believe that oxidative stress can phosphorylate IRS proteins by activating the IKKβ/NF-κB and JNK pathways, resulting in IRS degradation (86). Furthermore, some researchers believe that metabolic disturbances caused by peripheral IR may be the source of oxidative stress in brain tissue. IR increases FFA and promotes glucotoxicity and lipotoxicity and promotes NF-κB nuclear translocation to increase ROS production (87). Mitochondria are the main source of ROS, and in recent years, researchers have recently begun to study the relationship between mitochondrial function and IR. On the one side, researchers think in some cases, the reduced mitochondrial function may be a major cause of IR. They found that increased intracellular fatty acyl CoA and diglycerides in myocytes as a result of reduced β-oxidation due to mitochondrial dysfunction or increased plasma transport, activating serine/threonine kinases such as protein kinase C (PKC) in skeletal muscle. This, in turn, activates IRS1 serine residues, resulting in IR (88). Obesity is a common cause of IR. The 75-kDA glucose regulatory protein (GRP75) is downregulated in mice fed a high-fat diet (HFD); however, increasing GRP75 prevents HFD-induced obesity and IR. GRP75 is required for mitochondrial homeostasis because it is a component of the mitochondrial mass control system and mitochondrial-associated membranes. It can improve insulin sensitivity by regulating mitochondrial function by controlling the turnover of the mitochondrial-supercomplex (89). Furthermore, insulin is required for the maintenance of mitochondrial function. Researchers recently discovered that insulin deprivation not only resulted in decreased efficiency of ATP production by isolated muscle mitochondria but also increased degradation of mitochondrial proteins in streptozotocin (STZ)-induced diabetic mice treated continuously with insulin implants and mice that had their insulin implants removed after re-establishing healthy glycemic control. This increase in protein degradation could be attributed to increased expression of the mitochondrial autophagy marker protein (Beclin) (90). To better understand the connection between IR and mitochondrial function, researchers used IRS1 and IRS2 double-knockout mice to mimic IR. This study revealed increased expression of several forkhead box O1 (Foxo1) target genes in the liver of double knockout mice, including heme oxygenase-1 (Hmox1), which disrupts complexes III and IV in the respiratory chain and reduces the NAD+/NADH ratio and ATP production (91).
Neurons are the most fundamental structural and functional elements of the nervous system. There is widespread agreement that prompt and effective neuroprotection following an ischemic stroke can significantly improve patients’ prognoses. In clinical practice, drugs (e.g., gangliosides) are widely used as neuroprotective agents in patients with ischemic stroke. However, the actual mechanism of neuronal death after ischemic stroke remains unknown. We discuss the association of IR with these mechanisms based on a recent review of the mechanisms of neuronal death after ischemic stroke (92). Ischemic stroke causes a large release of the neuroexcitatory transmitter glutamate, which leads to excessive activation of NMDA receptors and allows a large inward flow of calcium ions. Excessive intracellular calcium ion accumulation activates many calcium-dependent proteases, lipases, and deoxyribonucleases, leading to cell death. Moreover, calcium overload also causes the release of apoptotic factors from the mitochondria, which induces apoptosis (93). Excitotoxicity and inflammatory responses can cause not only receptor-interacting protein kinase 1 (RIPK1) activation to promote neuronal cell necrosis, but also DNA damage. Once DNA is damaged, p53 can promote ferroptosis by inhibiting SLC7A11 expression or promoting spermidine/spermine N1‐acetyltransferase 1(SAT1) and glutaminase 2 (GLS2) expression (94, 95). After an ischemic stroke, oxygen and glucose transport are restricted, anaerobic oxidative metabolism takes over as the primary source of neuronal ATP, and energy supply exceeds demand, resulting in neuronal apoptosis (96). oxygen-glucose deprivation also inhibits the AKT pathway leading to decreased mTORC1 activity, a classical pathway that blocks autophagy activation (97). Excessive ROS accumulation due to mitochondrial dysfunction activates FOXO3, increasing the abundance of LC3 to promote autophagosome generation, and excessive autophagy can increase neuronal apoptosis (98). Autophagy response dysfunction causes adipocyte dysfunction, and the development of IR. IR induces glucotoxicity, which exacerbates oxidative stress, inflammation, and endoplasmic reticulum stress caused by lipotoxicity, further impairing the autophagy response (99). Furthermore, our team found that IR inhibits GLUT4 membrane translocation, leading to the apoptosis of neurons due to insufficient glucose uptake. Inhibiting neuronal IR attenuates neuronal apoptosis (100). All of this suggests that IR could cause neuronal death. According to research, ischemic stroke patients’ declining memory and cognitive function may be caused by neural synaptic plasticity dysfunction (101, 102). Synaptic plasticity is an activity-dependent change in the strength of neuronal connections and has long been recognized as an important component of learning and memory (103). Considering that insulin is required for neurosynaptic function, IR also has an impact on neuronal function. It has the ability to alter synaptic plasticity in neurons (104). Neuronal injury may be responsible for poor clinical outcomes such as worsening neurological function and a poorer functional outcome at 3 months in patients with ischemic stroke due to IR. A new study discovered that maternal HFD-dependent IR impairs multigenerational synaptic plasticity, learning, and memory (105). By inducing IR in female mice with HFD and evaluating hippocampus-dependent synaptic plasticity and memory in female offspring (no difference between females and males), researchers found that offspring novelty recognition experimental preference indices were lower than controls and that only BDNF was reduced in all three generations of offspring mice. Epigenetic inhibition of exon-specific BDNF expression in offspring may explain how HFD-induced IR multi-generationally impairs synaptic plasticity, learning, and memory. Furthermore, maternal administration of BDNF or lack of pro-IR gene p66Shc abrogated the transmission of HFD-dependent cognitive dysfunction to offspring.
T2D is an established risk factor for ischemic stroke, however, numerous research has revealed that many ischemic stroke patients who do not have T2D but do have IR also have a significantly worse prognosis than those who do not have IR. Insulin is important for maintaining the functional integrity of the brain, and peripheral and central insulin dysfunction due to IR may be an independent risk factor for stroke. IR treatment is an important component of the secondary prevention of ischemic stroke (66). Increasing CNS insulin concentrations or CNS insulin sensitivity may be effective not only in preventing ischemic stroke but also in improving the prognosis of ischemic stroke patients. We discuss the treatment of IR in terms of non-pharmacological modalities, insulin therapy, and increasing insulin sensitivity (Table 2).
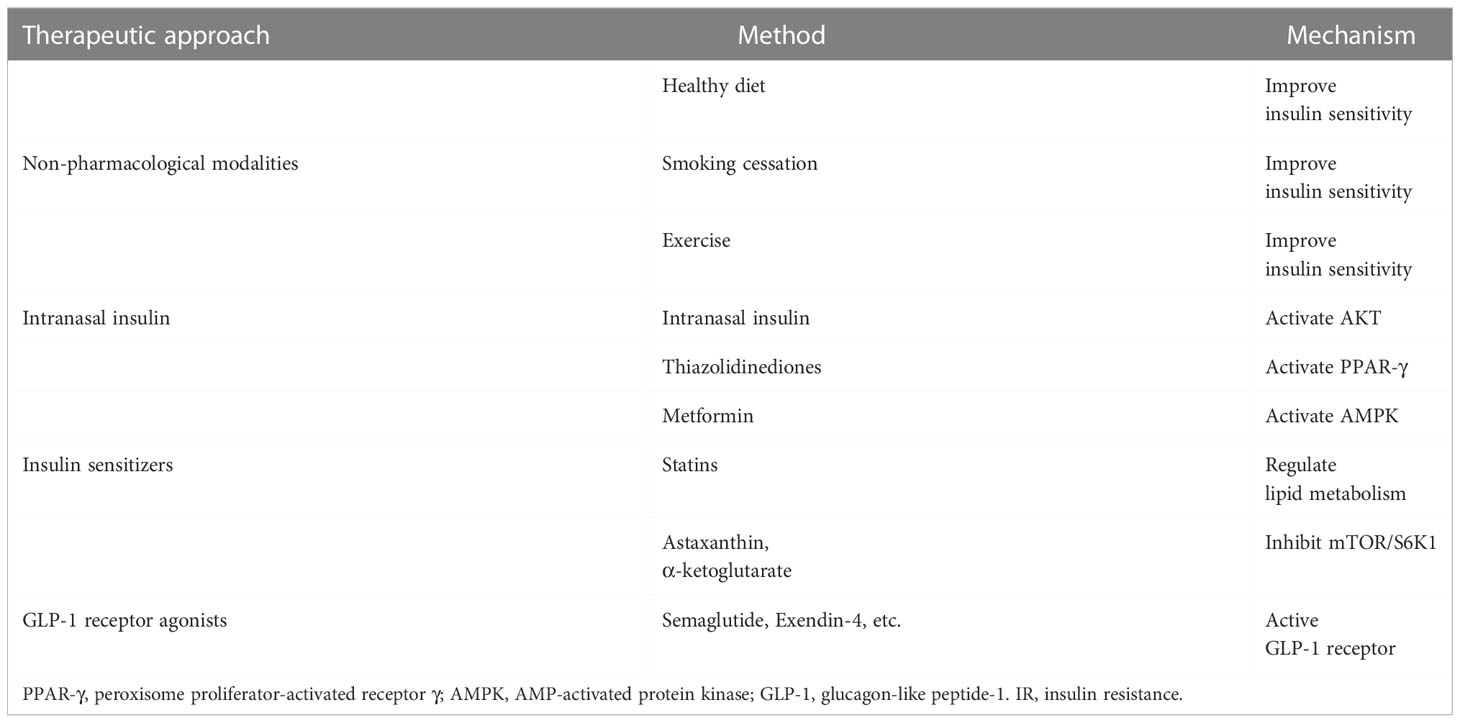
Table 2 The treatment of IR in ischemic stroke.
Lifestyle changes, including a healthy diet, weight loss, smoking cessation, and appropriate physical activity are well-known ways to improve peripheral insulin sensitivity and are considered primary prevention of ischemic stroke (66). Obesity is associated with an increased prevalence of vascular risk factors, and obesity is typically the primary cause of IR. In the INTERSTROKE research, obesity was responsible for 82% and 90% of the population-attributable risk for ischemic and hemorrhagic stroke, respectively (106). The researchers discovered that a healthy vegetarian diet was substantially associated with a lower risk of overall stroke and that vegetarian diet was unrelated to stroke in a follow-up of 3,015 ischemic strokes and 853 hemorrhagic strokes (107). Compared with the control condition, folic acid was associated with a lower risk of stroke [relative risk (RR)= 0.80, confidence interval (CI)=0.67-0.96], whereas combined calcium plus vitamin D intake was associated with an increased risk (RR=1.17, CI=1.05-1.30) (108). Dietary control in diabetic patients is a simple, effective, safe, and efficient way to improve IR (109). Researchers identified smoking and exposure to secondhand smoke as definite risk factors for stroke in a comprehensive review of the Global Burden of Disease Study 2019 (110). Researchers found that both male and female smokers (cigarettes + e-cigarettes and cigarettes or e-cigarettes) were more likely to develop IR than non-smokers (male, dual: odds ratio (OR)=2.19, CI=1.39–3.44; single: OR=1.78, CI=1.43–2.22; female, dual: OR=2.32, CI=1.01–5.34; single: OR=1.76, CI=1.28–2.42) (111). There is sufficient evidence to incorporate cardiopulmonary and mixed training involving walking into post-stroke rehabilitation programs. Cardiopulmonary training, as well as, to a lesser extent, mixed training, improves movement and balance and thus reduces disability during or after conventional stroke treatment (112). Exercise and IR have a well-established link. IRS1 and GLUT4 mRNA levels were lower in the proliferating endometrium of patients with polycystic ovaries compared with BMI-matched controls. Lifestyle changes combined with diet and exercise resulted in improved menstrual patterns in 65% of overweight/obese women with polycystic ovary syndrome, and significantly higher IRS1 and GLUT1 mRNA levels were found in the endometrium of these women with improved menstrual function, suggesting that lifestyle interventions can improve insulin sensitivity in patients (113). Recent studies have consistently shown that moderate repetitive exercise for 30 minutes or longer at least three times per week for at least eight weeks improves insulin sensitivity in patients with diabetes, obesity, and metabolic syndrome. Improved insulin sensitivity may be associated with weight loss (114). In summary, lifestyle changes are not only an important part of the primary prevention of ischemic stroke, but they can also play an important role in the treatment of IR in the secondary prevention of ischemic stroke.
IR is essentially a decrease in insulin cellular utilization, and exogenous insulin supplementation is also an effective way to increase intracellular insulin concentrations. Insulin administration in stroke patients has shifted from intravenous to intranasal. In a meta-analysis including 9 studies (1,491 patients), there was no statistically significant difference in the incidence of mortality (OR=1.16, CI= 0.89-1.49) and improvement in neurological function (OR=1.01, CI=0.81-1.26) in patients treated with intravenous insulin compared to controls. However, the odds of any hypoglycemia (OR=8.19, CI=5.60-11.98) and symptomatic hypoglycemia (OR=6.15, CI=1.88-20.15) were significantly higher in patients receiving intravenous insulin therapy (115). The reason for this phenomenon may be that although peripheral insulin injections are the classical way to treat T2D, they may induce hypoglycemia in stroke patients with only an insulin-resistant state that has not yet developed T2D, and this hypoglycemia can cause secondary brain damage, and peripheral insulin injections may be ineffective due to impaired insulin transport by BBB. As a result, researchers have concentrated on intranasal insulin administration as a neuroprotective treatment for ischemic stroke. Intranasal administration is a safe and effective method of bypassing the BBB and increasing distribution to the CNS without the downsides of systemic side effects or first-pass metabolism while reducing IR (116). Investigators discovered that the incidence of vascular disease was not significantly higher in the insulin group than in the placebo group in a clinical study examining the safety of intranasal insulin administration for the treatment of Alzheimer disease dementia, also demonstrating that intranasal insulin administration is safe (117). Intranasal insulin administration reduced cerebral infarction and neurological deficits and increased phosphorylation of Akt and endothelial nitric oxide synthase (eNOS) proteins in STZ-induced diabetic rats with focal cerebral ischemia-reperfusion injury. When insulin is combined with N-iminoethyl-L-ornithine, an eNOS inhibitor, the beneficial effects of insulin on infarct volume and neurological deficits in ischemia-reperfusion diabetic rats are inhibited, but the hypoglycemic effect of insulin is not affected (118). Another hemorrhagic stroke study found that administering 1 IU of intranasal insulin significantly reduced hematoma volume, brain edema, and BBB permeability in intracerebral hemorrhage-induced mice. The researchers discovered that after intranasal insulin administration, the expression of AKT (Ser473) and GSK3 (Ser9) in perihematomal tissue was significantly increased. Insulin binding to its receptor activated AKT through serine phosphorylation at position 437, and the activated AKT promoted GSK3β phosphorylation at Ser9, which inactivated GSK3β. GSK3β was associated with neuronal death, and GSK3β over-activation induces neuronal degeneration and reduces the expression of claudin-1 and claudin-3, the major components of BBB tight junctions. The activation of the AKT/GSK3 signaling pathway by intranasal insulin may be the mechanism by which intranasal insulin administration improves neurological function in mice with cerebral hemorrhage (119). The evidence for intranasal insulin administration in protecting neurological function is sufficient, and intranasal insulin administration is a potential therapeutic modality for improving IR and restoring neurological function in ischemic stroke. In short, AKT is an essential pathway for insulin to exert its physiological effects, and although clinical trials of intranasal insulin administration for ischemic stroke need to be supplemented, the evidence for intranasal insulin administration in protecting neurological function is sufficient.
Another viable strategy is to use insulin sensitizers to directly combat IR. Despite the fact that there have been few studies on insulin sensitizers in ischemic stroke, we have identified a number of useful insulin sensitizers based on previous research in T2D patients. Thiazolidinediones (TZDs) reduce insulin resistance directly by activating peroxisome proliferator-activated receptor γ (PPAR-γ), which promote mesenchymal stem cell differentiation into adipocytes, promote lipogenesis in peripheral adipocytes, lower hepatic and peripheral triglycerides, reduce visceral adipocyte activity, and enhance adiponectin (120). Two common TZDs are pioglitazone and rosiglitazone, both of which are PPAR-γ agonists. 3,876 patients who had recently experienced an ischemic stroke or TIA and had HOMA-IR > 3.0 but no T2D were split into two groups in a multicenter, double-blind experiment (pioglitazone or placebo). They discovered pioglitazone not only lowers the risk of T2D (hazard ratio (HR)=0.48, CI=0.33-0.69) but also the incidence of stroke and myocardial infarction (HR=0.76, CI=0.62-0.93) (121). 5,039 people with stroke or TIA participated in a meta-analysis of 5 RCTs, of which 4 evaluated the medication pioglitazone and 1 rosiglitazone (122). Additionally, it was discovered that PPAR-γ agonists, as opposed to a placebo, lower the incidence of recurrent stroke (RR=0.66, CI=0.44-0.99). Researchers discovered that PPAR-γ agonists decreased the composite outcome of major vascular events, such as cardiovascular death, non-fatal myocardial infarction, or non-fatal stroke total occurrences (RR=0.73, CI=0.54-0.99) in a single experiment with 984 patients. Since it doesn’t result in hypoglycemia, metformin, the most often used medication to treat T2D, is arguably the subject of the most research. Metformin exerts its activity through two main mechanisms: AMP-activated protein kinase (AMPK)-dependent and AMPK-independent modalities. When metformin activates AMPK, it can increase fatty acid oxidation by inhibiting the phosphorylation of acetyl coenzyme A carboxylase, ultimately improving lipid metabolism and insulin sensitivity (123). Metformin also has neuroprotective benefits in ischemic stroke by activating the AMPK pathway. Activated AMPK reduces neuroinflammation by inhibiting the release of inflammatory markers, decreasing ROS generation to reduce oxidative stress, and promoting autophagy. Furthermore, by reducing glutamate release, AMPK activation prevents glutamate excitotoxicity-induced apoptosis (124). Statins are lipid-lowering medications that are primarily used to treat cardiovascular disorders caused by high blood lipid levels, such as hyperlipidemia, hypertension, atherosclerotic heart disease, and others. Previous research has shown that long-term pravastatin plus captopril treatment improves the progression of IR and associated risk factors (hyperinsulinemia, hypercholesterolemia) (125). Another study came to the same conclusion, showing that in ob/ob mice—which developed obesity and IR due to a loss of functioning leptin, the infarct volume was considerably higher after middle cerebral artery occlusion (MCAO) than in wild-type or lean mice. Short-term treatment with rosuvastatin (10 mg/kg/day for 3 days) did not reduce infarct volume in wild-type and lean mice, but significantly reduced infarct volume in ob/ob mice with IR (126). However, ten weeks of intensive atorvastatin (40 mg/d) treatment increased the emergence of insulin resistance and insulin secretion in participants without diabetes (127). This conflicting outcome indicates that statins for insulin resistance still require additional experimental confirmation. Apart from this, several medicines (e.g., α-ketoglutarate, astaxanthin) can reduce IR and increase neuronal survival by decreasing aberrant serine phosphorylation of IRS1 directly via inhibiting mTOR/S6K1 (100, 128). In summary, there is substantial evidence that insulin sensitizers can boost brain insulin sensitivity, successfully treat ischemic stroke, and improve neurological function.
Glucagon-like peptide-1 (GLP-1) maintains glucose metabolism stability by increasing insulin production and suppressing glucagon secretion, enhancing cell proliferation and growth, and avoiding cell excitotoxicity and death (129). GLP-1 receptors (GLP-1Rs) are found throughout the CNS (130). GLP-1 receptor agonists (GLP-1RAs) not only be used to treat T2D by increasing peripheral insulin sensitivity but have also been found to increase neuronal sensitivity to insulin (131, 132). In a meta-analysis of 33,457 subjects, GLP-1RAs treatment was found to reduce the incidence of major adverse cardiovascular events (cardiovascular death, non-fatal myocardial infarction, and non-fatal stroke) compared to placebo (HR= 0.90, CI=0.82-0.99) and did not cause adverse effects such as severe hypoglycemia (133). Another meta-analysis, which included 35 preclinical studies, 11 retrospective database studies, 7 cardiovascular outcomes trials, and 4 prospective clinical studies, found that administration of GLP-1RAs after stroke reduced infarct volume, apoptosis, inflammatory responses, and oxidative stress, while also promoting neurogenesis, angiogenesis, and increased cerebral blood flow to exert neuroprotective effects. Furthermore, in cardiovascular outcome trials, dulaglutide and semaglutide were observed to minimize the risk of stroke (134). Exendin-4, another GLP-1RAs, reduces the activation of astrocyte-derived matrix metalloproteinase-9 (MMP-9), vascular endothelial growth factor (VEGF-A), MCP1, and chemokine C-X-C motif ligand 1 (CXCL-1) by oxygen-glucose deprivation (OGD), as well as the activation of the JAK2/STAT3 signaling pathway by OGD. Exendin-4 dramatically improved neurological function scores and reduced infarct volume in MCAO rats, yielding similar outcomes (135). Taken together, GLP-1RAs are a very promising medicine for the treatment of IR in ischemic stroke, but additional clinical trial evidence is required.
Insulin is essential for brain function. IR, which leads to insulin dysregulation, can cause neurological damage via a variety of mechanisms. Numerous studies have shown that IR is not just a feature of T2D, but it also plays a key role in the development and progression of ischemic stroke. IR is an independent risk factor for ischemic stroke, IR contributes to the development of ischemic stroke by promoting thrombosis and atherosclerosis formation. IR, which is associated with a poor prognosis in ischemic stroke patients, exacerbates the inflammatory response, oxidative stress, and neuronal damage. Current IR treatment in patients with ischemic stroke is based on medicine selection based on T2D treatment experiences, such as insulin, insulin sensitizers, and GLP-1RAs. In many studies, insulin sensitizers and GLP-1RAs had no major side effects in patients with ischemic stroke, however, the use of insulin has attracted controversy. Although peripheral insulin injection therapy is the preferred treatment for T2D, it has a number of negative effects in ischemic stroke patients, and safer and more effective intranasal insulin administration is becoming more common (Figure 3). However, these studies still have some limitations, the first of which is the dispute over how to identify IR. For instance, while some authors consider HOMA-IR >3 to validate the diagnosis of IR, others define IR as HOMA-IR ≥2.5. Although ethnicity may be a factor in the cut-off value heterogeneity, a consistent scoring standard can boost the credibility of experimental findings. Secondly, further research is needed to fully understand the variety of roles that IR plays in ischemic stroke. Aside from the inhibitory effects of insulin on the regulation of glucose metabolism, inflammatory response, oxidative stress, neuronal function, and vascular function, there could be additional mechanisms that modulate ischemic stroke that is independent of the insulin signaling pathway. Overall, there is mounting proof that IR is an independent risk factor for ischemic stroke and a major component in poor patient prognosis. The prevention and treatment of ischemic stroke may benefit from a systematic understanding of the mechanisms behind IR in ischemic stroke.
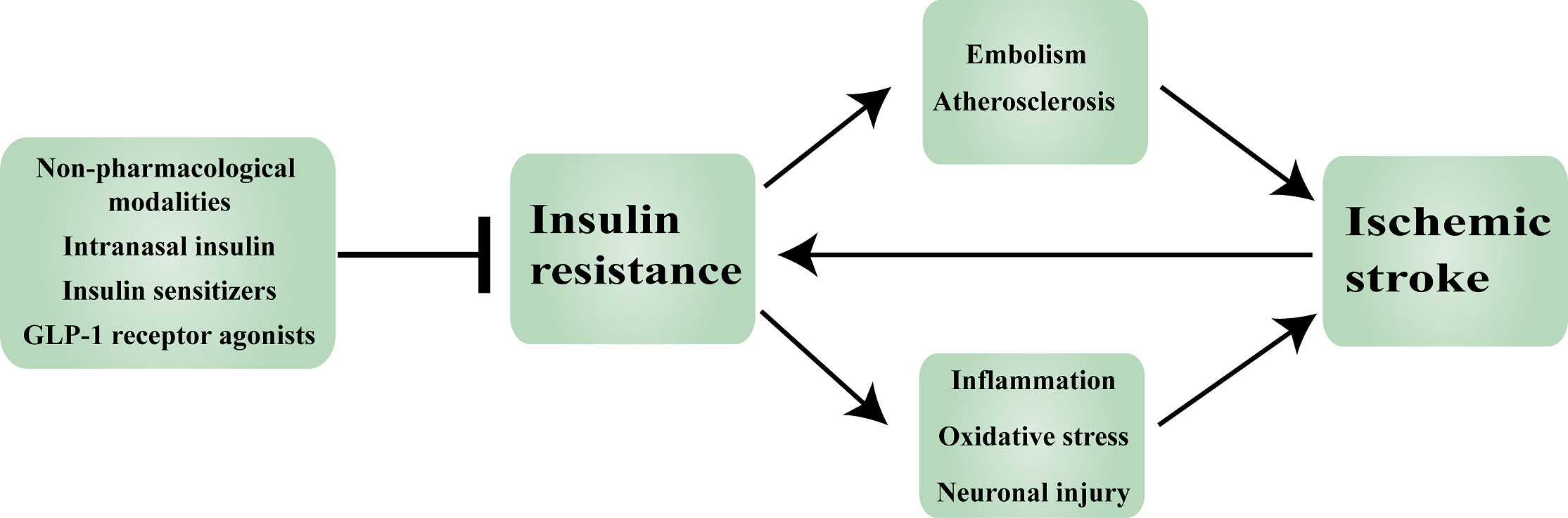
Figure 3 Insulin resistance in ischemic stroke: mechanisms and therapeutic approaches. GLP-1: Glucagon-like peptide-1.
P-FD wrote the manuscript. H-SZ, JW, C-HH and WL contributions to design of the work. Y-YG and J-NM made suggestions for some of the work. C-HH and WL revised the work. All authors contributed to the article and approved the submitted version.
This work was supported by grants from the National Natural Science Foundation of China (82130037, 81971122 for C.-H.H., NO. 82171323 for W.L., NO. 82101456 for Y.-Y.G.) and Natural Science Foundation of Jiangsu Province, China (BK20201113 for W.L.).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Johnson CO, Nguyen M, Roth GA, Nichols E, Alam T, Abate D, et al. Global, regional, and national burden of stroke, 1990–2016: A systematic analysis for the global burden of disease study 2016. Lancet Neurol (2019) 18(5):439–58. doi: 10.1016/s1474-4422(19)30034-1
2. Agrawal R, Reno CM, Sharma S, Christensen C, Huang Y, Fisher SJ. Insulin action in the brain regulates both central and peripheral functions. Am J Physiol Endocrinol Metab (2021) 321(1):E156–e63. doi: 10.1152/ajpendo.00642.2020
3. Saini V, Guada L, Yavagal DR. Global epidemiology of stroke and access to acute ischemic stroke interventions. Neurology (2021) 97(20 Supplement 2):S6. doi: 10.1212/WNL.0000000000012781
4. Wieberdink RG, Koudstaal PJ, Hofman A, Witteman JCM, Breteler MMB, Arfan Ikram M. Insulin resistance and the risk of stroke and stroke subtypes in the nondiabetic elderly. Am J Epidemiol (2012) 176(8):699–707. doi: 10.1093/aje/kws149
5. Ago T, Matsuo R, Hata J, Wakisaka Y, Kuroda J, Kitazono T, et al. Insulin resistance and clinical outcomes after acute ischemic stroke. Neurology (2018) 90(17):e1470–e7. doi: 10.1212/WNL.0000000000005358
6. Rehni AK, Cho S, Dave KR. Ischemic brain injury in diabetes and endoplasmic reticulum stress. Neurochem Int (2022) 152:105219. doi: 10.1016/j.neuint.2021.105219
7. Kullmann S, Kleinridders A, Small DM, Fritsche A, Häring H-U, Preissl H, et al. Central nervous pathways of insulin action in the control of metabolism and food intake. Lancet Diabetes Endocrinol (2020) 8(6):524–34. doi: 10.1016/S2213-8587(20)30113-3
8. Li M, Chi X, Wang Y, Setrerrahmane S, Xie W, Xu H. Trends in insulin resistance: Insights into mechanisms and therapeutic strategy. Signal Transduct Target Ther (2022) 7(1):216. doi: 10.1038/s41392-022-01073-0
9. Dienel GA. Brain glucose metabolism: Integration of energetics with function. Physiol Rev (2018) 99(1):949–1045. doi: 10.1152/physrev.00062.2017
10. Magistretti PJ, Allaman I. Lactate in the brain: From metabolic end-product to signalling molecule. Nat Rev Neurosci (2018) 19(4):235–49. doi: 10.1038/nrn.2018.19
11. Cunnane SC, Trushina E, Morland C, Prigione A, Casadesus G, Andrews ZB, et al. Brain energy rescue: An emerging therapeutic concept for neurodegenerative disorders of ageing. Nat Rev Drug Discov (2020) 19(9):609–33. doi: 10.1038/s41573-020-0072-x
12. Huang S, Czech MP. The Glut4 glucose transporter. Cell Metab (2007) 5(4):237–52. doi: 10.1016/j.cmet.2007.03.006
13. Ashrafi G, Wu Z, Farrell RJ, Ryan TA. Glut4 mobilization supports energetic demands of active synapses. Neuron (2017) 93(3):606–15.e3. doi: 10.1016/j.neuron.2016.12.020
14. Scherer T, Sakamoto K, Buettner C. Brain insulin signalling in metabolic homeostasis and disease. Nat Rev Endocrinol (2021) 17(8):468–83. doi: 10.1038/s41574-021-00498-x
15. Haeusler RA, McGraw TE, Accili D. Biochemical and cellular properties of insulin receptor signalling. Nat Rev Mol Cell Biol (2018) 19(1):31–44. doi: 10.1038/nrm.2017.89
16. Talbot K, Wang HY, Kazi H, Han LY, Bakshi KP, Stucky A, et al. Demonstrated brain insulin resistance in alzheimer’s disease patients is associated with igf-1 resistance, irs-1 dysregulation, and cognitive decline. J Clin Invest (2012) 122(4):1316–38. doi: 10.1172/JCI59903
17. Petersen MC, Shulman GI. Mechanisms of insulin action and insulin resistance. Physiol Rev (2018) 98(4):2133–223. doi: 10.1152/physrev.00063.2017
18. Lee SH, Park SY, Choi CS. Insulin resistance: From mechanisms to therapeutic strategies. Diabetes Metab J (2022) 46(1):15–37. doi: 10.4093/dmj.2021.0280
19. Matthews DR, Hosker JP, Rudenski AS, Naylor BA, Treacher DF, Turner RC. Homeostasis model assessment: Insulin resistance and B-cell function from fasting plasma glucose and insulin concentrations in man. Diabetologia (1985) 28(7):412–9. doi: 10.1007/BF00280883
20. Matsuda M, DeFronzo RA. Insulin sensitivity indices obtained from oral glucose tolerance testing: Comparison with the euglycemic insulin clamp. Diabetes Care (1999) 22(9):1462–70. doi: 10.2337/diacare.22.9.1462
21. Hong S, Han K, Park CY. The insulin resistance by triglyceride glucose index and risk for dementia: Population-based study. Alzheimers Res Ther (2021) 13(1):9. doi: 10.1186/s13195-020-00758-4
22. Radaelli T, Farrell KA, Huston-Presley L, Amini SB, Kirwan JP, McIntyre HD, et al. Estimates of insulin sensitivity using glucose and c-peptide from the hyperglycemia and adverse pregnancy outcome glucose tolerance test. Diabetes Care (2010) 33(3):490–4. doi: 10.2337/dc09-1463
23. Lim J, Kim J, Koo SH, Kwon GC. Comparison of triglyceride glucose index, and related parameters to predict insulin resistance in Korean adults: An analysis of the 2007-2010 Korean national health and nutrition examination survey. PloS One (2019) 14(3):e0212963. doi: 10.1371/journal.pone.0212963
24. Kahn CR. The molecular mechanism of insulin action. Annu Rev Med (1985) 36(1):429–51. doi: 10.1146/annurev.me.36.020185.002241
25. Kellar D, Craft S. Brain insulin resistance in alzheimer’s disease and related disorders: Mechanisms and therapeutic approaches. Lancet Neurol (2020) 19(9):758–66. doi: 10.1016/S1474-4422(20)30231-3
27. Maida CD, Daidone M, Pacinella G, Norrito RL, Pinto A, Tuttolomondo A. Diabetes and ischemic stroke: An old and new relationship an overview of the close interaction between these diseases. Int J Mol Sci (2022) 23(4). doi: 10.3390/ijms23042397
28. Kernan WN, Viscoli CM, Inzucchi SE, Brass LM, Bravata DM, McVeety JC, et al. Insulin resistance among patients with ischemic stroke and tia. STROKE (2002) 33(1):389–. doi: 10.1212/wnl.59.6.809
29. Katsumata T, Otori T, Nishiyama Y, Okubo S, Nishiyama Y, Nagayama H, et al. Correlation between insulin resistance and white matter lesions among non-diabetic patients with ischemic stroke. Neurological Res (2010) 32(7):743–7. doi: 10.1179/016164109X12608733393755
30. Georgakis MK, Harshfield EL, Malik R, Franceschini N, Langenberg C, Wareham NJ, et al. Diabetes mellitus, glycemic traits, and cerebrovascular disease: A mendelian randomization study. Neurology (2021) 96(13):e1732–e42. doi: 10.1212/wnl.0000000000011555
31. Jolugbo P, Ariëns RAS. Thrombus composition and efficacy of thrombolysis and thrombectomy in acute ischemic stroke. Stroke (2021) 52(3):1131–42. doi: 10.1161/strokeaha.120.032810
32. Kaur R, Kaur M, Singh J. Endothelial dysfunction and platelet hyperactivity in type 2 diabetes mellitus: Molecular insights and therapeutic strategies. Cardiovasc Diabetol (2018) 17(1):121. doi: 10.1186/s12933-018-0763-3
33. Hierons SJ, Marsh JS, Wu D, Blindauer CA, Stewart AJ. The interplay between non-esterified fatty acids and plasma zinc and its influence on thrombotic risk in obesity and type 2 diabetes. Int J Mol Sci (2021) 22(18). doi: 10.3390/ijms221810140
34. Alexander Y, Osto E, Schmidt-Trucksäss A, Shechter M, Trifunovic D, Duncker DJ, et al. Endothelial function in cardiovascular medicine: A consensus paper of the European society of cardiology working groups on atherosclerosis and vascular biology, aorta and peripheral vascular diseases, coronary pathophysiology and microcirculation, and thrombosis. Cardiovasc Res (2021) 117(1):29–42. doi: 10.1093/cvr/cvaa085
35. Muniyappa R, Chen H, Montagnani M, Sherman A, Quon MJ. Endothelial dysfunction due to selective insulin resistance in vascular endothelium: Insights from mechanistic modeling. Am J Physiol Endocrinol Metab (2020) 319(3):E629–e46. doi: 10.1152/ajpendo.00247.2020
36. Potenza AM, Gagliardi S, Nacci C, Carratu RM, Montagnani M. Endothelial dysfunction in diabetes: From mechanisms to therapeutic targets. Curr Medicinal Chem (2009) 16(1):94–112. doi: 10.2174/092986709787002853
37. Den Hartogh DJ, Vlavcheski F, Giacca A, Tsiani E. Attenuation of free fatty acid (Ffa)-induced skeletal muscle cell insulin resistance by resveratrol is linked to activation of ampk and inhibition of mtor and P70 S6k. Int J Mol Sci (2020) 21(14). doi: 10.3390/ijms21144900
38. Natali A, Toschi E, Baldeweg S, Ciociaro D, Favilla S, Saccà L, et al. Clustering of insulin resistance with vascular dysfunction and low-grade inflammation in type 2 diabetes. Diabetes (2006) 55(4):1133–40. doi: 10.2337/diabetes.55.04.06.db05-1076
39. Westergren HU, Svedlund S, Momo RA, Blomster JI, Wåhlander K, Rehnström E, et al. Insulin resistance, endothelial function, angiogenic factors and clinical outcome in non-diabetic patients with chest pain without myocardial perfusion defects. Cardiovasc Diabetol (2016) 15(1):36. doi: 10.1186/s12933-016-0353-1
40. Gao Y, Yu C, Pi S, Mao L, Hu B. The role of P2y12 receptor in ischemic stroke of atherosclerotic origin. Cell Mol Life Sci (2019) 76(2):341–54. doi: 10.1007/s00018-018-2937-2
41. Jing J, Meng X, Zhao X, Liu L, Wang A, Pan Y, et al. Dual antiplatelet therapy in transient ischemic attack and minor stroke with different infarction patterns: Subgroup analysis of the chance randomized clinical trial. JAMA Neurol (2018) 75(6):711–9. doi: 10.1001/jamaneurol.2018.0247
42. Hwang DL, Yen CF, Nadler JL. Insulin increases intracellular magnesium transport in human platelets. J Clin Endocrinol Metab (1993) 76(3):549–53. doi: 10.1210/jcem.76.3.8445010
43. Vinik AI, Erbas T, Park TS, Nolan R, Pittenger GL. Platelet dysfunction in type 2 diabetes. Diabetes Care (2001) 24(8):1476–85. doi: 10.2337/diacare.24.8.1476
44. Yang S, Zhao J, Chen Y, Lei M. Biomarkers associated with ischemic stroke in diabetes mellitus patients. Cardiovasc Toxicol (2016) 16(3):213–22. doi: 10.1007/s12012-015-9329-8
45. Kobiyama K, Ley K. Atherosclerosis. Circ Res (2018) 123(10):1118–20. doi: 10.1161/CIRCRESAHA.118.313816
46. Amarenco P, Denison H, Evans SR, Himmelmann A, James S, Knutsson M, et al. Ticagrelor added to aspirin in acute nonsevere ischemic stroke or transient ischemic attack of atherosclerotic origin. Stroke (2020) 51(12):3504–13. doi: 10.1161/strokeaha.120.032239
47. Di Pino A, DeFronzo RA. Insulin resistance and atherosclerosis: Implications for insulin-sensitizing agents. Endocrine Rev (2019) 40(6):1447–67. doi: 10.1210/er.2018-00141
48. Love KM, Liu Z. Dpp4 activity, hyperinsulinemia, and atherosclerosis. J Clin Endocrinol Metab (2021) 106(6):1553–65. doi: 10.1210/clinem/dgab078
49. Olefsky JM, Glass CK. Macrophages, inflammation, and insulin resistance. Annu Rev Physiol (2010) 72(1):219–46. doi: 10.1146/annurev-physiol-021909-135846
50. Coletta DK, Balas B, Chavez AO, Baig M, Abdul-Ghani M, Kashyap SR, et al. Effect of acute physiological hyperinsulinemia on gene expression in human skeletal muscle in vivo. Am J Physiology-Endocrinol Metab (2008) 294(5):E910–E7. doi: 10.1152/ajpendo.00607.2007
51. Fadini GP, Ciciliot S. Vascular smooth muscle cells and monocyte–macrophages accomplice in the accelerated atherosclerosis of insulin resistance states. Cardiovasc Res (2014) 103(2):194–5. doi: 10.1093/cvr/cvu144
52. Fu J, Yu MG, Li Q, Park K, King GL. Insulin’s actions on vascular tissues: Physiological effects and pathophysiological contributions to vascular complications of diabetes. Mol Metab (2021) 52:101236. doi: 10.1016/j.molmet.2021.101236
53. Mao L, Yin R, Yang L, Zhao D. Role of advanced glycation end products on vascular smooth muscle cells under diabetic atherosclerosis. Front Endocrinol (Lausanne) (2022) 13:983723. doi: 10.3389/fendo.2022.983723
54. Hoekstra JB, van Rijn HJ, Erkelens DW, Thijssen JH. C-peptide. Diabetes Care (1982) 5(4):438–46. doi: 10.2337/diacare.5.4.438
55. Alves MT, Ortiz MMO, Dos Reis G, Dusse LMS, Carvalho MDG, Fernandes AP, et al. The dual effect of c-peptide on cellular activation and atherosclerosis: Protective or not? Diabetes Metab Res Rev (2019) 35(1):e3071. doi: 10.1002/dmrr.3071
56. Walcher D, Babiak C, Poletek P, Rosenkranz S, Bach H, Betz S, et al. C-peptide induces vascular smooth muscle cell proliferation. Circ Res (2006) 99(11):1181–7. doi: 10.1161/01.RES.0000251231.16993.88
57. Schlessinger J. New roles for src kinases in control of cell survival and angiogenesis. Cell (2000) 100(3):293–6. doi: 10.1016/s0092-8674(00)80664-9
58. Aguilar-Ballester M, Herrero-Cervera A, Vinué Á, Martínez-Hervás S, González-Navarro H. Impact of cholesterol metabolism in immune cell function and atherosclerosis. Nutrients (2020) 12(7). doi: 10.3390/nu12072021
59. Athyros VG, Doumas M, Imprialos KP, Stavropoulos K, Georgianou E, Katsimardou A, et al. Diabetes and lipid metabolism. Hormones (2018) 17(1):61–7. doi: 10.1007/s42000-018-0014-8
60. Heeren J, Scheja L. Metabolic-associated fatty liver disease and lipoprotein metabolism. Mol Metab (2021) 50:101238. doi: 10.1016/j.molmet.2021.101238
61. Jia G, Sowers JR. Hypertension in diabetes: An update of basic mechanisms and clinical disease. Hypertension (2021) 78(5):1197–205. doi: 10.1161/hypertensionaha.121.17981
62. Ferrannini E, Buzzigoli G, Bonadonna R, Giorico MA, Oleggini M, Graziadei L, et al. Insulin resistance in essential hypertension. N Engl J Med (1987) 317(6):350–7. doi: 10.1056/NEJM198708063170605
63. Irsik DL, Chen JK, Brands MW. Chronic renal artery insulin infusion increases mean arterial pressure in Male sprague-dawley rats. Am J Physiol Renal Physiol (2018) 314(1):F81–f8. doi: 10.1152/ajprenal.00374.2017
64. Söderberg S, Ahrén B, Stegmayr B, Johnson O, Wiklund P-G, Weinehall L, et al. Leptin is a risk marker for first-ever hemorrhagic stroke in a population-based cohort. Stroke (1999) 30(2):328–37. doi: 10.1161/01.STR.30.2.328
65. da Silva AA, do Carmo JM, Li X, Wang Z, Mouton AJ, Hall JE. Role of hyperinsulinemia and insulin resistance in hypertension: Metabolic syndrome revisited. Can J Cardiol (2020) 36(5):671–82. doi: 10.1016/j.cjca.2020.02.066
66. Diener HC, Hankey GJ. Primary and secondary prevention of ischemic stroke and cerebral hemorrhage: Jacc focus seminar. J Am Coll Cardiol (2020) 75(15):1804–18. doi: 10.1016/j.jacc.2019.12.072
67. Chang Y, Kim CK, Kim M-K, Seo WK, Oh K. Insulin resistance is associated with poor functional outcome after acute ischemic stroke in non-diabetic patients. Sci Rep (2021) 11(1):1229. doi: 10.1038/s41598-020-80315-z
68. Jayaraj RL, Azimullah S, Beiram R, Jalal FY, Rosenberg GA. Neuroinflammation: Friend and foe for ischemic stroke. J Neuroinflamm (2019) 16(1):142. doi: 10.1186/s12974-019-1516-2
69. Geissmann F, Gordon S, Hume DA, Mowat AM, Randolph GJ. Unravelling mononuclear phagocyte heterogeneity. Nat Rev Immunol (2010) 10(6):453–60. doi: 10.1038/nri2784
70. Krady JK, Lin H-W, Liberto CM, Basu A, Kremlev SG, Levison SW. Ciliary neurotrophic factor and interleukin-6 differentially activate microglia. J Neurosci Res (2008) 86(7):1538–47. doi: 10.1002/jnr.21620
71. Kinuthia UM, Wolf A, Langmann T. Microglia and inflammatory responses in diabetic retinopathy. Front Immunol (2020) 11:564077. doi: 10.3389/fimmu.2020.564077
72. Perego C, Fumagalli S, Zanier ER, Carlino E, Panini N, Erba E, et al. Macrophages are essential for maintaining a M2 protective response early after ischemic brain injury. Neurobiol Dis (2016) 96:284–93. doi: 10.1016/j.nbd.2016.09.017
73. Buck BH, Liebeskind DS, Saver JL, Bang OY, Yun SW, Starkman S, et al. Early neutrophilia is associated with volume of ischemic tissue in acute stroke. Stroke (2008) 39(2):355–60. doi: 10.1161/STROKEAHA.107.490128
74. Shoelson SE, Lee J, Goldfine AB. Inflammation and insulin resistance. J Clin Invest (2006) 116(7):1793–801. doi: 10.1172/JCI29069
75. Shimobayashi M, Albert V, Woelnerhanssen B, Frei IC, Weissenberger D, Meyer-Gerspach AC, et al. Insulin resistance causes inflammation in adipose tissue. J Clin Invest (2018) 128(4):1538–50. doi: 10.1172/JCI96139
76. Rodriguez-Prados JC, Traves PG, Cuenca J, Rico D, Aragones J, Martin-Sanz P, et al. Substrate fate in activated macrophages: A comparison between innate, classic, and alternative activation. J Immunol (2010) 185(1):605–14. doi: 10.4049/jimmunol.0901698
77. Galvan-Pena S, O’Neill LA. Metabolic reprograming in macrophage polarization. Front Immunol (2014) 5:420. doi: 10.3389/fimmu.2014.00420
78. Chamorro Á, Dirnagl U, Urra X, Planas AM. Neuroprotection in acute stroke: Targeting excitotoxicity, oxidative and nitrosative stress, and inflammation. Lancet Neurol (2016) 15(8):869–81. doi: 10.1016/S1474-4422(16)00114-9
79. Ren JX, Li C, Yan XL, Qu Y, Yang Y, Guo ZN. Crosstalk between oxidative stress and Ferroptosis/Oxytosis in ischemic stroke: Possible targets and molecular mechanisms. Oxid Med Cell Longev (2021) 2021:6643382. doi: 10.1155/2021/6643382
80. Di Meo S, Iossa S, Venditti P. Skeletal muscle insulin resistance: Role of mitochondria and other ros sources. J Endocrinol (2017) 233(1):R15–r42. doi: 10.1530/joe-16-0598
81. Völz R, Harris W, Hirt H, Lee YH. Ros homeostasis mediated by Mpk4 and Summ2 determines synergid cell death. Nat Commun (2022) 13(1):1746. doi: 10.1038/s41467-022-29373-7
82. Matsuda S, Umeda M, Uchida H, Kato H, Araki T. Alterations of oxidative stress markers and apoptosis markers in the striatum after transient focal cerebral ischemia in rats. J Neural Transm (2009) 116(4):395–404. doi: 10.1007/s00702-009-0194-0
83. Bernstein AI, Garrison SP, Zambetti GP, O’Malley KL. 6-ohda generated ros induces DNA damage and P53- and puma-dependent cell death. Mol Neurodegeneration (2011) 6(1):2. doi: 10.1186/1750-1326-6-2
84. Niizuma K, Endo H, Nito C, Myer DJ, Chan PH. Potential role of puma in delayed death of hippocampal Ca1 neurons after transient global cerebral ischemia. Stroke (2009) 40(2):618–25. doi: 10.1161/strokeaha.108.524447
85. Yaribeygi H, Farrokhi FR, Butler AE, Sahebkar A. Insulin resistance: Review of the underlying molecular mechanisms. J Cell Physiol (2019) 234(6):8152–61. doi: 10.1002/jcp.27603
86. Evans JL, Maddux BA, Goldfine ID. The molecular basis for oxidative stress-induced insulin resistance. Antioxid Redox Signal (2005) 7(7-8):1040–52. doi: 10.1089/ars.2005.7.1040
87. Maciejczyk M, Żebrowska E, Chabowski A. Insulin resistance and oxidative stress in the brain: What’s new? Int J Mol Sci (2019) 20(4). doi: 10.3390/ijms20040874
88. Morino K, Petersen KF, Shulman GI. Molecular mechanisms of insulin resistance in humans and their potential links with mitochondrial dysfunction. Diabetes (2006) 55 Suppl 2(Suppl 2):S9–s15. doi: 10.2337/db06-S002
89. Zhao Q, Luo T, Gao F, Fu Y, Li B, Shao X, et al. Grp75 regulates mitochondrial-supercomplex turnover to modulate insulin sensitivity. Diabetes (2021) 71(2):233–48. doi: 10.2337/db21-0173
90. Robinson MM, Dasari S, Karakelides H, Bergen HR 3rd, Nair KS. Release of skeletal muscle peptide fragments identifies individual proteins degraded during insulin deprivation in type 1 diabetic humans and mice. Am J Physiol Endocrinol Metab (2016) 311(3):E628–37. doi: 10.1152/ajpendo.00175.2016
91. Cheng Z, Guo S, Copps K, Dong X, Kollipara R, Rodgers JT, et al. Foxo1 integrates insulin signaling with mitochondrial function in the liver. Nat Med (2009) 15(11):1307–11. doi: 10.1038/nm.2049
92. Tuo QZ, Zhang ST, Lei P. Mechanisms of neuronal cell death in ischemic stroke and their therapeutic implications. Med Res Rev (2022) 42(1):259–305. doi: 10.1002/med.21817
93. Zhou X, Chen H, Wang L, Lenahan C, Lian L, Ou Y, et al. Mitochondrial dynamics: A potential therapeutic target for ischemic stroke. Front Aging Neurosci (2021) 13:721428. doi: 10.3389/fnagi.2021.721428
94. Yuan J, Amin P, Ofengeim D. Necroptosis and Ripk1-mediated neuroinflammation in cns diseases. Nat Rev Neurosci (2019) 20(1):19–33. doi: 10.1038/s41583-018-0093-1
95. Kang R, Kroemer G, Tang D. The tumor suppressor protein P53 and the ferroptosis network. Free Radic Biol Med (2019) 133:162–8. doi: 10.1016/j.freeradbiomed.2018.05.074
96. Scheinberg P. Survival of the ischemic brain: A progress report. Circulation (1979) 60(7):1600–5. doi: 10.1161/01.cir.60.7.1600
97. Gu C, Yang J, Luo Y, Ran D, Tan X, Xiang P, et al. Znrf2 attenuates focal cerebral Ischemia/Reperfusion injury in rats by inhibiting Mtorc1-mediated autophagy. Exp Neurol (2021) 342:113759. doi: 10.1016/j.expneurol.2021.113759
98. Ahsan A, Liu M, Zheng Y, Yan W, Pan L, Li Y, et al. Natural compounds modulate the autophagy with potential implication of stroke. Acta Pharm Sin B (2021) 11(7):1708–20. doi: 10.1016/j.apsb.2020.10.018
99. Kitada M, Koya D. Autophagy in metabolic disease and ageing. Nat Rev Endocrinol (2021) 17(11):647–61. doi: 10.1038/s41574-021-00551-9
100. Ding PF, Zhu Q, Sheng B, Yang H, Xu HJ, Tao T, et al. Alpha-ketoglutarate alleviates neuronal apoptosis induced by central insulin resistance through inhibiting S6k1 phosphorylation after subarachnoid hemorrhage. Oxid Med Cell Longev (2022) 2022:9148257. doi: 10.1155/2022/9148257
101. Xing Y, Bai Y. A review of exercise-induced neuroplasticity in ischemic stroke: Pathology and mechanisms. Mol Neurobiol (2020) 57(10):4218–31. doi: 10.1007/s12035-020-02021-1
102. Cramer SC, Sur M, Dobkin BH, O’Brien C, Sanger TD, Trojanowski JQ, et al. Harnessing neuroplasticity for clinical applications. Brain (2011) 134(Pt 6):1591–609. doi: 10.1093/brain/awr039
103. Magee JC, Grienberger C. Synaptic plasticity forms and functions. Annu Rev Neurosci (2020) 43:95–117. doi: 10.1146/annurev-neuro-090919-022842
104. van der Heide LP, Kamal A, Artola A, Gispen WH, Ramakers GM. Insulin modulates hippocampal activity-dependent synaptic plasticity in a n-Methyl-D-Aspartate receptor and phosphatidyl-Inositol-3-Kinase-Dependent manner. J Neurochem (2005) 94(4):1158–66. doi: 10.1111/j.1471-4159.2005.03269.x
105. Fusco S, Spinelli M, Cocco S, Ripoli C, Mastrodonato A, Natale F, et al. Maternal insulin resistance multigenerationally impairs synaptic plasticity and memory via gametic mechanisms. Nat Commun (2019) 10(1):4799. doi: 10.1038/s41467-019-12793-3
106. Kleindorfer DO, Towfighi A, Chaturvedi S, Cockroft KM, Gutierrez J, Lombardi-Hill D, et al. Guideline for the prevention of stroke in patients with stroke and transient ischemic attack: A guideline from the American heart Association/American stroke association. Stroke (2021) 52(7):e364–467. doi: 10.1161/str.0000000000000375
107. Baden MY, Shan Z, Wang F, Li Y, Manson JE, Rimm EB, et al. Quality of plant-based diet and risk of total, ischemic, and hemorrhagic stroke. Neurology (2021) 96(15):e1940–e53. doi: 10.1212/wnl.0000000000011713
108. Khan SU, Khan MU, Riaz H, Valavoor S, Zhao D, Vaughan L, et al. Effects of nutritional supplements and dietary interventions on cardiovascular outcomes: An umbrella review and evidence map. Ann Intern Med (2019) 171(3):190–8. doi: 10.7326/m19-0341
109. Nesti L, Mengozzi A, Tricò D. Impact of nutrient type and sequence on glucose tolerance: Physiological insights and therapeutic implications. Front Endocrinol (Lausanne) (2019) 10:144. doi: 10.3389/fendo.2019.00144
110. Jeong SH, Joo HJ, Kwon J, Park E-C. Global, regional, and national burden of stroke and its risk factors, 1990-2019: A systematic analysis for the global burden of disease study 2019. Lancet Neurol (2021) 20(10):795–820. doi: 10.1016/s1474-4422(21)00252-0
111. Jeong SH, Joo HJ, Kwon J, Park EC. Association between smoking behavior and insulin resistance using triglyceride-glucose index among south Korean adults. J Clin Endocrinol Metab (2021) 106(11):e4531–e41. doi: 10.1210/clinem/dgab399
112. Saunders DH, Sanderson M, Hayes S, Johnson L, Kramer S, Carter DD, et al. Physical fitness training for stroke patients. Cochrane Database Syst Rev (2020) 3(3):Cd003316. doi: 10.1002/14651858.CD003316.pub7
113. Ujvari D, Hulchiy M, Calaby A, Nybacka Å, Byström B, Hirschberg AL. Lifestyle intervention up-regulates gene and protein levels of molecules involved in insulin signaling in the endometrium of Overweight/Obese women with polycystic ovary syndrome. Hum Reprod (2014) 29(7):1526–35. doi: 10.1093/humrep/deu114
114. Whillier S. Exercise and insulin resistance. Adv Exp Med Biol (2020) 1228:137–50. doi: 10.1007/978-981-15-1792-1_9
115. Ntaios G, Papavasileiou V, Bargiota A, Makaritsis K, Michel P. Intravenous insulin treatment in acute stroke: A systematic review and meta-analysis of randomized controlled trials. Int J Stroke (2014) 9(4):489–93. doi: 10.1111/ijs.12225
116. Hallschmid M. Intranasal insulin. J Neuroendocrinol (2021) 33(4):e12934. doi: 10.1111/jne.12934
117. Craft S, Raman R, Chow TW, Rafii MS, Sun CK, Rissman RA, et al. Safety, efficacy, and feasibility of intranasal insulin for the treatment of mild cognitive impairment and Alzheimer disease dementia: A randomized clinical trial. JAMA Neurol (2020) 77(9):1099–109. doi: 10.1001/jamaneurol.2020.1840
118. Huang SS, Lu YJ, Huang JP, Wu YT, Day YJ, Hung LM. The essential role of endothelial nitric oxide synthase activation in insulin-mediated neuroprotection against ischemic stroke in diabetes. J Vasc Surg (2014) 59(2):483–91. doi: 10.1016/j.jvs.2013.03.023
119. Zhu Y, Huang Y, Yang J, Tu R, Zhang X, He WW, et al. Intranasal insulin ameliorates neurological impairment after intracerebral hemorrhage in mice. Neural Regener Res (2022) 17(1):210–6. doi: 10.4103/1673-5374.314320
120. Lebovitz HE. Thiazolidinediones: The forgotten diabetes medications. Curr Diabetes Rep (2019) 19(12):151. doi: 10.1007/s11892-019-1270-y
121. Kernan WN, Viscoli CM, Furie KL, Young LH, Inzucchi SE, Gorman M, et al. Pioglitazone after ischemic stroke or transient ischemic attack. N Engl J Med (2016) 374(14):1321–31. doi: 10.1056/NEJMoa1506930
122. Liu J, Wang LN. Peroxisome proliferator-activated receptor gamma agonists for preventing recurrent stroke and other vascular events in people with stroke or transient ischaemic attack. Cochrane Database Syst Rev (2019) 10(10):Cd010693. doi: 10.1002/14651858.CD010693.pub5
123. He L. Metformin and systemic metabolism. Trends Pharmacol Sci (2020) 41(11):868–81. doi: 10.1016/j.tips.2020.09.001
124. Sharma S, Nozohouri S, Vaidya B, Abbruscato T. Repurposing metformin to treat age-related neurodegenerative disorders and ischemic stroke. Life Sci (2021) 274:119343. doi: 10.1016/j.lfs.2021.119343
125. Villa E, Rábano A, Albarrán OG, Ruilope LM, García-Robles R. Effects of chronic combined treatment with captopril and pravastatin on the progression of insulin resistance and cardiovascular alterations in an experimental model of obesity in dogs. Am J Hypertens (1998) 11(7):844–51. doi: 10.1016/s0895-7061(98)00053-3
126. Mayanagi K, Katakam PV, Gáspár T, Domoki F, Busija DW. Acute treatment with rosuvastatin protects insulin resistant (C57bl/6j Ob/Ob) mice against transient cerebral ischemia. J Cereb Blood Flow Metab (2008) 28(12):1927–35. doi: 10.1038/jcbfm.2008.81
127. Abbasi F, Lamendola C, Harris CS, Harris V, Tsai MS, Tripathi P, et al. Statins are associated with increased insulin resistance and secretion. Arterioscler Thromb Vasc Biol (2021) 41(11):2786–97. doi: 10.1161/atvbaha.121.316159
128. Li C, Ma B, Chen J, Jeong Y, Xu X. Astaxanthin inhibits P70 S6 kinase 1 activity to sensitize insulin signaling. Mar Drugs (2020) 18(10). doi: 10.3390/md18100495
129. Drucker DJ. Mechanisms of action and therapeutic application of glucagon-like peptide-1. Cell Metab (2018) 27(4):740–56. doi: 10.1016/j.cmet.2018.03.001
130. Brierley DI, Holt MK, Singh A, de Araujo A, McDougle M, Vergara M, et al. Central and peripheral glp-1 systems independently suppress eating. Nat Metab (2021) 3(2):258–73. doi: 10.1038/s42255-021-00344-4
131. Love KM, Liu J, Regensteiner JG, Reusch JEB, Liu Z. Glp-1 and insulin regulation of skeletal and cardiac muscle microvascular perfusion in type 2 diabetes. J Diabetes (2020) 12(7):488–98. doi: 10.1111/1753-0407.13045
132. Hanssen R, Kretschmer AC, Rigoux L, Albus K, Edwin Thanarajah S, Sitnikow T, et al. Glp-1 and hunger modulate incentive motivation depending on insulin sensitivity in humans. Mol Metab (2021) 45:101163. doi: 10.1016/j.molmet.2021.101163
133. Bethel MA, Patel RA, Merrill P, Lokhnygina Y, Buse JB, Mentz RJ, et al. Cardiovascular outcomes with glucagon-like peptide-1 receptor agonists in patients with type 2 diabetes: A meta-analysis. Lancet Diabetes Endocrinol (2018) 6(2):105–13. doi: 10.1016/s2213-8587(17)30412-6
134. Maskery MP, Holscher C, Jones SP, Price CI, Strain WD, Watkins CL, et al. Glucagon-like peptide-1 receptor agonists as neuroprotective agents for ischemic stroke: A systematic scoping review. J Cereb Blood Flow Metab (2021) 41(1):14–30. doi: 10.1177/0271678x20952011
Keywords: ischemic stroke, insulin resistance, atherosclerosis, embolism, therapeutic approaches
Citation: Ding P-F, Zhang H-S, Wang J, Gao Y-Y, Mao J-N, Hang C-H and Li W (2022) Insulin resistance in ischemic stroke: Mechanisms and therapeutic approaches. Front. Endocrinol. 13:1092431. doi: 10.3389/fendo.2022.1092431
Received: 08 November 2022; Accepted: 30 November 2022;
Published: 15 December 2022.
Edited by:
Yongjun Jiang, The Second Affiliated Hospital of Guangzhou Medical University, ChinaReviewed by:
Qianyan He, First Affiliated Hospital of Jilin University, ChinaCopyright © 2022 Ding, Zhang, Wang, Gao, Mao, Hang and Li. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Chunhua Hang, aGFuZ19uZXVyb3N1cmdlcnlAMTYzLmNvbQ==; Wei Li, d2VpLmxpQG5qdS5lZHUuY24=
†These authors have contributed equally to this work and share first authorship
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.