
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Endocrinol. , 09 January 2023
Sec. Obesity
Volume 13 - 2022 | https://doi.org/10.3389/fendo.2022.1052487
This article is part of the Research Topic Stress-Induced Weight Changes View all 6 articles
 Guanhao Wang1,2,3
Guanhao Wang1,2,3 Shuanqing Li1
Shuanqing Li1 Yingqi Li1,2,3Meihui Zhang1
Yingqi Li1,2,3Meihui Zhang1 Ting Xu1
Ting Xu1 Tianming Li1Lining Cao1*
Tianming Li1Lining Cao1* Jianfeng Lu1,4*
Jianfeng Lu1,4*Introduction: A vicious cycle ensues whereby prolonged exposure to social stress causes increased production of glucocorticoids (GCs), leading to obesity even further. Understanding the role of GCs, the key element in the vicious circle, might be helpful to break the vicious circle. However, the mechanism by which GCs induce obesity remains elusive.
Methods: Corticosterone (CORT) was administered to mice for 8 weeks. Food and water intake were recorded; obesity was analyzed by body-weight evaluation and magnetic resonance imaging (MRI); intestinal proliferation and survival were evaluated by H&E staining, EdU-progression test, TUNEL assay and immunofluorescence staining of Ki67 and CC3; RNA-seq was performed to analyze transcriptional alterations in small intestines and livers.
Results: Chronic CORT treatment induced obesity, longer small intestines, hepatic steatosis and elevated levels of serum insulin and leptin in mice; CORT-treated mice showed increased cell proliferation and decreased apoptosis of small intestines; RNA-seq results indicate that differentially expressed genes (DEGs) were enriched in several cell growth/death-associated signaling pathways.
Discussion: Herein we find that administration of CORT to mice promotes the proliferation and survival of intestinal cells, which might contribute to the longer small intestines and the elongated intestinal villi, thus leading to increased nutrient absorption and obesity in mice. Understanding CORT-induced alterations in intestines and associated signaling pathways might provide novel therapeutic clues for GCs or stress-associated obesity.
Obesity, which threatens the health of individuals, is a rapidly increasing pandemic. It is predicted that one fifth of adults worldwide will be expected to be obese by 2025, and the economic cost for chronic diseases due to obesity is estimated to be 990 billion US dollars per year globally (1). In the past few decades, excessive food intake or insufficient energy expenditure was considered to contribute to obesity. However, recent evidence is now mounting to suggest that stress-induced glucocorticoids (GCs) elevations play an important role in obesity (2, 3).
Accumulated evidence suggested that stress plays an important role in the development of obesity. Hypothalamic-pituitary-adrenocortical (HPA) axis, a well-known neuroendocrine system mediating stress response, stimulates the adrenal cortex to release GCs: cortisol in humans or corticosterone (CORT) in rodents into blood. Patients chronically exposed to high levels of GCs, like in Cushing’s syndrome (endogenous GCs) or when using high doses of exogenous GCs, are more likely to develop abdominal obesity (4, 5). Interestingly, in our modern society, the obesity pandemic coincides with an increase of factors that enhance GCs production, such as chronic stress, consumption of food with a high glycemic index, and a reduced amount of sleep. This thus indicated a vicious circle, within which the increased GCs action, obesity and stress interact and intensify with each other (6), leading inexorably to worsening of obesity and metabolic status. Understanding the role of GCs, the key element in the vicious circle, might be helpful to break the vicious circle. However, the mechanism by which GCs induce obesity is largely unknown.
Excessive food intake and nutrient absorption are likely to contribute to obesity. Small intestine is one of the major organs for nutrient absorption. Intestinal villi are tiny, finger-like structures which project into the intestinal cavity, greatly increasing the surface area for nutrients absorption. The remarkable length of the small intestines, determined by a balance between the rates of proliferation and death of intestinal cells, contributes greatly to its surface area for nutrient absorption (7–9). A recent study indicated that CORT-treated animals became obese, even when the food intake was restricted to the same amount as consumed by the control mice (10). This gives rise to the question whether GCs could regulate proliferation and survival of intestinal cells, thus leading to an increased gut absorptive capacity, which results in obesity.
In the present study, CORT was administered to mice, and the small intestines and the livers were evaluated. We found that CORT-treated mice displayed an obese phenotype along with longer small intestines and elongated intestinal villi, which might be the result of increased proliferation and decreased death of intestinal cells. RNA-seq analysis suggested that CORT-treatment significantly increased the expression levels of proliferation-related genes (such as Glul, Wee1 and Xiap) and decreased the expression levels of apoptosis-related genes (such as Casp1 and Casp7) in small intestines. Furthermore, in the liver, the expression levels of Fgf1 and Fgf21, which are known to regulate the length of intestines, were also up-regulated. We concluded that CORT induced longer small intestines and villi via promoting intestinal cell proliferation and survival, which contributed to obesity. This study would provide a novel therapeutic clue for the stress/GCs-related obesity.
After 8 weeks of treatment with CORT, mice showed remarkable increases in water intake, food intake, body weight and body size (Figures 1A–D). Magnetic resonance imaging (MRI) revealed that the CORT-treated mice showed remarkable fat accumulation (Figure 1E). According to the T1-weighted images, pseudo-color images, and fat distribution maps, CORT-treated mice showed a higher fat mass (10.10 ± 0.26 vs. 1.98 ± 0.50 g, p=0.0001) and a higher percentage of fat (31.37 ± 1.00 vs. 6.66 ± 1.69%, p = 0.0002) (Figures 1F, G). Histological analysis showed longer small intestines (Figures 1H, I) and hepatic steatosis (Figure 1J). Furthermore, we found a positive relationship between the body weight and the length of small intestines (Figure 1K). Chronic CORT exposure led to significantly elevated levels of serum leptin (31.91 ± 3.21 vs. 1.08 ± 0.12 ng/mL, p<0.0001) and insulin (28.90 ± 2.15 vs. 15.97 ± 3.31 μIU/mL, p=0.0179) (Figures 1L, M); however, there is no significant difference in the level of serum triglyceride (114.30 ± 43.00 vs. 252.10 ± 102.30 ng/μL, p=0.1939) between the two groups (Figure 1N). This result indicated that CORT-treatment increased the length of small intestines, which might contribute to more nutrients absorption, fat accumulation and weight gain.
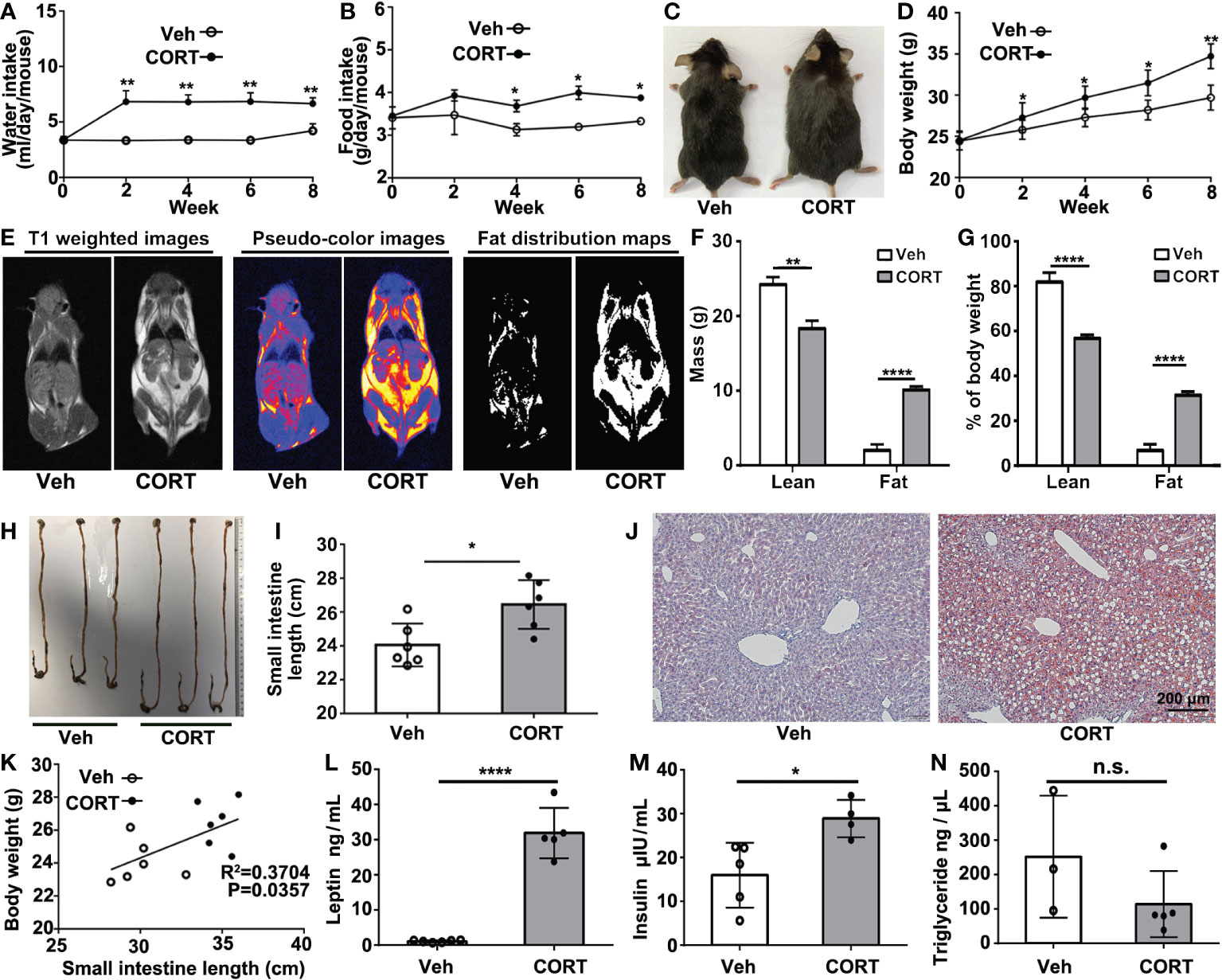
Figure 1 CORT induced obese phenotype, increased intestinal length, hepatic steatosis and changes in serum. (A) Water consumption and (B) food consumption of mice injected with CORT or Vehicle. (C) Representative images comparing the body size of a CORT-treated mouse with a vehicle-control mouse. (D) Body weight curves of mice treated with CORT or vehicle for 8 weeks. (E) Representative MRI images for body composition analysis. (F) Evaluation of fat mass. (G) Evaluation of fat percentage. (H) The representative images of the intestines of the mice treated with CORT or vehicle. (I) Quantification of the length of the small intestines from the mice treated with CORT or vehicle. (J) Representative images of Oil red O staining of the liver sections. (K) Correlation analysis between body weight and small intestine length. (L) Detection of the concentrations of serum leptin, (M) insulin and (N) triglyceride for the two groups of mice. Data are expressed as mean ± SEM, *p < 0.05, **p < 0.01, ****p < 0.0001. Veh: vehicle-treated group; CORT, corticosterone-treated group; n.s., not significant.
The tiny finger-like intestinal villi in the small intestines, projecting into the intestinal cavity, greatly increase the intestinal surface area for nutrients absorption. Several studies implied that the increase in villi length might contribute to increased nutrient absorption, weight gain and fat accumulation (7–9). In the present study, CORT-treated mice showed longer intestinal villi (605.10 ± 26.89 μm, n=12) than that in the control mice (444.80 ± 11.24 μm, n=14) (Figures 2A, B). The length of intestinal villus is determined by the balance between the rates of proliferation and death of intestinal cells, so we then evaluated cell proliferation and apoptosis in intestinal villi. EdU (ethynyl-2’-deoxyuridine), which could incorporate into newly synthesized DNA of dividing cells during the S phase (11), was administered by intraperitoneal injection to quantify intestinal cell proliferation and migration as previously described (12). In CORT-treated mice, small intestines showed remarkable increase in EdU progression (Figures 2C, D) and deeper crypt depth (Figures 2E, F; labeled with Ki67 and measured as the distance from the crypt mouth to the crypt base), indicating an increased proliferation rate. Furthermore, CORT-treated mice showed fewer TUNEL-positive (Figures 2G, H) or Cleaved Caspase 3 (CC3)-positive cells (Figures 2I, J) at the villus tip, indicating delayed shedding at the tip and decreased intestinal cell death by apoptosis. This result indicated that CORT-treatment regulated intestinal cell proliferation and survival.
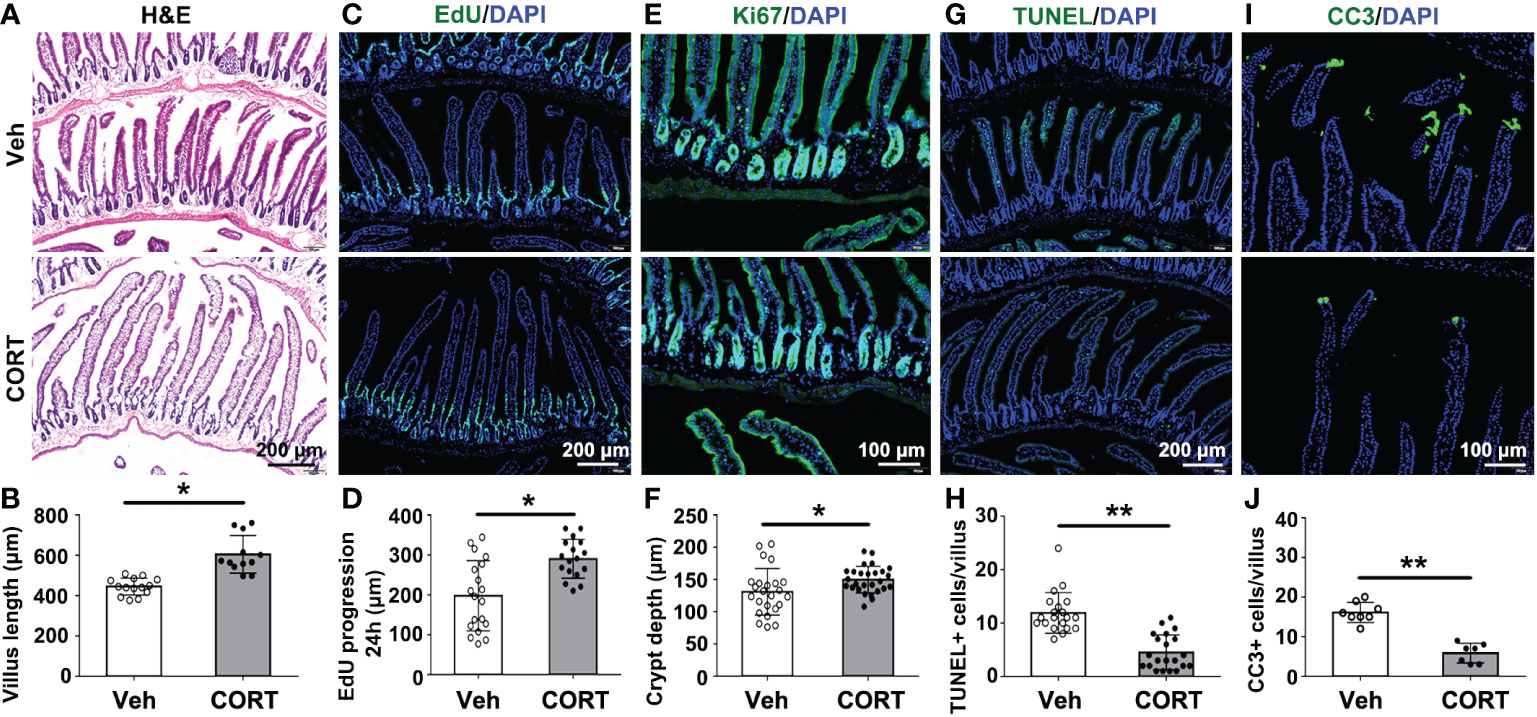
Figure 2 CORT increased intestinal villus length by breaking the balance between proliferation and death of intestinal cells. (A) Hematoxylin and eosin (H&E)-staining for duodenum sections. (B) Quantitative analysis of villi length for duodenum from Vehicle- or CORT-treated mice. (C) Immunofluorescence staining of EdU for the duodenum sections from Vehicle- or CORT-treated mice. (D) Quantification of the progression of EdU-labelled cells along the duodenum villi from Vehicle- or CORT-treated mice. (E) Immunofluorescence staining of Ki67 for the duodenum sections from Vehicle- or CORT-treated mice. (F) Quantification of the duodenal crypt depth indicated by Ki67-staining from Vehicle- or CORT-treated mice. (G) Immunofluorescence staining of TUNEL for the duodenum sections from Vehicle- or CORT-treated mice. (H) Quantification of TUNEL-positive cells of duodenal crypt from Vehicle- or CORT-treated mice. (I) Immunofluorescence staining of CC3 for the duodenum sections from Vehicle- or CORT-treated mice. (J) Quantification of CC3-positive cells of duodenal crypt from Vehicle- or CORT-treated mice. Data are expressed as mean ± SEM, *p < 0.05; **p < 0.01. CC3: Cleaved Caspase 3. Veh: vehicle-treated group; CORT: corticosterone-treated group.
To determine the transcriptional changes in the small intestines after CORT administration, we profiled the transcriptomics of small intestines by RNA-seq. The PCA (principal-component analysis) is shown in Figure 3A. After identifying the DEGs (genes with > 1.5-fold change in expression, p < 0.05), we found that there were 1155 genes with significant differences between the two groups (Figure 3B). Of the 1155 DEGs, 628 genes were up-regulated and 527 genes were down-regulated (Figure 3C). We further examined the functional roles of these genes. By KEGG analysis, 33 DEGs in the category of “Cell growth and death” were identified (Figure 3D). Enrichment analysis showed that the 33 DEGs were enriched in the pathways of Cell cycle, Apoptosis and Pathway in cancer (Figure 3E). The heatmap indicated that of the 33 genes, 11 genes were up-regulated and 22 genes were down-regulated in CORT-treated group. We identified 5 genes (Glul, Wee1, Xiap, Casp1 and Casp7) (Figure 3F), which were reported to mediate apoptosis resistance and cell proliferation (13–22). This result suggested that CORT-induced increased intestinal length might be due to activation of signaling pathways associated with cell proliferation and apoptosis resistance.
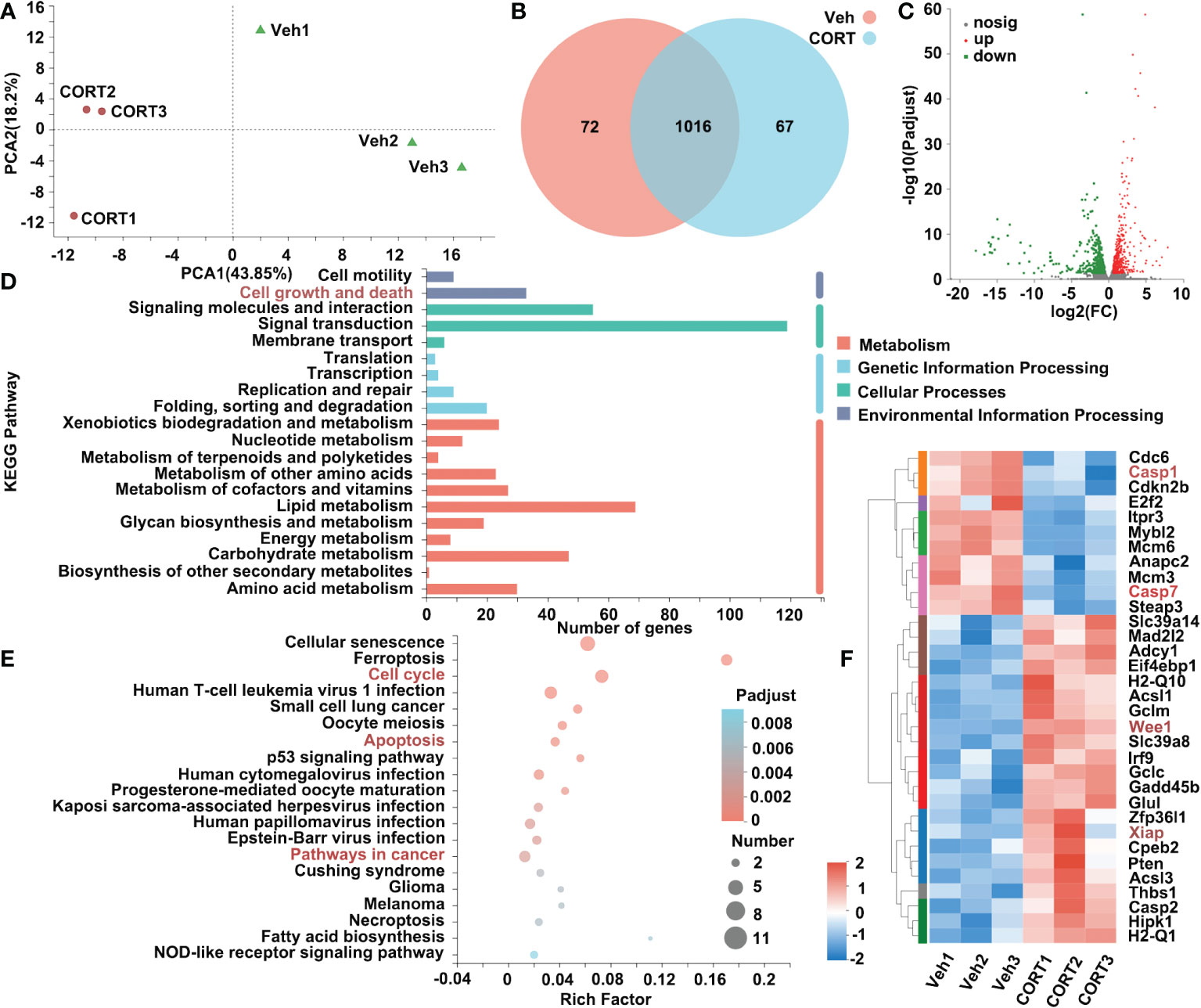
Figure 3 Identification of genes that were differentially expressed in intestines in response to CORT treatment. (A) Principal component analysis of the transcriptome of the intestine samples collected from the mice administrated with CORT or Vehicle. (B) Venn diagram displaying the distribution of DEGs (genes with > 1.5-fold change in expression) for the intestine samples of the two groups. If the TPM value of the gene was lower than 1, the gene was considered to be not expressed in the intestines of the group. (C) Volcano plots of the DEGs for intestines of the 2 groups. The up-regulated genes are presented by red dots, and the down-regulated genes are presented by green dots. (D) The functional classification of DEGs for intestines of the 2 groups based on first-category KEGG pathway analysis. (E) The functional classification of DEGs for intestines of the 2 groups based on second-category KEGG pathway analysis. (F) Heatmap of DEGs for intestines of the 2 groups in the category of “cell growth and death”. Veh: vehicle-treated group; CORT: corticosterone-treated group.
To better understand the impact of CORT, we also investigated the effects of CORT on the liver, the key organ for metabolism. After CORT treatment, the profile of transcriptomics changed dramatically. The PCA of RNA-seq is shown in Figure 4A. After identifying the DEGs (genes with > 1.5-fold change in expression, p < 0.05), we found that there were 2051 genes with significant differences between the CORT-treated and the control groups (Figure 4B). We further examined the functional roles of the 2051 genes. Of the 2051 DEGs, 1132 genes were up-regulated and 919 genes were down-regulated (Figure 4C). Among these genes, more than 220 genes are classified into Signal transduction (Figure 4D). Gene enrichment analysis showed signal transduction pathways (including MAPK, Rap1, PI3K-Akt and Ras signaling pathways) significantly changed in the liver when treated with CORT (Figure 4E). As shown in Figure 4F, 35 genes belonging to these pathways were significantly changed. Importantly, both fibroblast growth factor 1 (Fgf1) and fibroblast growth factor 21 (Ffg21), which could trigger these pathways, were increased after CORT treatment (Figure 4F). The increased expression levels of Fgf1 and Ffg21 in mice livers were also confirmed by quantitative real-time PCR (qPCR) (Figure S1A). This result indicated that the CORT-induced increased intestinal length and nutrients absorptive capability might be mediated by FGF signaling network, which functions to regulate cell growth and maintain the function and morphology of gastrointestinal tract.
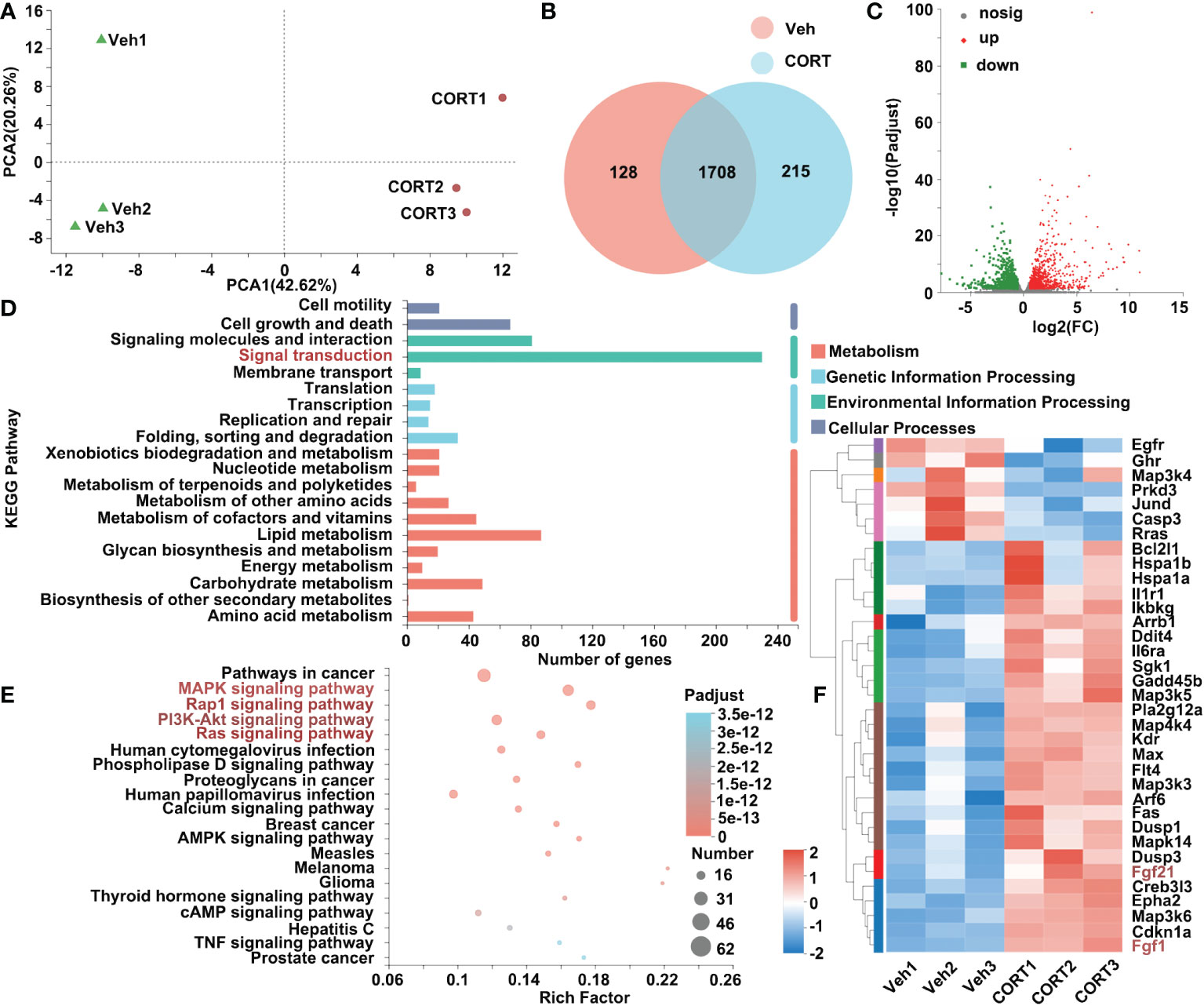
Figure 4 Identification of genes that were differentially expressed in livers in response to CORT treatment. (A) Principal component analysis of the transcriptome of the liver samples collected from the mice administrated with CORT or Vehicle. (B) Venn diagram displaying the distribution of DEGs (genes with > 1.5-fold change in expression) for the liver samples of the two groups. If the TPM value of the gene was lower than 1, the gene was considered to be not expressed in the livers of the group. (C) Volcano plots of the DEGs for the liver samples of the two groups. The up-regulated genes are presented by red dots, and the down-regulated genes are presented by green dots. (D) The functional classification of DEGs for livers of the 2 groups based on first-category KEGG pathway analysis. (E) The functional classification of DEGs for livers of the 2 groups based on second-category KEGG pathway analysis. (F) Heatmap of DEGs commonly expressed in the 4 pathways (labeled in red, 4E) for livers of the 2 groups in the category of “signal transduction”. Veh: vehicle-treated group; CORT: corticosterone-treated group.
The present study showed: (1) CORT treatment led to longer small intestines, which was then proved to be positively correlated to weight gain; (2) The CORT-treated mice displayed longer intestinal villi with enhanced proliferation and reduced apoptosis of intestinal cells; (3) RNA-seq analysis of the small intestines and the livers, indicating possible signaling pathways involved in the CORT-induced obesity.
Obesity is likely to be caused by calorie surpluses, in which energy intake initially exceeds energy expenditure (10). Accumulated evidence suggests that the increase of GCs plays an important role in the development of obesity (2). CORT-treated mice became obese, even when the food intake was restricted to the same amount as consumed by the control mice (6). This prompted us to raise a question whether CORT-treatment could cause higher absorptive capacity and improved nutrient absorption, thus leading to obesity or fat accumulation. In the present study, the CORT-treated mice exhibited obese phenotype (Figures 1C–G), longer small intestines (Figures 1H, I) and elongated intestinal villi (Figures 2A, B), which are reported to contribute greatly to the enormous absorptive surface area (7–9). Indeed, the unique features for the small intestines, such as circular folds and villi, can increase the absorptive surface area of the small intestine more than 600-fold, thus leading to more efficient nutrient absorption and obesity. In this study, male but not female mice were used as the experimental subjects. Inclusion of female mice in the scientific study is a controversial issue due to the fact that female mice go through an estrous cycle, during which their hormone concentrations change in a 4-to-5 day schedule (23). And most of the scientists argued that the response of female mice might vary too much to be used in the experiments if they are at different time points of their estrous cycle. Actually, accumulated evidence suggested that sex hormones could affect metabolism and distribution of adipose tissues and even obesity (24). Therefore, female mice were not included in the present study due to the reason that sex hormone might become a key variable and add undesirable noise for data collection and analysis.
The positive relationship between body weight and the intestinal length (Figure 1K) indicated that obesity might be attributed to CORT-induced intestinal cell proliferation and enormous absorptive surface area. However, we could not exclude the possibility that CORT-induced the central nervous system (CNS)-mediated hyperphagia might also cause obesity. Indeed, the feeding behavior is regulated by CNS to integrate peripheral hunger and satiety signals, subsequently regulating the energy balance and weight status. Feeding behavior is often distinguished by the separation of homeostatic and hedonic processes. Even in the presence of sufficient energy storage, the hedonic drive may override homeostatic control by enhancing the desire to consume delicious meals. Key CNS regions involved in the hedonic pathway include dopaminergic reward centers (nucleus accumbens, amygdala, ventral tegmental area) and the prefrontal impulsivity-control networks (25). In the central nucleus of the amygdala, long-term GC treatment stimulates the production of corticotropin-releasing factor (CRF). CRF enables the recruitment of a chronic stress network, which alters the normal output of autonomic, neuroendocrine and behavioral systems, leading to an increased HPA axis activity. Increased activation of this system leads to an increase in the consumption of high-energy foods (26), which in turn causes increased adipose tissue storage and even metabolism-associated disorders.
Consistent with the well-established role of CORT as a crucial regulator for glucose metabolism-associated hormones, such as leptin and insulin (27–29), the present study exposed that chronic CORT-treated mice showed remarkably increased levels for serum leptin and insulin (Figures 1L, M). This result is consistent with a previous study, which reported the elevated plasma leptin and insulin in chronic CORT-treated mice (30). In clinical studies: Newcomer et al. reported that short-term GCs treatment increased the plasma leptin concentration in a dose- and time-dependent manner (31); Plasma leptin levels in patients with Cushing’s syndrome were significantly higher than those in nonobese healthy participants and obese subjects without any metabolic or endocrine illnesses, indicating the role of GCs as a key regulator for leptin synthesis and secretion (32). The controversy remains about how GCs affect leptin secretion. One clinical study indicates that chronic exposure to endogenous GCs leads to insulin resistance, and GCs-induced hyperinsulinemia may act as a mediator of the steroid-induced elevation in leptin levels (33). However, a number of scientists suggest that the steroid-induced rise in leptin levels are not fully dependent on the hyperinsulinemia caused by GCs. According to the study from Veldman et al., patients with Cushing’s disease produced about twice as much leptin in an amplitude-specific manner, and this increased leptin concentration cannot be attributed to insulin production, indicating a cortisol-dependent change in adipose leptin secretion (34). Indeed, in vitro studies conducted on rodent adipocytes have demonstrated that GCs increased the mRNA level and secretion of leptin. These effects seem to be mediated by the direct transcriptional actions of GCs on glucocorticoid responsive elements (GRE) present in the leptin promoter (35).
Intestinal surface expansion depends on division of intestinal stem cells in the intestinal crypts and apoptosis of intestinal epithelial cell. Then we asked whether GCs could affect the proliferation or apoptosis of intestinal cells to enlarge intestinal surface area. The present study showed that CORT treatment enhanced the proliferation (Figures 2C–F) and inhibited the apoptosis of intestinal cells (Figures 2G–J). RNA-seq analysis showed that CORT up-regulated the expression of proliferation-related genes (such as Glul, Wee1 and Xiap) and down-regulated the expression of apoptosis-related genes (such as Casp1 and Casp7) (Figure 3F).
Glutamine plays a crucial role in maintaining a balance between proliferation and apoptosis of intestinal cells for normal functions (13). The expression level of glutamate-ammonia ligase (Glul, a gene of glutamine synthetase) was significantly up-regulated in CORT-treated small intestines, which is consistent with the previous studies which suggested that CORT could induce Glul expression in leukemia cells (14) and also in normal tissues (15). Although the function of CORT-induced Glul expression remained unclear, a previous study reported that inhibition of Glul decreased the proliferation of intestinal epithelial cells in vitro (16). Therefore, we speculated that CORT-induced Glul expression in mice small intestines might partly contribute to intestinal proliferation. High expression of Wee1 is positively corelated with cell proliferation and malignancy (17); and Wee1 inhibition could suppress gastrointestinal stromal tumor cell proliferation, and induce apoptosis and cell cycle arrest (18). Caspase family are essential for the initiation and execution of apoptosis (20). In murine model of intestinal ischemia-reperfusion (ICR) injury, the expression levels of Casp1 and Casp7 were significantly increased, while reducing Casp1 and Casp7 suppressed ICR-induced intestinal epithelial cell apoptosis (21, 22). Xiap showed its apoptosis resistance effect by repressing Casp7 using both an active site-directed mechanism and non-competitive mechanism (19).
All the blood leaving the stomach and intestines passes through the liver, which is a critical hub for numerous physiological processes, including macronutrient metabolism, lipid and cholesterol homeostasis, and the breakdown of xenobiotic drugs. As shown in Figure 4, RNA-seq analysis indicated the up-regulated Fgf1 and Fgf21 in CORT-treated livers. FGF signaling network plays a pivotal role in maintaining the function and morphology of gastrointestinal tract, such as WNT/FGF-dependent epithelial proliferation, FGF/RAS-MARK-mediated cell growth and differentiation, and development and repair processes of the gastrointestinal tract (36, 37). Dignass et al. showed that FGF1 (also known as acidic FGF, aFGF) enhanced epithelial cell proliferation (investigated on IEC-6, Caco-2, Caco-29 cell lines) (38). Interestingly, a mutant form of FGF1, which lost mitogenic activity, was proved to be more efficient at reducing cell apoptosis in the gut epithelium of rats following ICR injury (39). In addition, FGF1-treatment could attenuate ICR injury-induced excessive apoptosis in intestinal epithelium by promoting the proliferation of intestinal crypt cells (40, 41). Although the role of up-regulated Fgf21 in CORT-treated livers is largely unknown, several studies suggested that FGF21 might play a crucial role in maintaining the functions and morphology of intestines. Although FGF21 is primarily produced in the liver, a previous study reported a novel role of FGF21 as a hormonal factor contributing to neonatal intestinal function (42): the Fgf21-knockout (KO) pups fed with Fgf21-KO milk (lack of FGF21) showed a mild decreased body weight, whereas Fgf21-KO pups fed with WT milk (containing FGF21) showed up-regulated intestinal peptides, digestive enzymes, lactase enzymatic activity and normalized galactose levels and body weight. In addition, Sodium butyrate-treated mice showed increased proliferation of intestinal crypt cells, increased length of intestines and increased level of FGF21 in the liver, which might indicate the role of FGF21 in regulating intestinal cell proliferation (43). Further study should be carried out to explore the role of FGF21 in regulating intestinal function and morphology.
Obesity is a documented side effect of long-term GCs exposure, and GCs exert their physiological and pharmacological effect by binding to the nuclear receptor-glucocorticoid receptor (GR). A growing body of evidence suggests that GR responses to GCs to function as a regulator for adipose metabolism, including adipogenesis and lipolysis (44–46). Point-mutation (47) and polymorphisms (48–50) in GR gene are reported to be associated with obesity. We noticed the CORT-treated mice showed obesity and significantly decreased expression level of Nr3c1, a gene encodes GR, in the livers (Figure S1B). Consistent with the present study, a previous study reported that mice administered with CORT (4 weeks, drinking CORT in water) showed increased body weight and food intake, but decreased expression level for Nr3c1 in hypothalamus (51). According to clinical studies, long-term GC therapy usually causes the repression of Nr3c1 gene expression, which in turn causes a reduction in GC sensitivity and subsequently the development of acquired GC resistance (52). However, the precise factor controlling this process remains elusive. Indeed, the mechanisms for controlling Nr3c1 expression and translation are intricate and tightly regulated in tissues. Up to 13 variants have been identified in the untranslated exon 1 of human Nr3c1, functioning to regulate the transcription, the translation and the stability of mRNAs (53). In the present study, long-term CORT exposure-induced Nr3c1 down-regulation might be mediated by a negative feedback loop: one of the key factors affecting how cells respond to GCs is the GR expression level (54). Through a mechanism known as homologous down-regulation, GCs induce a decrease in GR levels in the majority of cells (55). As a result, the range of cellular responsiveness to GCs is constrained by this ligand-induced, receptor-mediated process. However, we could not exclude the possibility that alterations in GR expression/activation in brain or other tissues might also contribute to chronic CORT exposure-induced obesity. Further studies by genetic manipulation of GR expression in the interested organs of experimental animals might help to uncover the role of GR in CORT-induced obesity.
Above all, chronic CORT-treatment promotes intestine proliferation and inhibits apoptosis for intestinal cells via multiple metabolism-associated signaling pathways, thus leading to enlarged intestine-mediated more nutrient absorption and obesity. Understanding these altered metabolism-associated pathways might provide novel therapeutic clues for GCs or stress-associated obesity.
Male C57BL6/J mice (age: 8 weeks; weight: 23-25 g, n=32) purchased from Charles River Laboratories (Beijing, China) were maintained in a Specific Pathogen Free animal facility with standard conditions (room temperature: 24°C; humidity: 55%) under a 12:12 light/dark cycle. Food and water were provided ad libitum. All interventions and animal care procedures have been approved by the Animal Committee of School of Life Sciences and Technology, Tongji University, Shanghai, China.
Two groups of the weight-matched mice were injected with different solutions: either CORT or Vehicle. For drug trials, 34.7 mg corticosterone (Topscience, T0948L) was dissolved in 1ml DMSO, then 18.8 mL sterile water containing 200 μL Tween-80 was added into the solution to reach a final concentration of 1.734 mg/mL. Mice were administered with CORT (40 mg/kg) or vehicle (23.07 mL/kg) every day via subcutaneous (s.c.) injection for 8 weeks. Food intake (once a week), water intake (twice a week) and body weight (once a week) were recorded.
After 8 weeks of CORT injection, the mice were anesthetized and the body composition of mice was evaluated using a small animal in vivo MRI system (NM21-060H-I, Suzhou Niumag Analytical Instrument Corporation, Suzhou, China), according to the manufacturer’s instructions. Body composition was evaluated by T1-weighted images, and body fat was evaluated using the Meso NM21-060H-I sober small animal body composition analysis and imaging system (magnetic field strength, 0.5 T; Repetition Time (TR)/Echo Time (TE) = 360 ms/18.2 ms; matrix: 128 * 128; thickness: 3 mm).
At the end of the study, the whole-body perfusion and fixation was performed once the animal is under general anesthesia. Then the livers and intestines were isolated and transferred into 30% sucrose for dehydration. Then the completely dehydrated tissues were embedded in O.C.T. compound. Then freeze and store the embedded tissues at -80°C freezer until sectioning. The frozen tissue blocks were sectioned at 10-μm thickness using a cryostat (Lecia CM 1950), then the sections were stained for histological investigation using Hematoxylin-eosin (H&E) staining kit (Sangon Biotech, E607318-0200) and Oil Red O staining kit (Sangon Biotech, E607319-0010), following the manufacturer’s instructions. H&E image of swiss-rolled intestine was divided into 4 quadrants and 10 intact villi from each quadrant were randomly selected for measurement. Villus length was then determined as the distance from the distal edge of the crypt base to the villus apex.
In vivo proliferation assay was performed as previously described elsewhere (43) with some modifications. To visualize proliferative cells, mice were intraperitoneally injected with 5 mg/kg of EdU dissolved in sterile PBS and euthanized 24 hours later. Intestinal tissues were Swiss-rolled and dehydrated. Then the frozen sections were prepared as described above. Sections for intestinal rolls were stained for EdU using the Click-iT Alexa Flour 488 Imaging Kit (Thermo Fisher Scientific, C10337). The length from crypt to the EdU-labelled cells furthest from the crypt were measured from 40 villi in the duodenums of each mouse using ImageJ software.
Immunofluorescence staining was performed as we previously described (56) with some modifications. Slides were washed twice in PBS and incubated in blocking buffer (10% donkey/goat serum in PBST) for 1 hour at room temperature (RT). Then the slides were incubated with primary antibodies diluted in incubation buffer (5% donkey/goat serum in PBST) overnight at 4°C. After washing for 3 times, the slides were then incubated with Biotin-SP AffiniPure Goat Anti-Rabbit lgG (H+L) secondary antibody (Yesen, 33103ES60, dilution 1:200) for 2 hours at RT. Slides were then washed and incubated with Alexa Fluor 488 Streptavidin (Yesen, 35103ES60, dilution 1:400) and DAPI (Yesen, 40727ES10, dilution 1:1000). After washing with DPBS, the slides were mounted with anti-fade fluorescence mounting medium (Southern Biotech, 0100-01, Birmingham, USA). Imaging was performed using Olympus IX73 microscope system. Primary antibodies used for immunofluorescence: Ki67 (rabbit, Abcam ab16667, dilution 1:200), Cleaved Caspase 3 (CC3) (rabbit, Cell Signaling Technologies 9579S, dilution 1:200). The crypt depth was determined by Ki67-labeled cells to show the depth of the invagination between 2 villi. Deeper villi crypts indicate a higher metabolism state, allowing for the renewal and proliferation of the enterocytes. Immunofluorescence staining of CC3 was used to indicate enterocyte apoptosis. The number of CC3+ cells in each villus was counted using ImageJ software.
Terminal deoxynucleotidyl transferase dUTP nick end labeling (TUNEL) assay for detecting DNA fragments (apoptosis) was performed on frozen sections of duodenum using Click-IT TUNEL AlexaFluor647 kit (Roche, Cat. No. 11684795910), following the manufacturer’s instructions. TUNEL assay was used to indicate enterocyte apoptosis. The number of TUNEL+ cells in each villus was counted using ImageJ software.
The liver and intestine tissues (duodenum) of experimental mice were isolated and stored at -80°C until use. Then the frozen tissues (50 mg/sample) were send for RNA-extraction and RNA-seq analysis (Majorbio Bio-pharm Technology Co.,Ltd, Shanghai, China). Briefly, total RNA was extracted from different groups with three replicates. RNA-seq transcriptome library was prepared with 1 μg of total RNA using TruSeqTM RNA sample preparation Kit from Illumina (San Diego, CA). Then the paired-end RNA-seq library was sequenced with the Illumina HiSeq xten/NovaSeq 6000 sequencer (2 × 150 bp read length). The raw paired-end reads were trimmed and quality was controlled by SeqPrep (https://github.com/jstjohn/SeqPrep) and Sickle (https://github.com/najoshi/sickle), followed by aligning to the reference genome with orientation mode using HISAT2 software (57). The mapped reads of each sample were assembled by StringTie in a reference-based approach (58). The expression level of each transcript was calculated according to the transcripts per million reads (TPM) method. The TPM value of each DEG was shown in supplementary files. Transcript abundances were quantified using RSEM software tool (59). DEGs analysis was performed by DESeq2 with adjusted P value ≤ 0.05. KEGG pathway analysis were performed using KOBAS (http://kobas.cbi.pku.edu.cn/home.do).
The tissues were promptly isolated, placed into TRIzol and homogenized in a homogenizer (-50°C, 65 Hz) for 60 s. The residual genomic DNA was removed by DNase I treatment. The total RNA was diluted in RNAase free water, then 1 μg of RNA was reverse transcribed to cDNA using PrimeScriptTMRT reagent kit (TAKARA). qPCR was performed using the SYBR Green Mix (Bio-Rad) in a BioRad CFX96 Thermal Cycler. Housekeeping gene beta-actin was used to normalize mRNA levels between different samples. Primer sequences are listed as follows:
Fgf1-forward: GAGGATCCTTCCTGATGGC
Fgf1-reverse: CTCATTTGGTGTCTGCGAG
Fgf21-forward: GTCATTCAAATCCTGGGTGTC
Fgf21-reverse: AAAGTGAGGCGATCCATAGAG
Nr3c1-forward: AGAGGACAACCTGACTTCC
Nr3c1-reverse: ATTCCAGGGCTTGAATATCCA
beta-actin-forward: GCTATGTTGCTCTAGACTTCG
beta -actin-reverse: GGATTCCATACCCAAGAAGG
The experimental mice were anesthetized, and the skin around eye was pulled away to allow the eye to bulge out of the socket. Then a sterile capillary tube was penetrated into the corner of the eye socket underneath the eyeball to collect blood from orbital sinus into an Eppendorf tube. The samples were centrifuged (4°C, 3000 rpm, 15 min), then the supernatants were collected into Eppendorf tubes and stored at -80°C until use. The concentrations of leptin, insulin and triglycerides were detected by using the ELISA kits (leptin: KE10048, Proteintech; Insulin: ab277390, Abcam; Triglyceride: MAK266, Sigma-Aldrich.), according to the manufacturer’s instructions.
Statistical analysis was performed using GraphPad PRISM 8 (GraphPad Software, San Diego, CA, USA). Unpaired student’s t-test was used to compare the difference between two groups. Simple linear regression was used to analyze correlations. Pearson’s R2 coefficient, with confidence interval 95% and two-tailed P value were calculated to test the effectiveness of pairing. All data were expressed as the mean ± the standard error of the mean (SEM). *p < 0.05, **p < 0.01, ****p < 0.0001 were considered to be significant.
The data presented in the study are deposited in the Gene Expression Omnibus (GEO) repository, accession number GSE214009.
The animal study was reviewed and approved by Animal Committee of School of Life Sciences and Technology, Tongji University, Shanghai, China.
GW planned and performed molecular and animal experiments, analyzed data, produced the figures and wrote the manuscript. SL, YL, MZ and TX performed molecular experiments and collected the data. TL performed animal experiments. LC produced the figures, wrote the manuscript and provided financial support. JL designed and conceived this project, provided financial support, wrote the manuscript and approved the manuscript. All authors contributed to the article and approved the submitted version.
This work was supported by the National Key Research and Development Program of China [Grant number: 2021YFA1101300 and 2017YFA0104100]; the National Natural Science Foundation of China [Grant number: 31771644, 81801331 and 31930068]; Shanghai Yangzhi Rehabilitation Hospital (Shanghai Sunshine Rehabilitation Center) Talent introduction plan [Grant number: KYPT202204], and the Fundamental Research Funds for the Central Universities.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The reviewer CL declared a past co-authorship with the author JL to the handling editor.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fendo.2022.1052487/full#supplementary-material
1. Di Cesare M, Bentham J, Stevens GA, Zhou B, Danaei G, Lu Y, et al. Trends in adult body-mass index in 200 countries from 1975 to 2014: A pooled analysis of 1698 population-based measurement studies with 19.2 million participants. Lancet (2016) 387(10026):1377–96. doi: 10.1016/s0140-6736(16)30054-x
2. Dallman MF. Stress-induced obesity and the emotional nervous system. Trends Endocrinol Metab (2010) 21(3):159–65. doi: 10.1016/j.tem.2009.10.004
3. Adam TC, Epel ES. Stress, eating and the reward system. Physiol Behav (2007) 91(4):449–58. doi: 10.1016/j.physbeh.2007.04.011
4. Ip CK, Zhang L, Farzi A, Qi Y, Clarke I, Reed F, et al. Amygdala npy circuits promote the development of accelerated obesity under chronic stress conditions. Cell Metab (2019) 30(1):111–+. doi: 10.1016/j.cmet.2019.04.001
5. Rosmond R, Dallman MF, Bjorntorp P. Stress-related cortisol secretion in men: Relationships with abdominal obesity and endocrine, metabolic and hemodynamic abnormalities. J Clin Endocrinol Metab (1998) 83(6):1853–9. doi: 10.1210/jc.83.6.1853
6. van Rossum EFC. Obesity and cortisol: New perspectives on an old theme. Obesity (2017) 25(3):500–1. doi: 10.1002/oby.21774
7. Taylor SR, Ramsamooj S, Liang RJ, Katti A, Pozovskiy R, Vasan N, et al. Dietary fructose improves intestinal cell survival and nutrient absorption. Nature (2021) 597(7875):263–+. doi: 10.1038/s41586-021-03827-2
8. Stojanovic O, Altirriba J, Rigo D, Spiljar M, Evrard E, Roska B, et al. Dietary excess regulates absorption and surface of gut epithelium through intestinal ppar alpha. Nat Commun (2021) 12(1):7031. doi: 10.1038/s41467-021-27133-7
9. Walton KD, Freddo AM, Wang S, Gumucio DL. Generation of intestinal surface: An absorbing tale. Development (2016) 143(13):2261–72. doi: 10.1242/dev.135400
10. Bowles NP, Karatsoreos IN, Li X, Vemuri VK, Wood J-A, Li Z, et al. A peripheral endocannabinoid mechanism contributes to glucocorticoid-mediated metabolic syndrome. Proc Natl Acad Sci United States America (2015) 112(1):285–90. doi: 10.1073/pnas.1421420112
11. Gratzner HG. Monoclonal-antibody to 5-bromodeoxyuridine and 5-iododeoxyuridine - a new reagent for detection of DNA-replication. Science (1982) 218(4571):474–5. doi: 10.1126/science.7123245
12. Parker A, Maclaren OJ, Fletcher AG, Muraro D, Kreuzaler PA, Byrne HM, et al. Cell proliferation within small intestinal crypts is the principal driving force for cell migration on villi. FASEB J (2017) 31(2):636–49. doi: 10.1096/fj.201601002
13. Kim M-H, Kim H. The roles of glutamine in the intestine and its implication in intestinal diseases. Int J Mol Sci (2017) 18(5):1051. doi: 10.3390/ijms18051051
14. Harmon JM, Thompson EB. Glutamine-synthetase induction by glucocorticoids in the glucocorticoid-sensitive human-leukemic cell-line cem-C7. J Cell Physiol (1982) 110(2):155–60. doi: 10.1002/jcp.1041100208
15. Max SR, Mill J, Mearow K, Konagaya M, Konagaya Y, Thomas JW, et al. Dexamethasone regulates glutamine-synthetase expression in rat skeletal-muscles. Am J Physiol (1988) 255(3):E397–403. doi: 10.1152/ajpendo.1988.255.3.E397
16. DeMarco V, Dyess K, Strauss D, West CM, Neu J. Inhibition of glutamine synthetase decreases proliferation of cultured rat intestinal epithelial cells. J Nutr (1999) 129(1):57–62. doi: 10.1093/jn/129.1.57
17. Ghelli Luserna di Rora A, Cerchione C, Martinelli G, Simonetti G. A Wee1 family business: Regulation of mitosis, cancer progression, and therapeutic target. J Hematol Oncol (2020) 13(1):126. doi: 10.1186/s13045-020-00959-2
18. Liu W, Zeng X, Yin Y, Li C, Yang W, Wan W, et al. Targeting the Wee1 kinase strengthens the antitumor activity of imatinib Via promoting kit autophagic degradation in gastrointestinal stromal tumors. Gastric Cancer (2020) 23(1):39–51. doi: 10.1007/s10120-019-00977-1
19. Suzuki Y, Nakabayashi Y, Nakata K, Reed JC, Takahashi R. X-Linked inhibitor of apoptosis protein (Xiap) inhibits caspase-3 and-7 in distinct modes. J Biol Chem (2001) 276(29):27058–63. doi: 10.1074/jbc.M102415200
20. Fan TJ, Han LH, Cong RS, Liang J. Caspase family proteases and apoptosis. Acta Biochim Et Biophys Sin (2005) 37(11):719–27. doi: 10.1111/j.1745-7270.2005.00108.x
21. Xu M, Yang Y, Deng Q-W, Shen J-T, Liu W-F, Yang W-J, et al. Microarray profiling and functional identification of lncrna in mice intestinal mucosa following intestinal Ischemia/Reperfusion. J Surg Res (2021) 258:389–404. doi: 10.1016/j.jss.2020.08.066
22. Taha MO, Alexandre e Silva T, Ota KS, Vilela WJ, Simoes RS, Starzewski Junior A, et al. The role of atenolol in the modulation of the expression of genes encoding pro- (Caspase-1) and anti- (Bcl2l1) apoptotic proteins in endothelial cells exposed to intestinal ischemia and reperfusion in rats. Acta Cirurgica Bras (2018) 33(12):1061–6. doi: 10.1590/s0102-865020180120000003
23. Caligioni CS. Assessing reproductive Status/Stages in mice. Curr Protoc Neurosci (2009) Appendix 4:4I–Appendix I. doi: 10.1002/0471142301.nsa04is48
24. Mayes JS, Watson GH. Direct effects of sex steroid hormones on adipose tissues and obesity. Obes Rev (2004) 5(4):197–216. doi: 10.1111/j.1467-789X.2004.00152.x
25. Kessler RM, Hutson PH, Herman BK, Potenza MN. The neurobiological basis of binge-eating disorder. Neurosci Biobehav Rev (2016) 63:223–38. doi: 10.1016/j.neubiorev.2016.01.013
26. Dallman MF, Pecoraro N, Akana SF, la Fleur SE, Gomez F, Houshyar H, et al. Chronic stress and obesity: A new view of "Comfort food". Proc Natl Acad Sci United States America (2003) 100(20):11696–701. doi: 10.1073/pnas.1934666100
27. Beaupere C, Liboz A, Feve B, Blondeau B, Guillemain G. Molecular mechanisms of glucocorticoid-induced insulin resistance. Int J Mol Sci (2021) 22(2):623. doi: 10.3390/ijms22020623
28. Leal-Cerro A, Soto A, Martinez MA, Dieguez C, Casanueva FF. Influence of cortisol status on leptin secretion. Pituitary (2001) 4(1-2):111–6. doi: 10.1023/a:1012903330944
29. la Fleur SE, Akana SF, Manalo SL, Dallman MF. Interaction between corticosterone and insulin in obesity: Regulation of lard intake and fat stores. Endocrinology (2004) 145(5):2174–85. doi: 10.1210/en.2003-1359
30. Karatsoreos IN, Bhagat SM, Bowles NP, Weil ZM, Pfaff DW, McEwen BS. Endocrine and physiological changes in response to chronic corticosterone: A potential model of the metabolic syndrome in mouse. Endocrinology (2010) 151(5):2117–27. doi: 10.1210/en.2009-1436
31. Newcomer JW, Selke G, Melson AK, Gross J, Vogler GP, Dagogo-Jack S. Dose-dependent cortisol-induced increases in plasma leptin concentration in healthy humans. Arch Gen Psychiatry (1998) 55(11):995–1000. doi: 10.1001/archpsyc.55.11.995
32. Masuzaki H, Ogawa Y, Hosoda K, Miyawaki T, Hanaoka I, Hiraoka J, et al. Glucocorticoid regulation of leptin synthesis and secretion in humans: Elevated plasma leptin levels in cushing's syndrome. J Clin Endocrinol Metab (1997) 82(8):2542–7. doi: 10.1210/jc.82.8.2542
33. Widjaja A, Schurmeyer TH, Von zur Muhlen A, Brabant G. Determinants of serum leptin levels in cushing's syndrome. J Clin Endocrinol Metab (1998) 83(2):600–3. doi: 10.1210/jc.83.2.600
34. Veldman RG, Frolich M, Pincus SM, Veldhuis JD, Roelfsema F. Hyperleptinemia in women with cushing's disease is driven by high-amplitude pulsatile, but orderly and eurhythmic, leptin secretion. Eur J Endocrinol (2001) 144(1):21–7. doi: 10.1530/eje.0.1440021
35. Gong DW, Bi S, Pratley RE, Weintraub BD. Genomic structure and promoter analysis of the human obese gene. J Biol Chem (1996) 271(8):3971–4. doi: 10.1074/jbc.271.8.3971
36. Katoh M, Katoh M. Fgf signaling network in the gastrointestinal tract (Review). Int J Oncol (2006) 29(1):163–8. doi: 10.3892/ijo.29.1.163
37. Danopoulos S, Schlieve CR, Grikscheit TC, Al Alam D. Fibroblast growth factors in the gastrointestinal tract: Twists and turns. Dev Dynamics (2017) 246(4):344–52. doi: 10.1002/dvdy.24491
38. Dignass AU, Tsunekawa S, Podolsky DK. Fibroblast growth-factors modulate intestinal epithelial-cell growth and migration. Gastroenterology (1994) 106(5):1254–62. doi: 10.1016/0016-5085(94)90017-5
39. Fu X-B, Li X-K, Wang T, Cheng B, Sheng Z-Y. Enhanced anti-apoptosis and gut epithelium protection function of acidic fibroblast growth factor after cancelling of its mitogenic activity. World J Gastroenterol (2004) 10(24):3590–6. doi: 10.3748/wjg.v10.i24.3590
40. Chen W, Fu X-B, Ge S-L, Sun T-Z, Zhou G, Han B, et al. Intravenous acid fibroblast growth factor protects intestinal mucosal cells against ischemia-reperfusion injury Via regulating bcl-2/Bax expression. World J Gastroenterol (2005) 11(22):3419–25. doi: 10.3748/wjg.v11.i22.3419
41. Li H-H, Fu X-B, Sun T-Z, Cai C-L, Zhou G, Chen W, et al. Non-mitogenic acidic fibroblast growth factor reduces intestinal dysfunction induced by ischemia and reperfusion injury in rats. J Gastroenterol Hepatol (2007) 22(3):363–70. doi: 10.1111/j.1440-1746.2006.04457.x
42. Gavalda-Navarro A, Hondares E, Giralt M, Mampel T, Iglesias R, Villarroya F. Fibroblast growth factor 21 in breast milk controls neonatal intestine function. Sci Rep (2015) 5:13717. doi: 10.1038/srep13717
43. Kundu P, Lee HU, Garcia-Perez I, Tay EXY, Kim H, Faylon LE, et al. Neurogenesis and prolongevity signaling in young germ-free mice transplanted with the gut microbiota of old mice. Sci Trans Med (2019) 11(518):eaau4760. doi: 10.1126/scitranslmed.aau4760
44. Shen Y, Roh HC, Komori M, Rosen ED. Adipocyte glucocorticoid receptor is important in lipolysis and insulin resistance due to exogenous steroids, but not insulin resistance caused by high fat feeding. Mol Metab (2017) 6(10):1150–60. doi: 10.1016/j.molmet.2017.06.013
45. Peckett AJ, Wright DC, Riddell MC. The effects of glucocorticoids on adipose tissue lipid metabolism. Metabol-Clin Exp (2011) 60(11):1500–10. doi: 10.1016/j.metabol.2011.06.012
46. Lee MJ, Fried SK. The glucocorticoid receptor, not the mineralocorticoid receptor, plays the dominant role in adipogenesis and adipokine production in human adipocytes. Int J Obes (2014) 38(9):1228–33. doi: 10.1038/ijo.2014.6
47. Charmandari E, Ichijo T, Jubiz W, Baid S, Zachman K, Chrousos GP, et al. A novel point mutation in the amino terminal domain of the human glucocorticoid receptor (Hgr) gene enhancing hgr-mediated gene expression. J Clin Endocrinol Metab (2008) 93(12):4963–8. doi: 10.1210/jc.2008-0892
48. Di Blasio AM, van Rossum EFC, Maestrini S, Berselli ME, Tagliaferri M, Podesta F, et al. The relation between two polymorphisms in the glucocorticoid receptor gene and body mass index, blood pressure and cholesterol in obese patients. Clin Endocrinol (2003) 59(1):68–74. doi: 10.1046/j.1365-2265.2003.01798.x
49. van Rossum EFC, Lamberts SWJ. Polymorphisms in the glucocorticoid receptor gene and their associations with metabolic parameters and body composition. Recent Prog Hormone Res (2004) 59:333–57. doi: 10.1210/rp.59.1.333
50. Giordano R, Marzotti S, Berardelli R, Karamouzis I, Brozzetti A, D'Angelo V, et al. Bcli polymorphism of the glucocorticoid receptor gene is associated with increased obesity, impaired glucose metabolism and dyslipidaemia in patients with addison's disease. Clin Endocrinol (2012) 77(6):863–70. doi: 10.1111/j.1365-2265.2012.04439.x
51. Sefton C, Harno E, Davies A, Small H, Allen T-J, Wray JR, et al. Elevated hypothalamic glucocorticoid levels are associated with obesity and hyperphagia in Male mice. Endocrinology (2016) 157(11):4257–65. doi: 10.1210/en.2016-1571
52. Spies L-ML, Verhoog NJD, Louw A. Acquired glucocorticoid resistance due to homologous glucocorticoid receptor downregulation: A modern look at an age-old problem. Cells (2021) 10(10):2529. doi: 10.3390/cells10102529
53. Presul E, Schmidt S, Kofler R, Helmberg A. Identification, tissue expression, and glucocorticoid responsiveness of alternative first exons of the human glucocorticoid receptor. J Mol Endocrinol (2007) 38(1-2):79–90. doi: 10.1677/jme.1.02183
54. Vanderbilt JN, Miesfeld R, Maler BA, Yamamoto KR. Intracellular receptor concentration limits glucocorticoid-dependent enhancer activity. Mol Endocrinol (1987) 1(1):68–74. doi: 10.1210/mend-1-1-68
55. Oakley RH, Cidlowski JA. Homologous down regulation of the glucocorticoid receptor: The molecular machinery. Crit Rev Eukaryotic Gene Expression (1993) 3(2):63–88.
56. Cao L, Hu R, Xu T, Zhang Z-N, Li W, Lu J. Characterization of induced pluripotent stem cell-derived human serotonergic neurons. Front Cell Neurosci (2017) 11:131. doi: 10.3389/fncel.2017.00131
57. Kim D, Langmead B, Salzberg SL. Hisat: A fast spliced aligner with low memory requirements. Nat Methods (2015) 12(4):357–U121. doi: 10.1038/nmeth.3317
58. Pertea M, Pertea GM, Antonescu CM, Chang T-C, Mendell JT, Salzberg SL. Stringtie enables improved reconstruction of a transcriptome from rna-seq reads. Nat Biotechnol (2015) 33(3):290–+. doi: 10.1038/nbt.3122
Keywords: corticosterone, obesity, small intestines, cell proliferation, cell survival and death
Citation: Wang G, Li S, Li Y, Zhang M, Xu T, Li T, Cao L and Lu J (2023) Corticosterone induces obesity partly via promoting intestinal cell proliferation and survival. Front. Endocrinol. 13:1052487. doi: 10.3389/fendo.2022.1052487
Received: 24 September 2022; Accepted: 20 December 2022;
Published: 09 January 2023.
Edited by:
Hassan Heshmati, Endocrinology Metabolism Consulting, LLC, United StatesReviewed by:
Chen-Che Jeff Huang, Auburn University, United StatesCopyright © 2023 Wang, Li, Li, Zhang, Xu, Li, Cao and Lu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jianfeng Lu, THUuamlhbmZlbmdAdG9uZ2ppLmVkdS5jbg==; aGVhbHRoeWdvZF93b3JsZEBob3RtYWlsLmNvbQ==; Lining Cao, Y3J5c3RhbHZpdmluaW5nQGFsaXl1bi5jb20=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.