 Jia-Tong Ding
Jia-Tong Ding Kang-Ping Yang
Kang-Ping Yang Kong-Lan Lin2†
Kong-Lan Lin2† Fang Zou
Fang Zou- 1Department of Endocrinology and Metabolism, The Second Affiliated Hospital of Nanchang University, Nanchang, China
- 2The Second Clinical Medicine School, Nanchang University, Nanchang, China
- 3School of Ophthalmology & Optometry, Nanchang University, Nanchang, China
Background: Considered a significant risk to health and survival, type 1 diabetes (T1D) is a heterogeneous autoimmune disease characterized by hyperglycemia caused by an absolute deficiency of insulin, which is mainly due to the immune-mediated destruction of pancreatic beta cells.
Scope of review: In recent years, the role of immune checkpoints in the treatment of cancer has been increasingly recognized, but unfortunately, little attention has been paid to the significant role they play both in the development of secondary diabetes with immune checkpoint inhibitors and the treatment of T1D, such as cytotoxic T-lymphocyte antigen 4(CTLA-4), programmed cell death protein-1(PD-1), lymphocyte activation gene-3(LAG-3), programmed death ligand-1(PD-L1), and T-cell immunoglobulin mucin protein-3(TIM-3). Here, this review summarizes recent research on the role and mechanisms of diverse immune checkpoint molecules in mediating the development of T1D and their potential and theoretical basis for the prevention and treatment of diabetes.
Major conclusions: Immune checkpoint inhibitors related diabetes, similar to T1D, are severe endocrine toxicity induced with immune checkpoint inhibitors. Interestingly, numerous treatment measures show excellent efficacy for T1D via regulating diverse immune checkpoint molecules, including co-inhibitory and co-stimulatory molecules. Thus, targeting immune checkpoint molecules may exhibit potential for T1D treatment and improve clinical outcomes.
1. Introduction
Type 1 diabetes mellitus (T1D), which is regarded as an autoimmune disorder driven by T cells, causes a lack of insulin and exogenous insulin dependency as a result of the destruction of the patient’s islet cells by T cells (1–3). Despite exogenous insulin therapy representing an effective therapeutic strategy, the high morbidity and mortality of T1D cannot be ignored (4). Residual islet cells, which have received little attention, are still able to achieve glycemic control and reduce chronic inflammation, so immunomodulatory therapies targeting islet cells may be crucial for maintaining residual islet cells as the focus switches from exogenous insulin to endogenous insulin (5, 6). At the same time, the phase of remission (also known as honeymoon, partial remission, or PR) is increasingly being described as a phase of glycemic control and temporary recovery of islet β-cells that may occur after approximately 3 months of insulin therapy in T1D (7). PR may last 6-9 months, with a probability of occurrence of 35-43%, and this phase is likely to have a profound impact on the prognosis of T1D (8). Although the exact timing and mechanism of PR are not yet clear, there is still much research on how to personalize immunotherapy at this stage, such as the latest FDA approval of teplizumab, which is the first immunomodulator shown to significantly delay disease progression in high-risk individuals before a clinical episode (9–11).
It has been suggested that the absence of co-suppressive immune checkpoint ligands (e.g. PD-L1, HLA-E, CD86, and Gal-9) in β-cells in PR can significantly affect the development of T1D (12–14). Immune checkpoints (ICPs) are a series of molecules expressed on the surface of Treg cells and other immune cells that prevent the body from over-activating to the detriment of its normal cells (15). In early studies, immune checkpoint inhibitors (ICIs) (e.g. anti-CTLA-4, anti-PD-1, anti-PD-L1) were investigated in combination with ICPs, and ICPs could be used in the treatment of tumor immune escape through immune checkpoint blockade(ICB) therapy (15, 16). For T1D, activating the expression of ICPs to protect pancreatic islet β-cells from T-cell attack may have the potential to reverse early-onset T1D or to improve prognosis (9, 17, 18). ICPs can also protect human islet-like organ transplants from T-cell attack, induce antigen-specific immune tolerance and reverse early-onset hyperglycemia in bioengineered β-cells (6, 18). Notwithstanding, due to their endocrine toxicity, improper use of ICIs may raise the chance of developing T1D, and ICIs-induced diabetes mellitus, or ICI-associated diabetes, appears to be distinct from T1D, although this has to be proven by additional research (19–23). Interestingly, there is also evidence suggesting that T1D can affect the therapeutic effect of ICIs on tumors by altering the activity of ICPs (24).
The individual immunological checkpoints and co-regulators linked to autoimmune diabetes will be reviewed and discussed in detail in this study. Future research may be able to pinpoint ways to avoid T1D by examining the processes and pathways of ICPs to delay its onset or lessen its severity (25, 26). This article will also provide a summary of some of the benefits of using ICPs to treat secondary T1D as well as some possible clinical uses for ICPs.
2. Tregs cells in T1D
Regulatory T cells (Tregs), also known as Foxp3+Tregs, are a class of suppressor T cells associated with the induction and maintenance of immune tolerance (27). When measured in peripheral blood using CD25 (the alpha chain of the IL-2 receptor) as a Tregs marker, a reduced number of T1D was found compared to the normal group (28). However, there was no change in Treg number by FOXP3 expression-defined Tregs peripheral blood frequency, and observation of the phenotype of Treg intercompartment in T1D patients also revealed no significant size change, pointing to the hypothesis that the major alteration of Treg in T1D is not numerical but rather its function (29, 30). Loss of Treg function has been attributed to pathways known to be critical for optimal Treg inhibitory function pathways, including the IL-2 and T cell receptor (TCR) pathways. A recent clinical study attempting to treat T1D using low-dose IL-2 in combination with Treg found a significant effect on Treg function maintenance while also neglecting the amplification of NK or CD8 T cells, which may be ameliorated by modification of IL-2 in the future (31). Tregs can be produced by three pathways: thymus-derived nTregs, peripheral in vivo-induced pTregs, and in vitro-induced Tregs, and inhibit the immune response function of APCs mainly through cellular contact, where receptors such as PD-1 or CTLA-4 on their surface will competitively bind ligands such as PD-L1/PD-L2, CD80/CD86 on antigen-presenting cells (APCs). Reduced function of Tregs leads to the development of T1D, with IL-10-induced chronic systemic hypo-inflammation state and Teff-mediated immune attack on β-cells, which will lead to the development of T1D (32, 33). Clinical trials using Treg have shown improved but not as promising results as expected, with only a few clinical studies showing that higher levels of Tregs and IL2 appear to improve endogenous insulin secretion in T1D, and the exploration of insulinogenic-specific Tregs in the immune response of patients with T1DM needs to continue in-depth (34–37). Therefore, how to use Tregs to target the autoimmunity against islet β cells that occurs in T1D has become a hot topic in scientific research (38, 39). In many studies using a mouse model of autoimmune diabetes, the use of IL-2 to modulate Treg was found to reduce interferon production by pancreatic infiltrating T cells, increase beta-cell numbers, and mitigate other immune therapies that interfere with Treg homeostasis and prevent disease (40–42). As research progresses, the mechanisms involved will be uncovered and more precise therapeutic modalities that do not produce off-target effects will be proposed (Figure 1).

Figure 1 The regulatory roles of T cells in autoimmune reaction (created with Biorender).
2.1. Treg-cell transplantation
In earlier studies, nTregs were found to promote beta-cell regeneration through autologous transplantation, and patients could reduce the amount of exogenous insulin needed to maintain normal blood glucose levels (43). These studies point to the protective effect of Tregs on the islets, and the prolonged honeymoon period and reduced insulin dosage exhibited by patients confirm the effectiveness of Tregs (44, 45). Mechanistically, defects in Tr1 cells would lead to autoimmune diseases, that IL-10 prevents islet destruction and clinical symptoms of T1D through the production of cytokine pathways such as IFN-γ or IL-17, and early intervention of IL2, which can aid in the induction or maintenance of Foxp3, helps to re-establish the proper immune environment and slow down or even reverse the pathological process of T1D (33).
2.2. Induction of Tregs cells
In recent years, research has increasingly focused on increasing Tregs levels through stimulation of other cell subpopulations, cytokine interactions, or pharmacological treatments (46, 47). One way to increase Tregs is by stimulating other cell subsets using tolerogenic dendritic cells (DCs), which have an anti-inflammatory phenotype and can be induced by infusion of pathogenic DCs, or by using DCs to induce Tregs to proliferate in vivo and in vitro (48, 49). It has been demonstrated that the relay transfer of DCs exposed to GM-CSF to naive mice leads to a significant delay in Foxp3+ T cell expansion and T1D onset. GM-CSF acts mainly on DCs and leads to the expansion of Foxp3+ Tregs, thus delaying the onset of T1D in NOD mice, and this inhibition may be mediated by enhanced IL-10 and TGF-β1 production (50). As for drugs, liru1 dexamethasone, vitamin D3 and rapamycin; or exposure to CTLA-4 membrane receptors and oligonucleotides exerted a slowing of the decline in β-cell function and improved HbA1c in recent-onset T1D (46, 48, 51).
2.3. Islet transplantation
Islet transplantation is an experimental treatment for T1D. As an experimental procedure, islet transplantation can only be performed as part of a clinical trial permitted by the U.S. Food and Drug Administration (FDA). Patients undergoing transplantation are often required to take long-term immunosuppressive drugs, which can be extremely harmful, and implantation-related foreign body reactions (FBR) often induce necrosis of the transplanted islets and lead to failure of glycemic control. The use of valproic acid (VPA) in short-chain fatty acids was shown to successfully protect islet grafts, prolong islet graft survival after islet transplantation, and increase IL-4-producing CD4 and Treg cell populations in NOD receptors, and VPA-induced Treg differentiation from juvenile CD4 and Treg cells by increasing the expression of transcription factor STAT5 and histone 3(H3) acetylation. CD4 and Treg cells. However, the hepatotoxicity, hyperammonemia, weight gain, and insulin resistance side effects of VPA cannot be ignored. To avoid this, the authors observed the same effectiveness of in situ transplantation using in vitro VPA-induced regulatory T cells, which also prolonged islet transplantation survival (52). However, a recent study has attempted to prepare a series of amphoteric-coated core-shell microcapsules (including carboxy betaine methacrylate [CBMA]-coated gelatin methacrylate [GelMA] [CBMA-GelMA], sulfobetaine methacrylate [SBMA]-coated gelMA [SBMA-GelMA] and methacrylic acid phosphorylcholine [MPC]-coated gelMA [MPC-GelMA]) and demonstrated their effectiveness in preventing protein adsorption, cell adhesion and inflammation in vitro (53).
3. ICP, ICI, and T1D
3.1. ICP and ICI in T1D
With the rise and wide application of immunotherapy in the field of tumor treatment, ICP and ICI have become research hotspots. ICP, immunosuppressive small molecules on the surface of T lymphocytes, prevent T cells from being overactivated by inhibiting T cell activation and downregulating immune responses, thereby protecting normal tissues from accidental injury, which is equivalent to installing a brake function on T cells (54, 55). With their flexible regulation of the duration and magnitude of physiological immune responses, ICPs play an indelible role in maintaining autoimmune homeostasis and immune tolerance (22). Common immune checkpoints include cytotoxic T lymphocyte-associated protein-4 (CTLA-4), programmed death-1 (PD-1) and T cell immunoglobulin and mucin domain-containing protein 3 (Tim-3) (56, 57). ICPs negatively modulate the immune response by binding to their ligands, such as PD-1 and PD-L1, but also thereby increase the potential for tumor immune evasion (58). Normally expressed on chronically activated T cells in peripheral tissues, PD-1 is also expressed on pancreatic islet cells. PD-1 transmits negative signals to T cells by binding to the ligand PD-L1 or PD-L2, thereby promoting the suppression of immune responses (59–63). Blocking the PD-1/PD-L1 pathway accelerated the risk of diabetes in non-obese diabetic (NOD) mice. But conversely, increasing the expression of PD-1/PD-L1 or using drugs to restore the PD-1/PD-L1 pathway reversed the course of diabetes in mice (64–66). In normal populations, the surface of islet cells expresses PD-1 for self-protection, and the surface of T cells also helps islet B cells expressing PD-1 bypass the immune response through ICP (62, 67). Furthermore, interferon is also a major regulator of PDL1 expression in human pancreatic β cells (68). In early pancreatitis, depending on the regulation of signal transducer and activator of transcription 1 (STAT1) and STAT2 genes, IFN-α can greatly increase the expression level of PD-L1 in pancreatic islet B cells (69, 70). During islet inflammation, PDL1 expression in β-cells is upregulated by a mechanism thought to be induced by type I and type II IFNs (12, 68). This suggests that pancreatic islet β-cells try to downregulate the immune response by upregulating PDL1, thus avoiding further tissue damage. However, if the PD-1/PDL1 pathway is blocked, it will break immune tolerance, and, ultimately, lead to the development of T1D (71, 72). This was confirmed in animal experiments in NOD mice: higher levels of PDL1 were detected in B cells that survived the immune attack, and high levels of PDL1 were also found to reduce the incidence of diabetes in NOD mice (73, 74).
Immune checkpoint inhibitors work by inhibiting the “off-duty” signal from tumor cells, restoring the immune system to function normally, and then attacking the tumor cells. ICIs block the binding of the ICP to its ligand, to overcome the inhibitory effect, unleash the suppressive function, and reactivates the specific immune function of T lymphocytes against cancer (75, 76). After the binding of PD-1 expressed on the surface of T cells to the ligand is inhibited, those self-reactive T cells that target islet cells are activated to attack their islet cells, and the islet cells are destroyed, resulting in decreased insulin secretion (63). Anti-PD-1 drugs induced PD-1 reduction may also activate autoimmune T cells, leading to autoimmune inflammation targeting pancreatic islet cells (67, 77, 78). In summary, with PD-1/PD-L1 inhibitors, the expression level of PD-L1 on B cells will be greatly reduced, which makes B cells lose their armor against autoimmune attacks. With continued loss of cells, T1D will be the inevitable result. And such events may be more frequent in individuals with susceptibility genotype, HLA haplotypes for example (62, 79).
3.2. Immune checkpoint inhibitor-induced type 1 diabetes (ICIT1D)
When the suppressive effects of T cell immunity are removed, T cells become hyperactivated, and the body mounts an autoimmune response, leading to a unique set of immune-related adverse events (irAEs) (80–83). IAE often affects the endocrine system and results in a variety of endocrine disorders, including hypophysitis, thyroid dysfunction, insulin deficiency diabetes, PAI, etc (21, 84). Relatively speaking, immune checkpoint inhibitor-induced type 1 diabetes (ICIT1D) is a comparatively rare type of adverse reaction with the incidence of ICIT1D equaling 1% (54, 85). However, ICIT1D induces life-threatening diabetic ketoacidosis (DKA) without timely treatment in about 38% to 71% of patients and may serve as a high-risk factor for adrenal crisis (86, 87).
Understanding the pathogenesis of ICIT1D plays an important role in its prevention and treatment (Figure 2). ICIT1D can be divided into four distinct entities: acute autoimmune insulin−dependent diabetes, the clinical presentation of type 1 diabetes, the complication of autoimmune pancreatitis, and autoimmune lipoatrophy, the first of which is most frequently reported (60, 88). ICIT1D usually manifests as a rapid, sustained, severe drop in insulin, C-peptide, and blood glucose levels, with a higher age of onset, faster progression, and less antibody positive compared with traditional T1D (25, 86, 89–91). Immunological characteristics indicate that patients with ICIT1D have humoral and cellular autoimmunity, and some patients may have islet autoantibodies, providing evidence for the involvement of autoimmune mechanisms (26, 62, 92). Based on a colossal number of case studies, more than three-quarters of all ICIT1D cases are associated with treatment with PD-1 inhibitors. Combined use of PD-1 and CTLA-4 inhibitors accounted for 17%, PD-L1 inhibitors for 6%, and very few were associated with CTLA-4 monotherapy (3%) (25, 63, 89, 91, 93).
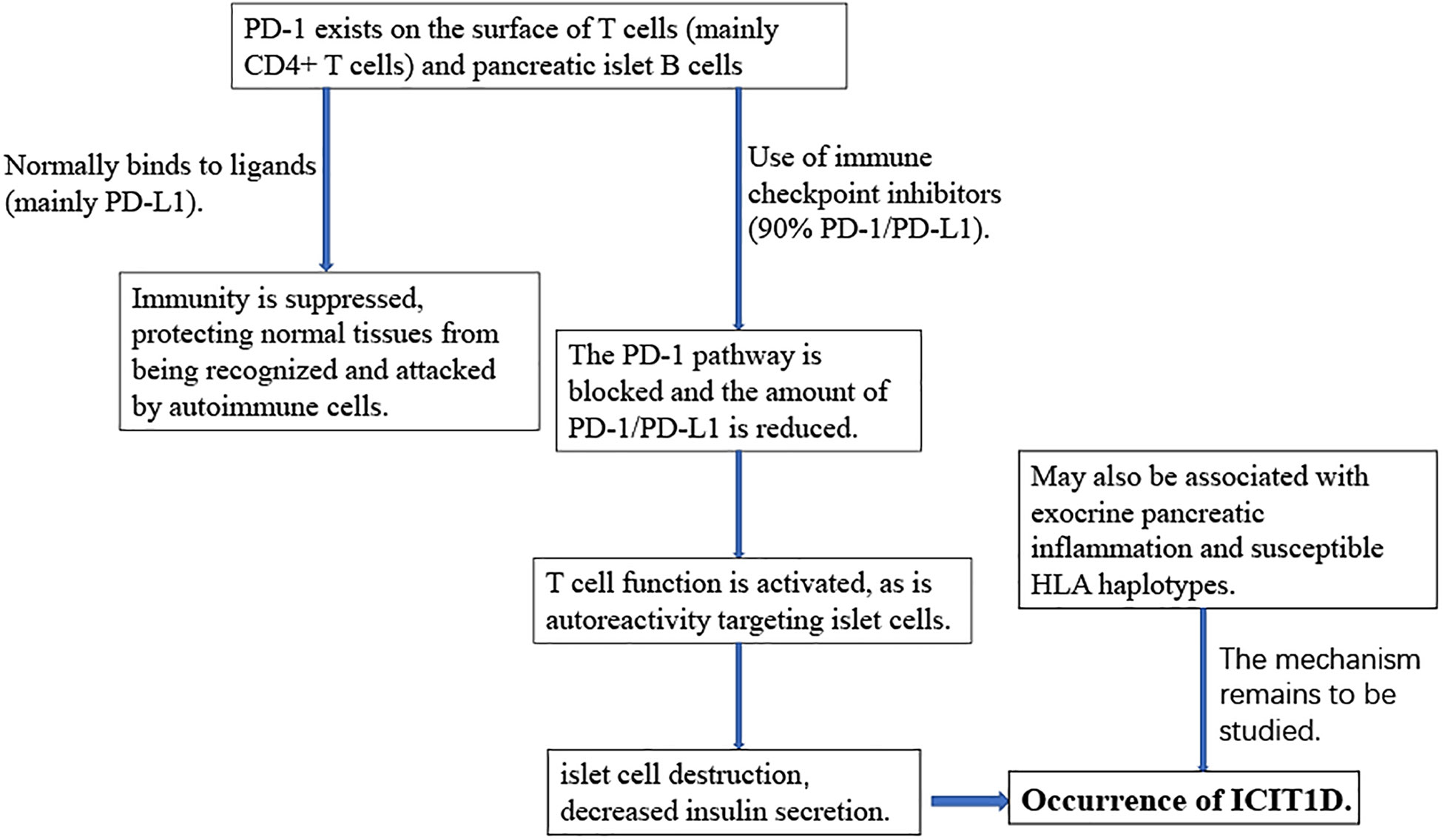
Figure 2 The roles of the PD-1/PD-L1 pathway in ICI-induced T1D.
4. Potential clinical application of immune checkpoint molecules in T1D
Although T1D is considered one of the annoying side effects induced by immune checkpoint inhibitors, immune checkpoint molecules exhibit the potential as a therapeutic maneuver for T1D control (20, 89, 94). In general, inhibition of autoreactive lymphocyte populations is considered an effective therapeutic strategy for the treatment of autoimmune diseases including T1D, but its clinical application is limited due to the massive suppression of lymphocytes involved in normal adaptive immunity (17, 95, 96). Therefore, targeted inhibition of pathogenic lymphocytes associated with autoimmune diseases generated considerable clinical interest (97, 98). Unlike most existing immune suppressants, inhibition of T-cell stimulation via blocking specific signaling molecules focuses on activated lymphocytes and preserves normal adaptive immunity (97). Immune checkpoint molecules perform as a strong immune regulator of self-tolerance and autoimmunity and regulate the response of various immune cells, including T cells, natural killer cells, dendritic cells, innate lymphoid cells, macrophages, and myeloid cells, and specific immune checkpoint mechanisms also participate in pathological processes of T1D (99–101). A recent cohort study also showed that higher levels of circulating immune checkpoint molecules, especially CD137/4-1BB and PD-1, may serve as prognostic biomarkers for new-onsets T1D and risk factors for the growth of an additional autoimmune disease (102). Considering the above characteristics, what follows in the passage reviews the possible therapeutic implications in T1D via regulating immune checkpoint molecules (Figure 3), including co-inhibitory molecules (Table 1) and co-stimulatory molecules (Table 2), especially CTLA-4, PD-1, LAG3, and TIGIT, which seems to be considered as pivotal regulatory molecules with excellent clinical application value.

Figure 3 The treatment measures for T1D via regulating T cells. The blue rectangles represent co-inhibitory molecules while the yellow rectangles represent co-stimulatory molecules (created with Biorender).
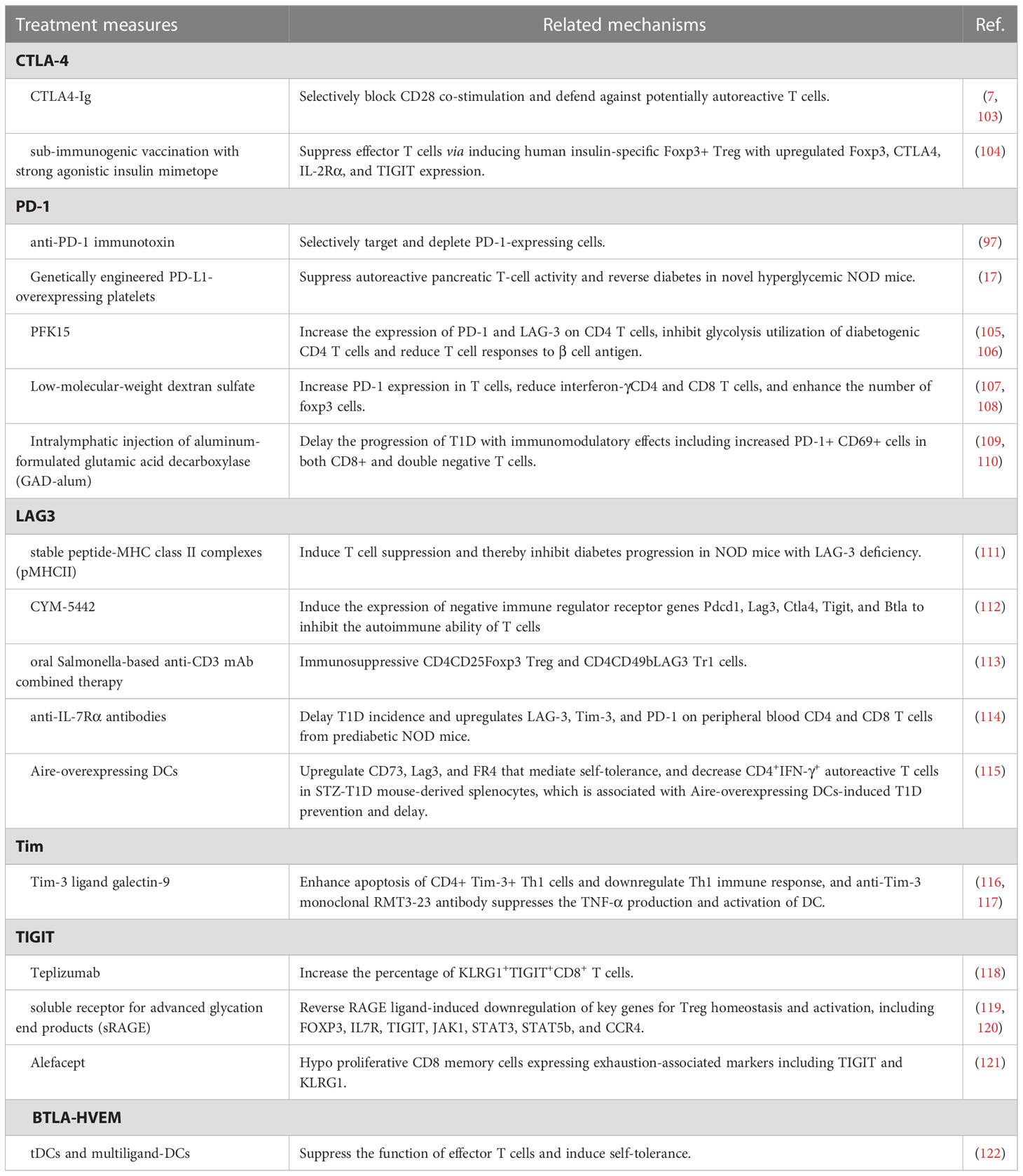
Table 1 The roles of treatment measures regulating co-inhibitory molecules for T1D.
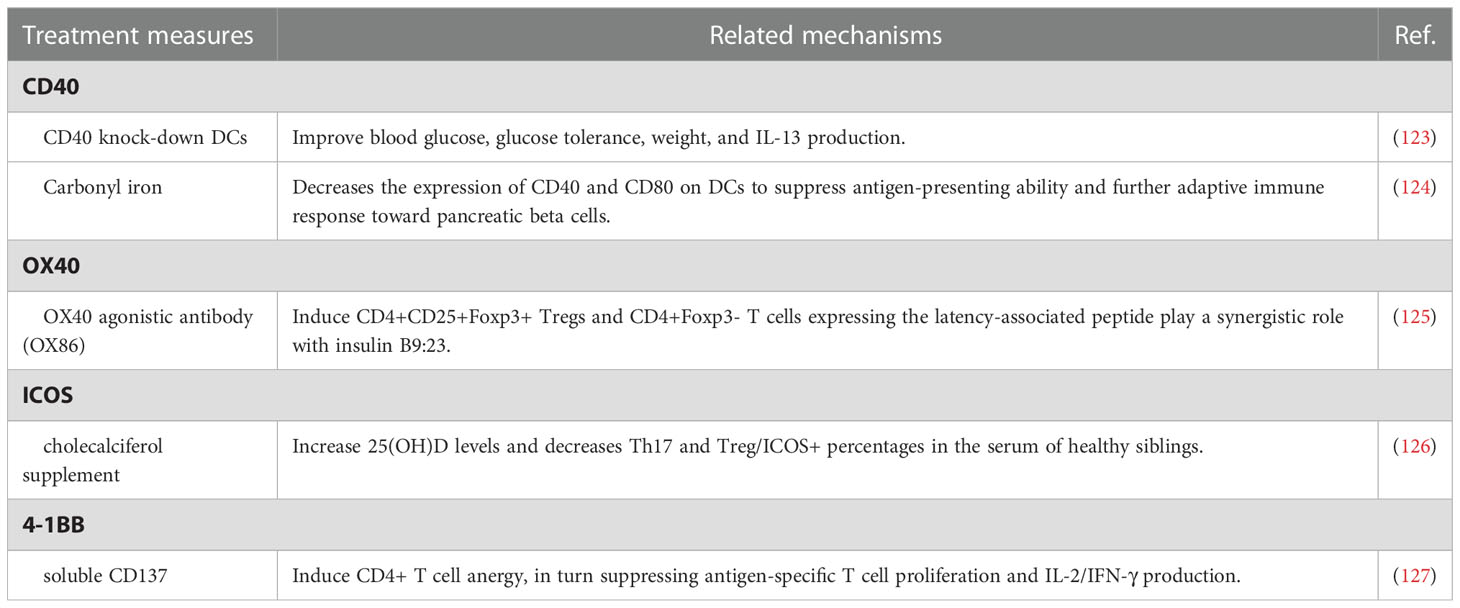
Table 2 The roles of treatment measures regulating co-stimulatory molecules for T1D.
4.1. Co-inhibitory molecules
CTLA-4 belongs to the immunoglobulin-related receptor family that is involved in multiple aspects of T cell immune regulation and peripheral tolerance and is considered to be one of the non-HLA genetic markers for T1D susceptibility (128–131). Due to a homolog structure to CD28, CTLA4 proteins share common ligands with CD28(CD80 and CD86) and even have higher affinity to CD80 and CD86 (132, 133). Therefore, CTLA4 proteins negatively regulate CD28-mediated T-cell co-stimulation (133, 134). Based on these characteristics, CTLA4-immunoglobulin (Ig), namely abatacept, selectively blocks CD28 co-stimulation and defends against potentially autoreactive T cells, thereby preventing T1D (7, 103, 132). Serr et al. have reported that sub-immunogenic vaccination with strong agonistic insulin mimetope efficiently suppresses effector T cells via inducing human insulin-specific Foxp3+ Treg with upregulated Foxp3, CTLA4, IL-2Rα and TIGIT expression, which provides a potential new drug target for prevention of islet autoimmunity of T1D (104).
PD-1(CD80), one member of the immunoglobulin superfamily, negatively regulates immune responses and mediates immune tolerance, impact on disease progression and aetiology of T1D (135–137). PD-1 deficiency specifically accelerates the development of subacute T1D in NOD mice (136, 138, 139). Downregulation of PD-1/PD-L1 on CD4+ and CD8+ T cells in patients with T1D is dynamically recovered in partial remission but decreased again after the partial remission phase (140, 141). Thus, the above-mentioned results suggest that PD-1/PD-L1 may be a potential target for T1D therapy. An anti-PD-1 immunotoxin selectively targeting and depleting PD-1-expressing cells delays disease onset in mouse models of autoimmune diabetes (97). Genetically engineered PD-L1-overexpressing platelets also suppress autoreactive pancreatic T-cell activity and reverse diabetes in novel hyperglycemic NOD mice (17). PFK15 treatment has been reported to increase the expression of PD-1 and LAG-3(lymphocyte-activation gene 3) on CD4 T cells and prevent the development of diabetes via inhibiting glycolysis utilization of diabetogenic CD4 T cells and reducing T cell responses to β cell antigen (105, 106). Low-molecular-weight dextran sulfate reduces the incidence of diabetes and even reverses diabetes in early-onset diabetic NOD mice, at least partly via increasing PD-1 expression in T cells, reducing interferon-γCD4 and CD8 T cells, and enhancing the number of FoxP3 cells (107, 108). Intralymphatic injection of aluminum-formulated glutamic acid decarboxylase (GAD-alum) delays the progression of T1D with immunomodulatory effects including increased PD-1+ CD69+ cells in both CD8+ and double negative T cells (109, 110).
Apart from CTLA-4 and PD-1, the next wave of co-inhibitory immune checkpoint receptor targets, including Lag-3, Tim-3, and TIGIT, are drawing increasing attention in clinical application.
LAG3 is an inhibitory immune checkpoint receptor regulating multiple immune functions, including T cell activation and proliferation, cytokine production, and cytolytic activity (142, 143). LAG-3 blockade promotes disease growth and progression in autoimmune-prone models. Corresponding to this, Jones et al. reported that T1D patients exhibited fewer LAG-3 CD4 and CD8 T cells compared with healthy controls (144). The interaction between LAG-3 and stable peptide-MHC class II complexes (pMHCII) induces T cell suppression and thereby inhibits diabetes progression in NOD mice with LAG-3 deficiency (111). Moreover, CYM-5442, a selective S1PR1 agonist, induces the expression of negative immune regulator receptor genes Pdcd1, Lag3, Ctla4, Tigit, and Btla to inhibit the autoimmune ability of T cells, leading to T1D prevention in the mouse Rip-LCMV T1D models (112). Similarly, oral Salmonella-based anti-CD3 mAb combined therapy tempts immunosuppressive CD4CD25Foxp3 Treg and CD4CD49bLAG3 Tr1 cells, then contributing to reversion of new-onset T1D in NOD mice (113). Treatment with anti-IL-7Rα antibodies for two weeks delays T1D incidence and upregulates LAG-3, Tim-3, and PD-1 on peripheral blood CD4 and CD8 T cells from prediabetic NOD mice (114). Autoimmune regulator (Aire)-overexpressing DCs upregulates CD73, Lag3, and FR4 that mediate self-tolerance, and decreases CD4+IFN-γ+ autoreactive T cells in STZ-T1D mouse-derived splenocytes, which is associated with Aire-overexpressing DCs induced T1D prevention and delay (115).
Tim-1, Tim-3, and Tim-4 are members of the T-cell immunoglobulin and mucin domain (Tim) molecule family in humans, and mediate peripheral immune tolerance via interacting with its ligands (145, 146). Compared with healthy controls, upregulated Tim-1 and downregulated Tim-3 led to imbalanced ratios of Tim-3/Tim-1 in T1D, in particular T1D patients with defective islet function (147). Another research focusing on Tregs reveals that Tim1 and Tim4 on CD4CD25 T cells decreased in peripheral blood mononuclear cells of patients with T1D than healthy volunteers (148). Tim-3 ligand galectin-9 enhances apoptosis of CD4+ Tim-3+ Th1 cells and downregulates Th1 immune response, and anti-Tim-3 monoclonal RMT3-23 antibody suppresses the TNF-α production and activation of DC, both exhibiting significant therapeutic effects on T1D (116, 117). Although a few studies addressing the therapeutic influence of Tim-related pathways for T1D in the last decade, we considered Tim as a novel target worthy of further exploration for treatment.
Expressed on Treg cells, T cell immunoglobulin and ITIM domain (TIGIT) is an inhibitory receptor that participates in the pathogenesis of T1D (149, 150). Higher percentage and expression levels of TIGIT are identified on CD4+CD25hi T cells, CD4+CD25- T cells, total CD4+ T cells, and non-CD4+ cells of peripheral blood mononuclear cells from T1D patients versus healthy controls (151). Teplizumab treatment increases the percentage of KLRG1+TIGIT+CD8+ T cells and suppresses disease progression to T1D in high-risk participants (118). A low circulating level of soluble receptors for advanced glycation end products(sRAGE) is representative of the high risk of T1D. sRAGE treatment reverses RAGE ligand-induced downregulation of key genes for Treg homeostasis and activation, including FOXP3, IL7R, TIGIT, JAK1, STAT3, STAT5b, and CCR4 (119, 120). Alefacept preserves endogenous insulin C-peptide production of T1D patients to a certain extent, which is related to hypo proliferative CD8 memory cells expressing exhaustion-associated markers including TIGIT and KLRG1 (121).
Another inhibitory immune checkpoint, BTLA (B- and T-lymphocyte attenuator)-HVEM (Herpesvirus entry mediator, namely TNFRSF14) complex, has been drawing increasing attention of the academic community as an important regulator in autoimmune reactions (152, 153). A lower expression of BTLA is identified in the peripheral blood of patients with young-onset T1D compared with adult-onset T1D patients (154). To explore a new therapeutic strategy, Gudi et al. conduct engineered tolerogenic dendritic cells (tDCs) expressing CTLA4 selective ligand and multiligand-DCs expressing a combination of CTLA4, PD1, and BTLA selective ligands, both of which present pancreatic β-cell antigen (BcAg). Both two types of engineered DCs, multiligand-DCs in particular, suppress the function of effector T cells and induce self-tolerance, thereby delaying the progression of T1D (122).
4.2. Co-stimulatory molecules
CD40 is a member of the tumor necrosis factor (TNF) receptor superfamily and interacts with CD40L to mediate the interaction between B and CD4+ T cells for germinal center responses and B cell activation (155, 156). Fully functional CD40 expression is not only required for hyperglycemia and insulitis in T1D but also induces relatively broad T‐cell receptor repertoire on CD40+ CD4+ cells (Th40 cells) during diabetogenesis (157). The number of Th40 cells significantly expands in the peripheral blood of T1D patients. Furthermore, Th40 cell levels also stratify pre-diabetic patients into two groups, with Th40-high subjects showing a higher percentage of disordered glucose tolerance, CD4/CD8 double-positive population, and T1D-associated HLA, including HLA DR4/DR4 and DQ8/DQ8 (158, 159). In the streptozotocin-induced diabetic mice model, CD40 knock-down DCs treatment improves blood glucose, glucose tolerance, weight, and IL-13 production (123). Highly expressed in B cells, Toll-like receptor 9(TLR9) is related to matrix metalloproteinases, tissue inhibitors of metalloproteinase-1, and CD40. B-cell-specific deletion of TLR9 near-completely protect NOD mice from T1D development (160). Adjuvant carbonyl iron inhibits the development of diabetes and decreases the expression of CD40 and CD80 on DCs to suppress antigen-presenting ability and further adaptive immune response toward pancreatic beta cells (124).
OX40, also named CD134 or TNFRSF4, a member of the TNF receptor family, serves as a co-stimulatory factor during T cell activation and controls effector and memory T cell responses (161–163). In T1D patients, soluble OX40 and OX40L expression in the serum is significantly upregulated and considered as potential indicators for disease progression, while membrane OX40 and OX40L expression on immune cells is significantly downregulated compared with the healthy controls (164). OX40 agonistic antibody (OX86) treatment induces CD4+CD25+Foxp3+ Tregs and CD4+Foxp3- T cells expressing the latency-associated peptide and reduces T1D incidence of NOD mice, which play a synergistic role with insulin B9:23. Interestingly, Tregs gathered from NOD mice treated with OX86 and insulin B9:23 also prevent T1D development when adoptively transferred into recipient mice (125).
Inducible co-stimulator (ICOS), a member of the CD28 superfamily, is expressed on activated T cells and specifically binds with its unique ligand ICOSL (165, 166). Children with impaired glucose tolerance and T1D exhibit a higher frequency of CXCR5+PD-1+ICOS+, CD4+CXCR5+, and CD4+CXCR5+ICOS+ circulating follicular helper T cells (Tfh). Interestingly, the expansion of CXCR5+PD-1+ICOS+ Tfh is more apparent in children with two or more biochemical autoantibodies (167, 168). Progressively reduction and suppression of ICOS+Foxp3+ Treg cells in islets are representing exacerbated T1D. Consistently, inhibited ICOS pathway also correlates with T1D progression in NOD.BDC2.5 mice (169). A recent cohort study exploring the potential association between blood serum 25 OH vitamin D(25[OH]D) levels and Th17 and Treg, and Treg/ICOS+ levels in healthy siblings of children with T1D reveals that Treg/ICOS+ percentages are higher in siblings with lower 25(OH)D levels and higher genetic risk for T1D. Furthermore, cholecalciferol supplement for 6 months increases 25(OH)D levels and decreases Th17 and Treg/ICOS+ percentages in the serum of healthy siblings (126). ICOS expression may also impact the effects of co-stimulation blockade administration. For example, higher frequencies of ICOS+ Tfh at baseline predict a poor clinical outcome following abatacept treatment (170).
As a member of the TNF receptor superfamily, 4-1BB, namely CD137 and TNFRSF9, is expressed on activated T cells and interacts with CD137L, the ligand of CD137, expressed by antigen-presenting cells (171). Interestingly, the impacts of CD137 on T1D progression in NOD mice associate with where it is expressed. CD137 in CD4 T cells suppresses T1D development, while CD137 expressed in CD8 T cells promotes disease progression (172). NOD.Tnfsf9-/- strain shows delayed T1D progression, less insulitis, and reduced β-cell-autoreactive CD8 T cells frequencies (173). Itoh et al. have reported that soluble CD137 induces CD4+ T cell anergy, in turn suppressing antigen-specific T cell proliferation and IL-2/IFN-γ production, thereby delaying progression to end-stage T1D in NOD mice (127).
5. Conclusion
ICIs block immune checkpoints and have emerged as a valuable alternative treatment for cancers with advanced stage, but endocrine toxicity, ICI-related DM, for example, limits their potential clinical application to some extent (20, 174). However, the similarity between ICI-related DM and T1D also suggests the potential feasibility of targeting immune checkpoint molecules for T1D treatment, which is also supported by higher circulating immune checkpoint molecule levels in T1D patients. Differing from massive immune inhibitors, targeted regulation of immune checkpoint molecules may specifically inhibit pathogenic lymphocytes associated with T1D (17, 96). Due to numerous co-inhibitory and co-stimulatory molecules involved in the treatment of T1D as mentioned above, it was valuable to explore novel therapeutic approaches regulating autoimmune-related T lymphocytes based on these targets for the management and treatment of T1D and may improve clinical outcomes.
Author contributions
All authors have discussed the proposed scope and content of the article before drafting. J-TD, K-PY, and K-LL wrote and revised the paper. Y-KC collected literature. FZ reviewed and edited the manuscript. All authors contributed to the article and approved the submitted version.
Funding
This work was supported by the National Natural Science Foundation of China (Grant Number: 82260169).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Ilonen J, Lempainen J, Veijola R. The heterogeneous pathogenesis of type 1 diabetes mellitus. Nat Rev Endocrinol (2019) 15(11):635–50. doi: 10.1038/s41574-019-0254-y
2. Gubitosi-Klug RA, Braffett BH, Hitt S, Arends V, Uschner D, Jones K, et al. Residual β cell function in long-term type 1 diabetes associates with reduced incidence of hypoglycemia. J Clin Invest (2021) 131(3). doi: 10.1172/JCI143011
3. Kupriyanova Y, Zaharia OP, Bobrov P, Karusheva Y, Burkart V, Szendroedi J, et al. Early changes in hepatic energy metabolism and lipid content in recent-onset type 1 and 2 diabetes mellitus. J Hepatol (2021) 74(5):1028–37. doi: 10.1016/j.jhep.2020.11.030
4. Singh AK, Khunti K. COVID-19 and diabetes. Annu Rev Med (2022) 73:129–47. doi: 10.1146/annurev-med-042220-011857
5. Brusko TM, Russ HA, Stabler CL. Strategies for durable β cell replacement in type 1 diabetes. Sci (New York NY). (2021) 373(6554):516–22. doi: 10.1126/science.abh1657
6. Yoshihara E, O'Connor C, Gasser E, Wei Z, Oh TG, Tseng TW, et al. Immune-evasive human islet-like organoids ameliorate diabetes. Nature (2020) 586(7830):606–11. doi: 10.1038/s41586-020-2631-z
7. Cabrera SM, Engle S, Kaldunski M, Jia S, Geoffrey R, Simpson P, et al. Innate immune activity as a predictor of persistent insulin secretion and association with responsiveness to CTLA4-ig treatment in recent-onset type 1 diabetes. Diabetologia (2018) 61(11):2356–70. doi: 10.1007/s00125-018-4708-x
8. Pollé OG, Delfosse A, Martin M, Louis J, Gies I, den Brinker M, et al. Glycemic variability patterns strongly correlate with partial remission status in children with newly diagnosed type 1 diabetes. Diabetes Care (2022) 45(10):2360–8. doi: 10.2337/dc21-2543
9. Au KM, Tisch R, Wang AZ. In vivo bioengineering of beta cells with immune checkpoint ligand as a treatment for early-onset type 1 diabetes mellitus. ACS Nano. (2021) 15(12):19990–20002. doi: 10.1021/acsnano.1c07538
10. Zhong T, Tang R, Gong S, Li J, Li X, Zhou Z. The remission phase in type 1 diabetes: Changing epidemiology, definitions, and emerging immuno-metabolic mechanisms. Diabetes Metab Res Rev (2020) 36(2):e3207. doi: 10.1002/dmrr.3207
11. Hirsch JS. FDA Approves teplizumab: A milestone in type 1 diabetes. Lancet Diabetes Endocrinol (2022) 11(1):18. doi: 10.1016/S2213-8587(22)00351-5
12. Colli ML, Ramos-Rodríguez M, Nakayasu ES, Alvelos MI, Lopes M, Hill JLE, et al. An integrated multi-omics approach identifies the landscape of interferon-α-mediated responses of human pancreatic beta cells. Nat Commun (2020) 11(1):2584. doi: 10.1038/s41467-020-16327-0
13. Pelgrom LR, Patente TA, Sergushichev A, Esaulova E, Otto F, Ozir-Fazalalikhan A, et al. LKB1 expressed in dendritic cells governs the development and expansion of thymus-derived regulatory T cells. Cell Res (2019) 29(5):406–19. doi: 10.1038/s41422-019-0161-8
14. Kresoja K-P, Rommel K-P, Wachter R, Henger S, Besler C, Klöting N, et al. Proteomics to improve phenotyping in obese patients with heart failure with preserved ejection fraction. Eur J Heart Fail (2021) 23(10):1633–44. doi: 10.1002/ejhf.2291
15. Franzin R, Netti GS, Spadaccino F, Porta C, Gesualdo L, Stallone G, et al. The use of immune checkpoint inhibitors in oncology and the occurrence of AKI: Where do we stand? Front Immunol (2020) 11:574271. doi: 10.3389/fimmu.2020.574271
16. Morad G, Helmink BA, Sharma P, Wargo JA. Hallmarks of response, resistance, and toxicity to immune checkpoint blockade. Cell (2021) 184(21):5309–37. doi: 10.1016/j.cell.2021.09.020
17. Zhang X, Kang Y, Wang J, Yan J, Chen Q, Cheng H, et al. Engineered PD-L1-Expressing platelets reverse new-onset type 1 diabetes. Adv Mater (2020) 32(26):e1907692. doi: 10.1002/adma.201907692
18. Au KM, Medik Y, Ke Q, Tisch R, Wang AZ. Immune checkpoint-bioengineered beta cell vaccine reverses early-onset type 1 diabetes. Advanced Materials (Deerfield Beach Fla). (2021) 33(25):e2101253. doi: 10.1002/adma.202101253
19. Gunjur A, Klein O, Kee D, Cebon J. Anti-programmed cell death protein 1 (anti-PD1) immunotherapy induced autoimmune polyendocrine syndrome type II (APS-2): A case report and review of the literature. J Immunother Cancer. (2019) 7(1):241. doi: 10.1186/s40425-019-0713-y
20. de Filette JMK, Pen JJ, Decoster L, Vissers T, Bravenboer B, van der Auwera BJ, et al. Immune checkpoint inhibitors and type 1 diabetes mellitus: A case report and systematic review. Eur J Endocrinol (2019) 181(3):363–74. doi: 10.1530/EJE-19-0291
21. Lin C, Li X, Qiu Y, Chen Z, Liu J. PD-1 inhibitor-associated type 1 diabetes: A case report and systematic review. Front Public Health (2022) 10:885001. doi: 10.3389/fpubh.2022.885001
22. Paschou SA, Stefanaki K, Psaltopoulou T, Liontos M, Koutsoukos K, Zagouri F, et al. How we treat endocrine complications of immune checkpoint inhibitors. ESMO Open (2021) 6(1):100011. doi: 10.1016/j.esmoop.2020.100011
23. Stamatouli AM, Quandt Z, Perdigoto AL, Clark PL, Kluger H, Weiss SA, et al. Collateral damage: Insulin-dependent diabetes induced with checkpoint inhibitors. Diabetes (2018) 67(8):1471–80. doi: 10.2337/dbi18-0002
24. Dai X, Bu X, Gao Y, Guo J, Hu J, Jiang C, et al. Energy status dictates PD-L1 protein abundance and anti-tumor immunity to enable checkpoint blockade. Mol Cell (2021) 81(11):2317–31.e6. doi: 10.1016/j.molcel.2021.03.037
25. Kotwal A, Haddox C, Block M, Kudva YC. Immune checkpoint inhibitors: an emerging cause of insulin-dependent diabetes. BMJ Open Diabetes Res Care (2019) 7(1):e000591. doi: 10.1136/bmjdrc-2018-000591
26. Zhang AL, Wang F, Chang LS, McDonnell ME, Min L. Coexistence of immune checkpoint inhibitor-induced autoimmune diabetes and pancreatitis. Front Endocrinol (Lausanne). (2021) 12:620522. doi: 10.3389/fendo.2021.620522
27. Chujo D, Nguyen TS, Foucat E, Blankenship D, Banchereau J, Nepom GT, et al. Adult-onset type 1 diabetes patients display decreased IGRP-specific Tr1 cells in blood. Clin Immunol (2015) 161(2):270–7. doi: 10.1016/j.clim.2015.08.014
28. Kukreja A, Cost G, Marker J, Zhang C, Sun Z, Lin-Su K, et al. Multiple immuno-regulatory defects in type-1 diabetes. J Clin Invest. (2002) 109(1):131–40. doi: 10.1172/JCI0213605
29. Brusko T, Wasserfall C, McGrail K, Schatz R, Viener HL, Schatz D, et al. No alterations in the frequency of FOXP3+ regulatory T-cells in type 1 diabetes. Diabetes (2007) 56(3):604–12. doi: 10.2337/db06-1248
30. Todd JA. Etiology of type 1 diabetes. Immunity (2010) 32(4):457–67. doi: 10.1016/j.immuni.2010.04.001
31. Dong S, Hiam-Galvez KJ, Mowery CT, Herold KC, Gitelman SE, Esensten JH, et al. The effect of low-dose IL-2 and treg adoptive cell therapy in patients with type 1 diabetes. JCI Insight (2021) 6(18). doi: 10.1172/jci.insight.147474
32. Zhou L, He X, Cai P, Li T, Peng R, Dang J, et al. Induced regulatory T cells suppress Tc1 cells through TGF-β signaling to ameliorate STZ-induced type 1 diabetes mellitus. Cell Mol Immunol (2021) 18(3):698–710. doi: 10.1038/s41423-020-00623-2
33. Yu H, Gagliani N, Ishigame H, Huber S, Zhu S, Esplugues E, et al. Intestinal type 1 regulatory T cells migrate to periphery to suppress diabetogenic T cells and prevent diabetes development. Proc Natl Acad Sci U S A. (2017) 114(39):10443–8. doi: 10.1073/pnas.1705599114
34. Song J. Development of auto antigen-specific regulatory T cells for diabetes immunotherapy. Immune Netw (2016) 16(5):281–5. doi: 10.4110/in.2016.16.5.281
35. Gliwiński M, Iwaszkiewicz-Grześ D, Wołoszyn-Durkiewicz A, Tarnowska M, Żalińska M, Hennig M, et al. Proinsulin-specific T regulatory cells may control immune responses in type 1 diabetes: Implications for adoptive therapy. BMJ Open Diabetes Res Care (2020) 8(1). doi: 10.1136/bmjdrc-2019-000873
36. Rosenzwajg M, Salet R, Lorenzon R, Tchitchek N, Roux A, Bernard C, et al. Low-dose IL-2 in children with recently diagnosed type 1 diabetes: A phase I/II randomised, double-blind, placebo-controlled, dose-finding study. Diabetologia (2020) 63(9):1808–21. doi: 10.1007/s00125-020-05200-w
37. Amouyal C, Klatzmann D, Tibi E, Salem JE, Halbron M, Popelier M, et al. Pregnant type 1 diabetes women with rises in c-peptide display higher levels of regulatory T cells: A pilot study. Diabetes Metab (2021) 47(3):101188. doi: 10.1016/j.diabet.2020.04.005
38. Ben-Skowronek I, Sieniawska J, Pach E, Wrobel W, Skowronek A, Tomczyk Z, et al. Potential therapeutic application of regulatory T cells in diabetes mellitus type 1. Int J Mol Sci (2021) 23(1):390. doi: 10.3390/ijms23010390
39. Zieliński M, Żalińska M, Iwaszkiewicz-Grześ D, Gliwiński M, Hennig M, Jaźwińska-Curyłło A, et al. Combined therapy with CD4(+) CD25highCD127(-) T regulatory cells and anti-CD20 antibody in recent-onset type 1 diabetes is superior to monotherapy: Randomized phase I/II trial. Diabetes Obes Metab (2022) 24(8):1534–43. doi: 10.1111/dom.14723
40. Marfil-Garza BA, Hefler J, Bermudez De Leon M, Pawlick R, Dadheech N, Shapiro AMJ. Progress in translational regulatory T cell therapies for type 1 diabetes and islet transplantation. Endocrine Rev (2020) 42(2):198–218. doi: 10.1210/endrev/bnaa028
41. Wang CJ, Petersone L, Edner NM, Heuts F, Ovcinnikovs V, Ntavli E, et al. Costimulation blockade in combination with IL-2 permits regulatory T cell sparing immunomodulation that inhibits autoimmunity. Nat Commun (2022) 13(1):6757. doi: 10.1038/s41467-022-34477-1
42. Nagy N, Kaber G, Kratochvil MJ, Kuipers HF, Ruppert SM, Yadava K, et al. Weekly injection of IL-2 using an injectable hydrogel reduces autoimmune diabetes incidence in NOD mice. Diabetologia (2021) 64(1):152–8. doi: 10.1007/s00125-020-05314-1
43. Kabelitz D, Geissler EK, Soria B, Schroeder IS, Fändrich F, Chatenoud L. Toward cell-based therapy of type I diabetes. Trends Immunol (2008) 29(2):68–74. doi: 10.1016/j.it.2007.11.001
44. Bluestone JA, Buckner JH, Fitch M, Gitelman SE, Gupta S, Hellerstein MK, et al. Type 1 diabetes immunotherapy using polyclonal regulatory T cells. Sci Transl Med (2015) 7(315):315ra189. doi: 10.1126/scitranslmed.aad4134
45. Marek-Trzonkowska N, Myśliwiec M, Dobyszuk A, Grabowska M, Derkowska I, Juścińska J, et al. Therapy of type 1 diabetes with CD4(+)CD25(high)CD127-regulatory T cells prolongs survival of pancreatic islets - results of one year follow-up. Clin Immunol (2014) 153(1):23–30. doi: 10.1016/j.clim.2014.03.016
46. Gibson VB, Nikolic T, Pearce VQ, Demengeot J, Roep BO, Peakman M. Proinsulin multi-peptide immunotherapy induces antigen-specific regulatory T cells and limits autoimmunity in a humanized model. Clin Exp Immunol (2015) 182(3):251–60. doi: 10.1111/cei.12687
47. Martinez NR, Augstein P, Moustakas AK, Papadopoulos GK, Gregori S, Adorini L, et al. Disabling an integral CTL epitope allows suppression of autoimmune diabetes by intranasal proinsulin peptide. J Clin Invest (2003) 111(9):1365–71. doi: 10.1172/JCI200317166
48. Orban T, Farkas K, Jalahej H, Kis J, Treszl A, Falk B, et al. Autoantigen-specific regulatory T cells induced in patients with type 1 diabetes mellitus by insulin b-chain immunotherapy. J Autoimmunity. (2010) 34(4):408–15. doi: 10.1016/j.jaut.2009.10.005
49. Russo F, Citro A, Squeri G, Sanvito F, Monti P, Gregori S, et al. InsB9-23 gene transfer to hepatocyte-based combined therapy abrogates recurrence of type 1 diabetes after islet transplantation. Diabetes (2020) 70(1):171–81. doi: 10.2337/db19-1249
50. Cheatem D, Ganesh BB, Gangi E, Vasu C, Prabhakar BS. Modulation of dendritic cells using granulocyte-macrophage colony-stimulating factor (GM-CSF) delays type 1 diabetes by enhancing CD4+CD25+ regulatory T cell function. Clin Immunol (2009) 131(2):260–70. doi: 10.1016/j.clim.2008.12.001
51. Alhadj Ali M, Liu Y-F, Arif S, Tatovic D, Shariff H, Gibson VB, et al. Metabolic and immune effects of immunotherapy with proinsulin peptide in human new-onset type 1 diabetes. Sci Trans Med (2017) 9(402). doi: 10.1126/scitranslmed.aaf7779
52. Lin JR, Huang SH, Wu CH, Chen YW, Hong ZJ, Cheng CP, et al. Valproic acid suppresses autoimmune recurrence and allograft rejection in islet transplantation through induction of the differentiation of regulatory T cells and can be used in cell therapy for type 1 diabetes. Pharm (Basel) (2021) 14(5):475. doi: 10.3390/ph14050475
53. Xiao Z, Wei T, Ge R, Li Q, Liu B, Ji Z, et al. Microfluidic production of zwitterion coating microcapsules with low foreign body reactions for improved islet transplantation. Small (2022) 18(29):e2202596. doi: 10.1002/smll.202202596
54. Chang LS, Barroso-Sousa R, Tolaney SM, Hodi FS, Kaiser UB, Min L. Endocrine toxicity of cancer immunotherapy targeting immune checkpoints. Endocr Rev (2019) 40(1):17–65. doi: 10.1210/er.2018-00006
55. Zhang Y, Zheng J. Functions of immune checkpoint molecules beyond immune evasion. Adv Exp Med Biol (2020) 1248:201–26. doi: 10.1007/978-981-15-3266-5_9
56. de Miguel M, Calvo E. Clinical challenges of immune checkpoint inhibitors. Cancer Cell (2020) 38(3):326–33. doi: 10.1016/j.ccell.2020.07.004
57. Lee JB, Ha SJ, Kim HR. Clinical insights into novel immune checkpoint inhibitors. Front Pharmacol (2021) 12:681320. doi: 10.3389/fphar.2021.681320
58. Lee HT, Lee SH, Heo YS. Molecular interactions of antibody drugs targeting PD-1, PD-L1, and CTLA-4 in immuno-oncology. Molecules (2019) 24(6):1190. doi: 10.3390/molecules24061190
59. Akturk HK, Kahramangil D, Sarwal A, Hoffecker L, Murad MH, Michels AW. Immune checkpoint inhibitor-induced type 1 diabetes: a systematic review and meta-analysis. Diabetes Med (2019) 36(9):1075–81. doi: 10.1111/dme.14050
60. Marchand L, Disse E, Dalle S, Reffet S, Vouillarmet J, Fabien N, et al. The multifaceted nature of diabetes mellitus induced by checkpoint inhibitors. Acta Diabetol (2019) 56(12):1239–45. doi: 10.1007/s00592-019-01402-w
61. Gillespie KM. Type 1 diabetes: Pathogenesis and prevention. Cmaj (2006) 175(2):165–70. doi: 10.1503/cmaj.060244
62. Clotman K, Janssens K, Specenier P, Weets I, De Block CEM. Programmed cell death-1 inhibitor-induced type 1 diabetes mellitus. J Clin Endocrinol Metab (2018) 103(9):3144–54. doi: 10.1210/jc.2018-00728
63. Mourad D, Azar NS, Eid AA, Azar ST. Immune checkpoint inhibitor-induced diabetes mellitus: Potential role of T cells in the underlying mechanism. Int J Mol Sci (2021) 22(4):2093. doi: 10.3390/ijms22042093
64. Ansari MJ, Salama AD, Chitnis T, Smith RN, Yagita H, Akiba H, et al. The programmed death-1 (PD-1) pathway regulates autoimmune diabetes in nonobese diabetic (NOD) mice. J Exp Med (2003) 198(1):63–9. doi: 10.1084/jem.20022125
65. Tan CL, Kuchroo JR, Sage PT, Liang D, Francisco LM, Buck J, et al. PD-1 restraint of regulatory T cell suppressive activity is critical for immune tolerance. J Exp Med (2021) 218(1). doi: 10.1084/jem.20182232
66. Kochupurakkal NM, Kruger AJ, Tripathi S, Zhu B, Adams LT, Rainbow DB, et al. Blockade of the programmed death-1 (PD1) pathway undermines potent genetic protection from type 1 diabetes. PloS One (2014) 9(2):e89561. doi: 10.1371/journal.pone.0089561
67. Tucker CG, Dwyer AJ, Fife BT, Martinov T. The role of programmed death-1 in type 1 diabetes. Curr Diabetes Rep (2021) 21(6):20. doi: 10.1007/s11892-021-01384-6
68. Colli ML, Hill JLE, Marroquí L, Chaffey J, Dos Santos RS, Leete P, et al. PDL1 is expressed in the islets of people with type 1 diabetes and is up-regulated by interferons-α and-γ via IRF1 induction. EBioMedicine (2018) 36:367–75. doi: 10.1016/j.ebiom.2018.09.040
69. Marroqui L, Dos Santos RS, Op de Beeck A, Coomans de Brachène A, Marselli L, Marchetti P, et al. Interferon-α mediates human beta cell HLA class I overexpression, endoplasmic reticulum stress and apoptosis, three hallmarks of early human type 1 diabetes. Diabetologia (2017) 60(4):656–67. doi: 10.1007/s00125-016-4201-3
70. Moore F, Naamane N, Colli ML, Bouckenooghe T, Ortis F, Gurzov EN, et al. STAT1 is a master regulator of pancreatic β-cell apoptosis and islet inflammation. J Biol Chem (2011) 286(2):929–41. doi: 10.1074/jbc.M110.162131
71. Colli ML, Szymczak F, Eizirik DL. Molecular footprints of the immune assault on pancreatic beta cells in type 1 diabetes. Front Endocrinol (Lausanne). (2020) 11:568446. doi: 10.3389/fendo.2020.568446
72. Zamani MR, Aslani S, Salmaninejad A, Javan MR, Rezaei N. PD-1/PD-L and autoimmunity: A growing relationship. Cell Immunol (2016) 310:27–41. doi: 10.1016/j.cellimm.2016.09.009
73. Rui J, Deng S, Arazi A, Perdigoto AL, Liu Z, Herold KC. β cells that resist immunological attack develop during progression of autoimmune diabetes in NOD mice. Cell Metab (2017) 25(3):727–38. doi: 10.1016/j.cmet.2017.01.005
74. Wang CJ, Chou FC, Chu CH, Wu JC, Lin SH, Chang DM, et al. Protective role of programmed death 1 ligand 1 (PD-L1)in nonobese diabetic mice: The paradox in transgenic models. Diabetes (2008) 57(7):1861–9. doi: 10.2337/db07-1260
75. Jia XH, Geng LY, Jiang PP, Xu H, Nan KJ, Yao Y, et al. The biomarkers related to immune related adverse events caused by immune checkpoint inhibitors. J Exp Clin Cancer Res (2020) 39(1):284. doi: 10.1186/s13046-020-01749-x
76. Xu Y, Fu Y, Zhu B, Wang J, Zhang B. Predictive biomarkers of immune checkpoint inhibitors-related toxicities. Front Immunol (2020) 11:2023. doi: 10.3389/fimmu.2020.02023
77. Byun DJ, Braunstein R, Flynn J, Zheng J, Lefkowitz RA, Kanbour S, et al. Immune checkpoint inhibitor-associated diabetes: A single-institution experience. Diabetes Care (2020) 43(12):3106–9. doi: 10.2337/dc20-0609
78. Zheng Z, Liu Y, Yang J, Tan C, Zhou L, Wang X, et al. Diabetes mellitus induced by immune checkpoint inhibitors. Diabetes Metab Res Rev (2021) 37(1):e3366. doi: 10.1002/dmrr.3366
79. Barroso-Sousa R, Barry WT, Garrido-Castro AC, Hodi FS, Min L, Krop IE, et al. Incidence of endocrine dysfunction following the use of different immune checkpoint inhibitor regimens: A systematic review and meta-analysis. JAMA Oncol (2018) 4(2):173–82. doi: 10.1001/jamaoncol.2017.3064
80. Puzanov I, Diab A, Abdallah K, Bingham CO 3rd, Brogdon C, Dadu R, et al. Managing toxicities associated with immune checkpoint inhibitors: consensus recommendations from the society for immunotherapy of cancer (SITC) toxicity management working group. J Immunother Cancer. (2017) 5(1):95. doi: 10.1186/s40425-017-0300-z
81. Wang M, Zhai X, Li J, Guan J, Xu S, Li Y, et al. The role of cytokines in predicting the response and adverse events related to immune checkpoint inhibitors. Front Immunol (2021) 12:670391. doi: 10.3389/fimmu.2021.670391
82. Das S, Johnson DB. Immune-related adverse events and anti-tumor efficacy of immune checkpoint inhibitors. J Immunother Cancer. (2019) 7(1):306. doi: 10.1186/s40425-019-0805-8
83. Weinmann SC, Pisetsky DS. Mechanisms of immune-related adverse events during the treatment of cancer with immune checkpoint inhibitors. Rheumatol (Oxford). (2019) 58(Suppl 7):vii59–67. doi: 10.1093/rheumatology/kez308
84. Iwama S. [Endocrine dysfunction associated with immune checkpoint blockade]. Gan To Kagaku Ryoho. (2020) 47(2):203–6.
85. Elia G, Ferrari SM, Galdiero MR, Ragusa F, Paparo SR, Ruffilli I, et al. New insight in endocrine-related adverse events associated to immune checkpoint blockade. Best Pract Res Clin Endocrinol Metab (2020) 34(1):101370. doi: 10.1016/j.beem.2019.101370
86. Wright JJ, Powers AC, Johnson DB. Endocrine toxicities of immune checkpoint inhibitors. Nat Rev Endocrinol (2021) 17(7):389–99. doi: 10.1038/s41574-021-00484-3
87. Rushworth RL, Torpy DJ, Falhammar H. Adrenal crisis. N Engl J Med (2019) 381(9):852–61. doi: 10.1056/NEJMra1807486
88. Farina KA, Kane MP. Programmed cell death-1 monoclonal antibody therapy and type 1 diabetes mellitus: A review of the literature. J Pharm Pract (2021) 34(1):133–40. doi: 10.1177/0897190019850929
89. Quandt Z, Young A, Anderson M. Immune checkpoint inhibitor diabetes mellitus: a novel form of autoimmune diabetes. Clin Exp Immunol (2020) 200(2):131–40. doi: 10.1111/cei.13424
90. Zhang R, Cai XL, Liu L, Han XY, Ji LN. Type 1 diabetes induced by immune checkpoint inhibitors. Chin Med J (Engl). (2020) 133(21):2595–8. doi: 10.1097/CM9.0000000000000972
91. Tsang VHM, McGrath RT, Clifton-Bligh RJ, Scolyer RA, Jakrot V, Guminski AD, et al. Checkpoint inhibitor-associated autoimmune diabetes is distinct from type 1 diabetes. J Clin Endocrinol Metab (2019) 104(11):5499–506. doi: 10.1210/jc.2019-00423
92. Yoneda S, Imagawa A, Hosokawa Y, Baden MY, Kimura T, Uno S, et al. T-Lymphocyte infiltration to islets in the pancreas of a patient who developed type 1 diabetes after administration of immune checkpoint inhibitors. Diabetes Care (2019) 42(7):e116–e8. doi: 10.2337/dc18-2518
93. Baden MY, Imagawa A, Abiru N, Awata T, Ikegami H, Uchigata Y, et al. Characteristics and clinical course of type 1 diabetes mellitus related to anti-programmed cell death-1 therapy. Diabetol Int (2019) 10(1):58–66. doi: 10.1007/s13340-018-0362-2
94. Orabona C, Mondanelli G, Puccetti P, Grohmann U. Immune checkpoint molecules, personalized immunotherapy, and autoimmune diabetes. Trends Mol Med (2018) 24(11):931–41. doi: 10.1016/j.molmed.2018.08.005
95. McNamara C, Sugrue G, Murray B, MacMahon PJ. Current and emerging therapies in multiple sclerosis: Implications for the radiologist, part 2-surveillance for treatment complications and disease progression. AJNR Am J Neuroradiol (2017) 38(9):1672–80. doi: 10.3174/ajnr.A5148
96. Wang JY, Wang WP. B7-H4, a promising target for immunotherapy. Cell Immunol (2020) 347:104008. doi: 10.1016/j.cellimm.2019.104008
97. Zhao P, Wang P, Dong S, Zhou Z, Cao Y, Yagita H, et al. Depletion of PD-1-positive cells ameliorates autoimmune disease. Nat BioMed Eng. (2019) 3(4):292–305. doi: 10.1038/s41551-019-0360-0
98. Arena A, Belcastro E, Accardo A, Sandomenico A, Pagliarosi O, Rosa E, et al. Preparation and In vitro evaluation of RITUXfab-decorated lipoplexes to improve delivery of siRNA targeting C1858T PTPN22 variant in b lymphocytes. Int J Mol Sci (2021) 23(1):408. doi: 10.3390/ijms23010408
99. Khan M, Arooj S, Wang H. NK cell-based immune checkpoint inhibition. Front Immunol (2020) 11:167. doi: 10.3389/fimmu.2020.00167
100. Riva A, Chokshi S. Immune checkpoint receptors: homeostatic regulators of immunity. Hepatol Int (2018) 12(3):223–36. doi: 10.1007/s12072-018-9867-9
101. Giat E, Ehrenfeld M, Shoenfeld Y. Cancer and autoimmune diseases. Autoimmun Rev (2017) 16(10):1049–57. doi: 10.1016/j.autrev.2017.07.022
102. Bruzzaniti S, Piemonte E, Mozzillo E, Bruzzese D, Lepore MT, Carbone F, et al. High levels of blood circulating immune checkpoint molecules in children with new-onset type 1 diabetes are associated with the risk of developing an additional autoimmune disease. Diabetologia (2022) 65(8):1390–7. doi: 10.1007/s00125-022-05724-3
103. Goenka R, Xu Z, Samayoa J, Banach D, Beam C, Bose S, et al. CTLA4-Ig-Based bifunctional costimulation inhibitor blocks CD28 and ICOS signaling to prevent T cell priming and effector function. J Immunol (2021) 206(5):1102–13. doi: 10.4049/jimmunol.2001100
104. Serr I, Furst RW, Achenbach P, Scherm MG, Gokmen F, Haupt F, et al. Type 1 diabetes vaccine candidates promote human Foxp3(+)Treg induction in humanized mice. Nat Commun (2016) 7:10991. doi: 10.1038/ncomms10991
105. Martins CP, New LA, O'Connor EC, Previte DM, Cargill KR, Tse IL, et al. Glycolysis inhibition induces functional and metabolic exhaustion of CD4(+) T cells in type 1 diabetes. Front Immunol (2021) 12:669456. doi: 10.3389/fimmu.2021.669456
106. Scharping NE, Menk AV, Moreci RS, Whetstone RD, Dadey RE, Watkins SC, et al. The tumor microenvironment represses T cell mitochondrial biogenesis to drive intratumoral T cell metabolic insufficiency and dysfunction. Immunity (2016) 45(3):701–3. doi: 10.1016/j.immuni.2016.08.009
107. Lu G, Rausell-Palamos F, Zhang J, Zheng Z, Zhang T, Valle S, et al. Dextran sulfate protects pancreatic beta-cells, reduces autoimmunity, and ameliorates type 1 diabetes. Diabetes (2020) 69(8):1692–707. doi: 10.2337/db19-0725
108. Itoh A, Ridgway WM. Targeting innate immunity to downmodulate adaptive immunity and reverse type 1 diabetes. Immunotargets Ther (2017) 6:31–8. doi: 10.2147/ITT.S117264
109. Barcenilla H, Pihl M, Wahlberg J, Ludvigsson J, Casas R. Intralymphatic GAD-alum injection modulates b cell response and induces follicular helper T cells and PD-1+ CD8+ T cells in patients with recent-onset type 1 diabetes. Front Immunol (2021) 12:797172. doi: 10.3389/fimmu.2021.797172
110. Ludvigsson J, Krisky D, Casas R, Battelino T, Castano L, Greening J, et al. GAD65 antigen therapy in recently diagnosed type 1 diabetes mellitus. N Engl J Med (2012) 366(5):433–42. doi: 10.1056/NEJMoa1107096
111. Maruhashi T, Sugiura D, Okazaki IM, Shimizu K, Maeda TK, Ikubo J, et al. Binding of LAG-3 to stable peptide-MHC class II limits T cell function and suppresses autoimmunity and anti-cancer immunity. Immunity (2022) 55(5):912–24 e8. doi: 10.1016/j.immuni.2022.03.013
112. Marro BS, Ware BC, Zak J, de la Torre JC, Rosen H, Oldstone MB. Progression of type 1 diabetes from the prediabetic stage is controlled by interferon-alpha signaling. Proc Natl Acad Sci USA (2017) 114(14):3708–13. doi: 10.1073/pnas.1700878114
113. Mbongue JC, Rawson J, Garcia PA, Gonzalez N, Cobb J, Kandeel F, et al. Reversal of new onset type 1 diabetes by oral salmonella-based combination therapy and mediated by regulatory T-cells in NOD mice. Front Immunol (2019) 10:320. doi: 10.3389/fimmu.2019.00320
114. Vazquez-Mateo C, Collins J, Fleury M, Dooms H. Broad induction of immunoregulatory mechanisms after a short course of anti-IL-7Ralpha antibodies in NOD mice. BMC Immunol (2017) 18(1):18. doi: 10.1186/s12865-017-0201-4
115. Li D, Li H, Fu H, Niu K, Guo Y, Guo C, et al. Aire-overexpressing dendritic cells induce peripheral CD4(+) T cell tolerance. Int J Mol Sci (2015) 17(1):38. doi: 10.3390/ijms17010038
116. Kanzaki M, Wada J, Sugiyama K, Nakatsuka A, Teshigawara S, Murakami K, et al. Galectin-9 and T cell immunoglobulin mucin-3 pathway is a therapeutic target for type 1 diabetes. Endocrinology (2012) 153(2):612–20. doi: 10.1210/en.2011-1579
117. Chou FC, Shieh SJ, Sytwu HK. Attenuation of Th1 response through galectin-9 and T-cell ig mucin 3 interaction inhibits autoimmune diabetes in NOD mice. Eur J Immunol (2009) 39(9):2403–11. doi: 10.1002/eji.200839177
118. Herold KC, Bundy BN, Long SA, Bluestone JA, DiMeglio LA, Dufort MJ, et al. An anti-CD3 antibody, teplizumab, in relatives at risk for type 1 diabetes. N Engl J Med (2019) 381(7):603–13. doi: 10.1056/NEJMoa1902226
119. Leung SS, Borg DJ, McCarthy DA, Boursalian TE, Cracraft J, Zhuang A, et al. Soluble RAGE prevents type 1 diabetes expanding functional regulatory T cells. Diabetes (2022) 71(9):1994–2008. doi: 10.2337/db22-0177
120. Salonen KM, Ryhanen SJ, Forbes JM, Harkonen T, Ilonen J, Simell O, et al. A drop in the circulating concentrations of soluble receptor for advanced glycation end products is associated with seroconversion to autoantibody positivity but not with subsequent progression to clinical disease in children en route to type 1 diabetes. Diabetes Metab Res Rev (2017) 33(4). doi: 10.1002/dmrr.2872
121. Diggins KE, Serti E, Muir V, Rosasco M, Lu T, Balmas E, et al. Exhausted-like CD8+ T cell phenotypes linked to c-peptide preservation in alefacept-treated T1D subjects. JCI Insight (2021) 6(3). doi: 10.1172/jci.insight.142680
122. Gudi RR, Perez N, Karumuthil-Melethil S, Li G, Vasu C. Activation of T cell checkpoint pathways during beta-cell antigen presentation by engineered dendritic cells promotes protection from type 1 diabetes. Immunology (2022) 166(3):341–56. doi: 10.1111/imm.13476
123. Mahmoudzadeh A, Pourfathollah AA, Karimi MH, Moazzeni SM. CD40 knocked down tolerogenic dendritic cells decrease diabetic injury. Iran J Immunol (2017) 14(4):270–80.
124. Vujicic M, Saksida T, Mostarica Stojkovic M, Djedovic N, Stojanovic I, Stosic-Grujicic S. Protective effects of carbonyl iron against multiple low-dose streptozotocin-induced diabetes in rodents. J Cell Physiol (2018) 233(6):4990–5001. doi: 10.1002/jcp.26338
125. Bresson D, Fousteri G, Manenkova Y, Croft M, von Herrath M. Antigen-specific prevention of type 1 diabetes in NOD mice is ameliorated by OX40 agonist treatment. J Autoimmun (2011) 37(4):342–51. doi: 10.1016/j.jaut.2011.10.001
126. Savastio S, Cadario F, D'Alfonso S, Stracuzzi M, Pozzi E, Raviolo S, et al. Vitamin d supplementation modulates ICOS+ and ICOS- regulatory T cell in siblings of children with type 1 diabetes. J Clin Endocrinol Metab (2020) 105(12). doi: 10.1210/clinem/dgaa588
127. Itoh A, Ortiz L, Kachapati K, Wu Y, Adams D, Bednar K, et al. Soluble CD137 ameliorates acute type 1 diabetes by inducing T cell anergy. Front Immunol (2019) 10:2566. doi: 10.3389/fimmu.2019.02566
128. Rowshanravan B, Halliday N, Sansom DM. CTLA-4: A moving target in immunotherapy. Blood (2018) 131(1):58–67. doi: 10.1182/blood-2017-06-741033
129. Ueda H, Howson JM, Esposito L, Heward J, Snook H, Chamberlain G, et al. Association of the T-cell regulatory gene CTLA4 with susceptibility to autoimmune disease. Nature (2003) 423(6939):506–11. doi: 10.1038/nature01621
130. Frommer L, Kahaly GJ. Type 1 diabetes and autoimmune thyroid disease-the genetic link. Front Endocrinol (Lausanne). (2021) 12:618213. doi: 10.3389/fendo.2021.618213
131. Zurawek M, Dzikiewicz-Krawczyk A, Izykowska K, Ziolkowska-Suchanek I, Skowronska B, Czainska M, et al. miR-487a-3p upregulated in type 1 diabetes targets CTLA4 and FOXO3. Diabetes Res Clin Pract (2018) 142:146–53. doi: 10.1016/j.diabres.2018.05.044
132. Hosseini A, Gharibi T, Marofi F, Babaloo Z, Baradaran B. CTLA-4: From mechanism to autoimmune therapy. Int Immunopharmacol. (2020) 80:106221. doi: 10.1016/j.intimp.2020.106221
133. Edner NM, Carlesso G, Rush JS, Walker LSK. Targeting co-stimulatory molecules in autoimmune disease. Nat Rev Drug Discovery (2020) 19(12):860–83. doi: 10.1038/s41573-020-0081-9
134. Wong C, Darby JM, Murphy PR, Pinfold TL, Lennard PR, Woods GM, et al. Tasmanian Devil CD28 and CTLA4 capture CD80 and CD86 from adjacent cells. Dev Comp Immunol (2021) 115:103882. doi: 10.1016/j.dci.2020.103882
135. Leung CS, Yang KY, Li X, Chan VW, Ku M, Waldmann H, et al. Single-cell transcriptomics reveal that PD-1 mediates immune tolerance by regulating proliferation of regulatory T cells. Genome Med (2018) 10(1):71. doi: 10.1186/s13073-018-0581-y
136. Gianchecchi E, Fierabracci A. Inhibitory receptors and pathways of lymphocytes: The role of PD-1 in treg development and their involvement in autoimmunity onset and cancer progression. Front Immunol (2018) 9:2374. doi: 10.3389/fimmu.2018.02374
137. Sur S. In silico analysis reveals interrelation of enriched pathways and genes in type 1 diabetes. Immunogenetics (2020) 72(8):399–412. doi: 10.1007/s00251-020-01177-3
138. Wang J, Yoshida T, Nakaki F, Hiai H, Okazaki T, Honjo T. Establishment of NOD-Pdcd1-/- mice as an efficient animal model of type I diabetes. Proc Natl Acad Sci USA (2005) 102(33):11823–8. doi: 10.1073/pnas.0505497102
139. Keir ME, Liang SC, Guleria I, Latchman YE, Qipo A, Albacker LA, et al. Tissue expression of PD-L1 mediates peripheral T cell tolerance. J Exp Med (2006) 203(4):883–95. doi: 10.1084/jem.20051776
140. Li X, Zhong T, Tang R, Wu C, Xie Y, Liu F, et al. PD-1 and PD-L1 expression in peripheral CD4/CD8+ T cells is restored in the partial remission phase in type 1 diabetes. J Clin Endocrinol Metab (2020) 105(6). doi: 10.1210/clinem/dgaa130
141. Shan Y, Kong Y, Zhou Y, Guo J, Shi Q, Li S, et al. Decreased expression of programmed death-1 on CD8(+) effector memory T lymphocytes correlates with the pathogenesis of type 1 diabetes. Acta Diabetol (2021) 58(9):1239–49. doi: 10.1007/s00592-021-01711-z
142. Zhang Q, Chikina M, Szymczak-Workman AL, Horne W, Kolls JK, Vignali KM, et al. LAG3 limits regulatory T cell proliferation and function in autoimmune diabetes. Sci Immunol (2017) 2(9). doi: 10.1126/sciimmunol.aah4569
143. Anderson AC, Joller N, Kuchroo VK. Lag-3, Tim-3, and TIGIT: Co-inhibitory receptors with specialized functions in immune regulation. Immunity (2016) 44(5):989–1004. doi: 10.1016/j.immuni.2016.05.001
144. Jones BE, Maerz MD, Bahnson HT, Somasundaram A, McCarthy LH, Speake C, et al. Fewer LAG-3(+) T cells in relapsing-remitting multiple sclerosis and type 1 diabetes. J Immunol (2022) 208(3):594–602. doi: 10.4049/jimmunol.2100850
145. Zhao L, Cheng S, Fan L, Zhang B, Xu S. TIM-3: An update on immunotherapy. Int Immunopharmacol. (2021) 99:107933. doi: 10.1016/j.intimp.2021.107933
146. Liu Y, Chen H, Chen Z, Qiu J, Pang H, Zhou Z. Novel roles of the Tim family in immune regulation and autoimmune diseases. Front Immunol (2021) 12:748787. doi: 10.3389/fimmu.2021.748787
147. Liu Y, Chen Z, Xiao Y, Chen H, Zhou Z. Altered expression of Tim family molecules and an imbalanced ratio of Tim-3 to Tim-1 expression in patients with type 1 diabetes. Front Endocrinol (Lausanne). (2022) 13:937109. doi: 10.3389/fendo.2022.937109
148. Guo H, Shen Y, Kong YH, Li S, Jiang R, Liu C, et al. The expression of Tim-1 and Tim-4 molecules in regulatory T cells in type 1 diabetes. Endocrine (2020) 68(1):64–70. doi: 10.1007/s12020-019-02173-8
149. Lee DJ. The relationship between TIGIT(+) regulatory T cells and autoimmune disease. Int Immunopharmacol. (2020) 83:106378. doi: 10.1016/j.intimp.2020.106378
150. Dominguez-Villar M, Hafler DA. Regulatory T cells in autoimmune disease. Nat Immunol (2018) 19(7):665–73. doi: 10.1038/s41590-018-0120-4
151. Hoseini-Aghdam M, Sheikh V, Eftekharian MM, Rezaeepoor M, Behzad M. Enhanced expression of TIGIT but not neuropilin-1 in patients with type 2 diabetes mellitus. Immunol Lett (2020) 225:1–8. doi: 10.1016/j.imlet.2020.06.003
152. Ning Z, Liu K, Xiong H. Roles of BTLA in immunity and immune disorders. Front Immunol (2021) 12:654960. doi: 10.3389/fimmu.2021.654960
153. Paluch C, Santos AM, Anzilotti C, Cornall RJ, Davis SJ. Immune checkpoints as therapeutic targets in autoimmunity. Front Immunol (2018) 9:2306. doi: 10.3389/fimmu.2018.02306
154. Pruul K, Kisand K, Alnek K, Metskula K, Reimand K, Heilman K, et al. Differences in B7 and CD28 family gene expression in the peripheral blood between newly diagnosed young-onset and adult-onset type 1 diabetes patients. Mol Cell endocrinology. (2015) 412:265–71. doi: 10.1016/j.mce.2015.05.012
155. Karnell JL, Rieder SA, Ettinger R, Kolbeck R. Targeting the CD40-CD40L pathway in autoimmune diseases: Humoral immunity and beyond. Adv Drug Delivery Rev (2019) 141:92–103. doi: 10.1016/j.addr.2018.12.005
156. Tang T, Cheng X, Truong B, Sun L, Yang X, Wang H. Molecular basis and therapeutic implications of CD40/CD40L immune checkpoint. Pharmacol Ther (2021) 219:107709. doi: 10.1016/j.pharmthera.2020.107709
157. Vaitaitis GM, Waid DM, Yussman MG, Wagner DH Jr. CD40-mediated signalling influences trafficking, T-cell receptor expression, and T-cell pathogenesis, in the NOD model of type 1 diabetes. Immunology (2017) 152(2):243–54. doi: 10.1111/imm.12761
158. Vaitaitis GM, Rihanek M, Alkanani AK, Waid DM, Gottlieb PA, Wagner DH, et al. Biomarker discovery in pre-type 1 diabetes; Th40 cells as a predictive risk factor. J Clin Endocrinol Metab (2019) 104(9):4127–42. doi: 10.1210/jc.2019-00364
159. Waid DM, Wagner RJ, Putnam A, Vaitaitis GM, Pennock ND, Calverley DC, et al. A unique T cell subset described as CD4loCD40+ T cells (TCD40) in human type 1 diabetes. Clin Immunol (Orlando Fla). (2007) 124(2):138–48. doi: 10.1016/j.clim.2007.05.003
160. Sha S, Pearson JA, Peng J, Hu Y, Huang J, Xing Y, et al. TLR9 deficiency in b cells promotes immune tolerance via interleukin-10 in a type 1 diabetes mouse model. Diabetes (2021) 70(2):504–15. doi: 10.2337/db20-0373
161. Fu Y, Lin Q, Zhang Z, Zhang L. Therapeutic strategies for the costimulatory molecule OX40 in T-cell-mediated immunity. Acta Pharm Sin B (2020) 10(3):414–33. doi: 10.1016/j.apsb.2019.08.010
162. Meylan F, Siegel RM. TNF superfamily cytokines in the promotion of Th9 differentiation and immunopathology. Semin Immunopathol (2017) 39(1):21–8. doi: 10.1007/s00281-016-0612-y
163. Fu N, Xie F, Sun Z, Wang Q. The OX40/OX40L axis regulates T follicular helper cell differentiation: Implications for autoimmune diseases. Front Immunol (2021) 12:670637. doi: 10.3389/fimmu.2021.670637
164. An J, Ding S, Li S, Sun L, Chang X, Huang Z, et al. Enhancement of the soluble form of OX40 and OX40L costimulatory molecules but reduction of the membrane form in type 1 diabetes (T1D). J Immunol Res (2019) 2019:1780567. doi: 10.1155/2019/1780567
165. Li DY, Xiong XZ. ICOS(+) tregs: A functional subset of tregs in immune diseases. Front Immunol (2020) 11:2104. doi: 10.3389/fimmu.2020.02104
166. Zhong JX, Chen J, Rao X, Duan L. Dichotomous roles of co-stimulatory molecules in diabetes mellitus. Oncotarget (2018) 9(2):2902–11. doi: 10.18632/oncotarget.23102
167. Viisanen T, Ihantola EL, Nanto-Salonen K, Hyoty H, Nurminen N, Selvenius J, et al. Circulating CXCR5+PD-1+ICOS+ follicular T helper cells are increased close to the diagnosis of type 1 diabetes in children with multiple autoantibodies. Diabetes (2017) 66(2):437–47. doi: 10.2337/db16-0714
168. Arab M, Razzaghy-Azar M, Salehi Z, Keshavarz M, Nasli-Esfahani E, Shekarabi M, et al. Increased circulating T follicular helper cells in Iranian children with type I diabetes. Iran J Allergy Asthma Immunol (2018) 17(6):557–63.
169. Kornete M, Mason E, Istomine R, Piccirillo CA. KLRG1 expression identifies short-lived Foxp3(+) treg effector cells with functional plasticity in islets of NOD mice. Autoimmunity (2017) 50(6):354–62. doi: 10.1080/08916934.2017.1364368
170. Edner NM, Heuts F, Thomas N, Wang CJ, Petersone L, Kenefeck R, et al. Follicular helper T cell profiles predict response to costimulation blockade in type 1 diabetes. Nat Immunol (2020) 21(10):1244–55. doi: 10.1038/s41590-020-0744-z
171. Wong HY, Schwarz H. CD137 / CD137 ligand signalling regulates the immune balance: A potential target for novel immunotherapy of autoimmune diseases. J Autoimmun (2020) 112:102499. doi: 10.1016/j.jaut.2020.102499
172. Forsberg MH, Ciecko AE, Bednar KJ, Itoh A, Kachapati K, Ridgway WM, et al. CD137 plays both pathogenic and protective roles in type 1 diabetes development in NOD mice. J Immunol (2017) 198(10):3857–68. doi: 10.4049/jimmunol.1601851
173. Foda BM, Ciecko AE, Serreze DV, Ridgway WM, Geurts AM, Chen YG. The CD137 ligand is important for type 1 diabetes development but dispensable for the homeostasis of disease-suppressive CD137(+) FOXP3(+) regulatory CD4 T cells. J Immunol (2020) 204(11):2887–99. doi: 10.4049/jimmunol.1900485
Keywords: immune checkpoints, immune checkpoint inhibitors, type 1 diabetes, lymphocyte, immunotherapy
Citation: Ding J-T, Yang K-P, Lin K-L, Cao Y-K and Zou F (2023) Mechanisms and therapeutic strategies of immune checkpoint molecules and regulators in type 1 diabetes. Front. Endocrinol. 13:1090842. doi: 10.3389/fendo.2022.1090842
Received: 06 November 2022; Accepted: 22 December 2022;
Published: 10 January 2023.
Edited by:
Manami Hara, The University of Chicago, United StatesReviewed by:
David Wagner, University of Colorado Anschutz Medical Campus, United StatesAnupam Kotwal, University of Nebraska Medical Center, United States
Line Jørgensen, Odense University Hospital, Denmark
Copyright © 2023 Ding, Yang, Lin, Cao and Zou. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Fang Zou, em91ZmFuZzkyMkAxMjYuY29t
†These authors have contributed equally to this work