 Francesca Miglietta1
Francesca Miglietta1 Luca Iamartino1
Luca Iamartino1 Gaia Palmini1
Gaia Palmini1 Francesca Giusti1
Francesca Giusti1 Francesca Marini2
Francesca Marini2 Teresa Iantomasi1
Teresa Iantomasi1 Maria Luisa Brandi2*
Maria Luisa Brandi2*- 1Department of Experimental Clinical and Biomedical Sciences “Mario Serio”, University of Florence, Florence, Italy
- 2Fondazione FIRMO Onlus (Italian Foundation for the Research on Bone Diseases), Florence, Italy
Hematopoietic stem cell transplantation (HSCT) is an established therapeutic strategy for the treatment of malignant (leukemia and lymphoma) and non-malignant (thalassemia, anemia, and immunodeficiency) hematopoietic diseases. Thanks to the improvement in patient care and the development of more tolerable conditioning treatments, which has extended the applicability of therapy to the elderly, a growing number of patients have successfully benefited from HSCT therapy and, more importantly, HSCT transplant-related mortality has consistently reduced in recent years. However, concomitantly to long term patient survival, a growing incidence of late HSCT-related sequelae has been reported, being variably associated with negative effects on quality of life of patients and having a non-negligible impact on healthcare systems. The most predominantly observed HSCT-caused complications are chronic alterations of the endocrine system and metabolism, which endanger post-operative quality of life and increase morbidity and mortality of transplanted patients. Here, we specifically review the current knowledge on HSCT-derived side-effects on the perturbation of mineral metabolism; in particular, the homeostasis of calcium, focusing on current reports regarding osteoporosis and recurrent renal dysfunctions that have been observed in a percentage of HSC-transplanted patients. Possible secondary implications of conditioning treatments for HSCT on the physiology of the parathyroid glands and calcium homeostasis, alone or in association with HSCT-caused renal and bone defects, are critically discussed as well.
1 Introduction
Hematopoietic stem cell transplantation (HSCT) is today a well-recognized treatment for many neoplastic [i.e. leukemias, lymphomas (1–5)] and non-neoplastic [i.e. thalassemia, anemia (6, 7)] hematological diseases (Table 1), performed by an infusion of hematopoietic stem cells (HSCs) that can be collected from bone marrow, cord blood, and peripheral blood (8–10).
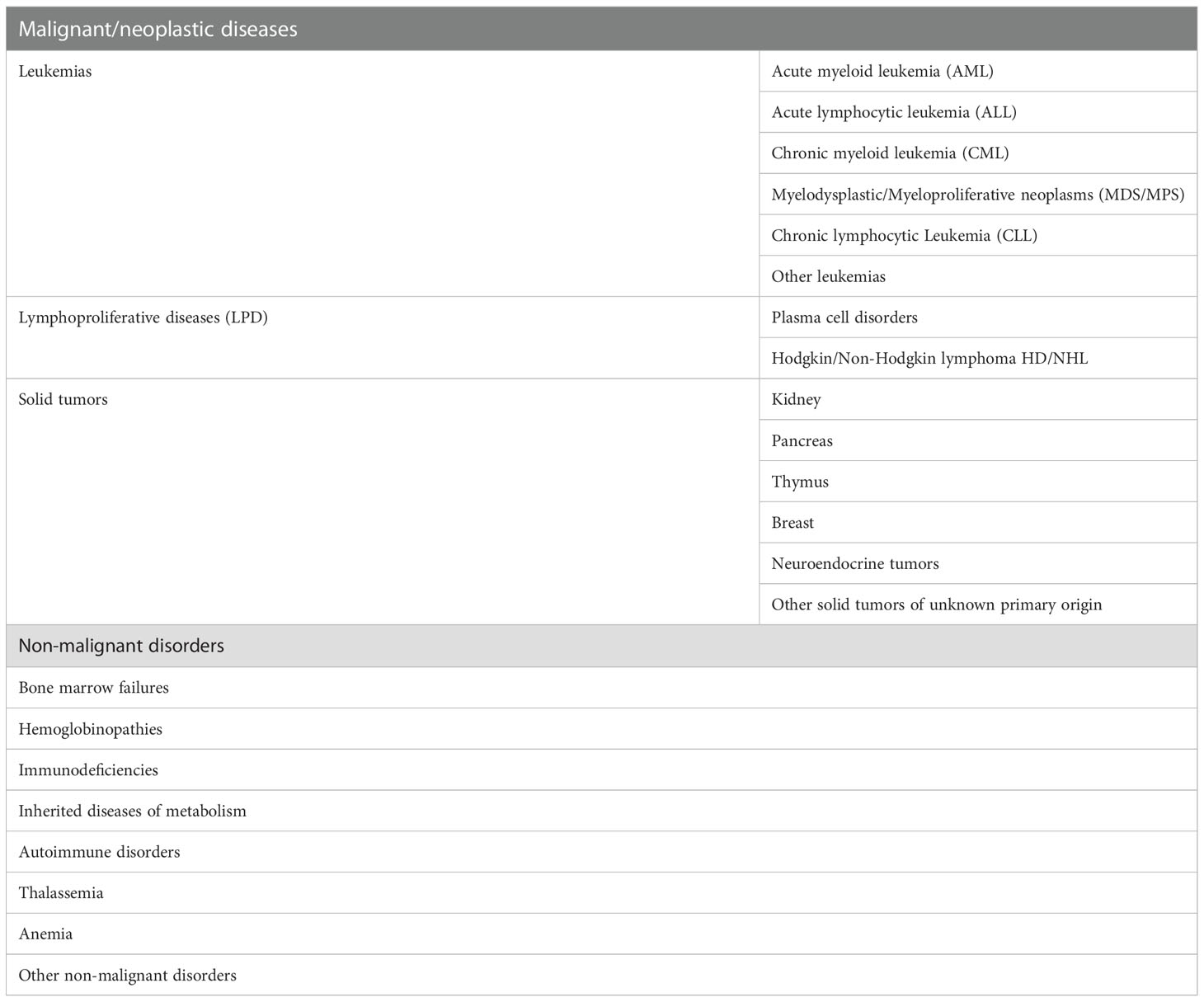
Table 1 Conditions for which patients may undergo HSCT.
Two types of HSCT can be performed, autologous or allogeneic transplantation, both leading to remission of bone marrow aplasia (11–13). In autologous HSCT, HSCs from the same recipient are collected and cryopreserved. After a conditioning treatment, the patient receives an infusion of his/her own HSCs (8). Allogeneic transplantation, instead, consists of the infusion of HSCs isolated from a healthy donor, who can, preferentially, be a half-HLA-matched biological parent or biological children, or, in absence of these first-degree relatives, a partially-HLA-matched family member or unrelated donor (14). Graft versus host disease (GVHD) represents a severe adverse reaction of the allogeneic transplantation, caused by the targeting of the host non-hematopoietic cells by the T lymphocytes of the donor. To minimize the risk of GVHD a stringent HLA matching between recipient and donor is strictly required. On the other hand, allogeneic transplanted HSCs have been showed to exert immunological effects against the malignant hematopoietic cells of the recipient, the so-called graft versus tumor GVT) reaction, due to donor’s polyclonal T-cell response targeting polymorphic antigens expressed on the recipient’s hematopoietic tissue. This characteristic made the allogeneic HSCT applicable also to elderly and clinically infirm patients, who were previously not candidates for the autologous transplantation because of the requirement of a high-dose of pre-transplantation conditioning treatment.
From 1957 to 2016 a total of 1,298,897 HSCTs were reported, of them 57.1% being autologous procedures (15). Approximately 90,000 first HSCTs are performed yearly worldwide and this number is estimated to increase by 10-20% annually. Autologous transplantations represent about 53% and allogeneic about 47%, of which about 53.6% are from related donors (15). In 2016, the World Health Organization reported a global increase of 6.2% in autologous HSCT and 7.0% in allogeneic HSCT (15). Today, HSCT is successfully performed in almost all age patients. A recent report by the European Society for Blood and Marrow Transplantation (EBMT) (16), resuming activity and trends on HSCT in 42 European and 11 collaborating countries, reported a total of 48,512 HSCTs (59% autologous and 41 allogeneic) of which 5,189 (10.7%) were performed in pediatric patients less than 18 years (76.9% allogeneic and 23.1% autologous). Main indications for allogeneic HSCT were myeloid malignancies, while for autologous transplantation were lymphoid malignancies.
Before receiving the intravenous infusion of HSCs, the recipient must undergo a conditioning treatment, consisting of chemotherapy and/or radiation therapy, in the 3-5 weeks prior to transplantation, leading to the ablation of the patient’s ailing bone marrow and the induction of sufficient immunosuppression to allow transplantation of HSCs, preventing the risk of rejection (8). The conditioning regimen can be myeloablative (full-dose) or non-myeloablative (reduced-intensity) (9).
The intensity and choice of conditioning regimen varies depending on the diagnosis and remission status of the disease, the presence or absence of comorbidities, the patient’s age and donor availability. The myeloablative regimen consist of therapy with alkylating agents, with or without total body irradiation (TBI), and it is designed to completely ablate bone marrow hemopoiesis, not allowing autologous hematological recovery. In contrast, non-ablative regimens, although causing cytopenia, do not require stem cell support (17).
Due to its immunosuppressive property, TBI is widely used as a conditioning regimen for patients with hematological malignancies undergoing HSCT. Generally, the regimen consists of a combination of 12 to 16 Gy TBI (usually hyperfractionated in order to decrease the risk of side effects, such as interstitial pneumonitis) (18, 19) with chemotherapeutic agents, such as cyclophosphamide (20–23), cytarabine (AraC) (24), etoposide (25), melphalan (26) and busulfan (27).
To avoid the short-term/long-term toxicity effects caused by high-dose TBI (especially in patients already treated with radiotherapy), alternative conditioning regimens can be used, having similar immunosuppressive efficacy (28, 29) based on the administration of chemotherapeutic agents, instead of TBI, such as alkylating agents. Busulfan is an alkylating agent with a strong toxic effect on non-dividing bone marrow cells, but it cannot be administered as the only treatment due to its low effect against mature lymphocytes (21). The regimen that is generally used on patients undergoing HSCT (both autologous and allogeneic) consists of high-dose busulfan (16 mg/kg total dose) and cyclophosphamide (120 mg/kg) (30). More recently, to further reduce regimen-dependent toxicity, cyclophosphamide has been replaced by treatment with fludarabine (a nucleoside analogue with immunosuppressive activity that synergizes with alkylating agents by inhibiting DNA repair (31, 32).
Reduced-intensity allogeneic HSCT results in a variable degree of initial donor-host mixed chimerism (21) that may vary depending on the intensity of the actual conditioning regimen, donor/host HLA disparity and transplant composition (33). The efficacy of GVT effects may also be influenced by tumor burden and proliferation rate of the neoplasm (33). To date, there are various types of non-ablative approaches based on a low-intensity TBI in combination with fluradabine or cloforabine followed by treatment with mycophenolate mofetil and a calcineurin inhibitor ± sirolimus, such as in the cohort studies published by Cooper et al. (34); or such as those developed at the MD Anderson Cancer Center (35), where a treatment approach based on a non-myeloablative regimen containing fludarabine (90 mg/m2) and cyclophosphamide (2,250 mg/m2) in combination with rituximab is suggested in order to prevent transplant rejection and increase host T-cell immunosuppression.
After HSCT, patients must follow immunosuppressive therapy consisting in corticosteroids and calcineurin inhibitors to prevent the occurrence of GVHD.
Currently, over 90,000 HSCTs are yearly performed worldwide, and, thanks to the development of more tolerable conditioning treatments, long-term survival of patients has been increasing along the year (36), with an estimated survival rate beyond 10 years after transplantation of approximately 80% (37). The mortality rate is due to various complications associated with the transplant itself, including acute and chronic GVHD, infections, and end-organ dysfunction (38). Moreover, as the post-transplant long-term survival has increased, there has been an increasing incidence of sequelae, impacting on mortality rate and morbidity, and worsening patient quality of life. One of the HSCT-derived main complications is the occurrence of endocrine alterations that impair the health of transplanted patients, with variable degrees of severity (39). Conditioning chemotherapy and radiotherapy, and post-operative immunosuppressive treatments are the principal causes of hormone dysfunctions, and subsequent development of endocrine syndromes, after transplantation (40, 41). As a consequence of these sequelae, transplanted patients require lifelong care for endocrine disease management.
Here, we review adverse events and sequelae caused by HSCT, and associated conditioning and post-transplantation immunosuppressive therapies, specifically altering local and systemic endocrine regulation of bone and mineral homeostasis.
2 HSCT-derived endocrine sequelae pathophysiology
2.1 Altered mineral homeostasis and renal dysfunction
Myeloablative and non-myeloablative conditioning media and cyclosporine treatment can create significant damages to patients by affecting electrolyte balance, through the alteration of the balance between intestinal absorption and renal excretion (42–44). Serum levels of calcium, magnesium, potassium, and phosphorus, in fact, tend to drop suddenly, thus creating a severe acid/base imbalance (44). Philibert et al. (43) observed that in 48 patients receiving HSCT from peripheral blood, 40% had serum potassium values less than 3.5 mmol/l, 33% had magnesium values less than 0.7 mmol/l, 43% had calcium values less than 2.14 mmol/l, and 35% had phosphorus values less than 0.6 mmol/l, immediately after HSCT. In total, 91% of patients had hypophosphatemia, while 49% had hypocalcemia after the transplantation. Crook et al. (44) reported severe hypophosphatemia in 77% of patients during the period of the conditioning regimen, or in the following week.
The hematopoiesis of transplanted cells may contribute to altering the phosphorus balance in patients undergoing HSCT, causing hypophosphatemia (45, 46); a diagnosis of severe hypophosphatemia after transplantation is directly associated with the proliferation of transplanted HSCs (44–48). Extensive phosphate consumption seen in HSCT patients is due to the need of transplanted HSCs of an energy source for the ex-novo proliferation of the hematopoietic cell compartment (44–48). Raanani et al. (45) observed a correlation between the release of the cytokines IL-6 and IL-8 and the severity of hypophosphatemia, defining the increase in these interleukins as a predictive index directly proportional to the severity of hypophosphatemia.
Hypomagnesemia observed in patients with HSCT may be one of the causes of altered electrolyte balance. Very low levels of magnesium cause a decrease in cAMP activity, which leads to prolonged opening of potassium channels, causing a decrease in serum levels of potassium itself (43, 49).
In addition, several studies (43, 44, 48, 50–53) have shown that intracellular magnesium depletion can compromise the functionality of parathyroid glands to secrete parathyroid hormone (PTH), through modulation of cAMP activity. Decreased PTH secretion, caused by hypomagnesemia, leads to hypocalcemia, and is associated with decreased active vitamin D (43, 49, 51, 52). Agus et al. observed that PTH-induced calcium release is severely impaired when the magnesium concentration is less than 0.8 mEq/l, and in severe hypomagnesemia there is resistance to PTH by target organs (49). It should be noted, however, that with magnesium replacement therapy, PTH serum levels are restored (42, 43, 52, 53).
Glucocorticoid therapy is the standard therapy against GVHD in allogeneic HSCT; these molecules alter PTH metabolism and calcium homeostasis. Among major side effects of glucocorticoids there are the suppression of osteocalcin (OCN) and alkaline phosphatase (ALP) expression, the induction of osteoclastogenesis and of bone resorption, and the impairment of osteoblastogenesis and osteoblastic activity, all causing severe bone loss and, consequently, osteoporosis (54). At the same time, glucocorticoids cause decreased intestinal calcium absorption and increased calciuria (Figure 1), causing a severe decrease in serum calcium levels (55, 56) and, thus, inducing a strong stimulation of PTH production and leading to the establishment of secondary hyperparathyroidism (55, 57, 58). However, studies related to this topic show mixed results. There are studies in which no correlation has been found between glucocorticoids (even at high concentrations) and PTH increase (59–64), whereas some reports have observed that there is a dose-dependent PTH increase during treatment with glucocorticoids. A study using Northern blot analysis was conducted to determine whether PTH release in humans was directly stimulated by glucocorticoids (65). Dexamethasone was found to increase the amount of preproPTH mRNA in cells by directly acting at gene level in parathyroid cells, increasing PTH synthesis through stimulation of PTH gene transcription (65).
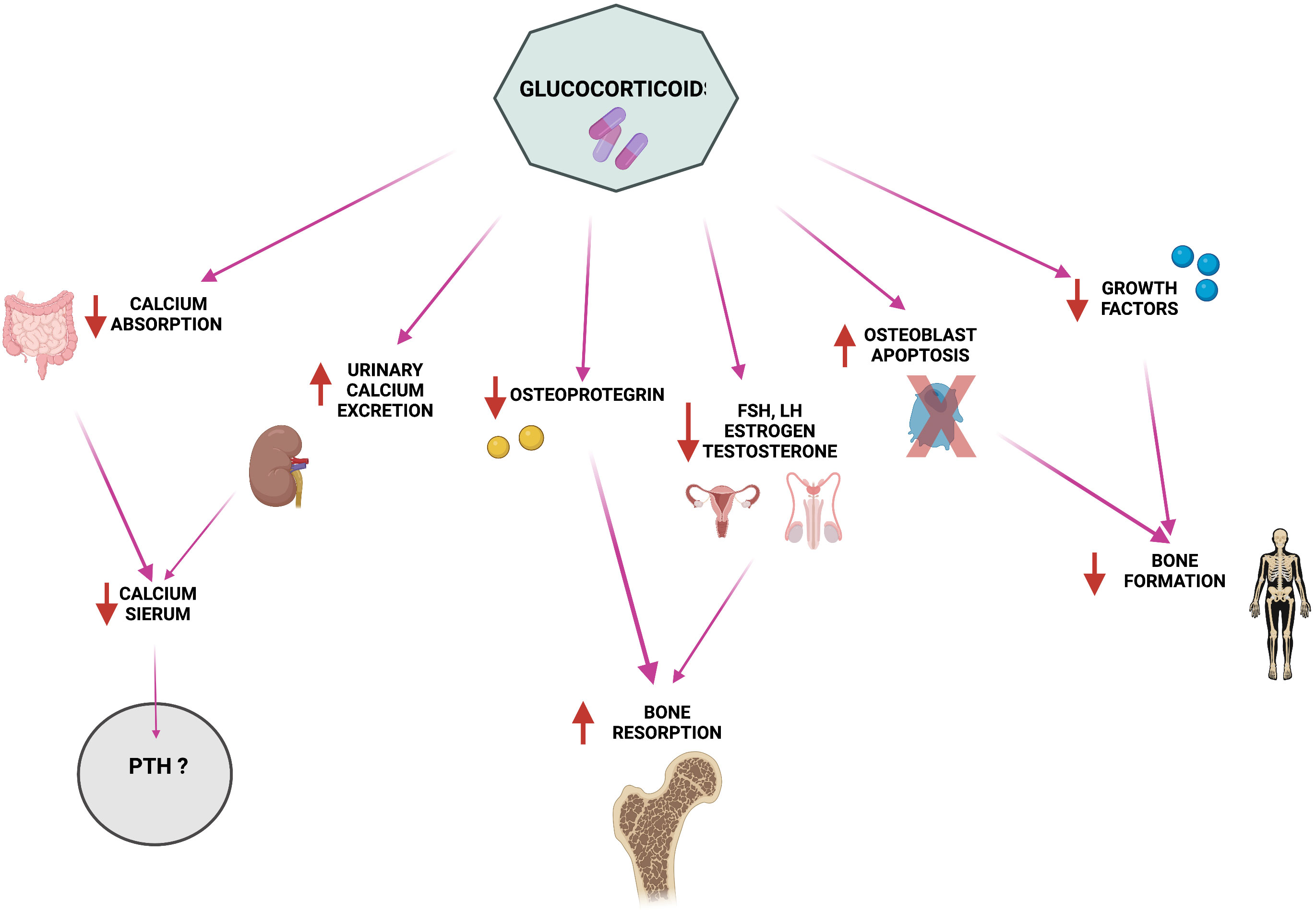
Figure 1 Pathophysiology of glucocorticoid-induced alterations in calcium homeostasis and bone metabolism. Created by BioRender.com.
The reduction in intestinal calcium absorption and the sharp increase in PTH serum levels (66) could be also synergistically caused by the interference of post-transplant long-term glucocorticoid therapy with vitamin D metabolism, creating a non-competitive effect on the vitamin itself (67, 68). Indeed, after transplantation, vitamin D levels drop further in 90% of patients, a severe decrease that was mainly related to the intake of glucocorticoids, rather than myeloablative conditioning treatment. Physiologically, vitamin D stimulates intestinal absorption of calcium and consequently promotes a decrease in PTH production and release by the parathyroid glands. In HSCT patients, concomitant lack of exposure to the sun, due to their disease status, and long-term therapy with immunosuppressive glucocorticoids induce an overall vitamin D deficiency that affects the correct calcium and phosphate homeostasis and favors the occurrence of osteopenia and osteoporosis (66).
Calcium and mineral metabolism may be further altered due to renal failure as a common complication following HSCT, causing profound effects on morbidity and long-term survival of patients. Chronic kidney disease (CKD), a late-stage gradual loss of kidney function, is commonly diagnosed after HSCT. In observational studies, CKD has shown a variable incidence in HSCT patients, varying between 18-66%, due to the different parameters used for CKD definition and the differences in the duration of follow-ups (69–71). In general, renal dysfunctions are multifactorial complications that can be caused by pre-transplant underlying diseases and comorbidities, conditioning regimens, transplant complications and prophylaxis treatments (72).
Similarly, acute kidney injury (AKI) is a severe renal complication characterized by defects in glomerular filtration. It is diagnosed in HSCT recipients with variable incidence depending on the type of HSCT [from 20% to 73% (73)], and on the administration of myeloablative and non-myeloablative conditioning treatments (70, 74–77).
Pre-existing renal pathologies, such as glomerulonephritis, develop from hematological malignancies for which HSCT intervention is planned, therefore, the underlying hematological diseases already provoke renal lesions before HSCT, increasing in this way the risk of severe renal failure in the long term HSCT survival (78). Moreover, comorbidities such as diabetes, hypertension, and arteriosclerosis have been shown to further contribute to renal failure after transplant (72).
Pre-operative conditioning regimes may damage the kidneys. Several chemotherapeutics have been reported to induce renal failure, such as pre-renal azotemia and acute tubular necrosis, due to their nephrotoxicity, so a reduction of their dosage in HSCT recipients has been recommended (72, 79, 80).
Several studies reported in HSCT patients undergone total body irradiation the occurrence of renal defects, diagnosed several years after the radiation exposure, due to the late effects of radiotherapy on kidney physiology (69, 72, 81–83). Typical histopathological features of radiation-induced nephropathy are vascular, glomerular, and tubulo-interstitial damages. Radiations cause DNA double-strand breaks resulting in apoptosis or necrosis of renal endothelial, tubular and glomerular cells (84).
Moreover, both chemotherapeutics and radiation therapy have been associated with hepatic sinusoidal obstruction syndrome, due their cytotoxic effects on hepatic sinusoidal endothelial cells. This HSCT complication has been seen to induce systemic detrimental effects, including AKI (85).
It has been observed that transplanted cells have nephrotoxic effects per se. This is evident in the “marrow infusion syndrome”, a dysfunction that is caused by the cryopreservants, (i.e., DMSO) that are commonly used for long-term storage of the hematopoietic stem cells before their transplantation. Residues of those reagents can be infused together with the stem cells, leading to hemolytic effects that eventually hamper renal functionality.
Another severe transplant complication observed in both autologous and allogeneic HSCT recipients is the transplant-associated thrombotic microangiopathy (TA-TMA), an endothelial injury syndrome, with a still unclear etiology, which leads to microangiophatic hemolytic anemia, intravascular platelet activation, and formation of thrombi within the microcirculation, with end-organ damage from ischemia, particularly in the kidneys. Among possible causes of TA-TMA, there are HSCT treatments, such as radiation, chemotherapy, calcineurin inhibitors, and some HSCT-associated complications, including viral infections, GVHD, and hepatic sinusoidal obstruction syndrome (72, 86–88). The recognition of the early stages of TA-TMA is often difficult and, if not promptly diagnosed and treated, it commonly leads to permanent renal injury and, possibly, death.
Patients undergoing transplant are at higher risk of infection due to the administration of immune suppressants. Sepsis has been ascribed as a HSCT complication that can damage renal functionality due to renal hypoperfusion, tubulointerstitial injury, and capillary thrombosis (89, 90). Patients who require treatment in intensive care unit after transplant have higher risks of developing AKI (76). Adenovirus infections and the reactivation of the BK (Polyomavirus hominis 1) virus, which can occur in HSCT recipients, can cause tubulointerstitial nephritis and cystitis (70, 91, 92).
Even prophylaxis measurements used to prevent infections can provoke renal failure, as has been reported in patients treated with antifungals, such as amphotericin B (78) and with aminoglycoside antibiotics (93).
Moreover, patients receiving allotransplant are at high risk of developing GVHD that can cause systemic detrimental effects, including nephrotic syndrome, transplant-associated microangiopathy and glomerulonephritis, most likely due to the excessive release of pro-inflammatory cytokines such as TNF-α, IL-6, and TGF-β (72, 88, 94, 95). To prevent GVHD and treat chronic GVHD, patients receive immunosuppressants, including steroids and calcineurin inhibitors, which sometimes need to be taken for prolonged periods of time, causing renal complications (71, 96, 97).
An impairment of kidney activity that is a quite common primary or secondary complication in HSCT patients is at the base of an altered systemic mineral homeostasis, and, indirectly, of bone metabolism, that could be further worsened if it is associated with malabsorptive syndromes.
2.2 Bone loss
Bone homeostasis is the result of a finely regulated and correctly balanced activity of two different types of cells: osteoblasts, which form new mineralized bone, and osteoclasts, which reabsorb old and/or damaged bone tissue. Alterations in this homeostasis lead to the development of two main classes of bone diseases: those characterized by an excess of new bone deposition and mineralization, and those characterized by increased bone mass loss and/or reduced mineralization. Ultimately, both these two classes of disease generate a structurally impaired bone tissue, with altered mechanical properties, prone to deformities and/or fragility fractures.
Recent studies have reported that alteration of bone homeostasis, with loss of bone mass (osteopenia, osteoporosis), is a common side effect of the HSCTs (98–100). Osteopenia is diagnosed in 50% of cases 4-6 years post HSCT, while osteoporosis is diagnosed in 20% (98). It has been reported that the loss of bone mass occurs already few months after HSCT, with patients showing a reduction in Bone Mineral Density (BMD) primarily at femoral neck and lumbar spine (101, 102). Transplanted patients have an overall higher fracture risk compared to the general population (103, 104). Pathogenesis of accelerated bone loss in HSCT recipients is a multifactorial dysfunction that is not yet completely understood due to its complexity and to the contribution of many factors that concomitantly impact bone homeostasis (Figure 1) (105–108). These multiple factors include the treatments that HSCT patients undergo before, during, and after transplantation (108). Underlying hematological diseases may also contribute to osteoporosis per se by inhibiting osteogenesis and promoting bone resorption, thus predisposing HSCT-patients to higher risks of BMD loss (109, 110). The age at transplantation appears to be a risk factor itself for bone mineral deficit in recipients of HSCT. Indeed, HSCT patients less than 10 years were showed to have, at an average age of 14 years after the transplantation, significantly lower Z-scores at lumbar spine and femur neck than HSCT patients transplanted at over 18 years and non-transplanted sibling controls (111).
Pre-transplant myeloablative treatments have been shown to negatively impact bone metabolism. Chemotherapeutic compounds, such as alkylating agents, methotrexate, and doxorubicin can deregulate bone remodeling by directly inhibiting osteoblast proliferation and osteoblastogenesis and, simultaneously, by stimulating osteoclast activity and bone resorption (112–116), or by indirectly damaging the reproductive system and sex hormone metabolism, which play a fundamental role in regulating skeletal modeling and life-long remodeling of bone tissue (117–120).
Total body irradiation, often coupled with chemotherapy as myeloablative conditioning treatment, dramatically reduces BMD by suppressing bone formation, enhancing bone resorption, and disrupting the inorganic components of the bone matrix (121–125). Moreover, irradiation has been associated with hypogonadism, hypopituitarism and growth hormone deficiency, thus leading to a decrease in trophic factors important for bone formation (105, 126, 127).
Chemotherapy- and/or radiotherapy-caused hypogonadism is one of the main causes of the occurrence of osteoporosis in HSCT patients. Indeed, under physiological conditions, sexual hormones are among the major regulators of bone metabolism in both females (estrogens and progesterone) and males (testosterone) (128).
Estrogens have a positive activity on bone formation, both by inhibiting osteoblast apoptosis and by increasing their lifespan (129). Consequently, the absence of these hormones in hypogonadal HSCT female patients promotes a bone metabolic framework oriented towards resorption. At the molecular level, estrogen absence leads to the non-activation of Src/Shc/ERK signaling and, consequently, the promotion of JNK, which, by modifying the activity of a series of transcription factors, including c-Jun/c-Fos, Elk-1, and CCAAT enhancer binding protein-β (C/EBPβ) (130, 131), promotes osteoblastic apoptosis. In addition, estrogen deficiency leads to a clear increase in NF-κB activity in osteoblastic cells (132), leading to a possible decrease in the expression of Fos-related antigen-1 (Fra-1), a transcription factor essential for bone matrix formation (131), contributing to a deficit in bone anabolism. At the same time, the estrogen deficiency leads to a fourfold increase in bone resorption activity (133, 134); the absence of estrogen causes a rapid decrease in bone mass due to the expansion of T-cells that produce high levels of TNFα in the bone microenvironment. TNFα, together with activation of macrophage colony-stimulating factor (M-CSF) and RANK, activates its p55 receptor, promoting osteoclastogenesis (135–138). Osteocytes respond to estrogen deficiency; the absence of estrogen increases osteocyte apoptosis, resulting in increased concentrations of RANKL, probably released by apoptotic osteocytes or as a result of cell-to-cell signaling between healthy and apoptotic osteocytes, which induces increased bone resorption through stimulation of the osteoclastic cell compartment (131, 139–142).
Testosterone deficiency is a known cause of bone loss and the onset of osteoporosis in men (143–153). Chronic testosterone deficiency, such as that occurring in hypogonadal HSCT male patients, could concur with the early-onset osteopenia/osteoporosis observed in these individuals.
Indeed, under physiological conditions, testosterone acts on bone metabolism in two different ways:
1) directly, in its active form, dihydrotestosterone (DTH), via interaction with androgenic receptors (ARs) present on osteoblasts, activating the TGF-β signaling pathway, and, thus, promoting osteoblastic proliferation and osteoblastogenesis (154, 155),
2) indirectly, after its conversion to estradiol (E2), by binding the estrogen receptor alpha (ERα), and exerting a local positive action on osteoblast function, exactly as systemic estrogens do in women.
Testosterone deficiency results in a decreased differentiation and activity of osteoblasts, leading to decreased new bone formation. Orchiectomized rodents showed that testosterone depletion induces a significant increase of RANKL and IL-6 expression in bone marrow-derived stromal cells, which strongly promote osteoclast differentiation, with consequently increased bone resorption and bone mass loss (156–158). Moreover, testosterone deficiency causes an indirect deficiency of estradiol (159, 160), which leads to an enhanced bone loss through the increased production of several inflammatory cytokines (IL-1 and TNFα, IL-6), which stimulate osteoclastic differentiation and activity and promote osteoblastic apoptosis (161–165).
Moreover, since testosterone plays a predominant role in male muscle strength and physical performance, it is conceivable that the absence of this hormone in hypogonadal HSCT male patients can cause sarcopenia, leading to greater weakness and increasing the risk of falling and fracturing (140, 166).
In relation to what has been described above regarding the importance of sex hormones in maintaining bone homeostasis, the onset of hypogonadism in HSCT patients obviously contributes to the loss of bone mass and the onset of osteoporosis; a greater relationship was found between individuals suffering from HSCT-derived medium/severe hypogonadism and decreased BMD and osteoporosis (167–169).
Several studies have reported that one early side effect of HSCT is the failure of bone morphogenesis, due to a severe reduction of osteoblast progenitor cells in bone marrow, causing post-transplant BMD loss (106, 108, 170–172). Moreover, the immune response, following the transplant, seems to play a pivotal role in altering bone homeostasis by promoting osteoclast maturation and activity (105, 106, 108). After HSCT, indeed, it has been observed that the balance between RANKL and osteoprotegerin (OPG), expressed/released by bone marrow stromal cells and osteoblasts, is shifted towards RANKL expression, causing an excessive induction of osteoclast differentiation and activity, with increased bone resorption. Simultaneously, it has been seen that certain cytokines typical of the immune and inflammatory response, such as granulocyte-macrophage colony-stimulating factor (GM-CSF), IL-6 and TNF-α can stimulate osteoclastic activity (173–175).
HSCT-derived immune derangement has been found to trigger bone loss, most frequently in allogeneic HSCT recipients, compared to autologous HSCT, due to the increased synthesis of specific inhibitors of the canonical Wnt/Beta Catenin pathway, Dickkopf-1, and Sclerostin (176), principally as a consequence of GVHD and/or administration of GVHD prophylactic treatments. It has been observed that GVHD occurs in 40-70% of allogeneic HSCT recipients and that the severity of GVHD is positively correlated with the degree of BMD reduction (106, 173, 174, 177–179). Anti-GVDH treatments, consisting in the administration of glucocorticoids and calcineurin inhibitors, also seem to play a major role in HSCT-associated osteoporosis (107, 108). High-dose glucocorticoid therapy is usually the first-line approach against GVDH, causing a rapid decrease of BMD in HSCT-recipients, by inhibiting new bone formation and promoting bone resorption (107, 180). Glucocorticoids can shift the ratio between OPG and RANKL towards the RANKL expression, with a consequent boost in bone turnover, in a time- and dose-dependent manner (102, 180–182). Similar to glucocorticoids, immune-suppressive calcineurin inhibitors, such as cyclosporin A and tacrolimus, seem to promote bone resorption and osteoclast activity, while both interfere with osteogenesis and osteoblasts activity (104, 183).
3 HSCT-derived endocrine sequelae management
3.1 Altered mineral homeostasis and renal dysfunction
3.1.1 Surveillance
General recommendations for the mineral homeostasis restoration include calcium intake and vitamin D supplementation (184, 185), together with a balanced diet, no smoking, alcohol taken in a controlled manner (≤2 alcohol units per day) and regular exercise. Recommended doses of calcium range from 1000 to 1200 mg/day of elemental calcium; if the dietary intake is insufficient, supplementation with calcium citrate or calcium carbonate is recommended.
Vitamin D deficiency can be treated with 50,000 IU of vitamin D3 once a week (or 7,000 IU/day) for 8-12 weeks to achieve serum vitamin D levels >20 ng/mL, followed by maintenance by intake of ≥1000 IU/day (186, 187). Management of CKD is based on slowing disease progression and minimizing possible further renal damage.
Screening by the primary care physician is essential for early diagnosis because he is aware of the patient’s comorbidities and has opportunities to assess changes in GFR levels. Hypertension, use of nephrotoxic agents, presence of cardiovascular disease, hyperglycemia, and increased albuminuria (188), along with age, obesity, smoking, and sex, are indices of increased risk factor for CKD. There are guidelines such as the KDIGO (189) or JNC8 (190) that provide information for screening for the disease.
Hypertension is important to keep under control in patients with C KD. Guidelines recommend keeping blood pressure below 140/90 mm Hg (190, 191), or 130/80mm Hg in the presence of albuminuria (if albuminuria levels exceed 300 mg/24, an angiotensin receptor blocker should also be taken) (191). Hemoglobin A1c should be around 7% to prevent hyperglycemia from infecting the kidney (191), sodium levels should not exceed 2000mg/d, and protein intake should be less than 0.8/kg/d (191).
Anemia can be a common complication of CKD. When GFR decreases, erythropoietin is most likely to decrease at the same time, since 80% of that protein, comes from renal interstitial cells, so with GFR values <60 and hemoglobin<10g/dL, subcutaneous erythropoietin treatment is administered (188).
Identification of risk factors, early diagnosis, and treatment of renal dysfunction are crucial for favorable outcomes. In addition, detection of subclinical renal dysfunction before transplantation can help in the choice of conditioning type so that it is less nephrotoxic.
3.1.2 Treatment
The study of individualized therapy, based on the calculation of body measurements, plasma-level drug metabolite concordances, and analysis of the patient’s renal function can lead to the decreased toxicity of conditioning regimens.
Cure of CKD after HSCT is very difficult to achieve, but if a specific cause is identified, it can be cured. The use of pamidronate, for example, can cause focal glomerulosclerosis; discontinuation of the drug can promote proteinuria decrease and halt disease progression (188).
The American Society for Blood and Marrow Transplantation has published recommendations on dose modification of chemotherapeutics used in conditioning regimens that are nephrotoxic or eliminated by the kidneys (79). Fludarabine, for instance, a drug commonly used in non-myeloablative regimens for patients with comorbidities, including renal failure, is not nephrotoxic, but being eliminated by the kidneys, it may lead to an increased risk of neurotoxic adverse effects in case of renal failure (72). Excessive plasma concentrations combined with low clearance of F-Ara-A, the active metabolite of fludarabine, have been shown to be linked to increased mortality without relapse in transplanted patients, while its high clearance leads to an increased susceptibility for GVHD (192). Consequently, a dosage reduction of 20-25% is recommended (72). Clofarabine, on the other hand, should be abolished for patients over 60 years with creatinine clearance <60 mL/min, while a dose reduction of 50% is recommended in patients with CrCl between 30 and 60mL/min (79). Prolonged mucositis has been observed with standard doses of melphalan in patients with CKD, therefore, the recommended dosage would be between 100 and 140 mg/m2 for dialysis-dependent patients with renal impairment (79). The bisulfan dosage is unmodified, in fact, its renal excretion is slight even for patients in dialysis (79).
Research is focusing on the study of TA-TMA pathology and renal GVHD through the study of specific non-invasive markers. Markers of endothelial damage such as thrombomodulin, plasminogen activator inhibitor type 1 and endothelial microparticles, and inflammatory cytokines IL-6, IL-15 and monocytin.
Other areas of research include understanding the pathology of TA-TMA and hypothesized renal GvHD and finding specific noninvasive markers and therapeutic options for both complications. Particles that are hypothesized to be important are markers of endothelial damage, such as thrombomodulin, plasminogen activator inhibitor type 1 and endothelial microparticles, as well as inflammatory cytokines: IL-6, IL-15, monocyte cytokine and monocyte chemoattractant protein-1 (193).
A prognostic marker for cutaneous GVHD, elafin, when present at the urinary level, has also been found to be a prognosis maker for AKI and CKD (193). It has been investigated that the decrease in renal vascular endothelial growth factor concentration, correlated with the subsequent serum elevation of neutrophil extracellular traps, may be related to TA-TMA risk (194), consequently, supplementation of this growth factor could be valid as a therapeutic strategy.
3.2 Bone loss
3.2.1 Surveillance
In 2006, guidelines for bone health screening and prevention of bone loss in HSCT survivors were published by the EBMT, the Centre for International Blood and Marrow Transplantation Research (CIBMTR), and the American Society for Blood and Marrow Transplantation (ASBMT) (108, 195), and successively revised and updated in 2012 (108, 196).
The general assessment of bone health status in patients suitable for HSTC should include a pre- transplant evaluation of: 1) patient and familial medical history, 2) physical examination, 3) evaluation of common modifiable osteoporosis risk factors to which the patient is exposed (197), 4) FRAX fracture risk index (184), 5) DXA at hip and lumbar spine (especially in adult women, and patients undergoing a long-term therapy with corticosteroids. In the latter ones a DXA evaluation should be planned by 3 months after the beginning of pharmacological treatment), 6) spine X-rays or vertebral fracture assessment by DXA (VFA) in older patients and high-risk patients, to identify vertebral fractures Risk factors for osteoporosis to be evaluated in HSCT patients include: age, female gender, smoking, alcohol, no physical activity, low weight, poor diet, hypogonadism, hyperparathyroidism, rheumatoid arthritis, liver and/or kidney disease, multiple myeloma, chemotherapy, glucocorticoid intake (dose- and time-dependent), and GVHD (Figure 2) (105, 196, 198–202). Post-transplant follow-up by DXA is recommended as early as 3 months after HSCT, and then annually, in patients at high risk (199), included those in treatment con corticosteroids, those having a pre-transplant low BMD, and/or those having experienced fragility fracture before HSCT (184, 203, 204). Lower risk patients may undergo DXA screening at one year after HSCT, and then on annual basis. DXA screening intervals may be reduced or extended based on post-transplant assessed BMD values, and according to the presence or absence of unmodifiable risk factors (older age, female gender, previous fragility fracture) and modifiable current risk factors.
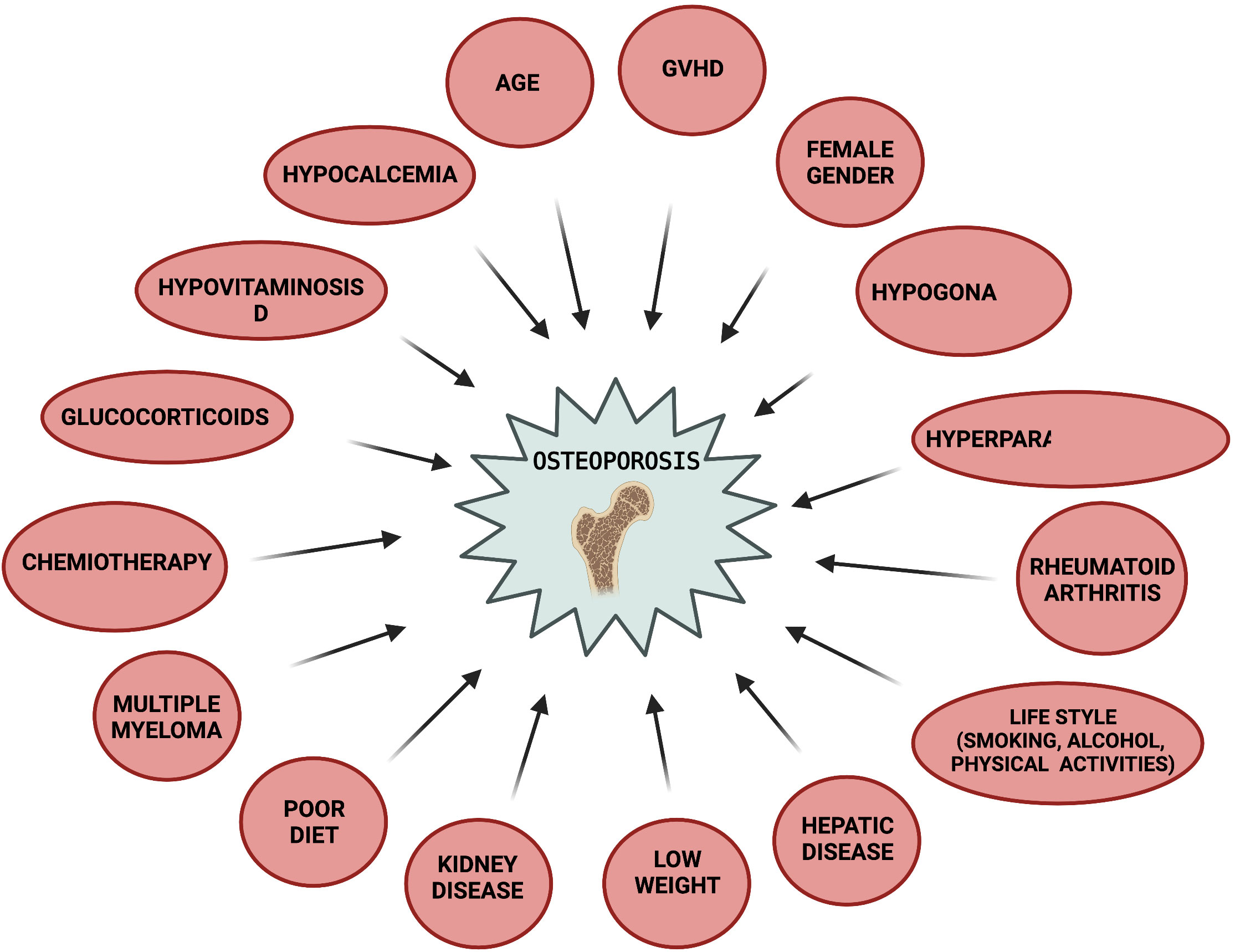
Figure 2 Risk factors for post-HSCT osteoporosis. Created by BioRender.com.
3.2.2 Treatment
Given the heterogeneity of HSCT patients in age, gender, and bone response to transplant and associated medical therapies, there are not standard treatment guidelines for bone in these patients (108, 205). Current therapeutic strategy for osteoporosis is, thus, the one indicated for all organ-transplanted patients (108). In general, the first 3 months after transplantation are the most at risk of complications and, consequently, any drug therapy not directly aimed at the success of the transplant is precluded to avoid further interactions and side effects, including anti-fracture drugs. After 3 months, if the patient’s medical conditions permit, and after a monitoring of renal and hepatic status, the guidelines allow treatment for the prevention/cure of bone loss (108, 205).
There are two types of approaches proposed for the prevention/treatment of bone loss. The first is called “treat to target” (205) and consists of treating patients with a therapy according to the target to be achieved; if this is not achieved, the therapeutic approach is changed until the most appropriate therapy is found for the patient (206). The second approach is usually applied to patients who cannot undergo the first approach (t-score <-3; medical complications; contraindications for a type of drug, etc.), and consists of a bisphosphonate oral therapy for 5 years, or intravenous administration for 3 years, followed by a discontinuation of treatment for at least 2 years, to be resumed if necessary (207). Oral treatment is generally the first to be prescribed, unless there is intestinal GVHD, esophageal dysmotility, or gastrointestinal reflux disease. Alternatively, intravenous zoledronate administration may be indicated, but must be strictly avoided in hypocalcemic patients or those with CDK stage 3B-5 (205). Bisphosphonate therapy has been shown to be safe for HSCT survivors (208–211), in improving BMD (208–211). One rare side effect of bisphosphonates, principally for Zoledronate (1% of treated cases), is the osteonecrosis of the jaw (212, 213). Since HSCT survivors with chronic oral GVHD are at increased risk (214), a complete oral examination and the complete elimination of oral infections, which represent major risk cofactors for this pathological manifestation, are strongly recommended prior to therapy with bisphosphonate (213, 215, 216).
Estrogen replacement therapy is considered the best treatment for bone loss associated with early menopause (217), like that experienced in hypogonadal HSCT female patients; this therapy should be limited only to subjects not at risk for breast cancer (218). Transdermal administration is preferred as it carries a lower risk of thromboembolism and stroke (219). Once the age of natural menopause has been reached, discontinuation of therapy is customized, as it has been shown that the benefits for the patient outweigh the risks in the first 10 years after menopause (220–223).
In hypogonadal HSCT male patients presenting low testosterone levels, testosterone replacement therapy is highly recommended, and it can also reduce the risk of early-onset osteoporosis occurrence (108, 144, 205, 224, 225).
Calcitonin nasal spray therapy reduces the risk of fractures but is not used as a routine drug because it is less effective than bisphosphonates (226, 227); it is only prescribed in patients who cannot receive bisphosphonates, or for patients with acute and painful fractures, because of the analgesic effect of calcitonin (227).
The beneficial effect of denosumab in HSCT patients is not yet well understood. There is one study in which 33 HSCT female patients with osteoporosis, who were naïve for anti-fracture drugs, were treated with denosumab (60 mg) three times every six months (0, 6, and 12 months). The BMD of the lumbar spine, femoral neck and total hip, measured 12 months after the first administration of denosumab, showed an improvement of 4.39 ± 6.63%, 3.11 ± 7.69% and 1.97 ± 6.01%, respectively, with respect to baseline, in the absence of serious adverse reactions to the point of discontinuing therapy. Data from this study suggested that denosumab could be an efficient therapy for the prevention/treatment of bone loss in HSCT recipients (228).
Currently, there are no conclusive data on safety and efficacy of teriparatide, raloxifene, or romosozumab for bone loss management in HSCT recipients.
Calcium and vitamin D supplementations are usually prescribed in combination with drug treatment (186, 187).
Medical therapy for bone loss in children surviving HSCT is similar to that performed for adults, but more oriented towards estrogen and growth hormone replacement therapy (229–233). Treatment with bisphosphonates is performed following the “treat to target” approach and is reserved for children with osteoporosis who do not respond to calcium and vitamin D supplementations, and estrogen and/or growth hormone replacement therapies (205). Teriparatide is not used, while calcitonin is administered as short-term adjunctive therapy (234).
In our clinical experience, we use the two most common anti-resorption drugs, either bisphosphonates or denosumab, for the prevention/treatment of early and/or increased bone loss in HSCT patients.
4 Discussion
Thanks to improvements in patient care and the implementation of more tolerable conditioning treatments and effective prophylaxis, long-term survival of HSCT has increased significantly in recent years. However, concomitant with the increase in survival after HSCT, complications, related to the transplant itself and/or to pre- and post-transplant required medical treatments, are emerging, and numerous retrospective and follow-up studies have highlighted the occurrence of multiple side effects that worsen patient quality of life and increase the risk of severe morbidity and mortality. Among these side effects, disorders affecting the endocrine systems are common after HSCT, including metabolic, growth, and sexual defects, and mineral metabolism alterations.
Among side effects of HSCT, the malfunctioning of calcium-tropic organs, such as kidneys, bones, and intestines, has proven implications for systemic mineral homeostasis, which can in turn affect the entire skeletal metabolism by causing increased bone resorption, leading to the onset of early osteoporosis and bones prone to fragility fracture in a wide percentage of HSCT survivors. Bone health management needs to be refined to better tailor osteoporosis therapies, specifically for HSCT recipients. The recent arrival in the market of romosozumab antibody will be an important addition in the pharmacological armamentarium of the physician taking care of bone fragility in these patients.
An interesting aspect that has been observed in few studies is that the alteration of the systemic mineral homeostasis following HSCT, also powered by post-transplant chronic glucocorticoid intake, affects the entire phosphate-calcium-PTH system by altering PTH secretion and causing the onset of secondary hyperparathyroidism, which, if not rebalanced, can lead to the development of chronic tertiary hyperparathyroidism. The presence of secondary or tertiary hyperparathyroidism further contributes to worsening both kidney defects and bone mass loss and fragility. Targeted studies evaluating a possible direct effect of HSCT on parathyroid glands/cells, and the occurrence of primary hyperparathyroidism, are currently lacking.
Author contributions
All authors listed have made a substantial, direct, and intellectual contribution to the work and approved it for publication.
Acknowledgments
This research was supported by Fondazione Italiana Ricerca sulle Malattie dell’Osso (Fondazione FIRMO Onlus).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Takami A. Hematopoietic stem cell transplantation for acute myeloid leukemia. Int J Hematol (2018) 107:513–8. doi: 10.1007/s12185-018-2412-8
2. Kassim AA, Savani BN. Hematopoietic stem cell transplantation for acute myeloid leukemia: A review. Hematol Oncol Stem Cell Ther (2017) 10(4):245–51. doi: 10.1016/j.hemonc.2017.05.021
3. Zahid U, Akbar F, Amaraneni A, Husnain M, Chan O, Riaz IB, et al. A review of autologous stem cell transplantation in lymphoma. Curr Hematol Malig Rep (2017) 12(3):217. doi: 10.1007/s11899-017-0382-1
4. Arora S, Majhail NS, Liu H. Hematopoietic progenitor cell mobilization for autologous stem cell transplantation in multiple myeloma in contemporary era. Clin Lymphoma Myeloma Leuk. (2019) 19(4):200–5. doi: 10.1016/j.clml.2018.12.010
5. Gaut D, Schiller GJ. Hematopoietic stem cell transplantation in primary central nervous system lymphoma: a review of the literature. Int J Hematol (2019) 109(3):260–77. doi: 10.1007/s12185-019-02594-1
6. Sanders JE, Buckner CD, Amos D, Levy W, Appelbaum FR, Doney K, et al. Ovarian function following marrow transplantation for aplastic anemia or leukemia. J Clin Oncol (1988) 6(5):813–8. doi: 10.1200/JCO.1988.6.5.813
7. Hematopoietic cell transplantation for solid tumors in adults - medical clinical policy bulletins | aetna. Available at: https://www.aetna.com/cpb/medical/data/800_899/0811.html.
8. Bazinet A, Popradi G. A general practitioner’s guide to hematopoietic stem-cell transplantation. Curr Oncol (2019) 26(3):187. doi: 10.3747/co.26.5033
9. AIL - associazione italiana contro le leucemie-linfomi e mieloma ONLUS . Available at: https://www.ail.it/.
10. Barriga F, Ramírez P, Wietstruck A, Rojas N. Hematopoietic stem cell transplantation: clinical use and perspectives. Biol Res (2012) 45:307–16. doi: 10.4067/S0716-97602012000300012
11. Rekers PE, Coulter M, Warren S. Effect of transplantation of bone marrow into irradiated animals. undefined (1950) 60(4):635. doi: 10.1001/archsurg.1950.01250010656001
12. Gilbar PJ, Pokharel K, Mangos HM. Aplastic anemia treated with daily transfusions and intravenous marrow; case report. Ann Intern Med (1939) 13(2):357. doi: 10.1177/1078155220967087
13. Thomas ED, Buckner CD, Banaji M, Clift RA, Fefer A, Flournoy N, et al. One hundred patients with acute leukemia treated by chemotherapy, total body irradiation, and allogeneic marrow transplantation. blood. 1977;49(4):511-533. Blood (2016) 128(20):2373.
14. Gladstone DE, Bettinotti MP. HLA donor-specific antibodies in allogeneic hematopoietic stem cell transplantation: challenges and opportunities. Hematol Am Soc Hematol Educ Program (2017) 2017(1):645–50. doi: 10.1182/asheducation-2017.1.645
15. Niederwieser D, Baldomero H, Bazuaye N, Bupp C, Chaudhri N, Corbacioglu S, et al. One and a half million hematopoietic stem cell transplants: continuous and differential improvement in worldwide access with the use of non-identical family donors. Haematologica (2022) 107(5):1045–53. doi: 10.3324/haematol.2021.279189
16. Passweg JR, Baldomero H, Chabannon C, Basak GW, de la Cámara R, Corbacioglu S, et al. Hematopoietic cell transplantation and cellular therapy survey of the EBMT: monitoring of activities and trends over 30 years. Bone Marrow Transplant (2021) 56(7):1651–64. doi: 10.1038/s41409-021-01227-8
17. Bacigalupo A, Ballen K, Rizzo D, Giralt S, Lazarus H, Ho V, et al. Defining the intensity of conditioning regimens: working definitions. Biol Blood Marrow Transplant (2009) 15(12):1628–33. doi: 10.1016/j.bbmt.2009.07.004
18. Shank B, O’Reilly RJ, Cunningham I, Kernan N, Yaholoml J, Brochstein J, et al. Total body irradiation for bone marrow transplantation: the memorial Sloan-Kettering cancer center experience. Radiother Oncol (1990) 18 Suppl 1(SUPPL. 1):68–81. doi: 10.1016/0167-8140(90)90180-5
19. Shank B, Hopfan S, Kim JH, Chu FCH, Grossbard E, Kapoor N, et al. Hyperfractionated total body irradiation for bone marrow transplantation: I. early results in leukemia patients. Int J Radiat Oncol Biol Phys (1981) 7(8):1109–15. doi: 10.1016/0360-3016(81)90170-X
20. Demirer T, Petersen FB, Appelbaum FR, Barnett TA, Sanders J, Deeg HJ, et al. Allogeneic marrow transplantation following cyclophosphamide and escalating doses of hyperfractionated total body irradiation in patients with advanced lymphoid malignancies: a phase I/II trial. Int J Radiat Oncol Biol Phys (1995) 32(4):1103–9. doi: 10.1016/0360-3016(95)00115-F
21. Gyurkocza B, Sandmaier BM. Conditioning regimens for hematopoietic cell transplantation: one size does not fit all. Blood (2014) 124(3):344–53. doi: 10.1182/blood-2014-02-514778
22. Petersen FB, Deeg HJ, Buckner CD, Appelbaum FR, Storb R, Clift RA, et al. Marrow transplantation following escalating doses of fractionated total body irradiation and cyclophosphamide–a phase I trial. Int J Radiat Oncol Biol Phys (1992) 23(5):1027–32. doi: 10.1016/0360-3016(92)90909-2
23. Brochstein JA, Kernan NA, Groshen S, Cirrincione C, Shank B, Emanuel D, et al. Allogeneic bone marrow transplantation after hyperfractionated total-body irradiation and cyclophosphamide in children with acute leukemia. N Engl J Med (1987) 317(26):1618–24. doi: 10.1056/NEJM198712243172602
24. Riddell S, Appelbaum FR, Buckner CD, Stewart P, Clift R, Sanders J, et al. High-dose cytarabine and total body irradiation with or without cyclophosphamide as a preparative regimen for marrow transplantation for acute leukemia. J Clin Oncol (1988) 6(4):576–82. doi: 10.1200/JCO.1988.6.4.576
25. Horning SJ, Chao NJ, Negrin RS, Hoppe RT, Kwak LW, Long GD, et al. The Stanford experience with high-dose etoposide cytoreductive regimens and autologous bone marrow transplantation in hodgkin’s disease and non-hodgkin’s lymphoma: preliminary data. Ann Oncol (1991) 2 Suppl 1(SUPPL. 1):47–50. doi: 10.1093/annonc/2.suppl_1.47
26. Moreau P, Facon T, Attal M, Hulin C, Michallet M, Maloisel F, et al. Comparison of 200 mg/m(2) melphalan and 8 gy total body irradiation plus 140 mg/m(2) melphalan as conditioning regimens for peripheral blood stem cell transplantation in patients with newly diagnosed multiple myeloma: final analysis of the intergroupe francophone du myélome 9502 randomized trial. Blood (2002) 99(3):731–5. doi: 10.1182/blood.V99.3.731
27. Scott B, Deeg HJ, Storer B, Chauncey T, Petersdorf S, Slattery J, et al. Targeted busulfan and cyclophosphamide as compared to busulfan and TBI as preparative regimens for transplantation in patients with advanced MDS or transformation to AML. Leuk Lymphoma (2004) 45(12):2409–18. doi: 10.1080/10428190412331283206
28. Nagler A, Rocha V, Labopin M, Unal A, Othman T, Campos A, et al. Allogeneic hematopoietic stem-cell transplantation for acute myeloid leukemia in remission: comparison of intravenous busulfan plus cyclophosphamide (Cy) versus total-body irradiation plus cy as conditioning regimen–a report from the acute leukemia working party of the European group for blood and marrow transplantation. J Clin Oncol (2013) 31(28):3549–56. doi: 10.1200/JCO.2013.48.8114
29. Litzow MR, Pérez WS, Klein JP, Bolwell BJ, Camitta B, Copelan EA, et al. Comparison of outcome following allogeneic bone marrow transplantation with cyclophosphamide-total body irradiation versus busulphan-cyclophosphamide conditioning regimens for acute myelogenous leukaemia in first remission. Br J Haematol (2002) 119(4):1115–24. doi: 10.1046/j.1365-2141.2002.03973.x
30. Tutschka PJ, Copelan EA, Klein JP. Bone marrow transplantation for leukemia following a new busulfan and cyclophosphamide regimen. Blood (1987) 70(5):1382–8. doi: 10.1182/blood.V70.5.1382.1382
31. Bornhäuser M, Storer B, Slattery JT, Appelbaum FR, Deeg HJ, Hansen J, et al. Conditioning with fludarabine and targeted busulfan for transplantation of allogeneic hematopoietic stem cells. Blood (2003) 102(3):820–6. doi: 10.1182/blood-2002-11-3567
32. Iravani M, Evazi MR, Mousavi SA, Shamshiri AR, Tavakoli M, Ashouri A, et al. Fludarabine and busulfan as a myeloablative conditioning regimen for allogeneic stem cell transplantation in high- and standard-risk leukemic patients. Bone Marrow Transplant (2007) 40(2):105–10. doi: 10.1038/sj.bmt.1705685
33. Baron F, Maris MB, Storer BE, Sandmaier BM, Panse JP, Chauncey TR, et al. High doses of transplanted CD34+ cells are associated with rapid T-cell engraftment and lessened risk of graft rejection, but not more graft-versus-host disease after nonmyeloablative conditioning and unrelated hematopoietic cell transplantation. Leukemia (2005) 19(5):822–8. doi: 10.1038/sj.leu.2403718
34. Cooper JP, Storer BE, Granot N, Gyurkocza B, Sorror ML, Chauncey TR, et al. Allogeneic hematopoietic cell transplantation with non-myeloablative conditioning for patients with hematologic malignancies: Improved outcomes over two decades. Haematologica (2021) 106(6):1599–607. doi: 10.3324/haematol.2020.248187
35. McSweeney PA, Niederwieser D, Shizuru JA, Sandmaier BM, Molina AJ, Maloney DG, et al. Hematopoietic cell transplantation in older patients with hematologic malignancies: replacing high-dose cytotoxic therapy with graft-versus-tumor effects. Blood (2001) 97(11):3390–400. doi: 10.1182/blood.V97.11.3390
36. Majhail NS. Long-term complications after hematopoietic cell transplantation. Hematol/Oncol Stem Cell Ther (2017) 10(4):220–7. doi: 10.1016/j.hemonc.2017.05.009
37. Bhatia S. Cause-specific late mortality after allogeneic stem cell transplantation. Hematology (2019) 2019(1):626. doi: 10.1182/hematology.2019000004
38. Mohty M, Apperley JF. Long-term physiological side effects after allogeneic bone marrow transplantation. Hematol Am Soc Hematol Educ Program (2010) 2010:229–36. doi: 10.1182/asheducation-2010.1.229
39. Mohty B, Mohty M. Long-term complications and side effects after allogeneic hematopoietic stem cell transplantation: an update. Blood Cancer J (2011) 1(4):e16. doi: 10.1038/bcj.2011.14
40. Tauchmanová L, Selleri C, de Rosa G, Pagano L, Orio F, Lombardi G, et al. High prevalence of endocrine dysfunction in long-term survivors after allogeneic bone marrow transplantation for hematologic diseases. Cancer (2002) 95(5):1076–84. doi: 10.1002/cncr.10773
41. Engelhardt BG, Savani U, Jung DK, Powers AC, Jagasia M, Chen H, et al. New-onset post-transplant diabetes mellitus after allogeneic hematopoietic cell transplant is initiated by insulin resistance, not immunosuppressive medications. Biol Blood Marrow Transplant (2019) 25(6):1225–31. doi: 10.1016/j.bbmt.2019.02.001
42. Steiner M, Steiner B, Wilhelm S, Freund M, Schuff-Werner P. Severe hypophosphatemia during hematopoietic reconstitution after allogeneic peripheral blood stem cell transplantation. Bone Marrow Transplant (2000) 25(9):1015–6. doi: 10.1038/sj.bmt.1702407
43. Philibert D, Desmeules S, Filion A, Poirier M, Agharazii M. Incidence and severity of early electrolyte abnormalities following autologous haematopoietic stem cell transplantation. Nephrol Dialysis Transplant (2008) 23(1):359–63. doi: 10.1093/ndt/gfm571
44. Crook M, Swaminathan R, Scheya S. Hypophosphataemia in patients undergoing bone marrow transplantation. Leuk Lymphoma (1996) 22;335–37. doi: 10.3109/10428199609051765
45. Raanani P, Levi I, Holzman F, Grotto I, Brok-Simoni F, Avigdor A, et al. Engraftment-associated hypophosphatemia - the role of cytokine release and steep leukocyte rise post stem cell transplantation. Bone Marrow Transplant (2001) 27(3):311–7. doi: 10.1038/sj.bmt.1702761
46. Oliveira DC, Nogueira-Pedro A, Santos EW, Hastreiter A, Silva GB, Borelli P, et al. A review of select minerals influencing the haematopoietic process. Nutr Res Rev (2018) 31(2):267–80. doi: 10.1017/S0954422418000112
47. Clark AND, Euwin RE, Lee S. Severe hypophosphataemia during stem cell harvesting in chronic myeloid leukaemia. Br J Haernatol (1995) 90, 450–52. doi: 10.1111/j.1365-2141.1995.tb05172.x
48. Uçkan D, Çetin M, Dida A, Batu A, Tuncer M, Tezcan I. Hypophosphatemia and hypouricemia in pediatric allogeneic bone marrow transplant recipients. Pediatr Transplant (2003) 7(2):98–101. doi: 10.1034/j.1399-3046.2003.00022.x
50. di Stefano A, Roinel N, de Rouffignac C, Wittner M. Transepithelial Ca2+ and Mg2+ transport in the cortical thick ascending limb of henle’s loop of the mouse is a voltage-dependent process. Ren Physiol Biochem (1993) 16(4):157–66. doi: 10.1159/000173762
51. Rude RK. Functional hypoparathyroidism and parathyroid hormone end-o r g a n resistance in human magnesium deficiency. Clin Endocrinol (1976) 5, 209–224. doi: 10.1111/j.1365-2265.1976.tb01947.x
52. Fatemi S, Ryzen E, Flores J, Endres DB, Rude RK. Effect of experimental human magnesium depletion on parathyroid hormone secretion and 1,25-dihydroxyvitamin d metabolism. J Clin Endocrinol Metab (1991) 73(5):1067–72. doi: 10.1210/jcem-73-5-1067
53. Chase LR, Slatopolsky E, Greenwalt A, Krinski T. Secretion and metabolic efficacy of parathyroid hormone in patients with severe hypomagnesemia. J Clin Endocrinol Metab (1974) 38(3):363–71. doi: 10.1210/jcem-38-3-363
54. Rubin MR, Bilezikian JP. The role of parathyroid hormone in the pathogenesis of glucocorticoid-induced osteoporosis: A re-examination of the evidence (2002). Available at: https://academic.oup.com/jcem/article/87/9/4033/2846373.
55. Suzuki Y, Ichikawa Y, Saito E, Homma M. Importance of increased urinary calcium excretion in the development of secondary hyperparathyroidism of patients under glucocorticoid therapy. Metabolism (1983) 32(2):151–6. doi: 10.1016/0026-0495(83)90221-4
56. Lund B, Storm TL, Lund B, Melsen F, Mosekilde L, Andersen RB, et al. Bone mineral loss, bone histomorphometry and vitamin d metabolism in patients with rheumatoid arthritis on long-term glucocorticoid treatment. Clin Rheumatol (1985) 4(2):143–9. doi: 10.1007/BF02032284
57. Lukert BP, Adams JS. Calcium and phosphorus homeostasis in man: Effect of corticosteroids. Arch Intern Med (1976) 136(11):1249–53. doi: 10.1001/archinte.1976.03630110025009
58. Gennari C, Imbimbo B, Montagnani M, Bernini M, Nardi P, Avioli LV. Effects of prednisone and deflazacort on mineral metabolism and parathyroid hormone activity in humans. Calcif Tissue Int (1984) 36(3):245–52. doi: 10.1007/BF02405325
59. Pearce G, Tabensky DA, Delmas PD, Baker HWG, Seeman E. Corticosteroid-induced bone loss in men. J Clin Endocrinol Metab (1998) 83(3):801–6. doi: 10.1210/jcem.83.3.4621
60. Hattersley AT, Meeran K, Burrin J, Hill P, Shiner R, Ibbertson HK. The effect of long- and short-term corticosteroids on plasma calcitonin and parathyroid hormone levels. Calcif Tissue Int (1994) 54(3):198–202. doi: 10.1007/BF00301678
61. Paz-Pacheco E, El-Hajj Fuleihan G, Leboff MS. Intact parathyroid hormone levels are not elevated in glucocorticoid-treated subjects. J Bone Miner Res (1995) 10(11):1713–8. doi: 10.1002/jbmr.5650101114
62. Seeman E, Kumar R, Hunder GG, Scott M, Heath H IIIrd, Riggs BL. Production, degradation, and circulating levels of 1,25-dihydroxyvitamin d in health and in chronic glucocorticoid excess. J Clin Invest (1980) 66(4):664–9. doi: 10.1172/JCI109902
63. Slovik DM, Neer RM, Ohman JL, Lowell FC, Clark MB, Segre GV, et al. Parathyroid hormone and 25-hydroxyvitamin d levels in glucocorticoid-treated patients. Clin Endocrinol (Oxf) (1980) 12(3):243–8. doi: 10.1111/j.1365-2265.1980.tb02706.x
64. Hahn TJ, Halstead LR, Baran DT. Effects off short term glucocorticoid administration on intestinal calcium absorption and circulating vitamin d metabolite concentrations in man. J Clin Endocrinol Metab (1981) 52(1):111–5. doi: 10.1210/jcem-52-1-111
65. Peraldi MN, Rondeau E, Jousset V, el M’selmi A, Lacave R, Delarue F, et al. Dexamethasone increases preproparathyroid hormone messenger RNA in human hyperplastic parathyroid cells in vitro. Eur J Clin Invest (1990) 20(4):392–7. doi: 10.1111/j.1365-2362.1990.tb01875.x
66. Sproat L, Bolwell B, Rybicki L, Dean R, Sobecks R, Pohlman B, et al. Vitamin d level after allogeneic hematopoietic stem cell transplant. Biol Blood Marrow Transplant (2011) 17(7):1079–83. doi: 10.1016/j.bbmt.2010.12.704
67. Rickers H, Deding, Christiansen C, Rødbro P, Næstoft J. Corticosteroid-induced osteopenia and vitamin d metabolism. effect of vitamin d2, calcium phosphate and sodium fluoride administration. Clin Endocrinol (Oxf) (1982) 16(4):409–15. doi: 10.1111/j.1365-2265.1982.tb00734.x
68. Cosman F, Nieves J, Herbert J, Shen V, Lindsay R. High-dose glucocorticoids in multiple sclerosis patients exert direct effects on the kidney and skeleton. J Bone Miner Res (1994) 9(7):1097–105. doi: 10.1002/jbmr.5650090718
69. Hingorani S. Chronic kidney disease in long-term survivors of hematopoietic cell transplantation: Epidemiology, pathogenesis, and treatment. J Am Soc Nephrol (2006) 17(7):1995–2005. doi: 10.1681/ASN.2006020118
70. Renaghan AD, Jaimes EA, Malyszko J, Perazella MA, Sprangers B, Rosner MH. Acute kidney injury and CKD associated with hematopoietic stem cell transplantation. Clin J Am Soc Nephrol (2020) 15(2):289–97. doi: 10.2215/CJN.08580719
71. Ellis MJ, Parikh CR, Inrig JK, Kambay M, Patel UD. Chronic kidney disease after hematopoietic cell transplantation: A systematic review. Am J Transplant (2008) 8(11):2378–90. doi: 10.1111/j.1600-6143.2008.02408.x
72. Jaguś D, Lis K, Niemczyk L, Basak GW. Kidney dysfunction after hematopoietic cell transplantation–etiology, management, and perspectives. Hematol/Oncol Stem Cell Ther (2018) 11(4):195–205. doi: 10.1016/j.hemonc.2018.07.004
73. Krishnappa V, Gupta M, Manu G, Kwatra S, Owusu OT, Raina R. Acute kidney injury in hematopoietic stem cell transplantation: A review. Int J Nephrol (2016) 2016;5163789. doi: 10.1155/2016/5163789
74. Sawinski D. The kidney effects of hematopoietic stem cell transplantation. Adv Chronic Kidney Dis (2014) 21(1):96–105. doi: 10.1053/j.ackd.2013.08.007
75. Parikh CR, Schrier RW, Storer B, Diaconescu R, Sorror ML, Maris MB, et al. Comparison of ARF after myeloablative and nonmyeloablative hematopoietic cell transplantation. Am J Kidney Dis (2005) 45(3):502–9. doi: 10.1053/j.ajkd.2004.11.013
76. Canet E, Lengline E, Zafrani L, Peraldi MN, Socié G, Azoulay E. Acute kidney injury in critically ill allo-HSCT recipients. Bone Marrow Transplant (2014) 49(8):1121–2. doi: 10.1038/bmt.2014.100
77. Ando M, Mori J, Ohashi K, Akiyama H, Morito T, Tsuchiya K, et al. A comparative assessment of the RIFLE, AKIN and conventional criteria for acute kidney injury after hematopoietic SCT. Bone Marrow Transplant (2010) 45(9):1427–34. doi: 10.1038/bmt.2009.377
78. Ganguli A, Sawinski D, Berns JS. Kidney diseases associated with haematological cancers. Nat Rev Nephrol (2015) 11(8):478–90. doi: 10.1038/nrneph.2015.81
79. Bodge MN, Reddy S, Thompson MS, Savani BN. Preparative regimen dosing for hematopoietic stem cell transplantation in patients with chronic kidney disease: Analysis of the literature and recommendations. Biol Blood Marrow Transplant (2014) 20(7):908–19. doi: 10.1016/j.bbmt.2014.02.013
80. Kersting S, Koomans HA, Hené RJ, Verdonck LF. Acute renal failure after allogeneic myeloablative stem cell transplantation: Retrospective analysis of incidence, risk factors and survival. Bone Marrow Transplant (2007) 39(6):359–65. doi: 10.1038/sj.bmt.1705599
81. Chappell ME, Keeling DM, Prentice HG, Sweny P. Haemolytic uraemic syndrome after bone marrow transplantation: an adverse effect of total body irradiation? Bone Marrow Transplant (1988) 3(4):339–47.
82. Lawton CA, Cohen EP, Murray KJ, Derus SW, Casper JT, Drobyski WR, et al. Long-term results of selective renal shielding in patients undergoing total body irradiation in preparation for bone marrow transplantation. Bone Marrow Transplant (1997) 20(12):1069–74. doi: 10.1038/sj.bmt.1701022
83. Cohen EP. Radiation nephropathy after bone marrow transplantation. Kidney Int (2000) 58(2):903–18. doi: 10.1046/j.1523-1755.2000.00241.x
84. Klaus R, Niyazi M, Lange-Sperandio B. Radiation-induced kidney toxicity: molecular and cellular pathogenesis. Radiat Oncol (2021) 16(1):1–11. doi: 10.1186/s13014-021-01764-y
85. McDonald GB, Hinds MS, Fisher LD, Schoch HG, Wolford JL, Banaji M, et al. Veno-occlusive disease of the liver and multiorgan failure after bone marrow transplantation: A cohort study of 355 patients. Ann Intern Med (1993) 118(4):255–67. doi: 10.7326/0003-4819-118-4-199302150-00003
86. Changsirikulchai S, Myerson D, Guthrie KA, McDonald GB, Alpers CE, Hingorani SR. Renal thrombotic microangiopathy after hematopoietic cell transplant: Role of GVHD in pathogenesis. Clin J Am Soc Nephrol (2009) 4(2):345–53. doi: 10.2215/CJN.02070508
87. Obut F, Kasinath V, Abdi R. Post-bone marrow transplant thrombotic microangiopathy. Bone Marrow Transplant (2016) 51(7):891–7. doi: 10.1038/bmt.2016.61
88. Laskin BL, Goebel J, Davies SM, Jodele S. Small vessels, big trouble in the kidneys and beyond: Hematopoietic stem cell transplantation-associated thrombotic microangiopathy. Blood (2011) 118(6):1452–62. doi: 10.1182/blood-2011-02-321315
89. Kogon A, Hingorani S. Acute kidney injury in hematopoietic cell transplantation. Semin Nephrol (2010) 30(6):615–26. doi: 10.1016/j.semnephrol.2010.09.009
90. Schrier RW, Wang W. Acute renal failure and sepsis. New Engl J Med (2004) 351(2):159–69. doi: 10.1056/NEJMra032401
91. Hirsch HH, Steiger J. Polyomavirus BK. Lancet Infect Dis (2003) 3(10):611–23. doi: 10.1016/S1473-3099(03)00770-9
92. Ito M, Hirabayashi N, Uno Y, Nakayama A, Asai J. Necrotizing tubulointerstitial nephritis associated with adenovirus infection. Hum Pathol (1991) 22(12):1225–31. doi: 10.1016/0046-8177(91)90104-W
93. Rougier F, Claude D, Maurin M, Maire P. Aminoglycoside nephrotoxicity. Curr Drug Targets Infect Disord (2004) 4(2):153–62. doi: 10.2174/1568005043340858
94. Sakellari I, Barbouti A, Bamichas G, Mallouri D, Kaloyannidis P, Fragidis S, et al. GVHD-associated chronic kidney disease after allogeneic haematopoietic cell transplantation. Bone Marrow Transplant (2013) 48(10):1329–34. doi: 10.1038/bmt.2013.55
95. Brukamp K, Doyle AM, Bloom RD, Bunin N, Tomaszewski JE, Cizman B. Nephrotic syndrome after hematopoietic cell transplantation: do glomerular lesions represent renal graft-versus-host disease? Clin J Am Soc Nephrol (2006) 1(4):685–94. doi: 10.2215/CJN.00380705
96. Naesens M, Kuypers DRJ, Sarwal M. Calcineurin inhibitor nephrotoxicity. Clin J Am Soc Nephrol (2009) 4(2):481–508. doi: 10.2215/CJN.04800908
97. da Silva JB, Lima MH de M, Secoli SR. Influence of cyclosporine on the occurrence of nephrotoxicity after allogeneic hematopoietic stem cell transplantation: A systematic review. Rev Bras Hematol Hemoter (2014) 36(5):363–8. doi: 10.1016/j.bjhh.2014.03.010
98. Bhatia S. Long-term health impacts of hematopoietic stem cell transplantation inform recommendations for follow-up. Expert Rev Hematol (2011) 4(4):437–54. doi: 10.1586/ehm.11.39
99. Bhatia S, Ramsay NKC, Weisdorf D, Griffiths H, Robison LL. Bone mineral density in patients undergoing bone marrow transplantation for myeloid malignancies. Bone Marrow Transplant (1998) 22(1):87–90. doi: 10.1038/sj.bmt.1701275
100. Kashyap A, Kandeel F, Yamauchi D, Palmer JM, Niland JC, Molina A, et al. Effects of allogeneic bone marrow transplantation on recipient bone mineral density: A prospective study. Biol Blood Marrow Transplant (2000) 6:344–51. doi: 10.1016/S1083-8791(00)70061-9
101. Buchs N, Helg C, Collao C, Chapuis B, Slosman D, Bonjour JP, et al. Allogeneic bone marrow transplantation is associated with a preferential femoral neck bone loss. Osteoporos Int (2001) 12(10):880–6. doi: 10.1007/s001980170041
102. Schulte CMS, Beelen DW. Bone loss following hematopoietic stem cell transplantation: A long-term follow-up. Blood (2004) 103(10):3635–43. doi: 10.1182/blood-2003-09-3081
103. Pundole XN, Barbo AG, Lin H, Champlin RE, Lu H. Increased incidence of fractures in recipients of hematopoietic stem-cell transplantation. J Clin Oncol (2015) 33(12):1364–70. doi: 10.1200/JCO.2014.57.8195
104. Stern JM, Sullivan KM, Ott SM, Seidel K, Fink JC, Longton G, et al. Bone density loss after allogeneic hematopoietic stem cell transplantation: A prospective study. Biol Blood Marrow Transplant (2001) 7(5):257–64. doi: 10.1053/bbmt.2001.v7.pm11400947
105. Weilbaecher KN. Mechanisms of osteoporosis after hematopoietic cell transplantation. Biol Blood Marrow Transplant (2000) 6(2 A):165–74. doi: 10.1016/S1083-8791(00)70039-5
106. Tauchmanovà L, Colao A, Lombardi G, Rotoli B, Selleri C. Review: Bone loss and its management in long-term survivors from allogeneic stem cell transplantation. J Clin Endocrinol Metab (2007) 92(12):4536–45. doi: 10.1210/jc.2006-2870
107. Khan Z, Agarwal NB, Bhurani D, Khan MA. Risk factors for hematopoietic stem cell transplantation-associated bone loss. Biol Blood Marrow Transplant (2020) 27(3):212–21. doi: 10.1016/j.bbmt.2020.10.002
108. Kendler DL, Body JJ, Brandi ML, Broady R, Cannata-Andia J, Cannata-Ortiz MJ, et al. Bone management in hematologic stem cell transplant recipients. Osteoporos Int (2018) 29(12):2597–610. doi: 10.1007/s00198-018-4669-4
109. Ng AC, Khosla S, Charatcharoenwitthaya N, Kumar SK, Achenbach SJ, Holets MF, et al. Bone microstructural changes revealed by high-resolution peripheral quantitative computed tomography imaging and elevated DKK1 and MIP-1α levels in patients with MGUS. Blood (2011) 118(25):6529–34. doi: 10.1182/blood-2011-04-351437
110. Ruchlemer R, Amit-Kohn M, Tvito A, Sindelovsky I, Zimran A, Raveh-Brawer D. Bone loss and hematological malignancies in adults: a pilot study. Support Care Cancer (2018) 26(9):3013–20. doi: 10.1007/s00520-018-4143-z
111. Petryk A, Polgreen LE, Zhang L, Hodges JS, Dengel DR, Hoffmeister PA, et al. Bone mineral deficits in recipients of hematopoietic cell transplantation: the impact of young age at transplant. Bone Marrow Transplant (2014) 49(2):258. doi: 10.1038/bmt.2013.156
112. Zhao D, Wang C, Zhao Y, Shu B, Jia Y, Liu S, et al. Cyclophosphamide causes osteoporosis in C57BL/6 male mice: Suppressive effects of cyclophosphamide on osteoblastogenesis and osteoclastogenesis. Oncotarget (2017) 8(58):98163–83. doi: 10.18632/oncotarget.21000
113. Wheeler DL, vander Griend RA, Wronski TJ, Miller GJ, Keith EE, Graves JE. The short- and long-term effects of methotrexate on the rat skeleton. Bone (1995) 16(2):215–21. doi: 10.1016/8756-3282(94)00032-U
114. May KP, West SG, Mcdermott MT, Huffer WE. The effect of low-dose methotrexate on bone metabolism and histomorphometry in rats. Arthritis Rheumatol (1994) 37(2):201–6. doi: 10.1002/art.1780370208
115. Hadji P, Ziller M, Maskow C, Albert U, Kalder M. The influence of chemotherapy on bone mineral density, quantitative ultrasonometry and bone turnover in pre-menopausal women with breast cancer. Eur J Cancer (2009) 45(18):3205–12. doi: 10.1016/j.ejca.2009.09.026
116. Buttiglieri S, Ruella M, Risso A, Spatola T, Silengo L, Avvedimento EV, et al. The aging effect of chemotherapy on cultured human mesenchymal stem cells. Exp Hematol (2011) 39(12):1171–81. doi: 10.1016/j.exphem.2011.08.009
117. Gradishar WJ, Schilsky RL. Effects of cancer treatment on the reproductive system. Crit Rev Oncol Hematol (1988) 8(2):153–71. doi: 10.1016/S1040-8428(88)80009-X
118. Rodriguez-Wallberg KA. Impact of Cancer Treatment on Reproductive Health and Options for Fertility Preservation. In “Cancer Treatment – Conventional and Innovative Approaches”, ed Letícia Rangel. (IntechOpen, London, UK) (2013) 519–37.
119. Hirbe A, Morgan EA, Uluçkan Ö, Weilbaecher K, Guise, Coleman, et al. Skeletal complications of breast cancer therapies. Clin Cancer Res (2006) 12(20 PART 2):6309–14. doi: 10.1158/1078-0432.CCR-06-0652
120. Michaud LB, Goodin S. Cancer-treatment-induced bone loss, part 1. Am J Health-Syst Pharmacy (2006) 63(5):419–30. doi: 10.2146/ajhp050045.p1
121. Sakurai T, Sawada Y, Yoshimoto M, Kawai M, Miyakoshi J. Radiation-induced reduction of osteoblast differentiation in C2C12 cells. J Radiat Res (2007) 48(6):515–21. doi: 10.1269/jrr.07012
122. Gal TJ, Munoz-Antonia T, Muro-Cacho CA, Klotch DW. Radiation effects on osteoblasts in vitro. Arch Otolaryngol Head Neck Surg (2000) 126(9):1124. doi: 10.1001/archotol.126.9.1124
123. Higham CE, Faithfull S. Bone health and pelvic radiotherapy. Clin Oncol (2015) 27(11):668–78. doi: 10.1016/j.clon.2015.07.006
124. Willey JS, Lloyd SAJ, Robbins ME, Bourland JD, Smith-Sielicki H, Bowman LC, et al. Early increase in osteoclast number in mice after whole-body irradiation with 2 gy X rays. Radiat Res (2008) 170(3):388–92. doi: 10.1667/RR1388.1
125. Green DE, Adler BJ, Chan ME, Lennon JJ, Acerbo AS, Miller LM, et al. Altered composition of bone as triggered by irradiation facilitates the rapid erosion of the matrix by both cellular and physicochemical processes. PloS One (2013) 8(5):e64952. doi: 10.1371/journal.pone.0064952
126. Bakker B, Massa GG, van Rijn AM, Mearadji A, van der Kamp HJ, Niemer-Tucker MMB, et al. Effects of total-body irradiation on growth, thyroid and pituitary gland in rhesus monkeys. Radiother Oncol (1999) 51(2):187–92. doi: 10.1016/S0167-8140(99)00059-6
127. Achermann JC, Hindmarsh PC, Brook CGD. The relationship between the growth hormone and insulin-like growth factor axis in long-term survivors of childhood brain tumours. Clin Endocrinol (Oxf) (1998) 49(5):639–45. doi: 10.1046/j.1365-2265.1998.00585.x
128. Khosla S, Melton LJ, Riggs BL. The unitary model for estrogen deficiency and the pathogenesis of osteoporosis: Is a revision needed? J Bone Miner Res (2011) 26(3):441. doi: 10.1002/jbmr.262
129. Kousteni S, Bellido T, Plotkin LI, O’Brien CA, Bodenner DL, Han L, et al. Nongenotropic, sex-nonspecific signaling through the estrogen or androgen receptors: Dissociation from transcriptional activity. Cell (2001) 104(5):719–30. doi: 10.1016/S0092-8674(02)08100-X
130. Kousteni S, Han L, Chen JR, Almeida M, Plotkin LI, Bellido T, et al. Kinase-mediated regulation of common transcription factors accounts for the bone-protective effects of sex steroids. J Clin Invest (2003) 111(11):1651. doi: 10.1172/JCI200317261
131. Khosla S, Oursler J, Monroe DG. Estrogen and the skeleton. Trends Endocrinol Metab (2012) 23(11):576–81. doi: 10.1016/j.tem.2012.03.008
132. Chang J, Wang Z, Tang E, Fan Z, McCauley L, Franceschi R, et al. Inhibition of osteoblastic bone formation by nuclear factor-kappaB. Nat Med (2009) 15(6):682–9. doi: 10.1038/nm.1954
133. Tomkinson A, Gevers EF, Wit JM, Reeve J, Noble BS. The role of estrogen in the control of rat osteocyte apoptosis. J Bone Miner Res (1998) 13(8):1243–50. doi: 10.1359/jbmr.1998.13.8.1243
134. Emerton KB, Hu B, Woo AA, Sinofsky A, Hernandez C, Majeska RJ, et al. Osteocyte apoptosis and control of bone resorption following ovariectomy in mice. Bone (2010) 46(3):577–83. doi: 10.1016/j.bone.2009.11.006
135. Roggia C, Gao Y, Cenci S, Weitzmann MN, Toraldo G, Isaia G, et al. Up-regulation of TNF-producing T cells in the bone marrow: a key mechanism by which estrogen deficiency induces bone loss in vivo. Proc Natl Acad Sci U.S.A. (2001) 98(24):13960–5. doi: 10.1073/pnas.251534698
136. Cenci S, Weitzmann MN, Roggia C, Namba N, Novack D, Woodring J, et al. Estrogen deficiency induces bone loss by enhancing T-cell production of TNF-alpha. J Clin Invest (2000) 106(10):1229–37. doi: 10.1172/JCI11066
137. Sass DA, Liss T, Bowman AR, Rucinski B, Popoff SN, Pan Z, et al. The role of the T-lymphocyte in estrogen deficiency osteopenia. J Bone Miner Res (1997) 12(3):479–86. doi: 10.1359/jbmr.1997.12.3.479
138. Lee SK, Kadono Y, Okada F, Jacquin C, Koczon-Jaremko B, Gronowicz G, et al. T Lymphocyte-deficient mice lose trabecular bone mass with ovariectomy. J Bone Miner Res (2006) 21(11):1704–12. doi: 10.1359/jbmr.060726
139. Ru J-y, Wang Y-f. Osteocyte apoptosis: the roles and key molecular mechanisms in resorption-related bone diseases. Cell Death Dis (2020) 11(10):846. doi: 10.1038/s41419-020-03059-8
140. Almeida M, Laurent MR, Dubois V, Claessens F, O’Brien CA, Bouillon R, et al. Estrogens and androgens in skeletal physiology and pathophysiology. Physiol Rev (2017) 97(1):135. doi: 10.1152/physrev.00033.2015
141. Xiong J, Onal M, Jilka RL, Weinstein RS, Manolagas SC, O’Brien CA. Matrix-embedded cells control osteoclast formation. Nat Med (2011) 17(10):1235–41. doi: 10.1038/nm.2448
142. Nakashima T, Hayashi M, Fukunaga T, Kurata K, Oh-Hora M, Feng JQ, et al. Evidence for osteocyte regulation of bone homeostasis through RANKL expression. Nat Med (2011) 17(10):1231–4. doi: 10.1038/nm.2452
143. Fink HA, Ewing SK, Ensrud KE, Barrett-Connor E, Taylor BC, Cauley JA, et al. Association of testosterone and estradiol deficiency with osteoporosis and rapid bone loss in older men. J Clin Endocrinol Metab (2006) 91(10):3908–15. doi: 10.1210/jc.2006-0173
144. Mellström D, Johnell O, Ljunggren Ö, Eriksson AL, Lorentzon M, Mallmin H, et al. Free testosterone is an independent predictor of BMD and prevalent fractures in elderly men: MrOS Sweden. J Bone Miner Res (2006) 21(4):529–35. doi: 10.1359/jbmr.060110
145. Szulc P, Claustrat B, Marchand F, Delmas PD. Increased risk of falls and increased bone resorption in elderly men with partial androgen deficiency: The MINOS study. J Clin Endocrinol Metab (2003) 88(11):5240–7. doi: 10.1210/jc.2003-030200
146. Kenny AM, Prestwood KM, Marcello KM, Raisz LG. Determinants of bone density in healthy older men with low testosterone levels. Journals Gerontol: Ser A (2000) 55(9):M492–7. doi: 10.1093/gerona/55.9.M492
147. Tuck SP, Scane AC, Fraser WD, Diver MJ, Eastell R, Francis RM. Sex steroids and bone turnover markers in men with symptomatic vertebral fractures. Bone (2008) 43(6):999–1005. doi: 10.1016/j.bone.2008.08.123
148. Meier C, v. NT, DJ H, Schindler C, MM K, AL R, et al. Endogenous sex hormones and incident fracture risk in older men: The dubbo osteoporosis epidemiology study. Arch Intern Med (2008) 168(1):47–54. doi: 10.1001/archinternmed.2007.2
149. Risto O, Hammar E, Hammar K, Fredrikson M, Hammar M, Wahlström O. Elderly men with a history of distal radius fracture have significantly lower calcaneal bone density and free androgen index than age-matched controls. Aging Male (2012) 15(1):59–62. doi: 10.3109/13685538.2011.593659
150. Woo J, Kwok T, Leung JCS, Ohlsson C, Vandenput L, Leung PC. Sex steroids and bone health in older Chinese men. Osteoporos Int (2012) 23(5):1553–62. doi: 10.1007/s00198-011-1552-y
151. Hsu B, Seibel MJ, Cumming RG, Blyth FM, Naganathan V, Bleicher K, et al. Progressive temporal change in serum SHBG, but not in serum testosterone or estradiol, is associated with bone loss and incident fractures in older men: The concord health and ageing in men project. J Bone Miner Res (2016) 31(12):2115–22. doi: 10.1002/jbmr.2904
152. Mellström D, Vandenput L, Mallmin H, Holmberg AH, Lorentzon M, Odén A, et al. Older men with low serum estradiol and high serum SHBG have an increased risk of fractures. J Bone Miner Res (2008) 23(10):1552–60. doi: 10.1359/jbmr.080518
153. Roddam AW, Appleby P, Neale R, Dowsett M, Folkerd E, Tipper S, et al. Association between endogenous plasma hormone concentrations and fracture risk in men and women: the EPIC-Oxford prospective cohort study. J Bone Miner Metab (2009) 27(4):485–93. doi: 10.1007/s00774-009-0060-z
154. Mohamad NV, Soelaiman IN, Chin KY. A concise review of testosterone and bone health. Clin Interv Aging (2016) 11:1317–24. doi: 10.2147/CIA.S115472
155. Gill RK, Turner RT, Wronski TJ, Bell NH. Orchiectomy markedly reduces the concentration of the three isoforms of transforming growth factor β in rat bone, and reduction is prevented by testosterone. Endocrinology (1998) 139(2):546–50. doi: 10.1210/endo.139.2.5717
156. Chin KY, Ima-Nirwana S. The effects of orchidectomy and supraphysiological testosterone administration on trabecular bone structure and gene expression in rats (2015). Available at: https://www.tandfonline.com/doi/abs/10.3109/13685538.2014.954995.
157. Li X, Ominsky MS, Stolina M, Warmington KS, Geng Z, Niu QT, et al. Increased RANK ligand in bone marrow of orchiectomized rats and prevention of their bone loss by the RANK ligand inhibitor osteoprotegerin. Bone (2009) 45(4):669–76. doi: 10.1016/j.bone.2009.06.011
158. Bellido T, Jilka RL, Boyce BF, Girasole G, Broxmeyer H, Dalrymple SA, et al. Regulation of interleukin-6, osteoclastogenesis, and bone mass by androgens. the role of the androgen receptor. J Clin Invest (1995) 95(6):2886–95. doi: 10.1172/JCI117995
159. Catalano A, Gaudio A, Agostino RM, Morabito N, Bellone F, Lasco A. Trabecular bone score and quantitative ultrasound measurements in the assessment of bone health in breast cancer survivors assuming aromatase inhibitors. J Endocrinol Invest (2019) 42(11):1337–43. doi: 10.1007/s40618-019-01063-0
160. Zitzmann M, Brune M, Vieth V, Nieschlag E. Monitoring bone density in hypogonadal men by quantitative phalangeal ultrasound. Bone (2002) 31(3):422–9. doi: 10.1016/S8756-3282(02)00831-1
161. Raisz LG. Pathogenesis of osteoporosis: concepts, conflicts, and prospects. J Clin Invest (2005) 115(12):3318–25. doi: 10.1172/JCI27071
162. Ding KH, Wang ZZ, Hamrick MW, Deng ZB, Zhou L, Kang B, et al. Disordered osteoclast formation in RAGE-deficient mouse establishes an essential role for RAGE in diabetes related bone loss. Biochem Biophys Res Commun (2006) 340(4):1091–7. doi: 10.1016/j.bbrc.2005.12.107
163. Xie J, Méndez JD, Méndez-Valenzuela V, Aguilar-Hernández MM. Cellular signalling of the receptor for advanced glycation end products (RAGE). Cell Signal (2013) 25(11):2185–97. doi: 10.1016/j.cellsig.2013.06.013
164. Lorenzo JA, Naprta A, Rao Y, Alander C, Glaccum M, Widmer M, et al. Mice lacking the type I interleukin-1 receptor do not lose bone mass after ovariectomy. Endocrinology (1998) 139(6):3022–5. doi: 10.1210/endo.139.6.6128
165. Kimble RB, Matayoshi AB, Vannice JL, Kung VT, Williams C, Pacifici R. Simultaneous block of interleukin-1 and tumor necrosis factor is required to completely prevent bone loss in the early postovariectomy period. Endocrinology (1995) 136(7):3054–61. doi: 10.1210/endo.136.7.7789332
166. Shigehara K, Izumi K, Kadono Y, Mizokami A. Testosterone and bone health in men: A narrative review. J Clin Med (2021) 10(3):1–12. doi: 10.3390/jcm10030530
167. Stanley HL, Schmitt BP, Poses RM, Deiss WP. Does hypogonadism contribute to the occurrence of a minimal trauma hip fracture in elderly men? J Am Geriatr Soc (1991) 39(8):766–71. doi: 10.1111/j.1532-5415.1991.tb02698.x
168. Rochira V, Antonio L, Vanderschueren D. EAA clinical guideline on management of bone health in the andrological outpatient clinic. Andrology (2018) 6(2):272–85. doi: 10.1111/andr.12470
169. Antonio L, Caerels S, Jardi F, Delaunay E, Vanderschueren D. Testosterone replacement in congenital hypogonadotropic hypogonadism maintains bone density but has only limited osteoanabolic effects. Andrology (2019) 7(3):302–6. doi: 10.1111/andr.12604
170. Lee WY, Cho SW, Oh ES, Oh KW, Lee JM, Yoon KH, et al. The effect of bone marrow transplantation on the osteoblastic differentiation of human bone marrow stromal cells. J Clin Endocrinol Metab (2002) 87(1):329–35. doi: 10.1210/jcem.87.1.8135
171. Tauchmanovà L, Serio B, del Puente A, Risitano AM, Esposito A, de Rosa G, et al. Long-lasting bone damage detected by dual-energy x-ray absorptiometry, phalangeal osteosonogrammetry, and in vitro growth of marrow stromal cells after allogeneic stem cell transplantation. J Clin Endocrinol Metab (2002) 87(11):5058–65. doi: 10.1210/jc.2002-020800
172. Banfi A, Podestà M, Fazzuoli L, Sertoli MR, Venturini M, Santini G, et al. High-dose chemotherapy shows a dose-dependent toxicity to bone marrow osteoprogenitors: A mechanism for post-bone marrow transplantation osteopenia. Cancer (2001) 92(9):2419–28. doi: 10.1002/1097-0142(20011101)92:9<2419::AID-CNCR1591>3.0.CO;2-K
173. Withold W, Rollbach S, Reinauer H, Wolf HH, Rollbach S, Heyll A, et al. Relationship between bone metabolism and plasma cytokine levels in patients at risk of post-transplantation bone disease after bone marrow transplantation. Clin Chem Lab Med (1996) 34(4):295–300. doi: 10.1515/cclm.1996.34.4.295
174. Lee WY, Baek KH, Rhee EJ, Tae HJ, Oh KW, Kang MI, et al. Impact of circulating bone-resorbing cytokines on the subsequent bone loss following bone marrow transplantation. Bone Marrow Transplant (2004) 34(1):89–94. doi: 10.1038/sj.bmt.1704535
175. Manolagas SC, Jilka R. Review articles. New Engl J Med (1995) 332(5):305–11. doi: 10.1056/NEJM199502023320506
176. Lane NE. Glucocorticoid induced osteoporosis: New insights into the pathophysiology and treatments. Curr Osteoporos Rep (2019) 17(1):1. doi: 10.1007/s11914-019-00498-x
177. Stern JM, Chesnut CH 3rd, Bruemmer B, Sullivan KM, Lenssen PS, Aker SN, et al. Bone density loss during treatment of chronic GVHD. Bone Marrow Transplant (1996) 17(3):395–400.
178. Ferrara JLM, Levine JE. Graft-versus-host disease in the 21st century: New perspectives on an old problem. Semin Hematol (2006) 43(1):1–2. doi: 10.1053/j.seminhematol.2005.11.028
179. Lee SJ, Vogelsang G, Flowers MED. Chronic graft-versus-host disease. Biol Blood Marrow Transplant (2003) 9(4):215–33. doi: 10.1053/bbmt.2003.50026
180. Canalis E. Mechanisms of glucocorticoid action in bone. Curr Osteoporos Rep (2005) 3(3):98–102. doi: 10.1007/s11914-005-0017-7
181. Kovvuru K, Kanduri SR, Vaitla P, Marathi R, Gosi S, Anton DFG, et al. Risk factors and management of osteoporosis post-transplant. Med (Lithuania) (2020) 56(6):1–18. doi: 10.3390/medicina56060302
182. O’Brien CA, Jia D, Plotkin LI, Bellido T, Powers CC, Stewart SA, et al. Glucocorticoids act directly on osteoblasts and osteocytes to induce their apoptosis and reduce bone formation and strength. Endocrinology (2004) 145(4):1835–41. doi: 10.1210/en.2003-0990
183. Kanda J, Izumo N, Furukawa M, Shimakura T, Yamamoto N, Takahashi HE, et al. Effects of the calcineurin inhibitors cyclosporine and tacrolimus on bone metabolism in rats. Biomed Res (Japan) (2018) 39(3):131–9. doi: 10.2220/biomedres.39.131
184. Cosman F, de Beur SJ, LeBoff MS, Lewiecki EM, Tanner B, Randall S, et al. Clinician’s guide to prevention and treatment of osteoporosis. Osteoporos Int (2014) 25(10):2359–81. doi: 10.1007/s00198-014-2794-2
185. Ross AC, Manson JAE, Abrams SA, Aloia JF, Brannon PM, Clinton SK, et al. The 2011 report on dietary reference intakes for calcium and vitamin d from the institute of medicine: What clinicians need to know. J Clin Endocrinol Metab (2011) 96(1):53–8. doi: 10.1210/jc.2010-2704
186. Välimäki MJ, Kinnunen K, Volin L, Tähtelä R, Löyttyniemi E, Laitinen K, et al. A prospective study of bone loss and turnover after allogeneic bone marrow transplantation: effect of calcium supplementation with or without calcitonin. Bone Marrow Transplant (1999) 23(4):355–61. doi: 10.1038/sj.bmt.1701586
187. Sunyecz JA. The use of calcium and vitamin d in the management of osteoporosis. Ther Clin Risk Manag (2008) 4(4):827. doi: 10.2147/TCRM.S3552
188. Charles C, Ferris AH. Chronic kidney disease. Primary Care: Clinics Office Practice (2020) 47(4):585–95. doi: 10.1016/j.pop.2020.08.001
189. KDIGO 2012 clinical practice guideline for the evaluation and management of chronic kidney disease . Available at: www.publicationethics.org.
190. James PA, Oparil S, Carter BL, Cushman WC, Dennison-Himmelfarb C, Handler J, et al. 2014 evidence-based guideline for the management of high blood pressure in adults: Report from the panel members appointed to the eighth joint national committee (JNC 8). JAMA (2014) 311(5):507–20. doi: 10.1001/jama.2013.284427
191. Inker LA, Astor BC, Fox CH, Isakova T, Lash JP, Peralta CA, et al. KDOQI US commentary on the 2012 KDIGO clinical practice guideline for the evaluation and management of CKD. Am J Kidney Dis (2014) 63(5):713–35. doi: 10.1053/j.ajkd.2014.01.416
192. Sanghavi K, Wiseman A, Kirstein MN, Cao Q, Brundage R, Jensen K, et al. Personalized fludarabine dosing to reduce nonrelapse mortality in hematopoietic stem-cell transplant recipients receiving reduced intensity conditioning. Transl Res (2016) 175:103–115.e4. doi: 10.1016/j.trsl.2016.03.017
193. Woywodt A, Haubitz M, Buchholz S, Hertenstein B. Counting the cost: markers of endothelial damage in hematopoietic stem cell transplantation. Bone Marrow Transplant (2004) 34(12):1015–23. doi: 10.1038/sj.bmt.1704733
194. Arai Y, Yamashita K, Mizugishi K, Watanabe T, Sakamoto S, Kitano T, et al. Serum neutrophil extracellular trap levels predict thrombotic microangiopathy after allogeneic stem cell transplantation. Biol Blood Marrow Transplant (2013) 19(12):1683–9. doi: 10.1016/j.bbmt.2013.09.005
195. Rizzo JD, Wingard JR, Tichelli A, Lee SJ, van Lint MT, Burns LJ, et al. Recommended screening and preventive practices for long-term survivors after hematopoietic cell transplantation: joint recommendations of the European group for blood and marrow transplantation, the center for international blood and marrow transplant research, and the American society of blood and marrow transplantation. Biol Blood Marrow Transplant (2006) 12(2):138–51. doi: 10.1016/j.bbmt.2005.09.012
196. Majhail NS, Rizzo JD, Lee SJ, Aljurf M, Atsuta Y, Bonfim C, et al. Recommended screening and preventive practices for long-term survivors after hematopoietic cell transplantation. Biol Blood Marrow Transplant (2012) 18(3):348–71. doi: 10.1016/j.bbmt.2011.12.519
197. Buckley L, Guyatt G, Fink HA, Cannon M, Grossman J, Hansen KE, et al. 2017 American College of rheumatology guideline for the prevention and treatment of glucocorticoid-induced osteoporosis. Arthritis Care Res (Hoboken) (2017) 69(8):1095–110. doi: 10.1002/acr.23279
198. Baumgartner A, Moesch M, Zumsteg M, Struja T, Bernet S, Medinger M, et al. Predictors of impaired bone health in long-term survivors after allogeneic stem cell transplantation. Bone Marrow Transplant (2019) 54(10):1651–61. doi: 10.1038/s41409-019-0484-6
199. Kanis JA, Borgstrom F, de Laet C, Johansson H, Johnell O, Jonsson B, et al. Assessment of fracture risk. Osteoporos Int (2005) 16(6):581–9. doi: 10.1007/s00198-004-1780-5
200. Schimmer AD, Minden MD, Keating A. Osteoporosis after blood and marrow transplantation: Clinical aspects. Biol Blood Marrow Transplant (2000) 6(2):175–81. doi: 10.1016/S1083-8791(00)70040-1
201. Melton LJ, Kyle RA, Achenbach SJ, Oberg AL, Rajkumar SV. Fracture risk with multiple myeloma: A population-based study. J Bone Miner Res (2005) 20(3):487–93. doi: 10.1359/JBMR.041131
202. Rizzoli R, Biver E. Glucocorticoid-induced osteoporosis: who to treat with what agent? Nat Rev Rheumatol (2014) 11(2):98–109. doi: 10.1038/nrrheum.2014.188
203. Schousboe JT, Shepherd JA, Bilezikian JP, Baim S. Executive summary of the 2013 international society for clinical densitometry position development conference on bone densitometry. J Clin Densitometry (2013) 16(4):455–66. doi: 10.1016/j.jocd.2013.08.004
204. Pulsipher MA, Skinner R, McDonald GB, Hingorani S, Armenian SH, Cooke KR, et al. National cancer institute, national heart, lung and blood Institute/Pediatric blood and marrow transplantation consortium first international consensus conference on late effects after pediatric hematopoietic cell transplantation: The need for pediatric-specific long-term follow-up guidelines. Biol Blood Marrow Transplant (2012) 18(3):334–47. doi: 10.1016/j.bbmt.2012.01.003
205. Bar M, Ott SM, Lewiecki EM, Sarafoglou K, Wu JY, Thompson MJ, et al. Bone health management after hematopoietic cell transplantation: An expert panel opinion from the American society for transplantation and cellular therapy. Biol Blood Marrow Transplant (2020) 26(10):1784–802. doi: 10.1016/j.bbmt.2020.07.001
206. Lewiecki EM, Kendler DL, Davison KS, Hanley DA, Harris ST, McClung MR, et al. Western Osteoporosis alliance clinical practice series: Treat-to-Target for osteoporosis. Am J Med (2019) 132(11):e771–7. doi: 10.1016/j.amjmed.2019.04.044
207. Curtis JR, Saag KG, Arora T, Wright NC, Yun H, Daigle S, et al. Duration of bisphosphonate drug holidays and associated fracture risk. Med Care (2020) 58(5):419–26. doi: 10.1097/MLR.0000000000001294
208. Tauchmanovà L, de Simone G, Musella T, Orio F, Ricci P, Nappi C, et al. Effects of various antireabsorptive treatments on bone mineral density in hypogonadal young women after allogeneic stem cell transplantation. Bone Marrow Transplant (2006) 37(1):81–8. doi: 10.1038/sj.bmt.1705196
209. Hari P, DeFor TE, Vesole DH, Bredeson CN, Burns LJ. Intermittent zoledronic acid prevents bone loss in adults after allogeneic hematopoietic cell transplantation. Biol Blood Marrow Transplant (2013) 19(9):1361–7. doi: 10.1016/j.bbmt.2013.06.015
210. Grigg AP, Shuttleworth P, Reynolds J, Schwarer AP, Szer J, Bradstock K, et al. Pamidronate reduces bone loss after allogeneic stem cell transplantation. J Clin Endocrinol Metab (2006) 91(10):3835–43. doi: 10.1210/jc.2006-0684
211. Pundole X, Cheema HI, Petitto GS, Lopez-Olivo MA, Suarez-Almazor ME, Lu H. Prevention and treatment of bone loss and fractures in patients undergoing a hematopoietic stem cell transplant: a systematic review and meta-analysis. Bone Marrow Transplant (2017) 52(5):663–70. doi: 10.1038/bmt.2016.312
212. Kyrgidis A, Vahtsevanos K, Koloutsos G, Andreadis C, Boukovinas I, Teleioudis Z, et al. Bisphosphonate-related osteonecrosis of the jaws: A case-control study of risk factors in breast cancer patients. J Clin Oncol (2008) 26(28):4634–8. doi: 10.1200/JCO.2008.16.2768
213. Ruggiero SL, Dodson TB, Fantasia J, Goodday R, Aghaloo T, Mehrotra B, et al. American Association of oral and maxillofacial surgeons position paper on medication-related osteonecrosis of the jaw–2014 update. J Oral Maxillofac Surgery (2014) 72(10):1938–56. doi: 10.1016/j.joms.2014.04.031
214. Schubert MM, Correa MEP. Oral graft-Versus-Host disease. Dent Clin North Am (2008) 52(1):79–109. doi: 10.1016/j.cden.2007.10.004
215. Yarom N, Shapiro CL, Peterson DE, van Poznak CH, Bohlke K, Ruggiero SL, et al. Medication-related osteonecrosis of the jaw: MASCC/ISOO/ASCO clinical practice guideline. J Clin Oncol (2019) 37(25):2270–90. doi: 10.1200/JCO.19.01186
216. Anderson K, Ismaila N, Flynn PJ, Halabi S, Jagannath S, Ogaily MS, et al. Role of bone-modifying agents in multiple myeloma: American society of clinical oncology clinical practice guideline update. J Clin Oncol (2018) 36(8):812–8. doi: 10.1200/JCO.2017.76.6402
217. Grossman DC, Curry SJ, Owens DK, Barry MJ, Davidson KW, Doubeni CA, et al. Hormone therapy for the primary prevention of chronic conditions in postmenopausal women US preventive services task force recommendation statement. JAMA - J Am Med Assoc (2017) 318(22):2224–33. doi: 10.1001/jama.2017.18261
218. Yeh MW, Zhou H, Adams AL, Ituarte PHG, Li N, Liu ILA, et al. The relationship of parathyroidectomy and bisphosphonates with fracture risk in primary hyperparathyroidism: An observational study. Ann Intern Med (2016) 164(11):715–23. doi: 10.7326/M15-1232
219. Canonico M, Carcaillon L, Plu-Bureau G, Oger E, Singh-Manoux A, Tubert-Bitter P, et al. Postmenopausal hormone therapy and risk of stroke: Impact of the route of estrogen administration and type of progestogen. Stroke (2016) 47(7):1734–41. doi: 10.1161/STROKEAHA.116.013052
220. Manson JE, Allison MA, Rossouw JE, Carr JJ, Langer RD, Hsia J, et al. Estrogen therapy and coronary-artery calcification. New Engl J Med (2007) 356(25):2591–602. doi: 10.1056/NEJMoa071513
221. Bhupathiraju SN, Grodstein F, Rosner BA, Stampfer MJ, Hu FB, Willett WC, et al. Hormone therapy use and risk of chronic disease in the nurses’ health study: A comparative analysis with the women’s health initiative. Am J Epidemiol (2017) 186(6):696–708. doi: 10.1093/aje/kwx131
222. Hodis HN, Mack WJ, Henderson VW, Shoupe D, Budoff MJ, Hwang-Levine J, et al. Vascular effects of early versus late postmenopausal treatment with estradiol. New Engl J Med (2016) 374(13):1221–31. doi: 10.1056/NEJMoa1505241
223. Nelson HD, Rizzo J, Harris E, Cauley J, Ensrud K, Bauer DC, et al. Osteoporosis and fractures in postmenopausal women using estrogen. Arch Intern Med (2002) 162(20):2278–84. doi: 10.1001/archinte.162.20.2278
224. Finkelstein JS, Lee H, Leder BZ, Burnett-Bowie SAM, Goldstein DW, Hahn CW, et al. Gonadal steroid–dependent effects on bone turnover and bone mineral density in men. J Clin Invest (2016) 126(3):1114–25. doi: 10.1172/JCI84137
225. Weber TJ. Battle of the sex steroids in the male skeleton: and the winner is…. J Clin Invest (2016) 126(3):829–32. doi: 10.1172/JCI85006
226. Chesnut CH, Silverman S, Andriano K, Genant H, Gimona A, Harris S, et al. A randomized trial of nasal spray salmon calcitonin in postmenopausal women with established osteoporosis: the prevent recurrence of osteoporotic fractures study. Am J Med (2000) 109(4):267–76. doi: 10.1016/S0002-9343(00)00490-3
227. Downs RW, Bell NH, Ettinger MP, Walsh BW, Favus MJ, Mako B, et al. Comparison of alendronate and intranasal calcitonin for treatment of osteoporosis in postmenopausal women. J Clin Endocrinol Metab (2000) 85(5):1783–8. doi: 10.1210/jcem.85.5.6606
228. Jeong C, Kim HJ, Lee S, Ha J. Effect of denosumab on bone mineral density of hematopoietic stem cell transplantation recipients. Int J Endocrinol (2020) 2020; 3410921. doi: 10.1155/2020/3410921
229. Sanders JE, Guthrie KA, Hoffmeister PA, Woolfrey AE, Carpenter PA, Appelbaum FR. Final adult height of patients who received hematopoietic cell transplantation in childhood. Blood (2005) 105(3):1348–54. doi: 10.1182/blood-2004-07-2528
230. Giavoli C, Porretti S, Ferrante E, Cappiello V, Ronchi CL, Travaglini P, et al. Recombinant hGH replacement therapy and the hypothalamus-pituitary-thyroid axis in children with GH deficiency: When should we be concerned about the occurrence of central hypothyroidism? Clin Endocrinol (Oxf) (2003) 59(6):806–10. doi: 10.1046/j.1365-2265.2003.01892.x
231. Portes ES, Oliveira JHA, Maccagnan P, Abucham J. Changes in serum thyroid hormones levels and their mechanisms during long-term growth hormone (GH) replacement therapy in GH deficient children. Clin Endocrinol (Oxf) (2000) 53(2):183–9. doi: 10.1046/j.1365-2265.2000.01071.x
232. Shevde NK, Bendixen AC, Dienger KM, Pike JW. Estrogens suppress RANK ligand-induced osteoclast differentiation via a stromal cell independent mechanism involving c-jun repression. Proc Natl Acad Sci U S A (2000) 97(14):7829–34. doi: 10.1073/pnas.130200197
233. Gohel A, McCarthy MB, Gronowicz G. Estrogen prevents glucocorticoid-induced apoptosis in osteoblasts in vivo and in vitro. Endocrinology (1999) 140(11):5339–47. doi: 10.1210/endo.140.11.7135
Keywords: hematopoietic stem cell transplantation, mineral homeostasis, bone metabolism, osteoporosis, bone resorption
Citation: Miglietta F, Iamartino L, Palmini G, Giusti F, Marini F, Iantomasi T and Brandi ML (2023) Endocrine sequelae of hematopoietic stem cell transplantation: Effects on mineral homeostasis and bone metabolism. Front. Endocrinol. 13:1085315. doi: 10.3389/fendo.2022.1085315
Received: 31 October 2022; Accepted: 26 December 2022;
Published: 12 January 2023.
Edited by:
Valentina Giudice, University of Salerno, ItalyReviewed by:
Smita Jha, National Institutes of Health (NIH), United StatesIlaria Roato, University of Turin, Italy
Copyright © 2023 Miglietta, Iamartino, Palmini, Giusti, Marini, Iantomasi and Brandi. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Maria Luisa Brandi, bWFyaWFsdWlzYS5icmFuZGlAdW5pZmkuaXQ=; bWFyaWFsdWlzYUBtYXJpYWx1aXNhYnJhbmRpLml0