
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Endocrinol. , 12 December 2022
Sec. Bone Research
Volume 13 - 2022 | https://doi.org/10.3389/fendo.2022.1033843
This article is part of the Research Topic Fish as Model Organism for Skeletal Diseases View all 15 articles
 Amanda Stenzel1†
Amanda Stenzel1† Abigail Mumme-Monheit1†
Abigail Mumme-Monheit1† Juliana Sucharov1
Juliana Sucharov1 Macie Walker2
Macie Walker2 Jennyfer M. Mitchell1
Jennyfer M. Mitchell1 Bruce Appel2
Bruce Appel2 James T. Nichols1*
James T. Nichols1*The Notch pathway is a cell-cell communication system which is critical for many developmental processes, including craniofacial development. Notch receptor activation induces expression of several well-known canonical targets including those encoded by the hes and her genes in mammals and zebrafish, respectively. The function of these genes, individually and in combination, during craniofacial development is not well understood. Here, we used zebrafish genetics to investigate her9 and her6 gene function during craniofacial development. We found that her9 is required for osteoblasts to efficiently mineralize bone, while cartilage is largely unaffected. Strikingly, gene expression studies in her9 mutants indicate that although progenitor cells differentiate into osteoblasts at the appropriate time and place, they fail to efficiently lay down mineralized matrix. This mineralization role of her9 is likely independent of Notch activation. In contrast, her9 also functions redundantly with her6 downstream of Jagged1b-induced Notch activation during dorsoventral craniofacial patterning. These studies disentangle distinct and redundant her gene functions during craniofacial development, including an unexpected, Notch independent, requirement during bone mineralization.
The Notch signaling pathway is conserved across metazoans. Notch signaling is highly pleiotropic and functions in the development, homeostasis and regeneration of many different cells and tissues, including skeletal cells (1–3). During canonical vertebrate Notch activation, membrane-bound Jagged or Delta ligands bind to membrane-bound Notch receptors to induce cell-cell communication (4). After binding, the Notch receptor is cleaved releasing the Notch intracellular domain (NICD) which translocates to the nucleus where it directly induces transcription of downstream target genes (5). Canonical Notch downstream targets include the homologs of the Drosophila genes hairy and enhancer of split. In mammals these genes are called hairy and enhancer of split (hes) (6). In zebrafish the homologous genes are called hairy and enhancer of split related (her) (7). The hes and her genes belong to a family of helix-loop-helix transcriptional repressors with Orange domain (8). The roles that these effectors play in various Notch signaling contexts are not yet clear. Further confounding our understanding, some her genes (like her9) have both Notch-dependent and Notch-independent expression in different situations (9–12). Thus, more work is needed to understand specific roles of the different hes/her genes, individually and in combination, and to explore contexts where their expression can be Notch dependent or independent.
The vertebrate craniofacial skeleton is largely derived from post-migratory neural crest cells. After these cells migrate into the pharyngeal arches, various signaling pathways act upon them to confer dorsal versus ventral cellular identity. These dorsal and ventral cells then give rise to the diversely shaped cartilages and bones of the jaw and jaw support structures. The Notch pathway is one signaling system that functions during craniofacial development to specify dorsal versus ventral identity in zebrafish (1, 13–16). Much of our understanding of this process comes from studies of genetic mutants, overexpression conditions, and pharmacological inhibitors in zebrafish. In particular, the Notch ligand jagged1b (jag1b) is critical for zebrafish craniofacial development, and jag1b mutant phenotypes are phenocopied by the Notch cleavage inhibitor dibenzazepine (DBZ) (13). In jag1b mutants, dorsal craniofacial structures are shaped more like ventral structures suggesting identity transformations. In support, in jag1b mutants, expression of ventral identity genes like dlx5a are expanded into dorsal domains (1). These zebrafish studies offer insight into the mechanisms behind human craniofacial disease phenotypes. Humans with mutations in the human ortholog JAG1 develop Alagille syndrome which is characterized by craniofacial abnormalities including a broad forehead, deep set eyes, small pointed chin, and midface hypoplasia (17). Midface hypoplasia, characterized by a reduction of bone, is observed in both humans and mouse models (18, 19). This dorsal bone reduction defect is similar to the reduced dorsal bone observed in zebrafish jag1b mutants. Thus, genetic studies in zebrafish inform the etiology of human genetic disease phenotypes.
Jag1b appears to be the most critical Notch ligand for zebrafish craniofacial development. In contrast, the roles of the various Notch target genes in craniofacial patterning are less clear. For example, her6 expression is regionalized to the dorsal domain of the pharyngeal arches, making it a strong candidate for functioning downstream of jag1b during craniofacial patterning. However, zebrafish her6 mutants are indistinguishable from wild types (14), indicating her6 function is not required for dorsal identity patterning, or may function redundantly with other genes. Mice mutant for Hes1, the rodent ortholog of her6, develop dramatic craniofacial phenotypes (20) at least in part due to neural crest cell survival defects (21). To our knowledge, a patterning function of Hes1 has not yet been demonstrated in mice. Another Notch target gene of this same family, her9, was reported to have overt craniofacial defects when mutagenized in zebrafish (22), but details of these defects were not analyzed, and no figures illustrating the skeletal phenotype were provided. Thus, the role of this gene in craniofacial development remains unknown. However, the human ortholog, HES4, is contained within a region deleted in chromosome 1p36 deletion syndrome, which is associated with a small, short and wide head, vision and hearing problems, abnormalities of the skeleton as well as abnormalities of the brain (23). Because there are many genes encompassed by this deletion, whether HES4/her9 is involved in craniofacial patterning remains unknown. Adding to the confusion, there is no HES4/her9 ortholog in the mouse genome. Therefore, the mouse system cannot inform the function of this gene.
Given that her6 is specifically expressed in cranial neural crest cells from the dorsal pharyngeal arches, and that her6 mutants do not develop overt phenotypes in zebrafish, we hypothesized that her6 functions redundantly with other her genes downstream of jag1b during zebrafish craniofacial patterning. Double and triple mutant genetic experiments are needed to test this hypothesis.
The Notch pathway also plays a vital role in skeletal development occurring after craniofacial patterning. After regional identity is specified, precursor cells differentiate into chondrocytes and osteoblasts that make cartilage and bone, respectively. Numerous studies implicate Notch signaling and Notch target genes in both chondrocyte and osteoblast cell differentiation (3). Chondrogenic differentiation assays in human cells indicate that HES/HER proteins bind to SOX9 binding sites in COL2A1 enhancers, thereby suppressing expression of genes critical for chondrocyte differentiation (24). Similarly, in mouse cells these genes suppress Sox9 and Col2a1 expression and chondrogenesis (25). Thus, Notch activation decreases chondrogenesis, at least in these experimental contexts. The role of Notch target genes during osteoblast differentiation is complicated and unclear. Specifically, some studies indicate Notch target genes, including HES4/her9, function to promote osteoblast formation (26). Meanwhile others indicate that Notch signaling, via hes/her genes, represses osteoblast differentiation (27, 28). Thus, hes/her genes are reported to have both an inductive and a suppressive effect on osteoblast differentiation. However, several of these studies were carried out with cell culture overexpression conditions, which may not inform in vivo function. A more complete understanding of the complicated role of these genes during osteoblast differentiation will likely arise from studies examining in vivo genetic models.
Here we used zebrafish genetics to discover that her9 function is crucial for osteoblasts to efficiently mineralize bone. Surprisingly, osteoblasts differentiate in the correct time and place in her9 mutants, yet mineralized matrix production is dramatically impaired. This mineralization role is independent of Notch cleavage. However, we did find a redundant role for her9 and her6 downstream of Jagged induced Notch activation during craniofacial patterning.
jag1b functions in cranial neural crest cells of the dorsal anterior pharyngeal arches (arches 1 and 2) (1). We, and others, demonstrated that developing zebrafish heads express her6, in a jag1b dependent manner (14, 15). Yet, her6 mutants do not produce overt craniofacial phenotypes (14). These results motivate the hypothesis that her6 functions redundantly with other her genes downstream of jag1b. To explore what other her genes might be acting downstream of jag1b during craniofacial development, we searched for genes expressed in the same cranial neural crest cell population as jag1b and her6. We examined a 24 hours post fertilization (hpf) single cell RNA sequencing (scRNA-seq) dataset from sorted cranial neural crest cells. This dataset is an unpublished replicate to test for reproducibility of our previously published experiment (29). Both replicates are highly similar, and the results of this comparison will be reported elsewhere. We found that 16 her genes are detectibly expressed in this dataset (Figure S1), however her9 and her6 are the family members with the strongest expression in the skeletogenic, pharyngeal arch neural crest cells. In contrast, many of the other her genes are only weakly, or not detectably, expressed in the anterior pharyngeal arches. In fact, most of the other her genes in this dataset are restricted to the melanocyte population. Further examining her9 and her6 revealed that they are expressed in a subpopulation of anterior arch cranial neural crest cells along with jag1b (Figure 1A). While her6 is broadly expressed among cranial neural crest cells, the jag1b expressing population is enriched for her6. her9 is broadly, but more weakly, expressed in most cranial neural crest cells but the strongest expression is in the jag1b population. We further analyzed these expression data with side-by-side comparisons in two-color plots to look for co-expression between jag1b, her9, and her6 (Figure S2). These plots further support our interpretation that jag1b, her9 and her6 are expressed in the same population, and in some cases in the same cells. We next re-clustered the anterior arch population to further analyze jag1b and her9 and her6 in these cells which give rise to the craniofacial skeleton. In these analyses we include UMAP plots with the expression of known “landmark” genes. These include the pan-anterior arch gene dlx2a, the intermediate and ventral anterior arch gene dlx5a and the extreme ventral anterior arch gene hand2. We observed in these studies, that jag1b, known to be expressed in the dorsal anterior arch population (1), is complementary to dlx5a, as expected. Moreover jag1b expression overlaps with her9 and her6 in these analyses. These data further support the finding that her9 and her6 are expressed in the same population of cells as jag1b. These results make her9 and her6 strong candidates for genes functioning downstream of jag1b during craniofacial development. To directly test if her9 is dependent upon jag1b function, we performed RT-qPCR in wild types and jag1b mutants at 28 and 48 hpf. We previously demonstrated that her6 is a transcriptional target of jag1b (15); expression is downregulated in jag1b mutants compared with wild-type controls at both 28 and 48 hpf (Figure 1B). Here, we find that her9 expression is significantly decreased in jag1b mutants at 48 hpf compared with wild types. There is no significant difference at 28 hpf. These findings indicate that her9 expression is jag1b independent at 28 hpf and jag1b dependent at 48 hpf. That both her6 and her9 expression are dependent upon jag1b in zebrafish heads at 48 hpf, motivates the hypothesis that they function redundantly downstream of Jag-induced Notch signaling at this stage.
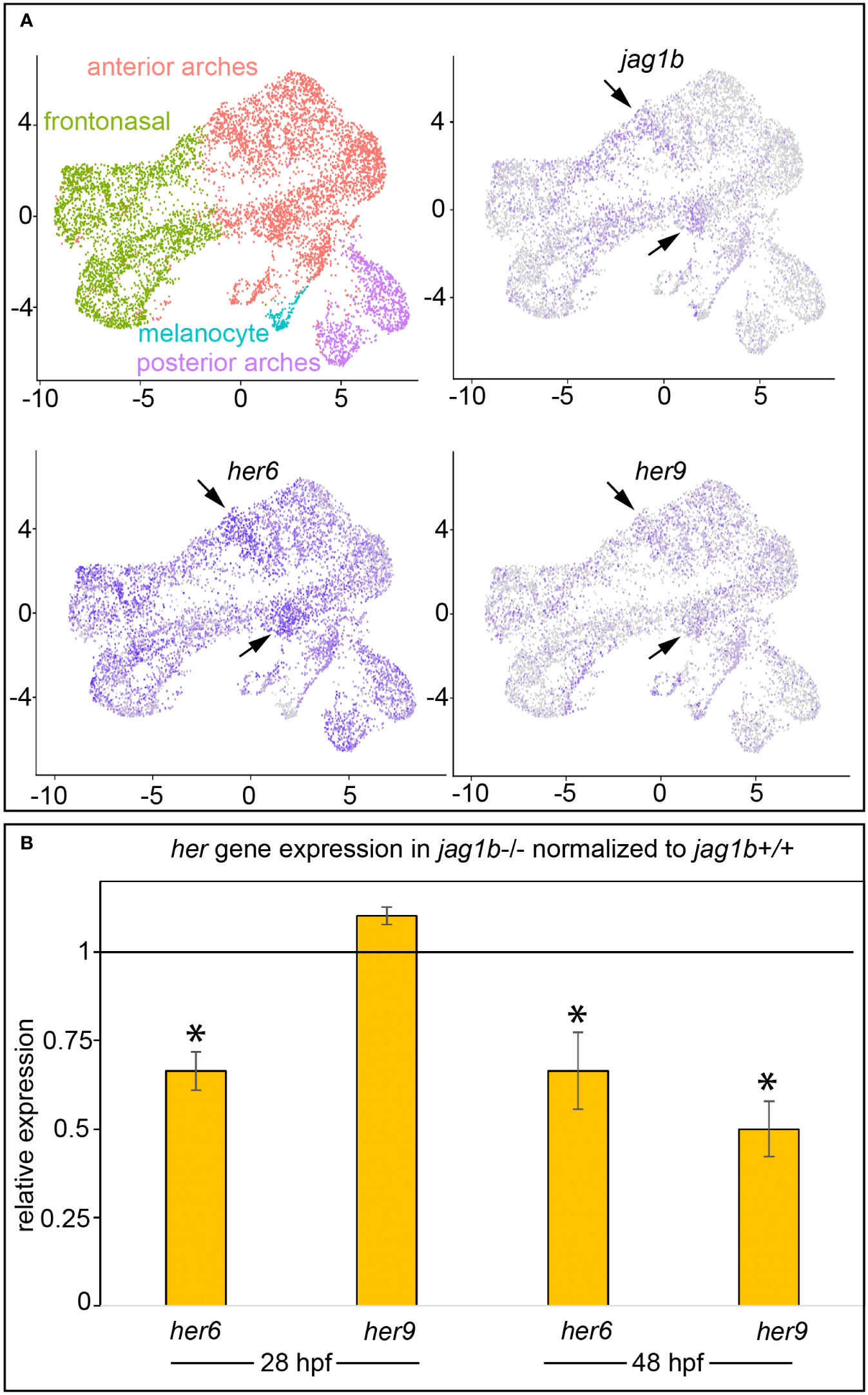
Figure 1 her6 and her9 are expressed in the same subpopulation of cranial neural crest cells as jag1b, and their expression is dependent upon jag1b function. (A) UMAPs from single cell RNA-sequencing on 24 hpf sorted cranial neural crest cells. As in our previous work (29), cranial neural crest cells can be clustered into four distinct populations. Based on our previous study and the landmark genes enriched in each population we can identify where the cells reside in the intact animal. Arrows indicate jag1b enrichment in a subset of anterior arch cells. Although her6 is broader, arrows indicate that some of the strongest expressing cells are the jag1b enriched populations. her9 is also broadly expressed among cranial neural crest cells, but at lower levels than her6. Arrows indicate her9 enrichment in the jag1b;her6 expressing populations. (B) We performed qPCR on 28 and 48 hpf genotyped heads from wild type and jag1b homozygous mutant siblings. Expression of each her gene was normalized to rps18, then normalized to the relevant wild-type control. Reactions were performed in technical triplicate and the results represent three to six biological replicates. Each biological replicate was pooled heads from 5–6 genotyped homozygous wild types or homozygous mutants. Error bars are standard deviation. Asterisks indicate significant differences from wild type controls at p < 0.05 by students t-test.
The jag1b-dependent her6;her9 expression predicts that loss of function mutations in these genes will produce similar phenotypes to jag1b mutants. To test this prediction, we first examined her9 individually. We recovered a her9 mutant allele from an ENU mutagenesis screen (see Methods for details). The mutant allele produces a premature termination codon at amino acid 148 of 291, near the end of the region predicted to encode the Orange domain (Figure 2A). We detected reduced her9 transcript levels in homozygous mutants suggesting that this lesion impacts her9 function (Figure S3). When we crossed adults heterozygous for this her9 mutation we failed to recover homozygous mutants from these crosses at 12 dpf, indicating that the mutation is homozygous lethal, as reported previously (22). We next stained offspring from these crosses with Alcian Blue and Alizarin Red at 6 dpf. Alcian Blue labels cartilage by interacting with mucopolysaccharides (30). Alizarin Red stains bone by complexing with calcium in mineralized bone (31). We observed that cartilage staining in her9 mutants was overtly normal (Figures 2B–I), although the overall size of the craniofacial skeleton, including the cartilage elements, was significantly smaller than wild type-controls (Figure S4). In contrast to the cartilage skeleton, which is strongly stained with Alcian Blue, we observed dramatic reductions in Alizarin Red-stained bone in these mutants. We found reduced mineralization in cranial neural crest cell-derived structures like the opercle bone, branchiostegal rays, maxilla, entopterygoid, ceratobranchial 5 (cb5) bones and teeth. The cleithrum, which is derived from mesoderm (32), was also weakly and variably stained in her9 homozygous mutants (Figure 2C). We also observed that notochord mineralization was defective in her9 mutants (Figures 2D, E). Thus, her9 functions in the mineralization of structures derived from both cranial neural crest cells as well as mesoderm. Scoring the individual bones for mineralization defect penetrance revealed that parasphenoid mineralization loss is 100% penetrant, while the cleithrum and the opercle mineralization losses are more variable (Figure 2J). Thus, all bones require her9 function, but some boney structures are more sensitive to her9 loss than others. Both scRNA-seq and in situ gene expression studies indicate that her9 is broadly expressed during the pharyngula period (Figures 1A,S3), consistent with the broad defects in bone mineralization.
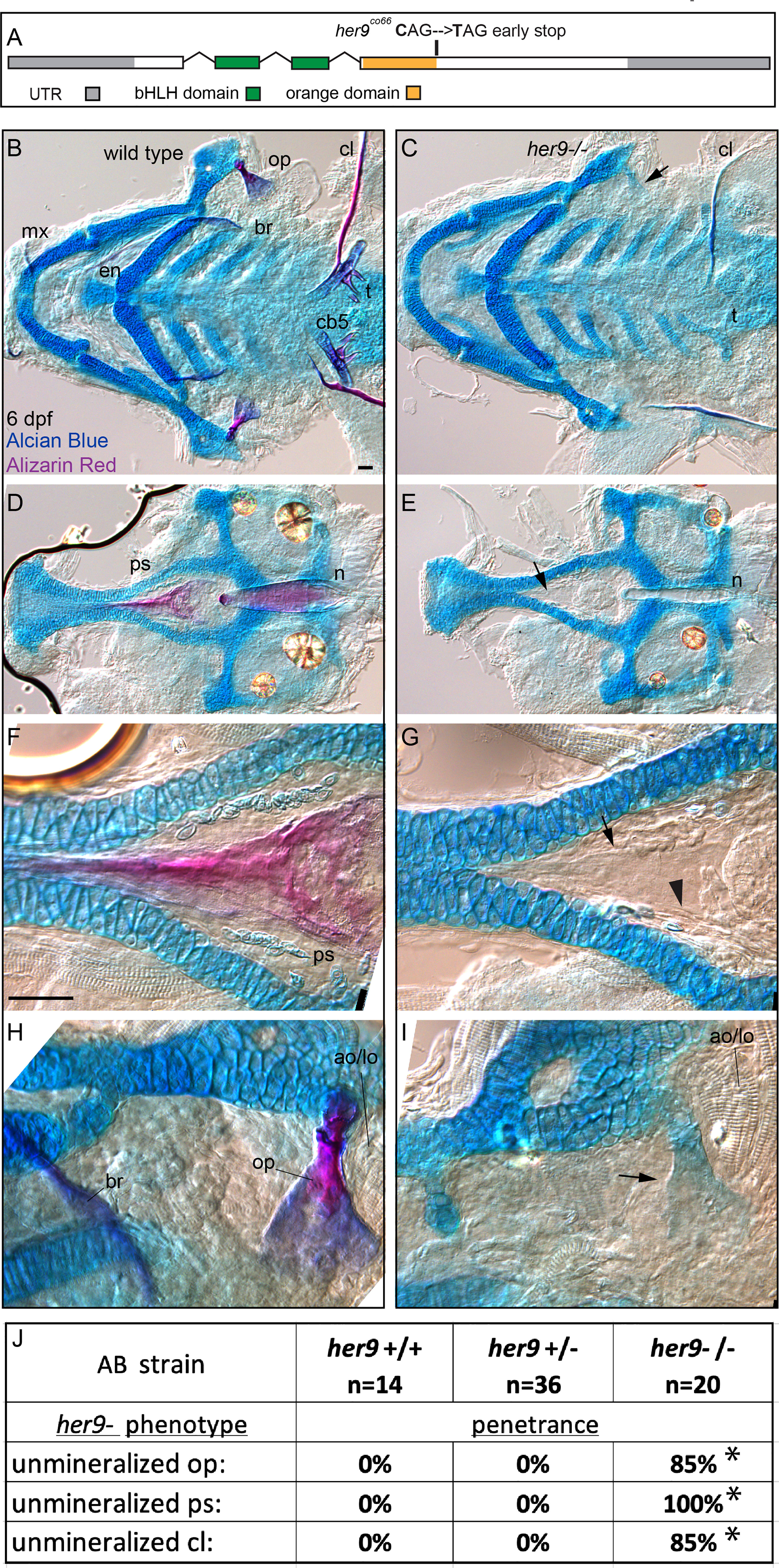
Figure 2 her9 is required for efficient mineralization. (A) Exon diagram for her9 indicating the untranslated regions (UTR) and the regions predicted to encode the basic helix-loop-helix (bHLH) and Orange domains. The location of the early stop encoded by the her9co66 allele is indicated. (B, C) Zebrafish heterozygous for her9 were pairwise intercrossed and six days post fertilization (dpf) larvae were stained with Alcian Blue and Alizarin Red to label cartilage and bone. The individuals were then genotyped, the viscerocrania were dissected, flat mounted, and then imaged. The following craniofacial skeletal elements are indicated in the wild-type individual: opercle bone (op), branchiostegal ray (br), maxilla (mx), entopterygoid (en), ceratobranchial 5 (cb5) bone, teeth (t), and the cleithrum (cl). Arrow in C indicates the unmineralized, Alcian Blue-positive opercle. Scale bar is 50 μm (D, E) The neurocrania from the same experiments as B and C were dissected, flat mounted, and then imaged. The parasphenoid bone (ps) and notochord (n) is indicated in the wild-type individual. Arrow in D indicates the shadow bone where the mineralized parasphenoid would be observable in wild types. (F, G) Enlargements from D and E. Arrow indicates the unmineralized shadow bone. Arrowhead denotes osteoblast-shaped cells surrounding the shadow bone. Scale bar is 50 μm (H, I) Enlargements from (B, C). The adductor operculi and the levetor operculi muscles (ao/lo) are indicated. Arrow in I indicates the unmineralized, Alcian Blue-positive opercle with the adductor operculi and the levetor operculi muscles (ao/lo) attached. (J) The penetrance of her9 mutant-associated phenotypes observed in 6 dpf larvae for each genotype are indicated. Asterisk indicates significant difference (p<0.05) in penetrance between the indicated genotype and wild type by Fishers exact test.
We next aimed to determine if mineralization phenotype in her9 mutants persists into later stages of skeletal development. To this end, we analyzed her9 mutant and wild-type sibling control skeletons in 8 dpf zebrafish (Figure 3). Strikingly we observed that while bone mineralization was still reduced in her9 mutants, some mineralization had occurred. Specifically, the opercle and parasphenoid had reduced, but clearly observable, Alizarin Red staining at this stage compared with wild-type controls. Thus, the early strong mineralization defects in her9 mutants can partially recover at later stages of skeletal development.
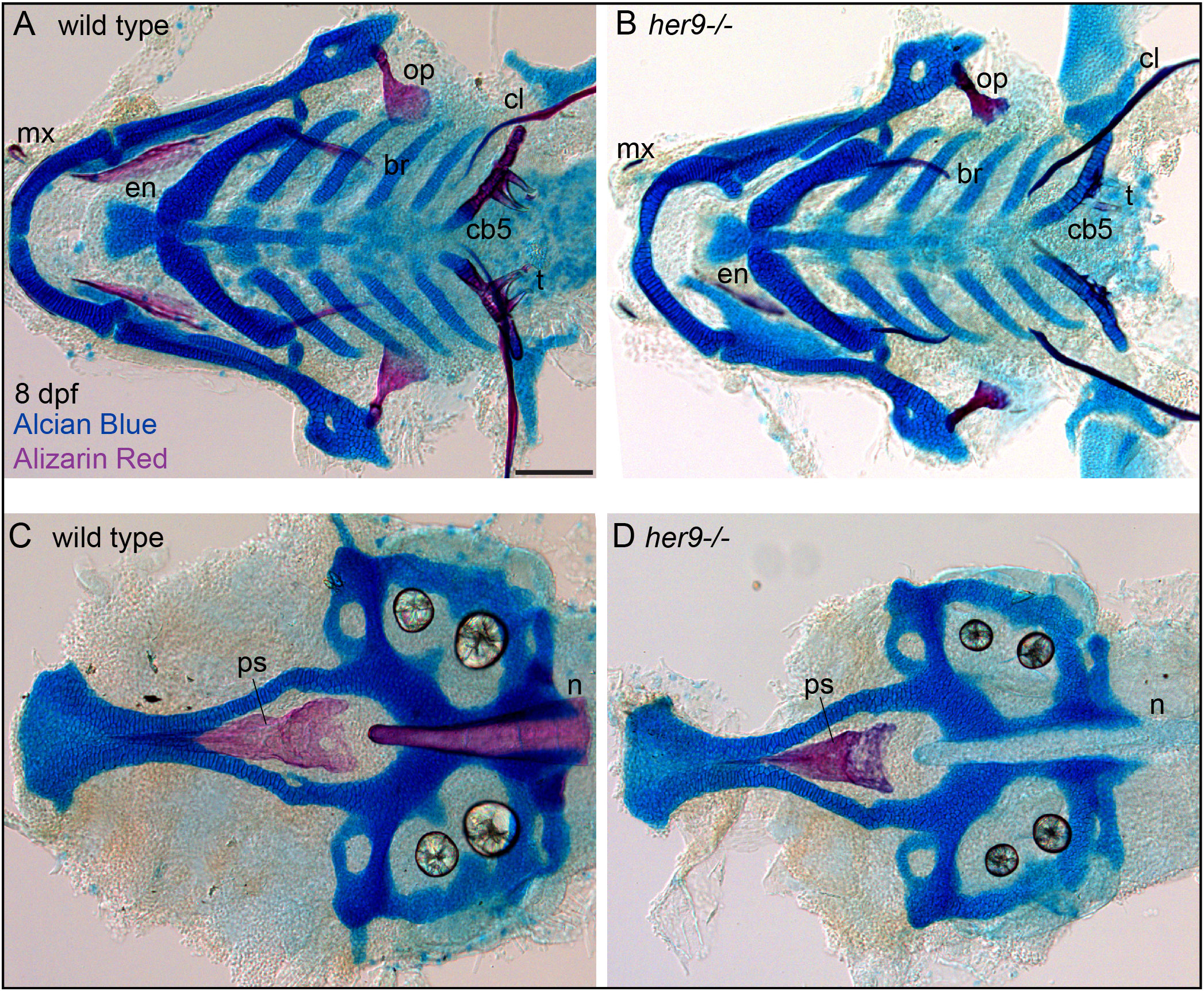
Figure 3 Mineralization defects in her9 homozygous mutants partially recover at late stages of larval development. Zebrafish heterozygous for her9 were pairwise intercrossed and at eight days post fertilization (dpf) larvae were stained with Alcian Blue and Alizarin Red to label cartilage and bone. The individuals were then genotyped, the viscerocrania and neurocrania were dissected, flat mounted, and then imaged. (A, B) Viscerocrania from wild-type and her9 homozygous mutant larvae. (C, D) Neurocrania from wild-type and her9 homozygous mutant larvae. The following craniofacial elements are indicated: opercle bone (op), branchiostegal ray (br), maxilla (mx), entopterygoid (en), ceratobranchial 5 (cb5) bone, teeth (t), cleithrum (cl), parasphenoid bone (ps), and notochord (n). Scale bar is 100 μm.
To determine if this bone mineralization phenotype was due to her9 function downstream of Notch signaling, we treated wild-type embryos with the pharmacological gamma secretase inhibitor DBZ, which inhibits Notch intramembranous cleavage and release of NICD, and phenocopies the jag1b mutant (13, 15). When we treated embryos during the craniofacial patterning stage (18-48 hpf), DBZ produced craniofacial phenotypes reminiscent of the jag1b mutant phenotype (Figure S5). Like jag1b mutants, there was no detectable bone mineralization phenotype in these treated animals. When we treated later stage animals during chondrocyte and osteoblast differentiation (48-72 hpf) with DBZ, there were no overt changes to the craniofacial skeleton. However, this treatment window produced a body-curvature phenotype indicating that DBZ is affecting Notch signaling outside the head during this stage. Finally, when we treated developing fish at later stages while the skeleton grows and becomes more elaborate (72-96 hpf) with DBZ, they were indistinguishable from untreated controls. Longer treatments from 18-72 hpf produce dramatic global defects and cannot be meaningfully interpreted for mineralization. These results indicate that inhibiting Notch cleavage and NICD release does not phenocopy the her9 phenotype but does phenocopy the jag1b phenotype. Thus, the role that her9 plays in bone mineralization is likely independent of Notch cleavage and jag1b function.
There was only weak or no Alizarin Red staining in 6 dpf her9 mutant craniofacial bony elements. However, when we closely examined these preps, we observed ‘shadow bones’ or vacant spaces shaped like the wild type structures where the elements would normally develop in wild types. For some of these, like the opercle, there was Alcian Blue staining of the unmineralized element (Figure 2C). Interestingly, the adductor operculi and levator operculi muscle groups which articulate with the posterior edge of the opercle at this stage appeared to be present and articulated with the unmineralized opercle. For some structures like the parasphenoid, there was no detectable Alcian Blue, just a shadow bone in the shape of the wild-type bone observable by Nomarski imaging (Figure 2E). High magnification images of the shadow bones revealed osteoblast-shaped cells in the expected position surrounding the element (Figure 2G). These findings motivate the hypothesis that the cells which normally produce the affected bony elements in wild types, may be present in the correct location but are variably unable to produce mineralized matrix with the correct timing.
To determine whether the cells we observed surrounding the shadow bones are differentiated osteoblasts, we crossed the her9 mutant into the sp7:EGFP transgenic line which faithfully labels active, bone-secreting osteoblasts (33). Fish with this transgene can be assayed with live Alizarin Red staining, which is a highly sensitive method of labeling mineralized bone (34). A strength of this approach is that the same bone can be longitudinally tracked in an individual over developmental time. We first examined the developing opercle at 3 dpf, when the initial opercle osteoblasts appear (35). In wild types, the first osteoblasts outline the early spicule-shaped opercle bone (Figure 4A) (36). By 6 dpf, the majority of osteoblasts are closely associated with the rapidly growing ventral edge. Examining the cells and bone matrix together at 6 dpf reveals that the early-forming spicule is the most mineralized and has the fewest osteoblasts, while the rapidly growing ventral edge, with the most osteoblasts, is more weakly mineralized. Similarly, the older, anterior end of the branchiostegal ray has fewer osteoblasts and more mineralized matrix, while the newly forming posterior end has the most osteoblasts and the least mineralized matrix. These observations indicate that we are tracking live, local mineralized matrix deposition by transgenic osteoblasts in areas of rapidly growing bone. In her9 mutants, the earliest opercle osteoblasts are observable in a similar pattern to wild type, outlining a stick shape (Figure 4B). These results support our hypothesis that the cells surrounding the shadow bones (Figure 2) are indeed osteoblasts. Like wild types, by 6 dpf most of the her9 mutant osteoblasts are located along the growing ventral edge. However, in contrast to wild types, the space outlined by osteoblasts is only very weakly stained with Alizarin Red at both 3 and 6 dpf (Figure 4B). Of note, the live fluorescent method of Alizarin Red staining was able to detect weakly mineralized matrix in her9 mutants which was not observable in fixed skeletal preps imaged with transmitted light (Figure 2). Previous reports also indicate that Alizarin Red fluoresecence can reveal mineralized structures that are difficult to observe with visible light (37). This discovery that there was some mineralized bone in homozygous mutants are consistent with our late-stage study above (Figure 3) demonstrating that mineralized structures can be found in her9 mutants at 8 dpf. Thus, mineralization is not completely absent in these mutants. Nevertheless, the amount of mineralized bone we are able to detect by both measures, at all stages, is severely reduced in her9 mutants. In contrast to the bone, the otoliths are strongly stained with Alizarin red in both wild types and mutants, serving as an internal staining control and demonstrating that otolith mineralization is not dependent upon her9 function. However, the otoliths are smaller in her9 mutants compared with wild-type controls. Like other zebrafish craniofacial mutants (15, 35, 38), there is variation among mutant phenotypes; the shape of the opercle varies among her9 mutant individuals (Figure 5). In spite of this variation, the her9 mutant opercle bones are consistently smaller than wild-type opercles. Together with our cartilage measurements described above (Figure S4), we conclude that the entire craniofacial complex is smaller in her9 mutants compared with wild types, and that her9 mutants produce osteoblasts, which appear with the same spatiotemporal dynamics as wild types, but her9 mutant osteoblasts are not able to secrete mineralized matrix with the same spatiotemporal dynamics.
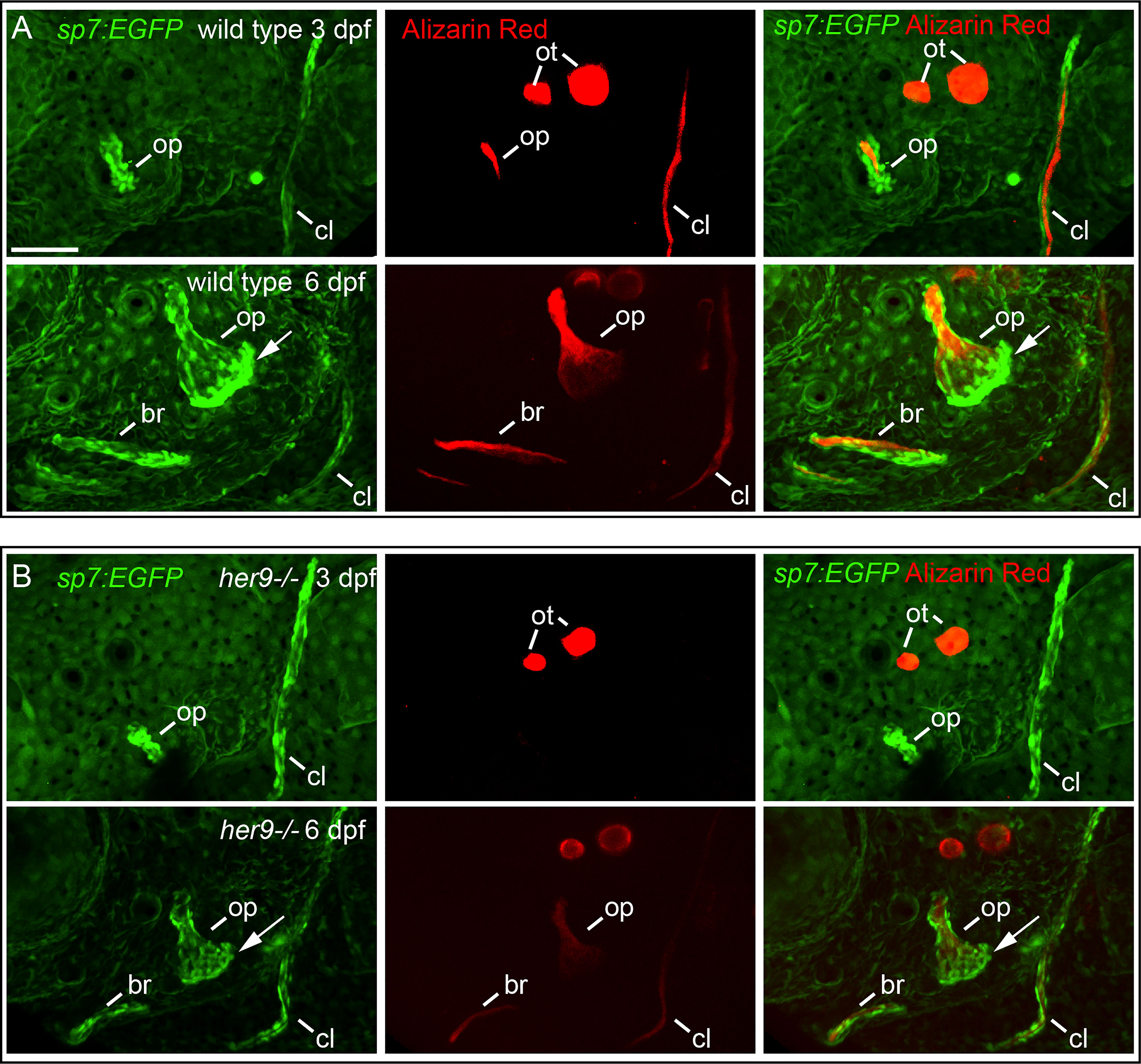
Figure 4 Osteoblasts differentiate in a spatiotemporal pattern similar to wild types in her9 mutants, but only weakly mineralize bone. sp7:EGFP;her9 heterozygotes were crossed to her9 heterozygotes and offspring were sorted for transgene expression. 16 live transgenic animals were labeled with Alizarin Red and imaged at 3 dpf. After imaging, animals were recovered and grown in individual wells until 6 dpf when they were labeled with Alizarin Red and imaged again. Individual animals were recovered for genotyping to identify homozygous wild types (A) and homozygous her9 mutants (B) that were imaged twice. The following structures are indicated: opercle bone (op), branchiostegal ray (br), cleithrum (cl), otoliths (ot). Arrows indicate osteoblasts prominently detected along the ventral edge of the opercle at 6 dpf. Scale bar is 50 μm.
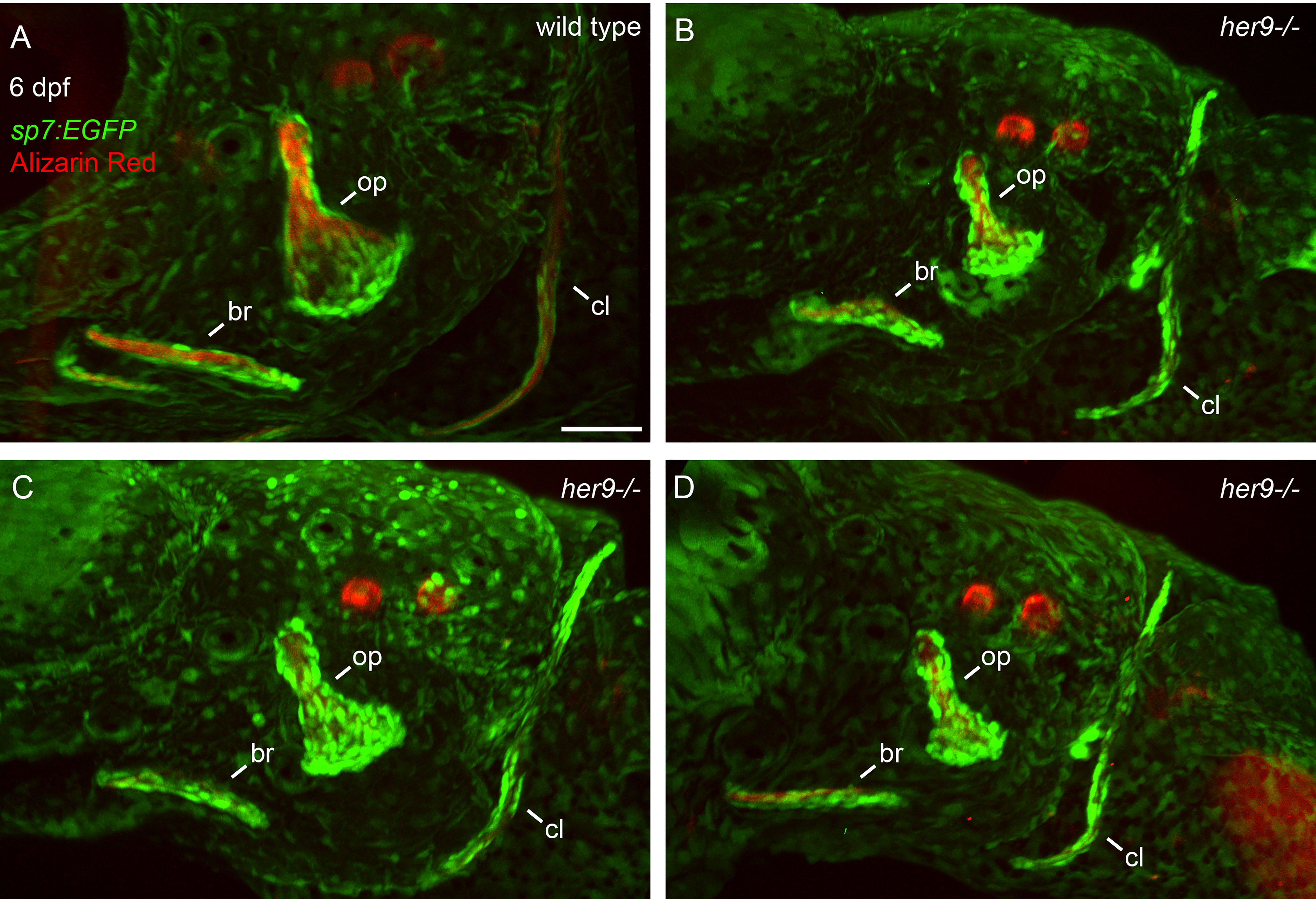
Figure 5 Opercle shapes are variable in her9 mutants. sp7:EGFP;her9 heterozygotes were crossed to her9 heterozygotes and offspring were sorted for transgene expression. 24 live transgenic animals were labeled with Alizarin Red and imaged at 6 dpf. Imaged animals were genotyped. (A) Representative opercle region from a wild-type larva. (B–D) Three different her9 mutant larvae are shown. The following structures are indicated: opercle bone (op), branchiostegal ray (br), cleithrum (cl). Scale bar is 50 μm.
her9 function is required for fish to efficiently mineralize bone. Therefore, we hypothesized that her9 would be strongly expressed in osteoblasts. We tested this hypothesis by performing in situ hybridization in the sp7:EGFP transgenic background to monitor her9 expression while labeling osteoblasts. We found that her9 is broadly expressed at 72 hpf, but expression was not concentrated in osteoblasts that are actively secreting matrix (Figure 6). Thus, while her9 expression at this stage was detected in the pectoral fin, and in the trunk, surprisingly her9 was not strongly expressed in the osteoblasts themselves.
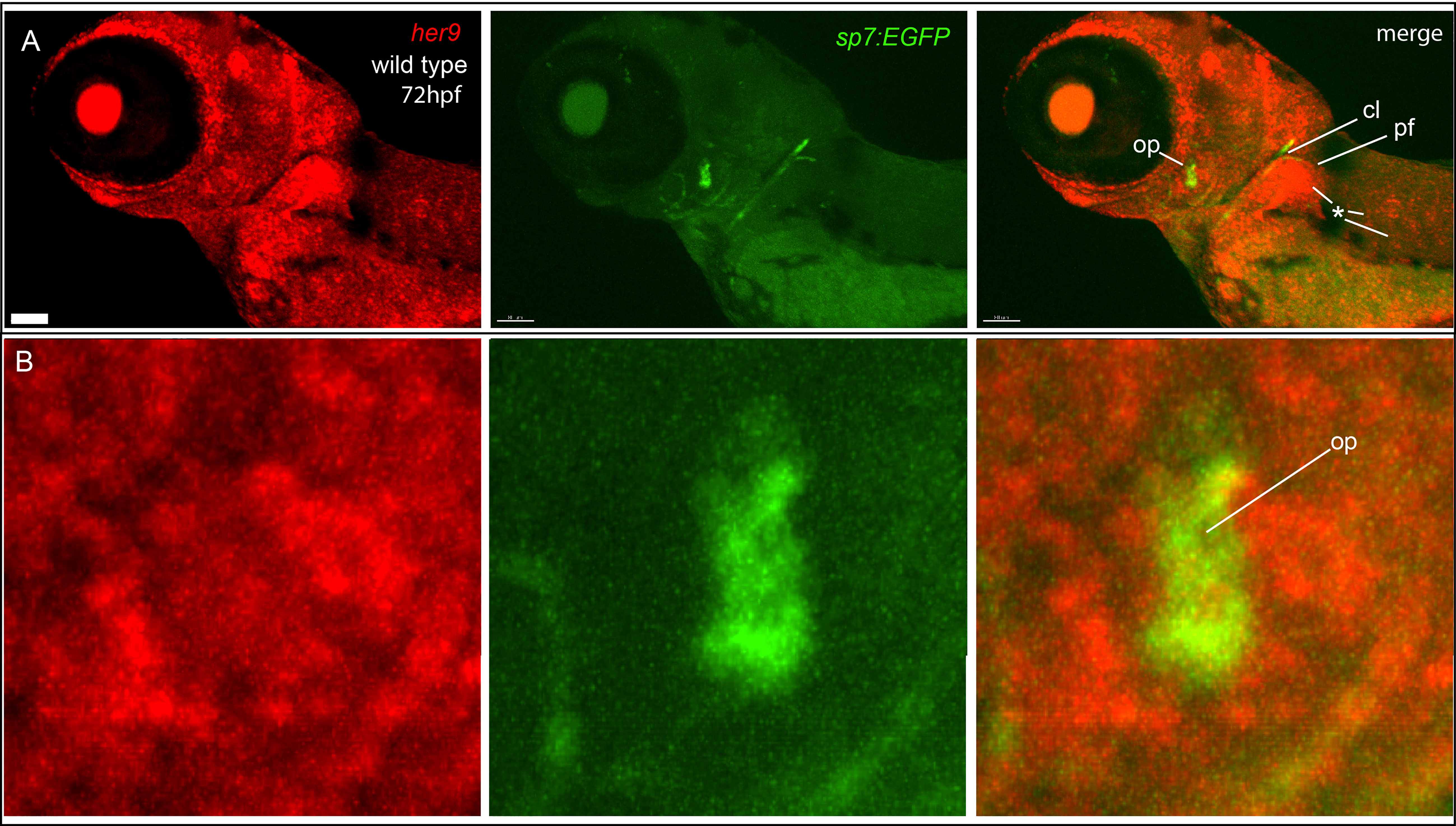
Figure 6 her9 is broadly expressed, but not enriched in osteoblasts. (A) In situ hybridization detects broad her9 expression at 72 hpf. Asterisks indicate areas of strong her9 expression. Opercle osteoblasts do not strongly express her9. (B) Enlargements of images in (A). Images in (B) are enlargements of (A). Scale bar is 80 μm.
By labeling osteoblasts and weakly mineralized bone with sp7:EGFP and live Alizarin Red staining, respectively, we are able to observe opercle shape even when mineralization defects are severe in her9 mutants (Figures 4, 5). Using this protocol, we observed divergence from the characteristic fan-shaped opercle in some individuals. We discovered that, occasionally, her9 mutant opercles at 6 dpf were less fan shaped, and instead were more stick-like, reminiscent of jag1b mutants (Figure 5). Similarly, the opercle bone was more stick shaped than wild-type controls at 8 dpf. These observations motivated the hypothesis that in addition to a Notch-independent role in osteoblast mineralization, her9 may also function to pattern the craniofacial skeleton downstream of jag1b. To test this hypothesis, we performed a genetic interaction experiment between jag1b and her9. We intercrossed jag1b;her9 double heterozygotes and stained offspring of these crosses with Alcian Blue and Alizarin red (Figure 7A). By scoring phenotypes in genotyped larvae at 6dpf, we discovered her9 and jag1b genetically interact. Removing both copies of her9 from jag1b homozygous mutants significantly increased the penetrance of an “dysmorphic foramen” phenotype in the dorsal hyomandibular cartilage (Figure 7B). This dysmorphic foramen class includes mildly affected animals with a larger foramen as well as more severe individuals no identifiable foramen. These more severe phenotypes were previously interpreted to be due to the dorsal hyomandibular cartilage transforming to be more rod shaped, partially resembling the ventral ceratohyal cartilage (1). However, we did not observe any significant changes in the penetrance of the dorsal arch 1 phenotype, a reduced palatoquadrate. We cannot meaningfully score double mutants for opercle bone phenotypes because the bone is not labeled with alizarin red in these fixed preps. We did not observe any changes in the penetrance of the her9-associated mineralization phenotype when jag1b was removed. These results indicate that her9 and jag1b genetically interact during patterning of the dorsal hyomandibular skeleton, but not during bone mineralization.
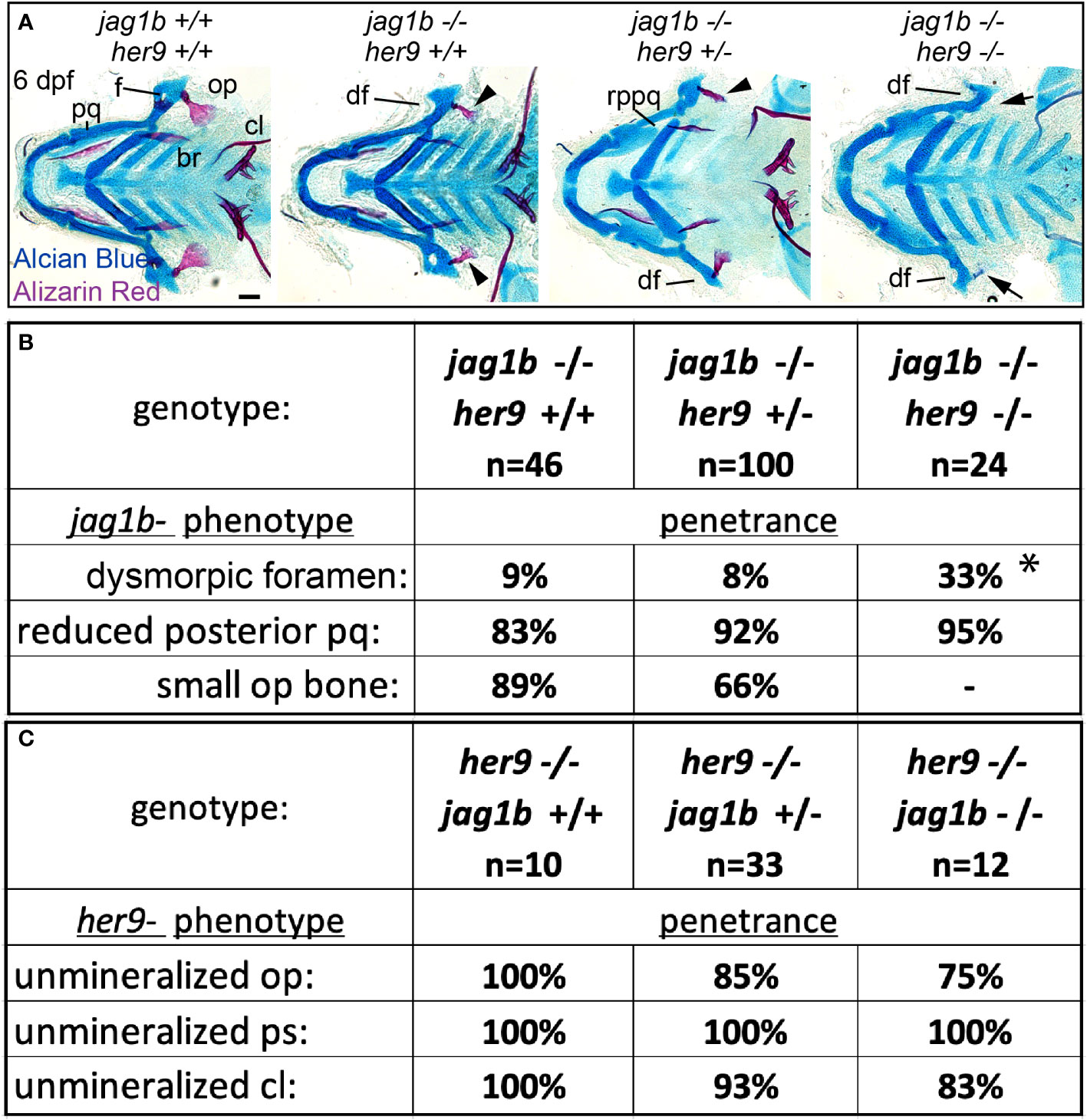
Figure 7 Mutations in her9 enhance jag1b mutant phenotypes. (A) jag1b;her9 double heterozygotes were intercrossed and six days post fertilization (dpf) larvae were stained with Alcian Blue and Alizarin Red to label cartilage and bone. The individuals were then genotyped, the viscerocrania were dissected and flat mounted then imaged. The following craniofacial skeletal elements are indicated in the wild-type individual: opercle bone (op), branchiostegal ray (br), cleithrum (cl), foramen (f) palatoquadrate (pq). The dysmorphic foramen (df) and reduced posterior pq (rppq) phenotypes are indicated, and the small op bone phenotype is indicated by an arrowhead. Arrows mark the opercle mineralization defect associated with her9 homozygous mutants. Poorly stained posterior arch derived cartilages and one missing entopterygoid bone in jag1b-/-;her9+/- are staining and mounting artifacts in this individual and not representative phenotypes associated with this genotype. Scale bar is 50 μm (B) Genotyped preps from A were scored for penetrance of jag1b mutant-associated phenotypes. Asterisk in dysmorphic foramen row indicates that jag1b-/-;her9-/- is significantly different from both jag1b-/-;her9+/+ and jag1b-/-;her9+/- using a Fisher’s exact test with p<0.05. (C) Genotyped preps from A were scored for penetrance of her9 mutant-associated phenotypes.
Removing her9 from jag1b mutants enhanced the jag1b craniofacial phenotype; double mutants had jag1b associated defects at higher penetrance than single mutants. However, penetrance was not increased to 100%. Therefore, we hypothesized that additional her genes might redundantly function downstream of jag1b during craniofacial patterning. her6 is a known Notch target, and its cranial neural crest cell expression pattern makes it a strong candidate for functioning downstream of jag1b (Figure 1). A her6 mutant was previously found to have a craniofacial skeleton indistinguishable from wild types (14). This mutant allele produces an eight-base pair deletion inducing a frameshift after amino acid 10 out of 270 then a premature termination codon after 12 missense amino acids. We speculated that this might not be a null allele, due to in-frame alternative start codons predicted in the transcript. Therefore, we generated a new mutation further downstream in her6 that is a 4 base pair deletion causing a frameshift at amino acid 51, four nonsense amino acids, and an early stop at amino acid 55 (Figure 8A). Similar to the previously generated allele, our her6 mutant also had a craniofacial skeleton that was indistinguishable from wild types (Figure 8B). We conclude that her6 is not essential for craniofacial development in an otherwise wild-type background. Next, we tested whether the her6 mutation genetically interacts with jag1b, like her9 does. We crossed jag1b;her6 double heterozygous mutants and stained offspring with Alcian Blue and Alizarin Red. When we scored genotyped, stained larvae, we found that no new phenotypes were produced. However, the penetrance of the jag1b mutant-associated reduced opercle bone was significantly increased bilaterally in jag1b;her6 double homozygous mutants compared with jag1b single mutants (Figures 8B, C).
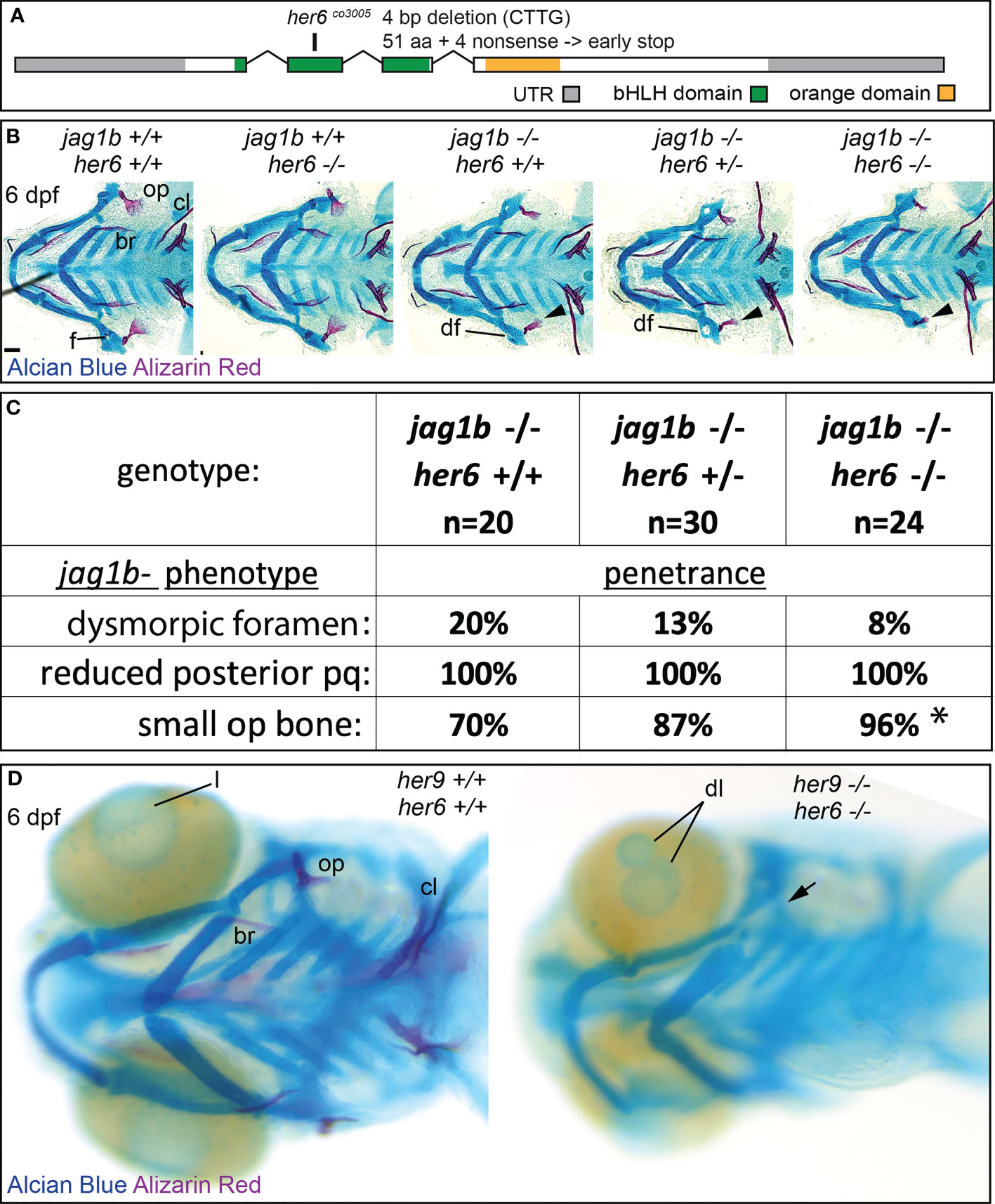
Figure 8 Mutations in her6 enhance the jag1b mutant phenotype, her6 and her9 are redundantly required for eye development. (A) Exon diagram for her6 indicating the untranslated regions (UTR) and the regions predicted to encode the basic helix-loop-helix (bHLH) and Orange domains. The location of the deletion encoded by the her6co3005 allele is indicated. (B) jag1b;her6 double heterozygotes were intercrossed and six days post fertilization (dpf) larvae were stained with Alcian Blue and Alizarin Red to label cartilage and bone. The individuals were then genotyped, the viscerocrania were dissected and flat mounted then imaged. The following craniofacial skeletal elements are indicated in the wild-type individual: opercle bone (op), branchiostegal ray (br), cleithrum (cl), foramen (f). The dysmorphic foramen phenotype (df) is indicated, and the small op bone phenotype is indicated by an arrowhead. Scale bar is 50 μm (C) Genotyped preps from B were scored for penetrance of jag1b mutant-associated phenotypes. Asterisk in small op bone row indicates that penetrance in jag1b-/-;her6-/- is significantly different from jag1b-/-;her6+/+ (p<0.05). (D) her6;her9 double heterozygotes were intercrossed and six days post fertilization (dpf) larvae were stained with Alcian Blue and Alizarin Red to label cartilage and bone. The individuals were then genotyped and whole-mount imaged. An arrow indicates unmineralized opercle. The lens (l) is indicated in the wild type and a double lens phenotype is indicated in double homozygous mutants (dl).
We next hypothesized that her6 and her9 might function redundantly during craniofacial patterning. To test this hypothesis, we combined the two mutations and intercrossed adults heterozygous for both her6 and her9, and stained offspring with Alcian Blue and Alizarin Red (Figure 8D). Analyzing genotyped individuals revealed that the ceratohyal is occasionally misaligned at the midline in double homozygous mutants which is not observed in either single mutant. Further, fish double homozygous for both her6 and her9 developed eye phenotypes that were not present in either single mutant (Figure 8D). However, her6;her9 double mutants did not phenocopy jag1b mutants, suggesting that other effectors provide further redundancy downstream of jag1b.
Our double mutant analyses demonstrated that jag1b genetically interacts with both her9 and her6, and that her9 and her6 genetically interact with each other (Figures 7, 8). While her6 enhanced the jag1b-associated opercle phenotype, her9 enhanced the jag1b-associated foramen phenotype. Based on these findings, we hypothesized that jag1b triple mutant combinations would produce novel craniofacial defects. Specifically, if her genes genetically interact with jag1b then dramatic phenotypes, more severe than the sum of the single mutant phenotypes, would be predicted in the triple mutants. When we intercrossed triple heterozygous fish and stained for Alcian Blue and Alizarin Red, we found that triple homozygous mutants were severely affected by general delay and edema producing secondary defects (Figure S6). This genotype could not be meaningfully scored for skeletal phenotypes. However, when we examined jag1b and her6 homozygous mutants in combination with her9 heterozygotes, we uncovered a new phenotype not seen in any other allelic combination in this study, the opercle and branchiostegal ray shapes were reversed (Figure 9A). Rather than a fan-shaped opercle and stick shaped branchiostegal ray, we observed a stick shaped opercle and a fan shaped branchiostegal ray. We found the branchiostegal ray fan phenotype in 46% of fish with this triple-mutant genotype, which was never present in any other genotype (Figure 9B). In some cases, the expanded branchiostegal ray was fused with the opercle bone forming a large plate-like structure (Figure 9A). These fusions were reminiscent of transformation phenotypes found when the Endothelin pathway is manipulated (39), motivating the hypothesis that these phenotypes are dorsoventral identity transformations. To directly test for molecular evidence of identity transformation, we performed in situ hybridization for the ventral arch gene dlx5a on these families (Figure 9C). Consistent with previous reports (1), we found that dlx5a expression was expanded dorsally in jag1b single mutants compared with wild-type controls. Strikingly, in the triple mutant genotype, we found that dlx5a expression was shifted dorsally. That is, dlx5a expression was present in the dorsal-most aspect of the second arch and absent ventrally in this arch.
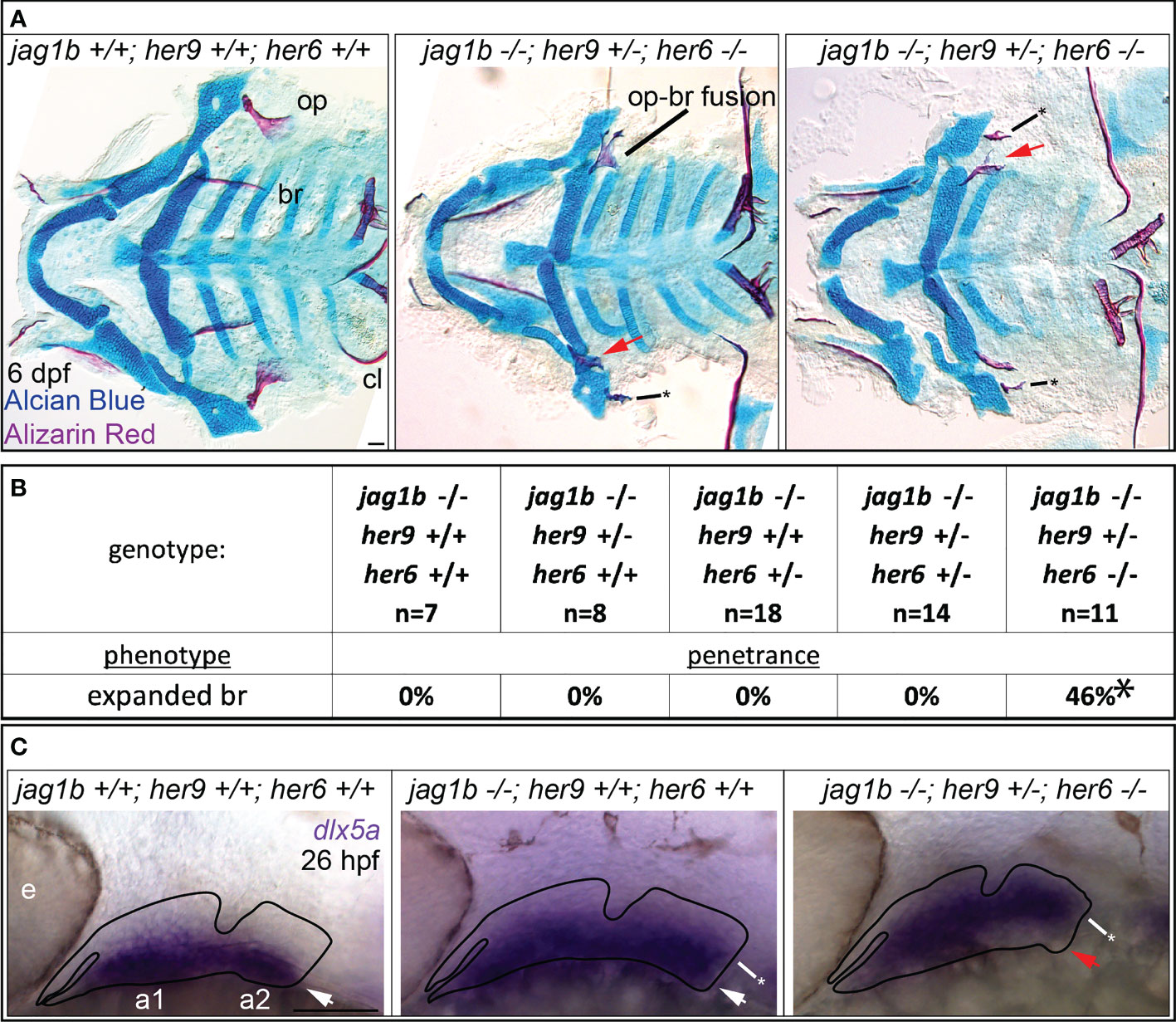
Figure 9 jag1b, her9, and her6 redundantly regulate dorsal versus ventral identity. (A) jag1b;her6;her9 triple heterozygotes were intercrossed and six days post fertilization (dpf) larvae were stained with Alcian Blue and Alizarin Red to label cartilage and bone. The individuals were then genotyped, the viscerocrania were dissected and flat mounted then imaged. The opercle (op), branchiostegal ray (br), and cleithrum (cl) are indicated in the wild type control. The bony fusion between the opercle and the branchiostegal ray is indicated. Red arrows indicate expanded, fan-shaped branchiostegal ray. Asterisk marks stick-shaped op phenotype (B) Genotyped preps from A were scored for penetrance of expanded branchiostegal ray. Asterisk indicates that penetrance of this phenotype in jag1b-/-;her9+/-;her6-/- is significantly different from all other genotypes, where it is never observed. (C) jag1b;her6;her9 triple heterozygotes were intercrossed and 26 hours post fertilization (hpf) larvae were fixed for in situ hybridization experiments to label dlx5a expression. White arrows indicate ventral dlx5a expression, asterisks mark dorsally expanded dlx5a expression. Red arrow indicates reduced ventral dlx5a expression in triple mutants suggesting ventral-versus-dorsal identity reversal. This phenotype was observed on both sides of in 3/3 embryos examined with this genotype. Scale bars are 50 μm.
We discovered that zebrafish her9 is critical for timely mineralization. This mineralization role is nonredundant with other genes since loss of her9 in an otherwise wild-type background produces fully penetrant mineralization defects, for at least some bony elements. We discovered that bones derived from neural crest cells, as well as those derived from mesoderm, both require her9 function for mineralization. However, some bones (like the parasphenoid) never mineralize in her9 mutants at 6 dpf. Meanwhile others (like the cleithrum) partially mineralize at this stage. These findings indicate that some bones are more sensitive to her9 loss than others. We propose that the ancestral role of her/hes genes is in both patterning and mineralization, but that these functions were subfunctionalized resulting in her9 primarily functioning in bone mineralization while retaining vestigial, partially redundant patterning function. In support, we recently proposed that vestigial, redundant paralog function can be retained and function to buffer against loss of one of the paralogs (40). It would be interesting to determine if there is a nonredundant patterning role of her9 later in development. Some mineralization mutants display correct patterning of early-forming skeletal elements, but then develop late-stage defects in skull shape (41). However, we are unable to test for this possibility since the her9 mutation is homozygous lethal. It is possible that generating new mutant her9 alleles, or an allelic series, might produce different phenotypes and be informative for a more complete understanding of her9 function.
It is interesting, and surprising, that her9 expression is not enriched in osteoblasts. In entpd5a (nob) mutants, a mineralization phenotype similar to her9 mutants develops (42). Unlike her9 however, entpd5a is expressed in osteoblasts. Although expression of entpd5a in cells beyond osteoblasts is sufficient to rescue entpd5a mutants. Our data suggest that the mineralization role for her9 is non cell-autonomous to the bone producing osteoblast cells. Determining the mechanism by which her9 broadly controls mineralization, and any interaction with entpd5a, is a major focus for future study. We conclude that the her9 cranial neural crest cell expression we detect by in situ and scRNAseq during early craniofacial development (24 hpf) functions in patterning downstream of jag1b, the mineralization function of her9 is likely due to this gene functioning in other tissues, as broad expression is observed in in situ hybridization.
Our study uncouples the mineralization and patterning roles of her9. In contrast to mineralization, the patterning role for her9 is redundant with other family members since her9 mutations do not affect patterning in an otherwise wild-type background, but do enhance patterning phenotypes when combined with other Notch pathway mutants. There are likely other Notch target genes, along with her6 and her9, functioning in a patterning role downstream of jag1b since her6;her9 double mutants do not phenocopy the jag1b phenotype. We found some evidence to suggest spatially distinct roles for different Notch target genes. Specifically, removing her6 from jag1b mutants increases the penetrance of the small opercle bone phenotype bilaterally. Meanwhile removing her9 from jag1b mutants increases penetrance of the foramen phenotype. Thus, different functions downstream of jag1b activation of Notch may have been subfunctionalized among her/hes genes. One caveat of this interpretation is that fixed skeletal preps from her9 mutants cannot be scored for bone shape phenotypes, so we don’t know if homozygous her9 mutants affect the penetrance of the stick opercle phenotype found in jag1b mutants. The her gene family is large, perhaps contributing to the extent of redundancy among family members. Future study disabling more her genes might reveal widespread redundancies during craniofacial development. Our previous work indicates that genes with stronger wild type expression are correlated with a larger buffering role when one family member is mutated (40). Therefore, our scRNA-seq dataset motivates examining her8a and her11 next, since transcripts for these genes are detected, albeit weakly, in anterior arch cranial neural crest cells whereas many other family members are undetectable.
When jag1b and her6 and her9 function are disabled in combination, a branchiostegal ray phenotype develops that is not observed in any other combination. In this genotype, the branchiostegal ray was expanded into an opercle-like fan shape. At the same time, the opercle was reduced into a branchiostegal-like stick shape. These findings suggest both dorsal-to-ventral and ventral-to-dorsal homeotic transformations occur in this genotype. Consistently, we also found individuals with this genotype where the opercle and branchiostegal were fused, similar fusion phenotypes are observed in mutations in the mef2ca transcription factor encoding gene and are characterized as dorsoventral homeotic transformations (38, 43). Finally, we observed molecular evidence of homeotic transformation by examining dlx5a expression. dlx5a expression, which is ventrally restricted in wild types, is dorsally shifted in triple mutants. We interpret these data to indicate that dorsoventral identity is reversed in these triple mutants.
Mutations in her6 do not produce an overt phenotype on their own but do genetically interact with other Notch pathway genes like jag1b and her9. Given this, it is curious that her6 has been retained in the zebrafish genome. The her/hes genes can clearly be lost from genomes since the mouse ortholog of her9, Hes4, was lost. We propose that the her/hes family is highly redundant and interchangeable, and is therefore prone to gene losses and changes in functional requirement through evolutionary time.
The nonredundant and redundant roles of her9 in mineralization and patterning, respectively, are also differentially dependent on Notch cleavage by gamma secretase. The mineralization role does not require Notch cleavage by gamma secretase since we are unable to phenocopy her9 mutant phenotypes by inhibiting this enzyme. But this inhibitor does phenocopy the patterning phenotypes seen in jag1b mutants. Thus, it is possible that the mineralization role of her9 is downstream of other signaling systems. For example, other reports indicate her9 is in the Nodal and Retinoic Acid pathways (9, 22). This could be tested by inhibiting some of the other pathways associated with her9 to test for phenocopy of the her9 mineralization phenotype. Alternatively, her9 may function downstream of gamma secretase-independent Notch activation (44). This alternative could be tested by mutating the relevant Notch receptors and ligands, or dominant-negative approaches to test for phenocopy of the mineralization phenotype, although previous studies utilizing these methods did not report a broad mineralization phenotype following Notch signaling blockade (16).
Normally, bones form from the stepwise production of mineralized matrix. As osteoblasts mature, they first make osteoid bone, then mineralized matrix. We find that her9 is required for the proper timing of the mineralization stages of bone formation. In her9 mutants, osteoblasts differentiate in the correct position, with the normal timing, but they don’t mineralize the matrix with normal timing. However, there is detectible unmineralized matrix in some cases, like the opercle. Alcian Blue, which stains osteoid bone but not mature bone matrix (45), reproducibly labeled her9 mutant opercles. Therefore, we interpret this her9 mutant-associated phenotype as an extended osteoid stage. Intriguingly, extended osteoid stage opercles may occur naturally in some fish species, serving as a mechanism for generating bone shape diversity (46). In this work, the authors proposed that there are regions of extended osteoid in sculpin opercles contributing to opercle shape variation among different sculpin species. Thus, it is tempting to speculate that evolutionary changes in her9 expression or function between different sculpin species underlie the extended osteoid observed in some sculpins. This would lead to naturally extended osteoid proposed to contribute to the diverse opercle bone shapes seen among sculpins. Further, that zebrafish her9 mutants apparently produce an extended osteoid stage may inform human health. Rickets in children or osteomalacia in adults are disorders that arise when osteoid does not mineralize properly.
In this study we discovered a non-redundant, Notch-independent, bone mineralization requirement for her9. We also found a redundant role for her9 and her6 during Jag-Notch craniofacial patterning. These studies help us to understand human HES4-associated skeletal disease in an in vivo setting, which is not possible in rodent models. Our work helps to clarify the complex roles of her/hes genes during vertebrate skeletal development.
All zebrafish were maintained and staged according to established protocols (47, 48). The jag1bb1105 allele and genotyping protocol has been previously described (1). The transgenic line Tg(sp7:EGFP)b1212 has been previously described (33).
The her9co66 mutant line was generated by ENU mutagenesis and has a point mutation in exon 4 which produces an early stop codon near the predicted end of the Orange domain. The transition mutation converts the wild-type sequence GGGACAGAT to GGGATAGAT. The mutagenesis and mutant allele identification was performed as described (49). To identify the causative lesion, co66 generated on the TU background was outcrossed to the AB strain for two generations and F3 embryos were screened. Heterozygous adults were intercrossed and 33 phenotypic wild type and 33 phenotypic mutant larvae were used to prepare pooled genomic DNA. Using 223 simple sequence-length polymorphism markers, we placed the co66 locus on chromosome 23 between z7550, (31.6 centimorgan (cM), 13.6 megabase (mb)) and z7973 (34 cM, 26.0 mb). We identified her9, located at 23.4 mb as a candidate gene and found the mutation by Sanger sequencing which was homozygous only in phenotypically mutant individuals. The mutation was then maintained in our closed colony by outcrossing to AB for over seven generations, genotyping for her9 heterozygotes at each generation. All individuals in this study were genotyped and the mineralization defect phenotype was 100% penetrant in her9 homozygous mutants, and never present in heterozygotes or homozygous wild types. To genotype her9co66, a DNA fragment was amplified using primers Her9-FW-exon2, 5’-TCTTCAAAGCCAATCATGGAAA-3’, and Her9-RV-exon4, 5’-GAAGAGGCTGAGCCAAATGA-3’, and then digested with BsmFI to produce a 604 bp fragment in mutants and 539/66 bp fragments in wild types. We generated the her6co3005 allele using CRISPR/Cas9 mutagenesis (50). We injected Cas9 mRNA along with two guide RNAs (5’- GCGAGAATCAACGAAAGCTT-3’ and 5’-CCGTTCTTGGGAAGTACCGA-3’) into single cell zebrafish embryos targeting exons 2 and 4 respectively. Injected zebrafish were screened for mutagenesis via PCR amplification using primers her6 FW exon1 5’-GGCGAGCATGAACACTACA-3’ and her6 RV exon4 5’-GGTTCCTGTCCACATGTGAA-3’. Sanger sequencing revealed a 4 bp deletion in exon 2 (AAAGGGTC) which is predicted to cause a frameshift after amino acid 10 of 270 and a premature stop codon after 12 missense amino acids. Sanger sequencing also showed a naturally occurring 5bp deletion in intron 2. Genotyping for her6 was done using PCR amplification with primers her6 geno FW 5’- GAGCATGAACACTACACCTGATA-3’ and her6 geno RV 5’- CGAAAGTCCGACTGAGTCTTT-3’. The PCR product was then digested with HindIII to produce a 264bp fragment in mutants and 187/77 bp fragments in wild types. All zebrafish procedures have been approved by the University of Colorado Institutional Animal Care and Use Committee (IACUC), Protocol # 00188. Genomic DNA for genotyping was obtained by tail amputation. Fish were anesthetized by adding 0.01-0.2 mg/ml Tricaine (MS-222) to embryo media. Animals were euthanized by hypothermic shock followed by 1.5% sodium hypochlorite.
Alcian Blue and Alizarin Red were used to stain cartilage and bone, respectively as previously described (30, 51). Live Alizarin Red staining was performed on zebrafish as described (35) at 3 dpf and 6 dpf. Zebrafish larvae were placed in 0.01% Alizarin Red and 0.01% HEPES solution in E2 media and allowed to swim for 30 minutes. The zebrafish were then transferred to clean E2 media and imaged.
Genotyped Alcian Blue and Alizarin Red stained zebrafish skeletons were scored for penetrance of relevant phenotypes. A Fisher’s Exact test was used to determine significant differences in penetrance between genotypes.
Notch cleavage inhibition using DBZ was performed similar to previously reported (13, 15). Wild-type zebrafish were treated with 10µM of DBZ or DMSO vehicle control from 18-48hpf, or 48-72hpf, or 72-96hpf. All treated zebrafish were raised to 6dpf and stained for bone and cartilage.
Whole mount in situ hybridization with fluorescence was performed as previously described (52). The her9 and dlx5a probes have been previously described (9, 53).
Gene expression studies were performed as previously described (15). Live individual 28 and 48 hpf embryos had their heads removed. Decapitated bodies were genotyped to identify homozygous wild-types and homozygous jag1b mutants. Heads from 5–6 identified homozygous wild types were pooled and total RNA was extracted with TRIzol Reagent from ThermoFischer Scientific. cDNA was prepared with Superscript III from Invitrogen. qPCR experiments utilized a Real-Time PCR StepOnePlus system from Applied Biosystems and SYBR green. A standard curve was generated from serially diluted (1:2:10) cDNA pools, and primers with a slope of -3.3 +- 0.3 were accepted. The relative quantity of target cDNA was calculated using Applied Biosystems StepOne V.2.0 software and the comparative Ct method. After surveying the expression of many housekeeping genes at multiple stages, we determined that rps18 expression was the most consistent across stages and genotypes. Target gene expression in all experiments was normalized to rps18. Reactions were performed in technical triplicate, and the results represent two to six biological replicates. The following primers were used: her9FW, 5’-GAGAATCAACGAGAGCCTTG-3’, her9REV 5’-CTCCAG AATATCAGCTTTCTCC-3’, her6FW 5’-AACGAAAGCT TGGGTCAG-3’, her6REV 5’-ACTGTCATCTCCAGGATGT-3’, rps18FW 5’-CTGAACAGACAGAAGGACATAA-3’, rps18REV 5’-AGCCTCTCCAGATCTTCTC-3’.
Cranial neural crest cells were sorted as described (29). Briefly, 60 wild-type 24 hpf flia:EGFP;sox10:mRFP double transgenic embryos were dissociated into single-cell suspension using cold-active protease from Bacillus licheniformis, DNase, EDTA, and trituration. Approximately 30,000 live, GFP/RFP double-positive cells were sorted into tubes pre-coated with fetal bovine serum with a MoFlo XDP100. Sorted cells at 2.08x10^6 cells/ml concentration with 70% viability were loaded into the 10x Chromium Controller aiming to capture 10000 cells. Using the NovaSEQ6000, 2×150 paired-end sequencing Chromium 10x Genomics libraries were sequenced at 100K reads per cell using 3prime NexGem V3.1(Dual Index) sequencing.
Recovered sequencing reads were mapped to a custom Cell Ranger reference based on the GRCz11/danRer11 genome assembly using the Cell Ranger pipeline (10X Genomics). The 10X Genomics Cell Ranger pipeline removed empty droplets from the dataset based on the distribution of cell barcodes. Additional analysis was performed in R using the Seurat package (54). Quality filtering of these data excluded cells with no genes detectable (>0 unique molecular identifiers (UMIs) per gene). Each gene in the dataset was required to be present in a minimum of five cells. Regarding the capture of dead or dying cells, a minimum presence of 250 genes (at <2.5% of mitochondrial UMIs detected) were required. After quality filtering, 7928 cells remained for analysis.
Alcian Blue and Alizarin Red skeletons were dissected, flat mounted, and then imaged on a Leica DMi8 inverted microscope equipped with a Leica DMC2900. All fluorescent imaging was performed on a Leica DMi8 equipped with an Andor Dragonfly 301 spinning disk confocal system. Live imaging was performed as previously described (55).
The sequencing dataset presented in this study can accessed from the GEO database (accession number GSE220081).
The animal study was reviewed and approved by University of Colorado IACUC.
AS, AM-M, JS, MW and JMM designed performed and interpreted experiments and edited the manuscript. BA designed experiments. JN interpreted experiments wrote and edited the manuscript. All authors contributed to the article and approved the submitted version.
This work was funded by NIH R01DE029193 to JN, NIH F32DE029995 to JMM, NSF GRFP 1938058 to AMM.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fendo.2022.1033843/full#supplementary-material
1. Zuniga E, Stellabotte F, Crump JG. Jagged-notch signaling ensures dorsal skeletal identity in the vertebrate face. Development. (2010) 137(11):1843–52. doi: 10.1242/dev.049056
2. Kraus JM, Giovannone D, Rydzik R, Balsbaugh JL, Moss IL, Schwedler JL, et al. Notch signaling enhances bone regeneration in the zebrafish mandible. Development (2022) 149(5):dev199995. doi: 10.1242/dev.199995
3. Pakvasa M, Haravu P, Boachie-Mensah M, Jones A, Coalson E, Liao J, et al. Notch signaling: its essential roles in bone and craniofacial development. Genes diseases. (2021) 8(1):8–24. doi: 10.1016/j.gendis.2020.04.006
4. Nichols JT, Miyamoto A, Weinmaster G. Notch signaling - constantly on the move. Traffic. (2007) 8(8):959–69. doi: 10.1111/j.1600-0854.2007.00592.x
5. Artavanis-Tsakonas S, Muskavitch MA. Notch: the past, the present, and the future. Curr topics Dev Biol (2010) 92:1–29. doi: 10.1016/S0070-2153(10)92001-2
6. Kageyama R, Ohtsuka T, Kobayashi T. The Hes gene family: repressors and oscillators that orchestrate embryogenesis. Development (Cambridge, England) (2007) 134.7:1243–51
7. Kuretani A, Yamamoto T, Taira M, Michiue T. Evolution of hes gene family in vertebrates: the hes5 cluster genes have specifically increased in frogs. Evol (2021) 21(1):1–15. doi: 10.1186/s12862-021-01879-6
8. Fisher AL, Ohsako S, Caudy M. The WRPW motif of the hairy-related basic helix-loop-helix repressor proteins acts as a 4-amino-acid transcription repression and protein-protein interaction domain. Mol Cell Biol (1996) 16(6):2670–7. doi: 10.1128/MCB.16.6.2670
9. Latimer AJ, Shin J, Appel B. her9 promotes floor plate development in zebrafish. Dev dynamics: an Off Publ Am Assoc Anatomists. (2005) 232(4):1098–104. doi: 10.1002/dvdy.20264
10. Radosevic M, Robert-Moreno À, Coolen M, Bally-Cuif L, Alsina B. Her9 represses neurogenic fate downstream of Tbx1 and retinoic acid signaling in the inner ear. Development. (2011) 138(3):397–408. doi: 10.1242/dev.056093
11. Bae Y-K, Shimizu T, Hibi M. Patterning of proneuronal and inter-proneuronal domains by hairy-and enhancer of split-related genes in zebrafish neuroectoderm. Development (Cambridge, England) (2005) 132.6:1375–85.
12. Leve C, Gajewski M, Rohr KB, Tautz D. Homologues of c-hairy1 (her9) and lunatic fringe in zebrafish are expressed in the developing central nervous system, but not in the presomitic mesoderm. Dev Genes evolution. (2001) 211(10):493–500. doi: 10.1007/s00427-001-0181-4
13. Barske L, Askary A, Zuniga E, Balczerski B, Bump P, Nichols JT, et al. Competition between jagged-notch and Endothelin1 signaling selectively restricts cartilage formation in the zebrafish upper face. PloS Genet (2016) 12(4):e1005967. doi: 10.1371/journal.pgen.1005967
14. Askary A, Xu P, Barske L, Bay M, Bump P, Balczerski B, et al. Genome-wide analysis of facial skeletal regionalization in zebrafish. Development. (2017) 144(16):2994–3005. doi: 10.1242/dev.151712
15. Sucharov J, Ray K, Brooks EP, Nichols JT. Selective breeding modifies mef2ca mutant incomplete penetrance by tuning the opposing notch pathway. PloS Genet (2019) 15(12):e1008507. doi: 10.1371/journal.pgen.1008507
16. Paudel S, Gjorcheska S, Bump P, Barske L. Patterning of cartilaginous condensations in the developing facial skeleton. Dev Biol (2022) 486:44–55. doi: 10.1016/j.ydbio.2022.03.010
17. Oda T, Elkahloun AG, Pike BL, Okajima K, Krantz ID, Genin A, et al. Mutations in the human Jagged1 gene are responsible for alagille syndrome. Nat Genet (1997) 16(3):235–42. doi: 10.1038/ng0797-235
18. Suzuki A, Sangani DR, Ansari A, Iwata J. Molecular mechanisms of midfacial developmental defects. Dev Dynamics. (2016) 245(3):276–93. doi: 10.1002/dvdy.24368
19. Humphreys R, Zheng W, Prince LS, Qu X, Brown C, Loomes K, et al. Cranial neural crest ablation of Jagged1 recapitulates the craniofacial phenotype of alagille syndrome patients. Hum Mol Genet (2012) 21(6):1374–83. doi: 10.1093/hmg/ddr575
20. Akimoto M, Kameda Y, Arai Y, Miura M, Nishimaki T, Takeda A, et al. Hes1 is required for the development of craniofacial structures derived from ectomesenchymal neural crest cells. J Craniofacial Surgery. (2010) 21(5):1443–9. doi: 10.1097/SCS.0b013e3181ebd1a0
21. Kameda Y, Saitoh T, Nemoto N, Katoh T, Iseki S, Fujimura T. Hes1 is required for the development of pharyngeal organs and survival of neural crest-derived mesenchymal cells in pharyngeal arches. Cell Tissue Res (2013) 353(1):9–25. doi: 10.1007/s00441-013-1649-z
22. Coomer CE, Wilson SG, Titialii-Torres KF, Bills JD, Krueger LA, Petersen RA, et al. Her9/Hes4 is required for retinal photoreceptor development, maintenance, and survival. Sci Rep (2020) 10(1):1–20. doi: 10.1038/s41598-020-68172-2
23. Shimada S, Shimojima K, Okamoto N, Sangu N, Hirasawa K, Matsuo M, et al. Microarray analysis of 50 patients reveals the critical chromosomal regions responsible for 1p36 deletion syndrome-related complications. Brain Dev (2015) 37(5):515–26. doi: 10.1016/j.braindev.2014.08.002
24. Grogan SP, Olee T, Hiraoka K, Lotz MK. Repression of chondrogenesis through binding of notch signaling proteins HES-1 and HEY-1 to n-box domains in the COL2A1 enhancer site. Arthritis Rheumatism. (2008) 58(9):2754–63. doi: 10.1002/art.23730
25. Dong Y, Jesse AM, Kohn A, Gunnell LM, Honjo T, Zuscik MJ, et al. RBPjκ-dependent notch signaling regulates mesenchymal progenitor cell proliferation and differentiation during skeletal development. Development. (2010) 137(9):1461–71. doi: 10.1242/dev.042911
26. Cakouros D, Isenmann S, Hemming SE, Menicanin D, Camp E, Zannetinno ACW, et al. Novel basic helix–loop–helix transcription factor Hes4 antagonizes the function of twist-1 to regulate lineage commitment of bone marrow stromal/stem cells. Stem Cells Dev (2015) 24(11):1297–308. doi: 10.1089/scd.2014.0471
27. Hilton MJ, Tu X, Wu X, Bai S, Zhao H, Kobayashi T, et al. Notch signaling maintains bone marrow mesenchymal progenitors by suppressing osteoblast differentiation. Nat Med (2008) 14(3):306–14. doi: 10.1038/nm1716
28. Zhang Y, Lian JB, Stein JL, Van Wijnen AJ, Stein GS. The notch-responsive transcription factor hes-1 attenuates osteocalcin promoter activity in osteoblastic cells. J Cell Biochem (2009) 108(3):651–9. doi: 10.1002/jcb.22299
29. Mitchell JM, Sucharov J, Pulvino AT, Brooks EP, Gillen AE, Nichols JT. The alx3 gene shapes the zebrafish neurocranium by regulating frontonasal neural crest cell differentiation timing. Development (2021) 148(7):dev197483. doi: 10.1242/dev.197483
30. Walker MB, Kimmel CB. A two-color acid-free cartilage and bone stain for zebrafish larvae. Biotech Histochem (2007) 82(1):23–8. doi: 10.1080/10520290701333558
31. Puchtler H, Meloan SN, Terry MS. On the history and mechanism of alizarin and alizarin red s stains for calcium. J Histochem Cytochemistry. (1969) 17(2):110–24. doi: 10.1177/17.2.110
32. Kague E, Gallagher M, Burke S, Parsons M, Franz-Odendaal T, Fisher S. Skeletogenic fate of zebrafish cranial and trunk neural crest. PloS One (2012) 7(11):e47394. doi: 10.1371/journal.pone.0047394
33. DeLaurier A, Eames BF, Blanco-Sanchez B, Peng G, He X, Swartz ME, et al. Zebrafish sp7:EGFP: a transgenic for studying otic vesicle formation, skeletogenesis, and bone regeneration. Genesis. (2010) 48(8):505–11. doi: 10.1002/dvg.20639
34. Eames BF, DeLaurier A, Ullmann B, Huycke TR, Nichols JT, Dowd J, et al. FishFace: interactive atlas of zebrafish craniofacial development at cellular resolution. BMC Dev Biol (2013) 13(1):23. doi: 10.1186/1471-213X-13-23
35. Nichols JT, Blanco-Sanchez B, Brooks EP, Parthasarathy R, Dowd J, Subramanian A, et al. Ligament versus bone cell identity in the zebrafish hyoid skeleton is regulated by mef2ca. Development. (2016) 143(23):4430–40. doi: 10.1242/dev.141036
36. Kimmel CB, DeLaurier A, Ullmann B, Dowd J, McFadden M. Modes of developmental outgrowth and shaping of a craniofacial bone in zebrafish. PloS One (2010) 5(3):e9475. doi: 10.1371/journal.pone.0009475
37. Bensimon-Brito A, Cardeira J, Dionísio G, Huysseune A, Cancela M, Witten P. Revisiting in vivo staining with alizarin red s-a valuable approach to analyse zebrafish skeletal mineralization during development and regeneration. BMC Dev Biol (2016) 16(1):1–10. doi: 10.1186/s12861-016-0102-4
38. DeLaurier A, Huycke TR, Nichols JT, Swartz ME, Larsen A, Walker C, et al. Role of mef2ca in developmental buffering of the zebrafish larval hyoid dermal skeleton. Dev Biol (2014) 385(2):189–99. doi: 10.1016/j.ydbio.2013.11.016
39. Kimmel CB, Walker MB, Miller CT. Morphing the hyomandibular skeleton in development and evolution. J Exp Zool B Mol Dev Evol (2007) 308(5):609–24. doi: 10.1002/jez.b.21155
40. Bailon-Zambrano R, Sucharov J, Mumme-Monheit A, Murry M, Stenzel A, Pulvino AT, et al. Variable paralog expression underlies phenotype variation. Elife. (2022) 11:e79247. doi: 10.7554/eLife.79247
41. Kague E, Roy P, Asselin G, Hu G, Simonet J, Stanley A, et al. Osterix/Sp7 limits cranial bone initiation sites and is required for formation of sutures. Dev Biol (2016) 413(2):160–72. doi: 10.1016/j.ydbio.2016.03.011
42. Huitema LF, Apschner A, Logister I, Spoorendonk KM, Bussmann J, Hammond CL, et al. Entpd5 is essential for skeletal mineralization and regulates phosphate homeostasis in zebrafish. Proc Natl Acad Sci (2012) 109(52):21372–7. doi: 10.1073/pnas.1214231110
43. Miller CT, Swartz ME, Khuu PA, Walker MB, Eberhart JK, Kimmel CB. mef2ca is required in cranial neural crest to effect Endothelin1 signaling in zebrafish. Dev Biol (2007) 308(1):144–57. doi: 10.1016/j.ydbio.2007.05.018
44. Huppert SS, Ilagan MXG, De Strooper B, Kopan R. Analysis of notch function in presomitic mesoderm suggests a γ-secretase-independent role for presenilins in somite differentiation. Dev Cell (2005) 8(5):677–88. doi: 10.1016/j.devcel.2005.02.019
45. Baylink D, Wergedal J, Thompson E. Loss of proteinpolysaccharides at sites where bone mineralization is initiated. J Histochem Cytochemistry. (1972) 20(4):279–92. doi: 10.1177/20.4.279
46. Cytrynbaum EG, Small CM, Kwon RY, Hung B, Kent D, Yan YL, et al. Developmental tuning of mineralization drives morphological diversity of gill cover bones in sculpins and their relatives. Evol letters. (2019) 3(4):374–91. doi: 10.1002/evl3.128
47. Westerfield M. The zebrafish book: A guide for the laboratory use of zebrafish (Brachydanio rerio). Eugene OR: University of Oregon Press (1993).
48. Kimmel CB, Ballard WW, Kimmel SR, Ullmann B, Schilling TF. Stages of embryonic development of the zebrafish. Dev Dynamics. (1995) 203(3):253–310. doi: 10.1002/aja.1002030302
49. Kearns CA, Walker M, Ravanelli AM, Scott K, Arzbecker MR, Appel B. Zebrafish spinal cord oligodendrocyte formation requires boc function. Genetics (2021) 218(4):iyab082. doi: 10.1093/genetics/iyab082
50. Jao L-E, Wente SR, Chen W. Efficient multiplex biallelic zebrafish genome editing using a CRISPR nuclease system. Proc Natl Acad Sci (2013) 110(34):13904–9. doi: 10.1073/pnas.1308335110
51. Brooks E, Nichols J. Shifting zebrafish lethal skeletal mutant penetrance by progeny testing. J visualized experiments: JoVE (2017) 127:p.e56200. doi: 10.3791/56200
52. Nichols JT, Pan L, Moens CB, Kimmel CB. barx1 represses joints and promotes cartilage in the craniofacial skeleton. Development. (2013) 140(13):2765–75. doi: 10.1242/dev.090639
53. Walker MB, Miller CT, Coffin Talbot J, Stock DW, Kimmel CB. Zebrafish furin mutants reveal intricacies in regulating Endothelin1 signaling in craniofacial patterning. Dev Biol (2006) 295(1):194–205. doi: 10.1016/j.ydbio.2006.03.028
54. Butler A, Hoffman P, Smibert P, Papalexi E, Satija R. Integrating single-cell transcriptomic data across different conditions, technologies, and species. Nat Biotechnol (2018) 36(5):411–20. doi: 10.1038/nbt.4096
Keywords: craniofacial, skeleton, her, mineralization, Notch, bone, osteoblast, patterning
Citation: Stenzel A, Mumme-Monheit A, Sucharov J, Walker M, Mitchell JM, Appel B and Nichols JT (2022) Distinct and redundant roles for zebrafish her genes during mineralization and craniofacial patterning. Front. Endocrinol. 13:1033843. doi: 10.3389/fendo.2022.1033843
Received: 01 September 2022; Accepted: 17 November 2022;
Published: 12 December 2022.
Edited by:
Erika Kague, University of Bristol, United KingdomReviewed by:
D’Juan Farmer, University of California, Los Angeles, United StatesCopyright © 2022 Stenzel, Mumme-Monheit, Sucharov, Walker, Mitchell, Appel and Nichols. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: James T. Nichols, SmFtZXMuTmljaG9sc0BjdWFuc2NodXR6LmVkdQ==
†These authors have contributed equally to this work and share first authorship
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.