 Myeong Jun Song
Myeong Jun Song Jong Young Choi
Jong Young Choi- Department of Internal Medicine, College of Medicine, The Catholic University of Korea, Seoul, South Korea
Non-alcoholic fatty liver disease (NAFLD) is the most common form of chronic liver disease in the world. It is linked mainly to insulin resistance and metabolic syndrome including obesity and dyslipidemia. In addition, various endocrine dysfunctions including polycystic ovary syndrome (PCOS) and hypogonadism are involved in the development and progression of NAFLD. We need to know the disease pathophysiology more accurately due to the heterogeneity of clinical presentation of fatty liver disease. The liver is the major metabolic organ with sexual dimorphism. Sexual dimorphism is associated not only with behavioral differences between men and women, but also with physiological differences reflected in liver metabolism. In men, normal androgen levels prevent hepatic fat accumulation, whereas androgen deficiency induce hepatic steatosis. In women, higher androgens can increase the risk of NAFLD in PCOS. Sex hormone binding globulin (SHBG) is involved in androgen regulation. Recently, SHBG may be reported as a surrogate marker for NAFLD. Therefore, this review will focus on the mechanism of androgen dysfunction in the regulation of hepatic metabolism, the risk of developing NAFLD, and the potential role of SHBG in the course of NAFLD.; Keywords: Non-alcoholic fatty liver disease, insulin resistance, sexual dimorphism, androgen, sex hormone binding globulin
Introduction
Nonalcoholic fatty liver disease (NAFLD) imposes a major public health burden with increasing incidence of comorbidities including obesity and dyslipidemia (1). The main histologic and imaging characteristic of NAFLD is the hepatic accumulation of lipids. Dysregulation of lipid metabolism in liver leads to the accumulation of toxic lipids, which may result in inflammation, hepatocellular injury, and fibrosis (2–4).
Recently, it was suggested that metabolic dysfunction associated fatty liver disease, which is not merely a differential diagnosis for NAFLD could reflect the current knowledge of the disease pathophysiology and aid risk stratification and management of fatty liver disease, which shows a heterogenous clinical presentation (5).
Obesity and metabolic syndrome lead to the development of NAFLD and may be accompanied by endocrine and hormonal disturbances. The liver is the major metabolic organ related to sexual dimorphism (6, 7). NAFLD prevalence is 2.0-3.5-fold higher in men than in women (8). Epidemiological studies have reported that NAFLD is also more severe in men, indicating a deleterious effect of androgens and, on the other hand, protective effect of estrogens in the pathogenesis of NAFLD (9–11). However, the sex-specific mechanisms underlying the development and progression of NAFLD remain to be elucidated.
Sex-differences in the prevalence, progression, outcomes and comorbidities of fatty liver might be considered as the result of gender differences indicative of the liver phenotype between males and females (8). The sexual dimorphism usually observed in NAFLD is reflected in polycystic ovary syndrome (PCOS) in women and hypogonadism in men. Furthermore, sex hormone binding globulin (SHBG) is related to androgen regulation (12). Recently SHBG is reported as a surrogate marker for NAFLD. Investigating the role of androgens in the development and progression of NAFLD in consideration of the gender differences is necessary. Therefore, this review will focus on the mechanism of androgen dysfunction in the regulation of hepatic metabolism, the risk of developing NAFLD, and the potential role of SHBG in the course of NAFLD.
Hepatic lipid metabolism in NAFLD
Hepatic fatty acids (FAs) are derived from two main sources including excess carbohydrates and FAs that are produced by diet and lipolysis in the adipose tissue. Uptake of circulating lipids are facilitated by fatty acid transporters (CD36, FATP2-5) in hepatocyte membrane and is regulated by Peroxisome proliferator-activated receptor-γ (PPAR-γ) (13). Fatty acid binding protein 1 facilitate the transport of hydrophobic FAs with the cytoplasm. De novo lipogenesis (DNL) is regulated by the sterol regulatory element binding proteins 1c (SREBP1c) that induces the expression of genes involved in de novo FA synthesis including acetyl-CoA carboxylase (ACC), fatty acid synthase, the long-chain elongase and stearolyl-CoA desaturase (14). In NAFLD, enhanced SREBP1c-mediated DNL is a key character. FA oxidation is controlled by PPAR-α and reduces intrahepatic fat levels by utilizing FAs as a source of energy in mitochondrial β-oxidation. Mitochondrial dysfunction in NAFLD results in liver injury through increased production of reactive oxygen species. Triglycerides (TGs) are secreted from the hepatocytes in the form of very low density lipoprotein (VLDL) via the fusion of apolipoprotein B-100 (ApoB100). Decreased level of microsomal triglyceride transfer protein (MTTP) and ApoB100 may limit VLDL export and facilitate fat accumulation. An imbalance of these processes leads to the progression of steatosis to nonalcoholic steatohepatitis (NASH).
Hepatic sexual dimorphism according to sex hormone dysfunction
Sex hormones, including estrogens and androgens play a key regulatory role in lipid metabolism and insulin sensitivity (15–17). Sex hormone dysfunction may contribute to the development of NAFLD, because a reduction in insulin sensitivity increases hepatic gluconeogenesis and lipogenesis, in turn, this may exacerbate hepatic steatosis (18).
Among the sex hormones, estrogens have critical metabolic actions in both women and men. The biologically active form of estrogens is 17β-estradiol (E2). In premenopausal women, the main source of E2 is from cholesterol, while in postmenopausal women and men, it is primarily derived from testosterone aromatization. Estrogen reversibly binds to SHBG and diffuse into liver to exert its functions by binding to estrogen receptors (ERs). Estrogen reduce TG accumulation in the liver via ERα. Estrogens play a key role in protecting against hepatic steatosis, by promoting lipolysis and improving FA oxidation in mitochondria via the induction of ACC (19). Therefore, an imbalance in estrogens may have a marked effect on hepatic lipid metabolism. Menopause, a physiological condition of estrogen deficiency, promotes the risk of development and progression of NAFLD (60% and 32% prevalence rates in menopausal and premenopausal women, respectively) (20). The incidence of NAFLD after menopause increases significantly (to that observed in men) due to the protective effect of estrogens, although gender-differences in the prevalence of NAFLD also depend on age (8). Menopause-related fat redistribution also increases the risk of insulin resistance (IR) and subsequently the risk of NAFLD (21). Accordingly, premature menopausal women are also at risk of severe liver fibrosis (22).
On the contrary, androgens showed different opposite effects by sex differences. The major circulating androgens are dehydroepiandrosterone (DHT), androstenedione, testosterone, and dihydrotestosterone, but only testosterone and DHT can bind to androgen receptors (18, 23). Testosterone plays an important role in lipid, and protein metabolism, and has a major influence on body composition including adipose fat and skeletal muscle in men (18, 24). Androgens are achieved by activation of androgen receptor (AR), followed by binding to androgen response element (ARE) or interacting with cytoplasmic signal transduction pathways, including PKA and MAPK/ERK (18). Overexpression of genes involved in lipid accumulation and down expression of FA oxidation is modulated by 5α reductase inhibition because it does not covert testosterone to DHT. Normal androgen levels prevent hepatic fat accumulation, in men, while androgen deficiency induces hepatic steatosis. In women with PCOS, higher androgen levels can increase the risk of NAFLD (25, 26). Androgen dysfunction is a significant contributor to hepatic sexual dimorphism. Therefore, determining the exact mechanisms underlying the sex dimorphism for androgens associated with the development and progression of NAFLD is necessary.
Hyperandrogenism in women with polycystic ovary syndrome and NAFLD
PCOS affects 6–15% of women of reproductive age (27). It is characterized by chronic anovulation, hyperandrogenism (HA), and polycystic ovaries, with women often exhibiting menstrual cycle disturbances and hirsutism or acne (27). Recent studies reported a higher prevalence of NAFLD in women with PCOS than healthy controls (34-70% vs. 14-34%) (26, 28–30). In addition, NAFLD prevalence was significantly higher in patients with PCOS than healthy subjects, with an overall odds ratio of 3.93 (95% CI: 2.17-7.11) (31, 32). Conversely, women with NAFLD are diagnosed with PCOS more often than those without NAFLD (43.7% vs. 23.1%) (33). Therefore, women with PCOS should be screened for NAFLD, while premenopausal women with fatty liver should be screened for PCOS (34).
The pathophysiological mechanism that increases the risk of NAFLD in subjects with PCOS is multifactorial. IR and HA may have a bidirectional relationship with PCOS and NAFLD (35). IR represents a major physiological imbalance in patients with PCOS. As a compensatory response to IR, hyperinsulinemia develops and subsequently interacts synergistically with luteinizing hormone (LH), acting as a co-gonadotrophin within the ovary (36). The resultant activation of CYP17 promotes the production and release of androgens (37). Hyperinsulinemia also exerts extraovarian pleiotropic effects including enhancement of LH pulse amplitude, stimulation of adrenal P450c17α activity, and suppression of hepatic SHBG synthesis, thus elevating the bioavailability of free androgens (37–40).. Low SHBG levels in women can lead to progression of the characteristic phenotype of PCOS. Low SHBG concentrations are also related to NAFLD and IR in women with PCOS, and may reflect activity within a new liver-ovarian axis (41). In a recent study, two distinct PCOS subtypes were distinguished based on SHBG levels: a “reproductive” type that presents with higher SHBG levels and relatively low body mass index (BMI) and insulin levels, and a “metabolic” type that is characterized by higher BMI, glucose, and insulin levels, and lower SHBG levels (42). This finding suggests that PCOS is a heterogeneous complex disorder with different biological mechanisms (43).
HA itself is the independent risk factor to affect NAFLD, although IR is associated with high androgen levels (34). HA directly affects LDL receptors in the liver, causing an increase in LDL, that renders women with PCOS more likely to develop dyslipidemia and NAFLD (44). Furthermore, androgens may affect the production of adipokines, including leptin, and adiponectin, which could be concerned with the metabolic characteristics of PCOS and the development of NAFLD (45). In PCOS mice model, DHT increased binding of AR to ARE in elevating the SCAP-SRBEP1 interaction, resulting in increased hepatic DNL (46, 47). Chronic androgen excess induces IR and hepatic fat accumulation through mitochondrial dysfunction and causing apoptosis, and autophagy imbalance (48). Androgens can induce mitochondria β-oxidation imbalance and DNL and can exacerbate liver inflammatory injury by the overexpression of interleukin-6 (IL-6), tumor necrosis factor-α (TNF-α), MCP-1, and IL-1β (49). Therefore, HA may be a key contributor to NAFLD development in women with PCOS. The causality of the relationship between NAFLD and PCOS requires further research.
Lifestyle interventions are the most effective treatments for women with PCOS due to improvements in insulin sensitivity. Although metformin has been widely used in women with PCOS, it showed limited efficacy for resolving NASH (50, 51). A randomized clinical trial showed that liraglutide, a glucagon like pepatide-1 receptor agonist, achieved body weight loss, of 5.6%, a 66% reduction in NAFLD prevalence, and an 18% reduction in visceral adipose tissue (VAT) when administered to women with PCOS (52). However, the evidence is insufficient to recommend liraglutide as a treatment regimen in women with PCOS. Further research should aim to determine whether novel therapeutics can improve insulin sensitivity and reduce the risk of NAFLD in women with PCOS.
Hypogonadism in men and NAFLD
Male hypogonadism is a clinical syndrome characterized by deficient or absent gonadal function that results in insufficient testosterone secretion (53). Obesity is one of the most important risk factors for secondary hypogonadism in men (54). Male obesity secondary hypogonadism (MOSH) impairs fertility, sexual function, bone mineralization, and fat metabolism, also leads to lower muscle mass and altered body composition (54). Although the prevalence of MOSH remains unclear, rates as high as 45.0–57.5% have been reported (54–56). 26% of men with NAFLD have low free testosterone in a study of 159 men randomly selected from the NASH clinical research network cohort (57). Men with low free testosterone were more likely to have NASH and advanced fibrosis than simple steatosis (88% vs. 67%, 27% vs. 14%, respectively) (57).
The mechanism that increases the risk of NAFLD in subjects with hypogonadism has been poorly described. Testosterone plays an important role in insulin sensitivity, body composition and lipid metabolism (58). There is a bidirectional relationship exists between low testosterone and IR (59). In preclinical study, low testosterone levels may cause hepatic fat accumulation through increased DNL via upregulation of hepatic SREBP-1 (60, 61). The upregulation of SREBP-2 and ACC-1 is apparently due to reduced AMP-activated protein kinase (AMPK) activity (60). Testosterone may promote the expression of hepatic scavenger receptor class B member 1, which is involved in selective uptake of cholesterol esters from circulating high-density lipoprotein (HDL) and facilitates reverse cholesterol transport. Furthermore, testosterone decreases MTTP expression, thus reducing apolipoprotein B-mediated VLDL secretion and cholesterol 7α-hydroxylase levels, which in turn leads to hepatic steatosis due to increased cholesterol uptake and decreased removal (62, 63). Testosterone also significantly increases the mRNA expression of insulin receptors, resulting in increased insulin binding, and elevated glucose oxidation (64). Serine phosphorylation of insulin receptor substrate 1, which attenuates insulin signaling by inhibiting tyrosine phosphorylation, was improved by testosterone treatment (65, 66). Testosterone deprivation leads to reduced glucose transporter type 4 expression in liver tissue and results in hyperglycemia, low insulin level, and diminished glucose uptake in adipose and skeletal muscle tissue (67).
Low testosterone and SHBG in men are independent predictors of metabolic syndrome (68). Low testosterone is related to the visceral fat distribution and exhibits sexual dimorphism due to the dependency on testosterone and E2 of men and women, respectively. Testosterone levels are inversely proportional to the amount of visceral fat. Testosterone and E2 regulate the expansion of visceral fat by activating ERs (ERα and ERβ) and ARs. ER is activated by E2 derived from testosterone aromatization; thus, testosterone deficiency, which leads to low estradiol levels, is a major cause of visceral fat deposition and IR in men (69–71). Efficient AR activation also lowers body fat and increases insulin activity (69, 72). Thus, testosterone exerts its anti-obesity effect (which inhibits the expansion of visceral fat deposition, as well as insulin and leptin resistance, leading to lipogenesis in liver and adipose tissue) by activating the AR pathway (73–75). In addition, SHBG is associated with the low testosterone levels in men with adult-onset hypogonadism (76). SHBG regulates testicular negative feedback either directly or by modulating the entry of testosterone or estradiol into cells in the hypothalamus and/or pituitary to control gonadotropin synthesis and secretion (76). Low total testosterone and SHBG were strongly associated with increased likelihood of having metabolic syndrome, independent of IR (77). Therefore, Hypogonadism is a risk factor for the development of NAFLD.
Testosterone replacement therapy in hypogonadal men with metabolic syndrome had favorable effects on hepatic steatosis, insulin sensitivity, and glucose control (78, 79). However, evidence is lacking to support the use of testosterone therapy in hypogonadal patients with NAFLD. Furthermore, the biochemical mechanisms of underlying the potential therapeutic benefits of testosterone in NAFLD remain to be elucidated, and further study is needed to understand the liver-specific role of testosterone. Studies with large cohorts are necessary to determine whether men with low androgens levels on long-term testosterone therapy are protected against prostate cancer and have a reduced risk of cardiovascular disease over time.
Sex hormone binding globulin status in the course of NAFLD
SHBG is a glycoprotein produced by the liver (80). The primary function of SHBG is to bind and transport circulating testosterone and estradiol to regulate their bioavailability and sequester circulating androgens and estrogens (43, 81). SHGB shows high affinity for testosterone and low affinity for estradiol (81). The free testosterone in plasma is strongly influenced by the SHBG concentration because only 1-2% of testosterone in plasma is free and active; 65% is bound to SHBG and the rest to albumin. Therefore, women with low SHBG levels can have normal total testosterone levels but increased bioavailability thereof, which leads to the progression of PCOS. In addition, IR subsequently results in reproductive dysfunction by diminishing SHGB synthesis. PCOS patients with NAFLD usually have lower SHBG levels and a higher free androgen index compared with those without NAFLD, although differences in circulating androgens are not apparent (26, 34, 82, 83). Furthermore, experimental studies indicated that sex hormones bound to SHBG may directly mediate cell surface signaling, cellular delivery, and the biologic action of sex hormones (84–88). Future research on the association between the biological activity of SHBG binding fractions and risk of NAFLD is warranted.
SHBG also acts as a signal transduction factor. An experimental study showed that thyroid and estrogenic hormones increase SHBG synthesis by upregulating the expression of hepatocyte nuclear factor-4α (HNF-4α), which performs as a key factor in regulating SHBG promoter activity in the liver (89, 90). Conversely, (PPAR-γ competes with HNF-4α for a binding site on the SHGB promoter, such that PPAR-γ inhibits SHBG expression (91). SHBG levels are inversely correlated with hepatic TG and ACC activity (92). SHBG may downregulate the phosphatidylinositol 3−kinase (PI3K)/protein kinase B (AKT) pathway, which is involved in the development of local and systemic IR (93). Increased hepatic lipogenesis or IR downregulates HNF-4α expression thereby diminishing hepatic SHBG synthesis and production. In addition, inflammatory status affects the expression of SHBG. During chronic inflammation diseases, patients show increased expression of inflammatory cytokines such as IL-1 and TNF-α that affect the production of SHBG. The action of IL-1 is mediated by the NF-κB factor which downregulate HNF-4 α transcription leading to the suppression of SHGB synthesis (94).
Low testosterone is associated with a suboptimal distribution of body fat and adipocyte IR, which impairs suppression of lipolysis, and leads to ectopic fat deposition and “lipotoxicity” (95). Adipose inflammatory cytokines, such as TNF-α, IL- 6, and C-reactive protein, can impair hepatic insulin signaling and promote hepatic fat accumulation, leading to inhibition of HNF-4α mRNA via the activation of NF-κB or activating the Methyl ethyl ketone-1/2 (MET-1/2) and c-Jun N-terminal kinase (JNK) mitogen-activated protein kinase (MAPK) pathways (96, 97). In contrast, adiponectin increases SHBG production by activating AMPK, which increases FA oxidation and HNF-4α levels (98). Therefore, low circulating SHBG is associated with a high risk of NAFLD in men with hypogonadism.
SHBG may serve as a biomarker of NAFLD. Low testosterone and SHBG concentrations are related with metabolic syndrome and fatty liver. In a recent meta-analysis, low total testosterone was positively associated with NAFLD in men but inversely in women, on the other hand, low SHBG concentration was reportedly associated with a high risk of development of NAFLD in both men and women (99). Furthermore, SHBG has anti-inflammatory and lipolytic effects on adipocytes and macrophages, which could explain its association with lower incidence rates of metabolic syndrome and its complications (100). In a biopsy proven NAFLD study, lower SHBG levels were inversely related to the severity of steatosis (101). Therefore, improving SHBG expression can be a potential therapeutic target for NAFLD.
Conclusion
An intricate relationship exists between androgens and NAFLD that may independently affect hepatic homeostasis. Gender differences in the effects of androgen have been observed, with low testosterone levels affecting liver function in men but not in young women, in whom HA presents a risk factor for NAFLD. An apparent bidirectional connection exists between sexual dimorphism of androgens and NAFLD. In addition, SHBGs participate in hormone regulation, acting as a buffer in the context of androgen homeostasis (Figure 1). Therefore, understanding the molecular mechanism of androgens in the liver would aid the development of mechanism-based therapeutic interventions for NAFLD.
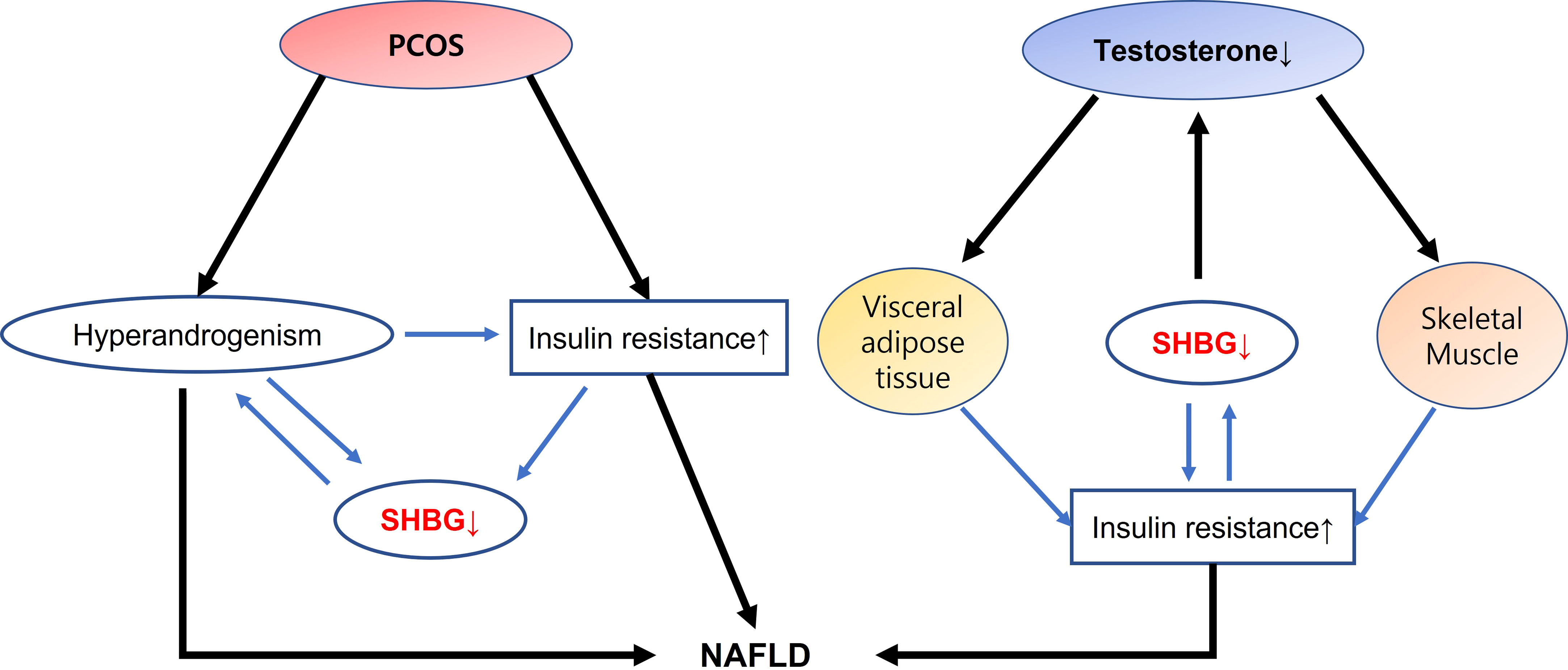
Figure 1 Interactions of SHBG with androgen dysfunction on NAFLD development In PCOS, HA and insulin resistance leads to downregulate SHBG production, and develops NAFLD. On the contrary, interaction of low testosterone with visceral adipose tissue and skeletal muscle may modulate insulin resistance that decreases SHBG expression and increase the risk of NAFLD. SHBG has a dual action in interacting insulin resistance as well as in acting as a buffer in the context of androgen homeostasis suggesting a role as a surrogate marker in the development of NAFLD. SHBG, sex hormone binding globulin; NAFLD, nonalcoholic fatty liver disease; PCOS, polycystic ovary syndrome; HA, hyperandrogenism.
Author contributions
MJS contributed to the selection and analysis of preview literature, and wrote the original draft for this mini review. JYC supervised the manuscript and contributed to the review and editing for this mini review. All authors contributed to the article and approved the submitted version.
Funding
This work was supported by the National Research Foundation of Korea (NRF) grant funded by the Korea government (MSIT) (No.2022R1F1A1063158).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Younossi Z, Tacke F, Arrese M, Chander Sharma B, Mostafa I, Bugianesi E, et al. Global perspectives on nonalcoholic fatty liver disease and nonalcoholic steatohepatitis. Hepatology (2019) 69:2672–82. doi: 10.1002/hep.30251
2. Marra F, Svegliati-Baroni G. Lipotoxicity and the gut-liver axis in NASH pathogenesis. J Hepatol (2018) 68:280–95. doi: 10.1016/j.jhep.2017.11.014
3. Lebeaupin C, Vallée D, Hazari Y, Hetz C, Chevet E, Bailly-Maitre B. Endoplasmic reticulum stress signalling and the pathogenesis of non-alcoholic fatty liver disease. J Hepatol (2018) 69:927–47. doi: 10.1016/j.jhep.2018.06.008
4. Volmer R, van der Ploeg K, Ron D. Membrane lipid saturation activates endoplasmic reticulum unfolded protein response transducers through their transmembrane domains. Proc Natl Acad Sci U S A (2013) 110:4628–33. doi: 10.1073/pnas.1217611110
5. Eslam M, Newsome PN, Sarin SK, Anstee QM, Targher G, Romero-Gomez M, et al. A new definition for metabolic dysfunction-associated fatty liver disease: An international expert consensus statement. J Hepatol (2020) 73:202–9. doi: 10.1016/j.jhep.2020.03.039
6. Roy AK, Chatterjee B. Sexual dimorphism in the liver. Annu Rev Physiol (1983) 45:37–50. doi: 10.1146/annurev.ph.45.030183.000345
7. Yang X, Schadt EE, Wang S, Wang H, Arnold AP, Ingram-Drake L, et al. Tissue-specific expression and regulation of sexually dimorphic genes in mice. Genome Res (2006) 16:995–1004. doi: 10.1101/gr.5217506
8. Della Torre S. Non-alcoholic fatty liver disease as a canonical example of metabolic inflammatory-based liver disease showing a sex-specific prevalence: Relevance of estrogen signaling. Front Endocrinol (Lausanne) (2020) 11:572490. doi: 10.3389/fendo.2020.572490
9. Della Torre S, Benedusi V, Fontana R, Maggi A. Energy metabolism and fertility: a balance preserved for female health. Nat Rev Endocrinol (2014) 10:13–23. doi: 10.1038/nrendo.2013.203
10. Buzzetti E, Parikh PM, Gerussi A, Tsochatzis E. Gender differences in liver disease and the drug-dose gender gap. Pharmacol Res (2017) 120:97–108. doi: 10.1016/j.phrs.2017.03.014
11. Lonardo A, Nascimbeni F, Ballestri S, Fairweather D, Win S, Than TA, et al. Sex differences in nonalcoholic fatty liver disease: State of the art and identification of research gaps. Hepatology (2019) 70:1457–69. doi: 10.1002/hep.30626
12. Hammond GL, Bocchinfuso WP. Sex hormone-binding globulin: gene organization and structure/function analyses. Horm Res (1996) 45:197–201. doi: 10.1159/000184787
13. Ipsen DH, Lykkesfeldt J, Tveden-Nyborg P. Molecular mechanisms of hepatic lipid accumulation in non-alcoholic fatty liver disease. Cell Mol Life Sci (2018) 75:3313–27. doi: 10.1007/s00018-018-2860-6
14. Song MJ, Malhi H. The unfolded protein response and hepatic lipid metabolism in non alcoholic fatty liver disease. Pharmacol Ther (2019) 203:107401. doi: 10.1016/j.pharmthera.2019.107401
15. Gupte AA, Pownall HJ, Hamilton DJ. Estrogen: an emerging regulator of insulin action and mitochondrial function. J Diabetes Res (2015) 2015:916585. doi: 10.1155/2015/916585
16. Rastrelli G, Filippi S, Sforza A, Maggi M, Corona G. Metabolic syndrome in Male hypogonadism. Front Horm Res (2018) 49:131–55. doi: 10.1159/000485999
17. Sanchez-Garrido MA, Tena-Sempere M. Metabolic dysfunction in polycystic ovary syndrome: Pathogenic role of androgen excess and potential therapeutic strategies. Mol Metab (2020) 35:100937. doi: 10.1016/j.molmet.2020.01.001
18. Shen M, Shi H. Sex hormones and their receptors regulate liver energy homeostasis. Int J Endocrinol (2015) 2015:294278. doi: 10.1155/2015/294278
19. Zhang H, Liu Y, Wang L, Li Z, Zhang H, Wu J, et al. Differential effects of estrogen/androgen on the prevention of nonalcoholic fatty liver disease in the male rat. J Lipid Res (2013) 54:345–57. doi: 10.1194/jlr.M028969
20. Gutierrez-Grobe Y, Ponciano-Rodríguez G, Ramos MH, Uribe M, Méndez-Sánchez N. Prevalence of non alcoholic fatty liver disease in premenopausal, posmenopausal and polycystic ovary syndrome women. role estrogens Ann Hepatol (2010) 9:402–9. doi: 10.1016/S1665-2681(19)31616-3
21. Von-Hafe M, Borges-Canha M, Vale C, Leite AR, Sérgio Neves J, Carvalho D, et al. Nonalcoholic fatty liver disease and endocrine axes-a scoping review. Metabolites (2022) 12:1–30. doi: 10.3390/metabo12040298
22. Klair JS, Yang JD, Abdelmalek MF, Guy CD, Gill RM, Yates K, et al. A longer duration of estrogen deficiency increases fibrosis risk among postmenopausal women with nonalcoholic fatty liver disease. Hepatology (2016) 64:85–91. doi: 10.1002/hep.28514
23. Charni-Natan M, Aloni-Grinstein R, Osher E, Rotter V. Liver and steroid hormones-can a touch of p53 make a difference? Front Endocrinol (Lausanne) (2019) 10:374. doi: 10.3389/fendo.2019.00374
24. Kelly DM, Jones TH. Testosterone: a metabolic hormone in health and disease. J Endocrinol (2013) 217:R25–45. doi: 10.1530/joe-12-0455
25. Schiffer L, Kempegowda P, Arlt W, O'Reilly MW. MECHANISMS IN ENDOCRINOLOGY: The sexually dimorphic role of androgens in human metabolic disease. Eur J Endocrinol (2017) 177:R125–r43. doi: 10.1530/eje-17-0124
26. Jones H, Sprung VS, Pugh CJ, Daousi C, Irwin A, Aziz N, et al. Polycystic ovary syndrome with hyperandrogenism is characterized by an increased risk of hepatic steatosis compared to nonhyperandrogenic PCOS phenotypes and healthy controls, independent of obesity and insulin resistance. J Clin Endocrinol Metab (2012) 97:3709–16. doi: 10.1210/jc.2012-1382
27. Conway G, Dewailly D, Diamanti-Kandarakis E, Escobar-Morreale HF, Franks S, Gambineri A, et al. The polycystic ovary syndrome: a position statement from the European society of endocrinology. Eur J Endocrinol (2014) 171:P1–29. doi: 10.1530/eje-14-0253
28. Vassilatou E, Lafoyianni S, Vryonidou A, Ioannidis D, Kosma L, Katsoulis K, et al. Increased androgen bioavailability is associated with non-alcoholic fatty liver disease in women with polycystic ovary syndrome. Hum Reprod (2010) 25:212–20. doi: 10.1093/humrep/dep380
29. Macut D, Tziomalos K, Božić-Antić I, Bjekić-Macut J, Katsikis I, Papadakis E, et al. Non-alcoholic fatty liver disease is associated with insulin resistance and lipid accumulation product in women with polycystic ovary syndrome. Hum Reprod (2016) 31:1347–53. doi: 10.1093/humrep/dew076
30. Cussons AJ, Watts GF, Mori TA, Stuckey BG. Omega-3 fatty acid supplementation decreases liver fat content in polycystic ovary syndrome: a randomized controlled trial employing proton magnetic resonance spectroscopy. J Clin Endocrinol Metab (2009) 94:3842–8. doi: 10.1210/jc.2009-0870
31. Vuppalanchi R, Noureddin M, Alkhouri N, Sanyal AJ. Therapeutic pipeline in nonalcoholic steatohepatitis. Nat Rev Gastroenterol Hepatol (2021) 18:373–92. doi: 10.1038/s41575-020-00408-y
32. Ramezani-Binabaj M, Motalebi M, Karimi-Sari H, Rezaee-Zavareh MS, Alavian SM. Are women with polycystic ovarian syndrome at a high risk of non-alcoholic fatty liver disease; a meta-analysis. Hepat Mon (2014) 14:e23235. doi: 10.5812/hepatmon.23235
33. Vassilatou E. Nonalcoholic fatty liver disease and polycystic ovary syndrome. World J Gastroenterol (2014) 20:8351–63. doi: 10.3748/wjg.v20.i26.8351
34. Paschou SA, Polyzos SA, Anagnostis P, Goulis DG, Kanaka-Gantenbein C, Lambrinoudaki I, et al. Nonalcoholic fatty liver disease in women with polycystic ovary syndrome. Endocrine (2020) 67:1–8. doi: 10.1007/s12020-019-02085-7
35. Petta S, Ciresi A, Bianco J, Geraci V, Boemi R, Galvano L, et al. Insulin resistance and hyperandrogenism drive steatosis and fibrosis risk in young females with PCOS. PloS One (2017) 12:e0186136. doi: 10.1371/journal.pone.0186136
36. Barber TM, Dimitriadis GK, Andreou A, Franks S. Polycystic ovary syndrome: insight into pathogenesis and a common association with insulin resistance. Clin Med (Lond) (2016) 16:262–6. doi: 10.7861/clinmedicine.16-3-262
37. Morin-Papunen LC, Vauhkonen I, Koivunen RM, Ruokonen A, Tapanainen JS. Insulin sensitivity, insulin secretion, and metabolic and hormonal parameters in healthy women and women with polycystic ovarian syndrome. Hum Reprod (2000) 15:1266–74. doi: 10.1093/humrep/15.6.1266
38. Diamanti-Kandarakis E, Dunaif A. Insulin resistance and the polycystic ovary syndrome revisited: an update on mechanisms and implications. Endocr Rev (2012) 33:981–1030. doi: 10.1210/er.2011-1034
39. Dunaif A. Insulin resistance and the polycystic ovary syndrome: mechanism and implications for pathogenesis. Endocr Rev (1997) 18:774–800. doi: 10.1210/edrv.18.6.0318
40. Yki-Järvinen H, Mäkimattila S, Utriainen T, Rutanen EM. Portal insulin concentrations rather than insulin sensitivity regulate serum sex hormone-binding globulin and insulin-like growth factor binding protein 1. vivo J Clin Endocrinol Metab (1995) 80:3227–32. doi: 10.1210/jcem.80.11.7593430
41. Chang RJ. The reproductive phenotype in polycystic ovary syndrome. Nat Clin Pract Endocrinol Metab (2007) 3:688–95. doi: 10.1038/ncpendmet0637
42. Dapas M, Lin FTJ, Nadkarni GN, Sisk R, Legro RS, Urbanek M, et al. Distinct subtypes of polycystic ovary syndrome with novel genetic associations: An unsupervised, phenotypic clustering analysis. PloS Med (2020) 17:e1003132. doi: 10.1371/journal.pmed.1003132
43. Qu X, Donnelly R. Sex hormone-binding globulin (SHBG) as an early biomarker and therapeutic target in polycystic ovary syndrome. Int J Mol Sci (2020) 21:1–17. doi: 10.3390/ijms21218191
44. Baranova A, Tran TP, Afendy A, Wang L, Shamsaddini A, Mehta R, et al. Molecular signature of adipose tissue in patients with both non-alcoholic fatty liver disease (NAFLD) and polycystic ovarian syndrome (PCOS). J Transl Med (2013) 11:133. doi: 10.1186/1479-5876-11-133
45. Xu A, Chan KW, Hoo RL, Wang Y, Tan KC, Zhang J, et al. Testosterone selectively reduces the high molecular weight form of adiponectin by inhibiting its secretion from adipocytes. J Biol Chem (2005) 280:18073–80. doi: 10.1074/jbc.M414231200
46. Wang D, He B. Current perspectives on nonalcoholic fatty liver disease in women with polycystic ovary syndrome. Diabetes Metab Syndr Obes (2022) 15:1281–91. doi: 10.2147/dmso.S362424
47. Seidu T, McWhorter P, Myer J, Alamgir R, Eregha N, Bogle D, et al. DHT causes liver steatosis via transcriptional regulation of SCAP in normal weight female mice. J Endocrinol (2021) 250:49–65. doi: 10.1530/joe-21-0040
48. Cui P, Hu W, Ma T, Hu M, Tong X, Zhang F, et al. Long-term androgen excess induces insulin resistance and non-alcoholic fatty liver disease in PCOS-like rats. J Steroid Biochem Mol Biol (2021) 208:105829. doi: 10.1016/j.jsbmb.2021.105829
49. Zhang Y, Meng F, Sun X, Sun X, Hu M, Cui P, et al. Hyperandrogenism and insulin resistance contribute to hepatic steatosis and inflammation in female rat liver. Oncotarget (2018) 9:18180–97. doi: 10.18632/oncotarget.24477
50. Ehrmann DA, Cavaghan MK, Imperial J, Sturis J, Rosenfield RL, Polonsky KS. Effects of metformin on insulin secretion, insulin action, and ovarian steroidogenesis in women with polycystic ovary syndrome. J Clin Endocrinol Metab (1997) 82:524–30. doi: 10.1210/jcem.82.2.3722
51. Rakoski MO, Singal AG, Rogers MA, Conjeevaram H. Meta-analysis: insulin sensitizers for the treatment of non-alcoholic steatohepatitis. Aliment Pharmacol Ther (2010) 32:1211–21. doi: 10.1111/j.1365-2036.2010.04467.x
52. Frøssing S, Nylander M, Chabanova E, Frystyk J, Holst JJ, Kistorp C, et al. Effect of liraglutide on ectopic fat in polycystic ovary syndrome: A randomized clinical trial. Diabetes Obes Metab (2018) 20:215–8. doi: 10.1111/dom.13053
54. Fernandez CJ, Chacko EC, Pappachan JM. Male Obesity-related secondary hypogonadism - pathophysiology, clinical implications and management. Eur Endocrinol (2019) 15:83–90. doi: 10.17925/ee.2019.15.2.83
55. Calderón B, Gómez-Martín JM, Vega-Piñero B, Martín-Hidalgo A, Galindo J, Luque-Ramírez M, et al. Prevalence of male secondary hypogonadism in moderate to severe obesity and its relationship with insulin resistance and excess body weight. Andrology (2016) 4:62–7. doi: 10.1111/andr.12135
56. Hofstra J, Loves S, van Wageningen B, Ruinemans-Koerts J, Jansen I, de Boer H. High prevalence of hypogonadotropic hypogonadism in men referred for obesity treatment. Neth J Med (2008) 66:103–9.
57. Sarkar M, Yates K, Suzuki A, Lavine J, Gill R, Ziegler T, et al. Low testosterone is associated with nonalcoholic steatohepatitis and fibrosis severity in men. Clin Gastroenterol Hepatol (2021) 19:400–2.e2. doi: 10.1016/j.cgh.2019.11.053
58. Kupelian V, Page ST, Araujo AB, Travison TG, Bremner WJ, McKinlay JB. Low sex hormone-binding globulin, total testosterone, and symptomatic androgen deficiency are associated with development of the metabolic syndrome in nonobese men. J Clin Endocrinol Metab (2006) 91:843–50. doi: 10.1210/jc.2005-1326
59. Marino L, Jornayvaz FR. Endocrine causes of nonalcoholic fatty liver disease. World J Gastroenterol (2015) 21:11053–76. doi: 10.3748/wjg.v21.i39.11053
60. Sakr HF, Hussein AM, Eid EA, AlKhateeb M. Possible mechanisms underlying fatty liver in a rat model of male hypogonadism: A protective role for testosterone. Steroids (2018) 135:21–30. doi: 10.1016/j.steroids.2018.04.004
61. Gariani K, Jornayvaz FR. Pathophysiology of NASH in endocrine diseases. Endocr Connect (2021) 10:R52–r65. doi: 10.1530/ec-20-0490
62. Schleich F, Legros JJ. Effects of androgen substitution on lipid profile in the adult and aging hypogonadal male. Eur J Endocrinol (2004) 151:415–24. doi: 10.1530/eje.0.1510415
63. Movérare-Skrtic S, Venken K, Andersson N, Lindberg MK, Svensson J, Swanson C, et al. Dihydrotestosterone treatment results in obesity and altered lipid metabolism in orchidectomized mice. Obes (Silver Spring) (2006) 14:662–72. doi: 10.1038/oby.2006.75
64. Parthasarathy C, Renuka VN, Balasubramanian K. Sex steroids enhance insulin receptors and glucose oxidation in Chang liver cells. Clin Chim Acta (2009) 399:49–53. doi: 10.1016/j.cca.2008.09.011
65. Aguirre V, Werner ED, Giraud J, Lee YH, Shoelson SE, White MF. Phosphorylation of Ser307 in insulin receptor substrate-1 blocks interactions with the insulin receptor and inhibits insulin action. J Biol Chem (2002) 277:1531–7. doi: 10.1074/jbc.M101521200
66. Muthusamy T, Murugesan P, Srinivasan C, Balasubramanian K. Sex steroids influence glucose oxidation through modulation of insulin receptor expression and IRS-1 serine phosphorylation in target tissues of adult male rat. Mol Cell Biochem (2011) 352:35–45. doi: 10.1007/s11010-011-0737-1
67. Muthusamy T, Murugesan P, Balasubramanian K. Sex steroids deficiency impairs glucose transporter 4 expression and its translocation through defective akt phosphorylation in target tissues of adult male rat. Metabolism (2009) 58:1581–92. doi: 10.1016/j.metabol.2009.05.010
68. Haffner SM, Shaten J, Stern MP, Smith GD, Kuller L. Low levels of sex hormone-binding globulin and testosterone predict the development of non-insulin-dependent diabetes mellitus in men. MRFIT Res Group Multiple Risk Factor Intervention Trial Am J Epidemiol (1996) 143:889–97. doi: 10.1093/oxfordjournals.aje.a008832
69. Bianchi VE, Locatelli V. Testosterone a key factor in gender related metabolic syndrome. Obes Rev (2018) 19:557–75. doi: 10.1111/obr.12633
70. Khaw KT, Barrett-Connor E. Lower endogenous androgens predict central adiposity in men. Ann Epidemiol (1992) 2:675–82. doi: 10.1016/1047-2797(92)90012-f
71. Zitzmann M, Faber S, Nieschlag E. Association of specific symptoms and metabolic risks with serum testosterone in older men. J Clin Endocrinol Metab (2006) 91:4335–43. doi: 10.1210/jc.2006-0401
72. Zitzmann M, Gromoll J, von Eckardstein A, Nieschlag E. The CAG repeat polymorphism in the androgen receptor gene modulates body fat mass and serum concentrations of leptin and insulin in men. Diabetologia (2003) 46:31–9. doi: 10.1007/s00125-002-0980-9
73. Fan W, Yanase T, Nomura M, Okabe T, Goto K, Sato T, et al. Androgen receptor null male mice develop late-onset obesity caused by decreased energy expenditure and lipolytic activity but show normal insulin sensitivity with high adiponectin secretion. Diabetes (2005) 54:1000–8. doi: 10.2337/diabetes.54.4.1000
74. Lin HY, Xu Q, Yeh S, Wang RS, Sparks JD, Chang C. Insulin and leptin resistance with hyperleptinemia in mice lacking androgen receptor. Diabetes (2005) 54:1717–25. doi: 10.2337/diabetes.54.6.1717
75. Singh R, Artaza JN, Taylor WE, Gonzalez-Cadavid NF, Bhasin S. Androgens stimulate myogenic differentiation and inhibit adipogenesis in C3H 10T1/2 pluripotent cells through an androgen receptor-mediated pathway. Endocrinology (2003) 144:5081–8. doi: 10.1210/en.2003-0741
76. Winters SJ. SHBG and total testosterone levels in men with adult onset hypogonadism: what are we overlooking? Clin Diabetes Endocrinol (2020) 6:17. doi: 10.1186/s40842-020-00106-3
77. Li C, Ford ES, Li B, Giles WH, Liu S. Association of testosterone and sex hormone-binding globulin with metabolic syndrome and insulin resistance in men. Diabetes Care (2010) 33:1618–24. doi: 10.2337/dc09-1788
78. Traish AM, Haider A, Doros G, Saad F. Long-term testosterone therapy in hypogonadal men ameliorates elements of the metabolic syndrome: an observational, long-term registry study. Int J Clin Pract (2014) 68:314–29. doi: 10.1111/ijcp.12319
79. Traish AM. Testosterone and weight loss: the evidence. Curr Opin Endocrinol Diabetes Obes (2014) 21:313–22. doi: 10.1097/med.0000000000000086
80. Lee IR, Dawson SA, Wetherall JD, Hahnel R. Sex hormone-binding globulin secretion by human hepatocarcinoma cells is increased by both estrogens and androgens. J Clin Endocrinol Metab (1987) 64:825–31. doi: 10.1210/jcem-64-4-825
81. Dunn JF, Nisula BC, Rodbard D. Transport of steroid hormones: binding of 21 endogenous steroids to both testosterone-binding globulin and corticosteroid-binding globulin in human plasma. J Clin Endocrinol Metab (1981) 53:58–68. doi: 10.1210/jcem-53-1-58
82. Nestler JE, Powers LP, Matt DW, Steingold KA, Plymate SR, Rittmaster RS, et al. A direct effect of hyperinsulinemia on serum sex hormone-binding globulin levels in obese women with the polycystic ovary syndrome. J Clin Endocrinol Metab (1991) 72:83–9. doi: 10.1210/jcem-72-1-83
83. Sarkar M, Wellons M, Cedars MI, VanWagner L, Gunderson EP, Ajmera V, et al. Testosterone levels in pre-menopausal women are associated with nonalcoholic fatty liver disease in midlife. Am J Gastroenterol (2017) 112:755–62. doi: 10.1038/ajg.2017.44
84. Ding EL, Song Y, Manson JE, Hunter DJ, Lee CC, Rifai N, et al. Sex hormone-binding globulin and risk of type 2 diabetes in women and men. N Engl J Med (2009) 361:1152–63. doi: 10.1056/NEJMoa0804381
85. Rosner W, Hryb DJ, Khan MS, Nakhla AM, Romas NA. Sex hormone-binding globulin mediates steroid hormone signal transduction at the plasma membrane. J Steroid Biochem Mol Biol (1999) 69:481–5. doi: 10.1016/s0960-0760(99)00070-9
86. Hammes A, Andreassen TK, Spoelgen R, Raila J, Hubner N, Schulz H, et al. Role of endocytosis in cellular uptake of sex steroids. Cell (2005) 122:751–62. doi: 10.1016/j.cell.2005.06.032
87. Fortunati N, Becchis M, Catalano MG, Comba A, Ferrera P, Raineri M, et al. Sex hormone-binding globulin, its membrane receptor, and breast cancer: a new approach to the modulation of estradiol action in neoplastic cells. J Steroid Biochem Mol Biol (1999) 69:473–9. doi: 10.1016/s0960-0760(99)00068-0
88. Kahn SM, Hryb DJ, Nakhla AM, Romas NA, Rosner W. Sex hormone-binding globulin is synthesized in target cells. J Endocrinol (2002) 175:113–20. doi: 10.1677/joe.0.1750113
89. Selva DM, Hammond GL. Thyroid hormones act indirectly to increase sex hormone-binding globulin production by liver via hepatocyte nuclear factor-4alpha. J Mol Endocrinol (2009) 43:19–27. doi: 10.1677/jme-09-0025
90. Pugeat M, Nader N, Hogeveen K, Raverot G, Déchaud H, Grenot C. Sex hormone-binding globulin gene expression in the liver: drugs and the metabolic syndrome. Mol Cell Endocrinol (2010) 316:53–9. doi: 10.1016/j.mce.2009.09.020
91. Selva DM, Hammond GL. Peroxisome-proliferator receptor gamma represses hepatic sex hormone-binding globulin expression. Endocrinology (2009) 150:2183–9. doi: 10.1210/en.2008-1289
92. Sáez-López C, Salcedo-Allende MT, Hernandez C, Simó-Servat O, Simó R, Selva DM. Sex hormone-binding globulin expression correlates with acetyl-coenzyme a carboxylase and triglyceride content in human liver. J Clin Endocrinol Metab (2019) 104:1500–7. doi: 10.1210/jc.2018-00740
93. Feng C, Jin Z, Chi X, Zhang B, Wang X, Sun L, et al. SHBG expression is correlated with PI3K/AKT pathway activity in a cellular model of human insulin resistance. Gynecol Endocrinol (2018) 34:567–73. doi: 10.1080/09513590.2017.1411474
94. Bourebaba N, Ngo T, Śmieszek A, Bourebaba L, Marycz K. Sex hormone binding globulin as a potential drug candidate for liver-related metabolic disorders treatment. BioMed Pharmacother (2022) 153:113261. doi: 10.1016/j.biopha.2022.113261
95. Longo M, Zatterale F, Naderi J, Parrillo L, Formisano P, Raciti GA, et al. Adipose tissue dysfunction as determinant of obesity-associated metabolic complications. Int J Mol Sci (2019) 20:1–23. doi: 10.3390/ijms20092358
96. Simó R, Barbosa-Desongles A, Sáez-Lopez C, Lecube A, Hernandez C, Selva DM. Molecular mechanism of TNFα-induced down-regulation of SHBG expression. Mol Endocrinol (2012) 26:438–46. doi: 10.1210/me.2011-1321
97. Simó R, Barbosa-Desongles A, Hernandez C, Selva DM. IL1β down-regulation of sex hormone-binding globulin production by decreasing HNF-4α via MEK-1/2 and JNK MAPK pathways. Mol Endocrinol (2012) 26:1917–27. doi: 10.1210/me.2012-1152
98. Simó R, Saez-Lopez C, Lecube A, Hernandez C, Fort JM, Selva DM. Adiponectin upregulates SHBG production: molecular mechanisms and potential implications. Endocrinology (2014) 155:2820–30. doi: 10.1210/en.2014-1072
99. Jaruvongvanich V, Sanguankeo A, Riangwiwat T, Upala S. Testosterone, sex hormone-binding globulin and nonalcoholic fatty liver disease: a systematic review and meta-analysis. Ann Hepatol (2017) 16:382–94. doi: 10.5604/16652681.1235481
100. Yamazaki H, Kushiyama A, Sakoda H, Fujishiro M, Yamamotoya T, Nakatsu Y, et al. Protective effect of sex hormone-binding globulin against metabolic syndrome: In vitro evidence showing anti-inflammatory and lipolytic effects on adipocytes and macrophages. Mediators Inflamm (2018) 2018:3062319. doi: 10.1155/2018/3062319
Keywords: non-alcholic fatty liver disease, insuline resistance, sexual dimorphism, androgen, sex hormone binding globhulin
Citation: Song MJ and Choi JY (2022) Androgen dysfunction in non-alcoholic fatty liver disease: Role of sex hormone binding globulin. Front. Endocrinol. 13:1053709. doi: 10.3389/fendo.2022.1053709
Received: 26 September 2022; Accepted: 08 November 2022;
Published: 22 November 2022.
Edited by:
Guglielmo Beccuti, University of Turin, ItalyReviewed by:
David Martinez Selva, Vall d’Hebron Research Institute (VHIR), SpainLynda Bourebaba, Wroclaw University of Environmental and Life Sciences, Poland
Copyright © 2022 Song and Choi. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jong Young Choi, anljaG9pQGNhdGhvbGljLmFjLmty