 Christina S. Ennis
Christina S. Ennis Pablo Llevenes
Pablo Llevenes Yuhan Qiu
Yuhan Qiu Ruben Dries
Ruben Dries Gerald V. Denis
Gerald V. Denis- 1Boston University-Boston Medical Center Cancer Center, Boston University School of Medicine, Boston, MA, United States
- 2Section of Hematology and Medical Oncology, Boston University School of Medicine, Boston, MA, United States
- 3Department of Pharmacology and Experimental Therapeutics, Boston University School of Medicine, Boston, MA, United States
- 4Division of Computational Biomedicine, Boston University School of Medicine, Boston, MA, United States
- 5Shipley Prostate Cancer Research Professor, Boston University School of Medicine, Boston, MA, United States
Obesity-driven (type 2) diabetes (T2D), the most common metabolic disorder, both increases the incidence of all molecular subtypes of breast cancer and decreases survival in postmenopausal women. Despite this clear link, T2D and the associated dysfunction of diverse tissues is often not considered during the standard of care practices in oncology and, moreover, is treated as exclusion criteria for many emerging clinical trials. These guidelines have caused the biological mechanisms that associate T2D and breast cancer to be understudied. Recently, it has been illustrated that the breast tumor microenvironment (TME) composition and architecture, specifically the surrounding cellular and extracellular structures, dictate tumor progression and are directly relevant for clinical outcomes. In addition to the epithelial cancer cell fraction, the breast TME is predominantly made up of cancer-associated fibroblasts, adipocytes, and is often infiltrated by immune cells. During T2D, signal transduction among these cell types is aberrant, resulting in a dysfunctional breast TME that communicates with nearby cancer cells to promote oncogenic processes, cancer stem-like cell formation, pro-metastatic behavior and increase the risk of recurrence. As these cells are non-malignant, despite their signaling abnormalities, data concerning their function is never captured in DNA mutational databases, thus we have limited insight into mechanism from publicly available datasets. We suggest that abnormal adipocyte and immune cell exhaustion within the breast TME in patients with obesity and metabolic disease may elicit greater transcriptional plasticity and cellular heterogeneity within the expanding population of malignant epithelial cells, compared to the breast TME of a non-obese, metabolically normal patient. These challenges are particularly relevant to cancer disparities settings where the fraction of patients seen within the breast medical oncology practice also present with co-morbid obesity and metabolic disease. Within this review, we characterize the changes to the breast TME during T2D and raise urgent molecular, cellular and translational questions that warrant further study, considering the growing prevalence of T2D worldwide.
Introduction
Obesity and metabolic disease poses a deepening challenge in the United States, where the burden of Type 2 diabetes (T2D) or pre-diabetes affects over 100 million adults (1–4). Critically, these diseases are implicated in a variety of cancers, both with an obesogenic environment, such as endometrium, colon, and kidney, as well as less common malignancies such as leukemia, multiple myeloma, and non-Hodgkin’s lymphoma (5). Obesity, while a potent risk factor outright, is highly associated with metabolic derangements that increase the incidence and mortality of such cancers (6). However, there are key groups of patients that exhibit counter-intuitive patterns of cancer development: metabolically-healthy obese and metabolically-obese normal weight, which are associated with a reduced and increased cancer prevalence, respectively, compared to metabolically-healthy normal-weight controls (7–10). The link between T2D and breast cancer is of particular significance, with diabetic women not only having a 40% increased risk of developing breast cancer compared to non-diabetic (ND) women, but also a 74% increase in overall mortality (2, 4). This high mortality rate is associated with more advanced stages and aggressive subtypes of breast cancer, such as estrogen receptor negative (ER-) and triple negative breast cancer (TNBC) (11, 12). Despite this significant risk factor, the cellular and molecular mechanisms underlying this comorbidity remain poorly understood and understudied. The combination of these illnesses is particularly challenging due to their heterogeneity and interconnectedness. Recent developments in spatial omics and multiplexed imaging technologies have revealed that cancers have complex spatial organization within their three-dimensional (3D) architectures that dictate a given cell’s spatial neighborhood, interactions, and phenotype to influence overall tumor behavior. The tumor microenvironment (TME), consisting of the cellular and extracellular structures surrounding cancer cells, has been identified to regulate essential tumor survival functions (13, 14). However, standard molecular tools, such as Oncotype, which are used in the clinic to assess personalized risk of recurrence do not account for the profound TME differences seen in T2D patients, with only one measure of mammary adipose inflammation (CD68) and no way to account for differing metabolism. Thus, patients with comorbid T2D may receive a dangerously low score that inaccurately estimates their true risk of progression and metastasis. Oncologists therefore urgently need improved diagnostic and therapeutic methods for patients with this comorbidity. Considering that diseased cells immediately adjacent to tumor are not passive structures but instead are active actors in tumor progression, we describe the impact of the TME on this complex yet increasingly common comorbidity (Figure 1).
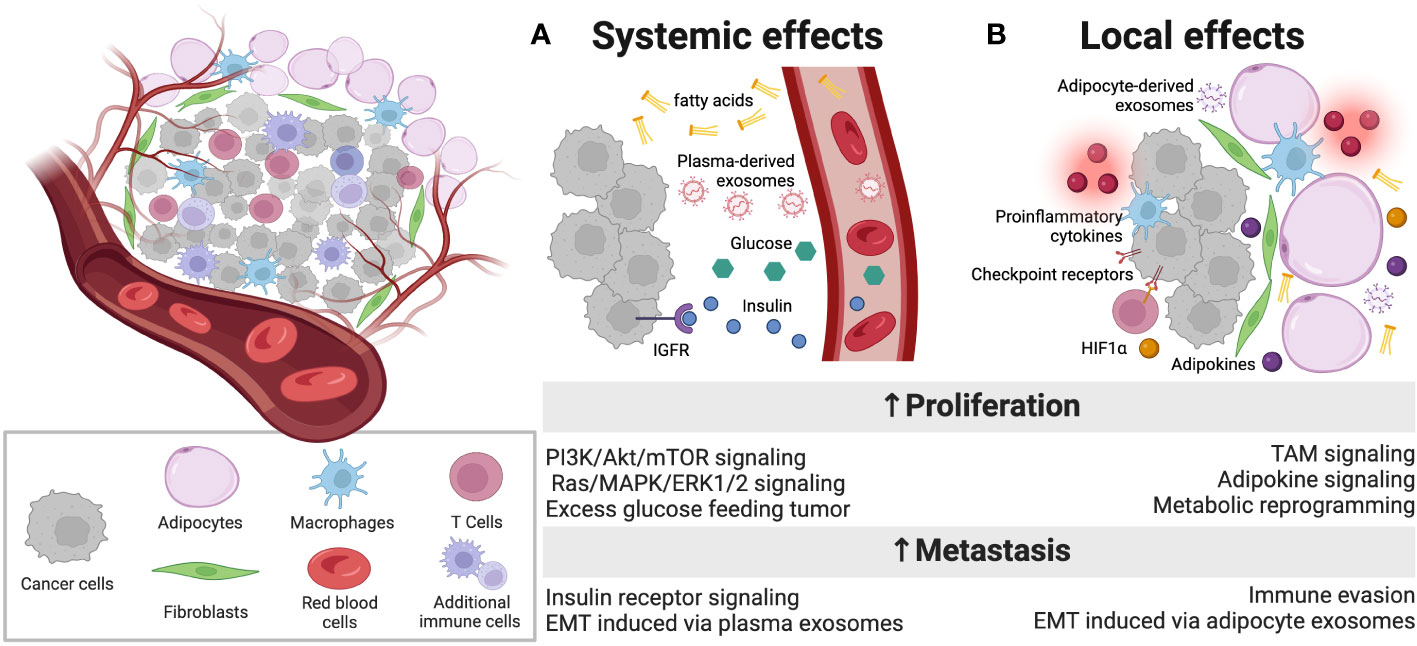
Figure 1 Overview of dysfunction within the diabetic breast TME. A complex array of cell types in the TME engages intercellular communication. (A) In addition to well described, systemic factors that are present at abnormally high levels in patients with obesity-driven diabetes, such as free fatty acids, glucose, insulin and IGF-1, which can promote proliferation, plasma exosomes are also altered in diabetes and carry intercellular instructions that promote tumor progression and metastasis. (B) Locally, adipocytes in the TME of diabetic patients are inflamed and dysfunctional, releasing proinflammatory cytokines that can alter the function of immune infiltrates, promoting T cell exhaustion through immune checkpoint engagement. These abnormal adipocytes also release adipokines and exosomes that carry payloads capable of reprogramming tumor cells to more aggressive and metastatic phenotypes. Additionally, fibroblasts respond to elevated levels of HIF1α in the local TME to further support the metabolic reprogramming of tumor cells. Figure created with Biorender.com.
Systemic effects
Insulin resistance-related metabolic reprogramming, a context-dependent and dynamic process that results from interactions between cancer cells and their local and systemic environments, includes three main aspects, 1) hyperinsulinemia, 2) hyperglycemia, and 3) dyslipidemia (15, 16).
Hyperinsulinemia is often seen in T2D as a result of insulin resistance, in which an impaired tissue response to insulin results in the pancreas increasing insulin levels to compensate and manage blood glucose levels. The significance of insulin signaling towards breast cancer progression has long been noted (17). Primarily, tumor cells widely overexpress insulin receptors (IRs) and insulin-like growth factor receptors (IGF-Rs) (18–21). These receptors can directly bind circulating insulin to activate downstream signaling pathways, such as PI3K/AKT/mTOR and the Ras/MAPK/ERK pathways, to increase mitosis and therefore cancer cell proliferation and invasion (22–29). Subsequent activation of the β-catenin signaling pathway via PI3K/AKT has been associated with cancer stemness and chemoresistance (30). Downregulation of IRs on tumor cells has been demonstrated to reduce tumor growth and lung metastasis in xenograft models of athymic mice (31). Moreover, though IR expression is highly expressed in the majority of early stage breast cancers, this expression is not clearly downregulated in the context of hyperinsulinemia (32). Additionally, IGF binding proteins, which limit the activity of IGF-1, are reduced in the presence of high levels of insulin (33, 34). Moreover, hyperinsulinemia can in turn increase IGF-1 expression in the liver and stimulate cell growth (18). Inhibition of IGF-IR has been shown to decrease growth of breast cancer in vitro (35, 36).
While hyperinsulinemia is seen as a primary causal factor for cancer, hyperglycemia has also been shown to positively associate with cancer incidence (37). It is well established that tumor cell proliferation needs glucose as an important source of fuel for ATP production as well as synthesis of DNA via the pentose-phosphate pathway (18). Further, hyperglycemia can promote epithelial-to-mesenchymal transition (EMT) to induce metabolic reprogramming by upregulating glucose uptake and lactate release (38, 39). As such, multiple large cohort and case-control studies have found that hyperglycemia is positively correlated with the risk of cancer (40–43). Critically, hyperglycemia does not exert a uniform effect on tumor growth in all in vivo models. For example, insulin-independent hyperglycemia increases the size of liver tumors and reduces apoptosis in a tumor-prone animal model, whereas in T1D in vivo models tumor growth is reduced by insulin (44, 45). However, improved glycemic control with compounds such as metformin has a mixed effect on cancer risk in diabetic patients, indicating that hyperglycemia may be an independent risk factor for cancer (46–51).
Dyslipidemia, characterized by elevated circulating levels of cholesterol, triglycerides, and free fatty acids, is also independently associated with an increased cancer prevalence (20, 52). Both elevated low-density lipoprotein and reduced high-density lipoprotein levels, the main transporters of cholesterol, have been demonstrated to be prognostic factors of breast cancer initiation, progression, and metastasis regardless of metabolic status (53–59). Cholesterol-lowering agents, such as lipophilic statins, have been shown to be protective against breast cancer recurrence and death (59). 27-hydroxycholesterol (27HC) is a primary metabolite of cholesterol, generated upon exposure to cytochrome P450 oxidase sterol 27-hydroxylase A1 (CYP27A1), a key enzyme in regulating cellular cholesterol homeostasis (59–61). 27HC acts as an ER agonist, activating the PI3K/AKT/mTOR and beta-catenin signaling pathways to stimulate cell proliferation and protein synthesis in ER-positive breast cancer (59, 61). Critically, high levels of CYP27A1 expression correlate with high-grade breast tumors, while inhibition of this enzyme reduces tumor growth in hormone-dependent breast cancer (61, 62). Cholesterol, triglycerides, and fatty acids are known to be critical lipid constituents of the cell, composing a majority of the cellular membrane. Highly proliferative cancer cells therefore benefit from the altered lipid metabolism associated with dyslipidemia to provide these essential building blocks (52, 63–66).
Local effects
In addition to global changes in signaling pathways, breast cancer with comorbid T2D also results in perturbations to the local TME. The breast TME is composed of several cell types that can all experience unique dysfunctions that work to promote tumor proliferation, invasion, and metastasis.
Adipocytes
Adipocytes are the most prevalent cell type by mass within the breast TME (67). Cancer-associated adipocytes (CAAs) have been demonstrated to promote breast cancer progression (68–70), as they function as an active endocrine tissue to release adipokines (e.g., IL-6, TNFα, leptin, and adiponectin) that can suppress an active immune response (71) and play critical roles in tumor cell proliferation, as well as matrix metalloproteinases that are important for tumor invasiveness (72). T2D and obesity are major contributing factors of inducing adipocyte abnormalities, which are known to promote cancer cell proliferation, invasion, and resistance to chemotherapy and radiotherapy (6). Breast cancer cells adapt to their unique TME to meet their needs for proliferation and cell survival via reprogramming their metabolic pathways (70, 73, 74). Accordingly, it has been reported that fatty acid oxidation and pathways required for formation of cell membranes and storage are upregulated in breast cancer (75, 76). In accordance with the need for fuel in proliferation, breast cancer cells develop ways to utilize FAs, such as de novo fatty acid synthesis (77). In human epidermal growth factor receptor 2 positive (HER2+) breast cancers, key enzymes involved in this process, fatty acid synthase and acetyl-CoA-carboxylase-a, are upregulated via the PI3K/Akt/mTOR pathway (65, 77). Breast cancer cells obtain fuel from its TME through an increased uptake of FAs from CAAs, which requires lipoprotein lipase and fatty acid binding protein 5 and 7, which are all overexpressed in TNBC (53, 77, 78). Despite the significance of CAA-mediated lipid transfer towards breast cancer progression, experiments utilizing primary CAAs from patients with comorbid T2D, who would be experiencing dyslipidemia and having these pathways further perturbed, is lacking. Muller and others have demonstrated that TNBC and ER+ breast tumor cells and nearby CAAs, particularly at the tumor’s invasive front, are likely to engage in crosstalk in a spatially organized manner that elicits tumor EMT and cancer stem-like cell (CSC) formation (79–81). Hursting and colleagues have demonstrated that obesity promotes these two pathways, thus suggesting a causal link between obesity and the associated metabolic derangement with TNBC development (82). However, the contribution of other key adipokines and cytokines in this obesity-associated CSC/EMT circuit must be further examined, as preliminary studies indicate that the leptin-adiponectin ratio imbalances do not fully account for all of the observed effects of diet-induced obesity on TNBC (83).
During an investigation into the role of CAAs in breast cancer progression, our group has recently implicated crosstalk from adipocyte-derived exosomes in driving EMT and cancer aggressiveness. Interest in exosomes, long ignored and thought to be merely cellular disposal systems, has recently been growing. Adipocyte-derived exosomes contain a significant payload of microRNAs that, when applied in a co-culture system with breast cancer cell lines, upregulate genes involved in CSC formation and invasion. Provocatively, fold-changes in these gene expression patterns were greater if the adipocytes had first been rendered insulin resistant or were isolated from patients with T2D (67). This finding supports the idea that the TME is likely more dangerous, leading to increased incidence and metastasis, when the patient has comorbid T2D, which is consistent with observations made in the Black Women’s Health Study (84). A similar phenomenon has been observed in mouse models of diet-induced obesity using breast cancer cell lines in E0771, where obesity causes insulin resistance and metabolic abnormalities in adipocytes that promote expansion of metastasis (85).
Fibroblasts
Fibroblasts, also known as cancer-associated fibroblasts (CAFs), are the most abundant cell type in the breast TME (86). These cells are derived from resident fibroblasts and a diverse population of mesenchymal cells upon exposure to proinflammatory cytokines such as TNFα and IL-1β (86, 87). Such cytokines are prevalent in the diabetic TME as they are secreted by diseased adipocytes, though little is known about how this may influence the development of CAF outgrowth (88). Provocatively, recent work from Zhu and colleagues report that adipocytes can de-differentiate into fibroblast-like precursor cells during breast tumor progression, with the ability to transform into functional pro-tumorigenic stromal cells such as myofibroblast- or macrophage-like cells (89). Though this phenomenon has not been identified in the context of T2D, the impact of metabolically impaired adipocytes on this mesenchymal transition and subsequent breast cancer progression demands further study. Once differentiated, CAFs exist as highly heterogeneous components of the TME that secrete a variety of soluble factors, such as chemokines and growth factors, that promote both tumor initiation, progression, and invasion (87, 90–92). A key pro-tumorigenic program of CAFs is their function as metabolic support for proliferating tumor cells (93, 94). Their catabolic phenotypes are induced by high levels of reactive oxygen species during oxidative stress upon hypoxia-inducible factor 1α (HIF1α) and nuclear factor κ B (NFκB) signaling within the TME (95). This metabolic shift towards lactate and pyruvate production works to fuel biosynthetic pathways of cancer cells, which can then rely on CAFs to provide a nutrient-rich TME (96). Critically, increased levels of lactate within the TME is known to acidify the area, thus inhibiting effector T cell function and potentially contributing to the failure of anti-tumor therapy in these patients (97–100). HIF1α stability and function are dysregulated by hyperglycemia, and disruption of this aberrant signaling can improve insulin sensitivity (101–104). Within the diabetic breast TME, higher levels of HIF1α and subsequent oxidative stress contribute to hypoxia (101, 103, 105). Hypoxia is a driving force of tumor progression as it stimulates vascularization (VEGF, ANG1, ANG2, MMPs, LOX, CAIX, CXCR4), upregulates EMT/CSC signatures (SNAI1, SNAI2, TWIST1, SOX9, SOX2, OCT4, NANOG), and contributes to drug resistance (106–109). Despite the importance of metabolic reprogramming in cancer biology, the role of CAFs in the diabetic TME have yet to be fully investigated.
Macrophages
Macrophages form a critical and diverse component of the breast TME (86, 87). They can exist as tissue resident cells or differentiate from circulating monocytes that are recruited to the tumor site via chemokines secreted by cancer and stromal cells (86, 87, 110). Once in the breast TME, macrophages can have both proinflammatory (M1-like) and anti-inflammatory (M2-like) functions. During normal immunological responses, most macrophages are leaning towards the M1-like phenotype and engage in responses to pathogens (111). The M2-like phenotype, however, is associated with T helper 2-type cytokines and is typically associated with wound healing and tissue remodeling (111). Most tumor-associated macrophages (TAMs) are leaning towards the M2-like phenotype as this class promotes cell proliferation and breast cancer progression via anti-inflammatory signaling pathways (IL10, CCL2, CCL17, CCL22, TGFB) (112). TAMs have been reported to support invasion and metastasis by secreting EGF1 and TNFα, which promote EMT and enhance the stemness and angiogenesis of cancers (113). Many studies have linked high TAM levels to a worse prognosis, suggesting that TAM depletion or reprogramming may serve as a key therapeutic target (114, 115). The common anti-diabetic drug metformin has been shown to modulate macrophage polarization, specifically by decreasing the percent of M2-like and increasing the percent of M1-like macrophages, through AMP-activated protein kinase (AMPK)-NFκB signaling (116). This suggests that metformin could be therapeutically advantageous in facilitating macrophage reprogramming.
Within the diabetic and obese TME, macrophages are known to play a critical role in local adipocyte inflammation. M1-like macrophages accumulate within adipose tissue and produce factors that deregulate adipocyte signaling processes, increase production of reactive oxygen species, and potentiate insulin resistance (110, 117–119). They can form a characteristic crown-like structure around the hypertrophied and dying adipocytes (110, 118, 119). This mammary adipose tissue inflammation is thought to contribute to the link between metabolic derangements and worse breast cancer prognosis (110, 117, 120–122). Critically, in patients with breast cancer and comorbid obesity, the presence of crown-like structure accumulation is associated with more aggressive, high-grade tumors (110, 114, 117). Despite these demonstrated functions, additional studies are needed to understand the potential clinical utility of crown-like structure accumulation as a biomarker of breast cancer risk or prognosis.
T cells
Another major component of the breast TME are tumor-infiltrating lymphocytes (TILs) (123–125). Importantly, although inflammation regulation in T2D has centered around macrophages, recent evidence suggests that T cells are vital for metabolic inflammation and insulin resistance associated with the disease (123, 126–131). The majority of TILs are T cells, which can be further subdivided into CD4+ helper and CD8+ cytotoxic T cells. The ratio between these two TIL populations is a critical prognostic indicator of breast cancer progression, with infiltration of CD8+ T cells associated with longer survival (124, 132–135). Miya and colleagues demonstrated that diabetic patients exhibit a decreased proportion of peripheral CD8+ T cells after glucose loading compared to nondiabetic control subjects (136). However, it is unclear how this finding may extend to the local TME of breast cancer patients with comorbid T2D. The effect of these cell populations is regulated by a balance between co-stimulatory and co-inhibitory signals at immune checkpoints. These regulatory pathways, involving molecules such as programmed death-1 (PD-1), typically work to inhibit T-cell function in order to prevent inappropriate immune reactions (137–139). However, these pathways are known to be hijacked by tumor cells to evade immune detection and clearance (137–139). Tumors are able to upregulate the expression of cognate ligands, such as programmed death-ligand 1 (PD-L1), on their cell surface, which reprogram local TILs towards an inhibitory state known as immune exhaustion (137–139). Immune exhaustion is a unique differentiation state for T cells, in which they become metabolically impaired and lose their effector functions to create an immunosuppressive TME (137–139). Both PD-1+ TILs and PD-L1+ tumors are associated with a worse prognosis for breast cancer patients (123, 124). Critically, circulating T cells in diabetic patients are known to have high surface expression of PD-1 (137, 140, 141). Immunotherapeutic strategies that aim to reverse this exhausted microenvironment have been gaining traction recently. Studies have shown that treatment with the anti-PD-1 agent pembrolizumab, in combination with chemotherapy as neoadjuvant therapy, resulted in a significant reduction in the risk of disease progression for patients with high-risk early-stage TNBC (142–145). However, little research has been done into whether breast cancer patients with comorbid T2D benefit from these same effects. Deepening our understanding of TIL biology and the role of exhaustion in T2D and breast cancer may be key in unraveling underlying disparities.
Spatial organization
Recent developments in spatial transcriptomic technologies have revealed that cancers have complex spatial organization within their three-dimensional architectures that dictate a given cell’s spatial neighborhood, interactions, and phenotype to influence overall tumor behavior (13, 14). Of note, immune responses, such as immunoregulatory pathways, are highly spatially organized processes within the breast TME (13, 146). Specifically, regulatory T and exhausted T cells co-occur in space with highly proliferative tumor cells, linking this spatially suppressed TME to poor patient outcomes (147). In addition, the adipocytes immediately adjacent to a breast tumor are known to be active actors in tumor progression, with investigators beginning to probe secretory and spatial relationships among breast adipocytes in invasive cancers with histological evidence of crosstalk (79–81). The spatial organization and transcriptional relationships among breast cancer cells and nearby adipocytes, particularly at the tumor invasive front, are likely to engage in exosome crosstalk that elicits tumor EMT and CSC formation. Taken together, the TME is likely more dangerous when a breast cancer patient has comorbid T2D. Deepening our understanding of how this increasingly common comorbidity may impact spatial heterogeneity, architecture, and signaling will be critical in improving therapeutic outcomes for these patients.
Treatment opportunities
The first-line medication for treatment of T2D is metformin, a biguanide drug that lowers glucose production by the liver through inhibiting the mitochondrial respiratory chain, activating AMPK, lowering cAMP, and reducing the expression of gluconeogenic enzymes, thus enhancing insulin sensitivity (148). Even though metformin has been used for more than 60 years in the clinic, several studies have demonstrated new indications and mechanisms of action for the drug as an anti-tumor agent (149). Given that metformin activates AMPK, it thereby inhibits mTOR pathways and decreases circulating insulin levels, with hyperinsulinemia being tied to worse breast cancer prognosis. It also inhibits the proliferation and invasion of cancer cells, which could limit metastatic spread (150–155). The Adjuvant Lapatinib and/or Trastuzumab Treatment Optimization trial tested metformin use in HER2+ breast cancer, showing an improvement in prognosis (156, 157). Other clinical studies have correlated metformin use with improved breast cancer-specific survival in HER2+ breast cancer, though not in TNBC (158). Goodwin and colleagues recently reported (159) a lack of survival benefit of metformin for either ER/PR+ or ER/PR- breast cancer patients. We consider that these results are skewed due to the exclusion of T2D patients and lack of stratification by patient BMI as improper exclusion criteria by considering all patients metabolically healthy, therefore the results are unsurprising. However, recent work has demonstrated that activation of AMPK upregulates the expression of EMT and stemness genes (NANOG, SOX2, BMI1), through the transcriptional upregulation of TWIST1 (160). This AMPK-driven stemness has been shown to play an important role in breast cancer drug resistance, thus complicating the effect metformin may have on breast cancer. This unexpected, potentially dangerous pathway of AMPK activation by metformin, which appears to increase survival of circulating metastatic breast cancer cells, demands further study.
Metformin has also been shown to have a beneficial effect in the regulation of T cell functions, providing a potential therapeutic for immune exhaustion via TSC1/mTOR (149). Many previous studies have linked the anticancer effects of metformin to the differentiation of T cells (149, 161). For instance, the use of metformin has been reported to increase the expression of CD8 and CD69 while decreasing PD-1 in TILs, thus increasing the number of CD8+ T cells while protecting them from apoptosis and exhaustion (161). Cytokines seem to be key in this process, as metformin upregulates the secretion of interferon γ, IL-2, and TNFα via AMPK (162, 163). Since the recent emergence of microRNAs as crucial regulators of T cell differentiation, there are reports showing metformin increasing miR-7 expression in an AMPK-dependent pathway and inhibiting the action of miR-107, which is linked to insulin sensitivity and the expression of PD-1 (161). Several ongoing clinical trials are examining whether metformin has benefit in the context of immune checkpoint blockade in treatment of solid tumor, such as NCT03048500 for non-small cell lung cancer, and NCT03800602 for refractory, microsatellite-stable colorectal cancer.
Overall, though most current studies that examine metformin’s use in breast cancer have reported a mixed picture on its efficacy, they demonstrate a potential therapeutic benefit of metformin in patients with breast cancer and comorbid T2D. It is clear that metformin holds considerable promise with regard to a potential antitumor agent.
Conclusions and future directions
Over 100 million Americans with T2D or pre-diabetes are predisposed to more aggressive tumors, but the mechanistic basis for this differential risk remains unclear and understudied (1). Herein, we have outlined the current state of knowledge on the relevant cell types and mechanisms underlying this comorbidity.
However, many questions remain on how cell diversity within the diabetic breast TME may impact tumorigenesis, proliferation, or even drug resistance in these patients. For example, it is unclear how related obesity, via an increase in dysfunctional adipocytes, may change the heterogeneity of the TME and how any subsequent changes may support a diversity of transcriptional states key in pro- and anti-tumorigenic processes. Furthermore, understanding the interplay between the TME and T2D is further complicated by the need to simultaneously understand how intercellular crosstalk is organized at the tissue level and how multiple regulatory layers of cellular identity might play distinct roles. As previously stated, these layers can encompass alterations in cellular composition, tissue architecture, RNA expression, cytokine expression, lipid content, metabolomics, and other types of molecular analytes that work in concert to create the underlying disease phenotypes. Nevertheless, with the advent of single cell RNA sequencing and, specifically spatial omics and multiplex imaging technologies, the field of oncology is entering a new era of technological innovation where the necessary multimodal spatial datasets can be created that will aid in providing the necessary systems-level understanding of these complex and multifaceted disease phenotypes and interactions. Thus, these approaches offer novel methods in studying breast cancer as well as other cancer types differentially impacted by T2D. Further investigating the effects of the metabolically disturbed TME cell types on intercellular communication and cancer pathogenesis will be critical in identifying biomarkers and novel therapeutic targets for patients with breast cancer and comorbid T2D. These patients have been excluded and clinical trial design should be adapted among cancer disparities consortia to include them in well-defined groups with sufficient statistical power. We propose that investigating the mechanisms of intercellular crosstalk with tumor cells in a T2D setting is crucial to fully understand how this comorbidity might be working, integrating metabolic and immune exhaustion signatures like tumor progression, metastasis, and immune checkpoint. Utilizing this holistic approach will be crucial in revealing novel insights into tumor progression and metastasis in T2D patients and adopting multiple levels and perspectives of metabolism and intercellular communication within the TME.
Author contributions
Conceptualization, CE, RD, and GD. Writing, CE, PL, and YQ. Revision and editing, CE, RD, and GD. All authors contributed to the article and approved the submitted version.
Funding
National Cancer Institute (U01CA182898, R01CA222170; GV Denis).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Abbreviations
27HC, 27-hydroxycholesterol; AMPK, AMP-activated protein kinase; CAA, cancer-associated adipocyte; CAF, cancer-associated fibroblast; CSC, cancer stem-like cell; CYP27A1, cytochrome P450 oxidase sterol 27-hydroxylase A1; EMT, epithelial-to-mesenchymal transition; ER, estrogen receptor; HER2, human epidermal growth factor receptor 2; HIF1α, hypoxia-inducible factor 1α; IGF-1R, insulin like growth factor 1 receptor; IR, insulin receptor; NFκB, nuclear factor κ B; PD-1, programmed death-1; PD-L1, programmed death-ligand 1; PI3K/Akt/mTOR, phosphoinositide 3 kinase/Akt/mammalian (or mechanistic) target of rapamycin; Ras/MAPK/ERK1/2, Ras/mitogen-activated protein kinase/extracellular signal-related kinase 1/2; T2D, type 2 diabetes; TAM, tumor associated macrophage; TIL, tumor infiltrating lymphocyte; TME, tumor microenvironment; TNBC, triple negative breast cancer.
References
1. CDC. National diabetes statistics report, 2020. Atlanta, GA: US Department of Health and Human Services (2020) p. 12–5. Available at: https://www.cdc.gov/diabetes/data/statistics-report/index.html.
2. Eketunde AO. Diabetes as a risk factor for breast cancer. Cureus (2020) 12(5):e8010. doi: 10.7759/cureus.8010
3. Giovannucci E, Harlan DM, Archer MC, Bergenstal RM, Gapstur SM, Habel LA, et al. Diabetes and cancer: A consensus report. CA Cancer J Clin (2010) 60(4):207–21. doi: 10.3322/caac.20078
4. Lee KN, Torres MA, Troeschel AN, He J, Gogineni K, McCullough LE. Type 2 diabetes, breast cancer specific and overall mortality: Associations by metformin use and modification by race, body mass, and estrogen receptor status. PloS One (2020) 15(5):e0232581. doi: 10.1371/journal.pone.0232581
5. Renehan AG, Roberts DL, Dive C. Obesity and cancer: Pathophysiological and biological mechanisms. Arch Physiol Biochem (2008) 114(1):71–83. doi: 10.1080/13813450801954303
6. Quail DF, Dannenberg AJ. The obese adipose tissue microenvironment in cancer development and progression. Nat Rev Endocrinol (2019) 15(3):139–54. doi: 10.1038/s41574-018-0126-x
7. Denis GV, Palmer JR. “Obesity-associated” breast cancer in lean women: Metabolism and inflammation as critical modifiers of risk. Cancer Prev Res (2017) 10(5):267–9. doi: 10.1158/1940-6207.CAPR-17-0083
8. Denis GV, Obin MS. ‘Metabolically healthy obesity’: Origins and implications. Mol Aspects Med (2013) 34(1):59–70. doi: 10.1016/j.mam.2012.10.004
9. Zheng X, Peng R, Xu H, Lin T, Qiu S, Wei Q, et al. The association between metabolic status and risk of cancer among patients with obesity: Metabolically healthy obesity vs. metabolically unhealthy obesity. Front Nutr (2022) 9:783660. doi: 10.3389/fnut.2022.783660
10. Liu B, Giffney HE, Arthur RS, Rohan TE, Dannenberg AJ. Cancer risk in normal weight individuals with metabolic obesity: A narrative review. Cancer Prev Res (2021) 14(5):509–20. doi: 10.1158/1940-6207.CAPR-20-0633
11. Palmer JR, Castro-Webb N, Bertrand K, Bethea TN, Denis GV. Type II diabetes and incidence of estrogen receptor negative breast cancer in African American women. Cancer Res (2017) 77(22):6462–9. doi: 10.1158/0008-5472.CAN-17-1903
12. Charlot M, Castro-Webb N, Bethea TN, Bertrand K, Boggs DA, Denis GV, et al. Diabetes and breast cancer mortality in black women. Cancer Cause Control (2017) 28(1):61–7. doi: 10.1007/s10552-016-0837-z
13. Keren L, Bosse M, Marquez D, Angoshtari R, Jain S, Varma S, et al. A structured tumor-immune microenvironment in triple negative breast cancer revealed by multiplexed ion beam imaging. Cell (2018) 174(6):1373–1387.e19. doi: 10.1016/j.cell.2018.08.039
14. Jackson HW, Fischer JR, Zanotelli VRT, Ali HR, Mechera R, Soysal SD, et al. The single-cell pathology landscape of breast cancer. Nature (2020) 578(7796):615–20. doi: 10.1038/s41586-019-1876-x
15. Sun G, Kashyap SR. Cancer risk in type 2 diabetes mellitus: Metabolic links and therapeutic considerations. J Nutr Metab (2011) 2011:708183. doi: 10.1155/2011/708183
16. Basha B, Samuel SM, Triggle CR, Ding H. Endothelial dysfunction in diabetes mellitus: Possible involvement of endoplasmic reticulum stress? Exp Diabetes Res (2012) 2012:481840. doi: 10.1155/2012/481840
17. Onitilo AA, Engel JM, Glurich I, Stankowski RV, Williams GM, Doi SA. Diabetes and cancer I: risk, survival, and implications for screening. Cancer Cause Control (2012) 23(6):967–81. doi: 10.1007/s10552-012-9972-3
18. Novosyadlyy R, LeRoith D. Hyperinsulinemia and type 2 diabetes: Impact on cancer. Cell Cycle (2010) 9(8):1449–50. doi: 10.4161/cc.9.8.11512
19. Lann D, LeRoith D. The role of endocrine insulin-like growth factor-I and insulin in breast cancer. J Mammary Gland Biol (2008) 13(4):371. doi: 10.1007/s10911-008-9100-x
20. Kang C, LeRoith D, Gallagher EJ. Diabetes, obesity, and breast cancer. Endocrinology (2018) 159(11):3801–12. doi: 10.1210/en.2018-00574
21. Belardi V, Gallagher EJ, Novosyadlyy R, LeRoith D. Insulin and IGFs in obesity-related breast cancer. J Mammary Gland Biol (2013) 18(3–4):277–89. doi: 10.1007/s10911-013-9303-7
22. Roberts DL, Dive C, Renehan AG. Biological mechanisms linking obesity and cancer risk: New perspectives. Annu Rev Med (2010) 61(1):301–16. doi: 10.1146/annurev.med.080708.082713
23. Chettouh H, Fartoux L, Aoudjehane L, Wendum D, Clapéron A, Chrétien Y, et al. Mitogenic insulin receptor-a is overexpressed in human hepatocellular carcinoma due to EGFR-mediated dysregulation of RNA splicing factors. Cancer Res (2013) 73(13):3974–86. doi: 10.1158/0008-5472.CAN-12-3824
24. Alvino CL, Ong SC, McNeil KA, Delaine C, Booker GW, Wallace JC, et al. Understanding the mechanism of insulin and insulin-like growth factor (IGF) receptor activation by IGF-II. PloS One (2011) 6(11):e27488. doi: 10.1371/journal.pone.0027488
25. Tzivion G, Dobson M, Ramakrishnan G. FoxO transcription factors; regulation by AKT and 14-3-3 proteins. Biochim Et Biophys Acta Bba - Mol Cell Res (2011) 1813(11):1938–45. doi: 10.1016/j.bbamcr.2011.06.002
26. Sarfstein R, Nagaraj K, LeRoith D, Werner H. Differential effects of insulin and IGF1 receptors on ERK and AKT subcellular distribution in breast cancer cells. Cells (2019) 8(12):1499. doi: 10.3390/cells8121499
27. Dupont J, Roith DL. Insulin-like growth factor 1 and oestradiol promote cell proliferation of MCF-7 breast cancer cells: new insights into their synergistic effects. Mol Pathol (2001) 54(3):149. doi: 10.1136/mp.54.3.149
28. Rostoker R, Abelson S, Bitton-Worms K, Genkin I, Ben-Shmuel S, Dakwar M, et al. Highly specific role of the insulin receptor in breast cancer progression. Endocr-relat Cancer (2015) 22(2):145–57. doi: 10.1530/ERC-14-0490
29. Periyasamy-Thandavan S, Takhar S, Singer A, Dohn MR, Jackson WH, Welborn AE, et al. Insulin-like growth factor 1 attenuates antiestrogen- and antiprogestin-induced apoptosis in ER+ breast cancer cells by MEK1 regulation of the BH3-only pro-apoptotic protein bim. Breast Cancer Res (2012) 14(2):R52. doi: 10.1186/bcr3153
30. Fleming HE, Janzen V, Celso CL, Guo J, Leahy KM, Kronenberg HM, et al. Wnt signaling in the niche enforces hematopoietic stem cell quiescence and is necessary to preserve self-renewal in vivo. Cell Stem Cell (2008) 2(3):274–83. doi: 10.1016/j.stem.2008.01.003
31. Zhang H, Fagan DH, Zeng X, Freeman KT, Sachdev D, Yee D. Inhibition of cancer cell proliferation and metastasis by insulin receptor downregulation. Oncogene (2010) 29(17):2517–27. doi: 10.1038/onc.2010.17
32. Mulligan AM, O’Malley FP, Ennis M, Fantus IG, Goodwin PJ. Insulin receptor is an independent predictor of a favorable outcome in early stage breast cancer. Breast Cancer Res Tr. (2007) 106(1):39–47. doi: 10.1007/s10549-006-9471-x
33. Qin L, Wang Y, Tao L, Wang Z. AKT down-regulates insulin-like growth factor-1 receptor as a negative feedback. J Biochem (2011) 150(2):151–6. doi: 10.1093/jb/mvr066
34. Levine AJ, Feng Z, Mak TW, You H, Jin S. Coordination and communication between the p53 and IGF-1–AKT–TOR signal transduction pathways. Gene Dev (2006) 20(3):267–75. doi: 10.1101/gad.1363206
35. Arteaga CL, Kitten LJ, Coronado EB, Jacobs S, Kull FC, Allred DC, et al. Blockade of the type I somatomedin receptor inhibits growth of human breast cancer cells in athymic mice. J Clin Invest (1989) 84(5):1418–23. doi: 10.1172/JCI114315
36. Arteaga CL, Osborne CK. Growth inhibition of human breast cancer cells in vitro with an antibody against the type I somatomedin receptor. Cancer Res (1989) 49(22):6237–41.
37. Giovannucci E. Insulin, insulin-like growth factors and colon cancer: A review of the evidence. J Nutr (2001) 131(11):3109S–20S. doi: 10.1093/jn/131.11.3109S
38. Zielinska HA, Holly JMP, Bahl A, Perks CM. Inhibition of FASN and ERα signalling during hyperglycaemia-induced matrix-specific EMT promotes breast cancer cell invasion via a caveolin-1-dependent mechanism. Cancer Lett (2018) 419:187–202. doi: 10.1016/j.canlet.2018.01.028
39. Qiu J, Zheng Q, Meng X. Hyperglycemia and chemoresistance in breast cancer: From cellular mechanisms to treatment response. Front Oncol (2021) 11:628359. doi: 10.3389/fonc.2021.628359
40. Saydah SH, Platz EA, Rifai N, Pollak MN, Brancati FL, Helzlsouer KJ. Association of markers of insulin and glucose control with subsequent colorectal cancer risk. Cancer Epidemiol Biomarkers Prev Publ Am Assoc Cancer Res Cosponsored Am Soc Prev Oncol (2003) 12(5):412–8.
41. Muti P, Quattrin T, Grant BJB, Krogh V, Micheli A, Schünemann HJ, et al. Fasting glucose is a risk factor for breast cancer: a prospective study. Cancer Epidemiol Biomarkers Prev Publ Am Assoc Cancer Res Cosponsored Am Soc Prev Oncol (2002) 11(11):1361–8.
42. Takahashi H, Mizuta T, Eguchi Y, Kawaguchi Y, Kuwashiro T, Oeda S, et al. Post-challenge hyperglycemia is a significant risk factor for the development of hepatocellular carcinoma in patients with chronic hepatitis c. J Gastroenterol (2011) 46(6):790–8. doi: 10.1007/s00535-011-0381-2
43. Stattin P, Bjoür O, Ferrari P, Lukanova A, Lenner P, Lindahl B, et al. Prospective study of hyperglycemia and cancer risk. Diabetes Care (2007) 30(3):561–7. doi: 10.2337/dc06-0922
44. Vigneri P, Frasca F, Sciacca L, Pandini G, Vigneri R. Diabetes and cancer. Endocr-relat Cancer (2009) 16(4):1103–23. doi: 10.1677/ERC-09-0087
45. Yamasaki K, Hayashi Y, Okamoto S, Osanai M, Lee G. Insulin-independent promotion of chemically induced hepatocellular tumor development in genetically diabetic mice. Cancer Sci (2010) 101(1):65–72. doi: 10.1111/j.1349-7006.2009.01345.x
46. Taubes G. Unraveling the obesity-cancer connection. Science (2012) 335(6064):28–32. doi: 10.1126/science.335.6064.28
47. Johnson JA, Bowker SL. Intensive glycaemic control and cancer risk in type 2 diabetes: a meta-analysis of major trials. Diabetologia (2011) 54(1):25–31. doi: 10.1007/s00125-010-1933-3
48. Moore LL, Chadid S, Singer MR, Kreger BE, Denis GV. Metabolic health reduces risk of obesity-related cancer in framingham study adults. Cancer Epidem Biomar (2014) 23(10):2057–65. doi: 10.1158/1055-9965.EPI-14-0240
49. Cheung YMM, Hughes M, Harrod J, Files J, Kirkner G, Buckley L, et al. The effects of diabetes and glycemic control on cancer outcomes in individuals with metastatic breast cancer. J Clin Endocrinol Metab (2022) 107(9):2511–21. doi: 10.1210/clinem/dgac375
50. Hershey DS. Importance of glycemic control in cancer patients with diabetes: Treatment through end of life. Asia-pacific J Oncol Nurs (2017) 4(4):313–8. doi: 10.4103/apjon.apjon_40_17
51. Dankner R, Boker LK, Boffetta P, Balicer RD, Murad H, Berlin A, et al. A historical cohort study on glycemic-control and cancer-risk among patients with diabetes. Cancer Epidemiol (2018) 57:104–9. doi: 10.1016/j.canep.2018.10.010
52. Bielecka-Dąbrowa A, Hannam S, Rysz J, Banach M. Malignancy-associated dyslipidemia. Open Cardiovasc Med J (2011) 5(1):35–40. doi: 10.2174/1874192401105010035
53. Scully T, Kase N, Gallagher EJ, LeRoith D. Regulation of low-density lipoprotein receptor expression in triple negative breast cancer by EGFR-MAPK signaling. Sci Rep-uk. (2021) 11(1):17927. doi: 10.1038/s41598-021-97327-y
54. Bell KE, Sebastiano KMD, Vance V, Hanning R, Mitchell A, Quadrilatero J, et al. A comprehensive metabolic evaluation reveals impaired glucose metabolism and dyslipidemia in breast cancer patients early in the disease trajectory. Clin Nutr (2014) 33(3):550–7. doi: 10.1016/j.clnu.2013.08.001
55. Schairer C, Gadalla SM, Pfeiffer RM, Moore SC, Engels EA. Diabetes, abnormal glucose, dyslipidemia, hypertension, and risk of inflammatory and other breast cancer. Cancer Epidemiol Prev Biomarkers (2017) 26(6):862–8. doi: 10.1158/1055-9965.EPI-16-0647
56. His M, Zelek L, Deschasaux M, Pouchieu C, Kesse-Guyot E, Hercberg S, et al. Prospective associations between serum biomarkers of lipid metabolism and overall, breast and prostate cancer risk. Eur J Epidemiol (2014) 29(2):119–32. doi: 10.1007/s10654-014-9884-5
57. Nowak C, Ärnlöv J. A mendelian randomization study of the effects of blood lipids on breast cancer risk. Nat Commun (2018) 9(1):3957. doi: 10.1038/s41467-018-06467-9
58. dos Santos CR, Fonseca I, Dias S, de Almeida JM. Plasma level of LDL-cholesterol at diagnosis is a predictor factor of breast tumor progression. BMC Cancer (2014) 14(1):132. doi: 10.1186/1471-2407-14-132
59. Cedó L, Reddy ST, Mato E, Blanco-Vaca F, Escolà-Gil JC. HDL and LDL: Potential new players in breast cancer development. J Clin Med (2019) 8(6):853.
60. Scully T, Ettela A, LeRoith D, Gallagher EJ. Obesity, type 2 diabetes, and cancer risk. Front Oncol (2021) 10:615375. doi: 10.3389/fonc.2020.615375
61. Kim DS, Scherer PE. Obesity, diabetes, and increased cancer progression. Diabetes Metab J (2021) 45(6):799–812. doi: 10.4093/dmj.2021.0077
62. Nelson ER, Chang C, McDonnell DP. Cholesterol and breast cancer pathophysiology. Trends Endocrinol Metab (2014) 25(12):649–55. doi: 10.1016/j.tem.2014.10.001
63. Samuel SM, Varghese E, Varghese S, Büsselberg D. Challenges and perspectives in the treatment of diabetes associated breast cancer. Cancer Treat Rev (2018) 70:98–111. doi: 10.1016/j.ctrv.2018.08.004
64. Santos CR, Schulze A. Lipid metabolism in cancer. FEBS J (2012) 279(15):2610–23. doi: 10.1111/j.1742-4658.2012.08644.x
65. Luo X, Cheng C, Tan Z, Li N, Tang M, Yang L, et al. Emerging roles of lipid metabolism in cancer metastasis. Mol Cancer (2017) 16(1):76. doi: 10.1186/s12943-017-0646-3
66. Szlasa W, Zendran I, Zalesińska A, Tarek M, Kulbacka J. Lipid composition of the cancer cell membrane. J Bioenerg Biomembr (2020) 52(5):321–42. doi: 10.1007/s10863-020-09846-4
67. Jafari N, Kolla M, Meshulam T, Shafran JS, Qiu Y, Casey AN, et al. Adipocyte-derived exosomes may promote breast cancer progression in type 2 diabetes. Sci Signal (2021) 14(710):eabj2807. doi: 10.1126/scisignal.abj2807
68. Liu Q, Dong Ht, Zhao T, Yao F, Xu Y, Chen B, et al. Cancer-associated adipocytes release FUCA2 to promote aggressiveness in TNBC. Endocr-relat Cancer (2022) 29(3):139–49. doi: 10.1530/ERC-21-0243
69. Zhao C, Wu M, Zeng N, Xiong M, Hu W, Lv W, et al. Cancer-associated adipocytes: emerging supporters in breast cancer. J Exp Clin Cancer Res Cr. (2020) 39(1):156. doi: 10.1186/s13046-020-01666-z
70. Attané C, Muller C. Drilling for oil: Tumor-surrounding adipocytes fueling cancer. Trends Cancer (2020) 6(7):593–604. doi: 10.1016/j.trecan.2020.03.001
71. Oshi M, Tokumaru Y, Angarita FA, Lee L, Yan L, Matsuyama R, et al. Adipogenesis in triple-negative breast cancer is associated with unfavorable tumor immune microenvironment and with worse survival. Sci Rep-uk. (2021) 11(1):12541. doi: 10.1038/s41598-021-91897-7
72. Kothari C, Diorio C, Durocher F. The importance of breast adipose tissue in breast cancer. Int J Mol Sci (2020) 21(16):5760. doi: 10.3390/ijms21165760
73. Hoy AJ, Balaban S, Saunders DN. Adipocyte–tumor cell metabolic crosstalk in breast cancer. Trends Mol Med (2017) 23(5):381–92. doi: 10.1016/j.molmed.2017.02.009
74. Balaban S, Shearer RF, Lee LS, van Geldermalsen M, Schreuder M, Shtein HC, et al. Adipocyte lipolysis links obesity to breast cancer growth: adipocyte-derived fatty acids drive breast cancer cell proliferation and migration. Cancer Metab (2017) 5(1):1. doi: 10.1186/s40170-016-0163-7
75. Samuel VT, Shulman GI. Mechanisms for insulin resistance: Common threads and missing links. Cell (2012) 148(5):852–71. doi: 10.1016/j.cell.2012.02.017
76. Zhao J, Xie F, Yang Y, Wang S. Reprogramming of fatty acid metabolism in breast cancer: a narrative review. Transl Breast Cancer Res (2021) 2(0):5–5. doi: 10.21037/tbcr-20-53
77. Monaco ME. Fatty acid metabolism in breast cancer subtypes. Oncotarget (2017) 8(17):29487–500. doi: 10.18632/oncotarget.15494
78. Hoy AJ, Nagarajan SR, Butler LM. Tumour fatty acid metabolism in the context of therapy resistance and obesity. Nat Rev Cancer (2021) 21(12):753–66. doi: 10.1038/s41568-021-00388-4
79. Duong MN, Geneste A, Fallone F, Li X, Dumontet C, Muller C. The fat and the bad: Mature adipocytes, key actors in tumor progression and resistance. Oncotarget (2017) 8(34):57622–41. doi: 10.18632/oncotarget.18038
80. Wang YY, Attané C, Milhas D, Dirat B, Dauvillier S, Guerard A, et al. Mammary adipocytes stimulate breast cancer invasion through metabolic remodeling of tumor cells. JCI Insight (2017) 2(4):e87489. doi: 10.1172/jci.insight.87489
81. Dirat B, Bochet L, Dabek M, Daviaud D, Dauvillier S, Majed B, et al. Cancer-associated adipocytes exhibit an activated phenotype and contribute to breast cancer invasion. Cancer Res (2011) 71(7):2455–65. doi: 10.1158/0008-5472.CAN-10-3323
82. Bowers LW, Rossi EL, McDonell SB, Doerstling SS, Khatib SA, Lineberger CG, et al. Leptin signaling mediates obesity-associated CSC enrichment and EMT in preclinical TNBC models. Mol Cancer Res (2018) 16(5):142–52. doi: 10.1158/1541-7786.MCR-17-0508
83. Andò S, Catalano S. The multifactorial role of leptin in driving the breast cancer microenvironment. Nat Rev Endocrinol (2012) 8(5):263–75. doi: 10.1038/nrendo.2011.184
84. Park NJ, Kang DH. Inflammatory cytokine levels and breast cancer risk factors: Racial differences of healthy Caucasian and African American women. Oncol Nurs Forum (2013) 40(5):490–500. doi: 10.1188/13.ONF.40-05AP
85. Pearce JV, Farrar JS, Lownik JC, Ni B, Chen S, Kan TW, et al. E0771 and 4T1 murine breast cancer cells and interleukin 6 alter gene expression patterns but do not induce browning in cultured white adipocytes. Biochem Biophys Rep (2019) 18:100624. doi: 10.1016/j.bbrep.2019.100624
86. Soysal SD, Tzankov A, Muenst SE. Role of the tumor microenvironment in breast cancer. Pathobiology (2015) 82(3–4):142–52. doi: 10.1159/000430499
87. Terceiro LEL, Edechi CA, Ikeogu NM, Nickel BE, Hombach-Klonisch S, Sharif T, et al. The breast tumor microenvironment: A key player in metastatic spread. Cancers (2021) 13(19):4798. doi: 10.3390/cancers13194798
88. Rakotoarivelo V, Lacraz G, Mayhue M, Brown C, Rottembourg D, Fradette J, et al. Inflammatory cytokine profiles in visceral and subcutaneous adipose tissues of obese patients undergoing bariatric surgery reveal lack of correlation with obesity or diabetes. Ebiomedicine (2018) 30:237–47. doi: 10.1016/j.ebiom.2018.03.004
89. Zhu Q, Zhu Y, Hepler C, Zhang Q, Park J, Gliniak C, et al. Adipocyte mesenchymal transition contributes to mammary tumor progression. Cell Rep (2022) 40(11):111362. doi: 10.1016/j.celrep.2022.111362
90. Fernández-Nogueira P, Fuster G, Gutierrez-Uzquiza Á, Gascón P, Carbó N, Bragado P. Cancer-associated fibroblasts in breast cancer treatment response and metastasis. Cancers (2021) 13(13):3146. doi: 10.3390/cancers13133146
91. Chen PY, Wei WF, Wu HZ, Fan LS, Wang W. Cancer-associated fibroblast heterogeneity: A factor that cannot be ignored in immune microenvironment remodeling. Front Immunol (2021) 12:671595. doi: 10.3389/fimmu.2021.671595
92. Costa A, Kieffer Y, Scholer-Dahirel A, Pelon F, Bourachot B, Cardon M, et al. Fibroblast heterogeneity and immunosuppressive environment in human breast cancer. Cancer Cell (2018) 33(3):463–479.e10. doi: 10.1016/j.ccell.2018.01.011
93. Martinez-Outschoorn UE, Lisanti MP, Sotgia F. Catabolic cancer-associated fibroblasts transfer energy and biomass to anabolic cancer cells, fueling tumor growth. Semin Cancer Biol (2014) 25:47–60. doi: 10.1016/j.semcancer.2014.01.005
94. Gentric G, Mechta-Grigoriou F. Tumor cells and cancer-associated fibroblasts: An updated metabolic perspective. Cancers (2021) 13(3):399. doi: 10.3390/cancers13030399
95. Wang L, Fan J, Yan CY, Ling R, Yun J. Activation of hypoxia-inducible factor-1α by prolonged in vivo hyperinsulinemia treatment potentiates cancerous progression in estrogen receptor-positive breast cancer cells. Biochem Bioph Res Co. (2017) 491(2):545–51. doi: 10.1016/j.bbrc.2017.03.128
96. Becker LM, O’Connell JT, Vo AP, Cain MP, Tampe D, Bizarro L, et al. Epigenetic reprogramming of cancer-associated fibroblasts deregulates glucose metabolism and facilitates progression of breast cancer. Cell Rep (2020) 31(9):107701. doi: 10.1016/j.celrep.2020.107701
97. de la Cruz-López KG, Castro-Muñoz LJ, Reyes-Hernández DO, García-Carrancá A, Manzo-Merino J. Lactate in the regulation of tumor microenvironment and therapeutic approaches. Front Oncol (2019) 9:1143. doi: 10.3389/fonc.2019.01143
98. Huber V, Camisaschi C, Berzi A, Ferro S, Lugini L, Triulzi T, et al. Cancer acidity: An ultimate frontier of tumor immune escape and a novel target of immunomodulation. Semin Cancer Biol (2017) 43:74–89. doi: 10.1016/j.semcancer.2017.03.001
99. Bellone M, Calcinotto A, Filipazzi P, Milito AD, Fais S, Rivoltini L. The acidity of the tumor microenvironment is a mechanism of immune escape that can be overcome by proton pump inhibitors. Oncoimmunology (2014) 2(1):e22058. doi: 10.4161/onci.22058
100. Navarro F, Casares N, Martín-Otal C, Lasarte-Cía A, Gorraiz M, Sarrión P, et al. Overcoming T cell dysfunction in acidic pH to enhance adoptive T cell transfer immunotherapy. Oncoimmunology (2022) 11(1):2070337. doi: 10.1080/2162402X.2022.2070337
101. Durrani IA, Bhatti A, John P. The prognostic outcome of ‘type 2 diabetes mellitus and breast cancer’ association pivots on hypoxia-hyperglycemia axis. Cancer Cell Int (2021) 21(1):351. doi: 10.1186/s12935-021-02040-5
102. Jiang C, Qu A, Matsubara T, Chanturiya T, Jou W, Gavrilova O, et al. Disruption of hypoxia-inducible factor 1 in adipocytes improves insulin sensitivity and decreases adiposity in high-fat diet–fed mice. Diabetes (2011) 60(10):2484–95. doi: 10.2337/db11-0174
103. Catrina SB, Zheng X. Hypoxia and hypoxia-inducible factors in diabetes and its complications. Diabetologia (2021) 64(4):709–16. doi: 10.1007/s00125-021-05380-z
104. Martinez-Outschoorn UE, Trimmer C, Lin Z, Whitaker-Menezes D, Chiavarina B, Zhou J, et al. Autophagy in cancer associated fibroblasts promotes tumor cell survival. Cell Cycle (2010) 9(17):3515–33. doi: 10.4161/cc.9.17.12928
105. Rojas A, Lindner C, Schneider I, Gonzàlez I, Araya H, Morales E, et al. Diabetes mellitus contribution to the remodeling of the tumor microenvironment in gastric cancer. World J Gastrointest Oncol (2021) 13(12):1997–2012. doi: 10.4251/wjgo.v13.i12.1997
106. Mathieu J, Zhang Z, Zhou W, Wang AJ, Heddleston JM, Pinna CMA, et al. HIF induces human embryonic stem cell markers in cancer cells. Cancer Res (2011) 71(13):4640–52. doi: 10.1158/0008-5472.CAN-10-3320
107. Shi R, Liao C, Zhang Q. Hypoxia-driven effects in cancer: Characterization, mechanisms, and therapeutic implications. Cells (2021) 10(3):678. doi: 10.3390/cells10030678
108. Muz B, de la Puente P, Azab F, Azab AK. The role of hypoxia in cancer progression, angiogenesis, metastasis, and resistance to therapy. Adv Exp Med Biol (2015) 3:83–92. doi: 10.2147/HP.S93413
109. Li D, Zhou L, Huang J, Xiao X. Effect of multidrug resistance 1/P-glycoprotein on the hypoxia-induced multidrug resistance of human laryngeal cancer cells. Oncol Lett (2016) 12(2):1569–74. doi: 10.3892/ol.2016.4749
110. Faria SS, Corrêa LH, Heyn GS, de Sant’Ana LP, Almeida R das N, Magalhães KG. Obesity and breast cancer: The role of crown-like structures in breast adipose tissue in tumor progression, prognosis, and therapy. J Breast Cancer (2020) 23(3):233–45. doi: 10.4048/jbc.2020.23.e35
111. Mills C. M1 and M2 macrophages: Oracles of health and disease. Crit Rev Immunol (2013) 32(6):463–88. doi: 10.1615/critrevimmunol.v32.i6.10
112. Mehta AK, Kadel S, Townsend MG, Oliwa M, Guerriero JL. Macrophage biology and mechanisms of immune suppression in breast cancer. Front Immunol (2021) 12:643771. doi: 10.3389/fimmu.2021.643771
113. Noy R, Pollard JW. Tumor-associated macrophages: From mechanisms to therapy. Immunity (2014) 41(1):49–61. doi: 10.1016/j.immuni.2014.06.010
114. Tiainen S, Masarwah A, Oikari S, Rilla K, Hämäläinen K, Sudah M, et al. Tumor microenvironment and breast cancer survival: combined effects of breast fat, M2 macrophages and hyaluronan create a dismal prognosis. Breast Cancer Res Tr. (2020) 179(3):565–75. doi: 10.1007/s10549-019-05491-7
115. Little AC, Pathanjeli P, Wu Z, Bao L, Goo LE, Yates JA, et al. IL-4/IL-13 stimulated macrophages enhance breast cancer invasion Via rho-GTPase regulation of synergistic VEGF/CCL-18 signaling. Front Oncol (2019) 9:456. doi: 10.3389/fonc.2019.00456
116. Chiang CF, Chao TT, Su YF, Hsu CC, Chien CY, Chiu KC, et al. Metformin-treated cancer cells modulate macrophage polarization through AMPK-NF-κB signaling. Oncotarget (2017) 8(13):20706–18. doi: 10.18632/oncotarget.14982
117. D’Esposito V, Ambrosio MR, Giuliano M, Cabaro S, Miele C, Beguinot F, et al. Mammary adipose tissue control of breast cancer progression: Impact of obesity and diabetes. Front Oncol (2020) 10:1554. doi: 10.3389/fonc.2020.01554
118. Wang L, Zhao RP, Song X, Wu W. Targeting ERβ in macrophage reduces crown-like structures in adipose tissue by inhibiting osteopontin and HIF-1α. Sci Rep-uk. (2019) 9(1):15762. doi: 10.1038/s41598-019-52265-8
119. Maliniak ML, Miller-Kleinhenz J, Cronin-Fenton DP, Lash TL, Gogineni K, Janssen EAM, et al. Crown-like structures in breast adipose tissue: Early evidence and current issues in breast cancer. Cancers (2021) 13(9):2222. doi: 10.3390/cancers13092222
120. Iyengar N, Brown K, Zhou X, Subbaramaiah K, Giri D, Gucalp A, et al. Metabolic obesity, adipose inflammation and elevated breast aromatase in women with normal body mass index. Cancer Prevention Res (2017) 10(4):235–43. doi: 10.1158/1940-6207.CAPR-16-0314
121. Au CC, Docanto MM, Zahid H, Raffaelli FM, Ferrero RL, Furness JB, et al. Des-acyl ghrelin inhibits the capacity of macrophages to stimulate the expression of aromatase in breast adipose stromal cells. J Steroid Biochem Mol Biol (2017) 170:49–53. doi: 10.1016/j.jsbmb.2016.07.005
122. Zahid H, Simpson ER, Brown KA. Inflammation, dysregulated metabolism and aromatase in obesity and breast cancer. Curr Opin Pharmacol (2016) 31:90–6. doi: 10.1016/j.coph.2016.11.003
123. Zhang Z, Liu S, Zhang B, Qiao L, Zhang Y, Zhang Y. T Cell dysfunction and exhaustion in cancer. Front Cell Dev Biol (2020) 8:17. doi: 10.3389/fcell.2020.00017
124. Wang J, Xu Y, Huang Z, Lu X. T Cell exhaustion in cancer: Mechanisms and clinical implications. J Cell Biochem (2018) 119(6):4279–86. doi: 10.1002/jcb.26645
125. Nicholas DA, Andrieu G, Strissel KJ, Nikolajczyk BS, Denis GV. BET bromodomain proteins and epigenetic regulation of inflammation: implications for type 2 diabetes and breast cancer. Cell Mol Life Sci (2017) 74(2):231–43. doi: 10.1007/s00018-016-2320-0
126. de Candia P, Prattichizzo F, Garavelli S, Rosa VD, Galgani M, Rella FD, et al. Type 2 diabetes: How much of an autoimmune disease? Front Endocrinol (2019) 10:451. doi: 10.3389/fendo.2019.00451
127. Nikolajczyk BS, Jagannathan-Bogdan M, Shin H, Gyurko R. State of the union between metabolism and the immune system in type 2 diabetes. Genes Immun (2011) 12(4):239–50. doi: 10.1038/gene.2011.14
128. Xia C, Rao X, Zhong J. Role of T lymphocytes in type 2 diabetes and diabetes-associated inflammation. J Diabetes Res (2017) 2017:1–6. doi: 10.1155/2017/6494795
129. Xia A, Zhang Y, Xu J, Yin T, Lu XJ. T Cell dysfunction in cancer immunity and immunotherapy. Front Immunol (2019) 10:1719. doi: 10.3389/fimmu.2019.01719
130. Qiao Yc, Shen J, He L, Hong Xz, Tian F, Pan Yh, et al. Changes of regulatory T cells and of proinflammatory and immunosuppressive cytokines in patients with type 2 diabetes mellitus: A systematic review and meta-analysis. . J Diabetes Res (2016) 2016:3694957. doi: 10.1155/2016/3694957
131. Bharath LP, Hart SN. Nikolajczyk BS. T-cell metabolism as interpreted in obesity-associated inflammation. Endocrinology (2022) 163(10):3694957. doi: 10.1210/endocr/bqac124
132. Mahmoud SMA, Paish EC, Powe DG, Macmillan RD, Grainge MJ, Lee AHS, et al. Tumor-infiltrating CD8+ lymphocytes predict clinical outcome in breast cancer. J Clin Oncol (2011) 29(15):1949–55. doi: 10.1200/JCO.2010.30.5037
133. Edechi CA, Ikeogu N, Uzonna JE, Myal Y. Regulation of immunity in breast cancer. Cancers (2019) 11(8):1080. doi: 10.3390/cancers11081080
134. Batalha S, Ferreira S, Brito C. The peripheral immune landscape of breast cancer: Clinical findings and In vitro models for biomarker discovery. Cancers (2021) 13(6):1305. doi: 10.3390/cancers13061305
135. Wang K, Shen T, Siegal GP, Wei S. The CD4/CD8 ratio of tumor-infiltrating lymphocytes at the tumor-host interface has prognostic value in triple-negative breast cancer. Hum Pathol (2017) 69:110–7. doi: 10.1016/j.humpath.2017.09.012
136. Miya A, Nakamura A, Miyoshi H, Takano Y, Sunagoya K, Hayasaka K, et al. Impact of glucose loading on variations in CD4+ and CD8+ T cells in Japanese participants with or without type 2 diabetes. Front Endocrinol (2018) 9:81. doi: 10.3389/fendo.2018.00081
137. Nojima I, Eikawa S, Tomonobu N, Hada Y, Kajitani N, Teshigawara S, et al. Dysfunction of CD8 + PD-1 + T cells in type 2 diabetes caused by the impairment of metabolism-immune axis. Sci Rep-uk. (2020) 10(1):14928. doi: 10.1038/s41598-020-71946-3
138. Wherry EJ, Kurachi M. Molecular and cellular insights into T cell exhaustion. Nat Rev Immunol (2015) 15(8):486–99. doi: 10.1038/nri3862
139. Yi JS, Cox MA, Zajac AJ. T-Cell exhaustion: characteristics, causes and conversion: T-cell exhaustion. Immunology (2010) 129(4):474–81. doi: 10.1111/j.1365-2567.2010.03255.x
140. Eikawa S, Nishida M, Mizukami S, Yamazaki C, Nakayama E, Udono H. Immune-mediated antitumor effect by type 2 diabetes drug, metformin. Proc Natl Acad Sci (2015) 112(6):1809–14. doi: 10.1073/pnas.1417636112
141. Jia Y, Zhao Y, Li C, Shao R. The expression of programmed death-1 on CD4+ and CD8+ T lymphocytes in patients with type 2 diabetes and severe sepsis. PloS One (2016) 11(7):e0159383. doi: 10.1371/journal.pone.0159383
142. Schmid P, Cortes J, Pusztai L, McArthur H, Kümmel S, Bergh J, et al. Pembrolizumab for early triple-negative breast cancer. New Engl J Med (2020) 382(9):810–21. doi: 10.1056/NEJMoa1910549
143. Luo C, Wang P, He S, Zhu J, Shi Y, Wang J. Progress and prospect of immunotherapy for triple-negative breast cancer. Front Oncol (2022) 12:919072. doi: 10.3389/fonc.2022.919072
144. Tarantino P, Corti C, Schmid P, Cortes J, Mittendorf EA, Rugo H, et al. Immunotherapy for early triple negative breast cancer: research agenda for the next decade. NPJ Breast Cancer (2022) 8(1):23. doi: 10.1038/s41523-022-00386-1
145. Deshpande RP, Sharma S, Watabe K. The confounders of cancer immunotherapy: Roles of lifestyle, metabolic disorders and sociological factors. Cancers (2020) 12(10):2983. doi: 10.3390/cancers12102983
146. Wu SZ, Al-Eryani G, Roden DL, Junankar S, Harvey K, Andersson A, et al. A single-cell and spatially resolved atlas of human breast cancers. Nat Genet (2021) 53(9):1334–47. doi: 10.1038/s41588-021-00911-1
147. Danenberg E, Bardwell H, Zanotelli VRT, Provenzano E, Chin SF, Rueda OM, et al. Breast tumor microenvironment structures are associated with genomic features and clinical outcome. Nat Genet (2022) 54(5):660–9. doi: 10.1038/s41588-022-01041-y
148. Rena G, Hardie DG, Pearson ER. The mechanisms of action of metformin. Diabetologia (2017) 60(9):1577–85. doi: 10.1007/s00125-017-4342-z
149. Xu L, Wang X, Chen Y, Soong L, Chen Y, Cai J, et al. Metformin modulates T cell function and alleviates liver injury through bioenergetic regulation in viral hepatitis. Front Immunol (2021) 12:638575. doi: 10.3389/fimmu.2021.638575
150. Zakikhani M, Blouin MJ, Piura E, Pollak MN. Metformin and rapamycin have distinct effects on the AKT pathway and proliferation in breast cancer cells. Breast Cancer Res Tr. (2010) 123(1):271–9. doi: 10.1007/s10549-010-0763-9
151. Hadad SM, Hardie DG, Appleyard V, Thompson AM. Effects of metformin on breast cancer cell proliferation, the AMPK pathway and the cell cycle. Clin Transl Oncol (2014) 16(8):746–52. doi: 10.1007/s12094-013-1144-8
152. De A, Kuppusamy G. Metformin in breast cancer: preclinical and clinical evidence. Curr Prob Cancer (2019) 44(1):100488. doi: 10.1016/j.currproblcancer.2019.06.003
153. Schexnayder C, Broussard K, Onuaguluchi D, Poché A, Ismail M, McAtee L, et al. Metformin inhibits migration and invasion by suppressing ROS production and COX2 expression in MDA-MB-231 breast cancer cells. Int J Mol Sci (2018) 19(11):3692. doi: 10.3390/ijms19113692
154. Koh M, Lee JC, Min C, Moon A. A novel metformin derivative, HL010183, inhibits proliferation and invasion of triple-negative breast cancer cells. Bioorgan Med Chem (2013) 21(8):2305–13. doi: 10.1016/j.bmc.2013.02.015
155. Chen YC, Li H, Wang J. Mechanisms of metformin inhibiting cancer invasion and migration. Am J Transl Res (2020) 12(9):4885–901.
156. Roshan MH, Shing YK, Pace NP. Metformin as an adjuvant in breast cancer treatment. SAGE Open Med (2019) 7:2050312119865114. doi: 10.1177/2050312119865114
157. Sonnenblick A, Agbor-Tarh D, Bradbury I, Cosimo SD, AzimJr HA, Fumagalli D, et al. Impact of diabetes, insulin, and metformin use on the outcome of patients with human epidermal growth factor receptor 2–positive primary breast cancer: Analysis from the ALTTO phase III randomized trial. J Clin Oncol (2017) 35(13):1421–29. doi: 10.1200/JCO.2016.69.7722
158. Kim HJ, Kwon H, Lee JW, Kim HJ, Lee SB, Park HS, et al. Metformin increases survival in hormone receptor-positive, HER2-positive breast cancer patients with diabetes. Breast Cancer Res (2015) 17(1):64. doi: 10.1186/s13058-015-0574-3
159. Goodwin PJ, Chen BE, Gelmon KA, Whelan TJ, Ennis M, Lemieux J, et al. Effect of metformin vs placebo on invasive disease–free survival in patients with breast cancer. Jama (2022) 327(20):1963–73. doi: 10.1001/jama.2022.6147
160. Saxena M, Balaji SA, Deshpande N, Ranganathan S, Pillai DM, Hindupur SK, et al. AMP-activated protein kinase promotes epithelial-mesenchymal transition in cancer cells through Twist1 upregulation. J Cell Sci (2018) 131(14):jcs208314. doi: 10.1242/jcs.208314
161. Zhang Z, Li F, Tian Y, Cao L, Gao Q, Zhang C, et al. Metformin enhances the antitumor activity of CD8 + T lymphocytes via the AMPK–miR-107–Eomes–PD-1 pathway. J Immunol (2020) 204(9):2575–88. doi: 10.4049/jimmunol.1901213
162. Denis GV, Sebastiani P, Bertrand KA, Strissel KJ, Tran AH, Slama J, et al. Inflammatory signatures distinguish metabolic health in African American women with obesity. PloS One (2018) 13(5):e0196755. doi: 10.1371/journal.pone.0196755
163. Amoani B, Sakyi SA, Mantey R, Laing EF, Ephraim RD, Sarfo-Katanka O, et al. Increased metformin dosage suppresses pro-inflammatory cytokine levels in systemic circulation and might contribute to its beneficial effects. J Immunoass Immunochem (2021) 42(3):252–64. doi: 10.1080/15321819.2020.1862861
Keywords: type II diabetes mellitus, intercellular communication, tumor microenvironment, metabolic reprogramming, exosomes
Citation: Ennis CS, Llevenes P, Qiu Y, Dries R and Denis GV (2022) The crosstalk within the breast tumor microenvironment in type II diabetes: Implications for cancer disparities. Front. Endocrinol. 13:1044670. doi: 10.3389/fendo.2022.1044670
Received: 14 September 2022; Accepted: 17 November 2022;
Published: 01 December 2022.
Edited by:
Pierre-Damien Denechaud, INSERM U1048 Institut des Maladies Métaboliques et Cardiovasculaires, FranceReviewed by:
Klara Brixius, German Sport University Cologne, GermanyCamille Attané, UMR5089 Institut de Pharmacologie et de Biologie Structurale (IPBS), France
Copyright © 2022 Ennis, Llevenes, Qiu, Dries and Denis. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Christina S. Ennis, ZW5uaXNjQGJ1LmVkdQ==