 Haiyang Du
Haiyang Du Xiaoyu Meng
Xiaoyu Meng Yu Yao1
Yu Yao1 Jun Xu
Jun Xu- 1Division of Orthopedics, Department of Orthopedics, The Second Affiliated Hospital of Harbin Medical University, Harbin, China
- 2Division of Endocrinology, Department of Internal Medicine, Tongji Hospital, Tongji Medical College, Huazhong University of Science and Technology, Wuhan, China
- 3Branch of National Clinical Research Center for Metabolic Diseases, Hubei, China
Since type 2 diabetes mellitus (T2DM) is a risk factor for Alzheimer’s disease (AD) and both have the same pathogenesis (e.g., insulin resistance), drugs used to treat T2DM have been gradually found to reduce the progression of AD in AD models. Of these drugs, glucagon-like peptide 1 receptor (GLP-1R) agonists are more effective and have fewer side effects. GLP-1R agonists have reducing neuroinflammation and oxidative stress, neurotrophic effects, decreasing Aβ deposition and tau hyperphosphorylation in AD models, which may be a potential drug for the treatment of AD. However, this needs to be verified by further clinical trials. This study aims to summarize the current information on the mechanisms and effects of GLP-1R agonists in AD.
Introduction
Alzheimer’s disease (AD), a global public health priority, is the most common neurodegenerative disease (1). AD is recognized as the leading cause of disability and death, and the progressive cognitive dysfunction in AD patients seriously affects the quality of life (2). According to previous research, there were 46.8 million people suffering from dementia worldwide in 2015 and AD is identified as the leading cause for dementia (3). Due to the severe cognitive impairment of AD patients, their treatment and care require substantial economic and financial support, causing serious damage to global economic development (3). The main pathological features of AD are amyloid plaques and neurofibrillary tangles (NFTs) as well as neuroinflammation and oxidative stress in the brain (2, 4, 5). Many studies have revealed that diabetes, insulin resistance and aging are major risk factors for AD (6–9). Interestingly, previous studies have shown brain insulin resistance in AD patients (10, 11). Therefore, AD is also called “type 3 diabetes” (12, 13). Unfortunately, so far, there is no effective treatment for AD.
Some human and animal studies show that insulin can accelerate the clearance level of Aβ in the brain and affects the phosphorylation of tau protein, meanwhile, enhances the synaptic activity and plasticity of neurons (14–16). It produces beneficial effects if administered for short periods, because raising peripheral insulin levels acutely increases insulin levels in the brain and cerebrospinal fluid (CSF) (17–19). However, sustained high levels of circulating insulin may conversely exert a negative influence on cognitive function, due to prolonged peripheral hyperinsulinemia down-regulates insulin receptors at the blood-brain barrier (BBB) and reduces insulin transport into the brain, leading to brain insulin resistance (BIR) (17–19).
Glucagon-like peptides 1 (GLP-1), derived from intestinal L cells, is an incretin hormone. GLP-1 is a target for treatment of diabetes because of the primary peripheral functions of inducing insulin secretion from pancreatic β cells, gut emptying and inhibiting glucagon secretion which results in lower blood glucose levels (20). Continuous administration of natural GLP-1 increases insulin levels and results in lower blood glucose and hemoglobin A1C (HbA1c) levels in patients with type 2 diabetes mellitus (T2DM) (20). GLP-1 has a short half-life of only two minutes, due to renal clearance and endogenous GLP-1 is degraded by the enzyme dipeptidyl peptidase IV (DPP-4) (20–22). For these reasons, GLP-1 analogues and DPP-4 inhibitors were synthesized to prolong the half-life of GLP-1.
GLP-1 can also be produced by neurons in central nervous system (CNS) (20, 23). GLP-1 receptors (GLP-1R) expression has been detected throughout the CNS including the hippocampus, neocortex, hypothalamus, and cerebellum (17, 20, 24). GLP-1R are expressed by neurons, glia cells do not express this receptor but induced expression when activated in an inflammatory response (17, 25). Recently, GLP-1 and its agonists have been found to have good neuro-regulation and protection effects in animal models (26–28). Therefore, this review summarizes the current information on the mechanisms and effects of GLP-1R agonists in AD.
Pathogenesis of AD
Amyloid β plaque
Amyloid beta (Aβ) is a peptide with a molecular weight of 4 KDa and a length of 40-42 amino acids (3, 29). Aβ-40 and Aβ-42, obtained by hydrolysis of transmembrane amyloid precursor protein (APP) by secretases (α, β and γ), are the main types of amyloid proteins and components of Aβ plaques through the aggregation of soluble oligomers (30). Aβ-42 aggregates and combines to Ca2+ channels and AMPA (α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid) receptors, to which the neurotransmitter glutamate binds, so Aβ is the most neurotoxic form (3, 31). Aβ plaques, which form and deposit in different regions of the brain, are recognized as foreign material by the brain and trigger an inflammatory and immune response by activating the microglia leading to neuronal degeneration and synaptic damage (2, 30). The amyloid hypothesis is the prevalent theory of AD pathogenesis, driven through an imbalance between Aβ production and Aβ clearance (2, 6, 21). Clinical studies have been reported that anti-Aβ protofibril antibody lecanemab has produced modest but highly statistically significant results in a trial. The drug met its primary endpoint of slowing cognitive decline in individuals with mild cognitive impairment (MCI), a clinical presentation thought to be a precursor to AD, and those with mild AD (32, 33).
Neurofibrillary tangles
Tau is a microtubule-associated protein and mainly found in the axonal compartment (34). Tau protein interacts with tubulin to stabilize the structure of neuronal microtubules through its isoforms and phosphorylation, supporting neurite differentiation and growth, as well as transporting motor proteins along the axons (29, 34). One of the defining pathological features of AD is the intraneuronal accumulation of NFTs (35). NFTs are primarily composed of paired helical filaments consisting of hyperphosphorylated tau protein (2, 35). The affinity of hyperphosphorylation of tau protein to microtubules decreased, and hyperphosphorylated tau protein can no longer perform the function of maintaining the structure of the neurons, which slows the axonal transport (30). The current studies suggest a reduced ability to clear out misfolded, oligomerized and aggregated tau proteins that increase with advancing age (36, 37).
Synaptic dysfunction and neurotransmitter imbalance
It has been well established that cholinergic transmission is essential for memory, learning, attention, and other higher brain functions, so cholinergic deficits play a key role in the neuropathology of AD (38). “Cholinergic hypothesis” states that the degeneration of basal forebrain neurons in AD patients leads to dysfunction and death of cholinergic neurons in the forebrain, followed by extensive presynaptic denervation occurs, and the loss of specific subtypes of acetylcholine receptors, leading to cognitive decline in AD patients (39). Acetylcholine is a major neurotransmitter in the brain, promoting experience-induced neuroplasticity, the synchronization of neuronal activity, and network connectivity (38). After depolarization of presynaptic neurons, acetylcholine is released into the synaptic cleft and binds to postsynaptic receptors such as acetylcholine muscarinic receptors (mAchRs) or acetylcholine nicotinic receptors (nAchRs) (40). Some facts attested that a reduction in the number of mAchRs and nAchRs in basal forebrain cholinergic neurons, and a 40%-50% decrease in cerebral acetylcholine level are considered pathogenic elements for dementia and AD (41, 42). Moreover, the levels of 5-hydroxytryptamine, γ-aminobutyric acid (GABA) and their receptors, as well as glutamate receptors are reduced in patients with AD (42–46). An imbalance of any of these neurotransmitters may lead to further deterioration of AD. Thus, homeostasis of multiple neurotransmitters is critical to keep cognitive integrity.
Neuroinflammation
Neuroinflammation, microglia activation in response to amyloid deposition, plays a central role in the pathogenesis of AD (30). During acute inflammation caused by Aβ build-up, microglia phagocytose Aβ and protect neurons from Aβ toxicity (47). Under normal circumstances, acute inflammation is followed by regression through the anti-inflammatory effects of microglia. In AD, Aβ accumulation still exists, resulting in chronic neuroinflammation (48). Chronic neuroinflammation is observed at relatively early stages of disease. Chronic activation of microglia is associated with protein degradation, mitochondria dysfunction, and defects of axonal transport and apoptosis, which adversely affect neuronal function and lead to cell death (49). In addition, neuroinflammation causes immune cells (monocytes and lymphocytes in the blood, such as T cells and B cells) to infiltrate the central nervous system from the periphery across the BBB, accelerating neuroinflammation and neurodegeneration (49, 50).
Insulin resistance
For the past few years, AD has also been considered as “type 3 diabetes” because of insulin resistance (IR) and dysregulation of insulin signaling in the brain (51). The majority of insulin in brain derives from pancreatic β-cells, which is mainly transported across the BBB. While some insulin molecules may be locally synthesized and released by neurons in the CNS (such as the hippocampus, prefrontal cortex, but not glial cells) (52). In the CNS, insulin contributes to synaptic maintenance, neuronal growth and survival, maintenance and regulation of learning and memory (53). Significantly decreased insulin and insulin receptor expression was observed in postmortem AD brain tissue, and changes in downstream insulin signaling molecules, including decreased levels of IRS-1/2, PI3K, p-Akt (51, 54). It has been reported that spatial memory improves after insulin injection in the hippocampus or intranasal administration of insulin (55, 56). Indeed, acute elevated peripheral insulin levels may increase insulin in cerebrospinal fluid, whereas, chronic peripheral hyperinsulinemia (such as insulin resistance or T2DM) may downregulate insulin receptors of the BBB, impair brain insulin uptake, and ultimately lead to learning, memory, and cognitive deficits (14, 52).
Metabolic impairment
The brain is an organ that requires a lot of glucose to produce energy, with almost 70 percent of the energy used by neurons. In patients with T2DM, cognitive deficits are classified into three broad stages according to severity: diabetes-related cognitive decline, mild cognitive impairment (MCI) and dementia (57). MCI is a high-risk condition for conversion to AD. A large proportion of patients progress to AD, while some MCI patients may remain stable (58). All neurodegenerative diseases exhibit significant metabolic impairment, including decreased glucose uptake or utilization, with a consequent diminution in ATP production (59). Reduced glucose metabolism in AD may be a consequence of reduced postsynaptic neurotransmission, since depolarizing agents trigger glucose uptake in the brain and the effect is reduced in AD (1). In the early pathological state of AD, glucose utilization (up to 45%) is reduced, which is closely related to the alteration of insulin signaling. It has been reported that poor glucose utilization and insulin resistance can enhance Aβ deposition and decrease its clearance (60). Besides, brains with AD have been found to have a higher incidence of abnormal lipid metabolism, which is the early risk factor for the development of amyloid pathology (61). Lipids (cholesterol and sphingolipids, especially) are the main structural components of the brain, and each type of lipid may have specific function (62). Evidence shows that the strongest genetic risk factor for late-onset AD is the E4 allele of the cholesterol transporter APOE (APOE4), besides, APOE4 could promote amyloid aggregation and impair clearance from the brain directly binding to Aβ (63). Furthermore, impaired brain cholesterol synthesis can also enhance insulin resistance in brain tissue by disrupting the conformation of insulin receptor in cell membranes and promoting aberrant receptor activation (64).
Oxidative stress and mitochondrial dysfunction
Oxidative stress is a serious imbalance between the production of reactive oxygen species (ROS) and reactive nitrogen species (RNS) and antioxidant defenses (65). Due to the active metabolism of neurons, the demand for oxygen is high and a large number of ROS are produced. Therefore, the neurons in brain are susceptible to oxidative damage and mitochondrial dysfunction (29, 66). In mouse models and autopsy analysis of AD patients, mitochondrial dysfunction and increased reactive oxygen species enhance Aβ aggregation. Moreover, the elevated markers of oxidative stress precede Aβ deposition and NFTs, suggesting that oxidative stress is an early event in the pathogenesis of AD (67). Oxidative stress raises intracellular free Ca2+ levels, which may have deleterious consequences. In addition, oxidative DNA damage can interfere with gene transcription and effect promoter function, resulting in transcription damage and mutation of key genes. Oxidative RNA damage can impair protein translation, and damaged RNA can degrade prematurely, further impairs the synthesis of essential proteins (65).
Regarding mitochondrial dysfunction involved in AD pathophysiology, it includes disturbances in oxidative phosphorylation (OXPHOS), and impaired energy metabolism as well as excess generation of ROS, altered mitochondrial biogenesis, transport and dynamics (68). The susceptibility of neurons to mitochondrial dysfunction may be explained by the high dependence of neurons on oxidative phosphorylation (69). In the early stages of AD, mitochondria are unable to produce enough energy due to Aβ peptides and phosphorylation of tau, and therefore impaired mitochondria eventually cause excessive production of ROS (70). Finally, it further promotes the progress of AD (71).
Autophagy
Autophagy is an important pathway in removing abnormal protein aggregates in cells and plays an essential role in protein homeostasis (72). The mammalian nervous system, especially for neurons, depends heavily on autophagy to clear large amounts of insoluble protein aggregates to maintain protein homeostasis (73). Data shown that dysfunction of autophagy lysosome system can affect the clearance of Aβ peptides and tau proteins, two major features of AD (74). Recently, increasing evidence indicates that functional autophagy is required for synaptic functions, including neurotransmission and synaptic plasticity (75). Moreover, impaired autophagy is associated with sustained inflammation in tissues and may contribute to the pathogenesis of chronic inflammation (76). Therefore, impaired autophagy may contribute to the AD pathogenesis (73). On the other hand, phosphorylated tau protein and Aβ disturb autophagy, which is a major event in AD pathogenesis (77).
Diagnostics and treatments of AD
Due to severe cognitive impairment in patients with advanced AD, the cost of treatment and care seriously affects economic development (2, 3). Therefore, early diagnosis and treatment are essential to stop the progression of the disease. Currently, the diagnosis of AD mainly depends on cognitive tests, imaging techniques and analysis of CSF protein. Imaging techniques are used as positive support to confirm the clinical diagnosis of AD, including MRI scans and positron emission tomography (PET) (78). Examination of CSF for p-tau, Aβ42 and total tau protein content has value in predicting AD (2, 79). However, the existing biomarker tests are either expensive or invasive, so, to develop the ideal biomarker tests for AD diagnosis is still needed (80).
Currently, there is only two classes of drugs approved for the treatment of AD: N-methyl D-aspartate (NMDA) antagonist (memantine), and cholinesterase inhibitors (tacrine, Donepezil, galantamine, rivastigmine). However, all these treatments are symptomatic and cannot prevent or reverse AD pathology (81). Therefore, the development of effective disease modification therapies is the focus of AD prevention and treatment research in the future. Due to the similar molecular mechanism between T2DM and AD, several drugs for the treatment of T2DM are increasingly being proposed for the treatment of AD (15, 82). Some drugs for the treatment of T2DM may cause systemic side-effects such as hypoglycemia, and the long-term administration of insulin could promote brain insulin resistance (17). In contrast, GLP-1R agonists are safer, and several studies have shown that GLP-1R agonists significantly improve cognitive impairment (Figure 1) (25, 26, 83–85).
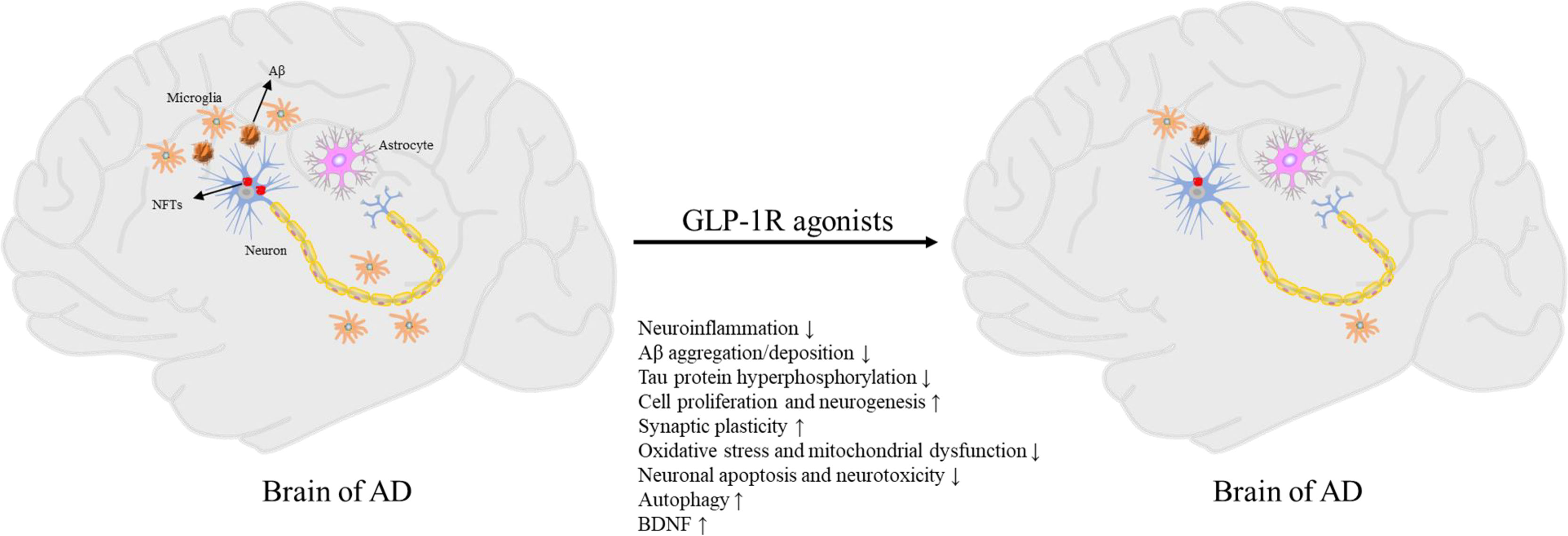
Figure 1 Pathogenesis of AD and neuroprotective effects of GLP-1R agonists.
Biological characteristics of GLP-1
In the 1980s, a new glucagon-like peptide, produced from proglucagon cleavage, was discovered to stimulate insulin secretion (86). Two glucagon-related peptides were identified in the proglucagon sequence, and were named glucagon-like peptides 1 and 2 (GLP-1 and GLP-2) (87). However, neither GLP-1 nor GLP-2 were active on insulin secretion, but a truncated version of GLP-1 was subsequently found to enhance insulin secretion in various experimental models and human studies (88–90). Ultimately, GLP-1 was identified as a potential incretin hormone (89, 91).
GLP-1, a 36-amino acid peptide, is produced in enteroendocrine L-cells of the distal small bowel and colon (92). Besides, GLP-1 can also be produced by neurons in CNS (20, 93). GLP-1 has different forms include GLP-1(1-37), GLP-1(7-36)amide and GLP-1(7-37) (22, 89). In humans, nearly all circulating GLP-1 is one of the latter two forms (89). While GLP-1(7-36)amide and GLP-1(7-37) are equally effective in stimulating insulin and C-peptide secretion, GLP-1(1-37) is much lower insulinotropic efficacy (88, 90, 94).
The effect of GLP-1 depends on blood glucose levels since it can only potentiate glucose-stimulated insulin secretion from islet beta cells in the hyperglycemic state rather than in the normal blood glucose state (95, 96). In addition, GLP-1 inhibits glucagon secretion in islet alpha cells, but only when blood glucose levels are higher than fasting (95, 96). Therefore, GLP-1 is save for use in T2DM and non-diabetic patients (97). Due to DPP-4 and renal clearance, native GLP-1 in humans has a half-life of about 1-2 minutes (21, 22, 89). In addition, DPP-4 cleaved GLP-1(7-36)amide and GLP-1(7-37) to form GLP-1(9-36)amide or GLP-1(9-37), low-affinity ligand of GLP-1 receptor and nonprimary role in regulating glucose metabolism (98–101). DPP-4 inhibitors, such as sitagliptin, alogliptin, linagliptin and saxagliptin, were synthesized to prolong the half-life of GLP-1 (102). Although DPP-4 inhibitors do not cross the BBB under normal conditions (the pharmacokinetic feature may prevent their repurposing use in neurodegenerative diseases), the permeability of BBB is increased in neurodegenerative diseases, so molecules otherwise unable to cross BBB could enter into CNS in these conditions (103). Several studies have showed the neuroprotective effects of DPP-4 inhibitors in animal models of AD (104, 105). Therefore, DPP-4 inhibitors, like GLP-1R agonists, may be a potential drug for the treatment of AD.
Tissue distribution of GLP-1R
GLP-1 mediates its effects by binding to its receptor, the GLP-1R, which is a sevenfold transmembrane G-coupled receptor that increases levels of cAMP by activating adenylate cyclase (89, 95, 96). GLP-1R is abundantly present in the pancreatic beta cells, gut, and the CNS, including the cerebral cortex, hippocampus, hypothalamus, thalamus, caudate-putamen and globus pallidum (89, 106–108). In addition, GLP-1R is moderately in the lung, heart, kidney, blood vessels, pancreatic alpha cells, and peripheral nervous system, with no expression of GLP-1R in liver, skeletal muscle or adipose tissue (109–112). However, the analysis of GLP-1R always confronted an obstacle due to the absence of antibodies with sufficient selectivity and availability. Further, it is worth noting that there was disputed information in the expression of GLP-1R in different cell types. The exact cellular localization of the GLP-1R remains equivocal due to the lack of selective antibodies and application of specific anti-GPCR antibodies (22). Recent studies identified the expression of GLP-1R in adipocytes (22, 113). In addition, low expression of GLP-1R in liver and muscle has been proposed (113).
In the peripheral system, GLP-1R mediates the actions of GLP-1 via the incretin axis, in which stimulation of the GLP-1R with GLP-1 primarily triggers insulin release from islet β cells in a glucose-dependent manner and inhibits glucagon secretion from islet α cells (110). The GLP-1R transduces signal mainly through Gαs coupling pathway (114). GLP-1R signaling enhances glucose-dependent insulin secretion by activating of Gαs, upregulating of cAMP, and subsequently activating of PKA (110). The cAMP/PKA pathway inhibits voltage-gated potassium channels that respond to depolarization, opens and allows K+ efflux, repolarizing the cell and allowing increased calcium influx through voltage-dependent calcium channels, resulting in the exocytosis of insulin from β-cells (115). GLP-1R activity also promotes the transactivation of epidermal growth factor receptor (EGFR), which then signals through phosphoinositide 3-kinase (PI3K) and insulin receptor substrate-2 (IRS-2), and subsequently activates extracellular-signal-regulated kinase 1 and 2 (ERK1/2) and nuclear translocation of PKC ξ to mediate β-cell proliferation and differentiation (110, 114).
In the CNS, GLP-1 also exert neuroprotective and neurotropic effects by binding to GLP-1R (20, 22, 116). GLP-1R expressed in the nucleus tractus solitarius signals to suppress appetite, delay gastric emptying and reduce body weight, and this is proposed to be mediated through PKA, decreasing phosphorylation of AMPK (22, 89, 110). In addition, GLP-1R overexpression results in improved cognitive function in mice and GLP-1R knockout severely impairs cognitive ability (5, 117, 118).
GLP-1R agonists
Several GLP-1R agonists, which mainly delay protease DPP-4 metabolism to overcome the problem with the rapid inactivation of GLP-1, have been developed (5, 119). Currently approved and commonly used GLP-1R agonists include exenatide (the synthetic form of Ex-4, a naturally occurring GLP-1R agonist), lixisenatide, dulaglutide liraglutide and semaglutide (Figure 2) (17, 110).
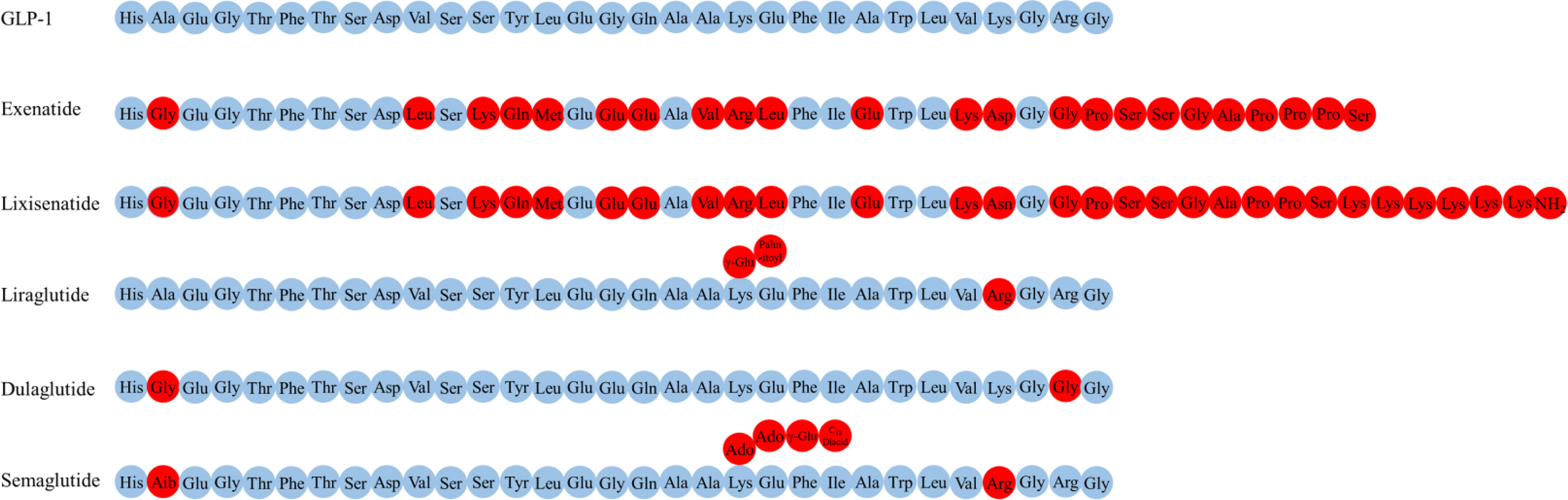
Figure 2 The amino acid structure of various GLP-1R agonists.
Exenatide, a peptide of 39 amino acids, has a much slower metabolism than endogenous GLP-1 (half-life of 3-4 h) (119, 120). Exenatide readily enters the brain when injected intravenously into mice (102). Elimination of exenatide is primarily achieved by glomerular filtration with subsequent proteolytic degradation (120–122). Increasing the dose of exenatide is not recommended in patients with an estimated glomerular filtration rate (eGFR) of 30-60 mL/min/1.73 m2, it is contraindicated for use in patients with an eGFR of less than 30 mL/min/1.73 m2 (123).
Lixisenatide comprises 44 amino acids and is based on the Ex-4 peptide sequence, omitting proline at position 36 and adding six lysine residues at the C-terminal (110). The binding affinity of lixisenatide to GLP-1R is fourfold higher than native GLP-1 (17). The circulating half-life of lixisenatide is approximately 3 h (124). Studies have shown that significant concentrations of lixisenatide were found in the brain of mice 30 minutes and 3 hours after intraperitoneal injection, suggesting that lixisenatide could cross the BBB (125). Lixisenatide is cleared by glomerular filtration, followed by tubular reabsorption and subsequent metabolic degradation (126). Dose adjustment is not recommended in patients with mild-severe renal impairment (eGFR = 15-89 mL/min/1.73 m2), but use is not recommended for patients with end-stage renal disease (127).
Liraglutide, 97% sequence identity to native GLP-1, is obtained by derivatizing GLP-1 with a fatty acid (128). This also facilitates albumin-binding and DPP-4 resistance, thereby allowing a half-life of 13 h (129). The main mechanisms of liraglutide protraction are as follows: (1) slow absorption after subcutaneous injection (130); (2) reduce clearance rate due to slowed metabolism and renal filtration (131). Significant levels of liraglutide were found in the brain of mice 30 minutes and 3 hours after intraperitoneal injection, indicating that liraglutide could cross the BBB (125). Liraglutide is approved in Europe for patients with T2DM and mild or moderate kidney damage, but is currently not recommended or should be used with caution in patients with severe renal impairment (130).
Dulaglutide is a long-acting GLP-1R agonist with a half-life of 4 days (124). The dulaglutide molecule consists of two modified DPP-4 resistant GLP-1(7-37) peptides fused to a modified IgG4 Fc fragment, which protect dulaglutide from proteolytic degradation by DPP-4 (132). In addition, its high molecular weight (57 kDa) prevents renal clearance and prolongs its half-life (133). No studies have addressed whether dulaglutide could cross the BBB. In patients with varying degrees of renal or hepatic impairment, no relevant change in dulaglutide exposure was observed relative to the degree of renal or hepatic impairment (132). Moreover, use of dulaglutide in people with T2DM is likely to confer additional renal benefits, and dulaglutide may be used in advanced chronic kidney disease at any eGFR level and without dose adjustment in contrast with most other noninsulin diabetes therapies (134, 135).
Semaglutide, a type of GLP-1R agonists with 94% sequence homology to GLP-1 and with an extended half-life of approximately 1 week, has been clinically approved to treat T2DM and is available in subcutaneous and oral dosage form (136, 137). Semaglutide interacted with circumventricular organs, where GLP-1R expression is abundant, and with regions protected by the BBB (lateral septal nucleus, nucleus tractus solitarius, hypothalamic arcuate nucleus) (102). The main elimination routs of semaglutide are through urine and feces. About 3% of the dose is emitted in the urine in an integral form (136).
Neuroprotective effects of GLP-1R agonists
GLP-1 and its mimetics can cross the BBB, therefore, are able to affect the CNS function such as cognition and neuroprotection (138). GLP-1R agonists were initially used to treat T2DM and soon after it was found that these drugs possess many other physiological properties, such as neuroprotection, neurotrophic, and anti-inflammatory effects, which may be useful to slow the progression of AD (Figure 3) (17).
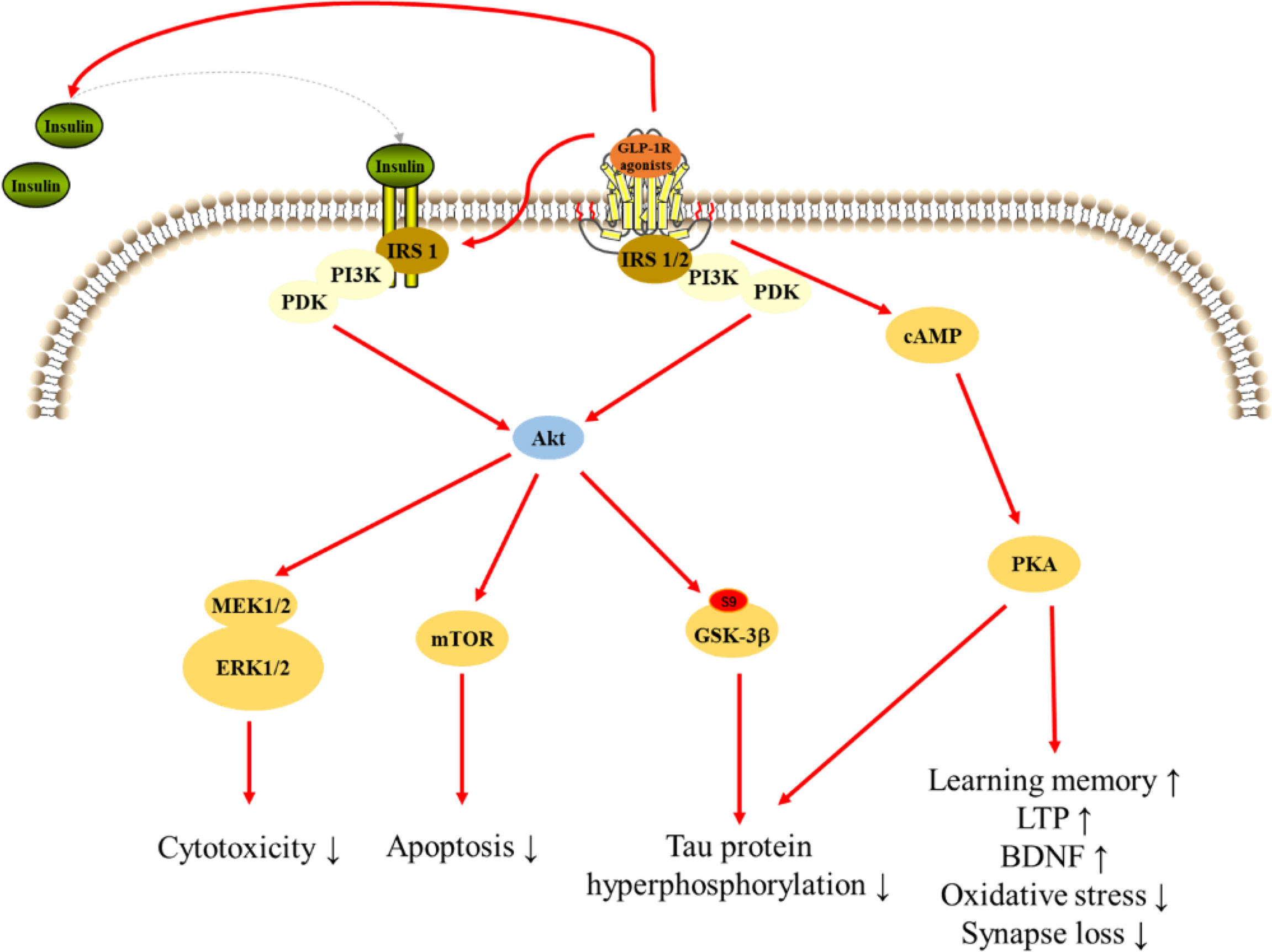
Figure 3 Signaling pathways for GLP-1R agonists in regulating cellular events in AD.
Anti-inflammatory effects
Previous studies have shown that GLP-1R mimics have anti-inflammatory effects in the CNS (139–141). Parthsarathy et al. found that liraglutide, an GLP-1R agonists, reduces the activated microglia load in the cortex and dentate gyrus region of hippocampus, and the activated astrocyte load in the cortex. Furthermore, the pro-inflammatory cytokine levels of IL-6, IL-12p70, IL-1β, and total nitrite concentration are reduced in the brains of mice treated with liraglutide (142). A study reports that prophylactic liraglutide treatment reduces chronic inflammation (activated microglia) in the cortex and prevents memory impairment in APP/PS1 mice (143). In a number of studies, GLP-1R agonists such as liraglutide, exenatide and lixisenatide can reduce neuroinflammation in AD models, thereby improving cognitive dysfunction (140, 141, 144–148). These data strongly suggest that GLP-1R agonists are beneficial to attenuate neuroinflammation-associated cognitive impairment and thereby improve cognition.
Reducing Aβ aggregation/deposition and tau protein hyperphosphorylation
Some studies have reported that GLP-1 can affect the pathological process of Aβ deposition and tau hyperphosphorylation (149–151). Perry et al. found that GLP-1 can reduce the levels of endogenous Aβ in the brain and reduce the levels of APP in cultured neuronal cells (152). Exendin-4 (an endogenous insulin releasing incretin, GLP-1) reduces Aβ accumulation and tau hyperphosphorylation in cellular and animal models of AD (85, 151, 153–156). In another study, (Val8)GLP-1 might prevent age-related neurodegenerative changes (such as AD) by preventing decline of learning and memory formation, reduction of tau hyperphosphorylation and protection of subcellular structures and morphology of neurons (149). Lixisenatide, a GLP-1R agonist, reduces neurofibrillary tangles and amyloid plaque load, and thereby ameliorates learning memory deficits in APP/PS1 mice (144, 157). Total brain APP and Aβ oligomer levels are reduced in Liraglutide-treated AD mice (147, 158–160), and intervention with liraglutide can prevent tau hyperphosphorylation (161–164). Dulaglutide, a novel long-acting GLP-1R agonist, ameliorates AD-like impairment of learning and memory ability by decreasing the hyperphosphorylation of tau and NFs proteins (150). Therefore, GLP-1R agonists might improve cognitive and memory function by reducing Aβ deposition and tau hyperphosphorylation in the brain.
Promoting cell proliferation and neurogenesis
In addition to its hormonal and neuropeptide activity, GLP-1 also is considered a growth factor that regulates cell growth and differentiation, and promotes interruption of pro-apoptotic processes (165). Hamilton et al. have found that progenitor cell division is enhanced after injected subcutaneously GLP-1 agonists exenatide (exendin-4) and liraglutide in the dentate gyrus of brain of mouse models of AD (166). In another study, intraperitoneal injection with GLP-1 analogue (Val)GLP-1 enhances neuronal stem cells and neurogenesis in the dentate gyrus of brain in wild type mice, so it may have potentially beneficial effects in the CNS (167). Moreover, chronic treatment with liraglutide showed an increase in stem cell proliferation and differentiation into mature neurons in APP/PS1 mice and controls at all ages, which may have beneficial effects in neurodegenerative disorders like AD (168). And total hippocampal CA1 pyramidal neuron numbers in senescence-accelerated mouse prone 8 mice are increased after received liraglutide, at the same time, the memory retention of mice is increased (169). Overall, GLP-1 may improve cognitive function by promoting neuronal stem cell proliferation and differentiation.
Enhancing synaptic plasticity
Evidence shows that the changes in synaptic function may be an early event in AD pathogenesis (170). Studies have found that (Val8)GLP-1 protects synapses from the detrimental effects of Aβ fragments on synaptic plasticity formation (171–173). In another study, McClean et al. reported that GLP-1R analogues enhance synaptic plasticity in area CA1 of the hippocampus (174). However, synaptic plasticity and memory formation are impaired in GLP-1 receptor knockout mice (118). Liraglutide was shown to prevent synapse loss and deterioration of synapse plasticity in the hippocampus of APP/PS1 mice, while enhance synaptic plasticity in the control mice (158, 175). And liraglutide protects synapse from Aβ oligomers-induced damage in hippocampal neurons (176, 177). Ohtake et al. found that exendin-4 increased the membrane protein level of the AMPA receptor GluR1 subunit and postsynaptic density protein-95, which were the critical mechanisms of long-term potentiation (LTP) as well as the formation of learning and memory (178). Moreover, lixisenatide and GLP-1 analogue CJC-1131 could protect against Aβ-induced suppression of hippocampal LTP (179, 180). These results suggest that one of neuroprotective effects of GLP-1R agonists is enhancing synaptic plasticity.
Attenuating oxidative stress and mitochondrial dysfunction
Oxidative stress plays a vital role in the pathogenesis and pathophysiology of AD (181). Chen et al. have reported that GLP-1/exendin-4 could ameliorate oxidative stress-induced injury in PC12 cells (182). GLP-1 also protects HT22 cells against oxidative stress-induced cell death (183). Moreover, Spielman et al. found that GLP-1 can reduce oxidative stress in BV2 microglia by inhibiting the accumulation of intracellular ROS and release of nitric oxide (NO), as well as by increasing the expression of the antioxidant glutathione peroxidase 1 (GPx1) and superoxide dismutase 1 (SOD1) (184). Studies have demonstrated that Liraglutide has neuroprotective effects on AD-like neurodegeneration induced by H2O2 in human neuroblastoma cell line SH-SY5Y (185, 186), and protects against brain Aβ accumulation by partially rescuing oxidative stress (146).
Mitochondria are involved in a series of biochemical events in cells, ordinarily, normal neurons have an intense energetic demand to support localized neuronal activities. However, dysfunctional mitochondria are associated with impaired neuronal function and associated neurodegenerative diseases (187). According to the report of An et al. GLP-1R agonists could repair mitochondrial damage in vitro, and promote mitochondrial biogenesis and antioxidant system peroxisome proliferator-activated receptor γ coactivator 1α (PGC-1α) signaling pathway in vivo (28). Xie et al. have reported that Liraglutide ameliorates mitochondrial dysfunction and prevents neuronal loss in the brain of 5×FAD mice (188). Likewise, another study shows that Exenatide alleviates mitochondrial dysfunction and cognitive impairment in 5×FAD mice (84). Moreover, Exendin-4 significantly increases Aβ-induced decrease in mitochondrial function, integrity and respiratory control rate in all brain regions (189).
These results suggest that GLP-1R agonists are able to improve cognitive function by attenuating the oxidative stress and mitochondria dysfunction in CNS.
Inhibiting neuronal apoptosis and neurotoxicity
Neuronal apoptosis, induced by Aβ and stress, is regard as a physiolopathologic marker in AD brain (190). Perry et al. found that GLP-1 and exendin-4 could completely protect cultured rat hippocampal neurons against glutamate-induced apoptosis (191). Moreover, exendin-4 protected PC12 cells from Aβ-induced apoptosis (192). During et al. reported that [Ser (2) exendin (1-9)], a GLP-1R agonist, significantly attenuated kainic acid-induced apoptosis in the CA3 region of the hippocampus (117). In another study, GLP-1 protected against methylglyoxal-induced pheochromocytoma (PC12) cell apoptosis though the PI3K/Akt/mTOR/GCLc/redox signaling pathway (193). Consistent with these data, GLP-1 attenuated apoptosis of PC12 cells induced by carboxymethyl lysine via peroxisome proliferation activated receptor-γ (PPAR-γ) (194). Chen et al. demonstrated that GLP-1 could significantly decrease the percentage of advanced glycation and products (AGEs)-induced SH-SY5Y cell apoptosis (195). And it also has been reported that liraglutide reduces cytotoxicity and apoptosis of SH-SY5Y cells during methylglyoxal, thapsigargin and Aβ stress (196–199). Likewise, in an AD model, SH-SY5Y cells were treated with Aβ, semaglutide inhibited apoptosis by inhibiting the expression of Bax induced by Aβ and increasing the expression of Bcl2 inhibited by Aβ (200). In summary, these data suggest one of neuroprotective effects of GLP-1R agonists is inhibiting neuronal apoptosis.
Enhancing autophagy
Autophagy deregulation may underlie the accumulation of neuropathological markers for AD, rendering it one of the main features in AD (201). Candeias et al. found that peripheral exendin-4 treatment promoted brain cortical autophagy upon type 2 diabetes rats (202). Liraglutide modulated the autophagy machinery homeostasis in SH-SY5Y cells (198), and attenuated Aβ42 generation in SH-SY5Y cells through enhancing autophagy (160). Another study showed that semaglutide protected against Aβ25-35 in SH-SY5Y cells by enhancing autophagy (200). These indicate autophagy is a key molecular event for GLP-1R agonists to protect neurons against damage.
Other effects
In addition to above effects, GLP-1R agonists have other neuroprotection mechanism on CNS, such as increasing the brain-derived neurotrophic factor (BDNF) levels and activity, regulating calcium homeostasis, promoting glycolysis, and reducing vascular damage. BDNF plays a crucial role in the pathophysiology of brain neurons. Ohtake et al. found exendin-4 increased the expression of BDNF in mouse neocortex (178). And several studies suggest that exenatide increased expression levels of BDNF in AD mice hippocampus and cortex (141, 203, 204). It has been shown that intracellular calcium overload induced by Aβ produces cytotoxicity, which cause a decrease in learning and memory as well as cognitive function. Recent studies reported that GLP-1R agonists, such as Val8-GLP-1(7-36), exendin-4 and liraglutide, attenuated Aβ1-42-induced calcium overload by regulating intracellular calcium homeostasis in cortical or hippocampal pyramidal cells (173, 205, 206). Glycolysis and oxidative phosphorylation, which break down glucose into the form of ATP to produce energy, is the main source of energy for the brain. Bomba et al. found that exenatide increased brain lactate dehydrogenase activity, enhancing anaerobic glucose catabolism in brains of PS1-KI mice (207). Zheng et al. revealed liraglutide improved aerobic glycolysis in astrocyte, and cortices of 5×FAD mice (208). Cerebral microvascular impairments occurring in AD may reduce Aβ clearance. Some studies found liraglutide reduced incidence of cerebral microanuerysms and leakage (209, 210).
Above all, GLP-1R agonists may improve cognitive function via different mechanism. These results suggest that GLP-1R agonists may have therapeutic and preventive effects on AD.
Signaling pathways underlying the neuroprotective effects of GLP-1R
PI3K signaling pathway
PI3K plays an important role in modulating cytoactivities. Data showed that GLP-1 significantly increased PI3K, Akt, and mammalian target of rapamycin (mTOR) phosphorylation without inducing the expression of PI3K, Akt, or mTOR. And the expression of downstream gene significantly reduced after treated inhibitors of PI3K, Akt, and mTOR. These results suggest PI3K/Akt/mTOR signaling mediated the protection of GLP-1 on apoptosis in neurons (193). Exendin-4 treatment reversed the intracerebroventricular-streptozotocin (ICV-STZ) -induced decline in the levels of phosphorylation of Akt at Ser473 and glycogen synthase kinase 3β (GSK-3β) at Ser9, a key kinase in AD, leading to decrease hyperphosphorylation of tau in rat hippocampus (182). And, exendin-4 significantly increased Aβ-induced decrease in the level of phosphorylated Akt in brain (189). However, lixisenatide inhibited the Aβ-induced activation of GSK-3β, with a significant increase in the phosphorylation of ser9 and a significant decrease in the phosphorylation of Y216, suggesting that lixisenatide can prevent Aβ-related impairments by affecting the PI3K-Akt-GSK3β pathway (179). Liraglutide administration prevented the decrease of AKT and GSK-3β phosphorylation after treated Aβ1-42 protein, which inhibiting tau hyperphosphorylation (164, 211). Moreover, liraglutide prevents Aβ and H2O2-induced neurotoxicity in SH-SY5Y cells via PI3K/Akt signaling pathway (185, 197). Lixisenatide relieved the Aβ25-35-induced suppression of the phosphorylation of Akt and MEK1/2 indicating the neuroprotection of lixisenatide might related to the Akt-MEK1/2 signaling pathway (206). A study reported that GLP-1(7-36) could protect HT22 cells against stressors via activation of survival signaling molecules, such as Akt and ERK1/2 (183). Dulaglutide decreased the hyperphosphorylation of tau and NFs proteins through improving the PI3K/Akt/GSK3β signaling pathway, which may be related to its protective effects on impaired of AD-like learning and memory (150).
cAMP/PKA pathway
The cAMP/PKA pathway, a ubiquitous cascade that modulates numerous cellular events within neurons (176). Pretreatment with liraglutide effectively and dose-dependently protected against the Aβ25-35-induced impairment of spatial memory and deficit of late-phase long-term potentiation (L-LTP), and also activated cAMP signal pathway in the rat brain (177). Furthermore, pretreatment of cultures with liraglutide attenuated measures of synapse induced by Aβ oligomers (AβOs), but liraglutide failed to prevent AβOs-induced synapse loss after treated PKA inhibitor, and liraglutide attenuated the AβOs-induced decrease of PKA activity, suggesting that activation of the cAMP/PKA pathway underlies the neuroprotective actions of liraglutide (176). Besides, GLP-1R agonists also regulated PC12 cells growth through regulating cAMP/PKA signaling pathway (212). In addition to acting on neuronal cells, GLP-1R agonists also exert effects on microglia and astrocytes through cAMP/PKA signaling pathway. GLP-1 reduced apoptotic death of BV2 microglia cells through the binding and activation of the GLP-1R, with subsequent activation of the PKA pathway. Moreover, GLP-1 upregulated BV2 microglia cells expression of BDNF in a PKA-dependent manner (184). In Aβ-treated astrocytes, GLP-1 prevented mitochondrial fragmentation, and improved the neuronal supportive ability via cAMP/PKA pathway (188).
Insulin signaling pathway
Insulin signaling in the brain is vital for the brain activity, and insulin resistance is one of key reasons of AD (213). Liraglutide treatment significantly decreased insulin receptor aberrations in conjunction with a concomitant decrease in amyloid plaque load and a highly significant reduction in astrocytosis and microglial number associated with both plaques and IR pathology (214). Liraglutide also restored neuronal insulin sensitivity in hyperinsulinemic conditions and reduced the Aβ formation and tau hyperphosphorylation in neuronal cells (215). The ERK and JNK are parts of the insulin signal pathway and closely associated with tau hyperphosphorylation. Liraglutide ameliorated the phosphorylated expressions of ERK and JNK interfered with by STZ and decreased the hyperphosphorylation of NFs (216). Exendin-4 significantly increased insulin level and phosphorylation of insulin receptor substrate 1 (IRS-1) in rat hippocampus, and could not change the status of tau phosphorylation without insulin in HT22 neurons, suggesting that insulin is required in reduction of tau hyperphosphorylation by GLP-1R agonists (154).
Application of GLP-1R agonists in AD
Several studies have reported that GLP-1 agonists could protect brain against various damage in AD models (Supplementary Table 1 and Table 1). Currently, GLP-1R agonists used in the treatment of AD models include (Val8)GLP-1, GLP-1(9-36)amide, GLP-1(7-36)amide, CJC-1131, Geniposide, Exendin(5-39), Exendin-4, NLY01 (engineered exendin-4), liraglutide, Lixisenatide, Dulaglutide, Exenatide, GLP-1, dual GLP-1/GIP receptor agonist (DA-JC4, DA-JC1, DA5-CH, DA-CH3), GLP-1/GIP/Gcg receptor triagonist (triagonist, TA), dual GLP-1 and Gcg receptor agonist (oxyntomodulin) (144, 147, 148, 150, 171, 180, 186, 189, 194, 218, 219, 228, 235, 237, 238, 241, 242).

Table 1 The effects of various GLP-1R agonists upon brain mechanisms in animal models for AD.
The treatment of (Val8)GLP-1 30 minutes prior to injection of Aβ fully reversed the impairment of LTP induced by Aβ (171, 217). Val8-GLP-1 also protected against Aβ1-40-induced impairment of learning and memory, and reduced total tau expression and hyperphosphorylated tau levels (149, 217). Likewise, GLP-1(9-36)amide could reverse AD-related alterations in hippocampal synaptic plasticity and memory deficits, but did not alter levels of APP and Aβ in APP/PS1 mice (218). And, GLP-1(7-36)amide significantly prevented LPS-, IL-1β-, H2O2-induced impairment in synaptic functions of hippocampal CA1 region (219). CJC-1131, a new chemical-modified GLP-1 agonist, was effective in resisting DPP-4 degradation. It is found that CJC-1311 effectively prevented Aβ1-42-induced impairments in spatial learning and memory, and also reversed Aβ1-42-induced suppression of hippocampal LTP (180). Geniposide, acting as a GLP-1R agonist, partially prevented STZ-induced learning and memory impairment via modulation of PI3K/GSK-3β signaling pathway and reduced STZ-induced hyperphosphorylation of tau protein (220). Besides, geniposide suppressed Aβ accumulation and alleviated cognitive dysfunction in APP/PS1 mice, attenuated Aβ-induced reduction of LTP in acute hippocampal slices, and attenuated Aβ-induced synaptic dysfunction in cultured hippocampal neurons (221, 222).
There are a large number of studies that have reported the application of exendin-4, liraglutide and exenatide in the treatment of cognitive dysfunction and pathological features in AD models. Exendin-4 treatment could reduce levels of Aβ in brain of mice and reduce levels of APP in cultured neuronal cells (152). Exendin-4 also could ameliorate brain levels of AβPP and Aβ, elevated by STZ-induced diabetes, in 3×Tg-AD mice (151). Behavioral measures of cognition in APP/PS1 mice treated with Exendin-4 was significantly improved. After administration of Exendin-4, Aβ oligomers-induced impaired axonal transport was prevented, and levels of soluble Aβ and amyloid plaque load were decreased in APP/PS1 mice (224, 227). In STZ-treatment rats, Exendin-4 improved learning and memory performance, protected hippocampal neurons against degeneration, and reversed STZ-induced tau hyperphosphorylation through downregulation of GSK-3β activity (154, 155, 182). For Aβ1-42-induced AD model, exendin-4 mitigated abnormal behavior and prevented Aβ1-42-induced impairment of LTP (85, 189, 226). Similar to exendin-4, liraglutide also improved cognitive and spatial memory, enhanced induction and maintenance of LTP, and reduced β-amyloid plaque formation as well as inflammatory response in APP/PS1 (143, 158, 159, 175, 214). Liraglutide decreased hyperphosphorylation of NFs and hyperphosphorylated tau in brain of STZ mice, and improved learning and memory impairment induced by STZ through ameliorating ERK and INK signaling pathways (216, 229). For Aβ25-35-treated mice, liraglutide dose-dependently prevented against Aβ25-35-induced impairment of spatial learning and memory, prevented depression of hippocampal L-LTP, and upregulated intracellular cAMP level (177). In db/db mice, Aβ1-42-treated mice or hTauP301L mice, liraglutide alleviated tau hyperphosphorylation, and prevented dysregulation of Akt and GSK-3β in brain (161, 163, 164). Liraglutide improved learning and memory performance, attenuated hyperphosphorylation of tau as well as NFs, and prevented neurodegeneration via improving JNK and ERK signaling in brain of 3×Tg mice (232). Liraglutide reduced astrocytes, microglia activity and amount of Aβ levels in cortical and hippocampal CA1 and CA3 regions in 5×FAD mice (147). While liraglutide improved spatial cognition, it also reduced astrocytes and microglia activity, ameliorated amount of Aβ levels and mitochondrial dysfunction in cortical and hippocampal CA1 and CA3 regions, and prevented neuron loss by activating cAMP/PKA pathway in brain of 5×FAD mice (188, 208). Lixisenatide and exenatide have the same effect as liraglutide in AD models (84, 141, 144, 148, 157, 179, 206, 213).
Moreover, dual GLP-1 and Gcg receptor agonist, dual GLP-1/GIP receptor agonist and GLP-1/GIP/Gcg receptor triagonist have also been successfully applied in the treatment of AD models. It is reported that both DA-JC4 and DA5-CH could improve STZ-induced learning and memory impairment, reduce levels of phosphorylated tau protein and STZ-induced chronic inflammation response in brain (233, 237). Besides, dual GLP-1/GIP receptor agonists (DA-JC4, DA5-CH, DA-JC1 and DA-CH3) reduced inflammation response, amyloid plaques and tau phosphorylation in brain, reversed memory loss, and enhanced synaptic plasticity in hippocampus of APP/PS1 mice (186, 234, 236, 238). In 3×Tg-AD treated with DA-JC4, the ability of recognize new object and spatial working memory was improved, hippocampal Aβ and tau pathology were alleviated, and hippocampal synaptic plasticity also was improved as well as levels of PSD95 and SYP were increased (235). After administration of Triple GLP-1/GIP/glucagon receptor agonist, learning and memory impairment of AD models (APP/PS1 mice and 3×Tg-AD mice) was improved, amyloid plaques, tau pathology, inflammation response and oxidative stress in brains of two AD models were ameliorated (239–241).
However, there are few clinical trials of GLP-1R agonists in AD patients. In a randomized placebo-controlled, double-blinded trial, although liraglutide did not affect Aβ load and cognition measures, 26 weeks of liraglutide treatment prevented expected decline of glucose metabolism that signifies cognitive impairment, synaptic dysfunction, and disease evolution (243). And the effect of liraglutide might be associated with improvement of BBB glucose transport capacity (244). Another double-blinded, placebo-controlled study reported that liraglutide increased intrinsic connectivity in bilateral hippocampal, medial frontal and lateral occipital regions, suggesting that liraglutide might reduce or delay AD progression (245). Patients with T2DM, persons at risk for AD, had a significantly lower associated risk of AD after administered exenatide, liraglutide and dulaglutide (83). These data indicate that GLP-1R agonists show great potential as a novel treatment for preventing AD processes.
Conclusion
Increasing evidence suggests that anti-diabetes medicine, GLP-1R agonists, have multiple neuroprotective mechanisms in AD models, such as anti-inflammation, anti-oxidative stress, reducing Aβ aggregation/deposition and tau protein hyperphosphorylation, reducing neuronal apoptosis and neurotoxicity, increasing cell proliferation and neurogenesis, increasing synaptic plasticity, and other beneficial effects. GLP-1R agonists might have the potential to be developed as a novel treatment of AD. Further clinical evidence is needed to verify the effects of GLP-1R agonists on cognitive function and pathology in AD patients.
Author contributions
JX and HD designed the study. HD wrote the paper. XM reviewed the paper and provided suggestions. YY collected references. JX is the guarantor of this work and had full access to all the data in the study and also takes responsibility for the integrity of the data. All authors contributed to the article and approved the submitted version.
Funding
This study was supported by grants from the Scientific Research Foundation of the Ministry of Education of China (Grant No.2021JH019) and Scientific Research Foundation of Postdoctoral Department of Heilongjiang Province (Grant No.LBH-Q21025).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fendo.2022.1033479/full#supplementary-material
References
1. Talbot K, Wang H-Y. The nature, significance, and glucagon-like peptide-1 analog treatment of brain insulin resistance in alzheimer’s disease. Alzheimer’s Dementia (2014) 10(1S):S12–25. doi: 10.1016/j.jalz.2013.12.007
2. Lane CA, Hardy J, Schott JM. Alzheimer’s disease. Eur J Neurol (2018) 25(1):59–70. doi: 10.1111/ene.13439
3. Abeysinghe A, Deshapriya R, Udawatte C. Alzheimer’s disease; a review of the pathophysiological basis and therapeutic interventions. Life Sci (2020) 256:117996. doi: 10.1016/j.lfs.2020.117996
4. Scheltens P, De Strooper B, Kivipelto M, Holstege H, Chételat G, Teunissen CE, et al. Alzheimer’s disease. Lancet (2021) 397(10284):1577–90. doi: 10.1016/S0140-6736(20)32205-4
5. Liu XY, Zhang N, Zhang SX, Xu P. Potential new therapeutic target for alzheimer’s disease: Glucagon-like peptide-1. Eur J Neurosci (2021) 54(10):7749–69. doi: 10.1111/ejn.15502
6. Li X, Song D, Leng SX. Link between type 2 diabetes and alzheimer’s disease: from epidemiology to mechanism and treatment. Clin Interv Aging (2015) 10:549–60. doi: 10.2147/CIA.S74042
7. Wang XF, Lin X, Li DY, Zhou R, Greenbaum J, Chen YC, et al. Linking alzheimer’s disease and type 2 diabetes: Novel shared susceptibility genes detected by cFDR approach. J Neurol Sci (2017) 380:262–72. doi: 10.1016/j.jns.2017.07.044
8. Li L, Cavuoto M, Biddiscombe K, Pike KE. Diabetes mellitus increases risk of incident dementia in APOEvarepsilon4 carriers: A meta-analysis. J Alzheimers Dis (2020) 74(4):1295–308. doi: 10.3233/JAD-191068
9. Miao Y, Guo D, Li W, Zhong Y. Diabetes promotes development of alzheimer’s disease through suppression of autophagy. J Alzheimers Dis (2019) 69(1):289–96. doi: 10.3233/JAD-190156
10. Kellar D, Craft S. Brain insulin resistance in alzheimer’s disease and related disorders: mechanisms and therapeutic approaches. Lancet Neurol (2020) 19(9):758–66. doi: 10.1016/S1474-4422(20)30231-3
11. Akhtar A, Sah SP. Insulin signaling pathway and related molecules: Role in neurodegeneration and alzheimer’s disease. Neurochem Int (2020) 135:104707. doi: 10.1016/j.neuint.2020.104707
12. Bae CS, Song J. The role of glucagon-like peptide 1 (GLP1) in type 3 diabetes: GLP-1 controls insulin resistance, neuroinflammation and neurogenesis in the brain. Int J Mol Sci (2017) 18(11):2493. doi: 10.3390/ijms18112493
13. Nguyen TT, Ta QTH, Nguyen TKO, Nguyen TTD, Giau VV. Type 3 diabetes and its role implications in alzheimer’s disease. Int J Mol Sci (2020) 21(9):3165. doi: 10.3390/ijms21093165
14. Dubey SK, Lakshmi KK, Krishna KV, Agrawal M, Singhvi G, Saha RN, et al. Insulin mediated novel therapies for the treatment of alzheimer’s disease. Life Sci (2020) 249:117540. doi: 10.1016/j.lfs.2020.117540
15. Munoz-Jimenez M, Zaarkti A, Garcia-Arnes JA, Garcia-Casares N. Antidiabetic drugs in alzheimer’s disease and mild cognitive impairment: A systematic review. Dement Geriatr Cognit Disord (2020) 49(5):423–34. doi: 10.1159/000510677
16. Kellar D, Lockhart SN, Aisen P, Raman R, Rissman RA, Brewer J, et al. Intranasal insulin reduces white matter hyperintensity progression in association with improvements in cognition and CSF biomarker profiles in mild cognitive impairment and alzheimer’s disease. J Prev Alzheimers Dis (2021) 8(3):240–8. doi: 10.14283/jpad.2021.14
17. Tramutola A, Arena A, Cini C, Butterfield DA, Barone E. Modulation of GLP-1 signaling as a novel therapeutic approach in the treatment of alzheimer’s disease pathology. Expert Rev Neurother (2017) 17(1):59–75. doi: 10.1080/14737175.2017.1246183
18. Schwartz MW, Figlewicz DF, Kahn SE, Baskin DG, Greenwood MR, Porte D Jr. Insulin binding to brain capillaries is reduced in genetically obese, hyperinsulinemic zucker rats. Peptides (1990) 11(3):467–72. doi: 10.1016/0196-9781(90)90044-6
19. Wallum BJ, Taborsky GJ Jr., Porte D Jr., Figlewicz DP, Jacobson L, Beard JC, et al. Cerebrospinal fluid insulin levels increase during intravenous insulin infusions in man. J Clin Endocrinol Metab (1987) 64(1):190–4. doi: 10.1210/jcem-64-1-190
20. Erbil D, Eren CY, Demirel C, Kucuker MU, Solaroglu I, Eser HY. GLP-1’s role in neuroprotection: a systematic review. Brain Inj (2019) 33(6):734–819. doi: 10.1080/02699052.2019.1587000
21. Laurindo LF, Barbalho SM, Guiguer EL, da Silva Soares de Souza M, de Souza GA, Fidalgo TM, et al. GLP-1a: Going beyond traditional use. Int J Mol Sci (2022) 23(2):739. doi: 10.3390/ijms23020739
22. Li QX, Gao H, Guo YX, Wang BY, Hua RX, Gao L, et al. GLP-1 and underlying beneficial actions in alzheimer’s disease, hypertension, and NASH. Front Endocrinol (Lausanne) (2021) 12:721198. doi: 10.3389/fendo.2021.721198
23. Calsolaro V, Edison P. Novel GLP-1 (Glucagon-like peptide-1) analogues and insulin in the treatment for alzheimer’s disease and other neurodegenerative diseases. CNS Drugs (2015) 29(12):1023–39. doi: 10.1007/s40263-015-0301-8
24. Muscogiuri G, DeFronzo RA, Gastaldelli A, Holst JJ. Glucagon-like peptide-1 and the Central/Peripheral nervous system: Crosstalk in diabetes. Trends Endocrinol Metab (2017) 28(2):88–103. doi: 10.1016/j.tem.2016.10.001
25. Holscher C. Central effects of GLP-1: new opportunities for treatments of neurodegenerative diseases. J Endocrinol (2014) 221(1):T31–41. doi: 10.1530/JOE-13-0221
26. Abd El-Rady NM, Ahmed A, Abdel-Rady MM, Ismail OI. Glucagon-like peptide-1 analog improves neuronal and behavioral impairment and promotes neuroprotection in a rat model of aluminum-induced dementia. Physiol Rep (2021) 8(24):e14651. doi: 10.14814/phy2.14651
27. Biswas SC, Buteau J, Greene LA. Glucagon-like peptide-1 (GLP-1) diminishes neuronal degeneration and death caused by NGF deprivation by suppressing bim induction. Neurochem Res (2008) 33(9):1845–51. doi: 10.1007/s11064-008-9646-4
28. An FM, Chen S, Xu Z, Yin L, Wang Y, Liu AR, et al. Glucagon-like peptide-1 regulates mitochondrial biogenesis and tau phosphorylation against advanced glycation end product-induced neuronal insult: Studies in vivo and in vitro. Neuroscience (2015) 300:75–84. doi: 10.1016/j.neuroscience.2015.05.023
29. Rojas M, Chavez-Castillo M, Bautista J, Ortega A, Nava M, Salazar J, et al. Alzheimer’s disease and type 2 diabetes mellitus: Pathophysiologic and pharmacotherapeutics links. World J Diabetes (2021) 12(6):745–66. doi: 10.4239/wjd.v12.i6.745
30. Khan S, Barve KH, Kumar MS. Recent advancements in pathogenesis, diagnostics and treatment of alzheimer’s disease. Curr Neuropharmacol (2020) 18(11):1106–25. doi: 10.2174/1570159X18666200528142429
31. Alberdi E, Wyssenbach A, Alberdi M, Sánchez-Gómez MV, Cavaliere F, Rodríguez JJ, et al. Ca2+-dependent endoplasmic reticulum stress correlates with astrogliosis in oligomeric amyloid β-treated astrocytes and in a model of alzheimer’s disease. Aging Cell (2013) 12(2):292–302. doi: 10.1111/acel.12054
32. Swanson CJ, Zhang Y, Dhadda S, Wang J, Kaplow J, Lai RYK, et al. A randomized, double-blind, phase 2b proof-of-concept clinical trial in early alzheimer’s disease with lecanemab, an anti-abeta protofibril antibody. Alzheimers Res Ther (2021) 13(1):80. doi: 10.1186/s13195-021-00813-8
33. Tahami Monfared AA, Tafazzoli A, Ye W, Chavan A, Zhang Q. Long-term health outcomes of lecanemab in patients with early alzheimer’s disease using simulation modeling. Neurol Ther (2022) 11(2):863–80. doi: 10.1007/s40120-022-00350-y
34. Barbier P, Zejneli O, Martinho M, Lasorsa A, Belle V, Smet-Nocca C, et al. Role of tau as a microtubule-associated protein: Structural and functional aspects. Front Aging Neurosci (2019) 11:204. doi: 10.3389/fnagi.2019.00204
35. Chesser AS, Pritchard SM, Johnson GV. Tau clearance mechanisms and their possible role in the pathogenesis of Alzheimer disease. Front Neurol (2013) 4:122. doi: 10.3389/fneur.2013.00122
36. Simic G, Babic Leko M, Wray S, Harrington C, Delalle I, Jovanov-Milosevic N, et al. Tau protein hyperphosphorylation and aggregation in alzheimer’s disease and other tauopathies, and possible neuroprotective strategies. Biomolecules (2016) 6(1):6. doi: 10.3390/biom6010006
37. Tsartsalis S, Xekardaki A, Hof PR, Kovari E, Bouras C. Early Alzheimer-type lesions in cognitively normal subjects. Neurobiol Aging (2018) 62:34–44. doi: 10.1016/j.neurobiolaging.2017.10.002
38. Hampel H, Mesulam MM, Cuello AC, Farlow MR, Giacobini E, Grossberg GT, et al. The cholinergic system in the pathophysiology and treatment of alzheimer’s disease. Brain (2018) 141(7):1917–33. doi: 10.1093/brain/awy132
39. Zhu C-C, Fu S-Y, Chen Y-X, Li L, Mao R-l, Wang J-Z, et al. Advances in drug therapy for alzheimer’s disease. Curr Med Sci (2021) 40(6):999–1008. doi: 10.1007/s11596-020-2281-2
40. Bekdash RA. The cholinergic system, the adrenergic system and the neuropathology of alzheimer’s disease. Int J Mol Sci (2021) 22(3):1273. doi: 10.3390/ijms22031273
41. Stanciu GD, Luca A, Rusu RN, Bild V, Beschea Chiriac SI, Solcan C, et al. Alzheimer’s disease pharmacotherapy in relation to cholinergic system involvement. Biomolecules (2019) 10(1):40. doi: 10.3390/biom10010040
42. Nyakas C, Granic I, Halmy LG, Banerjee P, Luiten PG. The basal forebrain cholinergic system in aging and dementia. rescuing cholinergic neurons from neurotoxic amyloid-beta42 with memantine. Behav Brain Res (2011) 221(2):594–603. doi: 10.1016/j.bbr.2010.05.033
43. Vakalopoulos C. Alzheimer’s disease: The alternative serotonergic hypothesis of cognitive decline. J Alzheimers Dis (2017) 60(3):859–66. doi: 10.3233/JAD-170364
44. Govindpani K, Turner C, Waldvogel HJ, Faull RLM, Kwakowsky A. Impaired expression of GABA signaling components in the alzheimer’s disease middle temporal gyrus. Int J Mol Sci (2020) 21(22):8704. doi: 10.3390/ijms21228704
45. Bi D, Wen L, Wu Z, Shen Y. GABAergic dysfunction in excitatory and inhibitory (E/I) imbalance drives the pathogenesis of alzheimer’s disease. Alzheimers Dement (2020) 16(9):1312–29. doi: 10.1002/alz.12088
46. Babaei P. NMDA and AMPA receptors dysregulation in alzheimer’s disease. Eur J Pharmacol (2021) 908:174310. doi: 10.1016/j.ejphar.2021.174310
47. Hansen DV, Hanson JE, Sheng M. Microglia in alzheimer’s disease. J Cell Biol (2018) 217(2):459–72. doi: 10.1083/jcb.201709069
48. Chew G, Petretto E. Transcriptional networks of microglia in alzheimer’s disease and insights into pathogenesis. Genes (Basel) (2019) 10(10):798. doi: 10.3390/genes10100798
49. Sochocka M, Diniz BS, Leszek J. Inflammatory response in the CNS: Friend or foe? Mol Neurobiol (2017) 54(10):8071–89. doi: 10.1007/s12035-016-0297-1
50. Waisman A, Liblau RS, Becher B. Innate and adaptive immune responses in the CNS. Lancet Neurol (2015) 14(9):945–55. doi: 10.1016/S1474-4422(15)00141-6
51. Zheng M, Wang P. Role of insulin receptor substance-1 modulating PI3K/Akt insulin signaling pathway in alzheimer’s disease. 3 Biotech (2021) 11(4):179. doi: 10.1007/s13205-021-02738-3
52. Sebastiao I, Candeias E, Santos MS, de Oliveira CR, Moreira PI, Duarte AI. Insulin as a bridge between type 2 diabetes and Alzheimer disease - how anti-diabetics could be a solution for dementia. Front Endocrinol (Lausanne) (2014) 5:110. doi: 10.3389/fendo.2014.00110
53. Bassil F, Fernagut PO, Bezard E, Meissner WG. Insulin, IGF-1 and GLP-1 signaling in neurodegenerative disorders: targets for disease modification? Prog Neurobiol (2014) 118:1–18. doi: 10.1016/j.pneurobio.2014.02.005
54. Steen E, Terry BM, Rivera EJ, Cannon JL, Neely TR, Tavares R, et al. Impaired insulin and insulin-like growth factor expression and signaling mechanisms in alzheimer’s disease–is this type 3 diabetes? J Alzheimers Dis (2005) 7(1):63–80. doi: 10.3233/JAD-2005-7107
55. McNay EC, Ong CT, McCrimmon RJ, Cresswell J, Bogan JS, Sherwin RS. Hippocampal memory processes are modulated by insulin and high-fat-induced insulin resistance. Neurobiol Learn Mem (2010) 93(4):546–53. doi: 10.1016/j.nlm.2010.02.002
56. Benedict C, Hallschmid M, Hatke A, Schultes B, Fehm HL, Born J, et al. Intranasal insulin improves memory in humans. Psychoneuroendocrinology (2004) 29(10):1326–34. doi: 10.1016/j.psyneuen.2004.04.003
57. Biessels GJ, Despa F. Cognitive decline and dementia in diabetes mellitus: mechanisms and clinical implications. Nat Rev Endocrinol (2018) 14(10):591–604. doi: 10.1038/s41574-018-0048-7
58. Batum K, Cinar N, Sahin S, Cakmak MA, Karsidag S. The connection between MCI and Alzheimer disease: neurocognitive clues. Turk J Med Sci (2015) 45(5):1137–40. doi: 10.3906/sag-1404-179
59. Cisternas P, Inestrosa NC. Brain glucose metabolism: Role of wnt signaling in the metabolic impairment in alzheimer’s disease. Neurosci Biobehav Rev (2017) 80:316–28. doi: 10.1016/j.neubiorev.2017.06.004
60. Poddar MK, Banerjee S, Chakraborty A, Dutta D. Metabolic disorder in alzheimer’s disease. Metab Brain Dis (2021) 36(5):781–813. doi: 10.1007/s11011-021-00673-z
61. Kulas JA, Weigel TK, Ferris HA. Insulin resistance and impaired lipid metabolism as a potential link between diabetes and alzheimer’s disease. Drug Dev Res (2020) 81(2):194–205. doi: 10.1002/ddr.21643
62. Cooper O, Hallett P, Isacson O. The role of upstream lipid and metabolic systems in alzheimer’s disease, parkinson’s disease and dementias. FEBS J (2022). doi: 10.1111/febs.16638
63. Huynh TV, Davis AA, Ulrich JD, Holtzman DM. Apolipoprotein e and alzheimer’s disease: the influence of apolipoprotein e on amyloid-beta and other amyloidogenic proteins. J Lipid Res (2017) 58(5):824–36. doi: 10.1194/jlr.R075481
64. Martin-Segura A, Ahmed T, Casadome-Perales A, Palomares-Perez I, Palomer E, Kerstens A, et al. Age-associated cholesterol reduction triggers brain insulin resistance by facilitating ligand-independent receptor activation and pathway desensitization. Aging Cell (2019) 18(3):e12932. doi: 10.1111/acel.12932
65. Butterfield DA, Halliwell B. Oxidative stress, dysfunctional glucose metabolism and Alzheimer disease. Nat Rev Neurosci (2019) 20(3):148–60. doi: 10.1038/s41583-019-0132-6
66. Thapa A, Carroll N. Dietary modulation of oxidative stress in alzheimer’s disease. Int J Mol Sci (2017) 18(7):1583. doi: 10.3390/ijms18071583
67. Ahmad W, Ijaz B, Shabbiri K, Ahmed F, Rehman S. Oxidative toxicity in diabetes and alzheimer’s disease: mechanisms behind ROS/RNS generation. J Biomed Sci (2017) 24(1):76. doi: 10.1186/s12929-017-0379-z
68. Cardoso S, Carvalho C, Correia SC, Seica RM, Moreira PI. Alzheimer’s disease: From mitochondrial perturbations to mitochondrial medicine. Brain Pathol (2016) 26(5):632–47. doi: 10.1111/bpa.12402
69. Perez Ortiz JM, Swerdlow RH. Mitochondrial dysfunction in alzheimer’s disease: Role in pathogenesis and novel therapeutic opportunities. Br J Pharmacol (2019) 176(18):3489–507. doi: 10.1111/bph.14585
70. Medala VK, Gollapelli B, Dewanjee S, Ogunmokun G, Kandimalla R, Vallamkondu J. Mitochondrial dysfunction, mitophagy, and role of dynamin-related protein 1 in alzheimer’s disease. J Neurosci Res (2021) 99(4):1120–35. doi: 10.1002/jnr.24781
71. Lim JW, Lee J, Pae AN. Mitochondrial dysfunction and alzheimer’s disease: prospects for therapeutic intervention. BMB Rep (2020) 53(1):47–55. doi: 10.5483/BMBRep.2020.53.1.279
72. Li Q, Liu Y, Sun M. Autophagy and alzheimer’s disease. Cell Mol Neurobiol (2016) 37(3):377–88. doi: 10.1007/s10571-016-0386-8
73. Zhang Z, Yang X, Song YQ, Tu J. Autophagy in alzheimer’s disease pathogenesis: Therapeutic potential and future perspectives. Ageing Res Rev (2021) 72:101464. doi: 10.1016/j.arr.2021.101464
74. Di Meco A, Curtis ME, Lauretti E, Pratico D. Autophagy dysfunction in alzheimer’s disease: Mechanistic insights and new therapeutic opportunities. Biol Psychiatry (2020) 87(9):797–807. doi: 10.1016/j.biopsych.2019.05.008
75. Lieberman OJ, Sulzer D. The synaptic autophagy cycle. J Mol Biol (2020) 432(8):2589–604. doi: 10.1016/j.jmb.2019.12.028
76. Eshraghi M, Adlimoghaddam A, Mahmoodzadeh A, Sharifzad F, Yasavoli-Sharahi H, Lorzadeh S, et al. Alzheimer’s disease pathogenesis: Role of autophagy and mitophagy focusing in microglia. Int J Mol Sci (2021) 22(7):3330. doi: 10.3390/ijms22073330
77. Hamano T, Enomoto S, Shirafuji N, Ikawa M, Yamamura O, Yen SH, et al. Autophagy and tau protein. Int J Mol Sci (2021) 22(14):7475. doi: 10.3390/ijms22147475
78. van Oostveen WM, de Lange ECM. Imaging techniques in alzheimer’s disease: A review of applications in early diagnosis and longitudinal monitoring. Int J Mol Sci (2021) 22(4):2110. doi: 10.3390/ijms22042110
79. Atri A. The alzheimer’s disease clinical spectrum: Diagnosis and management. Med Clin North Am (2019) 103(2):263–93. doi: 10.1016/j.mcna.2018.10.009
80. Chen Z, Zhong C. Decoding alzheimer’s disease from perturbed cerebral glucose metabolism: implications for diagnostic and therapeutic strategies. Prog Neurobiol (2013) 108:21–43. doi: 10.1016/j.pneurobio.2013.06.004
81. Breijyeh Z, Karaman R. Comprehensive review on alzheimer’s disease: Causes and treatment. Molecules (2020) 25(24):5789. doi: 10.3390/molecules25245789
82. Palleria C, Leporini C, Maida F, Succurro E, De Sarro G, Arturi F, et al. Potential effects of current drug therapies on cognitive impairment in patients with type 2 diabetes. Front Neuroendocrinol (2016) 42:76–92. doi: 10.1016/j.yfrne.2016.07.002
83. Akimoto H, Negishi A, Oshima S, Wakiyama H, Okita M, Horii N, et al. Antidiabetic drugs for the risk of Alzheimer disease in patients with type 2 DM using FAERS. Am J Alzheimers Dis Other Demen (2020) 35:1533317519899546. doi: 10.1177/1533317519899546
84. An J, Zhou Y, Zhang M, Xie Y, Ke S, Liu L, et al. Exenatide alleviates mitochondrial dysfunction and cognitive impairment in the 5xFAD mouse model of alzheimer’s disease. Behav Brain Res (2019) 370:111932. doi: 10.1016/j.bbr.2019.111932
85. Wang X, Wang L, Jiang R, Xu Y, Zhao X, Li Y. Exendin-4 antagonizes Abeta1-42-induced attenuation of spatial learning and memory ability. Exp Ther Med (2016) 12(5):2885–92. doi: 10.3892/etm.2016.3742
86. Patzelt C, Schug G. The major proglucagon fragment: an abundant islet protein and secretory product. FEBS Lett (1981) 129(1):127–30. doi: 10.1016/0014-5793(81)80772-7
87. Bell GI, Santerre RF, Mullenbach GT. Hamster preproglucagon contains the sequence of glucagon and two related peptides. Nature (1983) 302(5910):716–8. doi: 10.1038/302716a0
88. Holst JJ, Orskov C, Nielsen OV, Schwartz TW. Truncated glucagon-like peptide I, an insulin-releasing hormone from the distal gut. FEBS Lett (1987) 211(2):169–74. doi: 10.1016/0014-5793(87)81430-8
89. Muller TD, Finan B, Bloom SR, D’Alessio D, Drucker DJ, Flatt PR, et al. Glucagon-like peptide 1 (GLP-1). Mol Metab (2019) 30:72–130. doi: 10.1016/j.molmet.2019.09.010
90. Mojsov S, Weir GC, Habener JF. Insulinotropin: glucagon-like peptide I (7-37) co-encoded in the glucagon gene is a potent stimulator of insulin release in the perfused rat pancreas. J Clin Invest (1987) 79(2):616–9. doi: 10.1172/JCI112855
91. Kreymann B, Williams G, Ghatei MA, Bloom SR. Glucagon-like peptide-1 7-36: a physiological incretin in man. Lancet (1987) 2(8571):1300–4. doi: 10.1016/S0140-6736(87)91194-9
92. Sharma D, Verma S, Vaidya S, Kalia K, Tiwari V. Recent updates on GLP-1 agonists: Current advancements & challenges. BioMed Pharmacother (2018) 108:952–62. doi: 10.1016/j.biopha.2018.08.088
93. Yildirim Simsir I, Soyaltin UE, Cetinkalp S. Glucagon like peptide-1 (GLP-1) likes alzheimer’s disease. Diabetes Metab Syndr (2018) 12(3):469–75. doi: 10.1016/j.dsx.2018.03.002
94. Orskov C, Wettergren A, Holst JJ. Biological effects and metabolic rates of glucagonlike peptide-1 7-36 amide and glucagonlike peptide-1 7-37 in healthy subjects are indistinguishable. Diabetes (1993) 42(5):658–61. doi: 10.2337/diab.42.5.658
95. Reed J, Bain S, Kanamarlapudi V. Recent advances in understanding the role of glucagon-like peptide 1. F1000Res (2020) 9:F1000 Faculty Rev-239. doi: 10.12688/f1000research.20602.1
96. Drucker DJ. Mechanisms of action and therapeutic application of glucagon-like peptide-1. Cell Metab (2018) 27(4):740–56. doi: 10.1016/j.cmet.2018.03.001
97. Holscher C. Insulin signaling impairment in the brain as a risk factor in alzheimer’s disease. Front Aging Neurosci (2019) 11:88. doi: 10.3389/fnagi.2019.00088
98. Deacon CF, Johnsen AH, Holst JJ. Degradation of glucagon-like peptide-1 by human plasma in vitro yields an n-terminally truncated peptide that is a major endogenous metabolite in vivo. J Clin Endocrinol Metab (1995) 80(3):952–7. doi: 10.1210/jcem.80.3.7883856
99. Kieffer TJ, McIntosh CH, Pederson RA. Degradation of glucose-dependent insulinotropic polypeptide and truncated glucagon-like peptide 1 in vitro and in vivo by dipeptidyl peptidase IV. Endocrinology (1995) 136(8):3585–96. doi: 10.1210/endo.136.8.7628397
100. Vahl TP, Paty BW, Fuller BD, Prigeon RL, D’Alessio DA. Effects of GLP-1-(7-36)NH2, GLP-1-(7-37), and GLP-1- (9-36)NH2 on intravenous glucose tolerance and glucose-induced insulin secretion in healthy humans. J Clin Endocrinol Metab (2003) 88(4):1772–9. doi: 10.1210/jc.2002-021479
101. Zander M, Madsbad S, Deacon CF, Holst JJ. The metabolite generated by dipeptidyl-peptidase 4 metabolism of glucagon-like peptide-1 has no influence on plasma glucose levels in patients with type 2 diabetes. Diabetologia (2006) 49(2):369–74. doi: 10.1007/s00125-005-0098-y
102. Ferrari F, Moretti A, Villa RF. Incretin-based drugs as potential therapy for neurodegenerative diseases: current status and perspectives. Pharmacol Ther (2022) 239:108277. doi: 10.1016/j.pharmthera.2022.108277
103. Lin Z, Sur S, Liu P, Li Y, Jiang D, Hou X, et al. Blood-brain barrier breakdown in relationship to Alzheimer and vascular disease. Ann Neurol (2021) 90(2):227–38. doi: 10.1002/ana.26134
104. Chen S, Zhou M, Sun J, Guo A, Fernando RL, Chen Y, et al. DPP-4 inhibitor improves learning and memory deficits and AD-like neurodegeneration by modulating the GLP-1 signaling. Neuropharmacology (2019) 157:107668. doi: 10.1016/j.neuropharm.2019.107668
105. Siddiqui N, Ali J, Parvez S, Zameer S, Najmi AK, Akhtar M. Linagliptin, a DPP-4 inhibitor, ameliorates abeta (1-42) peptides induced neurodegeneration and brain insulin resistance (BIR) via insulin receptor substrate-1 (IRS-1) in rat model of alzheimer’s disease. Neuropharmacology (2021) 195:108662. doi: 10.1016/j.neuropharm.2021.108662
106. Alvarez E, Martinez MD, Roncero I, Chowen JA, Garcia-Cuartero B, Gispert JD, et al. The expression of GLP-1 receptor mRNA and protein allows the effect of GLP-1 on glucose metabolism in the human hypothalamus and brainstem. J Neurochem (2005) 92(4):798–806. doi: 10.1111/j.1471-4159.2004.02914.x
107. Zhang L, Zhang W, Tian X. The pleiotropic of GLP-1/GLP-1R axis in central nervous system diseases. Int J Neurosci (2021) p:1–38. doi: 10.1080/00207454.2021.1924707
108. Holst JJ. The incretin system in healthy humans: The role of GIP and GLP-1. Metabolism (2019) 96:46–55. doi: 10.1016/j.metabol.2019.04.014
109. Rajeev SP, Wilding J. GLP-1 as a target for therapeutic intervention. Curr Opin Pharmacol (2016) 31:44–9. doi: 10.1016/j.coph.2016.08.005
110. Graaf C, Donnelly D, Wootten D, Lau J, Sexton PM, Miller LJ, et al. Glucagon-like peptide-1 and its class b G protein-coupled receptors: A long march to therapeutic successes. Pharmacol Rev (2016) 68(4):954–1013. doi: 10.1124/pr.115.011395
111. Bullock BP, Heller RS, Habener JF. Tissue distribution of messenger ribonucleic acid encoding the rat glucagon-like peptide-1 receptor. Endocrinology (1996) 137(7):2968–78. doi: 10.1210/endo.137.7.8770921
112. Wei Y, Mojsov S. Tissue-specific expression of the human receptor for glucagon-like peptide-I: brain, heart and pancreatic forms have the same deduced amino acid sequences. FEBS Lett (1995) 358(3):219–24. doi: 10.1016/0014-5793(94)01430-9
113. Rowlands J, Heng J, Newsholme P, Carlessi R. Pleiotropic effects of GLP-1 and analogs on cell signaling, metabolism, and function. Front Endocrinol (Lausanne) (2018) 9:672. doi: 10.3389/fendo.2018.00672
114. Koole C, Pabreja K, Savage Emilia E, Wootten D, Furness Sebastian GB, Miller Laurence J, et al. Recent advances in understanding GLP-1R (glucagon-like peptide-1 receptor) function. Biochem Soc Trans (2013) 41(1):172–9. doi: 10.1042/BST20120236
115. MacDonald PE, Wang X, Xia F, El-kholy W, Targonsky ED, Tsushima RG, et al. Antagonism of rat beta-cell voltage-dependent k+ currents by exendin 4 requires dual activation of the cAMP/protein kinase a and phosphatidylinositol 3-kinase signaling pathways. J Biol Chem (2003) 278(52):52446–53. doi: 10.1074/jbc.M307612200
116. Batista AF, Bodart-Santos V, De Felice FG, Ferreira ST. Neuroprotective actions of glucagon-like peptide-1 (GLP-1) analogues in alzheimer’s and parkinson’s diseases. CNS Drugs (2019) 33(3):209–23. doi: 10.1007/s40263-018-0593-6
117. During MJ, Cao L, Zuzga DS, Francis JS, Fitzsimons HL, Jiao X, et al. Glucagon-like peptide-1 receptor is involved in learning and neuroprotection. Nat Med (2003) 9(9):1173–9. doi: 10.1038/nm919
118. Abbas T, Faivre E, Holscher C. Impairment of synaptic plasticity and memory formation in GLP-1 receptor KO mice: Interaction between type 2 diabetes and alzheimer’s disease. Behav Brain Res (2009) 205(1):265–71. doi: 10.1016/j.bbr.2009.06.035
119. Ahren B. GLP-1-based therapy of type 2 diabetes: GLP-1 mimetics and DPP-IV inhibitors. Curr Diabetes Rep (2007) 7(5):340–7. doi: 10.1007/s11892-007-0056-9
120. Yoo BK, Triller DM, Yoo DJ. Formulary forum: Exenatide: A new option for the treatment of type 2 diabetes. Ann Pharmacotherapy (2016) 40(10):1777–84. doi: 10.1345/aph.1H060
121. Heimburger SM, Bronden A, Johansen NJ, Dejgaard TF, Vilsboll T, Knop FK. The efficacy and safety of exenatide once weekly in patients with type 2 diabetes. Expert Opin Pharmacother (2019) 20(5):501–10. doi: 10.1080/14656566.2019.1571040
122. Genovese S, Mannucci E, Ceriello A. A review of the long-term efficacy, tolerability, and safety of exenatide once weekly for type 2 diabetes. Adv Ther (2017) 34(8):1791–814. doi: 10.1007/s12325-017-0499-6
123. Kalra S, Bhattacharya S, Kapoor N. Contemporary classification of glucagon-like peptide 1 receptor agonists (GLP1RAs). Diabetes Ther (2021) 12(8):2133–47. doi: 10.1007/s13300-021-01113-y
124. Sfairopoulos D, Liatis S, Tigas S, Liberopoulos E. Clinical pharmacology of glucagon-like peptide-1 receptor agonists. Hormones (Athens) (2018) 17(3):333–50. doi: 10.1007/s42000-018-0038-0
125. Hunter K, Holscher C. Drugs developed to treat diabetes, liraglutide and lixisenatide, cross the blood brain barrier and enhance neurogenesis. BMC Neurosci (2012) 13:33. doi: 10.1186/1471-2202-13-33
126. Scott LJ. Lixisenatide: a review of its use in patients with type 2 diabetes mellitus. BioDrugs (2013) 27(5):509–23. doi: 10.1007/s40259-013-0057-y
127. McCarty D, Coleman M, Boland CL. Lixisenatide: A new daily GLP-1 agonist for type 2 diabetes management. Ann Pharmacother (2017) 51(5):401–9. doi: 10.1177/1060028017689878
128. Knudsen LB, Lau J. The discovery and development of liraglutide and semaglutide. Front Endocrinol (Lausanne) (2019) 10:155. doi: 10.3389/fendo.2019.00155
129. Duarte AI, Candeias E, Correia SC, Santos RX, Carvalho C, Cardoso S, et al. Crosstalk between diabetes and brain: glucagon-like peptide-1 mimetics as a promising therapy against neurodegeneration. Biochim Biophys Acta (2013) 1832(4):527–41. doi: 10.1016/j.bbadis.2013.01.008
130. Jacobsen LV, Flint A, Olsen AK, Ingwersen SH. Liraglutide in type 2 diabetes mellitus: Clinical pharmacokinetics and pharmacodynamics. Clin Pharmacokinet (2016) 55(6):657–72. doi: 10.1007/s40262-015-0343-6
131. Knudsen LB, Nielsen PF, Huusfeldt PO, Johansen NL, Madsen K, Pedersen FZ, et al. Potent derivatives of glucagon-like peptide-1 with pharmacokinetic properties suitable for once daily administration. J Med Chem (2000) 43(9):1664–9. doi: 10.1021/jm9909645
132. Jendle J, Grunberger G, Blevins T, Giorgino F, Hietpas RT, Botros FT. Efficacy and safety of dulaglutide in the treatment of type 2 diabetes: a comprehensive review of the dulaglutide clinical data focusing on the AWARD phase 3 clinical trial program. Diabetes Metab Res Rev (2016) 32(8):776–90. doi: 10.1002/dmrr.2810
133. Glaesner W, Vick AM, Millican R, Ellis B, Tschang SH, Tian Y, et al. Engineering and characterization of the long-acting glucagon-like peptide-1 analogue LY2189265, an fc fusion protein. Diabetes Metab Res Rev (2010) 26(4):287–96. doi: 10.1002/dmrr.1080
134. Gerstein HC, Colhoun HM, Dagenais GR, Diaz R, Lakshmanan M, Pais P, et al. Dulaglutide and renal outcomes in type 2 diabetes: an exploratory analysis of the REWIND randomised, placebo-controlled trial. Lancet (2019) 394(10193):131–8. doi: 10.1016/S0140-6736(19)31150-X
135. Honigberg MC, Chang LS, McGuire DK, Plutzky J, Aroda VR, Vaduganathan M. Use of glucagon-like peptide-1 receptor agonists in patients with type 2 diabetes and cardiovascular disease: A review. JAMA Cardiol (2020) 5(10):1182–90. doi: 10.1001/jamacardio.2020.1966
136. Alorfi NM, Algarni AS. Clinical impact of semaglutide, a glucagon-like peptide-1 receptor agonist, on obesity management: A review. Clin Pharmacol (2022) 14:61–7. doi: 10.2147/CPAA.S374741
137. Mahapatra MK, Karuppasamy M, Sahoo BM. Therapeutic potential of semaglutide, a newer GLP-1 receptor agonist, in abating obesity, non-alcoholic steatohepatitis and neurodegenerative diseases: A narrative review. Pharm Res (2022) 39(6):1233–48. doi: 10.1007/s11095-022-03302-1
138. Yaribeygi H, Rashidy-Pour A, Atkin SL, Jamialahmadi T, Sahebkar A. GLP-1 mimetics and cognition. Life Sci (2021) 264:118645. doi: 10.1016/j.lfs.2020.118645
139. Lee CH, Jeon SJ, Cho KS, Moon E, Sapkota A, Jun HS, et al. Activation of glucagon-like peptide-1 receptor promotes neuroprotection in experimental autoimmune encephalomyelitis by reducing neuroinflammatory responses. Mol Neurobiol (2018) 55(4):3007–20. doi: 10.1007/s12035-017-0550-2
140. Solmaz V, Cinar BP, Yigitturk G, Cavusoglu T, Taskiran D, Erbas O. Exenatide reduces TNF-alpha expression and improves hippocampal neuron numbers and memory in streptozotocin treated rats. Eur J Pharmacol (2015) 765:482–7. doi: 10.1016/j.ejphar.2015.09.024
141. Bomba M, Granzotto A, Castelli V, Onofrj M, Lattanzio R, Cimini A, et al. Exenatide reverts the high-Fat-Diet-Induced impairment of BDNF signaling and inflammatory response in an animal model of alzheimer’s disease. J Alzheimers Dis (2019) 70(3):793–810. doi: 10.3233/JAD-190237
142. Parthsarathy V, Holscher C. The type 2 diabetes drug liraglutide reduces chronic inflammation induced by irradiation in the mouse brain. Eur J Pharmacol (2013) 700(1-3):42–50. doi: 10.1016/j.ejphar.2012.12.012
143. McClean PL, Jalewa J, Holscher C. Prophylactic liraglutide treatment prevents amyloid plaque deposition, chronic inflammation and memory impairment in APP/PS1 mice. Behav Brain Res (2015) 293:96–106. doi: 10.1016/j.bbr.2015.07.024
144. Cai HY, Yang JT, Wang ZJ, Zhang J, Yang W, Wu MN, et al. Lixisenatide reduces amyloid plaques, neurofibrillary tangles and neuroinflammation in an APP/PS1/tau mouse model of alzheimer’s disease. Biochem Biophys Res Commun (2018) 495(1):1034–40. doi: 10.1016/j.bbrc.2017.11.114
145. Yoon G, Kim YK, Song J. Glucagon-like peptide-1 suppresses neuroinflammation and improves neural structure. Pharmacol Res (2020) 152:104615. doi: 10.1016/j.phrs.2019.104615
146. Duarte AI, Candeias E, Alves IN, Mena D, Silva DF, Machado NJ, et al. Liraglutide protects against brain amyloid-beta1-42 accumulation in female mice with early alzheimer’s disease-like pathology by partially rescuing Oxidative/Nitrosative stress and inflammation. Int J Mol Sci (2020) 21(5):1746. doi: 10.3390/ijms21051746
147. Paladugu L, Gharaibeh A, Kolli N, Learman C, Hall TC, Li L, et al. Liraglutide has anti-inflammatory and anti-amyloid properties in streptozotocin-induced and 5xFAD mouse models of alzheimer’s disease. Int J Mol Sci (2021) 22(2):860. doi: 10.3390/ijms22020860
148. Zhang M, Wu Y, Gao R, Chen X, Chen R, Chen Z. Glucagon-like peptide-1 analogs mitigate neuroinflammation in alzheimer’s disease by suppressing NLRP2 activation in astrocytes. Mol Cell Endocrinol (2022) 542:111529. doi: 10.1016/j.mce.2021.111529
149. Li L, Zhang ZF, Holscher C, Gao C, Jiang YH, Liu YZ. (Val(8)) glucagon-like peptide-1 prevents tau hyperphosphorylation, impairment of spatial learning and ultra-structural cellular damage induced by streptozotocin in rat brains. Eur J Pharmacol (2012) 674(2-3):280–6. doi: 10.1016/j.ejphar.2011.11.005
150. Zhou M, Chen S, Peng P, Gu Z, Yu J, Zhao G, et al. Dulaglutide ameliorates STZ induced AD-like impairment of learning and memory ability by modulating hyperphosphorylation of tau and NFs through GSK3beta. Biochem Biophys Res Commun (2019) 511(1):154–60. doi: 10.1016/j.bbrc.2019.01.103
151. Li Y, Duffy KB, Ottinger MA, Ray B, Bailey JA, Holloway HW, et al. GLP-1 receptor stimulation reduces amyloid-β peptide accumulation and cytotoxicity in cellular and animal models of alzheimer’s disease. J Alzheimer’s Dis (2010) 19(4):1205–19. doi: 10.3233/JAD-2010-1314
152. Perry T, Lahiri DK, Sambamurti K, Chen D, Mattson MP, Egan JM, et al. Glucagon-like peptide-1 decreases endogenous amyloid-beta peptide (Abeta) levels and protects hippocampal neurons from death induced by abeta and iron. J Neurosci Res (2003) 72(5):603–12. doi: 10.1002/jnr.10611
153. Chen S, An FM, Yin L, Liu AR, Yin DK, Yao WB, et al. Glucagon-like peptide-1 protects hippocampal neurons against advanced glycation end product-induced tau hyperphosphorylation. Neuroscience (2014) 256:137–46. doi: 10.1016/j.neuroscience.2013.10.038
154. Yang Y, Ma D, Xu W, Chen F, Du T, Yue W, et al. Exendin-4 reduces tau hyperphosphorylation in type 2 diabetic rats via increasing brain insulin level. Mol Cell Neurosci (2016) 70:68–75. doi: 10.1016/j.mcn.2015.10.005
155. Xu W, Yang Y, Yuan G, Zhu W, Ma D, Hu S. Exendin-4, a glucagon-like peptide-1 receptor agonist, reduces Alzheimer disease-associated tau hyperphosphorylation in the hippocampus of rats with type 2 diabetes. J Investig Med (2015) 63(2):267–72. doi: 10.1097/JIM.0000000000000129
156. King MR, Anderson NJ, Deciu M, Guernsey LS, Cundiff M, Hajizadeh S, et al. Insulin deficiency, but not resistance, exaggerates cognitive deficits in transgenic mice expressing human amyloid and tau proteins. reversal by exendin-4 treatment. J Neurosci Res (2020) 98(11):2357–69. doi: 10.1002/jnr.24706
157. McClean PL, Holscher C. Lixisenatide, a drug developed to treat type 2 diabetes, shows neuroprotective effects in a mouse model of alzheimer’s disease. Neuropharmacology (2014) 86:241–58. doi: 10.1016/j.neuropharm.2014.07.015
158. McClean PL, Hölscher C. Liraglutide can reverse memory impairment, synaptic loss and reduce plaque load in aged APP/PS1 mice, a model of alzheimer’s disease. Neuropharmacology (2014) 76:57–67. doi: 10.1016/j.neuropharm.2013.08.005
159. Holubova M, Hruba L, Popelova A, Bencze M, Prazienkova V, Gengler S, et al. Liraglutide and a lipidized analog of prolactin-releasing peptide show neuroprotective effects in a mouse model of beta-amyloid pathology. Neuropharmacology (2019) 144:377–87. doi: 10.1016/j.neuropharm.2018.11.002
160. Kong J, Wan L, Wang Y, Zhang H, Zhang W. Liraglutide attenuates Abeta42 generation in APPswe/SH-SY5Y cells through the regulation of autophagy. Neuropsychiatr Dis Treat (2020) 16:1817–25. doi: 10.2147/NDT.S260160
161. Ma DL, Chen FQ, Xu WJ, Yue WZ, Yuan G, Yang Y. Early intervention with glucagon-like peptide 1 analog liraglutide prevents tau hyperphosphorylation in diabetic db/db mice. J Neurochem (2015) 135(2):301–8. doi: 10.1111/jnc.13248
162. Zhang Y, Xie JZ, Xu XY, Hu J, Xu T, Jin S, et al. Liraglutide ameliorates hyperhomocysteinemia-induced Alzheimer-like pathology and memory deficits in rats via multi-molecular targeting. Neurosci Bull (2019) 35(4):724–34. doi: 10.1007/s12264-018-00336-7
163. Hansen HH, Barkholt P, Fabricius K, Jelsing J, Terwel D, Pyke C, et al. The GLP-1 receptor agonist liraglutide reduces pathology-specific tau phosphorylation and improves motor function in a transgenic hTauP301L mouse model of tauopathy. Brain Res (2016) 1634:158–70. doi: 10.1016/j.brainres.2015.12.052
164. Qi L, Ke L, Liu X, Liao L, Ke S, Liu X, et al. Subcutaneous administration of liraglutide ameliorates learning and memory impairment by modulating tau hyperphosphorylation via the glycogen synthase kinase-3beta pathway in an amyloid beta protein induced alzheimer disease mouse model. Eur J Pharmacol (2016) 783:23–32. doi: 10.1016/j.ejphar.2016.04.052
165. Holst JJ, Burcelin R, Nathanson E. Neuroprotective properties of GLP-1: theoretical and practical applications. Curr Med Res Opin (2011) 27(3):547–58. doi: 10.1185/03007995.2010.549466
166. Hamilton A, Patterson S, Porter D, Gault VA, Holscher C. Novel GLP-1 mimetics developed to treat type 2 diabetes promote progenitor cell proliferation in the brain. J Neurosci Res (2011) 89(4):481–9. doi: 10.1002/jnr.22565
167. McGovern SF, Hunter K, Holscher C. Effects of the glucagon-like polypeptide-1 analogue (Val8)GLP-1 on learning, progenitor cell proliferation and neurogenesis in the C57B/16 mouse brain. Brain Res (2012) 1473:204–13. doi: 10.1016/j.brainres.2012.07.029
168. Parthsarathy V, Holscher C. Chronic treatment with the GLP1 analogue liraglutide increases cell proliferation and differentiation into neurons in an AD mouse model. PloS One (2013) 8(3):e58784. doi: 10.1371/journal.pone.0058784
169. Hansen HH, Fabricius K, Barkholt P, Niehoff ML, Morley JE, Jelsing J, et al. The GLP-1 receptor agonist liraglutide improves memory function and increases hippocampal CA1 neuronal numbers in a senescence-accelerated mouse model of alzheimer’s disease. J Alzheimers Dis (2015) 46(4):877–88. doi: 10.3233/JAD-143090
170. Perez-Cruz C, Nolte MW, van Gaalen MM, Rustay NR, Termont A, Tanghe A, et al. Reduced spine density in specific regions of CA1 pyramidal neurons in two transgenic mouse models of alzheimer’s disease. J Neurosci (2011) 31(10):3926–34. doi: 10.1523/JNEUROSCI.6142-10.2011
171. Gault VA, Holscher C. GLP-1 agonists facilitate hippocampal LTP and reverse the impairment of LTP induced by beta-amyloid. Eur J Pharmacol (2008) 587(1-3):112–7. doi: 10.1016/j.ejphar.2008.03.025
172. Gengler S, McClean PL, McCurtin R, Gault VA, Holscher C. Val(8)GLP-1 rescues synaptic plasticity and reduces dense core plaques in APP/PS1 mice. Neurobiol Aging (2012) 33(2):265–76. doi: 10.1016/j.neurobiolaging.2010.02.014
173. Wang XH, Yang W, Holscher C, Wang ZJ, Cai HY, Li QS, et al. Val(8)-GLP-1 remodels synaptic activity and intracellular calcium homeostasis impaired by amyloid beta peptide in rats. J Neurosci Res (2013) 91(4):568–77. doi: 10.1002/jnr.23181
174. McClean PL, Gault VA, Harriott P, Holscher C. Glucagon-like peptide-1 analogues enhance synaptic plasticity in the brain: a link between diabetes and alzheimer’s disease. Eur J Pharmacol (2010) 630(1-3):158–62. doi: 10.1016/j.ejphar.2009.12.023
175. McClean PL, Parthsarathy V, Faivre E, Holscher C. The diabetes drug liraglutide prevents degenerative processes in a mouse model of alzheimer’s disease. J Neurosci (2011) 31(17):6587–94. doi: 10.1523/JNEUROSCI.0529-11.2011
176. Batista AF, Forny-Germano L, Clarke JR, Lyra E Silva NM, Brito-Moreira J, Boehnke SE, et al. The diabetes drug liraglutide reverses cognitive impairment in mice and attenuates insulin receptor and synaptic pathology in a non-human primate model of alzheimer’s disease. J Pathol (2018) 245(1):85–100. doi: 10.1002/path.5056
177. Han WN, Holscher C, Yuan L, Yang W, Wang XH, Wu MN, et al. Liraglutide protects against amyloid-beta protein-induced impairment of spatial learning and memory in rats. Neurobiol Aging (2013) 34(2):576–88. doi: 10.1016/j.neurobiolaging.2012.04.009
178. Ohtake N, Saito M, Eto M, Seki K. Exendin-4 promotes the membrane trafficking of the AMPA receptor GluR1 subunit and ADAM10 in the mouse neocortex. Regul Pept (2014) 190-191:1–11. doi: 10.1016/j.regpep.2014.04.003
179. Cai HY, Holscher C, Yue XH, Zhang SX, Wang XH, Qiao F, et al. Lixisenatide rescues spatial memory and synaptic plasticity from amyloid beta protein-induced impairments in rats. Neuroscience (2014) 277:6–13. doi: 10.1016/j.neuroscience.2014.02.022
180. Zhang SX, Cai HY, Ma XW, Yuan L, Zhang J, Wang ZJ, et al. GLP-1 analogue CJC-1131 prevents amyloid β protein-induced impirments of spatial memory and synaptic plasticity in rats. Behav Brain Res (2017) 326:237–43. doi: 10.1016/j.bbr.2017.03.018
181. Tobore TO. On the central role of mitochondria dysfunction and oxidative stress in alzheimer’s disease. Neurol Sci (2019) 40(8):1527–40. doi: 10.1007/s10072-019-03863-x
182. Chen S, Liu AR, An FM, Yao WB, Gao XD. Amelioration of neurodegenerative changes in cellular and rat models of diabetes-related alzheimer’s disease by exendin-4. Age (Dordr) (2012) 34(5):1211–24. doi: 10.1007/s11357-011-9303-8
183. Yoshino Y, Ishisaka M, Tsujii S, Shimazawa M, Hara H. Glucagon-like peptide-1 protects the murine hippocampus against stressors via akt and ERK1/2 signaling. Biochem Biophys Res Commun (2015) 458(2):274–9. doi: 10.1016/j.bbrc.2015.01.098
184. Spielman LJ, Gibson DL, Klegeris A. Incretin hormones regulate microglia oxidative stress, survival and expression of trophic factors. Eur J Cell Biol (2017) 96(3):240–53. doi: 10.1016/j.ejcb.2017.03.004
185. Zheng C, Zhou M, Sun J, Xiong H, Peng P, Gu Z, et al. The protective effects of liraglutide on AD-like neurodegeneration induced by oxidative stress in human neuroblastoma SH-SY5Y cells. Chem Biol Interact (2019) 310:108688. doi: 10.1016/j.cbi.2019.06.001
186. Salles GN, Calio ML, Holscher C, Pacheco-Soares C, Porcionatto M, Lobo AO. Neuroprotective and restorative properties of the GLP-1/GIP dual agonist DA-JC1 compared with a GLP-1 single agonist in alzheimer’s disease. Neuropharmacology (2020) 162:107813. doi: 10.1016/j.neuropharm.2019.107813
187. Ke J, Tian Q, Xu Q, Fu Z, Fu Q. Mitochondrial dysfunction: A potential target for alzheimer’s disease intervention and treatment. Drug Discovery Today (2021) 26(8):1991–2002. doi: 10.1016/j.drudis.2021.04.025
188. Xie Y, Zheng J, Li S, Li H, Zhou Y, Zheng W, et al. GLP-1 improves the neuronal supportive ability of astrocytes in alzheimer’s disease by regulating mitochondrial dysfunction via the cAMP/PKA pathway. Biochem Pharmacol (2021) 188:114578. doi: 10.1016/j.bcp.2021.114578
189. Garabadu D, Verma J. Exendin-4 attenuates brain mitochondrial toxicity through PI3K/Akt-dependent pathway in amyloid beta (1-42)-induced cognitive deficit rats. Neurochem Int (2019) 128:39–49. doi: 10.1016/j.neuint.2019.04.006
190. Engidawork E, Gulesserian T, Seidl R, Cairns N, Lubec G. Expression of apoptosis related proteins in brains of patients with alzheimer’s disease. Neurosci Lett (2001) 303(2):79–82. doi: 10.1016/S0304-3940(01)01618-4
191. Perry T, Haughey NJ, Mattson MP, Egan JM, Greig NH. Protection and reversal of excitotoxic neuronal damage by glucagon-like peptide-1 and exendin-4. J Pharmacol Exp Ther (2002) 302(3):881–8. doi: 10.1124/jpet.102.037481
192. Qiu C, Wang YP, Pan XD, Liu XY, Chen Z, Liu LB. Exendin-4 protects Abeta(1-42) oligomer-induced PC12 cell apoptosis. Am J Transl Res (2016) 8(8):3540–8.
193. Kimura R, Okouchi M, Fujioka H, Ichiyanagi A, Ryuge F, Mizuno T, et al. Glucagon-like peptide-1 (GLP-1) protects against methylglyoxal-induced PC12 cell apoptosis through the PI3K/Akt/mTOR/GCLc/redox signaling pathway. Neuroscience (2009) 162(4):1212–9. doi: 10.1016/j.neuroscience.2009.05.025
194. Zhang H, Song B, Zhu W, Liu L, He X, Wang Z, et al. Glucagon-like peptide-1 attenuated carboxymethyl lysine induced neuronal apoptosis via peroxisome proliferation activated receptor-gamma. Aging (Albany NY) (2021) 13(14):19013–27. doi: 10.18632/aging.203351
195. Chen S, Yin L, Xu Z, An FM, Liu AR, Wang Y, et al. Inhibiting receptor for advanced glycation end product (AGE) and oxidative stress involved in the protective effect mediated by glucagon-like peptide-1 receptor on AGE induced neuronal apoptosis. Neurosci Lett (2016) 612:193–8. doi: 10.1016/j.neulet.2015.12.007
196. Sharma MK, Jalewa J, Holscher C. Neuroprotective and anti-apoptotic effects of liraglutide on SH-SY5Y cells exposed to methylglyoxal stress. J Neurochem (2014) 128(3):459–71. doi: 10.1111/jnc.12469
197. Liu X-Y, Wang L-X, Chen Z, Liu L-B. Liraglutide prevents beta-amyloid-induced neurotoxicity in SH-SY5Y cells via a PI3K-dependent signaling pathway. Neurological Res (2016) 38(4):313–9. doi: 10.1080/01616412.2016.1145914
198. Panagaki T, Michael M, Holscher C. Liraglutide restores chronic ER stress, autophagy impairments and apoptotic signalling in SH-SY5Y cells. Sci Rep (2017) 7(1):16158. doi: 10.1038/s41598-017-16488-x
199. Bianchi M, D’Oria V, Braghini MR, Petrini S, Manco M. Liraglutide treatment ameliorates neurotoxicity induced by stable silencing of Pin1. Int J Mol Sci (2019) 20(20):5064. doi: 10.3390/ijms20205064
200. Chang YF, Zhang D, Hu WM, Liu DX, Li L. Semaglutide-mediated protection against abeta correlated with enhancement of autophagy and inhibition of apotosis. J Clin Neurosci (2020) 81:234–9. doi: 10.1016/j.jocn.2020.09.054
201. Menzies FM, Fleming A, Rubinsztein DC. Compromised autophagy and neurodegenerative diseases. Nat Rev Neurosci (2015) 16(6):345–57. doi: 10.1038/nrn3961
202. Candeias E, Sebastião I, Cardoso S, Carvalho C, Santos MS, Oliveira CR, et al. Brain GLP-1/IGF-1 signaling and autophagy mediate exendin-4 protection against apoptosis in type 2 diabetic rats. Mol Neurobiol (2018) 55(5):4030–50. doi: 10.1007/s12035-017-0622-3
203. Bomba M, Granzotto A, Castelli V, Massetti N, Silvestri E, Canzoniero LMT, et al. Exenatide exerts cognitive effects by modulating the BDNF-TrkB neurotrophic axis in adult mice. Neurobiol Aging (2018) 64:33–43. doi: 10.1016/j.neurobiolaging.2017.12.009
204. Gumuslu E, Cine N, Ertan M, Mutlu O, Komsuoglu Celikyurt I, Ulak G. Exenatide upregulates gene expression of glucagon-like peptide-1 receptor and nerve growth factor in streptozotocin/nicotinamide-induced diabetic mice. Fundam Clin Pharmacol (2018) 32(2):174–80. doi: 10.1111/fcp.12329
205. Wang X, Wang L, Jiang R, Yuan Y, Yu Q, Li Y. Exendin-4 antagonizes Abeta1-42-induced suppression of long-term potentiation by regulating intracellular calcium homeostasis in rat hippocampal neurons. Brain Res (2015) 1627:101–8. doi: 10.1016/j.brainres.2015.09.015
206. Cai HY, Wang ZJ, Holscher C, Yuan L, Zhang J, Sun P, et al. Lixisenatide attenuates the detrimental effects of amyloid beta protein on spatial working memory and hippocampal neurons in rats. Behav Brain Res (2017) 318:28–35. doi: 10.1016/j.bbr.2016.10.033
207. Bomba M, Ciavardelli D, Silvestri E, Canzoniero LM, Lattanzio R, Chiappini P, et al. Exenatide promotes cognitive enhancement and positive brain metabolic changes in PS1-KI mice but has no effects in 3xTg-AD animals. Cell Death Dis (2013) 4:e612. doi: 10.1038/cddis.2013.139
208. Zheng J, Xie Y, Ren L, Qi L, Wu L, Pan X, et al. GLP-1 improves the supportive ability of astrocytes to neurons by promoting aerobic glycolysis in alzheimer’s disease. Mol Metab (2021) 47:101180. doi: 10.1016/j.molmet.2021.101180
209. Kelly P, McClean PL, Ackermann M, Konerding MA, Holscher C, Mitchell CA. Restoration of cerebral and systemic microvascular architecture in APP/PS1 transgenic mice following treatment with liraglutide. Microcirculation (2015) 22(2):133–45. doi: 10.1111/micc.12186
210. Carranza-Naval MJ, Del Marco A, Hierro-Bujalance C, Alves-Martinez P, Infante-Garcia C, Vargas-Soria M, et al. Liraglutide reduces vascular damage, neuronal loss, and cognitive impairment in a mixed murine model of alzheimer’s disease and type 2 diabetes. Front Aging Neurosci (2021) 13:741923. doi: 10.3389/fnagi.2021.741923
211. Qi L, Chen Z, Wang Y, Liu X, Liu X, Ke L, et al. Subcutaneous liraglutide ameliorates methylglyoxal-induced Alzheimer-like tau pathology and cognitive impairment by modulating tau hyperphosphorylation and glycogen synthase kinase-3beta. Am J Transl Res (2017) 9(2):247–60.
212. Li H, Cao L, Ren Y, Jiang Y, Xie W, Li D. GLP-1 receptor regulates cell growth through regulating IDE expression level in Abeta1-42-treated PC12 cells. Biosci Rep (2018) 38(4):BSR20171284. doi: 10.1042/BSR20171284
213. Robinson A, Lubitz I, Atrakchi-Baranes D, Licht-Murava A, Katsel P, Leroith D, et al. Combination of insulin with a GLP1 agonist is associated with better memory and normal expression of insulin receptor pathway genes in a mouse model of alzheimer’s disease. J Mol Neurosci (2019) 67(4):504–10. doi: 10.1007/s12031-019-1257-9
214. Long-Smith CM, Manning S, McClean PL, Coakley MF, O’Halloran DJ, Holscher C, et al. The diabetes drug liraglutide ameliorates aberrant insulin receptor localisation and signalling in parallel with decreasing both amyloid-beta plaque and glial pathology in a mouse model of alzheimer’s disease. Neuromolecular Med (2013) 15(1):102–14. doi: 10.1007/s12017-012-8199-5
215. Jantrapirom S, Nimlamool W, Chattipakorn N, Chattipakorn S, Temviriyanukul P, Inthachat W, et al. Liraglutide suppresses tau hyperphosphorylation, amyloid beta accumulation through regulating neuronal insulin signaling and BACE-1 activity. Int J Mol Sci (2020) 21(5):1725. doi: 10.3390/ijms21051725
216. Xiong H, Zheng C, Wang J, Song J, Zhao G, Shen H, et al. The neuroprotection of liraglutide on Alzheimer-like learning and memory impairment by modulating the hyperphosphorylation of tau and neurofilament proteins and insulin signaling pathways in mice. J Alzheimers Dis (2013) 37(3):623–35. doi: 10.3233/JAD-130584
217. Wang XH, Li L, Hölscher C, Pan YF, Chen XR, Qi JS. Val8-glucagon-like peptide-1 protects against Aβ1–40-induced impairment of hippocampal late-phase long-term potentiation and spatial learning in rats. Neuroscience (2010) 170(4):1239–48. doi: 10.1016/j.neuroscience.2010.08.028
218. Ma T, Du X, Pick JE, Sui G, Brownlee M, Klann E. Glucagon-like peptide-1 cleavage product GLP-1(9-36) amide rescues synaptic plasticity and memory deficits in alzheimer’s disease model mice. J Neurosci (2012) 32(40):13701–8. doi: 10.1523/JNEUROSCI.2107-12.2012
219. Iwai T, Sawabe T, Tanimitsu K, Suzuki M, Sasaki-Hamada S, Oka J. Glucagon-like peptide-1 protects synaptic and learning functions from neuroinflammation in rodents. J Neurosci Res (2014) 92(4):446–54. doi: 10.1002/jnr.23335
220. Gao C, Liu Y, Jiang Y, Ding J, Li L. Geniposide ameliorates learning memory deficits, reduces tau phosphorylation and decreases apoptosis via GSK3beta pathway in streptozotocin-induced alzheimer rat model. Brain Pathol (2014) 24(3):261–9. doi: 10.1111/bpa.12116
221. Lv C, Wang L, Liu X, Yan S, Yan SS, Wang Y, et al. Multi-faced neuroprotective effects of geniposide depending on the RAGE-mediated signaling in an Alzheimer mouse model. Neuropharmacology (2015) 89:175–84. doi: 10.1016/j.neuropharm.2014.09.019
222. Zhang Z, Wang X, Zhang D, Liu Y, Li L. Geniposide-mediated protection against amyloid deposition and behavioral impairment correlates with downregulation of mTOR signaling and enhanced autophagy in a mouse model of alzheimer’s disease. Aging (Albany NY) (2019) 11(2):536–48. doi: 10.18632/aging.101759
223. Oka J, Suzuki E, Kondo Y. Endogenous GLP-1 is involved in beta-amyloid protein-induced memory impairment and hippocampal neuronal death in rats. Brain Res (2000) 878(1-2):194–8. doi: 10.1016/S0006-8993(00)02741-4
224. Bomfim TR, Forny-Germano L, Sathler LB, Brito-Moreira J, Houzel JC, Decker H, et al. An anti-diabetes agent protects the mouse brain from defective insulin signaling caused by alzheimer’s disease- associated abeta oligomers. J Clin Invest (2012) 122(4):1339–53. doi: 10.1172/JCI57256
225. Wang X, Wang L, Xu Y, Yu Q, Li L, Guo Y. Intranasal administration of exendin-4 antagonizes Abeta31-35-induced disruption of circadian rhythm and impairment of learning and memory. Aging Clin Exp Res (2016) 28(6):1259–66. doi: 10.1007/s40520-016-0548-z
226. Jia XT, Ye T, Yuan L, Zhang GJ, Liu ZQ, Di ZL, et al. Exendin-4, a glucagon-like peptide 1 receptor agonist, protects against amyloid-beta peptide-induced impairment of spatial learning and memory in rats. Physiol Behav (2016) 159:72–9. doi: 10.1016/j.physbeh.2016.03.016
227. Wang Y, Chen S, Xu Z, Chen S, Yao W, Gao X. GLP-1 receptor agonists downregulate aberrant GnT-III expression in alzheimer’s disease models through the Akt/GSK-3beta/beta-catenin signaling. Neuropharmacology (2018) 131:190–9. doi: 10.1016/j.neuropharm.2017.11.048
228. Park JS, Kam TI, Lee S, Park H, Oh Y, Kwon SH, et al. Blocking microglial activation of reactive astrocytes is neuroprotective in models of alzheimer’s disease. Acta Neuropathol Commun (2021) 9(1):78. doi: 10.1186/s40478-021-01180-z
229. Yang Y, Zhang J, Ma D, Zhang M, Hu S, Shao S, et al. Subcutaneous administration of liraglutide ameliorates Alzheimer-associated tau hyperphosphorylation in rats with type 2 diabetes. J Alzheimers Dis (2013) 37(3):637–48. doi: 10.3233/JAD-130491
230. Spolcova A, Mikulaskova B, Holubova M, Nagelova V, Pirnik Z, Zemenova J, et al. Anorexigenic lipopeptides ameliorate central insulin signaling and attenuate tau phosphorylation in hippocampi of mice with monosodium glutamate-induced obesity. J Alzheimers Dis (2015) 45(3):823–35. doi: 10.3233/JAD-143150
231. Hansen HH, Fabricius K, Barkholt P, Kongsbak-Wismann P, Schlumberger C, Jelsing J, et al. Long-term treatment with liraglutide, a glucagon-like peptide-1 (GLP-1) receptor agonist, has no effect on beta-amyloid plaque load in two transgenic APP/PS1 mouse models of alzheimer’s disease. PloS One (2016) 11(7):e0158205. doi: 10.1371/journal.pone.0158205
232. Chen S, Sun J, Zhao G, Guo A, Chen Y, Fu R, et al. Liraglutide improves water maze learning and memory performance while reduces hyperphosphorylation of tau and neurofilaments in APP/PS1/Tau triple transgenic mice. Neurochem Res (2017) 42(8):2326–35. doi: 10.1007/s11064-017-2250-8
233. Shi L, Zhang Z, Li L, Holscher C. A novel dual GLP-1/GIP receptor agonist alleviates cognitive decline by re-sensitizing insulin signaling in the Alzheimer icv. STZ rat model. Behav Brain Res (2017) 327:65–74. doi: 10.1016/j.bbr.2017.03.032
234. Maskery M, Goulding EM, Gengler S, Melchiorsen JU, Rosenkilde MM, Holscher C. The dual GLP-1/GIP receptor agonist DA4-JC shows superior protective properties compared to the GLP-1 analogue liraglutide in the APP/PS1 mouse model of alzheimer’s disease. Am J Alzheimers Dis Other Demen (2020) 35:1533317520953041. doi: 10.1177/1533317520953041
235. Cai HY, Yang D, Qiao J, Yang JT, Wang ZJ, Wu MN, et al. A GLP-1/GIP dual receptor agonist DA4-JC effectively attenuates cognitive impairment and pathology in the APP/PS1/Tau model of alzheimer’s disease. J Alzheimers Dis (2021) 83(2):799–818. doi: 10.3233/JAD-210256
236. Cao Y, Hölscher C, Hu MM, Wang T, Zhao F, Bai Y, et al. DA5-CH, a novel GLP-1/GIP dual agonist, effectively ameliorates the cognitive impairments and pathology in the APP/PS1 mouse model of alzheimer’s disease. Eur J Pharmacol (2018) 827:215–26. doi: 10.1016/j.ejphar.2018.03.024
237. Li C, Liu W, Li X, Zhang Z, Qi H, Liu S, et al. The novel GLP-1/GIP analogue DA5-CH reduces tau phosphorylation and normalizes theta rhythm in the icv. STZ rat Model AD. Brain Behav (2020) 10(3):e01505. doi: 10.1002/brb3.1505
238. Panagaki T, Gengler S, Holscher C. The novel DA-CH3 dual incretin restores endoplasmic reticulum stress and autophagy impairments to attenuate Alzheimer-like pathology and cognitive decrements in the APPSWE/PS1DeltaE9 mouse model. . J Alzheimers Dis (2018) 66(1):195–218. doi: 10.3233/JAD-180584
239. Tai J, Liu W, Li Y, Li L, Holscher C. Neuroprotective effects of a triple GLP-1/GIP/glucagon receptor agonist in the APP/PS1 transgenic mouse model of alzheimer’s disease. Brain Res (2018) 1678:64–74. doi: 10.1016/j.brainres.2017.10.012
240. Li T, Jiao JJ, Holscher C, Wu MN, Zhang J, Tong JQ, et al. A novel GLP-1/GIP/Gcg triagonist reduces cognitive deficits and pathology in the 3xTg mouse model of alzheimer’s disease. Hippocampus (2018) 28(5):358–72. doi: 10.1002/hipo.22837
241. Li T, Jiao JJ, Su Q, Holscher C, Zhang J, Yan XD, et al. A GLP-1/GIP/Gcg receptor triagonist improves memory behavior, as well as synaptic transmission, neuronal excitability and Ca(2+) homeostasis in 3xTg-AD mice. Neuropharmacology (2020) 170:108042. doi: 10.1016/j.neuropharm.2020.108042
242. Wang ZJ, Han YF, Zhao F, Yang GZ, Yuan L, Cai HY, et al. A dual GLP-1 and gcg receptor agonist rescues spatial memory and synaptic plasticity in APP/PS1 transgenic mice. Horm Behav (2020) 118:104640. doi: 10.1016/j.yhbeh.2019.104640
243. Gejl M, Gjedde A, Egefjord L, Moller A, Hansen SB, Vang K, et al. In alzheimer’s disease, 6-month treatment with GLP-1 analog prevents decline of brain glucose metabolism: Randomized, placebo-controlled, double-blind clinical trial. Front Aging Neurosci (2016) 8:108. doi: 10.3389/fnagi.2016.00108
244. Gejl M, Brock B, Egefjord L, Vang K, Rungby J, Gjedde A. Blood-brain glucose transfer in alzheimer’s disease: Effect of GLP-1 analog treatment. Sci Rep (2017) 7(1):17490. doi: 10.1038/s41598-017-17718-y
Keywords: Alzheimer’s disease, GLP-1R agonists, cognitive function, amyloid beta, tau phosphorylation
Citation: Du H, Meng X, Yao Y and Xu J (2022) The mechanism and efficacy of GLP-1 receptor agonists in the treatment of Alzheimer’s disease. Front. Endocrinol. 13:1033479. doi: 10.3389/fendo.2022.1033479
Received: 31 August 2022; Accepted: 27 October 2022;
Published: 17 November 2022.
Edited by:
Jianbo Zhou, Beijing Tongren Hospital, Capital Medical University, ChinaReviewed by:
Finbarr P. M. O’Harte, Ulster University, United KingdomVenkateswarlu Kanamarlapudi, Swansea University Medical School, United Kingdom
Copyright © 2022 Du, Meng, Yao and Xu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jun Xu, eHVqdW4wMzA0QDE2My5jb20=