 Valeria Vezzoli1*
Valeria Vezzoli1* Faris Hrvat2
Faris Hrvat2 Giovanni Goggi2
Giovanni Goggi2 Silvia Federici2
Silvia Federici2 Biagio Cangiano1,2Richard Quinton3,4
Biagio Cangiano1,2Richard Quinton3,4 Luca Persani1,2
Luca Persani1,2 Marco Bonomi1,2
Marco Bonomi1,2- 1Department of Endocrine and Metabolic Diseases and Lab of Endocrine and Metabolic Research, IRCCS Istituto Auxologico Italiano, Milan, Italy
- 2Department of Medical Biotechnology and Translational Medicine, University of Milan, Milan, Italy
- 3Department of Endocrinology, Diabetes & Metabolism, Newcastle-upon-Tyne Hospitals, Newcastle-upon-Tyne, United Kingdom
- 4Translational & Clinical Research Institute, University of Newcastle-upon-Tyne, Newcastle-upon-Tyne, United Kingdom
Distinguishing between self limited delayed puberty (SLDP) and congenital hypogonadotropic hypogonadism (CHH) may be tricky as they share clinical and biochemical characteristics. and appear to lie within the same clinical spectrum. However, one is classically transient (SDLP) while the second is typically a lifetime condition (CHH). The natural history and long-term outcomes of these two conditions differ significantly and thus command distinctive approaches and management. Because the first presentation of SDLP and CHH is very similar (delayed puberty with low LH and FSH and low sex hormones), the scientific community is scrambling to identify diagnostic tests that can allow a correct differential diagnosis among these two conditions, without having to rely on the presence or absence of phenotypic red flags for CHH that clinicians anyway seem to find hard to process. Despite the heterogeneity of genetic defects so far reported in DP, genetic analysis through next-generation sequencing technology (NGS) had the potential to contribute to the differential diagnostic process between SLDP and CHH. In this review we will provide an up-to-date overview of the genetic architecture of these two conditions and debate the benefits and the bias of performing genetic analysis seeking to effectively differentiate between these two conditions.
Introduction
Definition and etiology
Delayed puberty (DP) is one of the most common clinical conditions evaluated by the pediatric endocrinologist, affecting over 2% of adolescents, and implies the lack of the first signs of pubertal development beyond the average expected age for the normal population or, when puberty has previously begun, the failure of appropriate progression. Due to the earlier onset of pubertal development in girls, puberty is generally defined to be delayed if there is absence of first pubertal signs at 13 years of age in females (breast budding or thelarche) and or 14 in males (testicular enlargement over 3-4 mL of volume), respectively. These limits of normality correspond to the mean age +2-2.5 SD of the healthy general population in Western Europe. This implies that 2.5-3% of the normal subjects will fall in the extreme tail of the normal gaussian curve, and will be classified as having DP although, as better explained below, they will likely fall in the self-limited (“benign”) form of this condition. Several underlying etiologies cause pubertal delay (Table 1), including chronic disease, energy-deficit and hypo- and (in girls) hyper- gonadotropic hypogonadism (1). To date, self-limited, or constitutional, DP (SLDP) is the highly common cause of pubertal delay in early adolescence, involving around 2/3 of male and 1/3 of female with DP; a para-physiological condition where individuals start puberty late, but eventually progressing to achieve full sexual maturation. By contrast, hypogonadism becomes the more common cause of DP by the late teenage years. Hypergonadotropic hypogonadism is due to intrinsic or primary gonadal failure and can easily be discerned from other forms of DP through unstimulated reproductive hormone levels, indicated by low sex steroids and high gonadotropins. It particularly concerns females (21% of DP cases), with Turner syndrome seen in 27% of girls with premature ovarian insufficiency (POI) (2, 3) but is a very uncommon cause of pubertal failure in males (vanishing testes syndrome). Conversely differential diagnosis between SLDP and hypogonadotropic hypogonadism (HH) is often challenging, as both conditions can exhibit overlapping clinical and hormonal features (2), albeit that more careful clinical ascertainment of CHH red flags could potentially identify around 50% of the CHH cases with a high degree of probability (4).
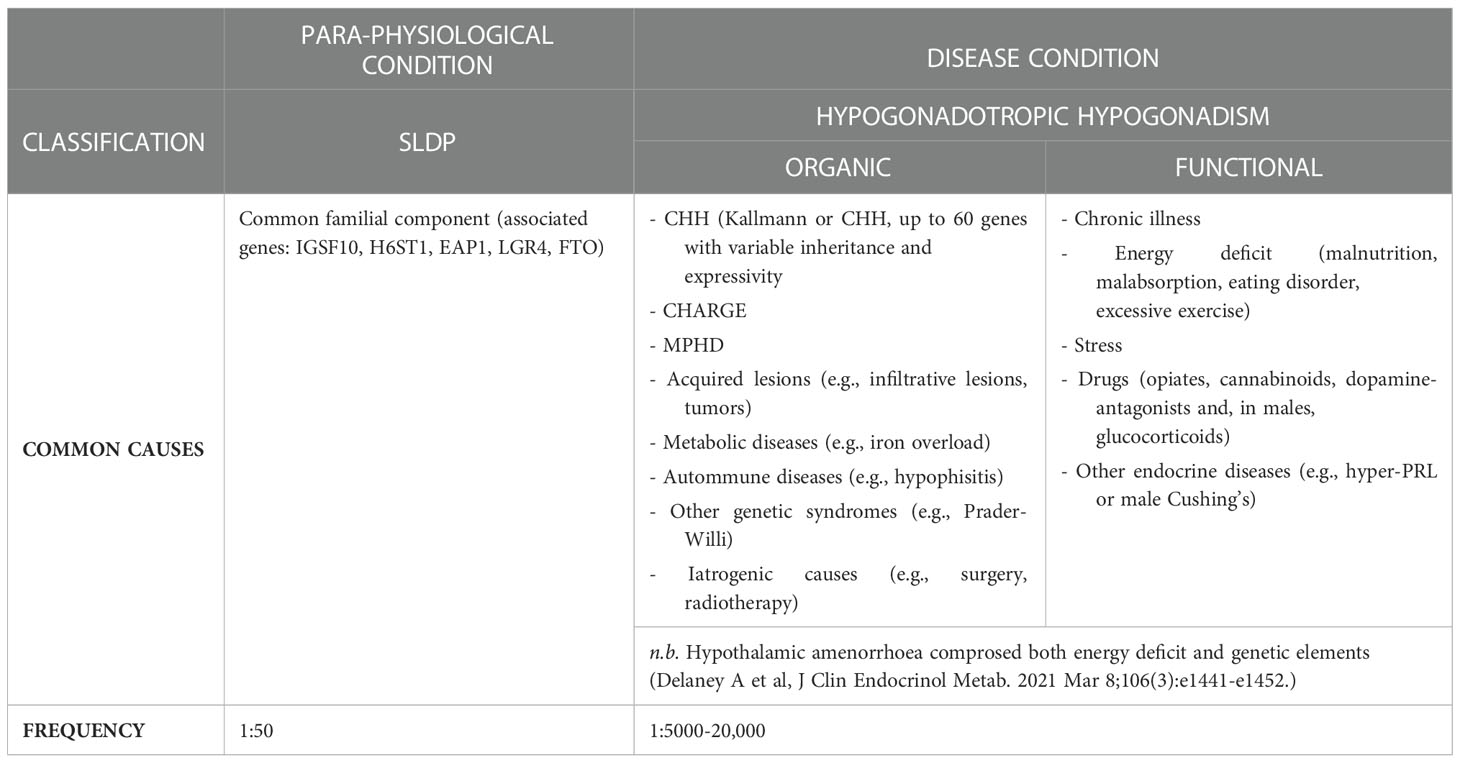
Table 1 List of conditions associated with CHH or SLDP.
Clinical features
Adolescents with CHH and SLDP present similar clinical traits and hormonal status at presentation but respond very differently to “expectative management” in terms of physical development and long-term psychosexual outcomes (5, 6). DP in females is established when breast budding development is absent by the age of 13 years, while in males when the testicular volume fails to surpass the threshold of 4 mL volume (evaluated by Prader orchidometer) by the age of 14 years (7). Family history of CHH versus SLDP can be helpful, but both conditions may occur within the same family (8, 9). Regarding phenotypic red flags for CHH, these comprise reproductive and non-reproductive defects, with the former only observed in males. A history of cryptorchidism, particularly the bilateral form, together with a possible neonatal underdeveloped penis (2.5 SD below average size for healthy subjects) and/or hypospadias in males is strongly suggests pre- and post-natal gonadotropin deficiency characteristic of CHH (absent minipuberty). Indeed, a definitive diagnosis can be made through basal biochemical evaluation of the HPG axis during the postnatal weeks corresponding to normal minipuberty (10, 11). However, there are no external signs postnatally in CHH female CHHs to suggest a congenital GnRH secretion defect. However, non-reproductive CHH red flags occur in both sexes, comprising clefting of the palate and/or lips, olfactory defects, alteration of digital bones, hearing loss, colorblindness, nystagmus and bimanual synkinesis, renal and/or dental agenesis or dysgenesis (10, 11). Anosmia or hyposmia are reported in around 50% of patients with CHH, thus olfactory defects (evaluated with T2-weighted coronal MRI of the olfactory bulbs and sulci and/or a quantitative olfactory test) should be evaluated and, whenever present, considered as a reliable clue towards CHH diagnosis (12). Slow growth rate and low weight tend to suggest functional HH or SLDP (7), especially when syndromic manifestations or red flags are absent. In contrast, children with CHH exhibit regular linear growth, although delayed bone maturation, osteopenia, and osteoporosis may be observed when CHH is diagnosed later in life (13, 14).
Hormone and stimulation test for differential diagnosis
Unlike the postnatal period, gonadotropin and sex hormone measurements cannot discriminate between SLDP and CHH in early adolescence, because gonadotropin concentrations are frequently borderline also in healthy subjects of this age (1). Frequent overnight blood sampling and LH pulse-analysis has been considered for the differential diagnosis, although similar pulsatile “fluctuation” was observed in patients with SLDP, CHH and in normal prepubertal children (15). Moreover, this technique can’t realistically be into routine clinical practice. In addition, the diagnostic efficacy of a single basal gonadotropin concentration it is inadequate (16, 17). A similarly low diagnostic power is offered by dynamic gonadotropin testing with GnRH or GnRH analogs (18). In fact, the lack of a gonadotropin reaction to GnRH stimulation can only confirm the absence of puberty onset, but it is not sufficient to provide a true differential diagnosis between SLDP and CHH (19). Measurement of Inhibin B, Anti-Mullerin Hormone, AMH, INSL3, and testosterone after hCG stimulation have been proposed to guide differential diagnosis (20). Although the data are promising (18, 21), they are not sufficient to support the clinical value of these newer endocrine markers. The use of new ultrasensitive methods (LC/MS) for determining hormone levels might, however, unravel new diagnostic perspectives. The testosterone response to long-term hCG stimulation and peak serum FSH response to GnRH were found to be significantly different in CHH patients (22), but there are potential long-term drawbacks to long-term hCG therapy in males who are FSH-naïve, in terms of promoting differentiation of a limited pool of Sertoli and germ cells before they have a chance to proliferate under FSH stimulation. Moreover, these methods are too inconvenient and expensive to be useful first-line approaches. Nevertheless, hormonal responsiveness to kisspeptin in boys with delayed puberty appears to be a promising new hormonal marker, although currently further studies are needed (23).
Low dose sex steroid “priming” test
Recent studies have examined the diagnostic value of testosterone priming to differentiate between SLDP from CHH. Short-term testosterone therapy (oral, injections or transdermal) in boys with SLDP would prompt HPG activation, with the result of enlargement of the testis and rise in the production of endogenous testosterone (24). This proposed “diagnostic test” would speed up diagnosis and consequent treatment of SLDP patients, with benefits on growth velocity and virilization (25). Subjects not responding to testosterone priming (i.e., CHH patients) (24) could be than efficiently analyzed with the more expensive tests described above or offered more sustained testosterone treatment. Estradiol priming has also been proposed for HPG activation in SLDP females (2). Although, as discussed in previous paragraphs, clinicians could benefit of multiple clinical and biochemical tests to produce the diagnosis, none of these could accurately discriminate between those patients who will naturally pass and progress normally during puberty (i.e. SLDP) and those who will likely require lifelong medical therapy (i.e. HH) (2, 3). The clinical distinction between SLDP and CHH in early adolescence is of crucial importance as if CHH is diagnosed, prompt drug treatment is mandatory for puberty induction (26–28), with combined gonadotropin therapy having the potential to optimize fertility potential and quality of life in males (29).
Genetics of CHH
CHH is defined by the diagnosis of gonadotropic deficiency throughout the infant mini- puberty or when puberty is absent or arrested in adolescence (30), although the median age at diagnosis and effective treatment of CHH remains unacceptably high around 19 years (5) and many patients present much later in adult life with sexual dysfunction, infertility, anaemia, myopathy or osteoporosis (31). CHH accounts for 24 to 85% of stable hypogonadotropic hypogonadism and includes normosmic subjects (nCHH) and subjects with olfactory defects identifying the Kallmann Syndrome (KS). KS results from mutations in genes acting in the development of olfactory neurons; nCHH can also be underpinned by the same “neurodevelopmental” genes but is more commonly associated with mutations of genes that regulate GnRH secretion. Currently, the utility of this dichotomous division in targeting genetic testing is reduced by the clinical and genetic overlap between the two conditions (32). In CHH, a genetic basis can be identified in around 50% of patients (14, 33, 34), although as more genes are identified it has become apparent in many patients that the genetic variant originally believed to fully explain their condition did not in fact represent the whole story. So far, mutations in more than 60 genes have been classified as genetic cause of CHH, whether nCHH, KS, or both (Table 2), with few rare loci also involved in complex syndromes such as CHARGE (35–38).These genes include ANOS1, FGF receptor 1 (FGFR1), FGF8, prokineticin 2 (PROK2), prokineticin 2 receptor (PROKR2), CHD7, NMDA receptor synaptonuclear signaling and neuronal migration factor (NSMF), GnRH1, GnRH receptor (GnRHR), KISS1, KISS1R, tacykinin 3 (TAC3), TACR3, semaphorin 3A (SEMA3A), SRY-box 10 (SOX10), IL-17 receptor D (IL17RD), FEZ family zinc finger 1 (FEZF1), WD repeat domain 11 (WDR11), heparin sulfate 6-O-sulfotransferase 1 (HS6ST1), and FGF17. These are key genes for regulating GnRH neuronal migration and differentiation, GnRH secretion, or its upstream or downstream pathways (Figure 1). GnRH neuroendocrine cells originate in the olfactory placode outside the central nervous system and subsequently migrate into the brain during embryonic development (39). This route offers a developmental connection between the sense of smell and the central control of reproduction, which are both affected in Kallmann syndrome. Evidence obtained in the past years (40) suggest that GnRH neurons originating from the neural crest and ectodermal progenitors migrate in tight association with growing axons of olfactory and terminal nerves. Reached the hypothalamus, GnRH neurons finally detach from their TN guiding fibers, disperse further into the brain parenchyma, and stop the migration. At birth, GnRH neurons project to the hypothalamic median eminence and release GnRH into the hypophyseal portal vasculature (41). GnRH acts via the GnRH receptor, which is expressed on gonadotropic cells in the anterior pituitary gland. This action elicits the secretion of the gonadotropins, luteinizing hormone and follicle-stimulating hormone which control gonadal maturation and adult reproductive physiology via the hypothalamic–pituitary–gonadal (HPG) axis. As with many other diseases of genetic origin, the application of next generation sequencing technology (NGS) to the diagnosis of CHH has boosted the discovery of new candidates involved in its etiology. All known forms of inheritance have been described: autosomal dominant, recessive and with variable penetrance; X-linked recessive; oligogenicity, and transmission linked to an imprinting locus. Moreover, as already reported for Fgf8 signaling system (42, 43), also interferences in the pre-hypothalamic epigenome (throught DNMTs/TETs proteins) could have major consequences on GnRH system neurodevelopment, resulting in CHH disorder. Due to the variable expressivity and incomplete penetrance of the genetic defects, together with the actual or potential impact of oligogenicity and epigenome modifications, there is a broad spectrum of phenotypes, whether with non-reproductive defects or pure neuroendocrine phenotype, and ranging from complete CHH, with LH/GnRH apulsatility and absent pubertal development (around 2/3 of cases), to partial hypogonadism with residual LH/GnRH pulsatility (low amplitude, low frequency, or nocturnal-only pattern) resulting in arrested early puberty (around 1/3 of cases), and even reversible CHH in 5 to 20% of cases patients (44). Crucially, the genetic architecture of CHH as is presently understood does not explain the 3-5-fold excess of affected males.

Table 2 List of genes implicated in CHH.
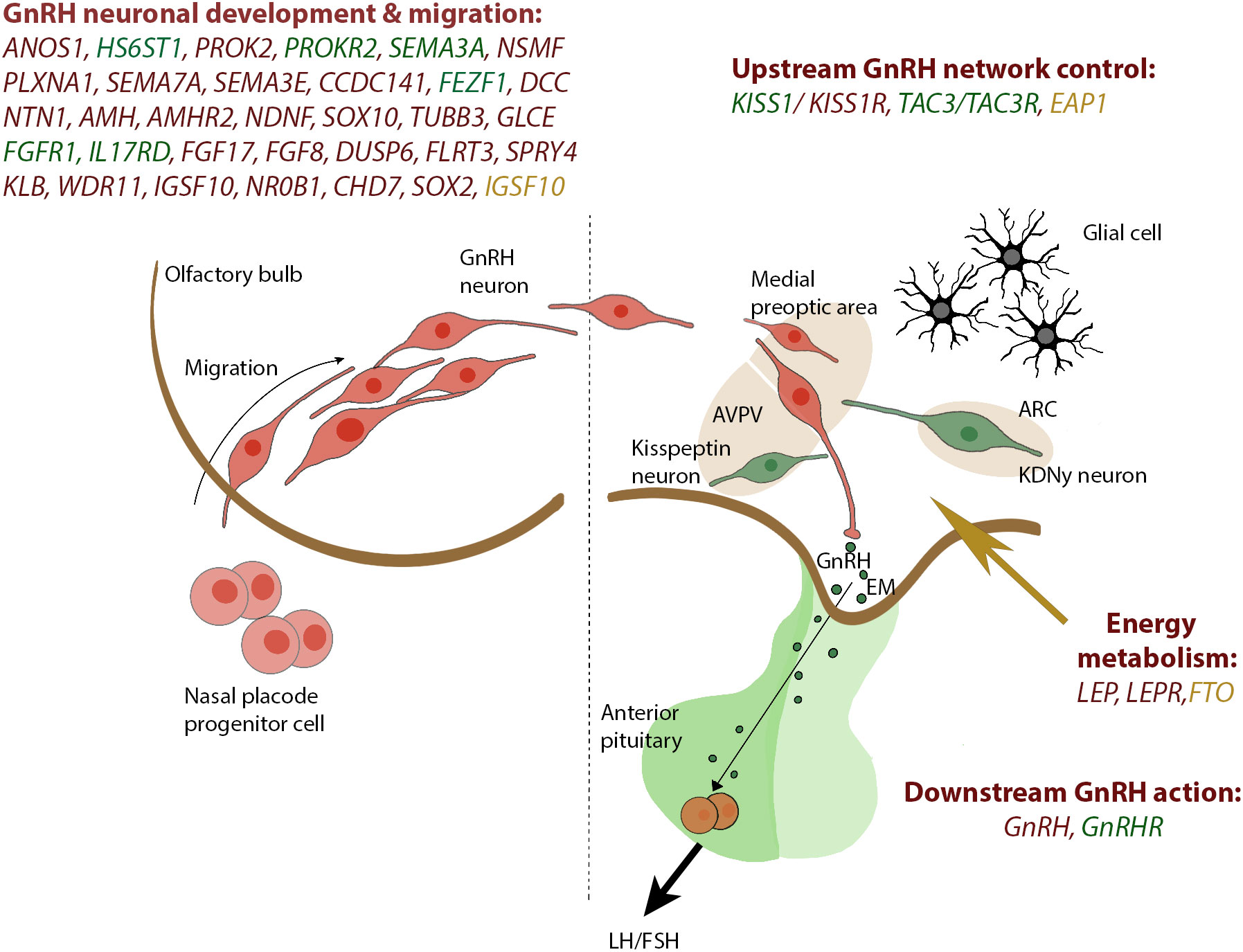
Figure 1 Genes reported in CHH and SLDP are related to GnRH development and GnRH function. The drawing on the left panel shows the migration of the GnRH neurons from the olfactory placode and into the hypothalamus which occurs within the first 10 to 12 weeks of rodent life. In the right panel, a concise view of our current understanding of the KISS1 neuronal networks governing GnRH secretion is depicted. Genes associated with CHH are depicted in red, shared genes between CHH and SLDP are depicted in green whereas genes related to SLDP are marked in orange. GnRH, gonadotropin-releasing hormone; KNDy, Kiss1/NKB/Dyn neuron; ARC, arcuate nucleus; AVPV, anteroventral periventricular nucleus; EM, median eminence.
Autosomal recessive forms
Isolated CHH is transmitted as an autosomal recessive trait due to mutations in three genes: GnRHR, for which over 60 families have been reported (45–49), KISS1R, with 27 identified families (45, 50) and TACR3, found in 20 families (45, 51–54). In these patients, the phenotype is a common nCHH, without any non-reproductive traits of disease (36). It is worth to report that for GnRHR mutations, responsible for about 40-50% of hereditary cases of nCHH (48), there is a broad phenotypic variability even in the same pedigree with the same mutation (55). On the other hand, biallelic mutations in GNRH1, KISS1, and TAC3, ligands of the above-mentioned receptors, are a rare cause of nCHH (36). Mutations in GNRHR represent a paradigm of impaired functionality of GnRH, in fact, GnRHR encodes a receptor coupled to G proteins which determines the release of gonadotropins in the pituitary (41). KISS1R, TACR3 and TAC3 genes, encoding for a G-protein-coupled receptor for Kisspeptin, for neurokinin B and its ligand, are all instead members of a complex network where the KDNY neurons exert regulatory action on GnRH function (37).
X-linked forms
ANOS1 (OMIM 300836), formerly KAL1, is characterized by an X-linked recessive inheritance pattern; intragenic microdeletions or pathological variations of this gene have been described in 10-20% of KS (33). For canonical clinical manifestations, such as CHH and anosmia, penetrance is complete (56–61). On the contrary, other clinical manifestations such as synkinesis (56) and renal agenesis show different expressions in individuals also carrying the same variant (57–68). About 144 families have been reported so far. It is worth mentioning a female phenotype observed in 10 patients, of which one case was linked to a biallelic variant of ANOS1 (69) and the other nine cases were traced to a mutation in another gene, indicationg a possible oligogenic inheritance. ANOS1 encodes Anosmin1, an extracellular protein that mediates cell adhesion and play a crucial role in the migration of GnRH neurons (41).
Autosomal dominant forms
Autosomal dominant (AD) transmission of CHH is seen more often in KS than in nCHH, with the most common genes (causing both nCHH and KS) being FGFR1 and CHD7. FGFR-1 encodes a tyrosine kinase receptor that regulates central developmental processes such as neuronal migration, fate, determination, and cell proliferation (41). FGFR1 plays a fundamental action in the proliferation and migration of GnRH neurons to the hypothalamus as well as directly promoting olfactory bulb development. Up to now, more than 140 mutations in this gene have been described, which generally lead to a loss of function with various mechanisms (splicing, nonsense, missense, frameshift and deletions) (56, 70, 71). FGFR1 mutations related to KS are exemplified by incomplete penetrance (30, 72, 73) and variable clinical expression even in the same family, with patients presenting anosmia, complete phenotype, or isolated pubertal delay (72–75). Additionally, it was reported that mutations in this gene cause also nCHH (73, 76–78). Other clinical traits of FGFR1 mutations, such as cleft lip and cleft palate, skeletal anomalies, and dental agenesis, are reported with variable frequency (30, 56, 66, 72, 79). With the discovery of other gene mutations in patients previously considered to have pure AD CHH due to FGFR1 mutations, it has become increasingly apparent that variable penetrance or oligogenicity are more common than AD inheritance with FGFR1 mutations. CHD7, located in 8q12.1, encodes chromodomain helicase DNA-binding protein 7, which is expressed in different fetal tissues including the developing brain. It is one of two CHH genetic loci shared with CHARGE syndrome, a rare disorder with autosomal dominant inheritances, characterized by congenital heart disease, coloboma, choanal atresia, genital and ear anomalies, and growth and developmental retardation. Its pattern of expression reflects a potential CHD7 contribution in the development of the olfactory bulb and GnRH neurons (41). Whereas CHARGE patients tend to harbor large de novo gene deletions, patients with KS and nCHH (80–82) tend to harbor missense mutations with partial loss of function that may either be de novo or inherited (83). Even in the case of CHD7 mutations there is broad phenotypic variability, going from KS through nCHH to isolated anosmia (82, 83). Other clinical traits associated with CHD7 mutations are deafness, anomalies of the outer ear and lip/cleft palate (83, 84). Indeed, it can sometimes be hard to distinguish between “mild CHARGE” and KS with multiple developmental defects. SOX10 is a transcription factor crucial for the early development of neural crest cells, which are multipotent precursor arising from the neural tube that differentiate into different cell types. SOX10 influence also hearing through its expression in the melanocytic intermediate cells of the cochlear stria vascularis during early development of the inner ear (85). AD SOX10 mutations have been described in nearly 40% of KS patients with deafness (86). PROKR2, a gene encoding a G protein-coupled receptor and its ligand PROK2 deserve to be reported in this section as a paradigm of mixed inheritance. The binding of Prok2 to its receptor activates a signaling cascade with effects on the migration of GnRH neurons (41). Mutation in PROKR2 were reported both in KS and nCHH. In 20% of cases AR inheritance pattern is reported for this gene, whereas the remaining cases are due to AD or oligogenic mechanisms (79, 87–94). PROK2 variants, usually less frequent, can present with AD or AR inheritance patterns. As with FGFR1, some cases of PROK2 and PROKR2 mutations that were originally believed to represent AD inheritance have since turned out to be more probably oligogenic. FGF8 encodes for a ligand of FGF1 receptor (36). Heterozygous mutations of FGF8 were identified both in KS and nCHH, also with oligogenic inheritance. The clinical signs include neurosensorial deafness camptodactyly and cleft lip/palate (3, 70, 95).
Oligogenic inheritance
Recently in several cases of nCHH and KS a mutation in two or more genes, with oligogenic inheritance, has been reported. In 2006 a case of KS harboring mutations of both PROKR2 and KAL1 was reported (79). However, the authors did not fully appreciate the significance of their finding and, consequently, it was not until the following year that a convincing report of two different genetic variants in FGFR1 and NSMF inherited within kindred causing KS only in the single individual carrying both (96). Subsequently, in 2010, Sykiotis (97) described oligogenic inheritance in 2.5% of subjects in a large series of CHH patients using a panel of just 8 genes. Successively an oligogenic mechanism in 7% (78) to 15% (32) of the subjects with CHH was documented by different groups. So far, an oligogenic mechanism of inheritance was documented at least for 16 genes (30). Reasonably, the application of NGS will increase the possibility of finding “oligogenicity” in CHH. However, the huge amount of data generated by NGS is now challenging the clear distinction between “oligogenicity” and the presence of benign variants that do not interfere with the phenotype that is determined by the variant that is principally responsible (36).
CNV contribution to the genetic architecture of CHH
Even if more than 60 candidates have been linked with the pathogenesis of CHH, approximately 50% of cases remain genetically undetermined (37). Part of this missing genetic heritability probably resides in new candidates that require larger cohorts for their discovery or in mutations not simply detectable by whole exome sequencing experiments (WES), such as copy number variants (CNVs) and variants in the non-coding portion of the genome. CNVs are structural variants that result in either gain (duplications) or loss (deletions) of genetic material (more than 50 bp of genomic DNA). Thus far, previous investigation of CNVs with chromosomal microarrays or karyotypes in CHH has led to essential genetic findings (98–100), showing an overall prevalence of ~1% in a subset of CHH-associated genes (101, 102). However, such a low prevalence of CNVs in previous CHH studies has been imputed to the low-resolution tools deployed to call CNVs (101). To date, complex analytic pipelines now available can detect CNVs of a smaller size compared to historic microarrays, allowing precise characterization of these structural variants. In 2022, Balasubramanian and colleagues employed new validated, high-resolution CNV capture technology to examine a large cohort of CHH patients, detecting a total CNV prevalence of 2% across 13/62 known CHH candidates (103). Although supporting the idea that CNVs in known CHH genes should be investigated in patients with CHH, this study indicates that the greater proportion of the missing heritability in CHH may relate to mutations of new coding/non-coding genes. It is hoped that, given the massive application of genome sequencing, these still elusive variants involving non-coding regions will illuminate the full genetic architecture of CHH.
Genetics of SLDP
Pondering the distribution of puberty timing in the normal population, SLDP can be assimilated to the extreme upper limit of normality. A clear diagnosis of this condition is often difficult to achieve, even if SLDP is frequently reported in multiple generations of the same family. Most commonly, the trait is inherited in an autosomal dominant pattern, often with complete penetrance, but autosomal recessive, X-linked and bilinear pedigrees have also been reported (104, 105). Epidemiological studies of twins in both sexes have shown that the time of puberty is a highly heritable trait and that genetics play a key role in determining when healthy individuals enter puberty (106, 107). It is clear from genome wide association studies (GWAS) that many different genetic signals are crucial in the discrepancy of pubertal timing observed in the normal population (108). In contrast, one or a small number of genetic variations in each family are generally described in delayed familial puberty, with a corresponding pattern of autosomal dominant inheritance. The recent application of NGS technology to self-limited DP have unraveled fascinating new mechanisms in the genetic control of puberty (Figure 1).
HS6ST1
Targeted and whole exome sequencing methods identified a mutation in HS6ST1 in a broad pedigree among a large cohort of patients with isolated familial delayed puberty, without associated CHH in their relatives (84). All the family members who carried the mutation exhibited a canonical SLDP phenotype, rather than CHH; the proband entering puberty spontaneously at 14.3 years. In parallel, a mouse heterozygous knockout model found the loss of an Hs6st1 allele to cause pubertal delay in females without impairment of adult reproductive capacity. Hs6st1 +/− mice showed no impairment of fertility, development of GnRH neurons, testes, or spermatogenesis. However, a substantial delay in the timing of vaginal opening (used to determine the onset of puberty in female rodents) was observed in females. Remarkably, the Hs6st1 +/- mice had normal olfactory bulbs without any reduction in the global number of GnRH neurons in the hypothalamus or projecting to the median eminence. Consequently, the pubertal delay observed in mice could be due to variation in GnRH neuron activity or other crucial downstream pathways, controlling the expression of Hs6st1 in both the arcuate and paraventricular nucleus (109, 110). In recent work involving 338 patients with GnRH deficiency, including 105 subjects with a positive family history, a variant in HS6ST1 gene was identified in almost 2% of patients with CHH (111). In this study, the inheritance model was complex, bypassing simple Mendelian transmission, and with substantial clinical heterogeneity suggesting a role for epigenetic mechanisms or mutations in other candidates to fully explain the observed phenotypes. In order to properly assign a role to HS6ST1 in SLDP, it is crucial to take in account the biological processes in which it takes part. Howard et al., 2018, found that reduced Hs6st1 expression and a consequent reduction of sulfotransferase activity in kisspeptin and other neurons can impact on their ability to regulate GnRH function and secretion (112). In addition, Hs6st1 activity is a prerequisite for the correct function of Anos1 and Fgfr1 (111). In summary, a functionally minor heterozygous mutation might cause SLDP, whereas a more severe mutation or the simultaneous effect of a second gene (i.e., oligogenicity) could lead to a more critical phenotype such as CHH or KS.
IGFS10
Targeted and whole exome sequencing methods identified deleterious mutations also in IGSF10 gene. Two N-terminal variants in IGSF10 were reported in 20 subjects with SLDP from six families, with an AD inheritance (8). Moreover, two C-terminal variants in the same gene were identified in 4 families of the same study. In one family, there was incomplete penetrance, whereas a de novo mutation was proposed for another family. All patients had a standard growth rate before puberty and a classic DP with a delayed pubertal spurt and normal (self-reported) sense of smell. IGFS10 gene had hitherto never been associated with any human pathology. During embryonic development mutations in IGFS10 impact the migration of GnRH neurons from the vomeronasal organ to the forebrain. Patients with mutations in this gene are characterized by isolated pubertal delay without retardation in growth (a pattern that is also observed in CHH). Abolition of GnRH neuronal migration due to anomalous IGSF10 signaling might determine reduced or deferred migration of GnRH neurons to the hypothalamus. This result into a functional defect in the GnRH network and a higher threshold for pubertal onset. In addition, IGSF10 loss-of-function mutations were found in subjects with hypothalamic amenorrhea-like phenotype, suggesting a common genetic origin of functional central hypogonadism with both CHH (113) and DP. Intriguingly, mutations in IGSF10 were recently reported in a pedigree with a Kallmann- like phenotype and in patients with both disorders of neuronal development and premature ovarian insufficiency (114). Studies on the role of the HS6ST1 and IGSF10 genes in PD hypothesize that developmental defects in the GnRH system during fetal life may regulate the timing of onset of puberty in adolescence, without determining other associated phenotypic characteristics. Whether these patients will have any shortcomings in their long-term reproductive capacity or sex life span remains to be evaluated.
FTO
FTO is considered the most impactful locus on BMI and the obesity risk (115). Recently, using NGS techniques, rare heterozygous variants in FTO gene associated with BMI and growth retardation in early childhood have been described in SLDP families (116). Moreover, mice lacking FTO had a significantly delayed onset of puberty (timing of vaginal opening) (116).
EAP1
EAP1 (Enhanced at puberty 1) encodes a nuclear transcription factor that trans-activates GnRH promoter, facilitating GnRH secretion, and parallelly inhibits the preproenkephalin promoter, which in turn represses GnRH secretion. Howard et al. first described human EAP1 mutations that appear to cause SLDP in 2 families (117). Affected subjects from these two families had canonical clinical and biochemical signs of SLDP, with delayed onset of Tanner stage 2 and delayed peak height velocity. Both subjects showed spontaneous pubertal development at age 18 without testosterone priming, thus excluding CHH. Two highly conserved variants, one rare missense variant in EAP1 and one in-frame deletion, were identified in subjects with familial delayed puberty. Compared to wild-type EAP1, mutants showed a biased ability to transactivate the GnRH promoter, imputable to the diminished protein levels caused by the in-frame deletion and the altered subcellular localization triggered by the missense mutation. The same work showed that in monkey hypothalamus Eap1 binding to the GnRH1 promoter rise at the onset of puberty. These recent findings suggest that genes that determine SLDP may play a role in the redundant mechanisms that regulate the onset of puberty (e.g., number of cells migrating from the olfactory placode to the hypothalamus and pre-optic areas or modulation of GnRH function and secretion), despite genes linked to CHH that directly control the migration or function of GnRH neurons. However, further studies are needed to uncover the strongness of this intuition.
LGR4
LGR4 encodes a receptor for R-spondins which, once activated, potentiates the canonical Wnt signaling pathway. Through GWAS analysis LGR4 had already been designed as a regulator of pubertal timing both in males (based on recalled age at voice breaking) and in females (based on the age of menarche (118, 119). However, mutations in LGR4 were not previously associated with actual human disease. A recent study from Dunkel’s group utilizing whole-exome sequencing of 160 individuals of 67 families in a well-characterized DP cohort identified 3 rare missense variants in LGR4 (120). All segregated with the DP trait with an AD pattern of transmission. Specific expression of Lgr4 at the site of GnRH neuron development has been reported. LGR4 mutants showed biased Wnt/β-catenin signaling, leading to consequences on protein expression, trafficking, and degradation. Lgr4-deficient mice showed a significantly delayed onset of puberty and lowered number of GnRH neurons compared to WT mice. In addition, we were demonstrating that lgr4 knockdown in zebrafish embryos impact development and migration of GnRH neurons. In addition, genetic lineage tracing displayed robust Lgr4-mediated Wnt/β-catenin signaling pathway stimulation during GnRH neuron development.
Shared genes between CHH and SLDP
It is well established the timing of puberty in normal populations has a strong genetic component (107, 119, 121, 122) even though general health, nutritional status and endocrine chemical disruptor can influence the expression of key regulators. To date, the knowledge about the genetic mechanisms that control HPG axis comes largely from reports on patients with GnRH deficiency, leading to the identification of rare variants underlying CHH. Zhu and collaborators recently addressed the hypothesis of a shared genetic basis between CHH and SLDP, performing WES analysis in 15 families with a CHH proband carrying a putative pathogenic variant in CHH genes and family members both with delayed and with normal puberty (9). A genetic origin was identified in half of relatives with delayed puberty and in in a small percentage (12%) of relatives with normal puberty. Moreover, they analyzed nearly fifty DP subjects without family history of CHH matched with controls from ExAC and identified mutations in CHH genes in 14.3% of DP subjects and in 5.6% of controls. The heterozygous allelic variants were in TAC3, TACR3, GnRHR, IL17RD and SEMA3A. Of note, control subjects also carried potentially pathogenic variants. Moerover, Cassatella et al., 2018 determined that the genetic architecture of SLDP is closer to that of normal controls than CHH probands. Exome sequencing showed potentially pathogenic variant in CHH genes (twenty-five genes with IGSF10) in 51% of CHH patients, in 7% of SLDP probands and in 18% of healthy subjects. Oligogenic inheritance was found in 15% of CHH patients and in only 1.4% of SLDP subjects and 2% of controls. To note, potentially pathogenic variants in SLDP patients were found in AXL, FGFR1, HST6ST1, PROKR2, FEZF1 and TAC3 genes.
Genetic evaluation supports differential diagnosis in patients with SLDP and CHH
As DP is a common condition in unaffected individuals, identifying a genetic cause in SLDP presents several pitfalls. Thus, a genetic variant likely to lead to this condition could have a quite high prevalence in the normal population. Furthermore, both the manifestation of pubertal delay in 10% of relatives of patients with CHH (9) and the likelihood of a spontaneous reversal in 10% of patients with CHH (123) remains consistent with a shared molecular basis of CHH and SLDP. Nevertheless, it remains true that genetic variants reported are distinct between the two diseases (32). This supports the hypothesis that NGS of a large panel of candidate genes might one day assist physicians to distinguish those adolescents with severe CHH from those with SLDP, enabling timely and correct treatment to CHH patients. Howard and collaborators studied the burden of genetic variants in an ethnically mixed cohort of adolescent patients with DP, with the purpose of validate the genetic analysis of known causative genes to confirm the diagnosis of CHH or SLDP. The results of this work show a fair correlation between patient genotype and clinical diagnosis, with a specificity of 100% and PPV of genetic tests for the diagnosis of patients with CHH. The authors also reported that subjects carrying homozygous or loss-of-function mutatons in CHH genes would probably have a final diagnosis of CHH. In contrast, patients presenting mutations reported only in SLDP with heterozygous inheritance were thought to be likely to have a final diagnosis of SLDP. These results confirm that WES testing can make a clear diagnosis of CHH for 17.4% of patients presenting with pubertal delay. This finding supports the implementation of genetic analysis in the clinical practice, in combination with clinical and biochemical observations, to validate the diagnosis of CHH in adolescents presenting with DP. It is worth mentioning that the same work identified in three patients with a clinical diagnosis of SLDP also pathogenic variants in other genes previously linked to CHH, namely DMXL2, OTUD4 and SEMA3.
Conclusions
Pubertal delay can be the presentation of a wide spectrum of clinical phenotypes ranging from CHH, which is a pathological condition and needs appropriate medical therapy, to SLDP, a possible para-physiological and benign condition mostly compatible with normal reproductive capacity post- puberty, to non-gonadal illness or energy deficit. Distinguishing between SLDP and CHH remains challenging and, thus far there is no clear and univocal evidence to assist clinicians in their differential diagnosis and management of SLDP and CHH. The availability of an accurate genetic test in clinical practice to discriminate between the two conditions has the potential for significant cost savings (preventing unnecessary investigations) and improvement of health and fertility outcomes for CHH patients.
Recently, the application of NGS technologies has shed light on the complex genetic mechanisms at the basis of CHH or SLDP, identifying new candidate genes and suggesting a different genetic backbone for these two conditions. This difference constitutes the main assumption on which to model a genetic tool that allows an early differential diagnosis between the two conditions. However, it should consider that so far only a few causal genes have been described in SLDP, leading to a lower pick-up rate for SLDP pathogenic variants in a putative diagnostic panel when compared with CHH mutations. Furthermore, variants contextualization in patients with oligogenic inheritance is difficult due to our lack of knowledge of variant–variant crosstalk. For these reasons, we suggest that so far only an integrated approach can increase the sensitivity and specificity in CHH diagnosis. Thus, we invite the readers to combine this type of genetic analysis with biochemical profiling (e.g., basal LH, FSH, inhibin B, AMH) and an accurate physical and anamnestic data collection to maximize the diagnostic accuracy. Nevertheless, in the future, as the knowledge of the genetic architecture of delayed puberty will be dramatically improved, genetic testing might offer a quick and precocious analytical tool also in clinical routine.
Author contributions
VV, FH, and MB wrote the draft. MB, GG, SF, BC, RQ and LP revised the manuscript. VV and MB finalized the manuscript. All authors contributed to the article and approved the submitted version.
Funding
Research funded by the Italian Ministry of Health (GR-2016-02362389) and partially supported by ‘Ricerca Corrente’ (O5C202_2012 -ICH-NGS).
Acknowledgments
The authors would like to acknowledge Amina Aleta for her contribution to Figure 1.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Persani L, Bonomi M, Cools M, Dattani M, Dunkel L, Gravholt CH, et al. ENDO-ERN expert opinion on the differential diagnosis of pubertal delay. Endocrine (2021) 71. doi: 10.1007/s12020-021-02626-z
2. Sedlmeyer I, Palmert M. Delayed puberty: Analysis of a Large case series from an academic center. J Of Clin Endocrinol Metab (2002) 87:1613–20. doi: 10.1210/jcem.87.4.8395
3. Varimo T, Miettinen P, K"ans"akoski J, Raivio T, Hero M. Congenital hypogonadotropic hypogonadism, functional hypogonadotropism or constitutional delay of growth and puberty? Anal large patient Ser single Palmert M Dunkel L (2016) 366:443–53. doi: 10.1056/nejmcp1109290
4. Giovanelli L, Quinton R. Letter to the Editor from giovanelli and Quinton: “Distinguishing self-limited delayed puberty from permanent hypogonadotropic hypogonadism: How and why?” J Clin Endocrinol Metab (2022) 107:5. doi: 10.1210/clinem/dgab925
5. Dwyer AA, Tiemensma J, Quinton R, Pitteloud N, Morin D. Adherence to treatment in men with hypogonadotrophic hypogonadism. Clin Endocrinol (Oxf) (2017) 86:377–83. doi: 10.1111/cen.13236
6. Dzemaili S, Tiemensma J, Quinton R, Pitteloud N, Morin D, Dwyer AA. Beyond hormone replacement: quality of life in women with congenital hypogonadotropic hypogonadism. Endocr Connect (2017) 6:404–12. doi: 10.1530/EC-17-0095
7. Palmert M, Dunkel L. Delayed puberty. New Engl J Of Med (2012) 366:443–53. doi: 10.1056/nejmcp1109290
8. Howard SR, Guasti L, Ruiz-Babot G, Mancini A, David A, Storr HL, et al. IGSF 10 mutations dysregulate gonadotropin-releasing hormone neuronal migration resulting in delayed puberty. EMBO Mol Med (2016) 8. doi: 10.15252/emmm.201606250
9. Zhu J, Choa REY, Guo MH, Plummer L, Buck C, Palmert MR, et al. A shared genetic basis for self-limited delayed puberty and idiopathic hypogonadotropic hypogonadism. J Clin Endocrinol Metab (2015) 100. doi: 10.1210/jc.2015-1080
10. Bonomi M, Vezzoli V, Krausz C, Guizzardi F, Vezzani S, Simoni M, et al. Characteristics of a nationwide cohort of patients presenting with isolated hypogonadotropic hypogonadism (IHH). Eur J Endocrinol (2018) 178. doi: 10.1530/EJE-17-0065
11. Bergadá I, Milani C, Bedecarrás P, Andreone L, Ropelato MG, Gottlieb S, et al. Time course of the serum gonadotropin surge, inhibins, and anti-müllerian hormone in normal newborn males during the first month of life. J Clin Endocrinol Metab (2006) 91. doi: 10.1210/jc.2006-1079
12. Lewkowitz-Shpuntoff HM, Hughes VA, Plummer L, Au MG, Doty RL, Seminara SB, et al. Olfactory phenotypic spectrum in idiopathic hypogonadotropic hypogonadism: Pathophysiological and genetic implications. J Clin Endocrinol Metab (2012) 97. doi: 10.1210/jc.2011-2041
13. Iolascon G, Frizzi L, Bianco M, Gimigliano F, Palumbo V, Sinisi A, et al. Bone involvement in males with kallmann disease. Aging Clin Exp Res (2015) 27:31–6. doi: 10.1007/s40520-015-0421-5
14. Young J, Xu C, Papadakis GE, Acierno JS, Maione L, Hietamäki J, et al. Clinical management of congenital hypogonadotropic hypogonadism. Endocr Rev (2019) 40. doi: 10.1210/er.2018-00116
15. Wu F, Butler G, Kelnar C, Stirling H, Huhtaniemi I. Patterns of pulsatile luteinizing hormone and follicle-stimulating hormone secretion in prepubertal (Midchildhood) boys and girls and patients with idiopathic hypogonadotropic hypogonadism (Kallmann’s syndrome): A study using an ultrasensitive time-resolved immunofluorometric assay. J Of Clin Endocrinol Metab (1991) 72:1229–37. doi: 10.1210/jcem-72-6-1229
16. Abitbol L, Zborovski S, Palmert M. Evaluation of delayed puberty: what diagnostic tests should be performed in the seemingly otherwise well adolescent? Arch Dis Child (2016) 101:767–71. doi: 10.1136/archdischild-2015-310375
17. Resende E, Lara B, Reis J, Ferreira B, Pereira G, Borges M. Assessment of basal and gonadotropin-releasing hormone-stimulated gonadotropins by immunochemiluminometric and immunofluorometric assays in normal children. J Of Clin Endocrinol Metab (2007) 92:1424–9. doi: 10.1210/jc.2006-1569
18. Binder G, Schweizer R, Blumenstock G, Braun R. Inhibin b plus LHvsGnRH agonist test for distinguishing constitutional delay of growth and puberty from isolated hypogonadotropic hypogonadism in boys. Clin Endocrinol (Oxf) (2014) 82:100–5. doi: 10.1111/cen.12613
19. Adulwahid NA, Armar NA, Morris DV, Adams J, Jacobs HS. Diagnostic tests with luteinising hormone releasing hormone should be abandoned. In: British Medical journal (Clinical research ed, vol. 291(6507. . p. 1471–2.
20. Albrethsen J, Ljubicic M, Juul A. Longitudinal increases in serum insulin-like factor 3 and testosterone determined by LC-MS/MS in pubertal Danish boys. J Of Clin Endocrinol Metab (2020) 105:3173–8. doi: 10.1210/clinem/dgaa496
21. Trabado S, Maione L, Bry-Gauillard H, Affres H, Salenave S, Sarfati J, et al. Insulin-like peptide 3 (INSL3) in men with congenital hypogonadotropic Hypogonadism/Kallmann syndrome and effects of different modalities of hormonal treatment: A single-center study of 281 patients. J Clin Endocrinol Metab (2014) 99:E268–75. doi: 10.1210/jc.2013-2288
22. Segal T, Mehta A, Anazodo A, Hindmarsh P, Dattani M. Role of gonadotropin-releasing hormone and human chorionic gonadotropin stimulation tests in differentiating patients with hypogonadotropic hypogonadism from those with constitutional delay of growth and puberty. J Of Clin Endocrinol Metab (2009) 94:780–5. doi: 10.1210/jc.2008-0302
23. Chan Y, Lippincott M, Kusa T, Seminara S. Divergent responses to kisspeptin in children with delayed puberty. JCI Insight (2018) 3:8. doi: 10.1172/jci.insight.99109
24. Sukumar SP, Bhansali A, Sachdeva N, Ahuja CK, Gorsi U, Jarial KDS, et al. Diagnostic utility of testosterone priming prior to dynamic tests to differentiate constitutional delay in puberty from isolated hypogonadotropic hypogonadism. Clin Endocrinol (Oxf) (2017) 86. doi: 10.1111/cen.13321
25. Galazzi E, Improda N, Cerbone M, Soranna D, Moro M, Fatti LM, et al. Clinical benefits of sex steroids given as a priming prior to GH provocative test or as a growth-promoting therapy in peripubertal growth delays: Results of a retrospective study among ENDO-ERN centres. Clin Endocrinol (Oxf) (2021) 94:219–28. doi: 10.1111/cen.14337
26. Rohayem J, Hauffa B, Zacharin M, Kliesch S, Zitzmann M. Testicular growth and spermatogenesis: new goals for pubertal hormone replacement in boys with hypogonadotropic hypogonadism? -a multicentre prospective study of hCG/rFSH treatment outcomes during adolescence-. Clin Endocrinol (Oxf) (2016) 86:75–87. doi: 10.1111/cen.13164
27. Federici S, Goggi G, Quinton R, Giovanelli L, Persani L, Cangiano B, et al. New and consolidated therapeutic options for pubertal induction in hypogonadism: In-depth review of the literature. Endocr Rev (2021) 43:824–51. doi: 10.1210/endrev/bnab043
28. Nordenström A, Ahmed SF, van den Akker E, Blair J, Bonomi M, Brachet C, et al. Pubertal induction and transition to adult sex hormone replacement in patients with congenital pituitary or gonadal reproductive hormone deficiency: an endo-ERN clinical practice guideline. Eur J Endocrinol (2022) 186:G9–G49. doi: 10.1530/EJE-22-0073
29. Howard S, Dunkel L. Sex steroid and gonadotropin treatment in Male delayed puberty. Puberty From Bench To Clinic (2015), 185–97. doi: 10.1159/000438891
30. Boehm U, Bouloux P-M, Dattani MT, de Roux N, Dodé C, Dunkel L, et al. European Consensus statement on congenital hypogonadotropic hypogonadism–pathogenesis, diagnosis and treatment. Nat Rev Endocrinol (2015) 11:547–64. doi: 10.1038/nrendo.2015.112
31. Pazderska A, Mamoojee Y, Artham S, Miller M, Ball SG, Cheetham T, et al. Safety and tolerability of one-year intramuscular testosterone regime to induce puberty in older men with CHH. Endocr Connect (2018) 7:133–8. doi: 10.1530/EC-17-0241
32. Cassatella D, Howard SR, Acierno JS, Xu C, Papadakis GE, Santoni FA, et al. Congenital hypogonadotropic hypogonadism and constitutional delay of growth and puberty have distinct genetic architectures. Eur J Endocrinol (2018) 178:377–88. doi: 10.1530/EJE-17-0568
33. Topaloğlu A. Update on the genetics of idiopathic hypogonadotropic hypogonadism. J Clin Res Pediatr Endocrinol (2018), 113–22. doi: 10.4274/jcrpe.2017.s010
34. Cangiano B, Swee D, Quinton R, Bonomi M. Genetics of congenital hypogonadotropic hypogonadism: peculiarities and phenotype of an oligogenic disease. Hum Genet (2020) 140:77–111. doi: 10.1007/s00439-020-02147-1
35. Seminara S, Hayes F, Crowley W. Gonadotropin-releasing hormone deficiency in the human (Idiopathic hypogonadotropic hypogonadism and kallmann’s syndrome): Pathophysiological and genetic considerations. Endocr Rev (1998) 19:521–39. doi: 10.1210/edrv.19.5.0344
36. Maione L, Dwyer A, Francou B, Guiochon-Mantel A, Binart N, Bouligand J, et al. GENETICS IN ENDOCRINOLOGY: Genetic counseling for congenital hypogonadotropic hypogonadism and kallmann syndrome: new challenges in the era of oligogenism and next-generation sequencing. Eur J Endocrinol (2018) 178:R55–80. doi: 10.1530/eje-17-0749
37. Stamou M, Cox K, Crowley W. Discovering genes essential to the hypothalamic regulation of human reproduction using a human disease model: Adjusting to life in the “-omics” era. Endocr Rev (2015) 2016:4–22. doi: 10.1210/er.2015-1045.2016.1
38. Sykiotis G, Pitteloud N, Seminara S, Kaiser U, Crowley W. Deciphering genetic disease in the genomic era: The model of GnRH deficiency. Sci Transl Med (2010) 2:32. doi: 10.1126/scitranslmed.3000288
39. Schwanzel-Fukuda M, Bick D, Pfaff DW. Luteinizing hormone-releasing hormone (LHRH)-expressing cells do not migrate normally in an inherited hypogonadal (Kallmann) syndrome. Mol Brain Res (1989) 6. doi: 10.1016/0169-328X(89)90076-4
40. Forni PE, Taylor-Burds C, Melvin VS, Williams T, Wray S. Neural crest and ectodermal cells intermix in the nasal placode to give rise to GnRH-1 neurons, sensory neurons, and olfactory ensheathing cells. J Neurosci (2011) 31. doi: 10.1523/JNEUROSCI.6087-10.2011
41. Bianco S, Kaiser U. The genetic and molecular basis of idiopathic hypogonadotropic hypogonadism. Nat Rev Endocrinol (2009) 5:569–76. doi: 10.1038/nrendo.2009.177
42. Linscott ML, Chung WCJ. TET1 regulates fibroblast growth factor 8 transcription in gonadotropin releasing hormone neurons. PloS One (2019) 14. doi: 10.1371/journal.pone.0220530
43. Linscott ML, Chung WCJ. Fibroblast growth factor 8 expression in GT1-7 GnRH-secreting neurons is androgen-independent, but can be upregulated by the inhibition of DNA methyltransferases. Front Cell Dev Biol (2016) 4:34. doi: 10.3389/fcell.2016.00034
44. Hutchins BI, Kotan LD, Taylor-Burds C, Ozkan Y, Cheng PJ, Gurbuz F, et al. CCDC141 mutation identified in anosmic hypogonadotropic hypogonadism (Kallmann syndrome) alters GnRH neuronal migration. Endocrinology (2016) 157:1956–66. doi: 10.1210/en.2015-1846
45. Francou B, Paul C, Amazit L, Cartes A, Bouvattier C, Albarel F, et al. Prevalence of KISS1 receptor mutations in a series of 603 patients with normosmic congenital hypogonadotrophic hypogonadism and characterization of novel mutations: a single-centre study. Hum Reprod (2016) 31:1363–74. doi: 10.1093/humrep/dew073
46. Tello J, Newton C, Bouligand J, Guiochon-Mantel A, Millar R, Young J. Congenital hypogonadotropic hypogonadism due to GNRH receptor mutations in three brothers reveal sites affecting conformation and coupling. PloS One (2012) 7:6. doi: 10.1371/journal.pone.0038456
47. de Roux N, Young J, Misrahi M, Genet R, Chanson P, Schaison G, et al. A family with hypogonadotropic hypogonadism and mutations in the gonadotropin-releasing hormone receptor. New Engl J Of Med (1997) 337:1597–603. doi: 10.1056/nejm199711273372205
48. Beranova M, Oliveira LMB, Bédécarrats GY, Schipani E, Vallejo M, Ammini AC, et al. Prevalence, phenotypic spectrum, and modes of inheritance of gonadotropin-releasing hormone receptor mutations in idiopathic hypogonadotropic hypogonadism 1. J Clin Endocrinol Metab (2001) 86:1580–8. doi: 10.1210/jcem.86.4.7395
49. de Roux N. GnRH receptor and GPR54 inactivation in isolated gonadotropic deficiency. Best Pract Res Clin Endocrinol Metab (2006) 20:515–28. doi: 10.1016/j.beem.2006.10.005
50. Seminara SB, Messager S, Chatzidaki EE, Thresher RR, Acierno JS, Shagoury JK, et al. The GPR54 gene as a regulator of puberty. New Engl J Med (2003) 349:1614–27. doi: 10.1056/NEJMoa035322
51. Topaloglu AK, Reimann F, Guclu M, Yalin AS, Kotan LD, Porter KM, et al. TAC3 and TACR3 mutations in familial hypogonadotropic hypogonadism reveal a key role for neurokinin b in the central control of reproduction. Nat Genet (2009) 41:354–8. doi: 10.1038/ng.306
52. Guran T, Tolhurst G, Bereket A, Rocha N, Porter K, Turan S. Others. (2009). hypogonadotropic hypogonadism due to a novel missense mutation in the first extracellular loop of the neurokinin b receptor. J Of Clin Endocrinol Metab 94:3633–9. doi: 10.1210/jc.2009-0551
53. Young J, Bouligand J, Francou B, Raffin-Sanson M-L, Gaillez S, Jeanpierre M, et al. TAC3 and TACR3 defects cause hypothalamic congenital hypogonadotropic hypogonadism in humans. J Clin Endocrinol Metab (2010) 95:2287–95. doi: 10.1210/jc.2009-2600
54. Gianetti E, Tusset C, Noel SD, Au MG, Dwyer AA, Hughes VA, et al. TAC3/TACR3 mutations reveal preferential activation of gonadotropin-releasing hormone release by neurokinin b in neonatal life followed by reversal in adulthood. J Clin Endocrinol Metab (2010) 95:2857–67. doi: 10.1210/jc.2009-2320
55. Vaaralahti K, Wehkalampi K, Tommiska J, Laitinen E, Dunkel L, Raivio T. The role of gene defects underlying isolated hypogonadotropic hypogonadism in patients with constitutional delay of growth and puberty. Fertil Steril (2011) 95:2756–8. doi: 10.1016/j.fertnstert.2010.12.059
56. Tsai P, Gill J. Mechanisms of disease: insights into X-linked and autosomal-dominant kallmann syndrome. Nat Clin Pract Endocrinol Metab (2006) 2:160–71. doi: 10.1038/ncpendmet0119
57. Dodé C, Hardelin J. Clinical genetics of kallmann syndrome. Ann Endocrinol (Paris) (2010) 71:149–57. doi: 10.1016/j.ando.2010.02.005
58. de Castro F, Seal R, Maggi R. ANOS1: a unified nomenclature for kallmann syndrome 1 gene (KAL1) and anosmin-1. Brief Funct Genomics (2016) 16:205–10. doi: 10.1093/bfgp/elw037
59. Gonçalves C, Fonseca F, Borges T, Cunha F, Lemos M. Expanding the genetic spectrum ofANOS1mutations in patients with congenital hypogonadotropic hypogonadism. Hum Reprod (2017).
60. Nie M, Xu H, Chen R, Mao J, Wang X, Xiong S, et al. Analysis of genetic and clinical characteristics of a Chinese kallmann syndrome cohort with ANOS1 mutations. Eur J Endocrinol (2017) 177:389–98. doi: 10.1530/EJE-17-0335
61. Quinton R, Duke VM, Robertson A, Kirk JMW, Matfin G, de Zoysa PA, et al. Idiopathic gonadotrophin deficiency: genetic questions addressed through phenotypic characterization*. Clin Endocrinol (Oxf) (2001) 55:163–74. doi: 10.1046/j.1365-2265.2001.01277.x
62. Legouis R, Hardelin J-P, Levilliers J, Claverie J-M, Compain S, Wunderle V, et al. The candidate gene for the X-linked kallmann syndrome encodes a protein related to adhesion molecules. Cell (1991) 67:423–35. doi: 10.1016/0092-8674(91)90193-3
63. Franco B, Guioli S, Pragliola A, Incerti B, Bardoni B, Tonlorenzi R, et al. A gene deleted in kallmann’s syndrome shares homology with neural cell adhesion and axonal path-finding molecules. Nature (1991) 353:529–36. doi: 10.1038/353529a0
64. Bick D, Franco B, Sherins RJ, Heye B, Pike L, Crawford J, et al. Intragenic deletion of the KALIG-1 gene in kallmann’s syndrome. New Engl J Med (1992) 326:1752–5. doi: 10.1056/NEJM199206253262606
65. Hardelin JP, Levilliers J, Young J, Pholsena M, Legouis R, Kirk J, et al. Xp22.3 deletions in isolated familial kallmann’s syndrome. J Clin Endocrinol Metab (1993) 76:827–31. doi: 10.1210/jcem.76.4.8473391
66. Costa-Barbosa FA, Balasubramanian R, Keefe KW, Shaw ND, Al-Tassan N, Plummer L, et al. Prioritizing genetic testing in patients with kallmann syndrome using clinical phenotypes. J Clin Endocrinol Metab (2013) 98:E943–53. doi: 10.1210/jc.2012-4116
67. Xu H, Li Z, Wang T, Wang S, Liu J, Wang DW. Novel homozygous deletion of segmental KAL1 and entire STS cause kallmann syndrome and X-linked ichthyosis in a Chinese family. Andrologia (2015) 47:1160–5. doi: 10.1111/and.12397
68. Rathi S, Madhu S, Kant S, Holla V, Arora R. Unusual presentation of kallmannn syndrome with contiguous gene deletion in three siblings of a family. Indian J Endocrinol Metab (2012) 16:326. doi: 10.4103/2230-8210.104077
69. Shaw ND, Seminara SB, Welt CK, Au MG, Plummer L, Hughes VA, et al. Expanding the phenotype and genotype of female GnRH deficiency. J Clin Endocrinol Metab (2011) 96:E566–76. doi: 10.1210/jc.2010-2292
70. Trarbach EB, Abreu AP, Silveira LFG, Garmes HM, Baptista MTM, Teles MG, et al. Nonsense mutations in FGF8 gene causing different degrees of human gonadotropin-releasing deficiency. J Clin Endocrinol Metab (2010) 95:3491–6. doi: 10.1210/jc.2010-0176
71. Pedersen-White J, Chorich L, Bick D, Sherins R, Layman L. The prevalence of intragenic deletions in patients with idiopathic hypogonadotropic hypogonadism and kallmann syndrome. Mol Hum Reprod (2008) 14:367–70. doi: 10.1093/molehr/gan027
72. Dodé C, Levilliers J, Dupont J-M, de Paepe A, le Dû N, Soussi-Yanicostas N, et al. Loss-of-function mutations in FGFR1 cause autosomal dominant kallmann syndrome. Nat Genet (2003) 33:463–5. doi: 10.1038/ng1122
73. Pitteloud N, Acierno JS, Meysing A, Eliseenkova AV, Ma J, Ibrahimi OA, et al. Mutations in fibroblast growth factor receptor 1 cause both kallmann syndrome and normosmic idiopathic hypogonadotropic hypogonadism. Proc Natl Acad Sci (2006) 103:6281–6. doi: 10.1073/pnas.0600962103
74. Salenave S, Chanson P, Bry H, Pugeat M, Cabrol S, Carel JC, et al. Kallmann’s syndrome: A comparison of the reproductive phenotypes in men carrying KAL1 and FGFR1/KAL2 mutations. J Clin Endocrinol Metab (2008) 93:758–63. doi: 10.1210/jc.2007-1168
75. Albuisson J, Pêcheux C, Carel J-C, Lacombe D, Leheup B, Lapuzina P, et al. Kallmann syndrome: 14 novel mutations in KAL1 and FGFR1 ( KAL2 ). Hum Mutat (2005) 25:98–9. doi: 10.1002/humu.9298
76. Raivio T, Sidis Y, Plummer L, Chen H, Ma J, Mukherjee A, et al. Impaired fibroblast growth factor receptor 1 signaling as a cause of normosmic idiopathic hypogonadotropic hypogonadism. J Clin Endocrinol Metab (2009) 94:4380–90. doi: 10.1210/jc.2009-0179
77. Trarbach EB, Teles MG, Costa EMF, Abreu AP, Garmes HM, Guerra G, et al. Screening of autosomal gene deletions in patients with hypogonadotropic hypogonadism using multiplex ligation-dependent probe amplification: detection of a hemizygosis for the fibroblast growth factor receptor 1. Clin Endocrinol (Oxf) (2010) 72:371–6. doi: 10.1111/j.1365-2265.2009.03642.x
78. Miraoui H, Dwyer AA, Sykiotis GP, Plummer L, Chung W, Feng B, et al. Mutations in FGF17, IL17RD, DUSP6, SPRY4, and FLRT3 are identified in individuals with congenital hypogonadotropic hypogonadism. Am J Hum Genet (2013) 92:725–43. doi: 10.1016/j.ajhg.2013.04.008
79. Dodé C, Teixeira L, Levilliers J, Fouveaut C, Bouchard P, Kottler M-L, et al. Kallmann syndrome: Mutations in the genes encoding prokineticin-2 and prokineticin receptor-2. PloS Genet (2006) 2:e175. doi: 10.1371/journal.pgen.0020175
80. Kim H, Layman L. The role of CHD7 and the newly identified WDR11 gene in patients with idiopathic hypogonadotropic hypogonadism and kallmann syndrome. Mol Cell Endocrinol (2011) 346:74–83. doi: 10.1016/j.mce.2011.07.013
81. Kim H-G, Kurth I, Lan F, Meliciani I, Wenzel W, Eom SH, et al. Mutations in CHD7, encoding a chromatin-remodeling protein, cause idiopathic hypogonadotropic hypogonadism and kallmann syndrome. Am J Hum Genet (2008) 83:511–9. doi: 10.1016/j.ajhg.2008.09.005
82. Marcos S, Sarfati J, Leroy C, Fouveaut C, Parent P, Metz C, et al. The prevalence of CHD7 missense versus truncating mutations is higher in patients with kallmann syndrome than in typical CHARGE patients. J Clin Endocrinol Metab (2014) 99:E2138–43. doi: 10.1210/jc.2014-2110
83. Balasubramanian R, Choi J-H, Francescatto L, Willer J, Horton ER, Asimacopoulos EP, et al. Functionally compromised CHD7 alleles in patients with isolated GnRH deficiency. Proc Natl Acad Sci (2014) 111:17953–8. doi: 10.1073/pnas.1417438111
84. Xu C, Cassatella D, van der Sloot AM, Quinton R, Hauschild M, de Geyter C, et al. Evaluating CHARGE syndrome in congenital hypogonadotropic hypogonadism patients harboring CHD7 variants. Genet Med (2018) 20:872–81. doi: 10.1038/gim.2017.197
85. Breuskin I, Bodson M, Thelen N, Thiry M, Borgs L, Nguyen L, et al. Sox10 promotes the survival of cochlear progenitors during the establishment of the organ of corti. Dev Biol (2009) 335:327–39. doi: 10.1016/j.ydbio.2009.09.007
86. Pingault V, Ente D, Dastot-Le Moal F, Goossens M, Marlin S, Bondurand N. Review and update of mutations causing waardenburg syndrome. Hum Mutat (2010) 31:391–406. doi: 10.1002/humu.21211
87. Cole LW, Sidis Y, Zhang C, Quinton R, Plummer L, Pignatelli D, et al. Mutations in Prokineticin 2 and Prokineticin receptor 2 genes in human gonadotrophin-releasing hormone deficiency: Molecular genetics and clinical spectrum. J Clin Endocrinol Metab (2008) 93:3551–9. doi: 10.1210/jc.2007-2654
88. Sarfati J, Fouveaut C, Leroy C, Jeanpierre M, Hardelin J, Dodé C. Greater prevalence of PROKR2 mutations in kallmann syndrome patients from the maghreb than in European patients. Eur J Endocrinol (2013) 169:805–9. doi: 10.1530/eje-13-0419
89. Sarfati J, Guiochon-Mantel A, Rondard P, Arnulf I, Garcia-Piñero A, Wolczynski S, et al. A comparative phenotypic study of kallmann syndrome patients carrying monoallelic and biallelic mutations in the prokineticin 2 or prokineticin receptor 2 genes. J Clin Endocrinol Metab (2010) 95:659–69. doi: 10.1210/jc.2009-0843
90. Cox K, Oliveira L, Plummer L, Corbin B, Gardella T, Balasubramanian R, et al. Modeling mutant/wild-type interactions to ascertain pathogenicity of PROKR2 missense variants in patients with isolated GnRH deficiency. Hum Mol Genet (2017) 27:338–50. doi: 10.1093/hmg/ddx404
91. Sinisi AA, Asci R, Bellastella G, Maione L, Esposito D, Elefante A, et al. Homozygous mutation in the prokineticin-receptor2 gene (Val274Asp) presenting as reversible kallmann syndrome and persistent oligozoospermia: Case report. Hum Reprod (2008) 23:2380–4. doi: 10.1093/humrep/den247
92. Tommiska J, Toppari J, Vaaralahti K, Känsäkoski J, Laitinen E-M, Noisa P, et al. PROKR2 mutations in autosomal recessive kallmann syndrome. Fertil Steril (2013) 99:815–8. doi: 10.1016/j.fertnstert.2012.11.003
93. Wang Y, Gong C, Qin M, Liu Y, Tian Y. Clinical and genetic features of 64 young male paediatric patients with congenital hypogonadotropic hypogonadism. Clin Endocrinol (Oxf) (2017) 87:757–66. doi: 10.1111/cen.13451
94. Libri DV, Kleinau G, Vezzoli V, Busnelli M, Guizzardi F, Sinisi AA, et al. Germline prokineticin receptor 2 (PROKR2) variants associated with central hypogonadism cause differental modulation of distinct intracellular pathways. J Clin Endocrinol Metab (2014) 99:E458–63. doi: 10.1210/jc.2013-2431
95. Falardeau J, Chung WCJ, Beenken A, Raivio T, Plummer L, Sidis Y, et al. Decreased FGF8 signaling causes deficiency of gonadotropin-releasing hormone in humans and mice. J Clin Invest (2008) 118:2822–31. doi: 10.1172/JCI34538
96. Andersen P. Nobelprisen i fysiologi og medisin 1991–små cellebiter–store fremskritt [The 1991 Nobel prize in physiology and medicine–small cellular parts–great progress]. Tidsskr Nor Laegeforen (1991) 111:3610–1.
97. Sykiotis GP, Plummer L, Hughes VA, Au M, Durrani S, Nayak-Young S, et al. Oligogenic basis of isolated gonadotropin-releasing hormone deficiency. Proc Natl Acad Sci (2010) 107:15140–4. doi: 10.1073/pnas.1009622107
98. Ballabio A, Parenti G, Tippett P, Mondello C, di Maio S, Tenore A, et al. X-Linked ichthyosis, due to steroid sulphatase deficiency, associated with kallmann syndrome (hypogonadotropic hypogonadism and anosmia): linkage relationships with xg and cloned DNA sequences from the distal short arm of the X chromosome. Hum Genet (1986) 72:237–40. doi: 10.1007/BF00291885
99. Vermeulen S, Messiaen L, Scheir P, de Bie S, Speleman F, de Paepe A. Kallmann syndrome in a patient with congenital spherocytosis and an interstitial 8p11.2 deletion. Am J Med Genet (2002) 108:315–8. doi: 10.1002/ajmg.10295
100. Young J, Metay C, Bouligand J, Tou B, Francou B, Maione L, et al. SEMA3A deletion in a family with kallmann syndrome validates the role of semaphorin 3A in human puberty and olfactory system development. Hum Reprod (2012) 27:1460–5. doi: 10.1093/humrep/des022
101. Amato LGL, Montenegro LR, Lerario AM, Jorge AAL, Guerra Junior G, Schnoll C, et al. New genetic findings in a large cohort of congenital hypogonadotropic hypogonadism. Eur J Endocrinol (2019) 181:103–19. doi: 10.1530/EJE-18-0764
102. Basaran Y, Bolu E, Unal H, Sagkan R, Taslipinar A, Ozgurtas T, et al. Multiplex ligation dependent probe amplification analysis of KAL1, GNRH1, GNRHR, PROK2 and PROKR2 in male patients with idiopathic hypogonadotropic hypogonadism. Endokrynol Pol (2013) 64:285–92. doi: 10.5603/ep.2013.0007
103. Stamou MI, Brand H, Wang M, Wong I, Lippincott MF, Plummer L, et al. Prevalence and phenotypic effects of copy number variants in isolated hypogonadotropic hypogonadism. J Clin Endocrinol Metab (2022) 107:2228–42. doi: 10.1210/clinem/dgac300
104. Sedlmeyer I, Hirschhorn J, Palmert M. Pedigree analysis of constitutional delay of growth and maturation: Determination of familial aggregation and inheritance patterns. J Of Clin Endocrinol Metab (2002) 87:5581–6. doi: 10.1210/jc.2002-020862
105. Wehkalampi K, Widén E, Laine T, Palotie A, Dunkel L. Patterns of inheritance of constitutional delay of growth and puberty in families of adolescent girls and boys referred to specialist pediatric care. J Of Clin Endocrinol Metab (2008) 93:723–8. doi: 10.1210/jc.2007-1786
106. Eaves L, Silberg J, Foley D, Bulik C, Maes H, Erkanli A, et al. Genetic and environmental influences on the relative timing of pubertal change. Twin Res (2004) 7:471–81. doi: 10.1375/1369052042335278
107. Morris D, Jones M, Schoemaker M, Ashworth A, Swerdlow A. Familial concordance for age at menarche: analyses from the breakthrough generations study. Paediatr Perinat Epidemiol (2011) 25:306–11. doi: 10.1111/j.1365-3016.2010.01183.x
108. Day F, Perry J, Ong K. Genetic regulation of puberty timing in humans. Neuroendocrinology (2015) 102:247–55. doi: 10.1159/000431023
109. Parkash J, Messina A, Langlet F, Cimino I, Loyens A, Mazur D, et al. Semaphorin7A regulates neuroglial plasticity in the adult hypothalamic median eminence. Nat Commun (2015) 6:6385. doi: 10.1038/ncomms7385
110. Pielecka-Fortuna J, Chu Z, Moenter S. Kisspeptin acts directly and indirectly to increase gonadotropin-releasing hormone neuron activity and its effects are modulated by estradiol. Endocrinology (2007) 149:1979–86. doi: 10.1210/en.2007-1365
111. Tornberg J, Sykiotis GP, Keefe K, Plummer L, Hoang X, Hall JE, et al. Heparan sulfate 6-o-sulfotransferase 1 , a gene involved in extracellular sugar modifications, is mutated in patients with idiopathic hypogonadotrophic hypogonadism. Proc Natl Acad Sci (2011) 108:11524–9. doi: 10.1073/pnas.1102284108
112. Howard SR, Oleari R, Poliandri A, Chantzara V, Fantin A, Ruiz-Babot G, et al. HS6ST1 insufficiency causes self-limited delayed puberty in contrast with other GnRH deficiency genes. J Clin Endocrinol Metab (2018) 103. doi: 10.1210/jc.2018-00646
113. Caronia LM, Martin C, Welt CK, Sykiotis GP, Quinton R, Thambundit A, et al. A genetic basis for functional hypothalamic amenorrhea. New Engl J Med (2011) 364:215–25. doi: 10.1056/NEJMoa0911064
114. Jolly A, Bayram Y, Turan S, Aycan Z, Tos T, Abali ZY, et al. Exome sequencing of a primary ovarian insufficiency cohort reveals common molecular etiologies for a spectrum of disease. J Clin Endocrinol Metab (2019) 104:3049–67. doi: 10.1210/jc.2019-00248
115. Yeo G. The role of the FTO (Fat mass and obesity related) locus in regulating body size and composition. Mol Cell Endocrinol (2014) 397:34–41. doi: 10.1016/j.mce.2014.09.012
116. Howard SR, Guasti L, Poliandri A, David A, Cabrera CP, Barnes MR, et al. Contributions of function-altering variants in genes implicated in pubertal timing and body mass for self-limited delayed puberty. J Clin Endocrinol Metab (2018) 103:649–59. doi: 10.1210/jc.2017-02147
117. Mancini A, Howard SR, Cabrera CP, Barnes MR, David A, Wehkalampi K, et al. EAP1 regulation of GnRH promoter activity is important for human pubertal timing. Hum Mol Genet (2019) 28:1357–68. doi: 10.1093/hmg/ddy451
118. Styrkarsdottir U, Thorleifsson G, Sulem P, Gudbjartsson DF, Sigurdsson A, Jonasdottir A, et al. Nonsense mutation in the LGR4 gene is associated with several human diseases and other traits. Nature (2013) 497:517–20. doi: 10.1038/nature12124
119. Day FR, Thompson DJ, Helgason H, Chasman DI, Finucane H, Sulem P, et al. Genomic analyses identify hundreds of variants associated with age at menarche and support a role for puberty timing in cancer risk. Nat Genet (2017) 49:834–41. doi: 10.1038/ng.3841
120. Mancini A, Howard SR, Marelli F, Cabrera CP, Barnes MR, Sternberg MJE, et al. LGR4 deficiency results in delayed puberty through impaired wnt/β-catenin signaling. JCI Insight (2020) 5:11. doi: 10.1172/jci.insight.133434
121. Parent A, Teilmann G, Juul A, Skakkebaek N, Toppari J, Bourguignon J. The timing of normal puberty and the age limits of sexual precocity: Variations around the world, secular trends, and changes after migration. Endocr Rev (2003) 24:668–93. doi: 10.1210/er.2002-0019
122. Wehkalampi K, Silventoinen K, Kaprio J, Dick D, Rose R, Pulkkinen L, et al. Genetic and environmental influences on pubertal timing assessed by height growth. Am J Of Hum Biol (2008) 20:417–23. doi: 10.1002/ajhb.20748
Keywords: delayed puberty, congenital hypogonadotropic hypogonadism, self-limited delayed puberty, genetic test, GnRH deficiency
Citation: Vezzoli V, Hrvat F, Goggi G, Federici S, Cangiano B, Quinton R, Persani L and Bonomi M (2023) Genetic architecture of self-limited delayed puberty and congenital hypogonadotropic hypogonadism. Front. Endocrinol. 13:1069741. doi: 10.3389/fendo.2022.1069741
Received: 14 October 2022; Accepted: 09 December 2022;
Published: 16 January 2023.
Edited by:
Anna Grandone, University of Campania Luigi Vanvitelli, ItalyReviewed by:
Wilson C. J. Chung, Kent State University, United StatesRosario Pivonello, University of Naples Federico II, Italy
Copyright © 2023 Vezzoli, Hrvat, Goggi, Federici, Cangiano, Quinton, Persani and Bonomi. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Valeria Vezzoli, di52ZXp6b2xpQGF1eG9sb2dpY28uaXQ=